
Abstract
Cellular senescence is commonly initiated in response to replicative or cell stress pathways. Senescent cells remain in a state of permanent cell cycle arrest, and although being metabolically active, they exhibit distinct senescence phenotypes. Though cellular senescence may be beneficial in tumour suppression and wound healing, it is commonly associated with age-related diseases. There are various mechanisms and drivers that contribute to ageing, but it is becoming increasingly apparent that processes related to chromatin and the epigenome are also important. Indeed, three of the nine hallmarks of ageing are genome specific including genomic instability, epigenetic alterations and telomere attrition. With the advent of new technologies like DNA adenine methyltransferase identification and chromosome conformation capture, the features and complexity of the ageing genome are being revealed. This chapter will address key characteristics of interphase nuclei during cellular senescence including the spatio-temporal organisation of chromosomes, chromatin remodelling and epigenome changes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
The Senescence Phenotype
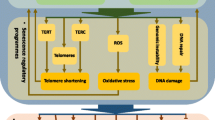
The term “cellular senescence” was originally coined by Hayflick in 1965 (Hayflick 1965). It was described as an important mechanism to suppress tumorigenicity. Senescence can be categorised into four types as shown in Fig. 5.1: (1) replicative senescence (RS) as a result of telomere dysfunction or shortening; (2) genotoxic stress-induced senescence due to endogenous stress, e.g. oxidative stress and severe or irreparable DNA damage; (3) oncogene-induced senescence (OIS) via the activation of aberrant signalling pathways caused by different mechanisms including natural endogenous processes such as mitogenic signalling or oxidative respiration, physical or chemical insults encountered during life or therapeutic treatment such as irradiation or chemotherapy; and (4) embryonic-senescence which occurs in a developmentally regulated manner (Coppé et al. 2010; Munoz-Espin et al. 2013; Storer et al. 2013; Graziano and Gonzalo 2017). The characteristics of senescence may vary depending on the mechanism by which it was induced; for instance, a senescence-associated secretory phenotype (SASP) that secretes proinflammatory mediators is present with some forms of senescence, but not others (Coppé et al. 2010). Regardless of the type of senescence, they each share the characteristic arrest in cell proliferation (Coppé et al. 2010). This chapter will concentrate on how the genome and its behaviour are altered during senescence.

The four main categories of senescence. Genomic stress-induced senescence can be induced via products from cellular metabolism, e.g. reactive oxygen species (ROS) produced by mitochondria or DNA damage due to errors in DNA replication, recombination or repair mechanisms. Activation of aberrant signalling pathways can lead to oncogene-induced senescence and may result from mitogenic signalling or genotoxic agents such as chemical mutagens and radioactivity. Embryonic senescence is important for developmentally regulated growth and patterning. Replicative senescence can lead to irreversible cell cycle arrest by telomere shortening or dysfunction
Organisation of Chromatin and the Epigenome During Ageing
DNA contains genetic information that, when expressed, ultimately codes for the synthesis of a range of proteins vital for the correct functioning of cells, tissues and the whole organism. The genome, when housed in cell nuclei, needs to be organised correctly so that this information can be safely conveyed during proliferation and cell division to daughter cells, be protected from damage and allow genes to be expressed or repressed depending on the protein requirements of the cell and differentiated tissue. The nucleosome is an octamer of histone proteins composed of two copies of histones H2A, H2B, H3 and H4. There are approximately 30 million nucleosomes within the genome, and DNA wraps around these nucleosome complexes to form chromatin (Xu and Liu 2019). Epigenetics involves heritable changes that alter the expression of genes but do not change the DNA sequence. These modifications can act directly on the DNA by adding methyl groups to cytosine; through post-translational modifications to histones including acetylation, methylation, phosphorylation, sumoylation or ubiquitination; or via non-coding RNAs such as microRNA (miRNA), Piwi-interacting RNA (piRNA) and small interfering RNA (siRNA) (Dupont et al. 2009; Wei et al. 2017). Deregulation of epigenetic mechanisms has been highlighted in disease aetiology and ageing. Epigenetic clocks predict the chronological age of individuals by studying the methylation status of cytosines in specific GC-rich regions of the genome, known as CpG islands (Horvath 2013). Mathematical algorithms are employed to determine DNA methylation levels (5-methylcytosine or 5mC) from sets of CpG islands to estimate the age of the DNA source (Horvath and Raj 2018). CpG islands are commonly found near promoter regions of the genome, are ≥0.5 kb long with a GC content of ≥55% and are generally unmethylated (Jeziorska et al. 2017). Global DNA hypomethylation, with hypermethylation of specific loci, is associated with physiological ageing (Gensous et al. 2017). Changes in DNA methylation have been demonstrated in a number of age-related diseases such as cancer (Xie et al. 2019), Parkinson’s disease (Miranda-Morales et al. 2017; Navarro-Sánchez et al. 2018) and Alzheimer’s disease (Levine et al. 2015) as well in cells derived from Hutchinson-Gilford progeria syndrome (HGPS) patients (Ehrlich 2019). However, epigenetic changes during ageing are complex. Although region-specific hypermethylation may be determined at specific CpG islands and gene loci, ageing is also associated with global hypomethylation across the genome (Gensous et al. 2017) and loss of heterochromatin (Goldman et al. 2004; Chandra et al. 2015).
The degree of chromatin compaction can vary in cells, with euchromatin being less compact and open in structure and heterochromatin being more condensed. Generally, euchromatin is rich in CpG islands, has a high GC content, is gene-dense and is associated with short interspersed elements (SINEs) and transcriptional activity (Medstrand et al. 2002; Elbarbary et al. 2016; Vanrobays 2017). Conversely, heterochromatin is AT-rich and gene-poor, associated with long interspersed elements (LINEs), and is inaccessible to transcription factors (Vanrobays 2017; Medstrand et al. 2002; Elbarbary et al. 2016). Epigenetically, histones in heterochromatin generally have methylated H3K9 and H3K27, whilst euchromatin has both acetylation and methylation of H3K4 and H3K36 (Ahringer and Gasser 2018). Heterochromatin can be further subdivided into constitutive heterochromatin and facultative heterochromatin. Constitutive heterochromatin is not transcribed, contains highly repetitive sequences and is H3K9 methylated to maintain a stable condensed state important for chromosome structure such as in centromeres and telomeres (Ahringer and Gasser 2018). Facultative heterochromatin is reversible and may adopt both open or compact conformations according to (1) spatial parameters, e.g. changes in nuclear localisation due to factors such as signalling; (2) temporal changes, e.g. within the cell cycle or development; or (3) heritable factors, e.g. chromosome X inactivation (Trojer and Reinberg 2007). Thus, euchromatin has the potential to be decondensed and express genes in certain tissues. Euchromatic and heterochromatic domains are established during embryogenesis and development to generate tissue-specific gene expression patterns (Villeponteau 1997). Commonly within interphase nuclei, heterochromatin is concentrated at the nuclear periphery, nucleoli, centromeres and telomeres (Goldman et al. 2002), whilst euchromatin is positioned within the nuclear interior (Romero-Bueno et al. 2019). However, ageing is associated with substantial changes in heterochromatin distribution and epigenetic modifications.
