
Abstract
Endothelium of blood vessels is continuously exposed to various hemodynamic forces. Flow-mediated epigenetic plasticity regulates vascular endothelial function. Recent studies have highlighted the significant role of mechanosensing-related epigenetics in localized endothelial dysfunction and the regional susceptibility for lesions in vascular diseases. In this article, we review the epigenetic mechanisms such as DNA de/methylation, histone modifications, as well as non-coding RNAs in promoting endothelial dysfunction in major arterial and venous diseases, consequent to hemodynamic alterations. We also discuss the current challenges and future prospects for the use of mechanoepigenetic mediators as biomarkers of early stages of vascular diseases and dysregulated mechanosensing-related epigenetic regulators as therapeutic targets in various vascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Mechanosensory transduction and epigenetic alterations are two significant extra-genomic components that substantially modify the inherent gene expression of vascular endothelial cells (ECs). Mechanical stimuli due to blood flow play a vital role in cell homeostasis in vascular ECs. Cellular response to atypical hemodynamic signals is now emerging as a significant pathological aspect especially pertinent to the cardiovascular system [1]. Mechanical forces control the morphogenesis of the vascular system and are rationally emerging as determinants in the pathophysiology of most of the vascular diseases.
Blood flow-induced epigenetic regulation has gained much attention as master regulators of vascular diseases in the last few years [2]. Epigenetic signatures define all the heritable gene expression changes other than the inherent nucleotide sequence and genome-wide alterations made to DNA and the supporting nucleoprotein scaffold. Epigenome landscape is dynamically regulated in spatiotemporal pattern, unlike DNA coding sequences and hence they possess a key role in determining phenotypic flexibility of cells in a context-dependent manner. Alteration of properties such as DNA de/methylation, histone acetylation/ methylation, and non-coding RNAs (ncRNAs) are some of the epigenetic gene expression altering mechanisms known so far [3]. The novel concept of ‘mechanoepigenetics’ was proposed by Missirlis in 2016 to describe the association of mechanical signaling with epigenetic regulation on chromatin [4]. Considering the extraordinary advances in the interactions between hemodynamics and epigenetic modulation hallmarking vascular diseases, our understanding of mechanotransduction-regulated epigenetics needs to be expanded, for the discovery of novel biomarkers and to enable new approaches for precision medicine.
Mechanosensing and signal transduction in human vasculature
Vascular beds are inconsistent and not uniformly organized through the body. The systemic vascular network system consists of the aorta which carries oxygenated blood from the heart. The smaller vessels, arterioles branch from the aorta and channel blood to the capillary networks. Through the capillaries exchange of products from the blood within tissues occurs. Venules, the smaller vein vessels, carry blood from the capillaries and transfer to the larger veins which return blood to the heart for pulmonary circulation.
The luminal endothelial layer in arterial and venous systems maintains homeostasis by sensing the flow forces and conducting mechanical, endocrine, and paracrine signals to underlying smooth muscle cells (SMCs), adventitial fibroblasts, extracellular matrix, and circulating monocytes [5]. The endothelium is the anatomical barrier between the vessel wall and blood. Endothelium acts as a dynamic organ that plays a key role in maintaining vascular integrity in response to hemodynamic cues. Natural pulsatility and viscosity of blood flow cause circumferential stretch and the frictional force exerted by blood flow causes shear stress, both of which are normal to the blood vessel [6]. Both fluid-wall shear stress (WSS) and circumferential stretch can stimulate the release of biochemical signaling molecules that maintain the normal physiological function of vessels. As expected, these forces change specifically in relation with vessel diameter [7]. Higher the diameter, lesser the physiological WSS would be. Shear stress on specific vessel wall is calculated using the Hagen–Poiseuille formula: τ = 4μQ/πr3, where τ is the shear stress, μ is the blood viscosity, Q is the volumetric flow rate, and r is the lumen radius. WSS is expressed in units of force per unit area [N/m2 or Pascal or dyn/cm2, where 1 N/m2 = 1 Pa = 10 dyn/cm2].
In this review, mechanobiology of vascular ECs and epigenetic alterations will be discussed, considering the pivotal role of ECs compared to any other vascular cell types, in shear sensing. Considering the scarcity of systematic reviews in this topic, reports about hemodynamic alterations induced epigenetic changes in vasculature that were published predominantly in last 20 years, and a few earlier pioneer papers were collected from databases such as PubMed, Medline, and Scopus. The major keywords used included hemodynamics, shear stress, hemodynamics in endothelial cells, shear induced epigenetic alterations, atherosclerosis, aneurysms, pulmonary vascular diseases, venous diseases, varicose veins, vein graft disease, and mechano-miRs. Mechanosensors on ECs can sense the slightest alterations in blood flow-related mechanical forces and transmit them by biochemical signaling to neighboring cells by a process called mechanotransduction [8]. Fluid mechanical forces in the physiological range result in vascular homeostasis, while pathophysiological mechanical forces result in vascular remodeling, prominent in several cardiovascular diseases.
A set of proteins has been proposed to orchestrate mechanosensing and signal transmission in vascular endothelium. Endothelial cells respond to changes of tension by activating or inactivating signaling pathways mediated by small GTPase, transcription factors, and ncRNAs.
The endothelium is phenotypically heterogeneous across the vascular network [9]. They exhibit a great range of structural and functional heterogeneity. The typical spatiotemporal differences in the extracellular microenvironment affect the homogeneity of the endothelium. Some of these EC heterogenous parameters seem to be inherently and epigenetically programmed; once set, they are independent of further cues from the extracellular environment. For instance, DNA microarray studies of arterial and venous ECs suggest an inherent capacity of arterial ECs to express genes associated with adhesion, proliferation, and apoptotic pathways in contrast to human saphenous vein ECs [10]. EC heterogeneity is all the more perceptible in arteries and veins due to the major difference between the blood flow rates and pressure parameters as well as due to distinct architecture [5]. Even the well-known vascular EC markers such as cell adhesion molecule PECAM-1 and protein tyrosine kinase Tie2 exhibit heterogeneity under different flow patterns [11, 12]. We have inadequate knowledge, however, of the mechanobiological alterations that cause vascular diseases. An improved understanding of the mechanobiology of the arterial and venous systems could lead to further insights on the flow-related epigenetic dysregulation in arterial and venous diseases.
Arterial mechanobiology
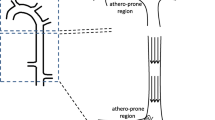
Arterial architecture greatly varies at sites of bifurcations, branch points, and curved regions. These terrains induce changes in flow patterns, which have vast effects on the vascular cells especially ECs. ECs in these regions experience low WSS due to complex flow patterns which include blood flow reversal [13]. This is associated with a high susceptibility for developing atherosclerotic lesions and plaque formation. Blood flow is pulsatile with distinct characteristics depending on the location of an artery. Flow can be laminar and unidirectional as well as laminar with oscillatory backflow. Some rare flow patterns such as multidirectional and turbulent also exist. Multidirectional flows occur at blood vessel bifurcations or other irregularities, while turbulence occurs near the aortic valve or in more severe disease conditions [14]. The possible role of hemodynamic forces in endothelial dysfunction was first documented in the 1960s by the observation that the initial lesions of atherosclerosis occur at arterial branches and curvatures where the blood flow is disturbed [15]. This led to a series of studies in flow mechanics in arterial vasculature [16,17,18].
Regardless of its relatively low magnitude compared to other forces, WSS is a key determinant of both physiological and pathological vascular remodeling. Arterial shear stresses range from 10 dyn/cm2 in the aorta to approximately 50 dyn/cm2 in smaller arterioles [19]. The physiological range of time-averaged WSS in straight arteries is considered as baseline WSS which is usually between 15 and 25 dyn/cm2 [20]. Steady pulsatile laminar flow in a physiological range in arteries stimulate the production of factors that support EC survival, quiescence and barrier function, and also prevalent coagulation, and proliferation of vascular VSMCs. The branch point and curvature of the artery experience cardiac cycle-dependent re-circulating non-unidirectional ‘disturbed’ or oscillatory flow with low WSS in the range of 1–4 dyn/cm2 [20, 21]. These areas are mostly found to be lesion-prone.
Investigations aimed at defining all major shear stress-responsive elements in vascular ECs are ongoing. Olesen et al. provided the first evidence for WSS modulation of ion channels in endothelia [22]. They found that shear stress activates a potassium channel in ECs. Alterations of ion channel activity represent an early response in the signaling cascade, at least in the case of endothelial mechanosensing. Ion channels in ECs control Ca2 + influx either by Ca2 + influx pathway or by regulating K + and Cl− channels. Caveolae, glycocalyx, occludin, focal adhesion complexes, etc., are the most studied mechanosensors. Caveolae are plasma membrane microdomains involved in Ca2 + signaling and mechanotransduction [23]. Endothelial cells of the vascular lumen are covered by endothelial glycocalyx. It is composed of membrane-bound negatively charged proteoglycans, glycoproteins, glycolipids, and glycosaminoglycans. Glycocalyx is known as the ‘gatekeeper of endothelial cells’ since it is the key determinant of vascular permeability while being a sensor plus transducer of WSS [24]. Occludin is another EC mechanosensor which is a type of tight junction protein that carries a rate-limiting transport within the intercellular cleft [25]. Focal adhesions are integrin containing flow sensors through which mechanical force and regulatory signals are transmitted between the endothelial cytoskeleton and extracellular matrix (ECM) [26]. The various mechanosensors in ECs are shown in Fig. 1.

Important mechanosensors in vascular endothelial cells. Primary mechanosensors on endothelial membrane such as Occludin, G-protein-coupled receptors (GPCRs), Caveolae, Primary cilium, Glycocalyx, Ion channels, PECAM-1 (CD31), VE-cadherin, VEGFR-2, and Integrins trigger upstream signal transducers and maintain endothelial homeostasis in response to unidirectional blood flow
Proteins such as Kruppel-like factor (KLF) 2 and 4, Hypoxia-inducible factor 1-alpha (HIF-1α), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Yes-associated protein (YAP)/transcriptional coactivator with a PDZ-binding domain (TAZ) are known to be involved in biochemical and biomechanical stimuli transduction and are the crucial mechanotransducers [27].
The most prominent among the mechanotransducers is transcription factor KLF2 which has a vascular protective role [28] and gets upregulated in response to laminar flow as shown in Fig. 2. Mechanosensing pathways in ECs are also reported to activate the master signaling MAPK and PI3K/Akt pathways in the face of unidirectional flow.

Regulation of KLF2 under laminar blood flow. KLF2 activates eNOS and antioxidant Nrf2 which prevent inflammatory signaling cascade. KLF2 is involved in the downregulation of pro-inflammatory, prothrombotic, and vasoconstrictive genes such as vascular cell adhesion molecule 1 (VCAM-1), Monocyte chemoattractant protein-1 (MCP-1), E-Selectin, endothelin-1 (ET-1), and plasminogen activator inhibitor-1(PAI-1)
Studies on how ECs perceive the direction of blood flow are very poorly understood. Recent studies suggest that plaque localization correlates more strongly with the transverse WSS than with oscillatory flow as thought earlier [29]. Substantiating this hypothesis, studies have shown that ECs in plaque prone arterial regions fail to align in the direction of blood flow, exposing themselves to perpendicular WSS, whereas in atheroresistant parts of arteries, flow is parallel to the cell axis [14].
