
Abstract
During an immune response, leukocytes undergo major changes in growth and function that are tightly coupled to dynamic shifts in metabolic processes. Immunometabolism is an emerging field that investigates the interplay between immunological and metabolic processes. The immune system has a key role to play in controlling cancer initiation and progression. Increasing evidence indicates the immunosuppressive nature of the local environment in tumor. In tumor microenvironment, immune cells collectively adapt in a dynamic manner to the metabolic needs of cancer cells, thus prompting tumorigenesis and resistance to treatments. Here, we summarize the latest insights into the metabolic reprogramming of immune cells in tumor microenvironment and their potential roles in tumor progression and metastasis. Manipulating metabolic remodeling and immune responses may provide an exciting new option for cancer immunotherapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
7.1 Introduction
As cancers edit and escape this initial immune detection, they also generate an immunosuppressive microenvironment which restricts T-cell infiltration, activation, and effector function both through direct repression (via cytokines, nutrients restriction, etc.) and the recruitment of immunosuppressive populations [1].
Tumors display altered metabolism relative to benign tissues [2, 3]. The common feature of this altered metabolism is the increased glucose uptake and fermentation of glucose to lactate. This phenomenon is observed even in the presence of completely functioning mitochondria and, together, is known as the “Warburg effect” [4]. These changes support abnormal survival and growth of malignant cells by providing energy, macromolecular precursors, and reducing equivalents [5]. Recently, increasing evidence indicates that cancer cells subvert the metabolic characteristics of the tumor microenvironment to shape immune responses within tumors [6]. Papers by Chang et al. [7] and Ho et al. [8] show that Warburg metabolism enables tumor cells to restrict glucose availability to T cells, suppressing antitumor immunity.
Immunometabolism is an emerging field of investigation dissecting the contribution of key metabolic pathways to immune cell development, fate, and behavior [9, 10]. The present article aims at understanding immune cells’ metabolism in tumor microenvironment and its potential role in tumor progression and metastasis. Characterizing the reciprocal metabolic interplay between immune and cancer cells will provide a better understanding of treatment efficacy and resistance and also help develop a new strategy for cancer immunotherapy.
7.2 Innate Cells in the Tumor Microenvironment
7.2.1 Macrophage
Tumor-associated macrophages (TAM) are among the most abundant inflammatory cells in tumors. Within the same tumor, the coexistence of two distinct TAM subpopulations has been shown, both derived from tumor-infiltrating inflammatory monocytes: M2-like MHC-IIlow TAM that reside in the hypoxic regions of the tumor and perform angiogenic, immunosuppressive, and protumoral activities and M1-like MHC-IIhigh TAM that are present in the normoxic tumor regions and possess proinflammatory and antitumoral characteristics [11, 12]. Recent studies on intracellular metabolism in macrophages provide new insights on the functioning of these critical controllers of innate and adaptive immunity [13, 14]. The metabolic reprogramming of M1 and M2 has been summarized in Fig. 7.1.

Metabolic reprogramming of macrophages in tumor microenvironment. Macrophages in a tumor microenvironment have been characterized as M1- and M2-polarized subtypes. In M1 macrophages, there is decreased respiration and a broken Krebs cycle, leading to accumulation of succinate and citrate, which act as signals to alter immune function. Downstream of TLR signaling, mitochondrial ROS (mROS) can also support the function of M1. In M2 macrophages, the Krebs cycle and oxidative phosphorylation are intact and FAO is utilized. Type 2 cytokine, such as IL-4, activates STAT6 transcription factor. Then STAT6 promotes the metabolic transition to oxidative metabolism by inducing genes of FAO and mitochondrial biogenesis. In addition, STAT6 transcriptionally induces PGC-1β, PPARγ, which synergize with STAT6 to enhance oxidative metabolism.GLUT1 glucose transporter 1, G6P glucose 6-phosphate, HIF-1α hypoxia-induced factor 1α, IL-1β interleukin-1β, IL-4 interleukin-4, OXPHOS oxidative phosphorylation, Pyr pyruvate, PGC-1β PPARγ coactivator-1β, PPAR peroxisome proliferation-activated receptor, ROS reactive oxygen species, STAT signal transducer and activator of transcription, TCA tricarboxylic acid cycle, TLR Toll-like receptor, TNF tumor necrosis factor