During ageing, altered histone modifications and the redistribution of heterochromatin is thought to be associated with changes in global gene expression and genomic instability. Whole-genome bisulfite sequencing (WGBS) and CpG DNA methylation microarrays have been used to examine the epigenetic profiles of samples derived from a newborn and centenarian (103-year-old) (Heyn et al. 2012). Overall, the centenarian sample had a lower DNA methylation content, with the most hypomethylated sequences in CpG-poor promoters and tissue-specific genes (Heyn et al. 2012). Interestingly, the methylation status in middle-aged adults showed an intermediate level of global DNA methylation, suggesting an accumulative change with advancing age (Heyn et al. 2012). This is not unique as loss of heterochromatin has also been linked to an ageing phenotype in model organisms including Caenorhabditis elegans and Drosophila (Haithcock et al. 2005; Larson et al. 2012; Maleszewska et al. 2016). Modifications to histones are made through histone-modifying enzymes including histone methyltransferases, histone demethylases, histone deacetylases and histone acetylases (Black et al. 2012). Therefore, changes in expression or activity of these enzymes may have a profound influence on the epigenetic landscape of the genome. This is observed in Arabidopsis thaliana whereby reduced transcription of methyltransferases and increased transcription of demethylases are associated with hypomethylation in ageing (Ogneva et al. 2016). Furthermore, mutations in a H3K4 methyltransferase in C. elegans and yeast have been shown to reduce longevity, whilst reduced levels of H3K36 demethylase increases lifespan (Sen et al. 2015; Ni et al. 2012).
Epigenetic changes during ageing and loss of heterochromatin also contribute to the derepression of previously silenced genes at those loci (Sturm et al. 2015). This can result in the activation and potential remobilisation of transposable elements (TEs) throughout the genome (Sturm et al. 2015). Given that nearly half of the human genome consists of TEs, this could lead to genomic instability if a TE were to relocate into a coding or regulatory sequence within the genome (Mills et al. 2006; de Koning et al. 2011; Sturm et al. 2015). Ultimately, the resulting DNA damage and instability may result in age-related diseases such as cancer (O’Donnell and Burns 2010), and there are data to demonstrate this mobility, enhancing senescence in humans (Baillie et al. 2011; De Cecco et al. 2013; Keyes 2013).
Nucleosome density has been shown to alter during ageing and is associated with a loss of histones (Hu et al. 2014; Song and Johnson 2018). Nucleosome density naturally varies across the genome with transcriptionally active regions having a lower density and more open chromatin and transcriptionally inactive regions being densely populated with nucleosomes (Boeger et al. 2003; Sidler et al. 2017). Loss of nucleosomes in yeast leads to an increase in transcriptional activity from previously repressed promoters and corresponds with extensive chromosomal alterations and elevation of DNA strand breaks (Hu et al. 2014). Changes in nucleosome density could be due to two mechanisms: (1) alterations in the activity of histone chaperones and (2) reduction in histone biogenesis within the cell (Booth and Brunet 2016). There is evidence that nucleosome assembly may be regulated by the histone chaperone ASF1 in both a DNA synthesis-dependent and DNA synthesis-independent manner along with other histone chaperones, chromatin assembly factor 1 (CAF-1) and histone repression A factor (HIRA) (Galvani et al. 2008). In yeast, loss of function of ASF1 may lead to aberrant heterochromatin formation and genomic instability (Tanae et al. 2012). Indeed, ASF1 expression decreases with increasing age in human cells (O’Sullivan et al. 2010). Here, the synthesis of histones in fibroblasts derived from an old individual was half that compared to those derived from a child (O’Sullivan et al. 2010). Histone biosynthesis was also altered in replicative senescent IMR90 and WI38 cells, leading to downregulation of the synthesis of histones H3 and H4 and post-translational modifications (O’Sullivan et al. 2010; Song and Johnson 2018). Thus, nucleosome density combined with changes in epigenetic post-translational modifications could be an important factor in the loss of heterochromatin observed in ageing.
Conversely , there are regions of the genome that become associated with heterochromatin during ageing (Tsurumi and Li 2012). Chromatin may be organised within senescence-associated heterochromatin foci (SAHF) (Morris et al. 2019; Lenain et al. 2017; Braig et al. 2005; Michaloglou et al. 2005; Haugstetter et al. 2010). SAHF share epigenetic features and characteristics commonly found in heterochromatin including late replicating DNA domains (Shah et al. 2013); epigenetic markers H3K9me3 and H3K27me3 (Chandra et al. 2012; Chandra et al. 2015; Chandra and Narita 2013); HP1 α, β and γ (Boumendil et al. 2019); heterochromatic proteins; histone variant macroH2A; and high-mobility group A (HMGA) proteins (Morris et al. 2019). SAHF structure encompasses a chromatin core that is compacted and enriched in H3K9m3 (a marker of constitutive heterochromatin) and an outer ring of chromatin containing H3K27me3 (a marker of facultative heterochromatin), which is protein rich but more relaxed (Lenain et al. 2017; Chandra et al. 2012; Sadaie et al. 2013). Over 90% of SAHF are commonly observed in cells that have undergone OIS (Chandra et al. 2015) with only a small proportion seen in replicative senescence in cultures (Chandra et al. 2015; Boumendil et al. 2019). SAHFs are not present in HGPS or senescent mouse cells, and it is unclear if they occur in vivo (Lazzerini Denchi et al. 2005; Shumaker et al. 2006; Scaffidi and Misteli 2006; Swanson et al. 2013). The formation of SAHF represses the expression of genes that are important for proliferation and the cell cycle such as cyclin A, proliferating nuclear antigen (PCNA), E2F target genes (Aird and Zhang 2013) and cyclin D1 (Zhang et al. 2007; Park et al. 2018) and thus leads to senescence. Evidence suggests that SAHF may result from an increase in nuclear pore density during OIS, with the nucleoporin TPR having a vital role in inducing the formation of SAHF and their maintenance (Boumendil et al. 2019).
Epigenetic modifications and heterochromatin distribution are altered in premature ageing syndromes. The majority of premature ageing syndromes are caused by either mutations leading to alterations in the nuclear lamina and matrix proteins or via defects in DNA repair systems (Musich and Zou 2009; Tiwari and Wilson 3rd 2019). Hutchinson-Gilford progeria syndrome (HGPS) is a premature ageing disease caused by a mutated lamin A protein. Here, there is a reduction in H3K9me3 and HP1 and loss of peripheral heterochromatin (Shumaker et al. 2006; Scaffidi and Misteli 2006). Werner syndrome (WS) is another progeroid syndrome that similarly has a loss of H3K9me3. WS is caused by mutations within the Werner helicase (WRN). Interestingly, WRN has been shown to associate with the methyltransferase SUV39H1 and HP1α and thus may be important in regulating heterochromatin during ageing (Zhang et al. 2015; Wang et al. 2016). Mesenchymal stem cells with an induced WRN deficiency show altered heterochromatin distribution and global loss of associated epigenetic methylation of histone H3 (Zhang et al. 2015; Shumaker et al. 2006). Loss of peripheral heterochromatin adjacent to the nuclear envelope (NE) (Goldman et al. 2004; Zhang et al. 2015) and a reduction in H3K9me3 and H3K27me3 levels but increase in H4K27me3 have been shown in HGPS cultured cells (Shumaker et al. 2006; Scaffidi and Misteli 2006).
