
Abstract
Newly synthesized proteins in eukaryotic cells can only function well after they are accurately transported to specific organelles. The establishment of protein databases and the development of programs have accelerated the study of protein subcellular locations, but their comparisons and evaluations of the prediction accuracy of subcellular location programs in plants are lacking. In this study, we built a random test set of maize proteins to evaluate the accuracy of six commonly used programs of subcellular locations: iLoc-Plant, Plant-mPLoc, CELLO, WoLF PSORT, SherLoc2, and Predotar. Our results showed that the accuracy of prediction varied greatly depending on the programs and subcellular locations involved. The programs using homology search methods (iLoc-Plant and Plant-mPLoc) performed better than those using feature search methods (CELLO, WoLF PSORT, SherLoc2, and Predotar). In particular, iLoc-Plant achieved an 84.9 % accuracy for proteins whose subcellular locations have been experimentally determined and a 74.3 % accuracy for all of the proteins in the test set. Regarding locations, the highest prediction accuracies for subcellular locations were obtained for the nucleus, followed by the cytoplasm, mitochondria, plastids, endoplasmic reticulum, and vacuoles, while the lowest were obtained for cell membrane, secreted, and multiple-location proteins. We discussed the accuracy of the six programs in this article. This study will assist plant biologists in choosing appropriate programs to predict the location of proteins and provide clues regarding their function, especially for hypothetical or novel proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In eukaryotic cells, proteins are encoded by DNA in the nucleus, synthesized in the cytoplasm and then sorted to different organelles to execute their biological tasks (Claros et al. 1997). Newly synthesized proteins can only function well in their proper subcellular locations. The subcellular location of a protein can provide insight for revealing the protein’s function and exploring protein-protein interactions in the cellular network system (Millar et al. 2009). Information of subcellular locations of proteins is useful for molecular cell biology, proteomics, system biology, and drug development and is important for the revolution of medicinal chemistry (Chou 2015). Determining the subcellular location of an unknown protein is the primary step in deducing its biological function (Jensen et al. 2002).
The approaches for determining the subcellular locations of proteins can be divided into two categories: experimental and computational methods. The experimental methods include fluorescent protein tagging (Kenri et al. 2004), immunofluorescence and immunoelectron microscopy (Kumar et al. 2000), the use of PhoA protein fusions (Bina et al. 1997), and Western/SDS-PAGE analysis of subcellular fractions (Hancock and Nikaido 1978). While such methods can provide high-quality location information, they can be costly and/or time-consuming.
Alternatively, numerous computational methods that provide fast and accurate localization predictions based on large-scale protein data are available. The computational prediction methods employed by these programs can be mainly separated into three categories: (1) predictions based on N-terminal sorting or signal sequences, as used by Predotar (Small et al. 2004) and TargetP (Emanuelsson et al. 2000); (2) predictions based on the amino acid composition, as used by ProtLock (Cedano et al. 1997); and (3) predictions based on various factors, such as N-terminal signal sequences, the amino acid composition, sequence homology, and Gene Ontology (GO) terms, as used by MultiLoc which employs N-terminal targeting sequences, sequence motifs, and amino acid compositions (Höglund et al. 2006) and SherLoc2 which employs the amino acid composition, sorting signals, functional motifs, homology similarity, and GO terms (Briesemeister et al. 2009).
The accuracy of the predictor is continuously increased due to constantly updated databases and improvement of the algorithm. In recent years, a large number of computational programs for predicting the subcellular localization of proteins have been developed (Emanuelsson et al. 2000; Small et al. 2004; Yu et al. 2006; Horton et al. 2007; Briesemeister et al. 2009; Shen and Chou 2010a, b; Wu et al. 2011). Especially, two series of web-servers are popular: PLoc series (consisting of six web-servers) and iLoc series (consisting of seven web-servers), for predicting the subcellular localization of proteins with both single and multiple sites in different species based on their sequences information alone (Chou 2015). However, there is a lack of comparisons and evaluation of the accuracy of such programs in plants.
In the present study, to evaluate the accuracy of subcellular location programs, we selected six current commonly used programs to test a selected dataset of maize sequences regarding the prediction of protein subcellular locations, including five programs employing comprehensive strategies (SherLoc2, WoLF PSORT, Plant-mPLoc, iLoc-Plant, and CELLO) and one single-strategy program (Predotar). The results will assist plant biologists in choosing appropriate programs to predict the locations of proteins and obtain clues regarding their function, especially for hypothetical or novel proteins.
