
Abstract
Photodynamic therapy has great potential to treat diverse types of cancer and infections. PDT involves the use of a photoactive drug called photosensitizer and visible light of the appropriate wavelength. The excited photosensitizer generates reactive oxygen species, which kill the cancer cells and microorganisms and destroy the tumor. PDT also generates immune responses by generating acute inflammation that activates the innate immune response. This is followed by priming of tumor-specific T lymphocytes that have the potential to destroy distant untreated tumor cells, besides developing a long-term memory “shield” that is effective in fighting possible recurrence of the cancer. Additionally, PDT can play a role in overcoming the escape mechanism used by progressing tumors trying to escape immune attack. Moreover, in cases of infections, PDT can have a beneficial effect by attracting and accumulating neutrophils into the infected area that can directly kill the bacterial cells.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Photodynamic therapy
- Antitumor immunity
- Tumor antigens
- Antigen-specific T-cells
- Damage-associated molecular patterns
- Regulatory T-cells
- Low-dose cyclophosphamide
- Beta-galactosidase
- Benzoporphyrin derivative
- Major histocompatibility complex
- Immunogenic cell death
- Myeloid-derived suppressor cells
- Dendritic cells
- Indoleamine 2,3-dioxygenase
- Checkpoint inhibitors
21.1 Introduction
Despite high investment in the field of cancer research, the overall results have been somewhat discouraging and have only produced marginal improvements in some types of cancer [1,2,3,4]. New-generation cancer drugs are now being tailored according to the patient and tumor genetic signatures and designed to exploit biochemical characteristics associated with tumors (such as ligands, receptors, and signaling pathways). But these approaches come with certain limitations, such as high cost, and more importantly, they are not applicable to a broad range of cancer patients and thus have limitations in comparison with older cheaper chemotherapeutic drugs [5]. Moreover, there are other difficulties, which arise due to the fact that the tumor often develops drug resistance and is often only detected at an advanced stage [6,7,8,9,10,11,12]. To complicate and worsen the situation further, some tumors appear to acclimatize and adapt to these initially active tailored drugs. Any time a specific pathway is blocked, the tumor tends to overcome this obstacle to its survival by developing an alternative pathway to continue its growth. Regardless of advances in cancer treatment, the conventional treatment package including surgery + radiation therapy + chemotherapy remains the most prevalent option for oncologists. In this chapter, we will discuss in detail an alternative antitumor technique called photodynamic therapy (PDT) and its ability to stimulate antitumor immune responses.
21.2 Photodynamic Therapy
There have been many preclinical and clinical studies carried out worldwide, showing that PDT has been proven to be a promising modality for the treatment of cancer and other malignancies [13,14,15,16]. PDT is now a clinically approved modality for the treatment and management of both nonmalignant and neoplastic diseases. It has the potential to overcome many of the shortcomings and problems associated with conventional cancer treatments. In photodynamic therapy a PS is the administered either systemically, locally, or topically to a patient bearing a lesion (mostly cancer), followed after some time by the illumination of the lesion with visible light of appropriate wavelength. In the presence of oxygen, the excited PS generates cytotoxic reactive oxygen species (ROS) and therefore leads to cell death [17,18,19,20,21] .
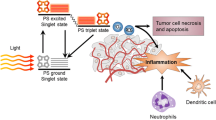
Since the lifetime of the ROS such as singlet oxygen is very short, approximately 10–320 ns, it has very limited cellular diffusion (10–55 nm), therefore PDT is highly localized [22], and the photodynamic damage only occurs in the vicinity of the PS molecular location. The PDT effect on the tumor occurs by three interrelated mechanisms: (1) killing of tumor cells directly; (2) tumor vasculature damage; and (3) induction of a strong inflammatory reaction that can lead to development of systemic immunity. The interaction between these three mechanisms and the tumor mass depends on factors such as the type and dose of the PS, the time frame of the PS administration (drug-light interval), the light characteristics (wavelength, total energy exposure or light dose, fluence rate, etc.), and the oxygen concentration in the tumor (Fig. 21.1).

PDT-induced antitumor effects. In tumors, cells loaded with PS absorb light and generate ROS species, which leads to predominantly apoptotic and necrotic cell death. Tumor cell death is accompanied with activation of the complement cascade, pro-inflammatory cytokine activation, rapid accumulation of neutrophils, followed by DCs and macrophages. Dying tumor cells and their debris are phagocytosed by phagocytic cells and DCs, which then migrate to the local lymph nodes and differentiate into antigen-presenting cells. Tumor antigen presentation is then followed by clonal expansion of tumor-specific lymphocytes that home to tumor sites and eliminate residual tumor cells
PDT has numerous advantages over other cancer treatment options presently in use. In addition to its selectivity and the possibility of repeated or multiple application, it is considered inexpensive (in comparison with some recent targeted agents) and has tolerable side effects. Moreover, tumors are rarely resistant to PDT [23, 24]. Several types of economical PS compounds are commercially available, and some are already approved to be used on patients. Most of the PS classes in common use are based on porphyrin or chlorin-type backbones or their derivatives. With the newer PS classes, problems such as prolonged skin photosensitization have been virtually eliminated [25]. In addition, these compounds absorb in the far-red region of the visible spectrum, optimal for deep tissue penetration. The list of benefits can be extended to include the absence of the adverse effects produced by radiation therapy and chemotherapy, lack of any significant change in tissue temperature during illumination, preservation of the connective tissue structures (collagen) at the site of PDT application, minimal induction of fibrosis compared to radiation therapy, and an improved cosmetic outcome. Therefore, PDT is a very promising treatment modality that needs further translational and clinical studies.
Studies have shown several and interconnected biological and physiological effects that occur during in vivo PDT. These effects depend on various factors such as the PS concentration, the location of PS in the organism/tumor site, and the dosage and rate of the applied irradiation. PDT effects include direct cell killing, occlusion of the tumor-associated vasculature, and modulation of the immune system, and sometimes all of these effects can be observed occurring simultaneously in a tumor model. At the cellular level, both necrosis and apoptosis have been observed to occur after PDT [14, 26,27,28,29]. It is a known fact that direct damage of the tumor cells and the nearby vasculature initiates several cell signaling cascades. Besides this, damage to endothelial cells leads to formation of thrombosis and consequently leads to occlusion of the tumor vasculature. In all these cases, the released fragments from the damaged cells and cytokines trigger a range of inflammatory mediators, which in turn activate the body’s defense mechanism, i.e., the innate immune response, which can also affect adaptive immunity. Thus, we can say that PDT generates a distinct systemic effect as well as working in sync with the body’s natural defense mechanisms. The overall success of PDT lies in the fact that it employs the body’s “natural pathways” of defense. PDT has been clinically applied to the treatment of early stage pulmonary, gastric, and esophageal carcinoma and has been examined for application to other diseases such as retinal diseases [30, 31] or cardiovascular disorders [32, 33].
21.3 DAMPs (Damage-Associated Molecular Patterns) and Tumor Ablative Therapies
The immunogenicity of cancer cells is an emerging determinant of anticancer immunotherapy [34]. One of the most attractive features of PDT is that besides destroying the tumor itself, it can also trigger an acute inflammatory reaction, thus activating the body’s immune system against the cancer cells as discussed above (Fig. 21.2). Thus, induction of a strong inflammatory reaction is a vital part of the antitumor effect of PDT. The local effect of PDT is localized edema and a strong acute inflammation reaction [35, 36]. PDT ends up generating an acute chemical insult within the tumor tissue which is recognized by the body as a type of localized trauma. After this trauma, there occurs a protective mechanism to reestablish tissue integrity and restore homeostasis at the damaged site. This includes removal of damaged cells, and then promoting the healing process at the affected area, in order to reinstate normal homeostasis. This elicited inflammation is initially nonspecific for the tumor antigens and is orchestrated by the innate immune system [37].