Venous mechanobiology
Due to their pivotal role in standing blood pressure, arteries and arterioles have received the major attention in earlier mechanobiological studies. Nevertheless, venous ECs also undergo dynamic changes due to mechanical cues. Veins such as saphenous veins and their tributaries which act against gravity require active one way-valves and skeletal muscle pumping systems to avoid backflow, blood stasis, and resultant turbulent flow. Veins near the heart do not need strict measures and have more or less laminar parallel flow. ECs in veins and venules are also hypothesized to have similar mechanosensory transduction pathways as in arteries [30]. Blood pressure on thin-walled veins causes circumferential stretch, while the regular passage of blood through the vessels results in the frictional force called WSS. Venous circulation has a non-pulsatile quasi-steady flow pattern and WSS is lower than in arteries, ranging from 1–6 dyn/cm2 in the veins of different diameters with as little as 1 dyn/cm2 in the vena cava [21]. Notably, shear stress in small venules is comparably high (20- 40 dyn/cm2) due to higher flow rates and small diameters of these vessels [7, 31]. Venous valves protrude from the luminal endothelium and extend as long thin cusps or flaps. They are generally bicuspid, although unicuspid and tricuspid venous valves are also there [32]. These valves allow the directional flow of blood toward heart, and prevent any backflow.
Studies on how circumferential stretch is detected by cells and transmitted in the vein wall have reported the presence of ion channels, glycocalyx, G-protein-coupled receptors (GPCR) and integrins in vein walls which respond to circumferential stretch [30]. Mechanical sensors in such as integrin α2, α5, αvβ3, normally present in arterioles have been identified in ECs from saphenous veins as well. Further, mechanical stretch on the thin vein wall induces the production of matrix metalloproteinases and associated vascular remodeling via integrin αv [33].
Hemodynamic variations in the venous system are considered as one of the inductive factors for inflammation in veins [34]. But the exact reason for the hemodynamic variation is still unknown. Disturbed flow in primary veins such as saphenous vein can occur at initial stages due to flow disturbance at vein valve sinuses [21]. Altered flow can also occur due to blood reflux through incompetent valves in patients carrying polymorphisms in genes coding for valve maintenance, as observed in our earlier studies on the FoxC2 gene [35].
Vein ECs are exposed to very low WSS compared to arterial ECs. Interestingly, endothelial homeostasis is still maintained in the venous system even under long-term exposure to low WSS. Earlier studies in atherogenesis using HUVEC cells indicated that low shear per se is pathophysiological. However, venous landscape indicates that unidirectional laminar flow at physiological shear always results in stable endothelial function, whereas disturbance or turbulence in the flow triggers a pathological cascade. Understanding the role of WSS-related sensors and signaling transducers in the vein wall will provide new molecular targets for the early diagnosis and treatment of venous diseases. Considering the spatiotemporal attributes of various vein diseases such as varicose veins and deep vein thrombosis, further studies on venous mechanobiology are warranted.
Mechanoepigenetic regulators in vascular ECs
Epigenetic programming by hemodynamic factors includes three highly interrelated mechanisms, such as DNA methylation, histone modifications and ncRNAs including microRNAs (Fig. 3). Epigenetic regulation results in multidimensional gene expression patterns. A major breakthrough in dissecting the epigenetic profile of the vascular endothelium was made by the development of the project ‘Encyclopedia of DNA elements’ (ENCODE). By comparative studies using epigenome datasets from ENCODE, several EC-specific promoters and enhancers sequences have been identified [36]. Most of such sequences are associated with genes that are enriched for angiogenesis and blood vessel morphogenesis.

Mechanoepigenetic regulators activated in vascular endothelial cells in response to disturbed blood flow. DNMT DNA methyltransferase, HDAC histone deacetylase, lncRNA long non-coding RNA, miR microRNA. Me denotes methylated CpG islands, Ac denotes acetylated histone proteins
DNA methylation
DNA methylation is the addition of a methyl group to the 5′ position of cytosine residues at cytosine preceding guanosine (CpG) islands. Methylation of CpG islands near gene transcription initiation sites results in the chromatin condensation and inactivation of gene transcription. DNA methylation is regulated by a family of DNMTs (DNA methyl transferases) including DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L [37].
In recent years, several research groups have reported that DNMTs are shear-responsive enzymes that tightly regulate flow-mediated EC gene expression [38, 39]. There is mounting evidence on the importance of shear-responsive DNMTs and global DNA methylation in controlling global gene expression during endothelial dysfunction associated with disturbed flow in arteries [38, 40, 41]. DNMT1 expression is induced by oscillatory WSS in cultured endothelial cells. Oscillation-related endothelial inflammation can be reversed by inhibition of DNMT with either 5-Aza-2′-deoxycytidine (5-Aza) or DNMT1 siRNA in disturbed flow terrains [39].
Disturbed blood flow suppresses atheroprotective shear-responsive KLF2 and KLF4 in vascular ECS [28]. KLF4 repression is mediated by hypermethylation of its promoter, in a DNMT3a-dependent method [38]. Systems biological, DNA methylome and transcriptome analysis have revealed that CRE methylation is regulated genome-wide, in a flow-sensitive mode. CREs (cAMP response elements) have a major role in the differential expression of several gene pathways. CREs located specifically in gene promoters in ECs are hypermethylated by the disturbed flow. These findings indicate that genome-wide CRE methylation is one of the prospective mechanisms by which disturbed flow regulates gene expression. Therefore, CRE-containing flow-sensitive genes such as HoxA5, Klf3, Cmklr1, Acvrl1, and Spry2 can serve as targets for further studies in EC flow sensing and reverse programming [39].
TET-mediated DNA demethylation
Demethylation counteracts DNMT functions and activates silenced genes. DNA demethylation is catalyzed by TET methylcytosine dioxygenases such as TET1, TET2, and TET3, which converts 5-methylcytosine (5-mc) into 5-hydroxymethylcytosine (5-hmc) [42]. TET2 (Tet Methyl cytosine Dioxygenase 2) expression in endothelial cells is downregulated by disturbed blood flow in vitro as well as in atherosclerotic lesions in vivo [43]. Expression of TET2-dependent autophagic markers is also reduced in EC exposed to disturbed flow. Investigations in this area are at a relatively nascent stage. Yet it is clear that there exists a very fine balance between TETs and DNMTs and that any imbalance will result in vascular endothelial dysfunction due to major changes in EC gene expression.
Histone post-translational modifications
Histone post-translational modifications (HPTMs) are essential for epigenetic regulation of transcriptional expression and they occur primarily at amino acid residues of the N-terminal tails of histones that protrude from the chromatin fiber. Histone modifications remodel chromatin, either by altering chromatin structure making it accessible to transcriptional factors as in euchromatin, or inactivating heterochromatin to which transcriptional factors do not get access to bind with DNA [44]. A myriad of HPTMs is currently known. The most studied and defined histone modifications are histone lysine methylation and acetylation. Histone acetylation is synchronized by the action of two enzymes, histone acetyltransferases (HATs) and deacetylases (HDACs). These enzymes add and take out acetyl groups from lysine residues present on histone N-terminal tails. Histone methylation is regulated by histone methyltransferases which have the ability to remove methyl groups from lysine residues. This reaction can happen in other basic residues, such as arginines and histidines but not as frequently as in lysine. Histone methylation is known to have stringent gene specificity and hence considered to be a more permanent epigenetic mark.
Over the past few years, several efforts have been made to characterize the histone modifications in ECs [45, 46]. It is presently known that specific HDACs can promote and inhibit angiogenesis [47, 48]. ECs that are exposed to disturbed fluid flow have an increased HDAC3 expression [49]. In ApoE−/− rats, HDAC3 is overexpressed in branching areas and the curvature of the aortic arch where blood flow is disturbed [50]. HDAC3 interacts with transcriptional factors such as Nrf2 and MEF2, preventing them from activating the expression of anti-inflammatory and anti-oxidative genes and thus resulting in endothelial dysfunction under disturbed flow [51]. NAD-dependent histone deacetylase, SIRT1 is downregulated in atheroprone regions of arteries [52]. SIRT1, when expressed appropriately can activate transcriptional factors such as FoxO3a and PGC-1α and increase the expression of antioxidant genes in ECs [53]. These findings indicate that differential expression of HDACs can have divergent effects on transcription.
Histone methylation is important for maintaining endothelial cell homeostasis. Earlier studies have reported that the functions of endothelial cells are regulated by several histone methyltransferases (Suv39h1, EZH2) and demethylases (Jmjd2B, Jmjd3, PHF8) [54]. The role of histone methylation in disturbed flow conditions has not been well described. Studies to understand their context-dependent role in vascular pathologies is needed to identify ways to reverse histone reprogramming in pathological conditions.
Non-coding RNAs
NcRNAs are a cluster of RNAs that are transcribed from DNA but do not encode functional proteins. ncRNAs have important roles in EC epigenetic regulation with involvement in transcription as well as translation. They act as cis-acting silencers as well as trans-acting regulators of site-specific modification and imprinted gene-silencing. Based on the size, ncRNAs can be subdivided into 2 major subgroups (Fig. 4). They regulate gene expression modulating at transcriptional, post-transcriptional, and translational levels [55].

Classification of non-coding RNAs based on size
MicroRNAs (miRNAs/miRs) have been implicated in the regulation of global gene expression and they act mainly as post-transcriptional repressors. MiRNAs interact with the 3′-untranslated region (3′UTR) of specific target mRNAs causing mRNA degradation and translational inhibition. It has been earlier demonstrated that physiological pulsatile laminar flow regulates the expression of miRNAs in ECs [56, 57]. These flow-sensitive miRNAs are generally known as ‘mechano-miRs’. In 2010, for the first time, Shu Chien and colleagues reported mechano-miRs (miR-19a and 23b) in cultured ECs, which were exposed to pulsatile unidirectional flow in vitro [58, 59]. Peter Davies and colleagues in the same year reported miR-10a, the first mechano-miR identified in endothelium in vivo [60]. Since then, several miRNAs that have both pro/anti- inflammatory pro/antiatherogenic roles have been identified.
Hitherto, there is no clear consensus on how WSS alterations influence miRNA expression secretion within the vascular wall. Several miRNAs are regulated by shear stress-inducible transcription factor KLF2 [61]. Interestingly, Wu et al. reported that KLF2 is directly regulated by miR-92a as well [62]. miR-92a is overexpressed during oscillatory flow and its action ultimately lowers the expression of KLF2 and KLF4 [63]. Table 1 shows major miRNAs associated with KLF2 and these observations suggest intricate mechanisms of mechanotransduction comprising WSS master regulator KLF2 and miRNAs. Overall, effective modulation of flow-induced EC signaling is dependent on a feedback mechanism involving mechanoresponsive genes and ncRNAs.
Very recent studies have unraveled a repertoire of unique EC-enriched lncRNAs (Table 2). Jeffrey Man and colleagues in 2018 described Spliced-transcript endothelial-enriched lncRNA (STEEL). This is the first identified lncRNA in ECs. STEEL enhances EC turnover and migration and has a prominent role in blood vessel formation and maturation. STEEL was found to be decreased in vascular ECs subjected to atheroprotective unidirectional pulsatile flow. STEEL is found to negatively regulate the major mechanosensors, KLF2 and eNOS [64].