7.2.1.1 Classically Activated Macrophages (M1)
M1 macrophage metabolism is characterized by aerobic glycolysis, fatty acid synthesis, and a truncated tricarboxylic acid (TCA) cycle. Although the importance of glycolysis in inflammatory activation of macrophages was first noted almost a century ago, its biochemical and bioenergetic importance had not been appreciated until recently. The activated M1macrophages have high rates of glucose and glutamine uptake and lactic acid production. However, neither glycolysis nor glutaminolysis are necessary for ATP generation. The decreased respiration and a broken TCA cycle of M1 macrophages lead to accumulation of succinate, citrate, and nitric oxide (NO), which support the production of key M1 cellular products or act as signals to alter immune function [15, 16]. The endogenous metabolites can adopt regulatory roles that govern specific aspects of inflammatory response, as recently shown for succinate, which regulates the proinflammatory interleukin-1β(IL-1β) hypoxia-inducible factor-1α(HIF-1α) axis. Furthermore, Lampropoulou V et al. [17] report that itaconate modulates macrophage metabolism and effector functions by inhibiting SDH-mediated oxidation of succinate. Collectively, these studies demonstrate that endogenous metabolite-derived signals might be important integrators and effectors of host immunity in tumor microenvironment. Both the increased mitochondrial oxidation of succinate via succinate dehydrogenase (SDH) and an elevation of mitochondrial membrane potential combine drive mitochondrial reactive oxygen species (ROS) production [13]. Blocking ROS production by uncoupling mitochondria inhibits this inflammatory phenotype. Therefore, remodeling of the TCA cycle is a metabolic adaptation accompanying inflammatory macrophage activation.
7.2.1.2 Alternatively Activated Macrophages (M2)
The metabolic signature of M2 macrophages is characterized by fatty acid oxidation (FAO) and an oxidative TCA cycle. Tumor-promoting M2 macrophages are induced under the influence of interleukin-4(IL-4), IL-13, IL-10, and macrophage colony-stimulating factor (M-CSF) and lack the cytotoxicity of M1 macrophages. M2 macrophages appear to contribute to immune suppression through the production of IL-10 and transforming growth factor-β (TGF-β) [18]. Myeloid-derived suppressor cells are immature myeloid-lineage cells that also can be immunosuppressive in the tumor microenvironment. Major mechanisms of suppression include the expression and functional activity of arginase [19] and the nitrosylation of surface proteins on infiltrating T cells, including the T-cell receptor (TCR) [20]. Gene expression profiling studies of human melanoma have revealed arginase transcripts expressed in a subset of non-T-cell-infiltrated tumors [21], so myeloid-derived suppressor cells may be a component of the phenotype of T-cell exclusion.
Processes that drive the glycolytic switch in M1 macrophages are downregulated in M2 macrophages. One example is that M1 macrophages express u-PFK2, an isoform of phosphofructokinase-2 that is highly active, promoting glycolysis [15]. In contrast, M2 macrophages express a different isoform, PFK FB1, which is much less active [22, 23]. Another example is that the activation of the key metabolic regulator pyruvate kinase M2 (PKM2) attenuated an LPS-induced proinflammatory M1 macrophage phenotype while promoting traits typical of an M2 macrophage [24]. The activation of PKM2 using two well-characterized small molecules, DASA-58 and TEPP-46, inhibited LPS-induced HIF-1α and IL-1β, the important genes involved in glycolysis. The mechanistic target of rapamycin complex 2(mTORC2) operated in parallel with the IL-4Rα-signal transducer and activator of transcription 6(Stat6) pathway to facilitate increased glycolysis during M2 activation via the induction of the transcription factor IRF4. IRF4 expression required both mTORC2 and Stat6 pathways, providing an underlying mechanism to explain how glucose utilization is increased to support M2 activation [25].
Since macrophage metabolism is certainly connected to its functionality, metabolic reprogramming of M2-like TAM might be a new strategy to repolarize TAM toward an antitumoral phenotype and thus dampen tumor growth and metastasis. Although a system-level understanding of TAM metabolism is currently absent and rather limited, there is an emerging evidence that unraveling the TAM phenotype might lead to the identification of alternative, novel targets for TAM-directed intervention.