PDT-induced inflammation. Damaging the endothelial cells (ECs) activates a cascade of events leading to local inflammation, vessel dilation, and platelet aggregation. Much of these effects are caused by the release of thromboxane (TBX), cytokines (such as interleukins IL1β, IL6, IL8, tumor necrosis factor-α), and infiltration of immune system cells (necrotic and apoptotic cells provide antigens to the DCs that migrate to lymph nodes)
PDT generates rapid and prolific “danger” signals, called damage-associated molecular patterns (DAMPs) or cell death-associated molecular patterns (CDAMPs), at the site of treatment, which are detected by the innate immune system [38,39,40,41,42]. The pattern of recognition receptors is responsible for detecting the PDT-caused localized insult perceived as “altered self” [37]. This response has probably developed over evolution to protect the host against pathogen invasion at sites of tissue damage. At the onset of inflammation, the tumor vasculature undergoes significant changes and becomes adhesive for inflammatory cells and permeable/leaky for blood proteins [37]. Numerous inflammatory cells, first neutrophils followed by mast cells, monocytes, and macrophages, infiltrate the PDT illumination site [43]. At this stage, the primary function of these cells is to “neutralize” the DAMPs/CDAMPs by eliminating cellular debris, compromised tissue components, etc. [37]. The vascular occlusion, observed after PDT illumination, effectively “walls off” the damaged area, until the damaged cells are removed by phagocytosis, thus preventing further spreading of the tissue damage [37]. Studies have shown that depletion of these inflammatory cells or inhibiting their activity diminishes the therapeutic effect of PDT [44,45,46,47]. Moreover, it has been shown that interleukins IL-1β and IL-6 are among the most critical cytokines in this process. Furthermore blocking the function of various adhesion molecules can render PDT ineffective [48, 49]. On the other hand, blocking the anti-inflammatory cytokines, IL-10 and TGF-β, can remarkably improve the outcome of PDT [37, 50].
In recent years a large volume of data has emerged on the effect of in situ tumor destruction (radiotherapy, chemical and biological ablation, PDT, cryoablation, high-temperature ablation (radiofrequency, microwave, laser, and ultrasound), and electrical-based techniques) on the inflammatory and immune components resulting in systemic antitumor immune responses. It is clear that in situ tumor ablation can allow release of tumor antigens, antigen cross-presentation, and the release of DAMPS, thus making the tumor act as its own cellular vaccine [51]. It is now clear that cancer cells can succumb to some anticancer therapies by undergoing a particular form of cell death that is characterized by an increased immunogenic potential, owing to the production of DAMPs. The release of DAMPs and other immunostimulatory factors by the cells gives rise to an immunogenic cell death (ICD) favoring the establishment of a productive interface with the immune system. ICD results in the elicitation of tumor-targeted immune responses associated with the elimination of residual, treatment-resistant cancer cells, as well as with the establishment of long-term immunological memory. Although ICD has been characterized with increased precision since its discovery, several questions remain to be addressed [52].
21.4 PDT and Adaptive Immunity Recognizing Specific Antigens
As discussed earlier, the long-term efficiency of the PDT treatment strongly depends on the initiation of antitumor immunity; and this response is reduced in immunocompromised mice [44, 53]. Moreover this reduced efficacy can be restored by transfer of bone marrow or T-cells, from immunocompetent mice. In this process, recognition of the major histocompatibility complex class I (MHC-I) is critical for activation of CD8+ T-cells; thus tumors that lack MHC-I expression are generally resistant to cell-mediated antitumor immune reactions [54, 55]. In a case in point, patients with vulvar intraepithelial neoplasia (VIN) who lacked high expression of MHC I molecules did not respond as well to PDT treatment, as did patients expressing high levels of MHC-I [56, 57]. Moreover, patients who responded well to PDT treatment had increased CD8+ T-cell infiltration into the treatment site as compared to nonresponders.
Research has shown that PDT treatment of cancer involves both innate and adaptive immune response by stimulating the release or expression of different pro-inflammatory mediators [35, 36, 49]. As a result, a powerful acute inflammatory response is launched causing accumulation of extensive numbers of neutrophils and other inflammatory cells at the PDT-treated site that can attack the cancer cells [36, 43]. The fact is that this initial reaction is not only a powerful tool to elicit direct antitumor effects [58,59,60], but as importantly, it stimulates the cells to release secondary inflammatory mediators (including the cytokines IL-1β, TNF-α, IL-6, and IL-10 and prostaglandins, histamines, leukotrienes, etc.) [61]. The one area that needed to be further explored was to study the local treatment effects on eliciting systemic immunological response, in particular, establishing the link between PDT-mediated immunity and tumor antigen recognition. Our laboratory was one of the first to recognize this effect. The authors designed a study in which a pair of equally lethal BALB/c colon adenocarcinomas were used: firstly, CT26 wild-type tumors (CT26WT), i.e., antigen negative, and, secondly, CT26.CL25 transduced with lacZ gene, thus expressing the tumor antigen β-galactosidase (β-gal). The idea was to study if PDT treatment would elicit a systemic antigen and epitope-specific antitumor immune response in otherwise identical cancer cells [62]. In this study, both used cell lines were equally lethal, and the level of β-gal expression in CT26.CL25 cells was low enough to allow the tumor to grow without triggering any clinically significant immune response (often seen in cancer patients). The PDT application could therefore generate significant differences in the therapeutic outcome and the observed elicitation of immune response.
The outcome was that PDT induced a local response in all β-gal antigen-negative CT26WT tumors, with clear reduction in size, but this lasted only until day 18 (Fig. 21.3) after that local regrowth occurred. The net result was that the growth was only stalled for 8–10 days. In the case of CT26.CL25 tumors, however, the difference was dramatic (Fig. 21.4); tumor reduction was not only complete after day 20, but most importantly, 100% of these β-gal antigen-positive tumors stayed in remission during the complete trial period of 90 days [62]. During the study, the PDT-induced immune response leading to elevated levels of released IFN-γ and TNF-α cytokines was also observed. Our study also showed that PDT can induce a very strong antigen-specific immune response, capable of generating memory immunity which allows mice to reject a rechallenge with the same antigen-positive cells. The induced immune response was potent enough to cause regression of a distant well-established antigen-positive tumor outside the treatment area (on the opposite flank) [62] (Fig. 21.5). The presence of activated antigen-specific and epitope-specific effector CTLs was also confirmed. During the study, it was found that regression of distant and untreated tumors took place in 70% of the treated mice.