LEENE is another flow-responsive lncRNA that enhances eNOS expression and is overexpressed during pulsatile shear stress when compared to during oscillatory shear stress. Studies have shown that there are binding sites for KLF2 and KLF4 around the transcriptional start site of LEENE [65]. MANTIS, LISPR1, and LASSIE are the other lncRNAs that have vital roles in EC alignment in response to vascular protective laminar flow [66,67,68]. Most such lncRNAs are regulated by forces induced by blood flow, and they modulate the expression of mechanotransductory KLF2. Taken together, it is clear that disturbed flow modulates master shear stress regulator KLF2 and thus affects endothelial function and inflammation (Fig. 5).

Altered hemodynamics in the epigenetic regulation of KLF2. Epigenetic factors that reduce endothelial KLF2 expression in response to disturbed flow. Downregulation of KLF2 negatively affects vascular tone and endothelial antioxidant system while result in the augmentation of endothelial inflammation
Epigenetic regulators of mechanosensing in vascular diseases
Even the slightest disturbance in laminar flow results in the activation of signaling cascades that causes vascular dysfunction, inflammation, and injury and thus the development of several vascular pathologies. Altered hemodynamics imparts changes in endothelial cell behavior and subsequently increases the susceptibility for several arterial and venous diseases. Most of our knowledge regarding hemodynamic alterations and their effects on endothelial function has accrued from studies on atherosclerosis and coronary artery disease (CAD). There has been some contribution from studies on other vascular diseases such as aneurysms, pulmonary vascular diseases and venous diseases.
Atherosclerosis
Atherosclerosis is a chronic condition in which arteries stiffen due to the formation of fibrofatty plaques in the intima. Atherosclerosis predominantly develops in hotspots such as vessel bifurcations and branch points, where the dysfunctional ECs initiate plaque accumulation. Arterial sites with low shear stress (< 5 dyn/cm2) are therefore affected more [20, 21]. Fluid mechanical measurements at atherosclerotic lesion-prone areas have revealed that mean WSS is 4 dyn/cm2 in arterial sites exposed to disturbed blood flow and 12 dyn/cm2 in the straight segments of the arterial tree, where there is steady laminar flow [20]. In a healthy individual, blood flow at arterial bifurcations as in the common carotid artery and its internal and external branches split into two high-velocity bloodstreams close to the inner walls of the arteries. Even a small intimal plaque in patients with atherosclerotic risk factors causes hemodynamic alterations and concomitant re-circulating turbulent flow. WSS in the regions of atherosclerotic plaque depends on the degree of luminal narrowing and measures > 50 dyn/cm2 in humans [69]. Birchall et al. with the aid of computational fluid dynamics (CFD) studies demonstrated in patients that instantaneous WSS at the point of stenosis ranged from 1.6 × 103 dyn/ cm2 to even as high as 4.5 × 103 dyn/ cm2 in atherosclerotic carotid arteries with 50% to 90% stenosis [70]. Their observation spurred interest in the interrelated roles of hemodynamics and epigenetic modifications in atherogenesis. Most studies on mechanobiological factors and related epigenetic modification mechanisms have been carried out in animal models for atherosclerosis.
Flow-induced DNA methylation in the pathogenesis of atherosclerosis
Doyon Won et al. in 2007 reported that disturbed hemodynamic forces induce in vascular ECs, a unique pattern of gene expression that predisposes vulnerable arterial regions to dysfunction of eNOS and atherosclerotic lesions [71]. Seven years later, three research groups independently reported that DNMTs expression is significantly regulated by shear stress [38, 39, 72]. They further demonstrated that disturbed flow-mediated endothelial gene expression is regulated in a DNMT-dependent manner. Disturbed blood flow induces atherosclerosis via DNMT1 and DNMT3a-dependent promoter methylation of mechanosensitive Homeobox protein A5, KLF3, and KLF4 [38]. HoxA5 modulation by disturbed flow further promotes the transcription of pro-inflammatory genes [39]. Repression of KLF4 by promoter methylation induced by disturbed flow, in turn, affects downstream molecules such as nitric oxide synthase 3 (NOS3), thrombomodulin, and MCP-1 [38].
DNMTs are crucial among the several factors that determine the fine balance between pro and anti-atherogenesis. Jo et al. in 2015 reported that undisturbed flow reduces DNMT activity and results in decreased DNA methylation and expression of antiatherogenic genes [39]. In a murine model with partial ligation of the carotid artery, they observed that disturbed blood flow increases the expression of DNMT1 and methylates promoters of mechanosensitive master switches such as HoxA5 and KLF3. Methylation patterns in these genes were restored to normal by both DNMT inhibitor 5-aza-2′-deoxycytidine (5-Aza) and siDNMT.
Flow associated histone post-translational modification in atherosclerosis
Several studies have confirmed the role of HDACs and HATs in atherosclerosis. As early as 2003, Illi et al. using an in vitro flow model described the effect of laminar shear stress on epigenetic histone modification and chromatin structure, providing the molecular basis for shear mediated gene regulation in ECs [73]. Both histone H3 acetylation and phosphorylation are activated in HUVECs exposed to laminar shear stress. Later, Wang et al. demonstrated that parallel uniform flow-induced phosphorylation of HDAC5 Ser259/498 and induced the expression of KLF2 [74]. Oscillatory shear stress induces the expression of both class 1 and 2 HDACs and downregulation of KLF2 [51, 75]. Oscillatory flow induces the binding of HDACs 1,2,3 with Nrf2 and binding of HDACs 3,5,7 with myocyte enhancer factor-2. Deacetylation of these genes results in the downregulation of antioxidant gene NQO1 and KLF2, which are reversed in the presence of HDAC siRNA. Low oscillatory WSS -induced HDAC signaling and EC responses are mediated by phosphatidylinositol 3-kinase/Akt pathway. Lee and his group were pioneers in demonstrating the role of specific HDACs in regulating the inflammatory and proliferative responses of cells to oscillatory disturbed flow.
Endothelial mechanosensing ncRNAs in atherosclerosis
The mechano-miRs involved in the atherogenesis, generally known as athero-miRs, play a vital role in the initiation and progression of atherosclerotic plaques. Cumulative evidence indicates a pivotal role for mechano-miRs in blood flow-dependent endothelial dysfunction and atherosclerosis [76,77,78]. As most of the evidence for the role of athero-miRs were identified using in vitro flow chamber-based assays in ECs, it is imperative to validate them in vivo given that several of their target flow-responsive genes which were identified in vivo are known to be lost or become dysfunctional during endothelial cell culture. Based on their presumed as well as defined roles in atherogenesis, athero-miRNAs are categorized into proatherogenic (proathero-miRs), antiatherogenic (antiathero-miRs) and dual role mechano-miRs (Table 3).
Proathero-miRs
MiR-17–92 cluster comprising miR-17, 18a, 19a, 19b, 20a, and 92a are proatherogenic in their function. They are highly expressed both in vitro cultured ECs exposed to disturbed flow as well as in ECs in the athero-susceptible regions of the aortic arch in pigs [59, 62, 63]. Son et al. with the help of a partially ligated carotid artery model has demonstrated that disturbed flow induces the expression of miR-712 and that inhibitory anti-miR-712 prevents plaque development in animals [77]. Tissue inhibitor of metalloproteinase-3 (TIMP3) is the target of miR-712 and its binding inactivates TIMP3 and activates matrix metalloproteinases leading to endothelial inflammation and atherosclerosis. As evident from Table 2, most of the mechano-miRs regulate the action of mechanotransductory as well as inflammatory genes in vascular ECs.
Antiathero-miRs
As described earlier, miR-19a and miR-23b are overexpressed in cells subjected to laminar unidirectional flow and are considered to be atheroprotective. Mechanistically, miR-19a targets cyclin D1 and induces endothelial quiescence [58]. miR-23b expression induced by pulsatile WSS suppresses EC proliferation [59]. These findings are interesting as the disturbed flow is known to increase endothelial proliferation in atheroprone regions. miR-10a, another important miRNA is relatively diminished in the lesser curvature regions of the porcine aortic arch when compared to atheroprotected areas in the thoracic aorta. These regions in the aortic arch have disturbed blood flow [60]. Studies indicate that miR-10a is an anti-inflammatory miRNA that targets MAP3K7 and βTRC, both of which promote inhibitor κB degradation and p65 translocation and thereby inhibit activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) in ECs.
Dual role mechano-miRs
Some known mechano-miRs have both anti- and proatherogenic effects in vivo. It is plausible since a single miRNA can target numerous target mRNAs, which may have contrasting roles in the pathogenesis of atherosclerosis. miR-21, miR-126, and miR-155 are some of the dual role mechano-miRs which through independent in vivo experiments have been identified to possess atheroprotective as well as atherogenic effects [79]. The phenotypic response of the cell may hence depend on the magnitude of mechanical forces and also on the specific anatomical location. More studies are necessary to delineate the intricate atherogenic mechanisms related to ambiguous mechano-miRs.
Another type of ncRNAs involved in the progression of atherosclerosis is lncRNAs. LncRNAs have been recently implicated in several biological processes and diseases. Several lncRNAs have been identified as epigenetic regulators in vascular diseases. LncRNAs such as STEEL, MANTIS, LASSIE, LISPR1, H19, SENCR, RNCR3, MEG3 are some of the EC regulators known to be associated with atherosclerosis. There is sparse information on their role in the response of ECS to alterations in ECs and resulting EC dysfunction and atherosclerosis.
STEEL is an EC-specific atherogenic lncRNA that can transcriptionally reduce the expression of eNOS and KLF2 genes by increasing chromatin accessibility and histone methylation in gene promoters [64]. In ECs, in response to atheroprotective unidirectional pulsatile flow, there is reduced STEEL expression, which in turn leads to the upregulation of both KLF2 and eNOS. MANTIS is another important lncRNA [66]. MANTIS is controlled by the histone demethylase JARID1B (Jumonji AT-rich interactive domain 1B). MANTIS gets enhanced by atheroprotective flow and favor angiogenic function. MANTIS modulates shear stress-induced EC alignment and angiogenic sprouting. Josipovic et al. reported LISPR1 another lncRNA enriched in ECS. LISPR1 is expressed in an atheroprotective laminar flow-dependent manner [67]. LISPR1 regulates S1PR1 which is an endothelial barrier-promoting receptor and thus negatively regulates atherosclerosis. Lyu and colleagues identified another important lncRNA, SENCR (smooth muscle and endothelial cell-enriched migration/differentiation-associated) whose expression is promoted by laminar shear. SENCR is flow-responsive and contributes to the integrity of endothelial cells through physical association with CKAP4, thereby stabilizing CDH5, which is associated with the cell membrane [80]. RNA sequencing studies have recently identified yet another atheroprotective lncRNA, LASSIE which similar to MANTIS is involved in EC alignment in response to laminar pulsatile flow [68].
Aneurysms
An aneurysm is an enlargement of an artery due to weakness in its wall. Aneurysms are irreversible and hence explorations in therapeutic interventions for them are important. Aneurysms in aorta increase the risk for aortic dissection, a serious condition resulting from a tear in the inner layer of the aorta. Aneurysms are generally associated with loss of the internal elastic lamina, thinning of the tunica media, and degeneration of the extracellular matrix of the vessel wall. The exact pathogenesis of aneurysm formation and rupture is yet unclear. Akin to atherosclerosis, aneurysms can occur throughout the arterial tree.
