
Abstract
Shear stress and cyclic stretch are mechanical forces on the vessel wall exerted by blood flow and luminal pressure. These forces regulate gene expression and function in vascular cells, including endothelial cells (ECs) and smooth muscle cells (SMCs), thus affecting vascular biology in health and pathobiology in disease. Epigenetics refers to the study of sequence-independent heritable DNA alterations that modulate gene expression, including DNA methylation, histone modification/chromatin remodeling, and RNA-based mechanisms. Recently, the roles of mechanical force-induced epigenetic modifications in vascular gene expression and function have been intensively investigated. This chapter presents a critical concept: vascular gene expression can be mechanically modulated without DNA sequence change. By elucidating the relationship between mechanical forces and epigenetic modifications in gene expression, cell proliferation, angiogenesis, migration, and pathological status, this review provides a conceptual framework for understanding how mechanical force-induced epigenetic modifications modulate gene expression and cellular function in vascular biology in health and pathobiology in disease. This review contributes to our knowledge of how the mechanical microenvironment affects epigenetic changes in vascular cells and modulates their functions and behaviors, with the consequent modulation in vascular diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
9.1 Introduction
The development of the normal vessel wall involves a highly regulated process of cell proliferation, migration, differentiation, and relaxation in vascular cells, which comprises endothelial cells (ECs) and smooth muscle cells (SMCs), that are constantly exposed to various types of hemodynamic forces [1]. Hemodynamic forces are generated by blood flow and luminal pressure, which can be characterized as shear stress, cyclic stretch, and hydrostatic pressure [2]. ECs are mainly exposed to shear stress resulting from blood flow parallel to the vessel wall, whereas SMCs and ECs are subjected to cyclic stretch caused by pulsatile blood flow and pressure [2]. Another mechanical force, i.e., hydrostatic pressure, exerted by a fluid at rest, usually affects the capillaries of the circulatory system and is less extensively studied. Mechanical force-induced signals are received by mechanoreceptors in the cell membrane, such as ion channels, integrins, receptors of tyrosine kinases, G protein-coupled receptors, junction proteins, membrane lipids, and primary cilia, and these are in turn transmitted to the interior of the cell. The mechanoreceptors act on adaptor molecules (e.g., Src homology 2 domain-containing protein and growth factor receptor-bound protein 2) and trigger a series of intracellular signaling cascades, which consequently modulate gene expression; cell proliferation, differentiation, and migration; and angiogenesis [3]. This process, known as mechanotransduction, eventually leads to functional and morphological changes that contribute to physiological homeostasis [4]. Unbalanced regulation of these cellular functions causes vascular cell dysfunction and leads to a pathological cell state, which consequently contributes to the development of cardiovascular disease (CVD) [5].
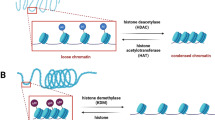
In recent decades, epigenetics, which is the study of sequence-independent heritable DNA changes, has made the connection between gene expression and environmental stimuli and linked their relationship to disease susceptibility [6, 7]. It provides a perspective new to the public by showing that gene function can be altered in ways other than by changes to the DNA sequence. The different epigenetic processes, including DNA methylation, histone-mediated chromatin remodeling, and RNA-based mechanisms, modulate gene expression to cause changes in cellular function [8]. These changes subsequently lead to the adaptability of the organism or disease. DNA methylation, the best-known epigenetic process, is the addition of a methyl group (CH3) from S-adenyl methionine (SAM) to the fifth carbon of a cytosine residue to form 5-methylcytosine (5mC) in the CpG pair [9]. The hypermethylation of CpG islands results in the recruitment of protein complexes that remove acetyl groups and repress gene expression [9]. DNA demethylation is a mechanism that reverses gene silencing and is involved in embryo development, germ cell differentiation, and neuronal functions [10]. Another important epigenetic process is histone-mediated chromatin remodeling. Chromatin, which is composed of DNA and histones, can be modified by histone acetyltransferase (HAT)-mediated acetylation and histone deacetylase (HDAC)-mediated deacetylation. Histone acetylation/deacetylation causes the conformation change of chromatin, thereby influencing gene transcription. Noncoding RNAs (ncRNAs), including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), have recently emerged as epigenetic regulators. MiRNAs, which consist of 18–22 nucleotide single-stranded RNAs, cause the degradation of messenger RNA (mRNA) by binding to the 3′-untranslated regions (3′-UTRs) of target genes or induce translational repression by binding to imperfectly complementary sequences [11]. LncRNAs, which are single-stranded RNAs containing more than 200 nucleotides, modulate gene expression by diverse mechanisms [12, 13]. Many cellular behaviors, such as cell proliferation and morphological changes, and the development of diseases, including cancer, CVD, and autoimmune disease, have been shown to be associated with epigenetic modulation [13,14,15,16,17,18]. Recently, the role of epigenetics in CVD has recently been intensively studied and has provided important insights into the diagnosis and therapeutic intervention of CVD [17]. Epigenetic factors, including HDACs [19], miRNAs [20, 21], and DNA methyltransferases (DNMTs) [22], have been shown to play vital roles in modulating vascular function and dysfunction and hence the development of atherosclerosis. In general, epigenetics offers a new perspective on gene regulation, which is not exerted by cis/trans-acting transcription but through multiple diverse processes that do not involve alterations to the DNA sequence.
In this chapter, the role and mechanism of epigenetics in regulating vascular biology and pathobiology in response to mechanical stimuli are discussed. This chapter shows the importance of mechanical forces (i.e., shear stress and cyclic stretch) and the corresponding signaling pathways in modulating vascular function and disease. Moreover, different epigenetic processes and their modulation of cellular responses, particularly those relating to vascular biology and pathobiology, are also described. This chapter summarizes the evidence that mechanical force-induced epigenetic modification influences homeostasis and pathology of the vessel wall. Such information provides new insights into the mechanisms by which epigenetic modification modulates gene expression, cellular function, and disease development in response to mechanical forces. Thus, epigenetic regulators have great potential as molecular targets or biomarkers that can be developed for the diagnosis and therapeutic intervention of vascular disorders associated with perturbed mechanical forces, such as atherosclerosis.
9.2 Vascular Mechanobiology
Blood vessels are subjected to sustained hemodynamic forces derived from blood flow and luminal pressure, which generate shear stress and cyclic circumferential stretch [2]. Another internal stress caused by both blood flow and pressure is known as hydrostatic pressure (i.e., normal stress), which arises from stationary fluid flow. Shear stress applied to vascular ECs is a frictional force parallel to the vessel wall, whereas normal stress is the force perpendicular to the vessel wall on which it acts. Blood flow-induced shear stress has been reported to be involved in atherosclerosis, as revealed by the tendency of atherosclerotic lesions to be localized in the curves and branches of arterial trees, where the local flow is usually disturbed due to low and oscillatory shear stress (OSS). Conversely, blood flow in the straight segments of the arteries is unidirectional and pulsatile, generating pulsatile shear stress (PSS), which has been shown to play an atheroprotective role in the vessel wall [23, 24]. Tensile force generated by pulsatile blood flow and luminal pressure leads to circumferential stretching of the vessel wall and affects gene expression in, and the function of, vascular cells. Stretch force acts on both vascular ECs and SMCs and play an important role in maintaining the contractile phenotype of SMCs and regulating vascular tone when the stretching is at the physiological level [25]. Abnormal stretching caused by hypertension or flow overload can disturb biochemical homeostasis, leading to vascular remodeling and altered vasorelaxation [26]. In contrast to its role in the SMCs, the role of cyclic stretch in vascular ECs has not been fully investigated. Hydrostatic pressure is determined by the density of liquid, the acceleration of gravity, and the height of fluid, which may play an important role in cellular physiology. However, it has received relatively less attention than shear stress and cyclic stretch in the field of vascular biology. In this section, the cellular responses of vascular ECs and SMCs to shear stress and cyclic stretch are discussed.
9.2.1 Shear Stress
9.2.1.1 Shear Stress Modulates Vascular Morphogenesis
Shear stress has been shown to modulate vascular morphogenesis and heart development during embryogenesis [27, 28]. The early formation of shear stress is produced by the heartbeat, which causes the reorganization and migration of ECs to form an efficient vascular network [28]. By using in vivo imaging and quantitative analyses of intracardiac flow forces in zebrafish embryos, a study showed that high-velocity vertical flow exists at two key stages in the developing heart [27]. Flow that is blocked at either the cardiac inflow or outflow tracts results in abnormal development of the third chamber and impaired valve formation in the heart [27]. The zinc finger-containing transcription factor Krüppel-like factor 2a (KLF2a), which is activated by the reverse flow in the atrioventricular canal, is highly associated with valve development. It has been reported that protein kinase D2 (PRKD2) causes the derepression of KLF2a through HDAC5 phosphorylation [28, 29]. Thus, KLF2a expression is greatly reduced in PRKD2 mutants, which do not form valves. Blood flow also activates the expression of miRNAs, such as miRNA-21 (miR-21) and miR-143, which leads to the cell proliferation that induces valve formation in the zebrafish heart [30]. In addition, there is evidence that fluid shear stress modulates vessel remodeling via endothelial nitric oxide synthase (eNOS) [31, 32]. Fernández-Varo et al. analyzed the vascular properties of cirrhotic rats with ascites and determined that they cause vascular remodeling [31]. Specifically, they found that the vessels in cirrhotic rats have higher levels of eNOS and a dramatic reduction in wall thickness and area compared to the vessel thickness and area in control rats, indicating that eNOS is required for the regulation of vessel compliance and vascular remodeling [31]. Another study also showed the critical role of eNOS in modulating ischemia-induced arteriogenesis, angiogenesis, and blood flow recovery in mice [32]. Taken together, these studies demonstrate the importance of mechanical forces in the modulation of epigenetic factor expression in embryonic cardiogenesis and vasculature development.