Materials and Methods
Datasets
The test set of maize protein sequences was selected from the UniProtKB database (http://www.uniprot.org/uniprot/). The UniProtKB database has been checked by experienced molecular biologists. The UniProtKB database currently includes 61,743 maize proteins, among which 747 proteins have been reviewed. Among the 747 reviewed maize proteins, only proteins with a full-length sequence and an identified subcellular location were selected for our test dataset. The selected proteins were divided into a single-location group and a multiple-location group based on whether the protein was located in one or more subcellular locations. According to the GO information of proteins in the UniProtKB database, the subcellular location of a protein was determined experimentally or assumed non-experimentally (by similarity).
Prediction Methods
We selected six commonly used protein subcellular localization programs to compare their prediction accuracy. The selected programs were iLoc-Plant, Plant-mPLoc, CELLO, WoLF PSORT, SherLoc2, and Predotar. These six programs possess the following characteristics: they are public resources available with a web server that predict eukaryotic proteins and accept large batches of sequences. The prediction features of these programs are listed in Table 1. The predictable subcellular locations include the cytoplasm, nucleus, mitochondria, vacuoles, peroxisomes, endoplasmic reticulum, Golgi apparatus, cell membrane, plastids/chloroplasts, secreted/extracellular, lysosomes, and the cell wall. According to the applied prediction method, these tools can be divided into those that employ homology search methods, including iLoc-Plant (http://www.jci-bioinfo.cn/iLoc-Plant), and Plant-mPLoc (http://www.csbio.sjtu.edu.cn/bioinf/plant-multi/), and feature search methods, including CELLO (http://cello.life.nctu.edu.tw/), WoLF PSORT (http://wolfpsort.org/), SherLoc2 (http://abi.inf.uni-tuebingen.de/Services/SherLoc2), and Predotar (http://urgi.versailles.inra.fr/predotar/predotar.html).
Data Analysis
When single-location proteins were predicted to show two or more locations, a positive result was accepted or rejected based on different criteria depending on the different programs. For the statistical programs iLoc-Plant, Plant-mPLoc, and CELLO, we accepted all predicted locations; for the scoring strategy predictor WoLF PSORT, we accepted those locations within 1 score from the highest score of the predicted location; and for the probability strategy predictor SherLoc2, we accepted those locations within a score of 0.2 from the highest score.
For the statistical analysis of the data, if the predicted programs only predicted one location for a single-location protein and the predicted result was consistent with the actual subcellular location indicated in the UniProtKB database, the prediction results were marked as “A.” If the programs predicted two subcellular locations for a single-location protein and either of the results was consistent with the real location, the results were marked as “B.” Accordingly, if there were three predicted results, the results were marked as “C,” and so on.
The match percentage was used to evaluate the accuracy of the programs, which were displayed as proteins with experimental evidence, non-experimental evidence, and all proteins. The prediction results were shown as the total match percentages for each predictor for all single-location proteins, the total match percentages for each predictor for each subcellular location, and the detailed match percentage of each subcellular location obtained by each predictor. The following related equations were employed: ‘Match score = 1× the number of A + 0.5 × the number of “B” + 1/3 × the number of “C”’; “Match percentage = match score / the number of proteins × 100 %”; “Total match score = match score for proteins with experimental evidence + match score for proteins with nonexperimental evidence”; and “Total match percentage = total match score / the total corresponding number of proteins × 100 %.” A match percentage exceeding 60 % was regarded as an acceptable match percentage, while a percentage exceeding 80 % was considered a reliable match percentage, which is more meaningful for guiding users to choose the appropriate programs.
For multiple-location maize proteins, the predicted results were relatively complicated. Such results were analyzed and checked using the methods described by Chou (2013). For the prediction accuracy, the arithmetic of “Accuracy” described by Chou (2013) was used.