Factors associated with the pathogenesis of aneurysms include genetic and environmental, more specifically, extracellular matrix defects and degeneration, hemodynamic stress and inflammation secondary to infection [81]. Recent reports suggest that WSS alterations may have a key role in the progression of an aneurysm. Castro et al. observed that mean WSS for unruptured aneurysms ranges from 10 to 230 dyn/cm2 compared to WSS ranging from 35 to as high as 1500 dyn/cm2 for ruptured aneurysms [82]. Earlier studies suggested that hemodynamic alterations in small arteries of the brain have a key role in the initiation and rupture of intracranial aneurysms [83].
Both high and low WSS have been found associated with aneurysm development and tear. Inflammation-mediated destructive remodeling or mural cells-mediated remodeling of the vessel wall can result in alterations in WSS and aneurysm progression [84]. Disturbed shear stress accelerates EC turnover, while low shear stress induces apoptosis in aneurysms. Initially, very high WSS, sensed by molecular sensors such as integrins and ion channels on arterial ECs induce MMP production that leads to ECM degeneration of arterial wall. This causes dilatation of lumina and degradation of the internal elastic lamina. As the diameter of the vessel increases, WSS decreases and matrix degradation stimuli are also reduced [85].
Irrespective of the cause, altered hemodynamics, including high WSS and arterial blood pressure cause dilation or rupture in aneurysms. Several histone modifications like upregulated HDACs, promoter methylations with enhanced flow-responsive DNMTs and ncRNAs are found associated with aneurysms. For instance in murine models, miR-126, a well-known laminar flow inducible miRNA improves cell survival and thereby significantly reduces the risk for abdominal aortic aneurysm formation [86]. Similar is the case with miR-29 reported by Chen et al. to be a stretch stress-responsive miRNA [87]. MiR-29 is involved in the loss of ECM in the aorta and formation of the aneurysm [88]. Bjork and colleagues recently reported that an oscillatory flow profile induces hypomethylation caused aberrant endothelial to mesenchymal transition in bicuspid aortic valves and increased the risk for aneurysm [89]. The tight regulatory mechanisms between DNMTs and TETs in response to oscillatory flow are yet to be recognized.
Cardenas et al. in their studies in thoracic aortic aneurysms, identified MALAT1, a shear-responsive lncRNA, which forms a chromatin-remodeling complex with histone deacetylase enzyme HDAC9 and the BRG1 (Brahma-related gene 1 protein) [90]. This complex regulates the promoters of key cytoskeletal and contractile genes and appears to be involved in pathological vascular smooth muscle cell dedifferentiation in aneurysm. The complex regulation of HDAC9/ MALAT1/ BRG1 is a perfect example of an interaction between multiple types of epigenetic regulatory mechanisms in vascular disease. In addition to the aforementioned role of HDAC9/MALAT1 in epigenetic regulation of thoracic aortic aneurysm, there is abundant evidence to link epigenetic mechanisms to aortic dissection, abdominal aortic aneurysms, and cerebral aneurysms. Unraveling the pathways through which histone modifications and ncRNA differential expression in the context of extreme hemodynamic alterations contribute to the pathophysiology of aneurysms may pave way for epigenome-based therapies and biomarkers in the future.
Pulmonary arterial hypertension
Pulmonary vascular disease is a broad term used to describe any condition that affects the blood vessels within the lungs. Pulmonary vascular disorder can lead to cardiovascular problems as well as impair the quality of a patient’s life. Pulmonary arterial hypertension (PAH), the most common pulmonary vascular disease and is associated with remodeling of pulmonary arteries [91]. Wall thickening due to medial hypertrophy and neointimal lesions, luminal narrowing and increased vascular resistance results in an increase in pulmonary arterial pressure. PAH is defined as a resting mean pulmonary artery pressure ≥ 25 mmHg in the absence of chronic lung disease, left heart disease, or venous thromboembolism. High WSS (> 80 dyn/cm2) has been observed in patients with a systemic-to-pulmonary communication leading to PAH [92]. Yet PAH patients are characterized by low blood flow and reduced shear stress (5–8 dyn/cm2) in proximal conduit vessels [93]. Both low and high WSS may exist in the pulmonary system and how the magnitude of flow regulates endothelial function in the pulmonary circulation has not been investigated deeply.
Recent studies are focusing on hemodynamic regulators of endothelial structure and function which perpetuate pulmonary vascular remodeling associated with PAH. Normally, vascular endothelium in response to high shear stress, lose their cobblestone morphology and the cells become elongated in the direction of blood flow. Failure in adapting to these morphological changes is an initial event associated with vascular remodeling. Interestingly, pressure off-loading during pulmonary artery banding prevents and even reverse occlusive vascular remodeling induced by hypoxia in rats exemplifying the role of hemodynamics in the disease pathogenesis [94]. Overexpression of DNMTs and HDACs has been found in animal models of PAH, but the role of WSS in their overexpression or global promoter methylation or histone acetylation has not yet been explored.
A year ago, Vanderlaan et al. reported that shear stress modulates the expression of KLF2 and KLF4 in pulmonary vein endothelial cells [95]. A missense mutation in KLF2 gene was recently reported in a family with autosomal heritable PAH, suggesting that KLF2 signaling may be affected in the disease. In patients with PAH, the levels of KLF2-dependent miR-150 in plasma exosomes were found to be reduced and the reduction in levels correlated with the duration of survival of patients [96]. Sindi et al. demonstrated the dysregulation of KLF2-induced miRNA signaling in endothelial cells in mice with PAH. They have also shown the anti-remodeling effects in pulmonary arteries by KLF2-induced miR-181a-5p and miR-324-5p through in vitro and in vivo studies. miR-181a-5p and miR-324-5p levels are less in patients in the early stages of PAH. The key target genes of miR-181a-5p, such as α-SMA, TNF-α, IL-1, Notch4 and MMP10, and miR-324-5p targets (MAPK, NFATC2, ETS-1) were also elevated in patients with PAH.
ECM remodeling is a major pathophysiological feature of PAH. This is tightly regulated by YAP/TAZ-miR-130/301 feedback circuit [97]. This regulatory cascade is induced by various stresses including fluid shear stress and hypoxia and considered as an early trigger for PAH.
Studies on the role of lncRNA in PAH are rare. Leisegang et al. found that MANTIS lncRNA which gets expressed by atheroprotective flow is reduced and its regulator JARID1B is upregulated in patients with idiopathic PAH [66]. LISPR1, a shear-responsive lncRNA, is also significantly decreased in the lung tissues of patients with idiopathic PAH. Su et al. reported that H19 induced proliferation of pulmonary artery SMCs isolated from animals with monocrotaline-induced PAH and that H19 knockout mice do not develop pulmonary artery remodeling [98]. The precise mechanism of MANTIS, H19 and LISPR1 involvement in PAH in the context of altered WSS needs to be further explored. Nevertheless, there is sufficient evidence to indicate the significant role of WSS in mediating epigenetic alterations in PAH.
PAH is mostly asymptomatic till pulmonary vascular lesions progress and hence early detection of the disease is difficult. An in-depth awareness of the molecular mechanisms underlying endothelial adaptation to high WSS will greatly enhance our knowledge on the pathogenesis of pulmonary vascular diseases and may aid in identifying new therapeutic strategies.
Vein graft disease
Vein graft occlusion is a major constraint to surgical revascularization in patients with coronary artery disease. Coronary artery bypass surgery is most often used to correct occlusive atherosclerotic lesions and to revascularize tissues in proximity to the lesions. The great saphenous vein is the most commonly used autologous conduit. Jose Goyanes of Madrid, Spain was the first one to use a vein graft (popliteal vein) to bridge an arterial defect during excision of a popliteal aneurysm [99]. More than five decades later, saphenous vein was used as a coronary artery bypass graft by the American Surgeon David Sabiston [100]. Vessel wall inflammation, intimal hyperplasia, and vessel occlusion accelerated atherosclerosis and graft rejection are serious complications of after grafting. Nearly one-third of patients develop graft stenosis in the conduit per se, anastomotic sites, or adjacent native artery segments [101]. While multiple factors contribute to graft stenosis, hemodynamic adaptability is considered the most important factor.
Vein wall remodeling is a key process during all the stages of venous graft disease. Vessel wall distension happens during the harvesting of the veins and on exposure to the arterial blood pressure after grafting. Due to distension injury, ECs become dysfunctional, smooth muscle cells in the venous wall are activated and ECM degradation ensues. ECM degradation products such as fibronectin, hyaluronic acid, and proteoglycans serve as endogenous ligands for toll-like receptors and inflammatory response is initiated in the vein graft [102]. Exposure to high shear stress (12–25dyn/cm2) in the arterial system is a challenge to the mechanotransduction process in the saphenous vein graft. The result is arterialization of the vein with SMC proliferation, intimal hyperplasia, and neointima formation. Platelets and fibrin deposition take place vessel wall is infiltrated by inflammatory cells [103]. In hypercholesterolemic conditions, uptake of lipids causes macrophages to turn into foam cells. Macrophage apoptosis leading to necrotic core formation result in atheromatous lesions, venous graft disease and vein graft failure in the long-term.
High flow and pressure in vein grafts increase apoptosis in the vessel wall. The effects of shear stress are mainly mediated by ECs in vein grafts. There will be elevated pressure, fenestration and also heightened endothelial denudation after engraftment. SMCs are also exposed to shear stress. The endothelial layer of the saphenous vein, structurally distinct from that of arteries, is highly vulnerable to cyclic stretch due to arterial blood flow. This exposure initiates the expression of adhesion factors such as MCP-1 and ICAM-1 [104].
Goossens et al. reported that hypermethylation was higher in venous grafts when compared to arterial grafts in patients undergoing coronary artery bypass grafting [105]. Whether this accounts for the difference in patency rates between arterial and venous grafts is unclear. The diverse responses exhibited by arterial and venous endothelial cells to arterial blood flow are probably dependent on the epigenetic memory in ECs determined by their origin.
Studies have shown that veins perfused for seven days under high blood pressure have high levels of transforming growth factor (TGF)-β1 and microRNAs-138/200b/200c. This change is associated with reduced TIMP1 and enhanced intimal hyperplasia [106, 107]. Shear-responsive miR-21 expression is elevated in ex vivo models of vein graft failure [108]. This enhanced expression is localized neointima in the failing grafts and is also associated with an intimal thickening.
Hunag et al. in 2020 reported that lncRNA NEAT1 (nuclear paraspeckle assembly transcript 1) is a mechanical stress factor in vein grafts [109]. NEAT1 expression is downregulated in endothelial cells subjected to mechanical stretch and is followed by an inflammation response and vein graft failure. New insights into flow driven epigenetic regulation in vein conduits exposed to arterial pressure may help in designing methods to prevent or retard vein graft occlusion.