In vivo PDT of tumor (one leg model). (a) Mean tumor volumes of CT26WT tumors and (b) CT26.CL25 tumors; means of 10–15 tumors. (c) Kaplan-Meier survival curves of % of mice cured from CT26.CL25 tumors and rechallenged either with CT26.CL25 or CT26WT tumor cells. (d) Mean level of cytokines TNF-α, INF-γ, IL-2, and IL-4; measured 5 days after PDT in CT26.CL25 and CT26WT tumor-bearing mice and control mice (Used with permission from Ref. [62])

In vivo PDT of tumors (two-leg model). Time courses of individual tumor volumes with two similar or mismatched bilateral tumors on the right and left legs. (a) Bilateral CT26WT tumors; right leg PDT treated. (b) Bilateral CT26WT tumors, untreated. (c) Bilateral CT26.CL25 tumors; right leg PDT treated. (d) Bilateral CT26.CL25 tumors, untreated. (e) Kaplan-Meier survival curves of % mice with tumor volumes smaller than 1 cm, in five groups: three groups with two similar bilateral CT26.CL25 tumors (one group untreated, one group with right leg tumor and PDT treated, and one group with right leg tumor surgically removed); two groups with two bilateral CT26WT tumors (one group untreated, one group with right leg tumor PDT treated). (f) Mismatched CT26.CL25 and CT26WT tumors; CT26WT treated with PDT. (g) Mismatched CT26.CL25 and CT26WT tumors; CT26.CL25 treated with PDT (Adapted from Mroz et al. open access [62])

(a) Tumor volumes of CT26.CL25 tumors PDT treated and untreated in BALB/c Nu/Nu immunocompromised mice. (b) Tumor volumes in bilateral CT26.CL25 tumors PDT treated and untreated in BALB/c Nu/Nu immunocompromised mice. (c) Kaplan-Meier survival curves of % surviving BALB/c and BALB/c Nu/Nu mice with either CT26.CL25 or CT26WT tumors, PDT treated. Non-treated BALB/c Nu/Nu mice with CT26.CL25 tumor is used as control (From Mroz et al. [62]; open access)
For the first time it was demonstrated that tumor cells may escape PDT-induced immunosurveillance due to loss of the tumor antigen. In clinical settings, it is known that some tumors escape from immune recognition and resist elimination; only now, we realized that this is occurs due to tumor antigen loss. We also demonstrated that PDT-induced antitumor effects are abrogated when there is no functional adaptive immune response as in athymic nude mice (Fig. 21.4). Clearly, effective vascular PDT treatment can not only destroy a local tumor but also induce systemic strong antigen-specific antitumor immune response. In addition, this immunity is so potent that it is able to induce regression and destruction of distant, antigen-positive tumors outside the irradiation field. The treatment also proved to be effective in inducing long-term immune memory effect, imparting a resistance to rechallenge. Our study was successful in proving that the observed tumor-destructive effect was mediated by tumor antigen-specific cytotoxic T-cells, induced after PDT, which are capable of recognizing the immuno-dominant epitope of the β-gal antigen.
To examine antigen-specific PDT-induced antitumor immune response in a more clinically relevant tumor model, the authors designed a different study, where a naturally occurring cancer antigen, namely, P1A, a mouse homologue of the human MAGE-type antigen, was employed [63]. We decided to use this specific cancer-testis antigen, since it is not only well-established, but more importantly, it is mostly expressed in testis and cancers and only at very low levels in other tissues [64,65,66,67]; P1A antigen-positive mouse mastocytoma P815 wild-type (parental) and P1A antigen-negative P1.204 (P815 derived) cell lines were compared.
Murine methylcholanthrene-induced mastocytoma P815 cancer cells are known to generate very interesting immunologic response patterns. The significance of P815 antigen arises from the fact that it shares many characteristics identified in TAA genes in human, such as those belonging to melanoma MAGE family and other tumors [68, 69]; these antigens are not expressed in most mature tissues with the exception of testis and placenta [70]. It is known that P815 can elicit CTL response against at least four distinct antigens: AB, C, D, and E [70,71,72,73,74,75,76,77,78,79]. It appears that the main CTL response against P815 tumor is geared toward AB and E antigens [73]. Also, it has been shown that T-cells isolated from DBA/2 mice implanted with P815 tumors primarily recognize either antigen AB or C-D-E, but not both [79]. Moreover, the two epitopes of the P815AB, P815A, and P815B are recognized by two different CTLs. Another gene codes for P815E and different CTLs recognize this antigen. On the other hand, the P815-derived P1.204 cell line is an immune system escape variant [80]; it has lost the P815AB antigen and only retains the P815E antigen.
During in vivo experiments performed by the authors, the majority of mice with P815 tumors demonstrated tumor regression after PDT irradiation and no recurrence during the trial period of 90 days. In stark contrast, mice with P1.204 tumors did not respond with tumor regression but rather with progression. The difference in response between the two tumor types was hypothesized to be due to differential triggering of immune response. To confirm the PDT-generated long-term immune system “activation” in this clinically relevant tumor model, we rechallenged the cured mice with the same tumor from which they were originally cured. Only mice cured for P1A antigen-positive P815 tumors rejected the rechallenge with P815, while all the naïve mice injected with either tumor cell type grew tumors. The implication of the finding is that P1A antigen-positive P815 tumors, after PDT treatment, develop strong and robust enough immune response that prevents tumor growth upon challenge with a tumorigenic dose of cells [80].
In the ex vivo study, the extent of induction of an antitumor immune response, as a result of PDT treatment of P1A expressing P815 tumors, and whether the antigen activated T-cells before and/or after PDT, was investigated. Cytokines secreted from CD4+ and CD8+ T-cells were measured upon stimulation. Our results showed that PDT of P1A antigen-positive tumors led to marked increase in IL-2 and TNF-α levels. Moreover, we were able to identify a population of CD8+ T-cells that were able to recognize the known epitope (LPYLGWLVF) of the P1A antigen using a pentamer approach and flow cytometry. In addition, when nude mice (lacking an adaptive immune system) bearing the P1A antigen-positive P815 tumors were treated with PDT, the antitumor effectiveness of PDT was curtailed to nil. Interestingly, the survival of these mice could be significantly prolonged by adoptive transfer of activated lymph node cells isolated from PDT-treated immunocompetent mice bearing the P815 tumor.
The initial escape of P815 tumors from immunosurveillance (and accordingly lack of response) has been documented to be due to antigenic loss [22, 38, 39]. It has been shown [74] that there are three different escape mechanisms employed by P1A tumors, presenting the peptide epitope LPYLGWLVF (expressed in different tumor models). In P815 tumors, all progressions occurred due to antigenic loss, while in J558 tumors (another P1A-positive tumor), all progressions took place due to antigenic drift (antigen mutation) [38], whereas all progressing methA tumors (a third P1A-positive tumor) developed resistance to CTLs.
Green fluorescent protein (GFP) is used as an optical reporter to noninvasively image the progression of mouse tumors (using whole-body fluorescence imaging) and, in addition, may act as a foreign (jellyfish) antigen. We asked whether GFP-expressing tumors could be used to monitor the response of tumor-bearing mice to PDT and whether the tumor response differed when a non-immunogenic tumor cell line was transduced with GFP. RIF-1 or RIF-1 EGFP (stably transduced with a retroviral vector) cells were injected in the leg of C3H/HeN mice and both cells and tumors grew equally well. We used PDT with benzoporphyrin derivative and a short drug-light interval. There were complete cures and 100% mouse survival of RIF-1 EGFP while RIF-1 wild-type tumors all recurred. Cured mice were resistant to rechallenge with RIF-1 EGFP cells and a rechallenge with wild-type RIF-1 cells grew significantly slower. There was also slower RIF-1 EGFP rechallenge growth but no rejection when RIF-1 EGFP tumors were surgically removed. There was a low rate of PDT cure of tumors when RIF-1 cells were transduced with an empty retroviral vector. The presence of antibodies against EGFP in mouse serum suggests EGFP can act as a foreign antigen and PDT can then stimulate a long-term memory immune response [81].
21.5 Cancer and Immunosuppression
Cancer often develops as a complication of severe immunosuppression. Tumor cells proliferate in an immunosuppressive microenvironment, which can be an obstacle in the immunotherapy of cancer. Cancers take advantage of the immune regulatory mechanism of the host that prevents autoimmunity, resulting in evasion of immunosurveillance and resistance to immune destruction. Regulatory T-cells, myeloid suppressor cells, inhibitory cytokines, and immune checkpoint receptors are the major components of the immunosuppression mechanisms in cancer progression [82]. Advances in the understanding of tumor immunology are opening up a new range of therapeutic targets, including overcoming immunosuppressive factors in the tumor microenvironment [83]. Manipulating immune responses may thus provide an exciting new option for cancer immunotherapy [84].