7.2.2 Dendritic Cell
Dendritic cells (DCs) display different phenotypes and activity in tumors and exhibit distinct pro-tumorigenic and anti-tumorigenic functions. DCs are supposed to play a key role in inducing and maintaining the antitumor immunity. However, their antigen-presenting function is jeopardized in tumor microenvironment. Under circumstance, DCs are polarized into tolerogenic phenotype with immunosuppressive function, which limit antitumor activity of effector T cells (TE). The metabolic switch of DCs from the anti-tumorigenic phenotype to the tolerogenic phenotype has been summarized in Fig. 7.2.

Metabolic reprogramming of dendritic cells in tumor microenvironment. Metabolic switch from anabolic metabolism to catabolic metabolisms consistent with DC function transferring from immunogenicity to tolerogenicity. (a) Demonstrates activated DCs that need metabolic reprogramming to provide the bioenergetic and biosynthetic support. The PI3K/Akt/mTOR pathway, which could be antagonized by the AMPK, is required for DCs maturation. Signaling via the kinases TBK1, IKKɛ, and Akt was essential for the TLR-induced increase in glycolysis by promoting the association of the glycolytic enzyme HK-II with mitochondria. Acetyl-CoA converted from citrate is a major acetyl donor for the acetylation pathway. (b) Illustrates that catabolic metabolism centered around OXPHOS is associated with cellular longevity and quiescence of DCs. Enhances catabolic metabolism through activation of the histone deacetylase (HDAC) sirtuin 1,inhibits the expression of hypoxia-inducible factor 1α(HIF1α), and upregulates PGC1α expression in DCs. In contrast to immunogenic DC, autophagy and the oxidation of fatty acids and glutamine render these cells more tolerogenic phenotypes. AMPK AMP-activated protein kinase, ATP adenosine triphosphate, ACC acetyl-CoA carboxylase, ACL ATP citrate lyase, AKT protein kinase B, FAS fatty acid synthesis, GLUT1 glucose transporter 1, G6P glucose 6-phosphate, HK hexokinase, LDH lactate dehydrogenase, mTOR mammalian target of rapamycin, PDH pyruvate dehydrogenase, PDK pyruvate dehydrogenase kinase, Pyr pyruvate, PGC-1 PPARγ coactivator-1, TBK1 TANK-binging kinase 1, IKKɛ inhibitor of nuclear factor-κB kinase subunit-ɛ, TCA tricarboxylic acid cycle
7.2.2.1 Immunogenic DCs
Catabolic metabolism centered around mitochondrial oxidative phosphorylation (OXPHOS) is associated with cellular longevity and quiescence of DCs, whereas cellular activation and proliferation are accompanied by a switch to glycolytic metabolism to support anabolic pathways needed for biosynthesis [26]. The phosphatidylinositol 3′-kinase/Akt(PI3K/Akt) pathway, which could be antagonized by the adenosine monophosphate (AMP)-activated protein kinase (AMPK), is required for DC maturation [27]. The switch from OXPHOS to glycolysis is a direct consequence of TLR-induced inducible nitric oxide synthase (iNOS) expression that through the production of NO poisons the mitochondrial respiratory chain in an autocrine fashion [28]. Bart Everts et al. [29] found that DC glycolytic flux increased within minutes of exposure to TLR agonists and that this served an essential role in supporting the de novo synthesis of fatty acids for the expansion of the endoplasmic reticulum and Golgi required for the production and secretion of proteins that are integral to DC activation. Signaling via the kinases TBK1, IKKɛ, and Akt was essential for the TLR-induced increase in glycolysis by promoting the association of the glycolytic enzyme HK-II with mitochondria [29] (Fig. 7.2a).
7.2.2.2 Tolerogenic DCs
In contrast to immunogenic DCs, tolerogenic DCs favor OXPHOS and FAO. Tolerogenic DCs, as opposed to immunogenic DCs, are maturation resistant and express increased levels of immunoregulatory factors, important for controlling regulatory T-cell (Treg) responses [30]. Proteomic analysis of human DCs treated with immunosuppressive drugs dexamethasone and vitamin D3 revealed increased expression of genes associated with mitochondrial metabolism and OXPHOS along with enhanced tolerogenic phenotypes [31,32,33](Fig. 7.2b). The direct inhibition of glycolysis in TLR-activated DCs favors the induction of forkhead box P3(Foxp3)-expressing helper T cells [29]. Consistent with a role for peroxisome proliferator-activated receptor gamma (PPARγ) coactivator 1α(PGC1α) in regulating DC activation, resveratrol, a drug favoring catabolic metabolism through activation of the histone deacetylase (HDAC) sirtuin 1, inhibits the expression of hypoxia-inducible factor 1α(HIF1α) and enhances PGC1α expression in DCs, which render these cells more tolerogenic phenotypes [34,35,36,37].