9.2.1.2 Shear Stress Regulates Physiological Functions
In addition to its functions in embryonic vascular morphogenesis and cardiogenesis, shear stress plays an important role in endothelial morphological changes and cellular functions, including cell proliferation, differentiation, and migration, through biochemical and biological events [26, 33]. It has been shown that the physiological level of PSS exerts protective functions by inducing nitric oxide (NO) production. Conversely, OSS-disturbed flow impairs biochemical homeostasis and leads to vascular remodeling and dysfunction (e.g., altered vasorelaxation, vascular tone, and stiffness). Shear stress-induced mechanotransduction in ECs has been investigated by using both in vitro and in vivo approaches. For in vitro studies, researchers have developed several devices to generate different types of shear stresses that stimulate blood flow in the human body. A parallel-plate flow channel is created by using a gasket with a thin silicon membrane that produces steady, unidirectional shear stress (USS, 12 dynes/cm2), PSS (12 ± 4 dynes/cm2), and reciprocating shear stress (i.e., OSS, 0.5 ± 4 dynes/cm2) on cultured ECs [33]. In vivo studies have been performed to evaluate the applicability and relevance of the in vitro findings to physiological and pathophysiological conditions. In nature, blood flow in the curves and branches of the arterial trees is disturbed, whereas flow in the straight segments of the arteries is pulsatile and unidirectional [34]. Transcription factor KLF2, which is an abundant molecule in ECs, promotes endothelial survival in response to oxidized low-density lipoprotein (ox-LDL) stimuli [35]. The continued application of PSS on ECs for 24 h led to a sustained expression of KLF2, which protected the ECs against oxidative stress stimuli [35]. In addition to KLF2, a number of genes have been shown to be induced by USS in ECs and are involved in EC survival, angiogenesis (e.g., Tie2 and fetal fiver kinase 1), and vascular remodeling (e.g., matrix metalloproteinase-1, MMP-1) [36]. Physiological levels of flow also maintain the gene expression profile in ECs in a nonproliferative state by increasing the expression of the growth arrest proteins GADD45, p21cip1, and p53 and inhibiting retinoblastoma protein (Rb) phosphorylation [3]. Application of USS to ECs induces a transient expression of monocyte chemotactic protein-1 (MCP-1) through the modulation of the Ras-mitogen activated protein kinases pathway [37]. Continuous application of USS causes downregulation of MCP-1 and that of various pro-inflammatory molecules such that their expression levels fall below the basal levels and the ECs remain in a noninflammatory state [37]. Shear stress can modulate intercellular junction proteins, such as VE-cadherin (VE-Cad), connexin19, and platelet endothelial cell adhesion molecule-1 [38], all of which play important roles in modulating the integrity and permeability of ECs. The continuous distribution of VE-Cad staining has been shown at the cell borders in the region affected by PSS [39, 40]. The cytoskeleton is reorganized via the Rac1, cdc42, and Rho signaling pathways, leading to cell morphological changes in response to shear stress stimuli [41,42,43]. EC exposure to USS or PSS induces the alignment of cytoskeletal fibers in the flow direction [44]. Taken together, the redistribution of cellular junctions and cytoskeletal proteins in regions subjected to PSS contributes to the maintenance of the cellular integrity and the physiological functions of ECs.
9.2.1.3 Shear Stress Is Involved in the Development of Vascular Pathologies
ECs are generally in a quiescent state unless stimulated by some pathophysiological conditions. Disturbed flow is involved in endothelial dysfunction and leads to a pathophysiological state, thereby contributing to the development of vascular disorders, including atherosclerosis and thrombosis and the complications related to them [2, 5]. In native circulation, disturbed flow generally occurs at arterial branches and curves, such as carotid bifurcations, branch points of the coronary, and the infrarenal and femoral arteries, as well as the inner curvature of the aortic arch, where atherosclerotic lesions preferentially develop [2]. ECs with a pathological status are changed structurally and functionally. Their morphology can be changed, such that they are enlarged and/or acquire an irregular shape. ECs can also lose the regulatory roles that are characterized by the endothelium layer becoming more permeable, by endothelial inflammation, and within the ECs, by oxidative stress. Disturbed flow can cause EC dysfunction through the expression of adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E-selectin; proinflammatory cytokines, such as interleukin (IL)-1, IL-6 and tumor necrosis factor-alpha (TNF-α); and proinflammatory chemokines, such as IL-8 and MCP-1 [2, 5]. These molecules attract leukocytes and monocytes to the surface of the activated ECs, thereby enabling lipoprotein penetration and inflammatory cell infiltration, which initiate a pro-inflammatory process within the vessel wall. This process is the first step of atherogenesis. KLF2, a key PSS-induced transcription factor, has been shown to have anti-inflammatory and anticoagulant roles in ECs [45]. Disturbed flow suppresses the expression of KLF2 and causes the dysfunction of ECs [35]. Disturbed flow also induces the sustained activation of transcription factor nuclear factor-κβ (NF-κβ), which induces the expression of proinflammatory genes in ECs [46]. Increased intracellular oxidative stress is another indicator of EC dysfunction. It has been shown that USS plays a protective role in vessels by inducing antioxidant enzymes, such as superoxide dismutase, heme oxygenase-1, and NADPH quinine oxidoreductase 1 (NQO1), which provide a homeostatic oxidative balance [47]. Homeostasis can be disrupted by disturbed flow, which causes the enhanced expression of reactive oxygen species (ROS), which damage the vessel wall [47]. Taken together, these studies show that disturbed shear stress induces differential cellular responses compared with that induced by PSS, leading to endothelial dysfunction and atherosclerotic lesion formation at preferential sites.
9.2.2 Stretch Force
9.2.2.1 Cyclic Stretch Regulates Physiological Functions in Vascular SMCs and ECs
Cyclic stretch is the circumferential stretching of the vessel wall, which plays an important role in regulating vascular tone and maintaining the contractile phenotype of SMCs under physiological conditions [48, 49]. Unlike shear stress, which is mainly sensed by ECs, both ECs and SMCs are subject to cyclic stretch [49, 50]. However, the role of cyclic stretch in endothelial function has not been fully investigated. Cellular responses may vary depending on whether the cell is subjected to physiological or pathological stretch. Normal stretching can establish homeostatic oxidative balance and maintain vascular integrity to support vessels in the physiological state. When cyclic stretching is perturbed, such as when stretching is excessive due to high blood pressure, homeostasis is disrupted, which leads to vascular remodeling, arterial stiffness, and calcification [51]. Although vascular cell roles in and responses to stretching are less clear than their roles in and responses to shear stress, these two forms of mechanical forces induce processes that share many similar features. Mechanoreceptors can sense stretch force and transmit mechanical stimuli to intracellular signaling pathways to regulate cellular behaviors [52]. Several studies have described the important role of stretching on SMC gene expression and cellular functions, such as proliferation/apoptosis, migration/alignment, and the phenotypic switching of SMCs [53, 54]. MMPs are calcium-dependent zinc-containing endopeptidases that can degrade extracellular matrix proteins and thus mediate the extracellular matrix (ECM) [55]. Applying cells with a physiological level of cyclic (1 Hz) uniaxial stretch was shown to repress the expression of MMP-2 and MMP-9 in human cultured SMCs [56]. This report demonstrated that SMCs responded to physiological stretching by altering MMPs, which led to the subsequent remodeling of the ECM surrounding the vasculature. Physiologic stretching also inhibits apoptosis and mitosis in vascular ECs and SMCs, respectively [57, 58]. The magnitude of the cyclic stretch can cause cytoskeletal rearrangement, which increases EC permeability, indicating an important role for cyclic stretch in the regulation of mass transport through the vessel wall [59].
9.2.2.2 Cyclic Stretch Regulates Pathophysiological Changes in Vascular SMCs and ECs
SMC hypertrophy, hyperplasia, and ECM remodeling are considered to play roles in the development of hypertension. Pathological stretching caused by hypertension disrupts vessel homeostasis and leads to vascular remodeling, phenotypic switching of SMCs (from the contractile type to the synthetic type), arterial stiffness, and calcification, which are involved in the pathogenesis of CVDs [51]. It has been suggested that hypertension reduces myocardin activity in SMCs, resulting in the initial switch from the contractile phenotype to the synthetic SMC phenotype [60]. Another study showed that cyclic stretch (30 cycles/min; 15% elongation) induces SMC proliferation [54]. These studies indicate a role for cyclic stretch in the modulation of SMC gene expression and phenotypic changes. Moreover, uniaxial cyclic stretch causes SMCs to align perpendicular to the direction of the stretching, which affects cytoskeleton rearrangement. However, the mechanism by which stretching induces cytoskeletal rearrangement remains unclear [53]. In addition to morphological changes, cyclic stretch also plays a role in SMC migration by promoting the translocation of a key intracellular signal transducer, protein kinase C-δ, to the cytoskeleton [61]. Chronic high-magnitude cyclic stretch also plays a role in the modulation of the inflammatory response in vascular ECs via vascular endothelial growth factor receptor 2 (VEGFR2) signaling and matrix remodeling [50, 62]. Gawlak et al. reported that human pulmonary ECs subjected to chronic cyclic stretch (18% cyclic stretch) had induced VEGFR2 expression and tyrosine phosphorylation, which led to an increase in the expression of ICAM-1 and VCAM-1. Collectively, these studies show that cyclic stretch plays an important role in SMC phenotypic switching, vessel homeostasis, and vascular functions.
9.3 Epigenetics
Epigenetics gradually emerged in the 1980s in the course of researchers studying many phenomena that were incompatible with classical Mendelian genetics. It has been defined as the study of heritable changes in gene expression and cellular phenotype and has provided a relatively new explanation for gene function that is altered in ways other than solely by changes in DNA sequence [8]. Epigenetics has been linked to many cellular behaviors, such as cell proliferation and morphological changes, and diseases, including cancers, CVDs, and reproductive, autoimmune, and neurobehavioral conditions [13,14,15,16,17,18]. Epigenetics includes two types of modifications: selective transcriptional regulation, such as DNA methylation and histone covalent modification/chromatin remodeling, and posttranscriptional regulation, including RNA-based mechanisms. The ncRNA-based mechanisms, which involve miRNAs, lncRNAs, and antisense RNAs, are the most recently recognized epigenetic modifications by which gene expression is regulated.