21.5.1 Regulatory T-Cells
CD4+ regulatory T-cells (Tregs) are a highly immunosuppressive subset of CD4+ T-cells that protect the host from developing autoimmune diseases and allergies, whereas in malignancies, they promote tumor progression by suppressing antitumor immunity. The elucidation of factors influencing Treg homeostasis and function has important implications for anticancer therapies. Thus, the manipulation of Tregs for up- or downregulation of their suppressive function is a new therapeutic strategy for treating cancer and autoimmune diseases [85]. Treg depletion augments antitumor immune responses in animal models. Additionally, increased numbers of Tregs and, in particular, decreased ratios of CD8(+) T-cells to Tregs among tumor-infiltrating lymphocytes are correlated with poor prognosis in various types of human cancers. Thus, implementation of a strategy restricting Treg-mediated immune suppression may expand the therapeutic spectrum of cancer immunotherapy, especially in patients with a lower number of neoantigens [86].
21.5.2 Myeloid Suppressor Cells
Tumor-associated myeloid cells comprise a heterogeneous population acting systemically (myeloid-derived suppressor cells/MDSCs) and/or locally in the tumor microenvironment (MDSCs and tumor-associated macrophages/TAMs). Both populations promote cancer cell proliferation and survival, angiogenesis, and lymphangiogenesis and elicit immunosuppression through different pathways, including the expression of immunosuppressive cytokines and checkpoint inhibitors. Several studies have demonstrated that myeloid cells can express different functional programs in response to different microenvironmental signals, a property defined as functional plasticity. Myeloid suppressor cells can on one hand support tumor growth and, on the other, limit autoimmune responses, indicating that their therapeutic reprogramming can generate opportunities in relieving immunosuppression in the tumor microenvironment or reinstating tolerance in autoimmune conditions [87].
Development of metastasis is determined by both the accretion of essential changes in cancerous cells and by their communication with different stromal elements in the tumor microenvironment. Specifically, the inflammatory response and emergence of immune regulatory cells, such as myeloid-derived suppressor cells (M2-activated macrophages, tolerogenic dendritic cells, neutrophils, myeloid-derived suppressor cells (MDSCs)) and lymphoid-derived regulatory cells (regulatory T, B, and NK cells) to the tumor site have all been reported to support tumor growth, in addition to tumor invasion and metastasis. Although the potential role for myeloid regulatory cells in tumor invasion and development of the pre-metastatic niche has been suggested, the concept still requires further supportive experimental and clinical evidence, as well as data related to specific factors and mechanisms responsible for myeloid regulatory cell functioning at malignant sites [88]. Different approaches are currently being explored to target MDSC with the aim to enhance immune-based therapies [89].
21.5.3 Immature Dendritic Cells
Dendritic cells (DCs) comprise a heterogeneous population of cells that play a key role in initiating, directing, and regulating adaptive immune responses, including those critically involved in tumor immunosurveillance. The efficiency of anticancer therapy exploiting dendritic cells depends upon the maturation status of the DCs and how it changes following their interaction with cancer cells. In a study, using mouse xenograft models of human tumors, it was shown that fast-growing “angiogenic” tumors were infiltrated by a more immature DC population than comparable dormant nonvascular tumors. Since immature DCs actively promote angiogenesis and tumor growth, strategies to promote DC maturation or methods for DC ablation suppresses this response. It was thus concluded that angiogenesis could be dependent on the presence of immature DCs. Thus, cancer immunotherapies that promote DC maturation may act by both augmenting the host immune response to the tumor and by suppressing tumor angiogenesis [90].
DCs are the sentinel antigen-presenting cells of the immune system, such that their productive interface with the dying cancer cells is crucial for proper communication of the “nonself” status of cancer cells to the adaptive immune system. The efficiency and the ultimate success of this communication depends upon the maturation status of the DCs and their interaction with cancer cells. Immature DCs facilitate tolerance toward cancer cells, while fully mature DCs that secrete the correct combinations of cytokines can strongly promote anticancer immunity [91].
21.5.4 Indoleamine 2,3-Dioxygenase
Indoleamine 2,3-dioxygenase (IDO) is an inducible enzyme that catalyzes the rate-limiting first step in tryptophan catabolism. This enzyme is overexpressed in response to IFN gamma in a variety of different malignancies. IDO causes immunosuppression through breakdown of tryptophan in the tumor microenvironment and the tumor-draining lymph nodes. The depletion of tryptophan and production of toxic catabolites renders effector T-cells inactive and dendritic cells immunosuppressive. Thus, the IDO pathway is an important mechanism for tumor-related immunosuppression, and blocking it could improve cancer immunotherapy outcomes. Preclinical data suggest that IDO inhibition can delay tumor growth, enhance dendritic cell vaccines, and synergize with chemotherapy through immune-mediated mechanisms [92]. IDO is an immunosuppressive enzyme, which mediates tumor immune escape in various cancers including hepatocellular carcinoma (HCC). Therefore, IDO inhibitors as adjuvant therapeutic agents may have clinical implications in HCC. This review proposes future prospects of IDO not only as a therapeutic target but also as a prognostic marker for HCC [93].
21.6 PDT and Immunostimulant Combinations
Treatment with PDT alone is often non-curative due to tumor-induced immune cell dysfunction and immune suppression. Motivated by this fact PDT can be combined with immunostimulants and other strategies designed overcome the tumor-induced immune suppressive mechanisms described above, in order to enhance antitumor immunity. There have been many studies reporting good results using this approach.
A study was performed in an animal model of metastatic cancer, to compare PDT alone with PDT combined with low-dose cyclophosphamide (CY). Low-dose CY is a treatment that has been suggested to deplete regulatory T-cells (T-regs) and augment the immune response to some tumors. We used J774 tumors (a highly metastatic reticulum cell sarcoma line) and PDT with benzoporphyrin derivative monoacid ring A, verteporfin for injection, and a short (15 min) drug-light interval. CY (50 or 150 mg/kg i.p.) was injected 48 h before light delivery. PDT alone led to tumor regressions and a survival advantage but no permanent cures were obtained. BPD-PDT in combination with low-dose CY (but not high-dose CY) led to 70% permanent cures. Low-dose CY alone gave no permanent cures but did provide a survival advantage and was shown to reduce CD4+FoxP3+ T-regs in lymph nodes, whereas high-dose CY reduced other lymphocyte classes as well. Cured animals were rechallenged with J774 cells, and the tumors were rejected in 71% of mice. Cured mice had tumor-specific T-cells in spleens as determined by a (51)Cr release assay (Fig. 21.6) [94].

Kaplan-Meier survival curves of mice treated with PDT combined with low-dose CY. (a) Plots represent no tumor treatment (as control), only PDT, low-dose CY, and low-dose CY + PDT. (b) Plots represent no tumor treatment (as control), only PDT, high-dose CY, and high-dose CY + PDT. Mice were killed in cases when the primary tumor diameter reached 1.5 cm or body weight dropped >15%
Our lab also investigated PDT mediated by verteporfin and 690 nm light delivered 15 min later, in combination with an immunomodulation approach using CpG oligodeoxynucleotide for the treatment of 4T1 metastatic breast cancer in a BALB/c immunocompetent mouse model. In vitro, CpG primed immature dendritic cells (DC) via toll-like receptor 9 to phagocytose PDT killed tumor cells leading to DC maturation and activation. Peritumoral injection of CpG after PDT in mice gave improved local tumor control and a survival advantage compared to either treatment alone (p < 0.05). CpG may be a valuable dendritic cell targeted immunoadjuvant to combine with PDT [95].