Plasmacytoid dendritic cells (pDCs) are a distinct lineage of DCs that are more specialized for cytokine production, particularly type I interferon (IFN) production. In vivo pDCs depletion delayed tumor growth, showing that tumor-associated pDC provides an immune-subversive environment, most likely through Treg activation, thus favoring tumor progression [38]. Wu et al. [39] recently report that through an autocrine type 1 IFN receptor-dependent pathway, induced changes in pDCs of cellular metabolism are characterized by increased FAO and OXPHOS. Direct inhibition of FAO and of pathways prevented full pDC activation [39].
How the effects of fatty acid synthesis differ so markedly in DCs isolated from tumors compared with those TLR-activated DCs is an important unanswered question. However, scientists speculate that accumulated fatty acids are supporting FAO and therefore tolerogenicity in the cancer setting [40]. These adaptations allow tolerogenic DCs a metabolic advantage in low-glucose, lactate-rich environments; they resist suppression of DCs function and proliferation in competition of nutrients.
7.3 Adaptive Cells in Tumor Microenvironment
Among the tumor-infiltrated lymphocytes (TILs),T cells are the most abundant cells [41]. CD8+ T cells have a central role in antitumor immunity, but their activity is suppressed in the tumor microenvironment. Reactivating the cytotoxicity of CD8+ T cells is of great clinical interest in cancer immunotherapy.
7.3.1 Effector T Cells(TE)
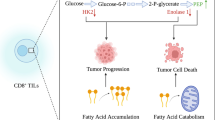
Most tumor cells express antigens that can mediate recognition by host CD8+ T cells [41]. Resting CD8+ T cells undergo dynamic shifts in cell metabolism and switch from an oxidative metabolism to aerobic glycolysis upon activation. This transition is essential to support growth and differentiation into cytotoxic T cells capable of dividing every 6–8 h and of producing inflammatory cytokines and the cytolytic granules perforin and granzyme-B [42]. Chang et al. [7] report that glucose consumption by tumors metabolically restricts T cells, leading to their dampened mTOR activity, glycolytic capacity, and IFN-γ production. Targeting aerobic glycolysis in the tumor has increased the supply of glucose to TILs, thus boosting their effector function. The checkpoint blockade antibodies against CTLA-4, PD-1, and PD-L1 restore glucose in tumor microenvironment, permitting T-cell glycolysis and IFN-γ production [7](Fig. 7.3). Recently, Ho et al. [8] uncovered the glycolytic metabolite phosphoenolpyruvate (PEP) as a new metabolic checkpoints for T-cell activity. PEP plays an important role in sustaining T-cell receptor-mediated Ca2+-nuclear factor of activated T cell (NFAT) signaling and effector functions by repressing sarco/ER Ca2+-ATPase (SERCA) activity. Together these data suggest that both nutrients and substrates concentration in a local microenvironment can have a marked impact on immune cell function.

Metabolic reprogramming of T lymphocytes in tumor microenvironment. Upon activation, lipid oxidation is downregulated in resting T cells, and glycolysis increases along with glutamine oxidation, in order to produce biosynthetic precursors required for rapid cell growth and proliferation. The checkpoint blockade antibodies against CTLA-4, PD-1, and PD-L1, which are used clinically, restore glucose in tumor microenvironment, permitting T-cell glycolysis and IFN-γ production. At the end of an immune response, the cells that survive to become memory T cells revert back to lipid oxidation with increased capacity for efficient energy generation. The mTOR pathway plays a key role in these metabolic shifts. Mechanistically, rapamycin treatment reduced mTORC1 activity and led to increased AMPK phosphorylation that correlated with an increased ability of CD8+ T cells to perform lipid oxidation. AMPK AMP-activated protein kinase, ATP adenosine triphosphate, CTLA-4 cytotoxic T-lymphocyte-associated protein 4, GLUT1 glucose transporter 1, G6P glucose 6-phosphate, mTOR mammalian target of rapamycin, PD-1 programmed cell death protein 1, PDL-1 programmed death-ligand 1, Pyr pyruvate, TCA tricarboxylic acid cycle
The lipid requirements of membrane synthesis are essential for TE function as well. Inhibiting cholesterol esterification in CD8+T cells led to potentiated effector function and enhanced proliferation [42, 43]. Inhibiting cholesterol esterification increases the plasma membrane cholesterol level of CD8+T cells, which causes enhanced T-cell receptor clustering and signaling as well as more efficient formation of the immunological synapse [43, 44]. Sterol regulatory element-binding protein (SREBP) is another critical target for meeting the heightened lipid requirements of membrane synthesis during blastogenesis [45].