Results
In our experiment, we separated all of the proteins in the test set into a single-location group and a multiple-location group. According to the annotation of proteins in UniProtKB, the single-location protein group included proteins located in the cytoplasm, nucleus, plastid, mitochondrion, vacuole, peroxisome, endoplasmic reticulum, Golgi apparatus, secreted, and cell membrane. The final test dataset consisted of 431 full-length maize proteins, including 411 single-location proteins (185 with experimental evidence and 226 with nonexperimental evidence) and 20 multiple-location proteins (Fig. 1). A total of 431 full-length maize proteins were predicted by the six programs (Table S1).

The distribution of subcellular locations of proteins used in the test set
Prediction Results for Single-location Proteins
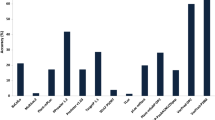
The results obtained for the 411 full-length single-location maize proteins using the six programs were evaluated (Table S1). The total match percentages for all of the single-location proteins reflected the general prediction accuracy of each predictor (Fig. 2a). The three most reliable programs were iLoc-Plant, Plant-mPLoc, and CELLO, followed by SherLoc 2 or WoLF PSORT, while Predotar was always the least reliable. The total match percentages for proteins with nonexperimental evidence were all lower compared with those with experimental evidence. Among the six programs, WoLF PSORT, Plant-mPLoc, and CELLO were able to predict the subcellular localizations of proteins relatively consistently, with less than a 10 % difference between the total match percentages for proteins for which experimental evidence and nonexperimental evidence were observed.

Comparison of the percentages of prediction accuracy by six predictors. a single-location proteins; b multiple-location proteins. Blue histogram, experimental proteins; red histogram, nonexperimental proteins; green histogram, all proteins
The acceptable match percentages obtained with the six programs were compared (Fig. 3). Because of the limited numbers of proteins showing peroxisome and Golgi apparatus locations, the match percentages for these two locations did not present sufficient credibility and will not be discussed further.

The acceptable match percentages obtained by six predictors. a All proteins, b experimental proteins, c nonexperimental proteins
For all of the proteins, iLoc-Plant, Plant-mPLoc, and CELLO indicated five subcellular locations with acceptable match percentages. Among these locations, iLoc-Plant and Plant-mPLoc showed five and four locations, respectively, with reliable match percentages, including the nucleus, vacuoles, plastids, and mitochondria in both and the cytoplasm in iLoc-Plant, while CELLO only showed one such location (cytoplasm). SherLoc 2 and WoLF PSORT both indicated three subcellular locations with acceptable match percentages, whereas SherLoc 2 only showed reliable match percentages for the cytoplasm and nucleus (Fig. 3a). The match percentages in all of the subcellular locations predicted by Predotar were lower than 60 %. With the exception of Predotar, the other programs all showed a greater than 70 % match percentage in the nucleus. iLoc-Plant performed best in predicting the subcellular localizations of all proteins.
For the proteins with experimental evidence, iLoc-Plant indicated four reliable match percentages, in the nucleus, vacuole, plastid, and cytoplasm. Plant-mPLoc showed three reliable match percentages, in the nucleus, vacuole, and plastid. CELLO and SherLoc indicated two locations, the cytoplasm and the nucleus. WoLF PSORT only performed reliably for the nucleus, and Predotar showed no reliable match percentages (Fig. 3b).
Compared with the results for proteins with experimental evidence, the accuracy of the prediction for proteins with nonexperimental evidence exhibited some variations. The reliable match percentages were consistent for the nucleus, vacuoles, and plastids in Plant-mPLoc, but only for the nucleus in iLoc-Plant. Among all of the programs, secreted proteins were first reliably predicted by CELLO (Fig. 3c). The match percentages for all subcellular locations obtained by Predotar were lower than 60 %, except for proteins with nonexperimental evidence in the endoplasmic reticulum. The results showed that iLoc-Plant and Plant-mPLoc presented the most acceptable and reliable match percentages for proteins with both experimental evidence and nonexperimental evidence. For all proteins and the proteins with nonexperimental evidence in some locations (nucleus, vacuoles, and plastids), Plant-mPloc showed a higher prediction consistency than iLoc-Plant.
Prediction Results for Multiple-location Proteins
Only iLoc-Plant, Plant-mPLoc, and WoLF PSORT have the capability to predict the subcellular locations of multiple-location proteins among the six programs. The match percentages for 20 multiple-location proteins were analyzed. It was found that iLoc-Plant performed best, showing prediction accuracies of 48.6 % for proteins with experimental evidence, 35.9 % for proteins with nonexperimental evidence, and 40.3 % for all proteins (Fig. 2b).