Varicose veins
Varicose veins are enlarged and twisted veins with impaired blood flow. Complications of this disease range from throbbing pain and severe cramps to hyperpigmentation, eczema, lipodermatosclerosis and unhealing ulcers. Despite advances in invasive as well as non-invasive treatment options for the obliteration of the affected veins, recurrence following treatment is relatively high. Genetic susceptibility and risk factors for orthostatism precipitate and aggravate the severity of varicose vein complications. Whether the vein wall or venous valve is involved, the end result is blood reflux and ensuing venous hypertension which triggers ECM modification in veins. Location of venous insufficiency in the lowermost extremities of the body suggests that the effects of WSS on vein ECs may be a significant factor in the pathogenesis of varicose veins [30]. The role of epigenetic mechanisms in venous disorders such as varicose veins or deep vein thrombosis has not been well studied.
High blood pressure in the veins of the lower extremity during prolonged standing causes circumferential stretch forces on the vein wall and causes stimulation on ECs as well as SMCs. Integrins, ion channels, glycocalyx, platelet endothelial cell adhesion factors and GPCRs are the major circumferential stretch associated sensors in vein ECs [30]. In conditions such as varicose veins with valve weakening, WSS could be the initial trigger and not pressure-driven circumferential stretch.
Mechanosensors in venous ECs are considered to be similar to those in arterial ECs. Whether the flow response signaling cascade is also similar in both the systems is debatable considering the very low pressure in veins compared to the pressure in arteries. We and others have shown that the shear receptive FoxC2 gene is mutated and overexpressed in vein samples from patients with varicose veins [35, 110, 111]. Mutations in Piezo1, a mechanosensitive ion channel, were recently reported in patients with lymphatic dysplasia, who also had varicose veins [112]. Jiang et al. reported that there is promoter hypomethylation in shear-sensitive osteopontin and flow-transducer integrin β3 genes in varicose veins [113]. This may be a key factor in the pathogenesis of varicose veins.
Cui et al. and Zalewsky et al. found overexpression of miR-34a in vein biopsies from patients with chronic venous insufficiency [114, 115]. miR-34a is an endothelial-enriched miRNA, which is downregulated when there is uniform laminar shear stress. miR-21 is a shear-sensitive miRNA, which is elevated in patients with varicose veins. Zambuzzi and colleagues recently observed that lncRNA HOTAIR is mechanosensitive in both arterial and venous ECs [116]. In an earlier study, reduced levels of HOTAIR have been found in varicose veins [117]. These evidence necessitate an investigation of mechanosensing and associated epigenomic alterations in venous diseases. Future mechanistic studies on mechanoepigenetic factors could lead to therapies focused on pathogenic factors, in place of the presently employed corrective therapies, for vein disorders.
Mechanoepigenetic drugs for vascular diseases
Mechanoepigenetics significantly impact gene expression in vascular ECs and the risk for vascular disease as well as disease severity. Current treatment strategies for vascular diseases aim to reverse the epigenetic changes through pharmacological approaches. The interrelated complexity of epigenetic signaling and alternative compensatory biological pathways complicate focused epigenetic therapies. Further, somatic mutations in epigenetic genes are sufficient to alter therapeutic responses in individuals [2]. Major classes of epigenetic drugs currently investigated are modulators that target DNMT, TET, HDAC, lncRNA, and miRNAs.
DNMT inhibition
Dunn et al. in mice with carotid ligation demonstrated that DNMT1 is associated with the pathogenesis of atherosclerosis and that treatment with 5-Aza, the well-known inhibitor of DNMT, can inhibit lesion formation. 5-Aza blocks DNMT interaction with gene promoters in the target DNA and promote gene transcription [39]. In their studies in rats with partially ligated carotid arteries and resulting disturbed flow, Zhou et al. found that 5-Aza can effectively reverse the atheroprone oscillatory shear flow-induced promoter hypermethylation [72]. Azanucleosides have limitations as they are prodrugs which can be irreversibly incorporated into DNA and at high doses have cytotoxic effects on normal cells [118]. DNMT silencing is a harmless alternative. Zhang and his colleagues found that DNMT1 silencing effectively reduces endothelial proliferation and inflammation in the arterial wall and thus prevent atherosclerosis [119].
TET2 modulation
TET2 in contrast to DNMT demethylates promoter DNA and aid increased gene transcription. TET2 as mentioned earlier is downregulated in response to disturbed flow [43]. There is cumulative evidence that deficiency of TET2 results in the formation of lesions of atherosclerosis [120]. Reduced expression of TET2 is associated with activation of NLRP3/IL-1β-dependent inflammasome as well as pro-inflammatory pathways, and atherosclerosis and heart failure in animals [121]. It is evident that small molecules that can increase TET2 expression and its demethylase activity can be exploited to develop novel drugs.
HDAC modulation
HDAC-based mechanisms are firmly associated with disturbed flow-mediated EC dysfunction and atherosclerosis. HDAC blockade can theoretically reverse this pathophysiological cascade. Yet, the rationale to use HDAC inhibitors in vascular diseases is debatable. This is because some HDAC inhibitors at high doses induce pro-inflammatory effects and at low doses have anti-inflammatory effects. Manea et al. have shown that the pan-HDAC inhibitor SAHA (Suberoylanilide hydroxamic acid), at relatively low dose, significantly reduces oxidative stress, inflammation, and progression of atherosclerotic lesions in mice [122]. SAHA was also shown to pharmacologically activate KLF2, a shear-responsive anti-inflammatory transcription factor [123]. The discovery of effective modulators to correct mechanoepigenetic aberrations can usher in advances in vascular medicine. Given that histone writers, readers and erasers interact with DNMTs and TETs to result in different patterns of gene transcription, utilizing these inhibitors in therapeutics is not going to be easy.
NcRNA regulators
Non-coding RNAs have a profound influence at all stages of transcription and protein translation (e.g., lncRNA) and hence these molecules offer immense therapeutic possibilities. LncRNAs have very low sequence conservation but have higher tissue specificity [55]. This characteristic makes it a promising target for drug development, as their interaction will help in causing less remote off-target effects. MALAT1/HDAC9/ BRG1, a lncRNA MALAT1-HDAC complex, as mentioned earlier is involved in the pathogenesis of thoracic aortic aneurysm [90]. Deletion of either MALAT1 or HDAC9 reduces the risk for aneurysm and its pathophysiological sequel. In pre-clinical studies, silencing lncRNA has been explored by the use of RNAi-based methods (siRNA) which silence lncRNA localized in the cytoplasm or by using LNA-GapmeR antisense oligonucleotides which induce RNase cleavage in nuclear-localized lncRNA [124].
Among all the ncRNAs classes, most of the screening therapeutic studies have been done using miRNAs probably because of insights on their mode of action and their potential to regulate multiple yet related molecular signaling pathways. One of the most investigated areas in epigenetic drug therapy is the local or systemic delivery of miRNA modulators for treating atherosclerosis. MiRNA modulators either as anti-miRs or as mimics antagonize or support the action of specific miRNAs and can cause a significant change in the gene profile of vascular ECs [77, 125]. Local delivery of mimics of miR-21 increases plaque stability and reduce the incidence of atherothrombosis in mice [126]. A targeted, lesion site-specific overexpression of this mechano-miR can prevent the rupture of vulnerable plaques. A schematic representation of possible therapeutic strategies for reversing epigenetic remodeling in vascular diseases is given in Fig. 6.

Schematic diagram representing mechanoepigenetic drugs for vascular diseases and their mechanism of action. These approaches are being studied in vitro as well as in animal models. iDNMT denotes DNMT inhibitors such as 5-Aza, iHDAC denotes HDAC inhibitors such as Suberoylanilide hydroxamic acid (SAHA)
In a recent study, Gongol et al. found that KLF2 and eNOS mRNAs contain a miR-93 seed sequence [127]. eNOS mRNA also has a miR-484 seed sequence indicating that the two flow-responsive genes are direct targets of respective miRNAs. They also found that atheroprone oscillatory shear stress regulates the biogenesis of miR-93 and miR-484. In vitro studies reveal that specific anti-miRs can restore KLF2 and eNOS expression as well as nitric oxide bioavailability in ECs exposed to disturbed oscillatory shear flow. In pulmonary arteries, KLF2 induces miR-181a-5p and miR-324-5p and can have anti-remodeling effects [96]. These findings are encouraging for efforts for the development of miRNA therapies for vascular diseases.
Inhibition of miR‐143/145 is found to reduce the progression of atherosclerotic plaques in mice [128]. Conversely, in vivo delivery of miR‐145 prevents intimal hyperplasia in vein graft disease [129]. These contrasting findings suggest that the therapeutic utility of miR‐145 could be context-dependent. Studies have, in animal models of vein graft failure demonstrated that anti-miR-21 inhibits shear-sensitive vascular remodeling and attenuates pathological neointima [108, 130].
For developing successful miRNA-based therapies, the following needs are to be addressed: (1) designing highly specific inhibitors and mimics that can specifically alter a unique mRNA-miRNA interaction without altering off-target genes, (2) strategies for cell type-specific targeted delivery by developing vectors for delivery of modulators to the desired vascular cell type, and (3) development of inducible miRNA modulators such as photoactivatable anti-miRs. CRISPR/Cas9 genome editing is another tool for gene expression modulation as it can manipulate all types of ncRNA. Investigations using this approach are at a nascent stage. With further understanding of the molecular intricacies in epigenetic actions modulated by blood flow, better and safer methods to revert abnormal vascular remodeling can be expected in the years to come.
Mechanoepigenetic biomarkers for vascular diseases
Identification of biological markers of early stages is crucial for preventing the development and progression of vascular diseases. Disturbed flow-induced endothelial dysfunction is a major characteristic of the early stages of any vascular disease. Therefore, epigenetic marks and variations associated with the disturbed flow may serve as biomarkers. A combined measurement of global DNA methylation and DNMT expression could provide a novel biomarker for vascular diseases.
Most of the biomarker investigations have been done in patients with atherosclerotic vascular diseases. Ma et al. in their study of 150 patients with atherosclerotic vascular disease and an equal number of healthy subjects, found that assessment of promoter methylation of ATP binding cassette subfamily A member 1 (ABCA1), TIMP1, and acetyl-CoA acetyl transferase 1 (ACAT1) is useful for early detection of atherosclerotic disease [131]. Wei et al. observed increased promoter methylation of the shear stress-responsive gene SMAD7 in the peripheral blood of patients with atherosclerotic vascular disease [132]. They suggested that methylated SMAD7 can be used as a predictive marker for atherosclerosis.
Ryer et al. in 2015 reported the potential predictive value of methylated CpG islands in the promoter of serpin peptidase inhibitor clade B (ovalbumin) member 9 (SERPINB9), in patients with abdominal aortic aneurysm [133].
Measurement of ncRNAs especially, miRNA expression in the blood sera or plasma is one of the most promising methods to assess early risk and prognosis in patients with vascular diseases. NcRNAs are mostly cell and tissue-specific. Their expression in serum would hence reflect pathophysiology at concerned anatomical sites. Endothelial cell-enriched miR-126, a mechano-miR, was recently investigated for its prognostic and diagnostic value in coronary artery disease (CAD) [134]. MiR-126 is expressed under laminar shear stress and affects syndecan-4 expression [135]. Fichtlscherer et al. nearly a decade ago reported reduced levels of expression of miR-126 in the serum of patients with CAD compared to healthy controls [136]. MiR-17-5p, a disturbed flow-induced pro-inflammatory miRNA [137] as well as KLF2 regulated miR-93 and miR-484 [127] has recently been reported as biomarkers with high sensitivity and specificity in patients with CAD. The levels of KLF2-dependent miR-150 are reduced in plasma exosomes from patients with PAH; the levels correlate with overall survival [96].