Nuclear Lamina and Nucleoskeleton
The nuclear lamina is located adjacent to the inner nuclear membrane (INM) and is composed of type V intermediate filaments proteins—lamins and lamina-associated proteins. Lamins are subdivided into the B-type lamins which are constitutively expressed within mammalian cells and A-type lamins that are developmentally regulated in differentiated cells. The nuclear lamina interacts with INM proteins, nuclear pore complexes and chromatin. It has a number of important roles including organising chromatin, involvement in DNA replication and gene expression and to support structurally the nucleus and its processes (Cau et al. 2014). The peripheral nuclear lamina is interconnected and part of a larger structural protein network known as the nuclear matrix (NM) (Cau et al. 2014) or nucleoskeleton. This structure is believed to be a filamentous meshwork of proteins (e.g. lamins A and C), DNA and RNA localised throughout the nucleoplasm that are resistant to high-salt treatment and nucleases during experiments. Similarly, the matrix structure is important for the structural integrity of nuclei and also supports gene expression, chromatin organisation, DNA replication and repair (Chattopadhyay and Pavithra 2007; Wilson and Coverley 2017; Bridger et al. 2014; Mehta et al. 2007; Elcock and Bridger 2008; Godwin et al. 2021). The NM interacts with chromatin typically via specialised AT-rich DNA sequences called scaffold/matrix attachment regions (S/MARs) (Barboro et al. 2012) and helps maintain the compartmentalisation of the nucleus and higher-order chromatin organisation important for the spatio-temporal dynamics of the cell.
Laminopathies include progeroid syndromes linked to A-type lamin mutations. These are typically characterised as having nuclear envelope deformities (Cau et al. 2014), with blebbing, herniations, invaginations and altered nuclear shape. These are caused by mutations that influence the post-translational processing of proteins, ultimately leading to defective protein function. For instance, in HGPS, there is a cryptic splice site that leads to a truncated form of lamin A which is permanently bound to a farnesyl moiety, termed “progerin” (Gilbert and Swift 2019). The build-up of progerin at the INM is toxic, leading to altered nuclear envelope integrity and perturbed chromatin organisation (Chandra et al. 2015; Stephens et al. 2018; Bikkul et al. 2018). Although progerin is primarily associated with HGPS, it has been suggested that a progerin-dependent mechanism may lead to natural ageing (Scaffidi and Misteli 2006; McClintock et al. 2007; Ashapkin et al. 2019). Evidence acquired by reverse transcription polymerase chain reaction (RT-PCR) has shown that fibroblasts obtained from naturally aged individuals expressed progerin mRNA, albeit at a low frequency of less than 50-fold (Scaffidi and Misteli 2006). Progerin has also been detected in cell lines derived from skin biopsies that had undergone prolonged cell culture, particularly in cells derived from older individuals (McClintock et al. 2007). However, it should be noted that the levels were very low. Senescence is frequently accompanied with profound changes to the INM organisation and accompanying processes.
The nuclear lamina interacts with the genome directly through lamina-associated domains (LADs) and via lamin-binding partners. DNA adenine methyltransferase identification (DamID) technology has been used to extensively map LADs throughout the nucleus. This technique is used to identify binding sites between DNA and chromatin-binding proteins. For instance, combining a nuclear lamin protein (e.g. lamin B1) to a bacterial DNA adenine methyltransferase (Dam) will highlight areas of DNA that have been in contact with the nuclear lamins as they will undergo adenine methylation. As adenine methylation does not naturally occur in eukaryotes, it acts as a detectable marker. LADs are of fundamental importance in anchoring transcriptionally silent heterochromatin to the nuclear lamina and maintaining the three-dimensional spatial arrangement of chromosomes (van Steensel and Belmont 2017; Romero-Bueno et al. 2019). However, lamins can be found throughout the nucleoplasm (Bridger et al. 1993) and not just at the nuclear envelope, and thus, this should be taken into consideration. LADs are also heterogeneous between cell types (Peric-Hupkes et al. 2010; Meuleman et al. 2013) but are associated with lamin B1 and lamin B1 receptor (LBR), which anchor heterochromatin to the nuclear lamina (Lukasova et al. 2018). However, during cellular senescence, LADs become extensively redistributed (Lochs et al. 2019). Normally, after DNA replication, DNA methyltransferase DNMT1 restores the histone methylation pattern; however, this appears to fail during senescence (Lochs et al. 2019) leading to hypomethylation. This hypomethylation, combined with the loss of lamin B1, leads to heterochromatin dissociating from the nuclear lamina (Lochs et al. 2019) and away from the nuclear periphery. This LAD rearrangement may also be associated with the accumulation of SAHF, which relocates heterochromatin to the nuclear interior (Lenain et al. 2017; Chandra et al. 2015).
Nucleolus
The nucleolus is also important for spatio-temporal regulation of the genome and is formed from chromosomes containing active nucleolar organiser regions (NORs) and other non-acrocentric but gene-rich chromosomes (van Koningsbruggen et al. 2010; Nemeth et al. 2010). Nucleoli are important in ribosome biogenesis and are initiated from the transcription of ribosomal RNA (rRNA) genes found in high copy number and arranged in tandem repeats within NORs (Bersaglieri and Santoro 2019). Genomic regions that are localised in close proximity to the nucleolus are termed nucleolus-associated domains (NADs) (Nemeth et al. 2010). Genome-wide mapping has demonstrated that NADs derived from HeLa, IMR90 and HT1080 human cell lines have a low gene density, low transcriptional levels, late-replicating loci and heterochromatin enriched with repressive histone modifications H4K20me3, H3K27me3 and H3K9me3 (van Koningsbruggen et al. 2010; Nemeth et al. 2010; Dillinger et al. 2017). During senescence, nucleoli may fuse and are associated with an increased size (Mehta et al. 2007). H3K9me3 modified heterochromatin localised at the nucleolus, is remodelled and is coupled with an observed dissociation of centromeric and pericentromeric satellite regions away from the nucleolus (Dillinger et al. 2017). Interestingly, mapping of NADs using Hi-C in senescent cells remains similar to that seen in proliferating cell lines although there are changes in sub-NADs association with nucleoli (subdomains smaller than 100 kb), which appear to correspond to transcriptional changes (Dillinger et al. 2017; Mehta et al. 2010).
Centromeres and Telomeres
Centromeres are heterochromatic regions and have satellite II and α-satellite repeat sequences that are normally constitutively repressed (De Cecco et al. 2013). However, in replicative senescent cells, the pericentric satellite has been shown to distend, and chromatin is reorganised becoming more accessible and hypomethylated (De Cecco et al. 2013; Cruickshanks et al. 2013). This centromere distension has been termed “senescence-associated distension of satellites” (SADS) and is associated with epigenetic modifications associated with early senescence (Criscione et al. 2016). Silencing of pericentric satellite DNA is helped and maintained by SIRT6, a histone deacetylase, which removes H3K18 acetylation in normal proliferative cells (Nagai et al. 2015; Tasselli et al. 2016). It is possible that SIRT6 depletion could lead to senescence (Tasselli et al. 2016; Nagai et al. 2015). Indeed, SIRT6 is an early factor sequestered to double-strand breaks, so prolonged recruitment to irreversibly damaged DNA associated with ageing may lead to depletion of SIRT6 at pericentric satellite DNA leading to the unravelling and SADS phenotype (Toiber et al. 2013; Nagai et al. 2015; Tasselli et al. 2016). SADS occurs as a common feature of senescence, irrespective of how senescence is induced and whether the p16 or p21 pathways are activated (Swanson et al. 2013). Unlike SAHF, SADS are found both during normal senescence and in progeria (Swanson et al. 2013).