Prediction Accuracy of the Six Programs for Individual Subcellular Locations
The prediction accuracy of the six programs varied among different subcellular locations. The detailed match percentages of each subcellular location obtained with each predictor were determined (Figure S1). The predictions for proteins located in the cytoplasm, nucleus, plastids, mitochondria, and vacuoles were relatively better than those in other subcellular locations, and reliable match percentages could be obtained with some of the programs.
For proteins located in the cytoplasm, SherLoc 2, CELLO, and iLoc-Plant were able to obtain reliable match percentages for all proteins and proteins with experimental evidence. SherLoc 2 performed the best, and Plant-mPLoc performed the worst (Figure S1 a). For proteins located in the nucleus, all of the programs displayed high match percentages, with the two most reliable programs being Plant-mPLoc and iLoc-Plant (Figure S1 b). The accuracy of prediction for all of the proteins located in the nucleus was also checked using Nuc-Ploc (Shen and Chou 2010a, b), a commonly used web server that specifically predicts the location of proteins found in the nucleus, and the accuracy of the prediction reached to 100 %. For proteins located in plastids, the two programs with the highest reliable match percentages were iLoc-Plant and Plant-mPLoc (Figure S1 c). For proteins located in the mitochondria, only iLoc-Plant and Plant-mPloc presented reliable match percentages for all proteins and proteins with nonexperimental evidence (Figure S1 d). For proteins located in vacuoles, Plant-mPLoc and iLoc-Plant could obtain reliable match percentages, even though the accuracy of prediction for proteins with nonexperimental evidence was 77.8 % for iLoc-Plant (Figure S1 e).
For proteins located in the cell membrane, no reliable match percentages were obtained with the six programs, and acceptable results could only be predicted with CELLO (Figure S1 f). Although CELLO showed a reliable match percentage for proteins with nonexperimental evidence located in secreted locations, the accuracy of prediction was quite low for proteins with experimental evidence (Figure S1 g). For proteins located in the endoplasmic reticulum, Plant-mPLoc performed with a consistently acceptable accuracy, for proteins with both experimental evidence and nonexperimental evidence (Figure S1 h).
At present, only two maize proteins located in peroxisomes or Golgi apparatus were retrieved from the UniProtKB database, so the accuracy of prediction was not analyzed for both locations (Figure S1 i and j).
Discussion
Prediction Accuracy Varies for Different Programs
The overall percentage of accurate predictions obtained using iLoc-Plant and Plant-mPLoc showed the best performance among all of the programs we selected. Both of these programs are reliable for the nucleus, vacuole, plastids, and mitochondria, showing very close match percentages, presumably due to their similar protein formulation and computational algorithms. iLoc-Plant and Plant-mPLoc were developed in the same laboratory and employ the BLAST homology search method to deduce the subcellular localizations of unknown proteins when they show significant homology to any protein (GO information not empty) in the UniProtKB database. Otherwise, the sequential evolution pattern will be used to predict the result. These tools employ the same prediction strategy, and both can generate predictions for multiple-location proteins. Compared with Plant-mPLoc, iLoc-Plant exhibits an improvement in the extraction of GO information and the involvement of an optimal threshold factor during the progress of prediction but removes a functional domain descriptor. Homology search methods rely mainly on BLAST results and the mining of GO information, based on the hypothesis of Nair and Rost (Nair and Rost 2002) that the information on the subcellular location of an unknown protein can be inferred if a close homolog with an experimentally verified localization can be found. Even for predicting multiple-location proteins, such strategy programs assume that the number of different subcellular locations of an unknown protein should be the same as its nearest neighbor protein. Based on the results of our experiment and other validation tests, the accuracy of programs employing this type of strategy is better than other approaches, when the query sequence shares high homology (≥30 %) with the curated protein with distinct subcellular localization. However, the prediction result lacks accuracy when the query protein presents no highly homologous relationships in the database because fewer features are extracted for protein formulation.