LncRNAs regulate gene expression at transcriptional, post-transcriptional and chromatin-remodeling stages. They have high tissue specificity. As described earlier, laminar shear stress both in cultured ECs as well as in aortic EC of animals induces lncRNA SENCR expression. As may be expected, SENCR expression is reduced in patients with atherosclerotic vascular disease and hence is of predictive value in the early stages of atherosclerotic disease [138]. H19, a shear stress-induced lncRNA, was found to be increased in the plasma samples of patients with CAD and hence suggested as a potential biomarker for CAD [139]. Large-scale multicentric studies are required to elucidate the potential of epigenetic modifiers as markers for the diagnosis of early stages of different vascular diseases.
Concluding remarks and future perspectives
In this review, we discuss all the epigenetic factors that are currently known to be involved in hemodynamics-directed EC dysfunction and epigenetic modulations that are associated with various vascular diseases. Atherosclerosis is the abundantly evaluated vascular disease with respect to mechanoepigenetics. Other vascular diseases studied to a lesser extent are pulmonary hypertension, aneurysm, and venous diseases.
Different forms of blood flow and diverse patterns of hemodynamic shear stress have a profound effect on endothelial function in various regions of the vascular system. Disturbed flow-based EC dysfunction is a major pathophysiological factor that determines disease phenotype, clinical complications, and mortality. The role of epigenetic regulators of endothelial cell response to hemodynamic alterations is not yet completely elucidated and warrant further investigation as they may also determine the pathogenesis of vascular diseases. Epigenetic factors such as DNMTs, TETs, HDACs, non-coding RNAs are involved in blood flow-regulated EC function. Epigenetic mediators, such as DNMTs and miRNAs, are effector biological molecules that can be finely modulated by targeted mechanisms. Epigenetic modifiers regulated by a disturbed flow can be altered in the early stages of disease pathogenesis. Unraveling mechanoepigenetic mechanisms could lead to the discovery of novel biomarkers for early stages and also new therapeutic strategies for vascular diseases. Most of the studies in mechanoepigenetics are now based on single-model systems in small rodents and the observations from them are unsuitable for translation into clinical evaluation. Appropriate cellular and animal models for mechanistic studies are to be identified. There is also a need for designing clinical studies to evaluate the potential of mechanoepigenetics to identify novel treatment options for vascular diseases.
References
Garoffolo G, Pesce M (2019) Mechanotransduction in the cardiovascular system: from developmental origins to homeostasis and pathology. Cells 8(12):1607
Zarzour A, Kim HW, Weintraub NL (2019) Epigenetic regulation of vascular diseases. Arterioscler Thromb Vasc Biol 39(6):984–990
Handy DE, Castro R, Oscalzo L, J, (2011) Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation 123(19):2145–2156
Missirlis YF (2016) Mechanoepigenetics. Front Cell Dev Biol 14:113
Kruger-Genge A, BlockiA FRP, Jung F (2019) Vascular endothelial cell biology: an update. Int J Mol Sci 20(18):4411
Hahn C, Schwartz MA (2009) Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10(1):53–62
Ballermann BJ, Dardik A, Eng E, Liu A (1998) Shear stress and the endothelium. Kidney Int Suppl 54(67):100–108
Trubelja A, Bao G (2018) Molecular mechanisms of mechanosensing and mechanotransduction in living cells. Extreme Mech Lett 20:91–98
Aird WC (2007) Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res 100(2):174–190
Deng DX-F, Tsalenko A, Vailaya A, Ben-Dor A, Kundu R, Estay I, Tabibiazar R, Kincaid R, Yakhini Z, Bruhn L (2006) Differences in vascular bed disease susceptibility reflect differences in gene expression response to atherogenic stimuli. Circ Res 98(2):200–208
Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437(7057):426–431
Obi S, Masuda H, Shizuno T, Sato A, Yamamoto K, Ando J, Abe Y, Asahara T (2012) Fluid shear stress induces differentiation of circulating phenotype endothelial progenitor cells. Am J Physiol Cell Physiol 303(6):C595–C606
Tarbell JM, Shi Z-D, Dunn J, Jo H (2014) Fluid mechanics, arterial disease, and gene expression. Annu Rev Fluid Mech 46:591–614
Baeyens N, Schwartz MA (2016) Biomechanics of vascular mechanosensation and remodeling. Mol Biol Cell 27(1):7–11
Schwartz C (1962) Observations on localization of arterial plaques. Circ Res 11:63–73
Fry DL (1969) Certain histological and chemical responses of the vascular interface to acutely induced mechanical stress in the aorta of the dog. Circ Res 24(1):93–108
Caro C, Fitz-Gerald J, Schroter R (1969) Arterial wall shear and distribution of early atheroma in man. Nature 223(5211):1159–1161
Friedman MH, Hutchins GM, Bargeron CB, Deters OJ, Mark FF (1981) Correlation between intimal thickness and fluid shear in human arteries. Atherosclerosis 39(3):425–436
Givens C, Tzima E (2016) Endothelial mechanosignaling: does one sensor fit all? Antioxid Redox Signal 25(7):373–388
Malek AM, Alper SL, Izumo S (1999) Hemodynamic shear stress and its role in atherosclerosis. JAMA 282(21):2035–2042
Chiu J-J, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91(1):327–387
Olesen S-P, Claphamt D, Davies P (1988) Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature 331(6152):168–170
Trouet D, Nilius B, Jacobs A, Remacle C, Droogmans G, Eggermont J (1999) Caveolin-1 modulates the activity of the volume-regulated chloride channel. J Physiol 520(Pt 1):113–119
Reitsma S, Slaaf DW, Vink H, Van Zandvoort MA, OudeEgbrink MG (2007) The endothelial glycocalyx: composition, functions, and visualization. Pflug Arch Eur J Phys 454(3):345–359
DeMaio L, Chang YS, Gardner TW, Tarbell JM, Antonetti DA (2001) Shear stress regulates occludin content and phosphorylation. Am J Physiol Heart Circ Physiol 281(1):105–113
Schwartz MA (2010) Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb Perspect Biol 2(12):a005066
Niu N, Xu S, Xu Y, Little PJ, Jin Z-G (2019) Targeting mechanosensitive transcription factors in atherosclerosis. Trends Pharmacol Sci 40(4):253–266
Nayak L, Lin Z, Jain MK (2011) “Go with the flow”: how Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid Redox Signal 15(5):1449–1461
Mohamied Y, Rowland EM, Bailey EL, Sherwin SJ, Schwartz MA, Weinberg PD (2015) Change of direction in the biomechanics of atherosclerosis. Ann Biomed Eng 43(1):16–25
Atta HM (2012) Varicose veins: role of mechanotransduction of venous hypertension. Int J Vasc Med 2012(2):538627
Lipowsky HH, Kovalcheck S, Zweifach BW (1978) The distribution of blood rheological parameters in the microvasculature of cat mesentery. Circ Res 43(5):738–749
Franklin KJ (1927) Valves in veins: an historical survey. Proc R Soc Med 21(1):1–33
Meng X, Mavromatis K, Galis ZS (1999) Mechanical stretching of human saphenous vein grafts induces expression and activation of matrix-degrading enzymes associated with vascular tissue injury and repair. Exp Mol Pathol 66(3):227–237
Reček Č (2006) Conception of the venous hemodynamics in the lower extremity. Angiology 57(5):556–563
Surendran S, Girijamma A, Nair R, Ramegowda KS, Nair DH, Thulaseedharan JV, Lakkappa RB, Kamalapurkar G, Kartha CC (2014) Forkhead box C2 promoter variant c.-512C> T is associated with increased susceptibility to chronic venous diseases. PLoS ONE 9(3):e90682
Consortium EP (2011) A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9(4):e1001046
Moore LD, Le T, Fan G (2013) DNA methylation and its basic function. Neuropsychopharmacology 38(1):23–38
Jiang Y-Z, Jiménez JM, Ou K, McCormick ME, Zhang L-D, Davies PF (2014) Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ Res 115(1):32–43
Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, Fan Y, Jordan IK, Jo H (2014) Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J Clin Invest 124(7):3187–3199
Castillo-Díaz SA, Garay-Sevilla ME, Hernández-González MA, Solís-Martínez MO, Zaina S (2010) Extensive demethylation of normally hypermethylated CpG islands occurs in human atherosclerotic arteries. Int J Mol Med 26(5):691–700
Jiang Y-Z, Manduchi E, Stoeckert CJ, Davies PF (2015) Arterial endothelial methylome: differential DNA methylation in athero-susceptible disturbed flow regions in vivo. BMC Genomics 16(1):506
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18(9):517–534
Yang Q, Li X, Li R, Peng J, Wang Z, Jiang Z, Tang X, Peng Z, Wang Y, Wei D (2016) Low shear stress inhibited endothelial cell autophagy through TET2 downregulation. Ann Biomed Eng 44(7):2218–2227
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395
Zampetaki A, Zeng L, Margariti A, Xiao Q, Li H, Zhang Z, Pepe Anna E, Wang G, Habi O, deFalco E, Cockerill G, Mason Justin C, Hu Y, Xu Q (2010) Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation 121(1):132–142
Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, Blacher S, Verdin E, Foidart J-M, Nusgens BV (2002) Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 21(3):427–436
Jin G, Bausch D, Knightly T, Liu Z, Li Y, Liu B, Lu J, Chong W, Velmahos GC, Alam HB (2011) Histone deacetylase inhibitors enhance endothelial cell sprouting angiogenesis in vitro. Surgery 150(3):429–435
Kaluza D, Kroll J, Gesierich S, Manavski Y, Boeckel J-N, Doebele C, Zelent A, Rössig L, Zeiher AM, Augustin HG (2013) Histone deacetylase 9 promotes angiogenesis by targeting the antiangiogenic microRNA-17–92 cluster in endothelial cells. Arterioscler Thromb Vasc Biol 33(3):533–543
Zampetaki A, Zeng L, Margariti A, Hu Y, Xu Q (2008) HDAC3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Am Heart Assoc 118(18):S409
Yan MS, Marsden PA (2015) Epigenetics in the vascular endothelium: looking from a different perspective in the epigenomics era. Arterioscler Thromb Vasc Biol 35(11):2297–2306