TE function has been regulated by transcriptional or posttranscriptional mechanisms. Aerobic glycolysis (the Warburg effect) is a metabolic hallmark of activated T cells and has been implicated in augmenting effector T-cell responses, including expression of the proinflammatory cytokine IFN-γ, via 3′ untranslated region (3′UTR)-mediated mechanisms [46]. Another study shows that lactate dehydrogenase A (LDHA) is induced in activated T cells to support aerobic glycolysis but promotes IFN-γ expression independently of its 3′UTR. Instead, LDHA maintains high concentrations of acetyl-coenzyme A to enhance histone acetylation and transcription of Ifng. Ablation of LDHA in T cells protects mice from immunopathology triggered by excessive IFN-γ expression or deficiency of regulatory T cells [47].
T-cell exhaustion is characterized by the stepwise and progressive loss of T-cell functions [1]. The exhausted T cells acquired a distinct metabolic profile from that of TE and TM cells. Two related studies by Bengsch et al. [48] and Scharping et al. [49] indicate dysfunctional mitochondria are identified as a key correlate of CD8+ T-cell exhaustion. PD-1 pathway blockade resulted in transcriptional rewiring and reengagement of effector circuitry in the exhausted CD8+ T cells’ epigenetic landscape [50]. Manipulating glycolytic and mitochondrial metabolism might enhance checkpoint blockade outcomes [51].
7.3.2 Memory T Cell (TM)
A successful immune response relies not only on the ability of T cells to extensively proliferate and attain effector function but also to form long-lived memory T cells that can respond again to future antigen encounter. There is intense interest in understanding how long-lived cellular immunity is generated. CD8+ cytotoxic T cells further differentiate into long-lived quiescent memory CD8+ T cells (TM). TM cells require efficient energy generation to support basic cellular functions and prevent cell death [52, 53]. Pearce et al. reported that CD8+TM cells possessed substantial mitochondrial spare respiratory capacity (SRC). SRC is the extra capacity available in cells to produce energy in response to increased stress or work and as such is associated with cellular survival. The mTOR pathway plays a key role in this metabolic remodeling. Tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), an adaptor protein in the TNF-receptor and interleukin-1R/Toll-like receptor superfamily, is known to be required for the transition from effector to TM cells [52, 53]. TM cells express high levels of the mitochondrial lipid transporter-carnitine palmitoyltransferase 1a (CPT1a), and retroviral CPT1a expression enhanced CD8+ TM generation in an adoptive transfer model.
Mitochondrial dynamics underlies TM fate. TE cells have punctate mitochondria, while TM cells possess fused networks. By altering cristae morphology, fusion protein Opa1 and fusion in TM cells are required for electron transport chain (ETC) complex associations favoring OXPHOS and FAO [54]. Enforcing fusion in TE cells imposes TM cell characteristics and enhances antitumor function. Thus, targeting mitochondrial dynamics may offer a novel adjuvant strategy to therapeutically influence cancer therapy.
Elevating L-arginine levels induced global metabolic changes including a shift from glycolysis to oxidative phosphorylation in activated T cells and promoted the generation of central memory-like cells endowed with higher survival capacity and, in a mouse model, antitumor activity [55]. During the process, three transcriptional regulators (BAZ1B, PSIP1, and TSN) sensed L-arginine levels and promoted T-cell survival [55].