In another study, we investigated whether the combination of PDT with low-dose CY could foster immunity against wild-type CT26 tumors expressing self-antigen (gp70) [96]. We had previously shown that CT26 wild-type tumors did not produce a long-term memory immune response when treated with PDT alone [62]. Administration of CY before PDT led to depletion of Treg and potentiated PDT-mediated immunity, leading to long-term survival. However the development of memory immunity (resistance to rechallenge) was only uncovered by a second round of Treg depletion using a second administration of low-dose CY [96].
It was recently reported that PDT can induce strong antitumour immunity toward tumor cells expressing the tumor-associated antigen P1A. Using four different mouse tumor models, we showed that antitumor immune response could be further improved when PDT is combined with a clinically approved epigenetic reversal agent that induces expression of an epigenetically silenced P1A antigen. Taken together these findings showed that PDT leads to strong specific antitumor immune responses and that epigenetic modification of tumor antigens levels may be a novel approach to further enhance the effectiveness of PDT providing a strong rationale for clinical development of this therapeutic approach [97].
The purpose of one of the studies was to determine if local PDT followed by intratumoral injection of naïve dendritic cells (IT-DC) could induce systemic antitumor immunity that could inhibit the growth of untreated tumors. It was concluded that PDT plus IT-DC administered to one tumor site led to tumor regression at distant sites, including multiple lung metastases. PDT + IT-DC induced potent systemic antitumor immunity in mice and should be evaluated in the treatment of human cancer [98].
21.7 PDT and Checkpoint Inhibitors
In recent years the introduction of checkpoint inhibitors has revolutionized the clinical treatment of many forms of advanced cancer [99]. Checkpoint inhibitors are particularly useful for potentiating T-cell-mediated immune attack against tumors. Ipilimumab (Yervoy), a monoclonal antibody targeting CTLA-4 receptor, is approved for the treatment of melanoma. Normally the CTLA-4 receptor antagonizes T-cell-mediated immunity; ipilimumab blocks this receptor leading to increased tumor killing by cytotoxic T-cells [100]. Another new anticancer drug is pembrolizumab (Keytruda), a monoclonal antibody, which targets the programmed cell death 1 (PD-1) receptor. Pembrolizumab is approved for the use against melanoma [101]. PD-1 is expressed on the surface of T-cells and B-cells and negatively regulates immune response. Inhibiting PD-1 prevents its cognate ligand PD-L1 (which is expressed on tumor cells) from binding to PD-1 and thereby killing the attacking T-cells. There are now other checkpoint inhibitors that target PD-1 or its cognate ligand PDL-1, such as nivolumab (Opdivo), atezolizumab, avelumab, and durvalumab.
There have recently been several papers that have explored the combination of PDT with checkpoint inhibitors in experimental animal tumor models. A study by Kleinovink et al. [102] studied PDT mediated by Bremachlorin and 660 nm light with a 6-h drug light interval on day 8 after MC38 tumors were implanted in C57BL/6 mice. PDT was combined with anti-CTLA4 antibody injected three times on days 7, 10, and 14 after tumor inoculation. The combination had an improved effect on double-tumor-bearing mice (only one tumor treated with PDT). Muchowicz et al. [103] tested the combination of BPD-PDT (15-min drug light interval) with anti-PDL-1 antibody injected every second day, in six doses, starting from 1 day before PDT in BALB/c mice with orthotopic 4T1 tumors. The combination led to 50% cures in this difficult model. A study by Gao et al. [104] looked at a combination of PDT using an integrin αvβ6-targeted phthalocyanine with an anti-PD-1 antibody in a 4T1 tumor model. The combination gave improved antitumor immunity and suppressed lung metastases metastasis.
The laboratory of Wenbin Lin at the University of Chicago has published a series of papers describing the combination of various nanotechnology-based PDT agents and checkpoint inhibitors in mice. One study [105] investigated the combination of nanoscale coordination polymer (NCP) core-shell nanoparticles loaded with oxaliplatin in the core and the PS pyropheophorbide attached to the shell, with anti PD-L1 antibody against CT26 tumors in BALB/c mice. They showed regression of both PDT treated primary tumors and nonirradiated distant tumors. Another study [106] used core-shell nanoparticles with zinc pyrophosphate and a lipid-conjugated pyropheophorbide PS in combination with anti PDL-1 antibody to produce antitumor immunity against 4T1 tumors. A third paper [107] reported PDT using a chlorin-based metal-organic framework (MOF) that also contained the indoleamine 2,3-dioxygenase (IDO) inhibitor (4-amino-N-(3-chloro-4-fluorophenyl)-N′-hydroxy-1,2,5-oxadiazole-3-carboximidamide) encapsulated in the channels of the MOF nanoparticles. PDT with this nanovehicle caused effective tumor regression of both primary, treated tumors and distant, untreated tumors in two syngeneic mouse models of colorectal cancer.
Xu and coworkers [108] constructed upconversion nanoparticles (UCNPs) loaded with the PS chlorin e6 and imiquimod (R837), a toll-like-receptor-7 agonist. PDT using NIR light excited the UCNP-Ce6-R837 nanoparticles when combined with anti-CTLA-4 antibody resulted in strong antitumor immune response to inhibit the growth of untreated distant tumors and produce memory immunity.
It should be noted that two very recent papers [109, 110] have reported that the response to checkpoint inhibitors has been shown to depend on the precise composition of the intestinal microbiome in both experimental models and also in patients. Apparently some bacteria in the gut encourage the development of antitumor immunity, while other bacterial species inhibit this response [111].
21.8 Concluding Remarks and Clinical Applications
There have been few reports as yet of antitumor immunity in patients treated with PDT. Abdel-Hady et al. [69] reported that high-risk HPV-infected premalignant genital lesions showed a poor response to ALA-PDT when the patients showed loss of HLA class I in the lesion, and when there was high CD8 infiltration in the lesion after PDT, the response was likely to be better. Kabingu et al. [112] reported that patients with cutaneous basal cell carcinomas (BCC) treated with ALA-PDT were more likely to have peripheral blood leukocytes that recognized Hip1, a transmembrane protein, which is overexpressed in BCC and can function as a tumor antigen, compared to patients that underwent surgery. Superficial lesions appeared to be especially susceptible to increased systemic antitumor immunity. Thong et al. showed [101] using Fotolon (a chlorin-based PS) in a single angiosarcoma patient that high fluence rate PDT showed success in local control, but only for up to 1 year. After recurrence, the tumor was treated again with low fluence rate PDT, but this time the treatment achieved tumor eradication, and spontaneous remission of non-treated distant lesions was observed, showing that an antitumor immune response had been activated.
Nevertheless, it is clear that antitumor systemic immunity after clinical PDT remains the exception rather than the rule. The reasons for this variability are many and diverse. The PDT parameters such as choice of PS, doses of both PS and light, fluence rate, and drug-light interval are all important in optimizing the immune response. The expression of the appropriate type and amount of antigens and neoantigens within the tumor is of critical importance. Another possible reason for this failure is the weakness of the immune system in older people as well as in patients with advanced tumor stages. Stage 4 cancer patients can often suffer from severe immunosuppression. Identifying and overcoming the immunosuppressive mechanisms that allow the tumor to grow in the first place provides a wealth of opportunities for combination treatments. These may include coadministration of various immunostimulatory adjuvants, strategies that involve dendritic cells, depletion of regulatory T-cells, and epigenetic reversal agents. In particular, the recent growth in popularity of checkpoint inhibitors, many of which are already approved for use in cancer patients, urgently suggests these agents should be clinically tested in patients who are receiving PDT. Future research will be able to test and optimize many of these PDT-based combinations.