The overall percentage of accurate predictions obtained using CELLO and SherLoc 2 ranged from 64 to 89 % for the cytoplasm, nucleus, plastid, membrane, mitochondria, and secreted subcellular locations. In contrast to iLoc-Plant and Plant-mPLoc, both CELLO and SherLoc 2 are established based on a support vector machine system that integrates features extracted directly from sequences and then uses a support vector machine classifier to generate prediction results. This strategy performs better for sequences with low homology because it takes mining information from the sequence itself into account, even though some programs, such as SherLoc 2, also combine an annotation-based prediction strategy. Although the accuracy of predictions obtained using this type of predictor is not good compared with homology search methods, the prediction results should still be acceptable due to putting a great deal of effort into predicting sequences with low homology.
WoLF PSORT presented a match percentage above 70 % in the plastid, cytoplasm, and nucleus locations in our experiment. This tool was created as a hybrid predictor, combining a feature selection progress and a KKN classifier for both simplex and multiple-location proteins. In the present study, its prediction of multiple-location proteins was poor.
The method employed by Predotar only involves N-terminal targeting sequences and covers three locations (plastids, mitochondria, and endoplasmic reticulum). The results obtained using Predotar were the poorest among all of the programs, even for its available predictive locations, due to its single feature extraction strategy and inappropriate algorithm.
We also used a feature-based predictor, Nuc-PLoc, to check the match percentage for proteins located in the nucleus, and the final match percentage was 100 %. This type of strategy predictor emphasizes sequence descriptions, and it performs comparably to homology search methods in high-homology and conserved regions, such as the nucleus, and performs better than homology-based methods in the case of low homology.
In this study, we showed that homology search methods perform better than feature search methods for maize proteins, but we strongly suggest the use of a practical guide combining programs employing both search methods for an unknown protein if it lacks a homology protein or GO information in the database. The same predictor or a different predictor may yield markedly different match percentages when tested using different benchmark datasets. This is because the more stringent a benchmark dataset is in excluding homologous sequences, the more difficult it is for a predictor to achieve a high match percentage. Additionally, the greater the number of subsets (subcellular locations) a benchmark dataset covers, the more difficult it is to achieve a high overall match percentage, as elaborated in a recent review.
Prediction Accuracy Varies for Different Subcellular Locations
Even though all of the programs were designed to identify more than one subcellular location, the match percentage varied for different subcellular locations due to different protein features, the number of reviewed proteins and GO information in the UniProtKB database.
The greatest number of match percentages and those of the highest reliability were obtained in the nucleus for all of the programs, except for Predotar. Such a high accuracy of prediction may mainly be due to the significantly highly conserved nuclear localization signal that is always included in the sequence of nucleus-located proteins (Boulikas 1993; Neufeld et al. 2000; Davidson et al. 2006). Some nucleoproteins do not include any nuclear location signal, and before being transported to the nucleus, all nuclear proteins are synthesized in the cytoplasm (Borer et al. 1989). Therefore, nucleus-located proteins can be predicted to be located in the cytoplasm, which is consistent with the findings of cell biological research (Su et al. 2007).
Proteins located in the cytoplasm could also be predicted reliably by SherLoc 2, CELLO, and iLoc-Plant for the set of all proteins. Most proteins are synthesized in the cytoplasm and include a specific sorting signal to guide their localization. There has been a great deal of research on cytoplasmic sorting signals, and the SH2 and SH3 domains are elements that control the interactions of cytoplasmic signaling proteins (Koch et al. 1991). SH2 plays a major role in mediating precise downstream signaling events (Wagner et al. 2013). Therefore, it is not surprising that a higher accuracy was obtained for the cytoplasm for proteins with experimental evidence by all of the programs.
In this study, reliable match percentages for plastid-located proteins were obtained when using Plant-mPLoc and iLoc-Plant, and the percentages were higher than previously reported (Shen and Chou 2010a, b). This high accuracy of prediction may due to the large volume of reviewed plastid-located proteins in the UniProtKB database, and there are many protein import receptors in plastids (Bauer et al. 2000; Kaundal et al. 2013). The accuracy of prediction for plastid-located proteins varies greatly between programs employing homology search methods versus feature search methods, which may result from a lack of conserved regions and ambiguous sequence features.