9.3.1 Methylation
DNA methylation, the most well-known epigenetic process, is the addition of a methyl group to the fifth carbon of a cytosine residue to form 5mC in the CpG pair [9]. CpGs tend to aggregate to form CpG islands and are quite rare in mammalian genomes (~1%), which are generally unmethylated. The hypermethylation of CpG islands results in the recruitment of protein complexes that remove acetyl groups and repress gene expression [9, 63]. DNA methylation is catalyzed by a family of DNMTs. DNMT1, DNMT3a, and DNMT3b have methyltransferase activity [64]. DNMT-3L is able to interact with DNMT3a and DNMT3b to methylate retrotransposons [65]. DNA methylation-associated proteins, including methyl CpG-binding domain proteins (MBDs) and ubiquitin-like PHD, and RING finger domain-containing proteins, are bound to DNA-containing methylated CpG dinucleotides and recruit repressor complexes to methylated promoter regions to inhibit transcription. DNA methylation plays roles in many illnesses and health conditions [66, 67]. During early embryonic development, CpG islands undergo differential methylation, which confers their totipotency or pluripotency [68]. In addition to embryonic development, CpG island methylation plays a crucial role in genomic imprinting [9]. In vascular ECs, the CpG islands are methylated at the promoters of eNOS and VEGFR2, from where they recruit MBD2 to suppress gene expression at these methylated CpG islands [69]. DNA demethylation catalyzed by Ten-eleven translocation (TET) methylcytosine dioxygenases is a mechanism for reversing DNMT-mediated gene repression. TETs, including TET1, TET2, and TET3, which convert 5mC into 5-hydroxymethylcytosine [10], has been reported to inhibit DNMT1 expression and global DNA methylation in atherosclerotic plaques [70, 71]. Moreover, TET2 has also been shown to regulate the phenotype changes of SMCs, contributing to endothelial dysfunction and macrophage-induced inflammation [72]. These studies demonstrate the importance of DNA methylation/demethylation in the modulation of vascular function.
9.3.2 Histone Modification and Chromatin Remodeling
Chromatin is composed of DNA and proteins, including four core histone proteins (H2A, H2B, H3, and H4) that are wrapped around 147 base pairs of DNA [73]. Histones are subjected to covalent posttranslational modifications (PTMs), including lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, lysine ubiquitylation, and lysine sumoylation on their tails [74]. Histone PTMs affect gene expression by recruiting histone modifiers and inducing chromatin remodeling. In general, tightly folded chromatin tends to inhibit gene expression, while more open chromatin leads to gene expression. Although gene transcription that is regulated by histone PTMs can be turned on and off, histone acetylation directly contributes to gene expression. Histone acetylation has been the most intensively studied modification, and the results have shown that this modification is caused by HATs transferring an acetyl group from acetyl-CoA to form ε-N-acetyllysine [74, 75]. HATs are divided into three families, namely, Gcn5-related N-acetyltransferases, MYSTs, and CREB-binding proteins (CBP/p300) [74, 75]. HDACs have an opposite action to that of HATs in that they remove acetyl groups from histones. There are three distinct families of HDACs: class I (HDAC1/2/3 and HDAC8), class II (HDAC4/5/6/7 and HDAC9/10), and class III [sirtuin (SIRT) family (SIRT1/2/3/4/5/6/7)] [76]. Class I HDACs are mostly expressed in the nucleus and display high enzymatic activity, whereas HDAC3, which is similar to the class IIA HDACs (HDAC4/5, HDAC7, and HDAC9), contributes to nuclear-cytoplasmic shuttling, providing a mechanism for linking extracellular signals with gene expression [77, 78]. Class IIB HDAC6 is the primary cytoplasmic deacetylase found in mammalian cells, whereas the functions of HDAC10 are poorly understood. Class III HDACs/SIRTs are the highly conserved protein family of nicotinamide adenine dinucleotide (NAD)-dependent histone deacetylases located in different subcellular compartments, such as the nucleus (SIRT1/3/6/7), cytoplasm (SIRT1/2), and the mitochondria (SIRT3/4/5) [79, 80]. SIRTs exert their functions by transferring an acetyl group to the cofactor NAD to generate O-acetyl ADP-ribose and nicotinamide, which serve as feedback inhibitors of enzymatic reactions [80]. Histone acetylation is a reversible process that is controlled by the antagonistic actions of HATs and HDACs. In general, hyperacetylation is involved in the upregulation of transcription, whereas hypoacetylation contributes to the downregulation of gene expression [81]. The balance between acetylation and deacetylation represents a critical regulatory mechanism for gene expression, developmental processes, and disease progression, such as those of CVDs [82]. It has been shown that HATs and inhibitors of HDAC attenuate CVDs by mediating certain cellular processes, including myocyte hypertrophy, apoptosis, oxidative stress, and inflammation [79, 82,83,84]. These results implicate HATs and other inhibitors of HDACs as novel agents useful for treating CVD patients [82, 85].
9.3.3 RNA-Based Mechanisms
Epigenetic modulation of gene expression through ncRNAs is a newly discovered regulatory mechanism [86]. ncRNAs, including miRNAs, small interfering RNAs, piwi-interacting RNAs, small nucleolar RNAs, and lncRNAs, are functional RNAs that are not translated into proteins [87, 88]. RNA-based epigenetic regulation of gene expression is divided into RNA interference (RNAi)-mediated transcriptional gene silencing and RNA-dependent histone methylation without RNAi. MiRNAs regulate chromatin structure and inhibit transcription via Argonaute (Ago) complexes that bind to the 3′-UTRs of complementary nascent RNAs or via the translational repression induced by the pairing of imperfect complementary sequences, leading to the degradation of the mRNA and the recruitment of histones and DNMTs [11, 89]. LncRNAs, distinct from other small ncRNAs, modulate gene expression by diverse mechanisms, which have been categorized into signaling, acting as molecular decoys, guiding ribonucleoprotein complexes to specific chromatin sites, and serving as scaffolds in the formation of transcriptional complexes. In general, many lncRNAs are able to bind to particular genomic sites, implying their ability to modulate chromatin activities. In addition, lncRNAs can also act posttranscriptionally by regulating translation and splicing and affecting mRNA stability. Xist, a 17 kb nuclear lncRNA, is expressed exclusively on the inactive X chromosome and mediates global inactivation of a randomly chosen X chromosome in an early developmental process in females [90]. This process is known as X chromosome inactivation, which transcriptionally silences the X chromosome coated by Xist, thereby providing equivalent gene expression between males and females. Xist may exert its function by directing the polycomb repressive complex (PRC) 2 to chromatin and catalyzing histone methylation to repress gene transcription [86]. Several studies have revealed that ncRNAs are involved in the regulation of various processes, such as metabolism; development, particularly vascular development; cell proliferation; and various diseases, including vascular diseases [88, 91, 92]. Chen et al. have shown that miR-146a plays a critical role in the inhibition of vascular inflammation and neointimal lesion formation in rat or mouse carotid artery [93] (Fig. 9.1). Shear- and synthetic SMC-induction of miR-146a in ECs via integrins/Nrf-2 targets IL-1 receptor-associated kinase (IRAK) to inhibit NF-κB signaling, which exerts negative feedback control in the biogenesis of itself. Moreover, EC miR-146a expression has been shown to modulate synthetic SMC phenotype toward a contractile state [93]. Taken together, atheroprotective shear stress-induced miR-146a expression inhibits EC inflammation and neointima formation in injured arteries. This regulation is highly correlated with clinical symptoms such as vascular remodeling after injury. MiR-126 has been reported to play roles in angiogenesis and anti-inflammation in vascular ECs. The angiogenic signaling and integrity of the vessel wall are regulated by miR-126 during embryogenesis [94]. MiR-126 has also been reported to attenuate TNF-α-induced VCAM-1 expression to affect leukocyte adhesion [95]. MiR-143/145 is involved in the modulation of SMC phenotypic switching in vessels [96]. The expression of the antisense noncoding RNA at the Ink4 locus (ANRIL) is regulated by DNA polymorphisms in this region, which is highly associated with the incidence of CADs [97]. The NF-κB-induced expression of ANRIL modulates the expression of IL-6 and IL-8 by recruiting the transcription factor Yin Yang 1, which also leads to endothelial inflammation [98]. In addition, ANRIL promotes the proliferation of SMCs by recruiting PRC complexes to cyclin-dependent kinase inhibitor 2A/B, which downregulates ANRIL [99]. In contrast to ANRIL, the expression of another lncRNA, lincRNA-p21, is decreased in the atherosclerotic plaques in ApoE−/− mice and humans, indicating its protective roles in vessels [100]. LincRNA-p21 inhibits SMC proliferation, neointima formation, and atherosclerosis by enhancing p53-mediated apoptosis in SMCs [100]. LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) is considered to be associated with vascular disease because its expression is low in atherosclerotic plaques of the coronary artery [101, 102]. Mice with Malat1 deficiency (Malat−/−) in an ApoE−/− background (ApoE−/− Malat−/−) possess an increased number of inflammatory cells and atherosclerotic lesions compared with the number in the ApoE−/− Malat1+/+ control mice [102]. Taken together, these studies demonstrate that ncRNAs are not genetic waste but play critical roles in the pathophysiology of vascular diseases.

Schematic diagram showing the mechanisms underlying the shear- and synthetic SMC-mediated miR-146a expressions and their inhibition in EC inflammation and neointimal lesion formation in rat or mouse carotid artery. Application of unidirectional shear stress (12 dynes/cm2) to ECs co-cultured with synthetic SMCs for 24 h induces EC miR-146a expression through integrins/Nrf-2 signaling cascade. miR-146a targets IRAK to inhibit NF-κB signaling, which exerts negative feedback control on the biogenesis of itself. In vivo, silencing either Nrf-2 or miR-146a leads to increased neointima formation of injured rat carotid artery under physiological flow. Overexpressing miR-146a inhibits neointima formation of rat or mouse carotid artery. Unidirectional shear stress applied to ECs can modulate synthetic SMC phenotype toward a contractile state through EC miR-146a induction. Taken together, atheroprotective shear stress-induced miR-146a expression inhibits EC inflammation and neointima formation of injured arteries, leading to vascular remodeling
9.4 Mechanical Force-Induced Epigenetic Modifications in Vascular Health and Disease
The roles of mechanical force-induced epigenetic modifications in vascular gene expression and various functional regulations have been intensively studied. In this section, the roles and related mechanisms of mechanical force-induced DNA methylation, histone acetylation/deacetylation, and miRNA/lncRNA expression in modulating vascular cell function are discussed (Tables 9.1, 9.2, and 9.3).