7.3.3 Regulatory T Cells (Treg)
Treg cells respond to immune and inflammatory signals to mediate immunosuppression. The evidences show maintaining lineage and survival integrity of Treg cells require simultaneous response to both environmental signals and metabolic homeostasis [56, 57]. Treg cell-specific deletion of Atg7 or Atg5, two essential genes in autophagy, leads to loss of Treg cells and greater tumor resistance. Mechanistically, autophagy deficiency upregulates metabolic regulators mTORC1 and c-Myc and glycolysis, which contribute to defective Treg function. Furthermore, mTORC1 acts as a fundamental “rheostat” in Treg cells to link immunological signals from TCR and IL-2 to lipogenic pathways and functional fitness and highlight a central role of metabolic programming of Treg cell suppressive activity in immune homeostasis and tolerance [57].
Tissues with low-glucose and high-lactate concentrations frequently require immune responses to be more pro-tolerant, avoiding unwanted reactions against self-antigens or commensal bacteria. Angelin et al. report that the Treg transcription factor Foxp3 reprograms T-cell metabolism by suppressing Myc and glycolysis, enhancing OXPHS, and increasing nicotinamide adenine dinucleotide oxidation [58]. These adaptations allow Tregs a metabolic advantage in low-glucose, lactate-rich environments. This metabolic phenotype may explain how Tregs promote peripheral immune tolerance during tissue injury but also how cancer cells evade immune destruction in the tumor microenvironment.
Why do TE, TM, and Treg cells adopt markedly different types of metabolism upon activation? One possibility is that the shift to glycolysis in TE may be optimally suited to the rapid, short-term bursts of activation that are required at sites of infection or inflammation, whereas FAO in TM may be better able to energetically support cell survival, as T cells continue to fight virus infection or tumors over a long time period. Understanding T cells metabolism may therefore lead to novel approaches for selective immune modulation in cancer and autoimmune diseases.
7.4 Remaining Questions and Bottleneck
Intense ongoing investigation of immune cell metabolism is yielding an exponentially growing amount of information. Armed with new information and a comprehensive understanding of how metabolism dictates immune cell fate, researchers may discover novel therapeutic strategies for treatment of tumor. For example, as we discussed in Sect. 7.3, the long-term survival and antitumor immunity of adoptively transferred CD8+T cells is dependent on their metabolic fitness. Scientists utilized a lipophilic cationic dye tetramethylrhodamine methyl ester (TMRM) to identify and isolate metabolically robust T cells based on their mitochondrial membrane potential (ΔΨm) [59]. Transfer of these low-ΔΨm T cells was associated with superior long-term in vivo persistence and an enhanced capacity to eradicate established tumors compared with high-ΔΨm cells. The use of ΔΨm-based sorting to enrich for cells with superior metabolic features was observed in CD8+ and CD4+ T-cell subsets.
At present, there are still obvious obstacles in reaching clinically effective conclusions about immunometabolism in tumor: the vast metabolic heterogeneity within a tumor and the lack of metabolic resemblance between tissue culture and in vivo conditions [60]. The in vitro studies performed cannot faithfully recapitulate the conditions of tumor local environment [61]. Therefore, the use of a reductionist approach to investigations, by focusing on specific nutrients, can produce misleading information which hampers reaching further conclusions. As a starting point, developing standardized techniques allowing assessment of the metabolism of human physiology is critical.