Telomeres , and their associated shelterin protein complex, are located at the ends of linear chromosomes and have a protective role in preventing genome instability by shielding exposed ends of DNA. During replication, DNA polymerases are unable to completely replicate the telomere region of the lagging strand leading to shortening due to the progressive loss of telomere repeats. This has been termed the “end-replication problem.” Consequently, the length of the telomeres shortens with each cell division leading to attrition. This has been extensively reported in ageing studies and is particularly pronounced within in vitro primary cells leading to a finite number of cell divisions or “replicative senescence” due to the shortened telomere lengths. The resulting exposure of the chromosome ends leads to the activation of DNA repair mechanisms and a persistent DNA damage response (DDR) (Victorelli and Passos 2017). Nevertheless, telomere dysfunction can occur irrespective of length with telomeric DNA damage being associated with an increase in senescence markers such as p16 (Victorelli and Passos 2017; Birch et al. 2015). Indeed, in postmitotic cardiomyocytes, there is an increase in DNA damage foci associated with telomeres during ageing (Anderson et al. 2019). Telomeres interact with a telomere repeat-binding factors 1 and 2 (TRF1 and TRF2, respectively) to form t-loops. TRF1 is thought to prevent fusion of telomere ends and regulate telomere length (van Steensel et al. 1998; Celli and de Lange 2005), whilst TRF2 forestalls the DNA damage response (Karlseder et al. 2004).
The positioning of telomeres in interphase nuclei appears to vary between species, cell type and disease status (Weierich et al. 2003; Chuang et al. 2004; Arnoult et al. 2010; Gilson et al. 2013). Nevertheless, positioning is non-random and integral to genomic stability (Chuang et al. 2004). There is evidence that telomeres are closely associated with the nucleoskeleton and A-type lamins (Ottaviani et al. 2009; de Lange 1992) in addition to reports that the telomeres of acrocentric chromosomes localise to perinucleolar regions (Ramirez and Surralles 2008). Loss of TRF2 has been linked to an increased DNA damage response and senescence (van Steensel et al. 1998; Okamoto et al. 2013). TRF2 may interact with lamin A/C, which are important proteins within the INM and nuclear matrix (Wood et al. 2014). In HGPS, there is a reduction in TRF2 (Wood et al. 2014) and apparent telomere loss. Interestingly, studies using human telomerase reverse transcriptase (hTERT) to evade replicative senescence on proliferating fibroblasts and HGPS cells have shown dramatic genome reorganisation with the mislocalisation of whole chromosomes 18 in the control cells and chromosome 18 and X in HGPS cells (Bikkul et al. 2019). Differences in telomere organisation have also been demonstrated in terminally differentiated cells or quiescent cells in culture that have been contact inhibited (Nagele et al. 2001). Here, interphase nuclei exhibit close telomeric associations within quiescent, non-cycling cells compared with proliferating cells (Nagele et al. 2001). Clustering of telomeres has also been demonstrated in mouse embryonic fibroblasts, as well as partial association with centromeric clusters and promyelocytic leukaemia bodies (PML) (Molenaar et al. 2003; Weierich et al. 2003; Uhlirova et al. 2010).
Epigenetic changes also accompany telomere maintenance during ageing (Uhlirova et al. 2010). Levels of H3K9me3, H4K20me3 and HP1 protein have been shown to decrease in HGPS (Scaffidi and Misteli 2006). Treatment of embryonic mouse fibroblast with the histone deacetylase inhibitor Trichostatin A (TSA) led to the repositioning of telomeres to the nuclear interior and centromeres towards the nuclear periphery (Uhlirova et al. 2010). This has been observed in another system where telomere and centromeres were often polarised to opposite ends of the chromosome territories (Amrichova et al. 2003). This may have further ramifications as genes located in close proximity to telomeric heterochromatin are often silenced due to the “telomeric position effect” (TPE) (Baur et al. 2001; Ning et al. 2003). There is evidence of expression changes in telomeric genes during senescence (Ning et al. 2003) with increased expression of the 16q telomeric genes MGC3101 and CPNE7 in senescence and GAS11 and CDK10 in both senescent/quiescent cells (Ning et al. 2003). Thus, change in the epigenetic status of constitutive heterochromatin could lead to senescent-specific expression patterns .
Chromosomes and Chromosome Territories
In interphase nuclei, whole chromosomes occupy specific non-random locations within the nuclear space called chromosome territories (CTs), occupying similar locations between cell types and in vitro compared to ex vivo (Foster et al. 2012). In proliferating human fibroblasts, CTs are functionally compartmentalised with gene-rich chromosomes occupying a central position within the nucleus and are generally characterised as having higher levels of gene expression, open chromatin conformations and early replication timing (Croft et al. 1999; Cremer and Cremer 2001; Foster and Bridger 2005; Bridger et al. 2014). Conversely, gene-poor chromosomes are commonly associated with the nuclear periphery or nucleoli and are synonymous with heterochromatin, repression of gene expression, repressive histone modifications and late replication timing (Chiang et al. 2018; Croft et al. 1999). These functionally different compartments have been shown to be of importance during ageing. There is evidence that whole chromosome territories can occupy different nuclear locations during senescence (Bridger et al. 2000; Mehta et al. 2007). For instance, human chromosome 18 has been shown to occupy a peripheral position within proliferating fibroblast nuclei; however, upon replicative senescence, chromosome 18 was shown to be repositioned away from the nuclear periphery (Bridger et al. 2000). Therefore, CTs appear to be repositioned from a gene-density radial distribution in proliferating fibroblasts to a size-correlated radial position within senescent fibroblasts whereby small chromosomes are positioned within the nuclear interior and large chromosomes are localised at the nuclear periphery (Mehta et al. 2007). Altered nuclear positioning of whole chromosomes 13 and 18 from the nuclear periphery towards the interior has also been shown in cells with A-type lamins (Meaburn et al. 2005; Meaburn et al. 2007). Indeed, genome reorganisation is more finely observed with changes in the Topologically Associated Domain (TADs) compartment re-positioning in replicative senescent cells (Criscione et al. 2016; Sun et al. 2018).
Advances in Technologies
New technologies including CRISPR-multicolour (Ma et al. 2015) and CRISPRainbow (Ma et al. 2016) enable the study of higher-order chromatin and nuclear architecture organisation. Here, a live-cell system that utilises super-resolution microscopy is used to track genomic loci that are labelled by different coloured fluorescent-tagged dCas9-sgRNAs (Ma et al. 2016). Changes in transcriptional activity upon a specific stimulus can also be investigated by tracking the dynamics of a promoter and its interaction with cis-/trans-acting regulatory elements (Lau and Suh 2017). New advances in interrogating Hi-C data from senescent cells are permitting chromosome territory postions to be extrapolated from these data sets (Das et al. 2020).
Summary
The complexity of the ageing genome and structural organisation of the nucleus during senescence are becoming increasingly apparent, especially with advances in microscopy and global analyses such as super-resolution microscopy and chromosome conformation capture. Generally, the ageing epigenome is characterised by global hypomethylation and loss of heterochromatin; however, on the contrary, some regions of the genome are packaged into heterochromatin, e.g. SAHF. Satellite sequences may also be altered in senescence with distension of centromeres, or SADS, and shortening or dysfunction of telomeres. Characteristic structural changes to the nucleus include an increased size in senescent cells, reorganisation of LADs and sub-NADs and nuclear envelope deformities associated with mutations in lamin and lamin-associated proteins. Together, these can lead to large-scale reorganisation of the genome with repositioning of whole chromosome territories. Overall, these fundamental changes to the epigenome lead to alterations in global gene expression and genomic instability associated with ageing (Fig. 5.2).