9.4.1 Methylation
Dysregulation of DNA methylation, including both hypermethylation and hypomethylation, has been reported to occur in various diseases, including CVDs [133, 134]. Genetically atherosclerosis-prone apolipoprotein E-deficient (ApoE–/–) mice show alterations in DNA methylation profiles in their aortas and monocytes before histologically detectable vascular lesions appear [133]. The hypermethylation of the miR-145 promoter and its decreased expression in SMCs are caused by DNMT1 upregulation or TET2 downregulation, leading to the activation of the nucleotide-binding oligomerization domain-like receptor protein 3, which facilitates the inflammatory response and induces plaque formation [135]. This study indicated that the DNA methylation dynamically regulated by DNMTs and TETs plays a crucial role in atherosclerosis. Furthermore, several studies have demonstrated that DNMTs and TETs are modulated by mechanical forces that regulate vascular cell function (Table 9.1). DNMT1 has been shown to play a role in the regulation of endothelial functions in response to blood flow and may contribute to atherogenesis [103, 104]. Disturbed flow induces DNMT1 expression via the integrin/focal adhesion kinase (FAK)/mammalian target of rapamycin (mTOR)/p70S6 kinase signaling pathways to induce global DNA methylation, including on HoxA5, KLF3, Cyclin A, and connective tissue growth factor (CTGF), both in vivo and in vitro [103, 104]. The downregulation of DNMT1 by using 5-aza-2′-deoxycytidine or by inhibiting mTOR reduced the disturbed flow-induced endothelial inflammation and proliferation, thereby attenuating plaque formation in the ApoE−/− mice [103, 104]. The expression of eNOS regulated by the basal transcription machinery in the core promoter plays an important role in modulating endothelial functions such as vascular tone via NO production. However, eNOS mRNA is destabilized when ECs are under pathological conditions such as inflammation, proliferation, ox-LDL treatment, or hypoxia [136]. DNMT3a stimulated by OSS binds to the promoter of the transcription factor KLF4 and causes DNA methylation of the KLF4 promoter. This process results in the inhibition of KLF4 transcription and eNOS expression, leading to inflammation in ECs [105]. The hypermethylation of the KLF4 promoter as well as the downregulation of KLF4 and eNOS are also observed in the endothelium of OSS-affected regions in the porcine aorta [105]. In addition to DNMTs, TETs have also been shown to participate in the initiation and progression of atherosclerosis [106]. TET2 levels and TET2-mediated endothelial autophagy are decreased in response to low shear stress (5 dyne/cm2), which may contribute to atherogenesis. Moreover, overexpressed TET2 enhances eNOS expression and reduces endothelin-1 levels. This result implies that TET2 may play a role in vessel constriction [106]. Taken together, these studies provide new insight into the mechanism by which shear stress-mediated DNA methylation in ECs influences vascular cell function and, hence, atherosclerosis.
9.4.2 Histone Modification
9.4.2.1 Class I HDACs
Mechanical force-induced histone modifications cause chromatin remodeling that regulates gene transcription, which is responsible for the modulation of endothelial function (Table 9.2, Fig. 9.2). An in vitro study showed that USS facilitated the association of p53 with HDAC1 to cause the deacetylation of p53 at Lys-320 and Lys-373 in ECs [107]. The USS-mediated deacetylation of p53 induced p21 expression, leading to cell cycle arrest. In our previous study, class I HDACs were found to play a role in modulating OSS-induced cell proliferation and oxidation in ECs [108]. OSS stimulus caused an increased nuclear accumulation of HDAC1/2/3 and thus induced cyclin A expression but inhibited p21 expression, leading to the upregulation of EC proliferation [108]. OSS stimulation also induced the association of HDAC1/2/3 with NF-E2-related factor 2 (Nrf2) and HDAC3 with myocyte enhancer factor 2 (MEF2), which resulted in the deacetylation of Nrf2 and MEF2 and the inhibition of NQO1 and KLF2 expression [108]. By using the in vivo rat stenosis model, in which a U-clip was applied to the abdominal aorta to produce a 65% constricted diameter [35], we found increased expression of HDAC2/3/5 and incorporation of BrdU in ECs located in downstream of stenosis where OSS occurs. This OSS-dependent BrdU incorporation was attenuated in the ECs by the class I-specific HDAC inhibitor valproic acid (VPA) [108]. In addition, HDAC3 expression and the OSS-stimulated phosphorylation of its serine/threonine residues were shown to play an essential role in the survival and integrity of cultured ECs [109]. Inhibition of HDAC3 expression via specific short hairpin RNA reduced EC survival, by reducing Akt activity, which led to vessel rupture and atherosclerosis in ApoE−/− mice.

Schematic diagram of HDACs, HATs, and related molecules involved in hemodynamic force-induced vascular cell function and dysfunction. Atheroprone flow, i.e., disturbed and oscillatory flow, induces the expression and nuclear accumulation of class I HDAC1/2/3 and class II HDAC5/7 in ECs. Subsequently, this accumulation causes HDAC1/2/3-mediated deacetylation of Nrf-2 to inhibit NQO1 expression, which contributes to antioxidation in ECs. Furthermore, HDAC1/2/3 are involved in oscillatory flow-induced cell cycle progression, endothelial proliferation, and cell survival. Disturbed flow also causes HDAC3/5/7-mediated deacetylation of MEF2 to inhibit KLF2, thereby promoting endothelial anti-inflammatory responses and antioxidation. In contrast, atheroprotective flow, i.e., regular and pulsatile flow, induces the phosphorylation-dependent nuclear export of HDAC5/7 in ECs, thereby increasing the expression of NQO-1 and KLF2. Regular flow also enhances the expression of HDAC6, SIRT1, and P300, which play roles in the modulation of vascular tone and EC cytoskeletal remodeling. Another mechanical force, cyclic stretch, mainly mediates the regulation of SMC hypertrophy and migration through HDAC4/5 and HDAC3/4/7 signaling
9.4.2.2 Class II HDACs
HAT p300 has been reported to cooperate with p65 to bind to the shear stress response B element of the eNOS promoter, leading to eNOS expression under laminar flow [115]. In addition to p300, other HDACs have been shown to play roles in mechanical force-induced eNOS expression [112]. USS stimuli induce the phosphorylation of class II HDAC5 and its nuclear export through a calcium/calmodulin-dependent pathway [112]. This export subsequently caused the dissociation of HDAC5 from MEF2 and promoted MEF2 transcription, resulting in KLF2 and eNOS expression. Moreover, class II HDAC6 has been reported to modulate USS-induced cytoskeletal remodeling in ECs co-cultured with SMCs [113]. Tubulin is an important cytoskeletal protein, and its acetylation stabilizes microtubules and retards cell migration. USS stimulus activates HDAC6 to inhibit tubulin acetylation, leading to cytoskeletal remodeling and cell migration in ECs co-cultured with SMCs [113]. Furthermore, we have identified the roles of class II HDACs in modulating endothelial oxidation and inflammation in response to OSS and PSS stimuli [108]. OSS stimuli induce the expression and nuclear accumulation of class II HDACs in ECs, which causes HDAC3/5/7 to interact and form a complex with MEF2 to suppress KLF2 expression contribute to anti-inflammatory responses [108]. Another mechanical force, cyclic stretch (1 Hz at 10% elongation), has been shown to inhibit SMC migration through the hyperacetylation of histone H3, increased expression of HDAC7, and downregulation of HDAC3/4 [110]. The role of cyclic stretch-mediated histone modifications in CVDs have been studied in vivo. Western blotting analysis of the proteins from the aortas and mesenteric arteries of spontaneously hypertensive rats (SHRs) and Wistar Kyoto rats (WKYs) showed that the expression of HDAC4 and HDAC5 were decreased in the SHRs compared to their expression in the WKYs [111]. The downregulation of HDACs caused by VPA greatly reduced the blood pressure, cytokines, ROS, and angiotensin II in the SHR mouse model [137].
9.4.2.3 Class III HDACs
The class III HDAC SIRT1, interacting with AMP-activated protein kinase (AMPK), has been shown to influence eNOS expression in vitro and in vivo [114]. The USS-induced mechanical stimulation enhances the association of SIRT1 and eNOS, resulting in eNOS deacetylation and expression through AMPK signaling [114]. In addition to eNOS, SIRT1 also deacetylates p65 at lysine 310 in macrophages and suppresses macrophage binding to aortic ECs, thereby inhibiting NF-κβ signaling and reducing the expression of the adhesion molecules ICAM-1 and VCAM-1 [138]. An in vivo study showed that overexpressed endothelial SIRT1 in ApoE−/− mice maintained vascular cells in a physiological state and hence attenuated the formation of atherosclerotic plaques [138]. Furthermore, SIRT1 also plays roles in the proliferation and inflammation of SMCs. SIRT1 promotes the mitosis of senescence-resistant cells by suppressing p21. Moreover, the inhibitor of MMP-3 in tissues is enhanced by SIRT1 overexpression, which causes the downregulation of MMPs and induces anti-inflammatory responses in SMCs. Taken together, these studies indicate that mechanical forces mediate the activation and expression of HDACs and hence contribute to vascular cell function as well as the development of atherosclerosis.