References
Bergh J. Quo vadis with targeted drugs in the 21st century? J Clin Oncol. 2009;27(1):2–5.
Simard EP, Ward EM, Siegel R, Jemal A. Cancers with increasing incidence trends in the United States: 1999 through 2008. CA Cancer J Clin. 2012;62(2):118–28.
Siegel RL, Ward EM, Jemal A. Trends in colorectal cancer incidence rates in the United States by tumor location and stage, 1992–2008. Cancer Epidemiol Biomark Prev. 2012;21(3):411–6.
Fojo T, Grady C. How much is life worth: cetuximab, non-small cell lung cancer, and the $440 billion question. J Natl Cancer Inst. 2009;101(15):1044–8.
Simon R. Lost in translation: problems and pitfalls in translating laboratory observations to clinical utility. Eur J Cancer. 2008;44(18):2707–13.
Monzani E, Shtil AA, La Porta CA. The water channels, new druggable targets to combat cancer cell survival, invasiveness and metastasis. Curr Drug Targets. 2007;8(10):1132–7.
La Porta CA. Mechanism of drug sensitivity and resistance in melanoma. Curr Cancer Drug Targets. 2009;9(3):391–7.
Laconi E, Pani P, Farber E. The resistance phenotype in the development and treatment of cancer. Lancet Oncol. 2000;1:235–41.
Bianco R, Damiano V, Gelardi T, Daniele G, Ciardiello F, Tortora G. Rational combination of targeted therapies as a strategy to overcome the mechanisms of resistance to inhibitors of EGFR signaling. Curr Pharm Des. 2007;13(33):3358–67.
Bianco R, Garofalo S, Rosa R, Damiano V, Gelardi T, Daniele G, et al. Inhibition of mTOR pathway by everolimus cooperates with EGFR inhibitors in human tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer. 2008;98(5):923–30.
Gelardi T, Caputo R, Damiano V, Daniele G, Pepe S, Ciardiello F, et al. Enzastaurin inhibits tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer. 2008;99(3):473–80.
Bianco R, Rosa R, Damiano V, Daniele G, Gelardi T, Garofalo S, et al. Vascular endothelial growth factor receptor-1 contributes to resistance to anti-epidermal growth factor receptor drugs in human cancer cells. Clin Cancer Res. 2008;14(16):5069–80.
Robertson CA, Evans DH, Abrahamse H. Photodynamic therapy (PDT): a short review on cellular mechanisms and cancer research applications for PDT. J Photochem Photobiol B. 2009;96(1):1–8.
Moan J, Peng Q. An outline of the hundred-year history of PDT. Anticancer Res. 2003;23(5A):3591–600.
Juarranz A, Jaen P, Sanz-Rodriguez F, Cuevas J, Gonzalez S. Photodynamic therapy of cancer. Basic principles and applications. Clin Transl Oncol. 2008;10(3):148–54.
Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61(4):250–81.
Gollmer A, Besostri F, Breitenbach T, Ogilby PR. Spatially resolved two-photon irradiation of an intracellular singlet oxygen photosensitizer: correlating cell response to the site of localized irradiation. Free Radic Res. 2013;47(9):718–30.
Pimenta FM, Jensen RL, Holmegaard L, Esipova TV, Westberg M, Breitenbach T, et al. Singlet-oxygen-mediated cell death using spatially-localized two-photon excitation of an extracellular sensitizer. J Phys Chem B. 2012;116(34):10234–46.
Ogilby PR. Singlet oxygen – introduction. Photochem Photobiol. 2006;82(5):1133–5.
Vatansever F, de Melo WC, Avci P, Vecchio D, Sadasivam M, Gupta A, et al. Antimicrobial strategies centered around reactive oxygen species—bactericidal antibiotics, photodynamic therapy, and beyond. FEMS Microbiol Rev. 2013;37(6):955–89.
Castano AP, Demidova TN, Hamblin MR. Mechanisms in photodynamic therapy: part one-photosensitizers, photochemistry and cellular localization. Photodiagn Photodyn Ther. 2004;1(4):279–93.
Moan J, Berg K, Kvam E, Western A, Malik Z, Ruck A, et al. Intracellular localization of photosensitizers. Ciba Found Symp. 1989;146:95–107; discussion 11.
Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, et al. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61(1):3–13.
Xue LY, Chiu SM, Oleinick NL. Atg7 deficiency increases resistance of MCF-7 human breast cancer cells to photodynamic therapy. Autophagy. 2010;6(2):248–55.
Boyle RW, Dolphin D. Structure and biodistribution relationships of photodynamic sensitizers. Photochem Photobiol. 1996;64(3):469–85.
Oleinick NL, Morris RL, Belichenko I. The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem Photobiol Sci. 2002;1(1):1–21.
Almeida RD, Manadas BJ, Carvalho AP, Duarte CB. Intracellular signaling mechanisms in photodynamic therapy. Biochim Biophys Acta. 2004;1704(2):59–86.
Granville DJ, McManus BM, Hunt DW. Photodynamic therapy: shedding light on the biochemical pathways regulating porphyrin-mediated cell death. Histol Histopathol. 2001;16(1):309–17.
Girotti AW. Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms. J Photochem Photobiol B. 2001;63(1–3):103–13.
Obana A, Gohto Y, Kaneda K, Nakajima S, Takemura T, Miki T. Selective occlusion of choroidal neovascularization by photodynamic therapy with a water-soluble photosensitizer, ATX-S10. Lasers Surg Med. 1999;24(3):209–22.
Kramer M, Miller JW, Michaud N, Moulton RS, Hasan T, Flotte TJ, et al. Liposomal benzoporphyrin derivative verteporfin photodynamic therapy. Selective treatment of choroidal neovascularization in monkeys. Ophthalmology. 1996;103(3):427–38.
Tang G, Hyman S, Schneider JH Jr, Giannotta SL. Application of photodynamic therapy to the treatment of atherosclerotic plaques. Neurosurgery. 1993;32(3):438–43; discussion 43.
Hsiang YN, Crespo MT, Machan LS, Bower RD, Todd ME. Photodynamic therapy for atherosclerotic stenoses in Yucatan miniswine. Can J Surg. 1994;37(2):148–52.
Garg AD, Agostinis P. Cell death and immunity in cancer: from danger signals to mimicry of pathogen defense responses. Immunol Rev. 2017;280(1):126–48.
Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, et al. Photodynamic therapy. J Natl Cancer Inst. 1998;90(12):889–905.
Cecic I, Stott B, Korbelik M. Acute phase response-associated systemic neutrophil mobilization in mice bearing tumors treated by photodynamic therapy. Int Immunopharmacol. 2006;6(8):1259–66.
Korbelik M. PDT-associated host response and its role in the therapy outcome. Lasers Surg Med. 2006;38(5):500–8.
Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805(1):53–71.
Garg AD, Nowis D, Golab J, Agostinis P. Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis. 2010;15(9):1050–71.
Manfredi AA, Capobianco A, Bianchi ME, Rovere-Querini P. Regulation of dendritic- and T-cell fate by injury-associated endogenous signals. Crit Rev Immunol. 2009;29(1):69–86.
Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5.
Wang X, Ji J, Zhang H, Fan Z, Zhang L, Shi L, et al. Stimulation of dendritic cells by DAMPs in ALA-PDT treated SCC tumor cells. Oncotarget. 2015;6(42):44688–702.