References
Jiang Y, Li Y, Zhu B (2015) T-cell exhaustion in the tumor microenvironment. Cell Death Dis 6:e1792
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Son J et al (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443):101–105
Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41(3):211–218
DeBerardinis RJ et al (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7(1):11–20
Sukumar M, Roychoudhuri R, Restifo NP (2015) Nutrient competition: a new axis of tumor immunosuppression. Cell 162(6):1206–1208
Chang CH et al (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162(6):1229–1241
Ho PC et al (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162(6):1217–1228
Mathis D, Shoelson SE (2011) Immunometabolism: an emerging frontier. Nat Rev Immunol 11(2):81
Murray PJ, Rathmell J, Pearce E (2015) SnapShot: immunometabolism. Cell Metab 22(1):190–190. e1
Mantovani A et al (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23(11):549–555
Mantovani A et al (2004) Infiltration of tumours by macrophages and dendritic cells: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Novartis Found Symp 256:137–145. discussion 146-8, 259-69
Mills EL, et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell
Mills EL, O’Neill LA (2016) Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol 46(1):13–21
Rodriguez-Prados JC et al (2010) Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185(1):605–614
Tannahill GM et al (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496(7444):238–242
Lampropoulou V et al (2016) Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 24(1):158–166
Quatromoni JG, Eruslanov E (2012) Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res 4(4):376–389
Rodriguez PC et al (2003) L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol 171(3):1232–1239
Nagaraj S et al (2007) Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13(7):828–835
Harlin H et al (2009) Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69(7):3077–3085
Kelly B, O'Neill LA (2015) Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 25(7):771–784
Kelly B et al (2015) Metformin inhibits the production of reactive oxygen species from NADH:ubiquinone oxidoreductase to limit induction of interleukin-1beta (IL-1beta) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem 290(33):20348–20359
Palsson-McDermott EM et al (2015) Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21(1):65–80
Huang SC et al (2016) Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 45(4):817–830
Pearce EJ, Everts B (2015) Dendritic cell metabolism. Nat Rev Immunol 15(1):18–29
Krawczyk CM et al (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115(23):4742–4749
Everts B et al (2012) Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120(7):1422–1431
Everts B et al (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15(4):323–332
Motz GT, Coukos G (2013) Deciphering and reversing tumor immune suppression. Immunity 39(1):61–73
Dong H, Bullock TN (2014) Metabolic influences that regulate dendritic cell function in tumors. Front Immunol 5:24
Sim WJ, Ahl PJ, Connolly JE (2016) Metabolism is central to Tolerogenic dendritic cell function. Mediat Inflamm 2016:2636701
Ferreira GB et al (2015) Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep 10:711–725
Houtkooper RH, Pirinen E, Auwerx J (2012) Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 13(4):225–238
Lagouge M et al (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 127(6):1109–1122
Zheng J et al (2012) Resveratrol improves insulin resistance of catch-up growth by increasing mitochondrial complexes and antioxidant function in skeletal muscle. Metabolism 61(7):954–965
Svajger U, Obermajer N, Jeras M (2010) Dendritic cells treated with resveratrol during differentiation from monocytes gain substantial tolerogenic properties upon activation. Immunology 129(4):525–535
Le Mercier I et al (2013) Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res 73(15):4629–4640
Wu D et al (2016) Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44(6):1325–1336
O’Neill LA, Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213(1):15–23
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022
MacIver NJ, Michalek RD, Rathmell JC (2013) Metabolic regulation of T lymphocytes. Annu Rev Immunol 31:259–283
Yang W et al (2016) Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531(7596):651–655
Dustin ML (2016) Cancer immunotherapy: killers on sterols. Nature 531(7596):583–584
Kidani Y et al (2013) Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 14(5):489–499
Chang CH et al (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153(6):1239–1251
Peng M et al (2016) Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 354(6311):481–484
Bengsch B et al (2016) Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity 45(2):358–373
Scharping NE et al (2016) The tumor microenvironment represses T cell mitochondrial biogenesis to drive Intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45(2):374–388
Pauken KE et al (2016) Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354(6316):1160–1165
Balmer ML, Hess C (2016) Feeling worn out? PGC1alpha to the rescue for dysfunctional mitochondria in T cell exhaustion. Immunity 45(2):233–235
Pearce EL et al (2009) Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460(7251):103–107
Prlic M, Bevan MJ (2009) Immunology: a metabolic switch to memory. Nature 460(7251):41–42
Buck MD et al (2016) Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166(1):63–76
Geiger R et al (2016) L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167(3):829–842. e13
Wei J et al (2016) Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol 17(3):277–285
Zeng H et al (2013) mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499(7459):485–490
Angelin A, et al (2017) Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab
Sukumar M et al (2016) Mitochondrial membrane potential identifies cells with enhanced Stemness for cellular therapy. Cell Metab 23(1):63–76
Schug ZT, Vande Voorde J, Gottlieb E (2016) The nurture of tumors can drive their metabolic phenotype. Cell Metab 23(3):391–392
Davidson SM et al (2016) Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab 23(3):517–528
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Wu, D. (2017). Innate and Adaptive Immune Cell Metabolism in Tumor Microenvironment. In: Li, B., Pan, F. (eds) Immune Metabolism in Health and Tumor. Advances in Experimental Medicine and Biology, vol 1011. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-1170-6_7
Download citation
DOI: https://doi.org/10.1007/978-94-024-1170-6_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-1168-3
Online ISBN: 978-94-024-1170-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)