9.4.3 MicroRNA
9.4.3.1 miRNAs Are Regulated by Shear Stress
The functions of mechanical force-induced miRNAs in modulating cellular angiogenesis, inflammation, proliferation, and migration in vascular biology and disease have been extensively studied (Table 9.3, Fig. 9.3). It has been shown that KLF2 plays a critical role in flow-induced angiogenesis in zebrafish embryos through the miR-126/VEGF signaling pathway [116]. To elucidate the mechanisms by which miRNAs regulate cellular functions in response to mechanical stimulation, the miRNA expression profiles of cultured ECs subjected to differential flow were analyzed [117]. miR-663 was found to play a role in endothelial inflammation but not in apoptosis upon OSS stimulation. miR-21 activated by OSS in cultured ECs also triggered an inflammatory response by downregulating peroxisome proliferator-activated receptor α (PPARα) [118]. The role of miRNAs in the regulation of endothelial inflammation has been studied in vivo. The decreased expression of miR-10a in atherosclerosis-susceptible regions (i.e., the inner curvature of the aortic arch and aorta-renal branches) in swine vessels indicates that miR-10a may play a protective role during atherosclerosis [119]. miR-10a confers protection to the vascular system through its anti-inflammatory effect in ECs [119]. Moreover, miR-10a can be induced by PPS and OSS stimuli via HDAC signaling to modulate anti-inflammatory and inflammatory responses, respectively, through GATA-binding factor 6 and VCAM-1 expression [120, 121]. In addition to endothelial inflammation, miRNAs have been shown to participate in KLF2-mediated eNOS expression in response to shear stress [122, 139]. USS (12 dyne/cm2) but not OSS (0.5 ± 4 dyne/cm2) stimuli can downregulate miR-92a expression to increase KLF2 expression and thus facilitate eNOS induction and NO production, which contributes to the modulation of vascular tone [122]. Moreover, it has been shown that USS can induce miR-19a, miR-23b, and miR-101 expressions to inhibit the expressions of cyclin D, E2F, and mTOR, as well as the phosphorylation of Rb, which leads to cell cycle arrest and the inhibition of EC mitosis [123,124,125].

Schematic diagram of ncRNAs and related molecules involved in hemodynamic force-modulated vascular cell function and dysfunction. Disturbed flow induces the expression of miR-21 and miR-663 to downregulate PPAR-α expression, leading to an inflammatory response in ECs. Moreover, disturbed flow inhibits miR-10 expression to upregulate the expression of GATA6 and VCAM-1, leading to an inflammatory response in ECs. Conversely, regular flow induces the expression of miR-10a, miR-19a, and miR-23b, causing the downregulation of mTOR, cyclin D, and E2F and contributing to endothelial proliferation. Disturbed flow and regular flow also play roles in SMC phenotype switching via the secretion of miR-126-3p and miR-143/145 from ECs. In addition to miRNAs, regular flow also induces the expression of lncRNA MANTIS, which inhibits VCAM-1 levels and inflammation in ECs. Another mechanical force, cyclic stretch, mainly regulates SMC hypertrophy and proliferation. Pathological stretching causes the upregulation of miR-126 and miR-130 but downregulation of let-7d and miR-33, resulting in the inhibition of GSK-3β and GAX but increased expression of K-RAS and BMP3. These processes promote hypertrophy and proliferation in SMCs
9.4.3.2 miRNAs Are Regulated by Cyclic Stretch
Stretch force has also been shown to cause miR-26a expression, which serves as a hypertrophic gene in SMCs [126]. miR-26a subsequently downregulates glycogen synthase kinase-3β (GSK-3β), an anti-hypertrophic protein, to enhance SMC hypertrophy. The roles of miRNAs in modulating cellular functions during cyclic stretching have been studied in vivo. miR-130a and let-7d have been shown to be correlated with vascular remodeling in SHRs [127, 128]. The expression of miR-130a is upregulated, which suppresses the expression of growth arrest-specific homeobox (GAX) to promote SMC proliferation in these hypertensive rats. Conversely, let-7d is significantly downregulated in the SMCs of the SHRs. Let-7d can bind oncogene K-RAS to downregulate it, leading to the suppression of SMC mitosis. Recently, Huang et al. proposed that miRNA-33 protects against venous SMC proliferation in response to arterial mechanical stretch in a grafted vein [129]. The expression of miR-33 in venous SMCs subjected to a 10% 1.25 Hz arterial stretch in vitro is decreased, which upregulates bone morphogenetic protein 3 (BMP3), and increases phosphorylation of smad2 and smad5. These modulations and modification result in venous SMC proliferation and neointimal hyperplasia.
9.4.3.3 Extracellular miRNAs
Although active in cells, few miRNAs are found in extracellular biofluids, such as serum and saliva, or in cultured media [140,141,142,143,144]. In contrast to RNA, these released miRNAs are relatively stable in the extracellular environment, where they are in the continual presence of RNases, suggesting that they may have a protective mechanism that enables them to bypass areas with high RNase activity [145, 146]. Extracellular miRNAs might be conjugated with proteins, included in the lipid complexes, or wrapped with membrane vesicles to avoid degradation [147, 148]. The roles and mechanisms of miRNA exported to the extracellular environment have been studied. Biofluid miRNAs can be detected in several vesicles and complexes, indicating that the release of miRNAs to the extracellular region is mediated by extracellular vesicles (EVs), including exosomes, microvesicles, and apoptotic bodies (ABs), and by high-density lipoproteins and the Ago2 protein complex [140, 144, 147, 148]. These released miRNAs are transported to the recipient cells via specific pathways through which they modulate the expression of target genes/molecules by serving as signal transducers in cell–cell communications. The role and function of extracellular miRNAs in vascular biology have been investigated [131, 149, 150]. Application of unidirectional shear stress (12 dynes/cm2) to ECs co-cultured with synthetic SMCs for 24 h induces EC miR-146a expression to inhibit neointima formation and modulate synthetic SMC phenotype toward a contractile state [93]. This study provides evidence that miR-146a is secreted from ECs to act on the adjacent SMCs. ABs containing miR-126 have been reported to deliver cargo to recipient vascular cells and reduce atherogenesis in mice [149]. Human umbilical vein endothelial cells (HUVECs) were transfected with the KLF2 plasmid or subjected to PSS to generate miR-143/145-abundant EVs that contributed to SMC phenotypes. Coculturing HUVECs with SMCs led to the reduction of target gene expression in recipient SMCs, which attenuated the development of atherosclerosis [130]. Conversely, disturbed flow-induced expression and secretion of endothelial miR-126-3p promoted the phenotypic switch of SMCs, causing SMC hyperplasia and, hence, atherogenesis [131]. Taken together, these studies demonstrate that shear stress-mediated extracellular miRNAs play critical roles in EC–SMC interactions and vascular diseases. Thus, extracellular miRNAs may have the potential to be developed as noninvasive clinical biomarkers for atherosclerosis.
9.4.4 Long Noncoding RNA
LncRNAs have been shown to modulate vascular cell function and CVDs [151, 152]. Studies regarding mechanical force-mediated lncRNA function in vascular biology have recently emerged [132, 153]. The role of spliced-transcript endothelial-enriched lncRNA (STEEL) in angiogenesis has been identified [153]. STEEL transcriptionally upregulates eNOS and KLF2 expression via STEEL-mediated recruitment of the poly-ADP-ribosylase to the KLF2 promoter. Moreover, STEEL receives inhibitory feedback from both eNOS and KLF2 in response to USS stimulus [153]. Leisegang et al. demonstrated that the beneficial effects of HMG-CoA-reductase inhibitors (statins) and laminar flow on ECs are conferred by lncRNA MANTIS (also known as lncRNA n342419) [132] (Table 9.3, Fig. 9.3). Laminar flow and statins have been shown to activate MANTIS expression via the transcription factors KLF2 and KLF4, which causes a reduction in the association of the SWI/SNF chromatin remodeling factor BRG1 and the ICAM-1 promoter, thereby preventing the development of atherosclerosis [132]. Sphingosine-1-phosphate (S1P) is a potent signaling lipid activated by the S1P receptor (S1PR). Recently, long intergenic noncoding RNA antisense to S1PR1 (LISPR1), which is highly expressed in ECs and lung tissue but expressed at low levels in human lung diseases, plays an essential role in S1P signaling by regulating S1PR1 expression, thereby regulating endothelial migration and sprouting [154]. Downregulated LISPR1 inhibits S1P-induced migration and sprouting in ECs. Moreover, LISPR1 and S1PR1 expression are upregulated by the increased association of KLF2 with the S1PR1/LISPR1 shared promoter in response to USS and statins. Taken together, these studies suggest the possibility that lncRNAs can be developed as clinical biomarkers of vascular diseases and may be potential therapeutic drugs for CVDs.
9.5 Conclusions and Future Perspectives
Mechanical forces, including shear stress and cyclic stretch, can modulate gene expression, cellular functions, and morphological changes in vascular health and disease. Normal shear stress and cyclic stretch maintain vascular homeostasis, whereas disturbed flow and pathological cyclic stretch cause vascular cell dysfunction and thus promote the occurrence of vascular diseases. Epigenetic factors, including DNMTs, HDACs, miRNAs, and lncRNAs can modulate gene expression without altering the DNA sequence. Mechanisms by which mechanical forces act on vascular gene expression and cellular functions via differential intracellular signaling pathways are widely explored. However, studies investigating the role of mechanical force-induced epigenetic modifications in vascular biology have emerged only in recent decades. In this chapter, in vitro and in vivo studies on mechanical force-induced DNA methylation, histone modification/chromatin remodeling, and ncRNA-dependent modification in the regulation of gene expression, cellular function, and pathology are summarized. Studies regarding the mechanical regulation of vascular gene expression, proliferation/migration, angiogenesis, antioxidation, inflammation, and vascular disorders are discussed. Furthermore, the roles and regulations of critical vascular molecules such as eNOS, KLF2, ICAM-1, VCAM-1, NF-κβ, p21, and p53 are also described.