The protein formulation for mitochondrion-located proteins may be more complicated. Mitochondria are organelles that contain their own genetic system. Precursor forms of mitochondrial proteins include targeting and sorting signals that are essential to direct them to the mitochondria. Recently, a great deal of experimental evidence concerning the nature of these signals has been obtained (Pfanner et al. 1992; Verner 1993). Most of the hundreds of proteins that function in the mitochondria are encoded by nuclear DNA, synthesized in the cytoplasm and then transferred to the mitochondria, which may result in multi-labeling and a lack of a sorting signal during the transportation progress. In this study, iLoc-Plant and Plant-mPLoc were both able to obtain reliable match percentages in this location, and the percentages were slightly higher than reported previously (Shen and Chou 2010a, b).
An acceptable match percentage for ER-located proteins with experimental evidence was generated by iLoc-Plant, which was consistent with a previous study (Shen and Chou 2010a, b), as more ER-located proteins were selected in our test set. However, the results differed greatly when programs employing the feature search method were used. Endoplasmic reticulum proteins contain a special sequence at the beginning of the protein. Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum (Nilsson et al. 1989). Proteins that permanently reside in the lumen of the endoplasmic reticulum must somehow be distinguished from newly synthesized secretory proteins, and it has been proposed that a carboxy-terminal sequence marks proteins that are to be retained in the endoplasmic reticulum (Munro and Pelham 1987). Several soluble proteins that reside in the lumen of the ER contain a specific C-terminal sequence that prevents their secretion. Therefore, such results may arise from an insufficient number of ER-located proteins in the database and a lack of feature extraction.
The match percentage was lower than 32 % for all of the programs except for iLoc-Plant and Plant-mPLoc, which performed well in this location, presenting a match percentage above 80 %. Vacuoles play central roles in plant growth, development, and stress responses. However, there has been little research on the plant vacuole proteome. In Arabidopsis thaliana, 89 vacuole-localized proteins of unknown function have been identified, which could represent potential cargo of the N- and C-terminal propeptide sorting pathways or be related to the association of the vacuole with the cytoskeleton (Carter et al. 2004).
Proteins located in peroxisomes always contain a specific peroxisomal targeting signal, such as peroxisomal targeting signal 1 (Gould et al. 1989) or peroxisomal targeting signal 2 (Osumi et al. 1991; Swinkels et al. 1991). These special signals can readily transport newly synthesized peroxisomal proteins to the peroxisome. Peroxisomes do not possess their own genome, and the functional conversion of these organelles requires the transport of newly synthesized peroxisomal proteins from the cytosol into peroxisomes. Furthermore, certain proteins in plant peroxisomes lack both the typical peroxisomal targeting signal 1 and peroxisomal targeting signal 2, such as catalase (Esaka et al. 1997) and ascorbate peroxidase (Bunkelmann and Trelease 1996). The match percentage obtained for 21 proteins located in peroxisomes reached 66.7 % when using Plant-mPLoc, and the match percentage obtained for 21 proteins located in the Golgi apparatus was 76.2 % when using iLoc-Plant.
Acceptable results for cell membrane-located proteins and multiple-location proteins were not obtained by any of the programs because current methods perform very poorly in these locations (Sprenger et al. 2006). For multiple-location proteins, most prediction programs can currently only predict one location correctly. This low accuracy for multiple-location proteins is not surprising, as the prediction of these proteins relies not only on improvement of the algorithm but also on the experimental information in the database.
Most existing prediction tools available were established based on the assumption that a protein resides at only one subcellular location and cannot be used to deal with multiplex proteins. Only a few programs for specific species can predict well for multiplex proteins, such as, iLoc-Gpos (Wu et al. 2012) for gram-positive proteins, Euk-mPLoc 2.0 (Chou and Shen 2010) and iLoc-Euk (Chou et al. 2011) for eukaryotic proteins. However, proteins with multiple location sites are very important to both basic research and drug development because they may have some unique biological functions. This area remains challenging, and we propose the use of several types of programs employing comprehensive strategies to predict multiple-location proteins to obtain hints regarding their real locations.