Krosl G, Korbelik M, Dougherty GJ. Induction of immune cell infiltration into murine SCCVII tumour by photofrin-based photodynamic therapy. Br J Cancer. 1995;71(3):549–55.
Korbelik M, Dougherty GJ. Photodynamic therapy-mediated immune response against subcutaneous mouse tumors. Cancer Res. 1999;59(8):1941–6.
de Vree WJ, Essers MC, Koster JF, Sluiter W. Role of interleukin 1 and granulocyte colony-stimulating factor in photofrin-based photodynamic therapy of rat rhabdomyosarcoma tumors. Cancer Res. 1997;57(13):2555–8.
Chen WR, Huang Z, Korbelik M, Nordquist RE, Liu H. Photoimmunotherapy for cancer treatment. J Environ Pathol Toxicol Oncol. 2006;25(1–2):281–91.
Kousis PC, Henderson BW, Maier PG, Gollnick SO. Photodynamic therapy enhancement of antitumor immunity is regulated by neutrophils. Cancer Res. 2007;67(21):10501–10.
Sun J, Cecic I, Parkins CS, Korbelik M. Neutrophils as inflammatory and immune effectors in photodynamic therapy-treated mouse SCCVII tumours. Photochem Photobiol Sci. 2002;1(9):690–5.
Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Vaughan L, et al. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br J Cancer. 2003;88(11):1772–9.
Gollnick SO, Liu X, Owczarczak B, Musser DA, Henderson BW. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res. 1997;57(18):3904–9.
Keisari Y. Tumor abolition and antitumor immunostimulation by physico-chemical tumor ablation. Front Biosci (Landmark Ed). 2017;22:310–47.
Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:588.
Korbelik M, Krosl G, Krosl J, Dougherty GJ. The role of host lymphoid populations in the response of mouse EMT6 tumor to photodynamic therapy. Cancer Res. 1996;56(24):5647–52.
Maeurer MJ, Gollin SM, Martin D, Swaney W, Bryant J, Castelli C, et al. Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-a antigen. J Clin Invest. 1996;98(7):1633–41.
Maeurer MJ, Gollin SM, Storkus WJ, Swaney W, Karbach J, Martin D, et al. Tumor escape from immune recognition: loss of HLA-A2 melanoma cell surface expression is associated with a complex rearrangement of the short arm of chromosome 6. Clin Cancer Res. 1996;2(4):641–52.
Daayana S, Winters U, Stern PL, Kitchener HC. Clinical and immunological response to photodynamic therapy in the treatment of vulval intraepithelial neoplasia. Photochem Photobiol Sci. 2011;10(5):802–9.
Zawislak A, Donnelly RF, McCluggage WG, Price JH, McClelland HR, Woolfson AD, et al. Clinical and immunohistochemical assessment of vulval intraepithelial neoplasia following photodynamic therapy using a novel bioadhesive patch-type system loaded with 5-aminolevulinic acid. Photodiagn Photodyn Ther. 2009;6(1):28–40.
Stott B, Korbelik M. Activation of complement C3, C5, and C9 genes in tumors treated by photodynamic therapy. Cancer Immunol Immunother. 2007;56(5):649–58.
Cecic I, Korbelik M. Deposition of complement proteins on cells treated by photodynamic therapy in vitro. J Environ Pathol Toxicol Oncol. 2006;25(1–2):189–203.
Korbelik M, Cecic I. Complement activation cascade and its regulation: relevance for the response of solid tumors to photodynamic therapy. J Photochem Photobiol B. 2008;93(1):53–9.
Cecic I, Korbelik M. Mediators of peripheral blood neutrophilia induced by photodynamic therapy of solid tumors. Cancer Lett. 2002;183(1):43–51.
Mroz P, Szokalska A, Wu MX, Hamblin MR. Photodynamic therapy of tumors can lead to development of systemic antigen-specific immune response. PLoS One. 2010;5(12):e15194.
Mroz P, Vatansever F, Muchowicz A, Hamblin MR. Photodynamic therapy of murine mastocytoma induces specific immune responses against the cancer/testis antigen P1A. Cancer Res. 2013;73(21):6462–70.
Sharma A, Bode B, Wenger RH, Lehmann K, Sartori AA, Moch H, et al. Gamma-radiation promotes immunological recognition of cancer cells through increased expression of cancer-testis antigens in vitro and in vivo. PLoS One. 2011;6(11):e28217.
Chiriva-Internati M, Pandey A, Saba R, Kim M, Saadeh C, Lukman T, et al. Cancer testis antigens: a novel target in lung cancer. Int Rev Immunol. 2012;31(5):321–43.
Smith HA, McNeel DG. The SSX family of cancer-testis antigens as target proteins for tumor therapy. Clin Dev Immunol. 2010;2010:150591.
Pandey A, Kurup A, Shrivastava A, Radhi S, Nguyen DD, Arentz C, et al. Cancer testes antigens in breast cancer: biological role, regulation, and therapeutic applicability. Int Rev Immunol. 2012;31(5):302–20.
Brandle D, Bilsborough J, Rulicke T, Uyttenhove C, Boon T, Van den Eynde BJ. The shared tumor-specific antigen encoded by mouse gene P1A is a target not only for cytolytic T lymphocytes but also for tumor rejection. Eur J Immunol. 1998;28(12):4010–9.
Abdel-Hady ES, Martin-Hirsch P, Duggan-Keen M, Stern PL, Moore JV, Corbitt G, et al. Immunological and viral factors associated with the response of vulval intraepithelial neoplasia to photodynamic therapy. Cancer Res. 2001;61(1):192–6.
Uyttenhove C, Godfraind C, Lethe B, Amar-Costesec A, Renauld JC, Gajewski TF, et al. The expression of mouse gene P1A in testis does not prevent safe induction of cytolytic T cells against a P1A-encoded tumor antigen. Int J Cancer. 1997;70(3):349–56.
Van den Eynde B, Lethe B, Van Pel A, De Plaen E, Boon T. The gene coding for a major tumor rejection antigen of tumor P815 is identical to the normal gene of syngeneic DBA/2 mice. J Exp Med. 1991;173(6):1373–84.
Ramarathinam L, Sarma S, Maric M, Zhao M, Yang G, Chen L, et al. Multiple lineages of tumors express a common tumor antigen, P1A, but they are not cross-protected. J Immunol. 1995;155(11):5323–9.
Bilsborough J, Van Pel A, Uyttenhove C, Boon T, Van den Eynde BJ. Identification of a second major tumor-specific antigen recognized by CTLs on mouse mastocytoma P815. J Immunol. 1999;162(6):3534–40.
Bai XF, Liu JQ, Joshi PS, Wang L, Yin L, Labanowska J, et al. Different lineages of P1A-expressing cancer cells use divergent modes of immune evasion for T-cell adoptive therapy. Cancer Res. 2006;66(16):8241–9.
Lethe B, van den Eynde B, van Pel A, Corradin G, Boon T. Mouse tumor rejection antigens P815A and P815B: two epitopes carried by a single peptide. Eur J Immunol. 1992;22(9):2283–8.
Levraud JP, Pannetier C, Langlade-Demoyen P, Brichard V, Kourilsky P. Recurrent T cell receptor rearrangements in the cytotoxic T lymphocyte response in vivo against the p815 murine tumor. J Exp Med. 1996;183(2):439–49.
Markiewicz MA, Fallarino F, Ashikari A, Gajewski TF. Epitope spreading upon P815 tumor rejection triggered by vaccination with the single class I MHC-restricted peptide P1A. Int Immunol. 2001;13(5):625–32.