Functional roles of shear-induced DNMTs, TETs, HDACs, HAT, miRNAs, and lncRNAs in the regulation of gene expression and vascular cell function and dysfunction are well studied. However, the mechanisms by which shear stress and stretch force induce DNMTs, TETs, HDACs, and HAT expressions remain unclear and warrant further investigation. Mechanoreceptor integrin and its downstream FAK/mTOR/p70S6 signaling pathway have been shown to be involved in the disturbed flow-induced DNMT1 expression [103, 104]. In comparison to shear stress, the role of another mechanical force, cyclic stretch, in the epigenetic regulation of vascular physiology and pathology remains unclear. Recently, the effect of cyclic stretch on vascular ECs has been investigated [50]. There is increasing evidence that cyclic stretch serves as a potential trigger for the induction of the inflammatory response of ECs and inflammatory cells, leading to ECM remodeling [50]. Mechanical force-mediated endothelial function and its interplay with ECM are highly associated with the programming of abdominal aortic aneurysm [50]. Unlike shear stress caused by blood flow, the generation of mechanical stretch is more complicated. Matrix remodeling, which is involved in the interaction of the ECM with MMPs, alters the mechanical properties of vessels and therefore causes altered stretching [155]. The stretch force caused by ECM remodeling may play roles in epigenetic-mediated vascular health and disease, but these roles need to be elucidated. Extracellular miRNAs and lncRNAs are emerging epigenetic regulators that modulate vascular function in response to mechanical forces. Although extracellular miRNAs constitute a small portion of all miRNAs, their characteristics, such as circulating in biofluids and mounting resistance to RNAase, make them potential targets to develop as noninvasive clinical biomarkers of atherosclerosis and other CVDs. LncRNAs, distinct from small ncRNAs, modulate gene expression by diverse mechanisms. In addition to modulating gene transcription, lncRNAs can also regulate translation and RNA splicing and affect mRNA stability. However, studies on mechanical force-induced lncRNAs suggest that they rarely regulate vascular biology. Therefore, the role of these ncRNA-mediated epigenetic modifications in modulating vascular gene expression and the corresponding cellular functions need to be further investigated.
In conclusion, epigenetic studies increase our knowledge of mechanical forces that transcriptionally and posttranscriptionally regulate gene expression in vascular physiology and pathology without altering DNA sequences. These studies provide new insights into the dynamic regulation of vascular functions and the ways they alter the vascular biology or pathobiology, findings that are expected to lead to the development of diagnostic and therapeutic approaches for treating vascular diseases.
Abbreviations
- 3′-UTR:
-
3′-Untranslated region
- 5mC:
-
5-Methylcytosine
- ABs:
-
Apoptotic bodies
- Ago:
-
Argonaute
- AMPK:
-
AMP-activated protein kinase
- ANRIL:
-
Antisense noncoding RNA at the Ink4 locus
- ApoE–/–:
-
Apolipoprotein E-deficient genotype
- BMP3:
-
Bone morphogenetic protein 3
- CTGF:
-
Connective tissue growth factor
- CVD:
-
Cardiovascular disease
- DNMT:
-
DNA methyltransferase
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- EV:
-
Extracellular vesicle
- eNOS:
-
Endothelial nitric oxide synthase
- FAK:
-
Focal adhesion kinase
- GAX:
-
Growth arrest-specific homeobox
- GSK-3β:
-
Glycogen synthase kinase-3β
- H:
-
Histone
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- HUVEC:
-
Human umbilical vein endothelial cell
- ICAM-1:
-
Intercellular adhesion molecule-1
- IL:
-
Interleukin
- IRAK:
-
IL-1 receptor-associated kinase
- KLF:
-
Krüppel-like factor
- LISPR1:
-
Long intergenic noncoding RNA antisense to S1PR1
- LncRNA:
-
Long noncoding RNA
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- MBD:
-
Methyl CpG-binding domain protein
- MCP-1:
-
Monocyte chemotactic protein-1
- MEF2:
-
Myocyte enhancer factor 2
- MiRNA:
-
MicroRNA
- MiR-21:
-
MicroRNA-21
- MMP:
-
Matrix metalloproteinase
- mRNA:
-
Messenger RNA
- mTOR:
-
Mammalian target of rapamycin
- NAD:
-
Nicotinamide adenine dinucleotide
- NcRNA:
-
Noncoding RNA
- NF-κβ:
-
Nuclear factor-κβ
- NO:
-
Nitric oxide
- NQO1:
-
NADPH quinine oxidoreductase 1
- Nrf2:
-
NF-E2-related factor 2
- OSS:
-
Oscillatory shear stress
- Ox-LDL:
-
Oxidized low-density lipoprotein
- PPARα:
-
Peroxisome proliferator-activated receptor α
- PRC:
-
Polycomb repressive complex
- PRKD2:
-
Protein kinase D2
- PSS:
-
Pulsatile shear stress
- PTM:
-
Posttranslational modification
- Rb:
-
Retinoblastoma protein
- RNAi:
-
RNA interference
- ROS:
-
Reactive oxygen species
- S1P:
-
Sphingosine-1-phosphate
- S1PR:
-
Sphingosine-1-phosphate receptor
- SAM:
-
S-adenyl methionine
- SHR:
-
Spontaneously hypertensive rat
- SIRT:
-
Sirtuin
- SMC:
-
Smooth muscle cell
- STEEL:
-
Spliced-transcript endothelial-enriched lncRNA
- TET:
-
Ten-eleven translocation methylcytosine dioxygenase
- TNF-α:
-
Tumor necrosis factor
- USS:
-
Unidirectional shear stress
- VCAM-1:
-
Vascular cell adhesion molecule-1
- VE-Cad:
-
VE-cadherin
- VEGF:
-
Vascular endothelial growth factor
- VEGFR2:
-
Vascular endothelial growth factor receptor 2
- WKY:
-
Wistar Kyoto rat
References
Butcher DT, Alliston T, Weaver VM (2009) A tense situation: forcing tumour progression. Nat Rev Cancer 9:108–122
Chiu JJ, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91:327–387
Li YS, Haga JH, Chien S (2005) Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech 38:1949–1971
Jaalouk DE, Lammerding J (2009) Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10:63–73
Hahn C, Schwartz MA (2009) Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10:53–62
Van Speybroeck L (2002) From epigenesis to epigenetics: the case of C. H. Waddington. Ann N Y Acad Sci 981:61–81
Singh S, Sonkar SK, Sonkar GK, Mahdi AA (2019) Diabetic kidney disease: a systematic review on the role of epigenetics as diagnostic and prognostic marker. Diabetes Metab Res Rev 35:e3155
Goldberg AD, Allis CD, Bernstein E (2007) Epigenetics: a landscape takes shape. Cell 128:635–638
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38:23–38
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18:517–534
Winter J, Jung S, Keller S, Gregory RI, Diederichs S (2009) Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 11:228–234
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159
Sun M, Kraus WL (2015) From discovery to function: the expanding roles of long non-coding RNAs in physiology and disease. Endocr Rev 36:25–64
Man HJ, Yan MS, Lee JJ, Marsden PA (2016) Epigenetic determinants of cardiovascular gene expression: vascular endothelium. Epigenomics 8:959–979
Zoghbi HY, Beaudet AL (2016) Epigenetics and human disease. Cold Spring Harb Perspect Biol 8:a019497
Alexander MR, Owens GK (2012) Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74:13–40
Xu S, Pelisek J, Jin ZG (2018) Atherosclerosis is an epigenetic disease. Trends Endocrinol Metab 29:739–742
Mazzone R, Zwergel C, Artico M, Taurone S, Ralli M, Greco A, Mai A (2019) The emerging role of epigenetics in human autoimmune disorders. Clin Epigenetics 11:34
Zhou B, Margariti A, Zeng L, Xu Q (2011) Role of histone deacetylases in vascular cell homeostasis and arteriosclerosis. Cardiovasc Res 90:413–420
Condorelli G, Latronico MV, Cavarretta E (2014) microRNAs in cardiovascular diseases: current knowledge and the road ahead. J Am Coll Cardiol 63:2177–2187
Kumar S, Kim CW, Simmons RD, Jo H (2014) Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol 34:2206–2216
Dunn J, Simmons R, Thabet S, Jo H (2015) The role of epigenetics in the endothelial cell shear stress response and atherosclerosis. Int J Biochem Cell Biol 67:167–176
Ku DN, Giddens DP, Zarins CK, Glagov S (1985) Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis 5:293–302
Davies PF (1995) Flow-mediated endothelial mechanotransduction. Physiol Rev 75:519–560
Jufri NF, Mohamedali A, Avolio A, Baker MS (2015) Mechanical stretch: physiological and pathological implications for human vascular endothelial cells. Vasc Cell 7:8
Lu D, Kassab GS (2011) Role of shear stress and stretch in vascular mechanobiology. J R Soc Interface 8:1379–1385
Hove JR, Koster RW, Forouhar AS, Acevedo-Bolton G, Fraser SE, Gharib M (2003) Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421:172–177
Boselli F, Vermot J (2016) Live imaging and modeling for shear stress quantification in the embryonic zebrafish heart. Methods 94:129–134
Huynh QK, McKinsey TA (2006) Protein kinase D directly phosphorylates histone deacetylase 5 via a random sequential kinetic mechanism. Arch Biochem Biophys 450:141–148
Banjo T, Grajcarek J, Yoshino D, Osada H, Miyasaka KY, Kida YS, Ueki Y, Nagayama K, Kawakami K, Matsumoto T et al (2013) Haemodynamically dependent valvulogenesis of zebrafish heart is mediated by flow-dependent expression of miR-21. Nat Commun 4:1978
Fernandez AP, Serrano J, Castro S, Salazar FJ, Lopez JC, Rodrigo J, Nava E (2003) Distribution of nitric oxide synthases and nitrotyrosine in the kidney of spontaneously hypertensive rats. J Hypertens 21:2375–2388
Yu J, deMuinck ED, Zhuang Z, Drinane M, Kauser K, Rubanyi GM, Qian HS, Murata T, Escalante B, Sessa WC (2005) Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A 102:10999–11004
Chien S (2007) Mechanotransduction and endothelial cell homeostasis: the wisdom of the cell. Am J Physiol Heart Circ Physiol 292:H1209–H1224
Karino T, Goldsmith HL, Motomiya M, Mabuchi S, Sohara Y (1987) Flow patterns in vessels of simple and complex geometries. Ann N Y Acad Sci 516:422–441