Differences in nuclear structure organisation within proliferating/young cells and senescent cells. Senescence is associated with chromatin remodelling, loss of peripheral nuclear heterochromatin and an increase in hypomethylation. In addition, satellite DNA becomes unravelled to form SADs, and LADs and NADs are redistributed in senescent cells. Nucleoli may fuse and often have an increased size. SAHF formation is apparent in some senescence cells with a chromatin core enriched in H3K9me3 surrounded by an outer rim rich in H3K27me3. The nuclear lamina contains A- and B-type lamins and lamina-associated proteins that play a role in organising the genome. Within senescent and progeroid cells, the INM organisation is altered and often associated with nuclear envelope deformities
References
Ahringer J, Gasser SM (2018) Repressive chromatin in Caenorhabditis elegans: establishment, composition, and function. Genetics 208(2):491–511. https://doi.org/10.1534/genetics.117.300386
Aird KM, Zhang R (2013) Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol Biol (Clifton, NJ) 965:185–196. https://doi.org/10.1007/978-1-62703-239-1_12
Amrichova J, Lukasova E, Kozubek S, Kozubek M (2003) Nuclear and territorial topography of chromosome telomeres in human lymphocytes. Exp Cell Res 289(1):11–26. https://doi.org/10.1016/s0014-4827(03)00208-8
Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D, Proctor C, Correia-Melo C, Victorelli S, Fielder E, Berlinguer-Palmini R, Owens A, Greaves LC, Kolsky KL, Parini A, Douin-Echinard V, LeBrasseur NK, Arthur HM, Tual-Chalot S, Schafer MJ, Roos CM, Miller JD, Robertson N, Mann J, Adams PD, Tchkonia T, Kirkland JL, Mialet-Perez J, Richardson GD, Passos JF (2019) Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J 38(5):e100492. https://doi.org/10.15252/embj.2018100492
Arnoult N, Schluth-Bolard C, Letessier A, Drascovic I, Bouarich-Bourimi R, Campisi J, S-h K, Boussouar A, Ottaviani A, Magdinier F, Gilson E, Londoño-Vallejo A (2010) Replication timing of human telomeres is chromosome arm–specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet 6(4):e1000920. https://doi.org/10.1371/journal.pgen.1000920
Ashapkin VV, Kutueva LI, Kurchashova SY, Kireev II (2019) Are there common mechanisms between the Hutchinson-Gilford progeria syndrome and natural aging? Front Genet 10:455. https://doi.org/10.3389/fgene.2019.00455
Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, Talbot RT, Gustincich S, Freeman TC, Mattick JS, Hume DA, Heutink P, Carninci P, Jeddeloh JA, Faulkner GJ (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479(7374):534–537. https://doi.org/10.1038/nature10531
Barboro P, Repaci E, D’Arrigo C, Balbi C (2012) The role of nuclear matrix proteins binding to matrix attachment regions (Mars) in prostate cancer cell differentiation. PLoS One 7(7):e40617. https://doi.org/10.1371/journal.pone.0040617
Baur JA, Zou Y, Shay JW, Wright WE (2001) Telomere position effect in human cells. Science 292(5524):2075–2077. https://doi.org/10.1126/science.1062329
Bersaglieri C, Santoro R (2019) Genome organization in and around the nucleolus. Cell 8(6):579. https://doi.org/10.3390/cells8060579
Bikkul MU, Clements CS, Godwin LS, Goldberg MW, Kill IR, Bridger JM (2018) Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology 19(6):579–602. https://doi.org/10.1007/s10522-018-9758-4
Bikkul MU, Faragher RGA, Worthington G, Meinke P, Kerr ARW, Sammy A, Riyahi K, Horton D, Schirmer EC, Hubank M, Kill IR, Anderson RM, Slijepcevic P, Makarov E, Bridger JM (2019) Telomere elongation through hTERT immortalization leads to chromosome repositioning in control cells and genomic instability in Hutchinson-Gilford progeria syndrome fibroblasts, expressing a novel SUN1 isoform. Genes Chromosomes Cancer 58(6):341–356. https://doi.org/10.1002/gcc.22711
Birch J, Anderson RK, Correia-Melo C, Jurk D, Hewitt G, Marques FM, Green NJ, Moisey E, Birrell MA, Belvisi MG, Black F, Taylor JJ, Fisher AJ, De Soyza A, Passos JF (2015) DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 309(10):L1124–L1137. https://doi.org/10.1152/ajplung.00293.2015
Black JC, Van Rechem C, Whetstine JR (2012) Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 48(4):491–507. https://doi.org/10.1016/j.molcel.2012.11.006
Boeger H, Griesenbeck J, Strattan JS, Kornberg RD (2003) Nucleosomes unfold completely at a transcriptionally active promoter. Mol Cell 11(6):1587–1598
Booth LN, Brunet A (2016) The aging epigenome. Mol Cell 62(5):728–744. https://doi.org/10.1016/j.molcel.2016.05.013
Boumendil C, Hari P, Olsen KCF, Acosta JC, Bickmore WA (2019) Nuclear pore density controls heterochromatin reorganization during senescence. Genes Dev 33(3–4):144–149. https://doi.org/10.1101/gad.321117.118
Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA (2005) Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436(7051):660–665. https://doi.org/10.1038/nature03841
Bridger JM, Kill IR, O’Farrell M, Hutchison CJ (1993) Internal Lamin structures within G1 nuclei of human dermal fibroblasts. J Cell Sci 104(Pt 2):297–306
Bridger JM, Boyle S, Kill IR, Bickmore WA (2000) Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr Biol 10(3):149–152
Bridger JM, Arican-Gotkas HD, Foster HA, Godwin LS, Harvey A, Kill IR, Knight M, Mehta IS, Ahmed MH (2014) The non-random repositioning of whole chromosomes and individual gene loci in interphase nuclei and its relevance in disease, infection, aging, and cancer. Adv Exp Med Biol 773:263–279. https://doi.org/10.1007/978-1-4899-8032-8_12
Cau P, Navarro C, Harhouri K, Roll P, Sigaudy S, Kaspi E, Perrin S, De Sandre-Giovannoli A, Levy N (2014) Nuclear matrix, nuclear envelope and premature aging syndromes in a translational research perspective. Semin Cell Dev Biol 29:125–147. https://doi.org/10.1016/j.semcdb.2014.03.021
Celli GB, de Lange T (2005) DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7(7):712–718. https://doi.org/10.1038/ncb1275
Chandra T, Narita M (2013) High-order chromatin structure and the epigenome in SAHFs. Nucleus (Austin, Tex) 4(1):23–28. https://doi.org/10.4161/nucl.23189
Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, Stark R, Janky R, Narita M, Xue L, Chicas A, Nunez S, Janknecht R, Hayashi-Takanaka Y, Wilson MD, Marshall A, Odom DT, Babu MM, Bazett-Jones DP, Tavare S, Edwards PA, Lowe SW, Kimura H, Gilbert DM, Narita M (2012) Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47(2):203–214. https://doi.org/10.1016/j.molcel.2012.06.010
Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret J-Y, Andrews S, Fraser P, Reik W (2015) Global reorganization of the nuclear landscape in senescent cells. Cell Rep 10(4):471–483. https://doi.org/10.1016/j.celrep.2014.12.055
Chattopadhyay S, Pavithra L (2007) MARs and MARBPs: key modulators of gene regulation and disease manifestation. Subcell Biochem 41:213–230
Chiang M, Michieletto D, Brackley CA, Rattanavirotkul N, Mohammed H, Marenduzzo D, Chandra T (2018) Lamina and heterochromatin direct chromosome organisation in senescence and progeria. bioRxiv:468561. https://doi.org/10.1101/468561