Conclusion
To evaluate the accuracy of most types of prediction programs, we selected six commonly used programs to test a requested dataset of maize sequences selected from the UniProtKB database for protein subcellular location prediction. The prediction results varied greatly among the programs employing different strategies and among subcellular locations. Programs using homology search methods, such as iLoc-Plant and Plant-mPLoc, can achieve more accurate results for protein sequences showing high homology, while programs using the feature search method perform better in predicting low-homology sequences. For proteins located in the nucleus, cytoplasm, plastids and mitochondria, programs can provide more reliable results due to the relatively large number of reviewed proteins from these locations with clear GO information in the UniProtKB database showing conserved or well-studied sequence features that are consistent with the information obtained from cell biology research and the theory of biological evolution. For proteins located in other subcellular locations or in multiple locations, iLoc-Plant performs relatively better than the other programs, even though it cannot provide reliable results or acceptable results in some locations. These results can guide researchers in choosing appropriate programs for predicting hypothetical or novel maize proteins, and we propose that programs employing both the homology and feature search methods should be used to predict unknown proteins to obtain more accurate and reliable hints about their actual subcellular locations.
References
Bauer J, Chen K, Hiltbunner A, Wehrli E, Eugster M, Schnell D, Kessler F (2000) The major protein import receptor of plastids is essential for chloroplast biogenesis. Nature 403:203–207
Bina JE, Nano F, Hancock RE (1997) Utilization of alkaline phosphatase fusions to identify secreted proteins, including potential efflux proteins and virulence factors from Helicobacter pylori. FEMS Microbiol Lett 148:63–68
Borer RA, Lehner CF, Eppenberger HM, Nigg EA (1989) Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56:379–390
Boulikas T (1993) Nuclear locations signals (NLS). Crit Rev EGE 3:193–227
Briesemeister S, Blum T, Brady S, Lam Y, Kohlbacher O, Shatkay H (2009) SherLoc2: a high-accuracy hybrid method for predicting subcellular localization of proteins. J Proteome Res 8:5363–5366
Bunkelmann JR, Trelease RN (1996) Ascorbate peroxidase. A prominent membrane protein in oilseed glyoxysomes. Plant Physiol 110:589–598
Carter C, Pan S, Zouhar J, Avila EL, Girke T, Raikhel NV (2004) The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins. Plant Cell 16:3285–3303
Cedano J, Aloy P, Pérez-Pons JA, Querol E (1997) Relation between amino acid composition and cellular location of proteins. J Mol Biol 266:594–600
Chou KC (2013) Some remarks on predicting multi-label attributes in molecular biosystems. Mol Biosyst 9:1092–1100
Chou KC (2015) Impacts of bioinformatics to medicinal chemistry. Med Chem 11:218–234
Chou KC, Shen HB (2010) A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mPLoc 2.0. PLoS ONE 5:e9931
Chou KC, Wu ZC, Xiao X (2011) iLoc-Euk: a multi-label classifier for predicting the subcellular localization of singleplex and multiplex eukaryotic proteins. PLoS ONE 6:e18258
Claros MG, Brunak S, von Heijne G (1997) Prediction of N-terminal protein sorting signals. Curr Opin Struct Biol 7:394–398
Davidson PJ, Li SY, Lohse AG, Vandergaast R, Verde E, Pearson A, Patterson RJ, Wang JL, Arnoys EJ (2006) Transport of galectin-3 between the nucleus and cytoplasm. I. Conditions and signals for nuclear import. Glycobiology 16:602–611
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular locations of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Esaka M, Yamada N, Kitabayashi M, Setoguchi Y, Tsugeki R, Kondo M, Nishimura M (1997) cDNA cloning and differential gene expression of three catalases in pumpkin. Plant Mol Biol 33:141–155
Gould SJ, Keller GA, Hosken N, Wilkinson J, Subramani S (1989) A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol 108:1657–1664
Hancock RE, Nikaido H (1978) Outer membranes of gram-negative bacteria. XIX. Isolation from Pseudomonas aeruginosa PAO1 and use in reconstitution and definition of the permeability barrier. J Bacteriol 136:381–390
Höglund A, Dönnes P, Blum T, Adolph HW, Kohlbacher O (2006) MultiLoc: prediction of protein subcellular localization using N-terminal targeting sequences, sequence motifs and amino acid composition. Bioinformatics 22:1158–1165