Wang N, Miao H, Li YS, Zhang P, Haga JH, Hu Y, Young A, Yuan S, Nguyen P, Wu CC et al (2006) Shear stress regulation of Kruppel-like factor 2 expression is flow pattern-specific. Biochem Biophys Res Commun 341:1244–1251
Raffetto JD, Khalil RA (2008) Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol 75:346–359
Shyy JY, Li YS, Lin MC, Chen W, Yuan S, Usami S, Chien S (1995) Multiple cis-elements mediate shear stress-induced gene expression. J Biomech 28:1451–1457
Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437:426–431
Chiu JJ, Wang DL, Chien S, Skalak R, Usami S (1998) Effects of disturbed flow on endothelial cells. J Biomech Eng 120:2–8
Chien S (2008) Effects of disturbed flow on endothelial cells. Ann Biomed Eng 36:554–562
Matthews BD, Overby DR, Mannix R, Ingber DE (2006) Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J Cell Sci 119:508–518
Li S, Chen BP, Azuma N, Hu YL, Wu SZ, Sumpio BE, Shyy JY, Chien S (1999) Distinct roles for the small GTPases Cdc42 and Rho in endothelial responses to shear stress. J Clin Invest 103:1141–1150
Tzima E (2006) Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ Res 98:176–185
Galbraith CG, Skalak R, Chien S (1998) Shear stress induces spatial reorganization of the endothelial cell cytoskeleton. Cell Motil Cytoskeleton 40:317–330
Boon RA, Horrevoets AJ (2009) Key transcriptional regulators of the vasoprotective effects of shear stress. Hamostaseologie 29, 39–40, 41–43
Mohan S, Mohan N, Sprague EA (1997) Differential activation of NF-kappa B in human aortic endothelial cells conditioned to specific flow environments. Am J Physiol 273:C572–C578
Hosoya T, Maruyama A, Kang MI, Kawatani Y, Shibata T, Uchida K, Warabi E, Noguchi N, Itoh K, Yamamoto M (2005) Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J Biol Chem 280:27244–27250
Halka AT, Turner NJ, Carter A, Ghosh J, Murphy MO, Kirton JP, Kielty CM, Walker MG (2008) The effects of stretch on vascular smooth muscle cell phenotype in vitro. Cardiovasc Pathol 17:98–102
Qiu J, Zheng Y, Hu J, Liao D, Gregersen H, Deng X, Fan Y, Wang G (2014) Biomechanical regulation of vascular smooth muscle cell functions: from in vitro to in vivo understanding. J R Soc Interface 11:20130852
Ramella M, Bertozzi G, Fusaro L, Talmon M, Manfredi M, Catoria MC, Casella F, Porta CM, Boldorini R, Fresu LG et al (2019) Effect of cyclic stretch on vascular endothelial cells and abdominal aortic aneurysm (AAA): role in the inflammatory response. Int J Mol Sci 20:287
Lemarie CA, Tharaux PL, Lehoux S (2010) Extracellular matrix alterations in hypertensive vascular remodeling. J Mol Cell Cardiol 48:433–439
Li C, Xu Q (2000) Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal 12:435–445
Haga JH, Li YS, Chien S (2007) Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech 40:947–960
Birukov KG, Bardy N, Lehoux S, Merval R, Shirinsky VP, Tedgui A (1998) Intraluminal pressure is essential for the maintenance of smooth muscle caldesmon and filamin content in aortic organ culture. Arterioscler Thromb Vasc Biol 18:922–927
Verma RP, Hansch C (2007) Matrix metalloproteinases (MMPs): chemical-biological functions and (Q)SARs. Bioorg Med Chem 15:2223–2268
Asanuma K, Magid R, Johnson C, Nerem RM, Galis ZS (2003) Uniaxial strain upregulates matrix-degrading enzymes produced by human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 284:H1778–H1784
Chapman GB, Durante W, Hellums JD, Schafer AI (2000) Physiological cyclic stretch causes cell cycle arrest in cultured vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 278:H748–H754
Liu XM, Ensenat D, Wang H, Schafer AI, Durante W (2003) Physiologic cyclic stretch inhibits apoptosis in vascular endothelium. FEBS Lett 541:52–56
Birukov KG, Jacobson JR, Flores AA, Ye SQ, Birukova AA, Verin AD, Garcia JG (2003) Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol 285:L785–L797
Pfisterer L, Feldner A, Hecker M, Korff T (2012) Hypertension impairs myocardin function: a novel mechanism facilitating arterial remodelling. Cardiovasc Res 96:120–129
Li C, Wernig F, Leitges M, Hu Y, Xu Q (2003) Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J 17:2106–2108
Gawlak G, Son S, Tian Y, O’Donnell JJ 3rd, Birukov KG, Birukova AA (2016) Chronic high-magnitude cyclic stretch stimulates EC inflammatory response via VEGF receptor 2-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 310:L1062–L1070
Esteller M (2002) CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21:5427–5440
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28:1057–1068
Deplus R, Brenner C, Burgers WA, Putmans P, Kouzarides T, de Launoit Y, Fuks F (2002) Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res 30:3831–3838
Esteller M, Herman JG (2002) Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J Pathol 196:1–7
Weinhold B (2006) Epigenetics: the science of change. Environ Health Perspect 114:A160–A167
Reik W, Dean W, Walter J (2001) Epigenetic reprogramming in mammalian development. Science 293:1089–1093
Rao X, Zhong J, Zhang S, Zhang Y, Yu Q, Yang P, Wang MH, Fulton DJ, Shi H, Dong Z et al (2011) Loss of methyl-CpG-binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation 123:2964–2974
Greissel A, Culmes M, Napieralski R, Wagner E, Gebhard H, Schmitt M, Zimmermann A, Eckstein HH, Zernecke A, Pelisek J (2015) Alternation of histone and DNA methylation in human atherosclerotic carotid plaques. Thromb Haemost 114:390–402
Wierda RJ, Rietveld IM, van Eggermond MC, Belien JA, van Zwet EW, Lindeman JH, van den Elsen PJ (2015) Global histone H3 lysine 27 triple methylation levels are reduced in vessels with advanced atherosclerotic plaques. Life Sci 129:3–9
Liu Y, Peng W, Qu K, Lin X, Zeng Z, Chen J, Wei D, Wang Z (2018) TET2: a novel epigenetic regulator and potential intervention target for atherosclerosis. DNA Cell Biol 37:517–523
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Sterner DE, Berger SL (2000) Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64:435–459
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42
Witt O, Deubzer HE, Milde T, Oehme I (2009) HDAC family: what are the cancer relevant targets? Cancer Lett 277:8–21
Delcuve GP, Khan DH, Davie JR (2012) Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics 4:5
Wang GG, Allis CD, Chi P (2007) Chromatin remodeling and cancer, Part I: Covalent histone modifications. Trends Mol Med 13:363–372
Haigis MC, Guarente LP (2006) Mammalian sirtuins–emerging roles in physiology, aging, and calorie restriction. Genes Dev 20:2913–2921
Wade PA (2001) Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum Mol Genet 10:693–698
Wang Y, Miao X, Liu Y, Li F, Liu Q, Sun J, Cai L (2014) Dysregulation of histone acetyltransferases and deacetylases in cardiovascular diseases. Oxid Med Cell Longev 2014:641979
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–784
Xu WS, Parmigiani RB, Marks PA (2007) Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 26:5541–5552
Kim HJ, Bae SC (2011) Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res 3:166–179
Holoch D, Moazed D (2015) RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet 16:71–84
Stefani G, Slack FJ (2008) Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 9:219–230
Mattick JS (2009) The genetic signatures of noncoding RNAs. PLoS Genet 5:e1000459
Holoch D, Moazed D (2015) Small-RNA loading licenses Argonaute for assembly into a transcriptional silencing complex. Nat Struct Mol Biol 22:328–335
Erwin JA, Lee JT (2008) New twists in X-chromosome inactivation. Curr Opin Cell Biol 20:349–355
Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469:336–342
Weber C, Schober A, Zernecke A (2010) MicroRNAs in arterial remodelling, inflammation and atherosclerosis. Curr Drug Targets 11:950–956
Chen LJ, Chuang L, Huang YH, Zhou J, Lim SH, Lee CI, Lin WW, Lin TE, Wang WL, Chen L, Chien S, Chiu JJ (2015) MicroRNA mediation of endothelial inflammatory response to smooth muscle cells and its inhibition by atheroprotective shear stress. Circ Res 116:1157–1169
Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D (2008) miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 15:272–284
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ (2008) MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci U S A 105:1516–1521
Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460:705–710
Holdt LM, Beutner F, Scholz M, Gielen S, Gabel G, Bergert H, Schuler G, Thiery J, Teupser D (2010) ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol 30:620–627
Zhou X, Han X, Wittfeldt A, Sun J, Liu C, Wang X, Gan LM, Cao H, Liang Z (2016) Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-kappaB pathway. RNA Biol 13:98–108
Congrains A, Kamide K, Ohishi M, Rakugi H (2013) ANRIL: molecular mechanisms and implications in human health. Int J Mol Sci 14:1278–1292
Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y et al (2014) LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation 130:1452–1465
Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S et al (2014) Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res 114:1389–1397
Cremer S, Michalik KM, Fischer A, Pfisterer L, Jae N, Winter C, Boon RA, Muhly-Reinholz M, John D, Uchida S et al (2019) Hematopoietic deficiency of the long noncoding RNA MALAT1 promotes atherosclerosis and plaque inflammation. Circulation 139:1320–1334
Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C et al (2014) Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest 124:3187–3199
Zhang YP, Huang YT, Huang TS, Pang W, Zhu JJ, Liu YF, Tang RZ, Zhao CR, Yao WJ, Li YS et al (2017) The mammalian target of rapamycin and DNA methyltransferase 1 axis mediates vascular endothelial dysfunction in response to disturbed flow. Sci Rep 7:14996