Lee D-Y, Lee C-I, Lin T-E, Lim SH, Zhou J, Tseng Y-C, Chien S, Chiu J-J (2012) Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc Natl Acad Sci U S A 109(6):1967–1972
Stein S, Schäfer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U (2010) SIRT1 reduces endothelial activation without affecting vascular function in ApoE-/-mice. Aging (Albany NY) 2(6):353–360
Olmos Y, Sánchez-Gómez FJ, Wild B, García-Quintans N, Cabezudo S, Lamas S, Monsalve M (2013) SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid Redox Signal 19(13):1507–1521
Wei X, Yi X, Zhu XH, Jiang DS (2020) Histone methylation and vascular biology. Clin Epigenet 12(1):30
Surendran S, Kartha C (2017) Role of non-coding RNAs in vascular complications of diabetes mellitus. Mechanisms of vascular defects in diabetes mellitus. Springer, Berlin, pp 341–357
Weber M, Baker MB, Moore JP, Searles CD (2010) MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun 393(4):643–648
Chen K, Fan W, Wang X, Ke X, Wu G, Hu C (2012) MicroRNA-101 mediates the suppressive effect of laminar shear stress on mTOR expression in vascular endothelial cells. Biochem Biophys Res Commun 427(1):138–142
Qin X, Wang X, Wang Y, Tang Z, Cui Q, Xi J, Li Y-SJ, Chien S, Wang N (2010) MicroRNA-19a mediates the suppressive effect of laminar flow on cyclin D1 expression in human umbilical vein endothelial cells. Proc Natl Acad Sci U S A 107(7):3240–3244
Wang K-C, Garmire LX, Young A, Nguyen P, Trinh A, Subramaniam S, Wang N, Shyy JY, Li Y-S, Chien S (2010) Role of microRNA-23b in flow-regulation of Rb phosphorylation and endothelial cell growth. Proc Natl Acad Sci U S A 107(7):3234–3239
Fang Y, Shi C, Manduchi E, Civelek M, Davies PF (2010) MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc Natl Acad Sci U S A 107(30):13450–13455
Marin T, Gongol B, Chen Z, Woo B, Subramaniam S, Chien S, Shyy JY (2013) Mechanosensitive microRNAs-role in endothelial responses to shear stress and redox state. Free Radic Biol Med 64:61–68
Wu W, Xiao H, Laguna-Fernandez A, Villarreal G Jr, Wang K-C, Geary GG, Zhang Y, Wang W-C, Huang H-D, Zhou J, Li Y-S, Chien S, Garcia-Cardena G, Shyy JYJ (2011) Flow-dependent regulation of kruppel-like factor 2 is mediated by microRNA-92a. Circulation 124(5):633–641
Fang Y, Davies PF (2012) Site-specific microRNA-92a regulation of Krüppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Biol 32(4):979–987
Man HSJ, Sukumar AN, Lam GC, Turgeon PJ, Yan MS, Ku KH, Dubinsky MK, Ho JJD, Wang JJ, Das S, Mitchell N, Oettgen P, Sefton MV, Marsden PA (2018) Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc Natl Acad Sci USA 115(10):2401–2406
Miao Y, Ajami NE, Huang T-S, Lin F-M, Lou C-H, Wang Y-T, Li S, Kang J, Munkacsi H, Maurya MR, Gupta S, Chien S, Subramaniam S, Chen Z (2018) Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat Commun 9(1):292
Leisegang M, Fork C, Josipovic I, Richter F, Preussner J, Hu J, Miller M, Epah J, Hofmann P, Günther S, Moll F, Valasarajan C, Heidler J, Ponomareva Carvalho Y, Freiman T, Maegdefessel L, Plate K, Mittelbronn M, Uchida S, Brandes R (2017) Long noncoding RNA MANTIS facilitates endothelial angiogenic function. Circulation 136(1):65–79
Josipovic I, Pflüger B, Fork C, Vasconez AE, Oo JA, Hitzel J, Seredinski S, Gamen E, HeringdorfDMz CW, Looso M, Pullamsetti SS, Brandes RP, Leisegang MS (2018) Long noncoding RNA LISPR1 is required for S1P signaling and endothelial cell function. J Mol Cell Cardiol 116:57–68
Stanicek L, Lozano-Vidal N, Bink DI, Hooglugt A, Yao W, Wittig I, van Rijssel J, van Buul JD, van Bergen A, Klems A, Ramms AS, Le Noble F, Hofmann P, Szulcek R, Wang S, Offermanns S, Ercanoglu MS, Kwon HB, Stainier D, Huveneers S, Kurian L, Dimmeler S, Boon RA (2020) Long non-coding RNA LASSIE regulates shear stress sensing and endothelial barrier function. Commun Biol 3(1):265
Wang Y, Qiu J, Luo S, Xie X, Zheng Y, Zhang K, Ye Z, Liu W, Gregersen H, Wang G (2016) High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis. Regen Biomater 3(4):257–267
Birchall D, Zaman A, Hacker J, Davies G, Mendelow D (2006) Analysis of haemodynamic disturbance in the atherosclerotic carotid artery using computational fluid dynamics. Eur Radiol 16(5):1074–1083
Won D, Zhu S-N, Chen M, Teichert A-M, Fish JE, Matouk CC, Bonert M, Ojha M, Marsden PA, Cybulsky MI (2007) Relative reduction of endothelial nitric-oxide synthase expression and transcription in atherosclerosis-prone regions of the mouse aorta and in an in vitro model of disturbed flow. Am J Pathol 171(5):1691–1704
Zhou J, Li Y-S, Wang K-C, Chien S (2014) Epigenetic mechanism in regulation of endothelial function by disturbed flow: induction of DNA hypermethylation by DNMT1. Cell Mol Bioeng 7(2):218–224
Illi B, Nanni S, Scopece A, Farsetti A, Biglioli P, Capogrossi M, Gaetano C (2003) Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ Res 93(2):155–161
Wang W, Ha CH, Jhun BS, Wong C, Jain MK, Jin Z-G (2010) Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115(14):2971–2979
Fan Y, Lu H, Liang W, Hu W, Zhang J, Chen YE (2017) Krüppel-like factors and vascular wall homeostasis. J Mol Cell Biol 9(5):352–363
Ni C-W, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, Jo H (2010) Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 116(15):66–73
Son DJ, Kumar S, Takabe W, Kim CW, Ni C-W, Alberts-Grill N, Jang I-H, Kim S, Kim W, Won Kang S, Baker AH, WoongSeo J, Ferrara KW, Jo H (2013) The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat Commun 4:3000
Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJ, Zeiher AM, Scheffer MP, Frangakis AS, Yin X, Mayr M (2012) Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol 14(3):249–256
Kumar S, Kim CW, Simmons RD, Jo H (2014) Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitiveathero-miRs. Arterioscler Thromb Vasc Biol 34(10):2206–2216
Lyu Q, Xu S, Lyu Y, Choi M, Christie CK, Slivano OJ, Rahman A, Jin Z-G, Long X, Xu Y (2019) SENCR stabilizes vascular endothelial cell adherens junctions through interaction with CKAP4. Proc Natl Acad Sci USA 116(2):546–555
Ailawadi G, Eliason JL, Upchurch GR (2003) Current concepts in the pathogenesis of abdominal aortic aneurysm. J Vasc Surg 38(3):584–588
Castro M, Putman C, Sheridan M, Cebral J (2009) Hemodynamic patterns of anterior communicating artery aneurysms: a possible association with rupture. Am J Neuroradiol 30(2):297–302
Diagbouga MR, Morel S, Bijlenga P, Kwak BR (2018) Role of hemodynamics in initiation/growth of intracranial aneurysms. Eur J Clin Invest 48(9):e12992
Meng H, Tutino VM, Xiang J, Siddiqui A, High WSS or low WSS? (2014) Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. Am J Neuroradiol 35(7):1254–1262
Staarmann B, Smith M, Prestigiacomo CJ (2019) Shear stress and aneurysms: a review. Neurosurg Focus 47(1):E2
Shen G, Sun Q, Yao Y, Li S, Liu G, Yuan C, Li H, Xu Y, Wang H (2020) Role of ADAM9 and miR-126 in the development of abdominal aortic aneurysm. Atherosclerosis 297:47–54
Chen Y, Mohammed A, Oubaidin M, Evans CA, Zhou X, Luan X, Diekwisch TG, Atsawasuwan P (2015) Cyclic stretch and compression forces alter microRNA-29 expression of human periodontal ligament cells. Gene 566(1):13–17
Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, Van Heijningen P (2011) MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109(10):1115–1119
Björck HM, Du L, Pulignani S, Paloschi V, Lundströmer K, Kostina AS, Österholm C, Malashicheva A, Kostareva A, Evangelista A, Teixidó-Tura G (2018) Altered DNA methylation indicates an oscillatory flow mediated epithelial-to-mesenchymal transition signature in ascending aorta of patients with bicuspid aortic valve. Sci Rep 8(1):1–5
Cardenas CLL, Kessinger CW, Cheng Y, MacDonald C, MacGillivray T, Ghoshhajra B, Huleihel L, Nuri S, Ashish SY, Jaffer FA (2018) An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun 9(1):1–14
Rose-Jones LJ, Mclaughlin V (2015) Pulmonary hypertension: types and treatments. Curr Cardiol Rev 11(1):73–79
Hunter KS, Lanning CJ, Chen SY, Zhang Y, Garg R, Ivy DD, Shandas R (2006) Simulations of congenital septal defect closure and reactivity testing in patient-specific models of the pediatric pulmonary vasculature: a 3D numerical study with fluid-structure interaction. J Biomech Eng 128(4):564–572
Ben Driss A, Devaux C, Henrion D, Duriez M, Thuillez C, Levy BI, Michel JB (2000) Hemodynamic stresses induce endothelial dysfunction and remodeling of pulmonary artery in experimental compensated heart failure. Circulation 101(23):2764–2770
Abe K, Shinoda M, Tanaka M, Kuwabara Y, Yoshida K, Hirooka Y, McMurtry IF, Oka M, Sunagawa K (2016) Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc Res 111(1):16–25
Vanderlaan RD, Fu YY, Wang Y, Caldarone C (2019) Shear stress regulates Klf2 and Klf4 in pulmonary vein endothelial cells. Circulation 140(Suppl_1):A16370
Sindi HA, Russomanno G, Satta S, Abdul-Salam VB, Jo KB, Qazi-Chaudhry B, Ainscough AJ, Szulcek R, Bogaard HJ, Morgan CC (2020) Therapeutic potential of KLF2-induced exosomal microRNAs in pulmonary hypertension. Nat Commun 11(1):1–17
Bertero T, Cottrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB (2015) Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep 13(5):1016–1032
Su H, Xu X, Yan C, Shi Y, Hu Y, Dong L, Ying S, Ying K, Zhang R (2018) LncRNA H19 promotes the proliferation of pulmonary artery smooth muscle cells through AT1R via sponging let-7b in monocrotaline-induced pulmonary arterial hypertension. Respir Res 19(1):254
Harrison LH Jr (1976) Historical aspects in the development of venous autografts. Ann Surg 183(2):101–106
Chaikhouni A (2010) The magnificent century of cardiothoracic surgery. Heart views 11(1):31–37
Bandyk DF, Scabrook GR, Moldenhauer P, Lavin J, Edwards J, Cato R, Towne JB (1988) Hemodynamics of vein graft stenosis. J Vasc Surg 8(6):688–695
De Vries MR, Quax PH (2018) Inflammation in vein graft disease. Front Cardiovasc Med 5:3
Mitra AK, Gangahar DM, Agrawal DK (2006) Cellular, molecular and immunological mechanisms in the pathophysiology of vein graft intimal hyperplasia. Immunol Cell Biol 84(2):115–124