Ni B, Lin Z, Zhou L, Wang L, Jia Z, Zhou W, et al. Induction of P815 tumor immunity by DNA-based recombinant Semliki Forest virus or replicon DNA expressing the P1A gene. Cancer Detect Prev. 2004;28(6):418–25.
Brichard VG, Warnier G, Van Pel A, Morlighem G, Lucas S, Boon T. Individual differences in the orientation of the cytolytic T cell response against mouse tumor P815. Eur J Immunol. 1995;25(3):664–71.
Uyttenhove C, Maryanski J, Boon T. Escape of mouse mastocytoma P815 after nearly complete rejection is due to antigen-loss variants rather than immunosuppression. J Exp Med. 1983;157(3):1040–52.
Castano AP, Liu Q, Hamblin MR. A green fluorescent protein-expressing murine tumour but not its wild-type counterpart is cured by photodynamic therapy. Br J Cancer. 2006;94(3):391–7.
Cottier H, Hess MW, Walti ER. Immunodeficiency and cancer: mechanisms involved. Schweiz Med Wochenschr. 1986;116(34):1119–26.
Tremble LF, Forde PF, Soden DM. Clinical evaluation of macrophages in cancer: role in treatment, modulation and challenges. Cancer Immunol Immunother. 2017;66:1509.
Wu D. Innate and adaptive immune cell metabolism in tumor microenvironment. Adv Exp Med Biol. 2017;1011:211–23.
Togashi Y, Nishikawa H. Regulatory T cells: molecular and cellular basis for Immunoregulation. Curr Top Microbiol Immunol. 2017;410:3–27.
Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol. 2016;28(8):401–9.
Sica A, Massarotti M. Myeloid suppressor cells in cancer and autoimmunity. J Autoimmun. 2017;85:117.
Keskinov AA, Shurin MR. Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology. 2015;220(2):236–42.
Haile LA, Greten TF, Korangy F. Immune suppression: the hallmark of myeloid derived suppressor cells. Immunol Investig. 2012;41(6–7):581–94.
Fainaru O, Almog N, Yung CW, Nakai K, Montoya-Zavala M, Abdollahi A, et al. Tumor growth and angiogenesis are dependent on the presence of immature dendritic cells. FASEB J. 2010;24(5):1411–8.
Dudek AM, Martin S, Garg AD, Agostinis P. Immature, semi-mature, and fully mature dendritic cells: toward a DC-Cancer cells Interface that augments anticancer immunity. Front Immunol. 2013;4:438.
Soliman H, Mediavilla-Varela M, Antonia S. Indoleamine 2,3-dioxygenase: is it an immune suppressor? Cancer J. 2010;16(4):354–9.
Asghar K, Farooq A, Zulfiqar B, Rashid MU. Indoleamine 2,3-dioxygenase: as a potential prognostic marker and immunotherapeutic target for hepatocellular carcinoma. World J Gastroenterol. 2017;23(13):2286–93.
Castano AP, Mroz P, Wu MX, Hamblin MR. Photodynamic therapy plus low-dose cyclophosphamide generates antitumor immunity in a mouse model. Proc Natl Acad Sci U S A. 2008;105(14):5495–500.
Xia Y, Gupta GK, Castano AP, Mroz P, Avci P, Hamblin MR. CpG oligodeoxynucleotide as immune adjuvant enhances photodynamic therapy response in murine metastatic breast cancer. J Biophotonics. 2014;7(11–12):897–905.
Reginato E, Mroz P, Chung H, Kawakubo M, Wolf P, Hamblin MR. Photodynamic therapy plus regulatory T-cell depletion produces immunity against a mouse tumour that expresses a self-antigen. Br J Cancer. 2013;109(8):2167–74.
Wachowska M, Gabrysiak M, Muchowicz A, Bednarek W, Barankiewicz J, Rygiel T, et al. 5-Aza-2′-deoxycytidine potentiates antitumour immune response induced by photodynamic therapy. Eur J Cancer. 2014;50(7):1370–81.
Saji H, Song W, Furumoto K, Kato H, Engleman EG. Systemic antitumor effect of intratumoral injection of dendritic cells in combination with local photodynamic therapy. Clin Cancer Res. 2006;12(8):2568–74.
Sharon E, Streicher H, Goncalves P, Chen HX. Immune checkpoint inhibitors in clinical trials. Chin J Cancer. 2014;33(9):434–44.
Rotte A, Jin JY, Lemaire V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann Oncol. 2017;29(1):71–83.
Dagogo-Jack I, Lanfranchi M, Gainor JF, Giobbie-Hurder A, Lawrence DP, Shaw AT, et al. A retrospective analysis of the efficacy of Pembrolizumab in melanoma patients with brain metastasis. J Immunother. 2017;40(3):108–13.
Kleinovink JW, Fransen MF, Lowik CW, Ossendorp F. Photodynamic-immune checkpoint therapy eradicates local and distant tumors by CD8(+) T cells. Cancer Immunol Res. 2017;5(10):832–8.
Muchowicz A, Wachowska M, Stachura J, Tonecka K, Gabrysiak M, Wolosz D, et al. Inhibition of lymphangiogenesis impairs antitumour effects of photodynamic therapy and checkpoint inhibitors in mice. Eur J Cancer. 2017;83:19–27.
Gao L, Zhang C, Gao D, Liu H, Yu X, Lai J, et al. Enhanced anti-tumor efficacy through a combination of integrin alphavbeta6-targeted photodynamic therapy and immune checkpoint inhibition. Theranostics. 2016;6(5):627–37.
Zecha JA, Raber-Durlacher JE, Nair RG, Epstein JB, Elad S, Hamblin MR, et al. Low-level laser therapy/photobiomodulation in the management of side effects of chemoradiation therapy in head and neck cancer: part 2: proposed applications and treatment protocols. Support Care Cancer. 2016;24(6):2793–805.
Duan X, Chan C, Guo N, Han W, Weichselbaum RR, Lin W. Photodynamic therapy mediated by nontoxic Core-Shell nanoparticles synergizes with immune checkpoint blockade to elicit antitumor immunity and Antimetastatic effect on breast Cancer. J Am Chem Soc. 2016;138(51):16686–95.
Lu K, He C, Guo N, Chan C, Ni K, Weichselbaum RR, et al. Chlorin-based nanoscale metal-organic framework systemically rejects colorectal cancers via synergistic photodynamic therapy and checkpoint blockade immunotherapy. J Am Chem Soc. 2016;138(38):12502–10.
Xu J, Xu L, Wang C, Yang R, Zhuang Q, Han X, et al. Near-infrared-triggered photodynamic therapy with multitasking upconversion nanoparticles in combination with checkpoint blockade for immunotherapy of colorectal cancer. ACS Nano. 2017;11(5):4463–74.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2017;359(6371):91–7.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2017;359(6371):97–103.
Marinelli L, Tenore GC, Novellino E. Probiotic species in the modulation of the anticancer immune response. Semin Cancer Biol. 2017;46:182–90.
Kabingu E, Oseroff AR, Wilding GE, Gollnick SO. Enhanced systemic immune reactivity to a basal cell carcinoma associated antigen following photodynamic therapy. Clin Cancer Res. 2009;15(13):4460–6.
Acknowledgments
Research in the Hamblin laboratory is supported by US NIH Grant R01AI050875.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sharma, S.K., Hamblin, M.R. (2021). Photodynamic Therapy and Antitumor Immune Response. In: Rezaei, N. (eds) Cancer Immunology. Springer, Cham. https://doi.org/10.1007/978-3-030-50287-4_21
Download citation
DOI: https://doi.org/10.1007/978-3-030-50287-4_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-50286-7
Online ISBN: 978-3-030-50287-4
eBook Packages: MedicineMedicine (R0)