Jiang YZ, Jimenez JM, Ou K, McCormick ME, Zhang LD, Davies PF (2014) Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res 115:32–43
Yang Q, Li X, Li R, Peng J, Wang Z, Jiang Z, Tang X, Peng Z, Wang Y, Wei D (2016) Low shear stress inhibited endothelial cell autophagy through TET2 downregulation. Ann Biomed Eng 44:2218–2227
Zeng L, Zhang Y, Chien S, Liu X, Shyy JY (2003) The role of p53 deacetylation in p21Waf1 regulation by laminar flow. J Biol Chem 278:24594–24599
Lee DY, Lee CI, Lin TE, Lim SH, Zhou J, Tseng YC, Chien S, Chiu JJ (2012) Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc Natl Acad Sci U S A 109:1967–1972
Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe AE, Wang G, Habi O, deFalco E et al (2010) Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation 121:132–142
Yan ZQ, Yao QP, Zhang ML, Qi YX, Guo ZY, Shen BR, Jiang ZL (2009) Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J Biomech 42:945–948
Usui T, Okada M, Hara Y, Yamawaki H (2011) Exploring calmodulin-related proteins, which mediate development of hypertension, in vascular tissues of spontaneous hypertensive rats. Biochem Biophys Res Commun 405:47–51
Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin ZG (2010) Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115:2971–2979
Wang YH, Yan ZQ, Qi YX, Cheng BB, Wang XD, Zhao D, Shen BR, Jiang ZL (2010) Normal shear stress and vascular smooth muscle cells modulate migration of endothelial cells through histone deacetylase 6 activation and tubulin acetylation. Ann Biomed Eng 38:729–737
Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY (2010) Shear stress, SIRT1, and vascular homeostasis. Proc Natl Acad Sci U S A 107:10268–10273
Chen W, Bacanamwo M, Harrison DG (2008) Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J Biol Chem 283:16293–16298
Nicoli S, Standley C, Walker P, Hurlstone A, Fogarty KE, Lawson ND (2010) MicroRNA-mediated integration of haemodynamics and Vegf signalling during angiogenesis. Nature 464:1196–1200
Ni CW, Qiu H, Jo H (2011) MicroRNA-663 upregulated by oscillatory shear stress plays a role in inflammatory response of endothelial cells. Am J Physiol Heart Circ Physiol 300:H1762–H1769
Zhou J, Wang KC, Wu W, Subramaniam S, Shyy JY, Chiu JJ, Li JY, Chien S (2011) MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc Natl Acad Sci U S A 108:10355–10360
Fang Y, Shi C, Manduchi E, Civelek M, Davies PF (2010) MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci U S A 107:13450–13455
Lee DY, Lin TE, Lee CI, Zhou J, Huang YH, Lee PL, Shih YT, Chien S, Chiu JJ (2017) MicroRNA-10a is crucial for endothelial response to different flow patterns via interaction of retinoid acid receptors and histone deacetylases. Proc Natl Acad Sci U S A 114:2072–2077
Lee DY, Yang TL, Huang YH, Lee CI, Chen LJ, Shih YT, Wei SY, Wang WL, Wu CC, Chiu JJ (2018) Induction of microRNA-10a using retinoic acid receptor-alpha and retinoid x receptor-alpha agonists inhibits atherosclerotic lesion formation. Atherosclerosis 271:36–44
Wu W, Xiao H, Laguna-Fernandez A, Villarreal G Jr, Wang KC, Geary GG, Zhang Y, Wang WC, Huang HD, Zhou J et al (2011) Flow-dependent regulation of Kruppel-like factor 2 is mediated by microRNA-92a. Circulation 124:633–641
Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, Li YS, Chien S, Wang N (2010) MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci U S A 107:3240–3244
Wang KC, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, Wang N, Shyy JY, Li YS, Chien S (2010) Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc Natl Acad Sci U S A 107:3234–3239
Chen K, Fan W, Wang X, Ke X, Wu G, Hu C (2012) MicroRNA-101 mediates the suppressive effect of laminar shear stress on mTOR expression in vascular endothelial cells. Biochem Biophys Res Commun 427:138–142
Mohamed JS, Lopez MA, Boriek AM (2010) Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3beta. J Biol Chem 285:29336–29347
Wu WH, Hu CP, Chen XP, Zhang WF, Li XW, Xiong XM, Li YJ (2011) MicroRNA-130a mediates proliferation of vascular smooth muscle cells in hypertension. Am J Hypertens 24:1087–1093
Yu ML, Wang JF, Wang GK, You XH, Zhao XX, Jing Q, Qin YW (2011) Vascular smooth muscle cell proliferation is influenced by let-7d microRNA and its interaction with KRAS. Circ J 75:703–709
Huang K, Bao H, Yan ZQ, Wang L, Zhang P, Yao QP, Shi Q, Chen XH, Wang KX, Shen BR et al (2017) MicroRNA-33 protects against neointimal hyperplasia induced by arterial mechanical stretch in the grafted vein. Cardiovasc Res 113:488–497
Hergenreider E, Heydt S, Treguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M et al (2012) Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol 14:249–256
Zhu JJ, Liu YF, Zhang YP, Zhao CR, Yao WJ, Li YS, Wang KC, Huang TS, Pang W, Wang XF et al (2017) VAMP3 and SNAP23 mediate the disturbed flow-induced endothelial microRNA secretion and smooth muscle hyperplasia. Proc Natl Acad Sci U S A 114:8271–8276
Leisegang MS, Bibli SI, Gunther S, Pfluger-Muller B, Oo JA, Hoper C, Seredinski S, Yekelchyk M, Schmitz-Rixen T, Schurmann C et al (2019) Pleiotropic effects of laminar flow and statins depend on the Kruppel-like factor-induced lncRNA MANTIS. Eur Heart J 40:2523–2533
Lund G, Andersson L, Lauria M, Lindholm M, Fraga MF, Villar-Garea A, Ballestar E, Esteller M, Zaina S (2004) DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 279:29147–29154
Hiltunen MO, Turunen MP, Hakkinen TP, Rutanen J, Hedman M, Makinen K, Turunen AM, Aalto-Setala K, Yla-Herttuala S (2002) DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med 7:5–11
Zhong W, Li B, Xu Y, Yang P, Chen R, Wang Z, Shao C, Song J, Yan J (2018) Hypermethylation of the micro-RNA 145 promoter is the key regulator for NLRP3 inflammasome-induced activation and plaque formation. JACC Basic Transl Sci 3:604–624
Tai SC, Robb GB, Marsden PA (2004) Endothelial nitric oxide synthase: a new paradigm for gene regulation in the injured blood vessel. Arterioscler Thromb Vasc Biol 24:405–412
Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM, Francis J (2010) HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension 56:437–444
Stein S, Matter CM (2011) Protective roles of SIRT1 in atherosclerosis. Cell Cycle 10:640–647
Balligand JL, Feron O, Dessy C (2009) eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol Rev 89:481–534
Hunter MP, Ismail N, Zhang X, Aguda BD, Lee EJ, Yu L, Xiao T, Schafer J, Lee ML, Schmittgen TD et al (2008) Detection of microRNA expression in human peripheral blood microvesicles. PLoS One 3:e3694
Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A et al (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A 105:10513–10518
Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL et al (2011) Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A 108:5003–5008
Noferesti SS, Sohel MM, Hoelker M, Salilew-Wondim D, Tholen E, Looft C, Rings F, Neuhoff C, Schellander K, Tesfaye D (2015) Controlled ovarian hyperstimulation induced changes in the expression of circulatory miRNA in bovine follicular fluid and blood plasma. J Ovarian Res 8:81
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659
Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X et al (2008) Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res 18:997–1006
Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, Benjamin H, Kushnir M, Cholakh H, Melamed N et al (2008) Serum microRNAs are promising novel biomarkers. PLoS One 3:e3148
Sisco KL (2001) Is RNA in serum bound to nucleoprotein complexes? Clin Chem 47:1744–1745
El-Hefnawy T, Raja S, Kelly L, Bigbee WL, Kirkwood JM, Luketich JD, Godfrey TE (2004) Characterization of amplifiable, circulating RNA in plasma and its potential as a tool for cancer diagnostics. Clin Chem 50:564–573
Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh MN, Lutgens E et al (2009) Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal 2:ra81
Pfeifer P, Werner N, Jansen F (2015) Role and function of microRNAs in extracellular vesicles in cardiovascular biology. Biomed Res Int 2015:161393
Khyzha N, Alizada A, Wilson MD, Fish JE (2017) Epigenetics of atherosclerosis: emerging mechanisms and methods. Trends Mol Med 23:332–347
Arslan S, Berkan O, Lalem T, Ozbilum N, Goksel S, Korkmaz O, Cetin N, Devaux Y, Cardiolinc n (2017) Long non-coding RNAs in the atherosclerotic plaque. Atherosclerosis 266:176–181
Man HSJ, Sukumar AN, Lam GC, Turgeon PJ, Yan MS, Ku KH, Dubinsky MK, Ho JJD, Wang JJ, Das S et al (2018) Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc Natl Acad Sci U S A 115:2401–2406
Josipovic I, Pfluger B, Fork C, Vasconez AE, Oo JA, Hitzel J, Seredinski S, Gamen E, Heringdorf DMZ, Chen W et al (2018) Long noncoding RNA LISPR1 is required for S1P signaling and endothelial cell function. J Mol Cell Cardiol 116:57–68
Raines EW (2000) The extracellular matrix can regulate vascular cell migration, proliferation, and survival: relationships to vascular disease. Int J Exp Pathol 81:173–182
Acknowledgments
This work was supported by the Taiwan Ministry of Science and Technology grants MOST-108-2321-B-400-001 and 106-2320-B-400-030-MY3.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors declare that there are no conflicts of interest.
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wei, SY., Chiu, JJ. (2021). Mechanical Regulation of Epigenetic Modifications in Vascular Biology and Pathobiology. In: Hecker, M., Duncker, D.J. (eds) Vascular Mechanobiology in Physiology and Disease. Cardiac and Vascular Biology, vol 8. Springer, Cham. https://doi.org/10.1007/978-3-030-63164-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-63164-2_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-63163-5
Online ISBN: 978-3-030-63164-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)