Chuang TCY, Moshir S, Garini Y, Chuang AY-C, Young IT, Vermolen B, van den Doel R, Mougey V, Perrin M, Braun M, Kerr PD, Fest T, Boukamp P, Mai S (2004) The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol 2:12–12. https://doi.org/10.1186/1741-7007-2-12
Coppé J-P, Desprez P-Y, Krtolica A, Campisi J (2010) The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144
Cremer T, Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2(4):292–301. https://doi.org/10.1038/35066075
Criscione SW, De Cecco M, Siranosian B, Zhang Y, Kreiling JA, Sedivy JM, Neretti N (2016) Reorganization of chromosome architecture in replicative cellular senescence. Sci Adv 2(2):e1500882. https://doi.org/10.1126/sciadv.1500882
Croft JA, Bridger JM, Boyle S, Perry P, Teague P, Bickmore WA (1999) Differences in the localization and morphology of chromosomes in the human nucleus. J Cell Biol 145(6):1119–1131. https://doi.org/10.1083/jcb.145.6.1119
Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, Berger SL, Adams PD (2013) Senescent cells harbour features of the cancer epigenome. Nat Cell Biol 15(12):1495–1506. https://doi.org/10.1038/ncb2879
Das P, Shen T, McCord RP (2020) Inferring chromosome radial organization from Hi-C data. BMC Bioinf 21(1):511
De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM (2013) Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12(2):247–256. https://doi.org/10.1111/acel.12047
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD (2011) Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7(12):e1002384. https://doi.org/10.1371/journal.pgen.1002384
de Lange T (1992) Human telomeres are attached to the nuclear matrix. EMBO J 11(2):717–724
Dillinger S, Straub T, Németh A (2017) Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS One 12(6):e0178821. https://doi.org/10.1371/journal.pone.0178821
Dupont C, Armant DR, Brenner CA (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 27(5):351–357. https://doi.org/10.1055/s-0029-1237423
Ehrlich M (2019) DNA hypermethylation in disease: mechanisms and clinical relevance. Epigenetics:1–23. https://doi.org/10.1080/15592294.2019.1638701
Elbarbary RA, Lucas BA, Maquat LE (2016) Retrotransposons as regulators of gene expression. Science 351(6274):aac7247. https://doi.org/10.1126/science.aac7247
Elcock LS, Bridger JM (2008) Exploring the effects of a dysfunctional nuclear matrix. Biochem Soc Transactions 36:1378–83
Foster HA, Bridger JM (2005) The genome and the nucleus: a marriage made by evolution. Genome organisation and nuclear architecture. Chromosoma 114(4):212–229. https://doi.org/10.1007/s00412-005-0016-6
Foster HA, Griffin DK, Bridger JM (2012) Interphase chromosome positioning in in vitro porcine cells and ex vivo porcine tissues. BMC Cell Biol 13:30. https://doi.org/10.1186/1471-2121-13-30
Galvani A, Courbeyrette R, Agez M, Ochsenbein F, Mann C, Thuret J-Y (2008) In vivo study of the nucleosome assembly functions of ASF1 histone chaperones in human cells. Mol Cell Biol 28(11):3672–3685. https://doi.org/10.1128/MCB.00510-07
Gensous N, Bacalini MG, Pirazzini C, Marasco E, Giuliani C, Ravaioli F, Mengozzi G, Bertarelli C, Palmas MG, Franceschi C, Garagnani P (2017) The epigenetic landscape of age-related diseases: the geroscience perspective. Biogerontology 18(4):549–559. https://doi.org/10.1007/s10522-017-9695-7
Gilbert HTJ, Swift J (2019) The consequences of ageing, progeroid syndromes and cellular senescence on mechanotransduction and the nucleus. Exp Cell Res 378(1):98–103. https://doi.org/10.1016/j.yexcr.2019.03.002
Gilson E, Giraud-Panis M-J, Pisano S, Bennaroch D, Ledu M-H, Pei B (2013) One identity or more for telomeres? Front Oncol 3(48). https://doi.org/10.3389/fonc.2013.00048
Godwin LS, Bridger JM, Foster HA (2021) Fluorescence in situ hybridization on DNA Halo preparations to reveal whole chromosome, telomeres and gene loci. Jove, in press
Goldman RD, Gruenbaum Y, Moir RD, Shumaker DK, Spann TP (2002) Nuclear lamins: building blocks of nuclear architecture. Genes Dev 16(5):533–547. https://doi.org/10.1101/gad.960502
Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 101(24):8963–8968. https://doi.org/10.1073/pnas.0402943101
Graziano S, Gonzalo S (2017) Mechanisms of oncogene-induced genomic instability. Biophys Chem 225:49–57. https://doi.org/10.1016/j.bpc.2016.11.008
Haithcock E, Dayani Y, Neufeld E, Zahand AJ, Feinstein N, Mattout A, Gruenbaum Y, Liu J (2005) Age-related changes of nuclear architecture in Caenorhabditis elegans. Proc Natl Acad Sci U S A 102(46):16690–16695. https://doi.org/10.1073/pnas.0506955102
Haugstetter AM, Loddenkemper C, Lenze D, Gröne J, Standfuß C, Petersen I, Dörken B, Schmitt CA (2010) Cellular senescence predicts treatment outcome in metastasised colorectal cancer. Br J Cancer 103:505. https://doi.org/10.1038/sj.bjc.6605784
Hayflick L (1965) The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37:614–636
Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, Puca AA, Sayols S, Pujana MA, Serra-Musach J, Iglesias-Platas I, Formiga F, Fernandez AF, Fraga MF, Heath SC, Valencia A, Gut IG, Wang J, Esteller M (2012) Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci U S A 109(26):10522–10527. https://doi.org/10.1073/pnas.1120658109
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14(10):R115. https://doi.org/10.1186/gb-2013-14-10-r115
Horvath S, Raj K (2018) DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet 19(6):371–384. https://doi.org/10.1038/s41576-018-0004-3
Hu Z, Chen K, Xia Z, Chavez M, Pal S, Seol J-H, Chen C-C, Li W, Tyler JK (2014) Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev 28(4):396–408. https://doi.org/10.1101/gad.233221.113
Jeziorska DM, Murray RJS, De Gobbi M, Gaentzsch R, Garrick D, Ayyub H, Chen T, Li E, Telenius J, Lynch M, Graham B, Smith AJH, Lund JN, Hughes JR, Higgs DR, Tufarelli C (2017) DNA methylation of intragenic CpG islands depends on their transcriptional activity during differentiation and disease. Proc Natl Acad Sci 114(36):E7526. https://doi.org/10.1073/pnas.1703087114
Karlseder J, Hoke K, Mirzoeva OK, Bakkenist C, Kastan MB, Petrini JH, de Lange T (2004) The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol 2(8):E240. https://doi.org/10.1371/journal.pbio.0020240
Keyes WM (2013) Rearranging senescence: transposable elements become active in aging cells (comment on DOI 10.1002/bies.201300097). Bioessays 35(12):1023–1023. https://doi.org/10.1002/bies.201300157
Larson K, Yan S-J, Tsurumi A, Liu J, Zhou J, Gaur K, Guo D, Eickbush TH, Li WX (2012) Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet 8(1):e1002473. https://doi.org/10.1371/journal.pgen.1002473
Lau CH, Suh Y (2017) Genome and epigenome editing in mechanistic studies of human aging and aging-related disease. Gerontology 63(2):103–117. https://doi.org/10.1159/000452972
Lazzerini Denchi E, Attwooll C, Pasini D, Helin K (2005) Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol 25(7):2660–2672. https://doi.org/10.1128/mcb.25.7.2660-2672.2005