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai NK (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35:585–587
Jensen LJ, Gupta R, Blom N, Devos D, Tamames J, Kesmir C, Nielsen H, Staerfeld HH, Rapacki K, Workman C, Andersen CA, Knudsen S, Krogh A, Valencia A, Brunak S (2002) Prediction of human protein function from post-translational modifications and localization features. J Mol Biol 319:1257–1265
Kaundal R, Sahu SS, Verma R, Weirick T (2013) Identification and characterization of plastid-type proteins from sequence-attributed features using machine learning. BMC Bioinf 14:S7
Kenri T, Seto S, Horino A, Sasaki Y, Sasaki T, Miyata M (2004) Use of fluorescent-protein tagging to determine the subcellular locations of mycoplasma pneumoniae proteins encoded by the cytadherence regulatory locus. J Bacteriol 186:6944–6955
Koch CA, Anderson D, Moran MF, Ellis C, Pawson T (1991) SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 252:668–674
Kumar RB, Xie YH, Das A (2000) Subcellular locations of the Agrobacterium tumefaciens T-DNA transport pore proteins: VirB8 is essential for the assembly of the transport pore. Mol Microbiol 6:608–617
Millar AH, Carrie C, Pogson B, Whelan J (2009) Exploring the function-location nexus: using multiple lines of evidence in defining the subcellular location of plant proteins. Plant Cell 21:1625–1631
Munro S, Pelham HR (1987) A C-terminal signal prevents secretion of luminal ER proteins. Cell 48:899–907
Nair R, Rost B (2002) Sequence conserved for subcellular localization. Protein Sci 11:2836–2847
Neufeld KL, Nix DA, Bogerd H, Kang Y, Beckerle MC, Cullen BR, White RL (2000) White adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc Natl Acad Sci U S A 97:12085–12090
Nilsson T, Jackson M, Peterson PA (1989) Short cytoplasmic sequences serve as retention signals for transmembrane proteins in the endoplasmic reticulum. Cell 58:707–718
Osumi T, Tsukamoto T, Hata S, Yokota S, Miura S, Fujiki Y, Hijikata M, Miyazawa S, Hashimoto T (1991) Amino-terminal presequence of the precursor of peroxisomal 3-ketoacyl-CoA thiolase is a cleavable signal peptide for peroxisomal targeting. Biochem Biophys Res Commun 181:947–954
Pfanner N, Rassow J, van der Klei IJ, Neupert W (1992) A dynamic model of the mitochondrial protein import machinery. Cell 68:999–1002
Shen HB, Chou KC (2010a) Nuc-PLoc:a new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM. Protein Eng Des Sel 9:561–567
Shen HB, Chou KC (2010b) Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 5:e11335
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4:1581–1590
Sprenger J, Fink JL, Teasdale RD (2006) Evaluation and comparison of mammalian subcellular localization prediction methods. BMC Bioinf 7:S3
Su EC, Chiu HS, Lo A, Hwang JK, Sung TY, Hsu WL (2007) Protein subcellular localization prediction based on compartment-specific features and structure conservation. BMC Bioinf 8:330
Swinkels BW, Gould SJ, Bodnar AG, Rachubinski RA, Subramani S (1991) A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase. EMBO J 10:3255–3262
Verner K (1993) Co-translational protein import into mitochondria: an alternative view. Trends Biochem Sci 18:366–371
Wagner MJ, Stacey MM, Liu BA, Pawson T (2013) Molecular mechanisms of SH2- and PTB-domain-containing proteins in receptor tyrosine kinase signaling. Cold Spring Harb Perspect Biol 5:a008987
Wu ZC, Xiao X, Chou KC (2011) iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Mol Biosyst 2:3287–3297
Wu ZC, Xiao X, Chou KC (2012) iLoc-Gpos: a multi-layer classifier for predicting the subcellular localization of singleplex and multiplex Gram-positive bacterial proteins. Protein Pept Lett 19:4–14
Yu CS, Lin CJ, Hwang JK (2006) Prediction of protein subcellular localization. Protein Sci 64:643–651
Acknowledgments
We acknowledge the National Natural Science Foundation of China (Grant No. 31371543), the Plan for Scientific Innovation Talent of Henan Province (Grant No. 144200510012), and the Program for Innovative Research Team (in Science and Technology) in University of Henan Province (Grant No. 15IRTSTHN015) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Erhui Xiong and Chenyu Zheng contributed equally to this work.
Electronic Supplementary Material
Rights and permissions
About this article

Cite this article
Xiong, E., Zheng, C., Wu, X. et al. Protein Subcellular Location: The Gap Between Prediction and Experimentation. Plant Mol Biol Rep 34, 52–61 (2016). https://doi.org/10.1007/s11105-015-0898-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-015-0898-2