Methe H, Balcells M, del Carmen AM, Santacana M, Molins B, Hamik A, Jain MK, Edelman ER (2007) Vascular bed origin dictates flow pattern regulation of endothelial adhesion molecule expression. Am J Physiol-Heart C 292(5):H2167–H2175
Goossens EA, De Vries MR, Simons KH, Putter H, Quax PH, Nossent AY (2019) miRMap: profiling of 14q32 microRNA expression and DNA methylation throughout the human vasculature. Front Cardiovasc Med 6:113
Berard X, Déglise S, Alonso F, Saucy F, Meda P, Bordenave L, Corpataux JM, Haefliger JA (2013) Role of hemodynamic forces in the ex vivo arterialization of human saphenous veins. J Vasc Surg 57(5):1371–1382
Prandi F, Piola M, Soncini M, Colussi C, D’Alessandra Y, Penza E, Agrifoglio M, Vinci MC, Polvani G, Gaetano C, Fiore GB (2018) Adventitial vessel growth and progenitor cells activation in an ex vivo culture system mimicking human saphenous vein wall strain after coronary artery bypass grafting. PLoS ONE 10(2):e0117409
McDonald RA, White KM, Wu J, Cooley BC, Robertson KE, Halliday CA, McClure JD, Francis S, Lu R, Kennedy S, George SJ (2013) miRNA-21 is dysregulated in response to vein grafting in multiple models and genetic ablation in mice attenuates neointima formation. Eur Heart J 34(22):1636–1643
Huang Z, Winata WA, Zhang K, Zhao Y, Li Y, Zhou N, Zhou S, Fu W, Qiao B, Li G, Shao Y (2020) Reconstruction of a lncRNA-associated cerna network in endothelial cells under circumferential stress. Cardiol Res Pract 2020:1481937
Mellor RH, Brice G, Stanton AW, French J, Smith A, Jeffery S, Levick JR, Burnand KG, Mortimer PS, Lymphoedema Research Consortium (2007) Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation 115(14):1912–1920
Surendran S, Ramegowda KS, Suresh A, Raj SB, Lakkappa RK, Kamalapurkar G, Radhakrishnan N, Kartha CC (2016) Arterialization and anomalous vein wall remodeling in varicose veins is associated with upregulated FoxC2-Dll4 pathway. Lab Invest 96(4):399–408
Fotiou E, Martin-Almedina S, Simpson MA, Lin S, Gordon K, Brice G, Atton G, Jeffery I, Rees DC, Mignot C, Vogt J (2015) Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat Commun 6:8085
Jiang H, Lun Y, Wu X, Xia Q, Zhang X, Xin S, Zhang J (2014) Association between the hypomethylation of osteopontin and integrin β3 promoters and vascular smooth muscle cell phenotype switching in great saphenous varicose veins. Int J Mol Sci 15(10):18747–18761
Cui C, Liu G, Huang Y, Lu X, Lu M, Huang X, Li W, Jiang M (2012) MicroRNA profiling in great saphenous vein tissues of patients with chronic venous insufficiency. Tohoku J Exp Med 228(4):341–350
Zalewski DP, Ruszel KP, Stępniewski A, Gałkowski D, Bogucki J, Komsta Ł, Kołodziej P, Chmiel P, Zubilewicz T, Feldo M (2020) Dysregulations of microRNA and gene expression in chronic venous disease. J Clin Med 9(5):1251
da Silva RA, Ferreira MR, Gomes AM, Zambuzzi WF (2020) LncRNA HOTAIR is a novel endothelial mechanosensitive gene. J Cell Physiol 235(5):4631–4642
Biranvand AS, Khosravi M, Esfandiari G, Poursaleh A, Hosseini-Fard SR, Amirfarhangi A, Najafi M (2018) Associations between miR-661, miR-1202, lncRNA-HOTAIR, lncRNA-GAS5 and MMP9 in differentiated M2-macrophages of patients with varicose veins. Int Angiol 37(6):451–456
Covey JM, D'Incalci M, Tilchen EJ, Zaharko DS, Kohn KW (1986) Differences in DNA damage produced by incorporation of 5-aza-2′-deoxycytidine or 5, 6-dihydro-5-azacytidine into DNA of mammalian cells. Cancer Res 46(11):5511–5517
Zhang Y-P, Huang Y-T, Huang T-S, Pang W, Zhu J-J, Liu Y-F, Tang R-Z, Zhao C-R, Yao W-J, Li Y-S (2017) The mammalian target of rapamycin and DNA methyltransferase 1 axis mediates vascular endothelial dysfunction in response to disturbed flow. Sci Rep 7(1):1–12
Peng J, Yang Q, Li AF, Li RQ, Wang Z, Liu LS, Ren Z, Zheng XL, Tang XQ, Li GH, Tang ZH (2016) Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE−/− mice. Oncotarget 7(47):76423–76436
Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, Fuster JJ (2018) Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J Am Coll Cardiol 71(8):875–886
Manea S-A, Vlad M-L, Fenyo IM, Lazar A-G, Raicu M, Muresian H, Simionescu M, Manea A (2020) Pharmacological inhibition of histone deacetylase reduces NADPH oxidase expression, oxidative stress and the progression of atherosclerotic lesions in hypercholesterolemic apolipoprotein E-deficient mice; potential implications for human atherosclerosis. Redox Biol 28:101338
Xu Y, Xu S, Liu P, Koroleva M, Zhang S, Si S, Jin ZG (2017) Suberanilohydroxamicacid as a pharmacological kruppel-like factor 2 activator that represses vascular inflammation and atherosclerosis. J Am Heart Assoc 6(12):e007134
Lennox KA, Behlke MA (2016) Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res 44(2):863–877
Du F, Yu F, Wang Y, Hui Y, Carnevale K, Fu M, Lu H, Fan D (2014) MicroRNA-155 deficiency results in decreased macrophage inflammation and attenuated atherogenesis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 34(4):759–767
Jin H, Li DY, Chernogubova E, Sun C, Busch A, Eken SM, Saliba-Gustafsson P, Winter H, Winski G, Raaz U, Schellinger IN (2018) Local delivery of miR-21 stabilizes fibrous caps in vulnerable atherosclerotic lesions. Mol Ther 26(4):1040–1055
Gongol B, Marin T, Zhang J, Wang S-C, Sun W, He M, Chen S, Chen L, Li J, Liu J-H (2019) Shear stress regulation of miR-93 and miR-484 maturation through nucleolin. Proc Natl Acad Sci USA 116(26):12974–12979
Boettger T, Beetz N, Kostin S, Schneider J, Krüger M, Hein L, Braun T (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119(9):2634–2647
Nishio H, Masumoto H, Sakamoto K, Yamazaki K, Ikeda T, Minatoya K (2019) MicroRNA-145-loaded poly (lactic-co-glycolic acid) nanoparticles attenuate venous intimal hyperplasia in a rabbit model. J Thorac Cardiovasc 157(6):2242–2251
Wang XW, Zhang C, Lee KC, He XJ, Lu ZQ, Huang C, Wu QC (2017) Adenovirus-mediated gene transfer of microRNA-21 sponge inhibits neointimal hyperplasia in rat vein grafts. Int J Biol Sci 13(10):1309–1319
Ma SC, Zhang HP, Kong FQ, Zhang H, Yang C, He YY, Wang YH, Yang AN, Tian J, Yang XL, Zhang MH, Xu H, Jiang YD, Yu Z (2016) Integration of gene expression and DNA methylation profiles provides a molecular subtype for risk assessment in atherosclerosis. Mol Med Rep 13(6):4791–4799
Wei L, Zhao S, Wang G, Zhang S, Luo W, Qin Z, Bi X, Tan Y, Meng M, Ja Q, Qin H, Tian D, Aw Z (2018) SMAD7 methylation as a novel marker in atherosclerosis. Biochem Biophys Res Commun 496(2):700–705
Ryer E, Ronning K, Erdman R, Schworer C, Elmore J, Peeler T, Nevius C, Lillvis J, Garvin R, Franklin D, Kuivaniemi H, Tromp G (2015) The potential role of DNA methylation in abdominal aortic aneurysms. Int J Mol Sci 16(5):11259–11275
Wang X, Lian Y, Wen X, Guo J, Wang Z, Jiang S, Hu Y (2017) Expression of miR-126 and its potential function in coronary artery disease. Afr Health Sci 17(2):474–480
Mondadori dos Santos A, Metzinger L, Haddad O, M’baya-Moutoula E, Taïbi F, Charnaux N, Massy ZA, Hlawaty H, Meuth ML (2015) miR-126 is involved in vascular remodeling under laminar shear stress. Biomed Res Int 2015(2):497280
Fichtlscherer S, De Rosa S, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm C, Röxe T, Müller-Ardogan M, Bonauer A, Zeiher A, Dimmeler S (2010) Circulating microRNAs in patients with coronary artery disease. Circ Res 107:677–684
Zhong Z, Hou J, Zhang Q, Zhong W, Li B, Li C, Liu Z, Yang M, Zhao P (2018) Circulating microRNA expression profiling and bioinformatics analysis of dysregulated microRNAs of patients with coronary artery disease. Medicine 97(27):e11428
Boulberdaa M, Scott E, Ballantyne M, Garcia R, Descamps B, Angelini GD, Brittan M, Hunter A, McBride M, McClure J, Miano JM (2016) A role for the long noncoding RNA SENCR in commitment and function of endothelial cells. Mol Ther 24(5):978–990
Zhang Z, Gao W, Long QQ, Zhang J, Li YF, Yan JJ, Yang ZJ, Wang LS (2017) Increased plasma levels of lncRNA H19 and LIPCAR are associated with increased risk of coronary artery disease in a Chinese population. Sci Rep 7(1):1–9
Menghini R, Casagrande V, Cardellini M, Martelli E, Terrinoni A, Amati F, Vasa-Nicotera M, Ippoliti A, Novelli G, Melino G (2009) MicroRNA 217 modulates endothelial cell senescence via silent information regulator 1. Circulation 120(15):1524–1532
Li D, Yang P, Xiong Q, Song X, Yang X, Liu L, Yuan W, Rui YC (2010) MicroRNA-125a/b-5p inhibits endothelin-1 expression in vascular endothelial cells. J Hypertens 28(8):1646–1654
Soh J, Iqbal J, Queiroz J, Fernandez-Hernando C, Hussain MM (2013) Microrna-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat Med 19(7):892–900
Horie T, Baba O, Kuwabara Y, Chujo Y, Watanabe S, Kinoshita M, Horiguchi M, Nakamura T, Chonabayashi K, Hishizawa M (2012) Micro RNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/− mice. J Am Heart Assoc 1(6):e003376
Fu Y, Zhang Y, Wang Z, Wang L, Wei X, Zhang B, Wen Z, Fang H, Pang Q, Yi F (2010) Regulation of NADPH oxidase activity is associated with miRNA-25-mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol 32(6):581–589
Acknowledgements
We gratefully acknowledge Professor M Radhakrishna Pillai, Director, Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram for providing approval for this manuscript. We also acknowledge the funding given to us by KSCSTE-KBC, Government of Kerala (Grant File No: 050/YIPB/KBC/2017/KSCSTE). We acknowledge Dr N. Radhakrishnan Foundation Trust for the research support.
Author information
Authors and Affiliations
Contributions
SS, KCL, and AS conceived the theme, designed and prepared the manuscript, SS, RN, and CCK critically reviewed the manuscript and contributed to the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declared no potential conflicts of interest concerning the research, authorship and publication of this research article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Karthika, C.L., Ahalya, S., Radhakrishnan, N. et al. Hemodynamics mediated epigenetic regulators in the pathogenesis of vascular diseases. Mol Cell Biochem 476, 125–143 (2021). https://doi.org/10.1007/s11010-020-03890-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-020-03890-9