Lenain C, de Graaf CA, Pagie L, Visser NL, de Haas M, de Vries SS, Peric-Hupkes D, van Steensel B, Peeper DS (2017) Massive reshaping of genome-nuclear lamina interactions during oncogene-induced senescence. Genome Res 27(10):1634–1644. https://doi.org/10.1101/gr.225763.117
Levine ME, Lu AT, Bennett DA, Horvath S (2015) Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY) 7(12):1198–1211. https://doi.org/10.18632/aging.100864
Lochs SJA, Kefalopoulou S, Kind J (2019) Lamina associated domains and gene regulation in development and cancer. Cell 8(3):271
Lukasova E, Kovarik A, Kozubek S (2018) Consequences of Lamin B1 and Lamin B receptor downregulation in senescence. Cell 7(2). https://doi.org/10.3390/cells7020011
Ma H, Naseri A, Reyes-Gutierrez P, Wolfe SA, Zhang S, Pederson T (2015) Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci U S A 112(10):3002–3007. https://doi.org/10.1073/pnas.1420024112
Ma H, Tu L-C, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T (2016) Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nat Biotechnol 34(5):528–530. https://doi.org/10.1038/nbt.3526
Maleszewska M, Mawer JSP, Tessarz P (2016) Histone modifications in ageing and lifespan regulation. Curr Mol Biol Rep 2(1):26–35. https://doi.org/10.1007/s40610-016-0031-9
McClintock D, Ratner D, Lokuge M, Owens DM, Gordon LB, Collins FS, Djabali K (2007) The mutant form of Lamin a that causes Hutchinson-Gilford progeria is a biomarker of cellular aging in human skin. PLoS One 2(12):e1269. https://doi.org/10.1371/journal.pone.0001269
Meaburn KJ, Levy N, Toniolo D, Bridger JM (2005) Chromosome positioning is largely unaffected in lymphoblastoid cell lines containing emerin or A-type Lamin mutations. Biochem Soc Trans 33(Pt 6):1438–1440
Meaburn KJ, Cabuy E, Bonne G, Levy N, Morris GE, Novelli G, Kill IR, Bridger JM (2007) Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell 6(2):139–153. https://doi.org/10.1111/j.1474-9726.2007.00270.x
Medstrand P, van de Lagemaat LN, Mager DL (2002) Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res 12(10):1483–1495. https://doi.org/10.1101/gr.388902
Mehta IS, Figgitt M, Clements CS, Kill IR, Bridger JM (2007) Alterations to nuclear architecture and genome behavior in senescent cells. Ann N Y Acad Sci 1100:250–263. https://doi.org/10.1196/annals.1395.027
Mehta IS, Bridger JM, Kill IR (2010) Progeria, the nucleolus and farnesyltransferase inhibitors. Biochem Soc Trans 38(Pt 1):287–291. https://doi.org/10.1042/bst0380287
Meuleman W, Peric-Hupkes D, Kind J, Beaudry J-B, Pagie L, Kellis M, Reinders M, Wessels L, van Steensel B (2013) Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res 23(2):270–280. https://doi.org/10.1101/gr.141028.112
Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, Majoor DM, Shay JW, Mooi WJ, Peeper DS (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436(7051):720–724. https://doi.org/10.1038/nature03890
Mills RE, Bennett EA, Iskow RC, Luttig CT, Tsui C, Pittard WS, Devine SE (2006) Recently mobilized transposons in the human and chimpanzee genomes. Am J Hum Genet 78(4):671–679. https://doi.org/10.1086/501028
Miranda-Morales E, Meier K, Sandoval-Carrillo A, Salas-Pacheco J, Vázquez-Cárdenas P, Arias-Carrión O (2017) Implications of DNA methylation in Parkinson’s disease. Front Mol Neurosci 10:225–225. https://doi.org/10.3389/fnmol.2017.00225
Molenaar C, Wiesmeijer K, Verwoerd NP, Khazen S, Eils R, Tanke HJ, Dirks RW (2003) Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J 22(24):6631–6641. https://doi.org/10.1093/emboj/cdg633
Morris BJ, Willcox BJ, Donlon TA (2019) Genetic and epigenetic regulation of human aging and longevity. Biochim Biophys Acta (BBA) – Mol Basis Dis 1865(7):1718–1744. https://doi.org/10.1016/j.bbadis.2018.08.039
Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M (2013) Programmed cell senescence during mammalian embryonic development. Cell 155(5):1104–1118. https://doi.org/10.1016/j.cell.2013.10.019
Musich PR, Zou Y (2009) Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY) 1(1):28–37. https://doi.org/10.18632/aging.100012
Nagai K, Matsushita T, Matsuzaki T, Takayama K, Matsumoto T, Kuroda R, Kurosaka M (2015) Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes. Osteoarthr Cartil 23(8):1412–1420. https://doi.org/10.1016/j.joca.2015.03.024
Nagele RG, Velasco AQ, Anderson WJ, McMahon DJ, Thomson Z, Fazekas J, Wind K, Lee H (2001) Telomere associations in interphase nuclei: possible role in maintenance of interphase chromosome topology. J Cell Sci 114(2):377
Navarro-Sánchez L, Águeda-Gómez B, Aparicio S, Pérez-Tur J (2018) Epigenetic study in Parkinson’s disease: a pilot analysis of DNA methylation in candidate genes in brain. Cell 7(10):150. https://doi.org/10.3390/cells7100150
Nemeth A, Conesa A, Santoyo-Lopez J, Medina I, Montaner D, Peterfia B, Solovei I, Cremer T, Dopazo J, Langst G (2010) Initial genomics of the human nucleolus. PLoS Genet 6(3):e1000889. https://doi.org/10.1371/journal.pgen.1000889
Ni Z, Ebata A, Alipanahiramandi E, Lee SS (2012) Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11(2):315–325. https://doi.org/10.1111/j.1474-9726.2011.00785.x
Ning Y, Xu J-f, Li Y, Chavez L, Riethman HC, Lansdorp PM, N-p W (2003) Telomere length and the expression of natural telomeric genes in human fibroblasts. Hum Mol Genet 12(11):1329–1336. https://doi.org/10.1093/hmg/ddg139
O’Donnell KA, Burns KH (2010) Mobilizing diversity: transposable element insertions in genetic variation and disease. Mob DNA 1(1):21–21. https://doi.org/10.1186/1759-8753-1-21
O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J (2010) Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17(10):1218–1225. https://doi.org/10.1038/nsmb.1897
Ogneva ZV, Dubrovina AS, Kiselev KV (2016) Age-associated alterations in DNA methylation and expression of methyltransferase and demethylase genes in Arabidopsis thaliana. Biol Plant 60(4):628–634. https://doi.org/10.1007/s10535-016-0638-y
Okamoto K, Bartocci C, Ouzounov I, Diedrich JK, Yates JR 3rd, Denchi EL (2013) A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 494(7438):502–505. https://doi.org/10.1038/nature11873
Ottaviani A, Schluth-Bolard C, Rival-Gervier S, Boussouar A, Rondier D, Foerster AM, Morere J, Bauwens S, Gazzo S, Callet-Bauchu E, Gilson E, Magdinier F (2009) Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J 28(16):2428–2436. https://doi.org/10.1038/emboj.2009.201
Park J-W, Kim JJ, Bae Y-S (2018) CK2 downregulation induces senescence-associated heterochromatic foci formation through activating SUV39h1 and inactivating G9a. Biochem Biophys Res Commun 505(1):67–73. https://doi.org/10.1016/j.bbrc.2018.09.099
Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SWM, Solovei I, Brugman W, Gräf S, Flicek P, Kerkhoven RM, van Lohuizen M, Reinders M, Wessels L, van Steensel B (2010) Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 38(4):603–613. https://doi.org/10.1016/j.molcel.2010.03.016
Ramirez MJ, Surralles J (2008) Laser confocal microscopy analysis of human interphase nuclei by three-dimensional FISH reveals dynamic perinucleolar clustering of telomeres. Cytogenet Genome Res 122(3–4):237–242. https://doi.org/10.1159/000167809
Romero-Bueno R, Ruiz DP, Artal-Sanz M, Askjaer P, Dobrzynska A (2019) Nuclear Organization in Stress and Aging. Cell 8(7). https://doi.org/10.3390/cells8070664
Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young AR, Narita M, Perez-Mancera PA, Bennett DC, Chong H, Kimura H, Narita M (2013) Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 27(16):1800–1808. https://doi.org/10.1101/gad.217281.113
Scaffidi P, Misteli T (2006) Lamin A-dependent nuclear defects in human aging. Science 312(5776):1059–1063. https://doi.org/10.1126/science.1127168
Sen P, Dang W, Donahue G, Dai J, Dorsey J, Cao X, Liu W, Cao K, Perry R, Lee JY, Wasko BM, Carr DT, He C, Robison B, Wagner J, Gregory BD, Kaeberlein M, Kennedy BK, Boeke JD, Berger SL (2015) H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev 29(13):1362–1376. https://doi.org/10.1101/gad.263707.115
Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, Gregory BD, Adams PD, Berger SL (2013) Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27(16):1787–1799. https://doi.org/10.1101/gad.223834.113
Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD (2006) Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A 103(23):8703–8708. https://doi.org/10.1073/pnas.0602569103
Sidler C, Kovalchuk O, Kovalchuk I (2017) Epigenetic regulation of cellular senescence and aging. Front Genet 8:138–138. https://doi.org/10.3389/fgene.2017.00138
Song S, Johnson FB (2018) Epigenetic mechanisms impacting aging: a focus on histone levels and telomeres. Genes (Basel) 9(4):201. https://doi.org/10.3390/genes9040201
Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, Adam SA, Goldman RD, Marko JF (2018) Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell 29(2):220–233. https://doi.org/10.1091/mbc.E17-06-0410
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155(5):1119–1130. https://doi.org/10.1016/j.cell.2013.10.041
Sturm A, Ivics Z, Vellai T (2015) The mechanism of ageing: primary role of transposable elements in genome disintegration. Cell Mol Life Sci 72(10):1839–1847. https://doi.org/10.1007/s00018-015-1896-0
Sun L, Yu R, Dang W (2018) Chromatin architectural changes during cellular senescence and aging. Genes (Basel) 9(4):pii: E211. https://doi.org/10.3390/genes9040211
Swanson EC, Manning B, Zhang H, Lawrence JB (2013) Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol 203(6):929–942. https://doi.org/10.1083/jcb.201306073
Tanae K, Horiuchi T, Matsuo Y, Katayama S, Kawamukai M (2012) Histone chaperone Asf1 plays an essential role in maintaining genomic stability in fission yeast. PLoS One 7(1):e30472. https://doi.org/10.1371/journal.pone.0030472
Tasselli L, Xi Y, Zheng W, Tennen RI, Odrowaz Z, Simeoni F, Li W, Chua KF (2016) SIRT6 deacetylates H3K18ac at pericentric chromatin to prevent mitotic errors and cellular senescence. Nat Struct Mol Biol 23(5):434–440. https://doi.org/10.1038/nsmb.3202
Tiwari V, Wilson DM 3rd (2019) DNA damage and associated DNA repair defects in disease and premature aging. Am J Hum Genet 105(2):237–257. https://doi.org/10.1016/j.ajhg.2019.06.005
Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, D’Urso A, Naar AM, Kingston R, Rippe K, Mostoslavsky R (2013) SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell 51(4):454–468. https://doi.org/10.1016/j.molcel.2013.06.018
Trojer P, Reinberg D (2007) Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28(1):1–13. https://doi.org/10.1016/j.molcel.2007.09.011
Tsurumi A, Li WX (2012) Global heterochromatin loss: a unifying theory of aging? Epigenetics 7(7):680–688. https://doi.org/10.4161/epi.20540
Uhlirova R, Horakova AH, Galiova G, Legartova S, Matula P, Fojtova M, Varecha M, Amrichova J, Vondracek J, Kozubek S, Bartova E (2010) SUV39h- and A-type lamin-dependent telomere nuclear rearrangement. J Cell Biochem 109(5):915–926. https://doi.org/10.1002/jcb.22466
van Koningsbruggen S, Gierliński M, Schofield P, Martin D, Barton GJ, Ariyurek Y, Dunnen JTD, Lamond AI (2010) High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol Biol Cell 21(21):3735–3748. https://doi.org/10.1091/mbc.e10-06-0508
van Steensel B, Belmont AS (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169(5):780–791. https://doi.org/10.1016/j.cell.2017.04.022
van Steensel B, Smogorzewska A, de Lange T (1998) TRF2 protects human telomeres from end-to-end fusions. Cell 92(3):401–413. https://doi.org/10.1016/s0092-8674(00)80932-0
Vanrobays E (2017) Heterochromatin positioning and nuclear architecture. Annu Plant Rev 46:33. https://doi.org/10.1002/9781119312994.apr0502
Victorelli S, Passos JF (2017) Telomeres and cell senescence – size matters not. EBioMedicine 21:14–20. https://doi.org/10.1016/j.ebiom.2017.03.027
Villeponteau B (1997) The heterochromatin loss model of aging. Exp Gerontol 32(4–5):383–394
Wang J, Jia ST, Jia S (2016) New insights into the regulation of heterochromatin. Trends Genet: TIG 32(5):284–294. https://doi.org/10.1016/j.tig.2016.02.005
Wei JW, Huang K, Yang C, Kang CS (2017) Non-coding RNAs as regulators in epigenetics (review). Oncol Rep 37(1):3–9. https://doi.org/10.3892/or.2016.5236
Weierich C, Brero A, Stein S, von Hase J, Cremer C, Cremer T, Solovei I (2003) Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res 11(5):485–502
Wilson RHC, Coverley D (2017) Transformation-induced changes in the DNA-nuclear matrix interface, revealed by high-throughput analysis of DNA halos. Sci Rep 7(1):6475. https://doi.org/10.1038/s41598-017-06459-7
Wood AM, Rendtlew Danielsen JM, Lucas CA, Rice EL, Scalzo D, Shimi T, Goldman RD, Smith ED, Le Beau MM, Kosak ST (2014) TRF2 and lamin A/C interact to facilitate the functional organization of chromosome ends. Nat Commun 5:5467–5467. https://doi.org/10.1038/ncomms6467
Xie W, Baylin SB, Easwaran H (2019) DNA methylation in senescence, aging and cancer. Oncoscience 6(1–2):291–293. https://doi.org/10.18632/oncoscience.476
Xu J, Liu Y (2019) A guide to visualizing the spatial epigenome with super-resolution microscopy. FEBS J 0 (0). https://doi.org/10.1111/febs.14938
Zhang R, Chen W, Adams PD (2007) Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27(6):2343. https://doi.org/10.1128/MCB.02019-06
Zhang W, Li J, Suzuki K, Qu J, Wang P, Zhou J, Liu X, Ren R, Xu X, Ocampo A, Yuan T, Yang J, Li Y, Shi L, Guan D, Pan H, Duan S, Ding Z, Li M, Yi F, Bai R, Wang Y, Chen C, Yang F, Li X, Wang Z, Aizawa E, Goebl A, Soligalla RD, Reddy P, Esteban CR, Tang F, Liu G-H, Belmonte JCI (2015) Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348(6239):1160–1163. https://doi.org/10.1126/science.aaa1356
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bridger, J.M., Foster, H.A. (2020). Senescence and the Genome. In: Iourov, I., Vorsanova, S., Yurov, Y. (eds) Human Interphase Chromosomes. Springer, Cham. https://doi.org/10.1007/978-3-030-62532-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-62532-0_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-62531-3
Online ISBN: 978-3-030-62532-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)