
Abstract
Neuregulins, members of the largest subclass of growth factors of the epidermal growth factor family, mediate a myriad of cellular functions including survival, proliferation, and differentiation in normal tissues through binding to receptor tyrosine kinases of the ErbB family. However, aberrant neuregulin signaling in the tumor microenvironment is increasingly recognized as a key player in initiation and malignant progression of human cancers. In this chapter, we focus on the role of neuregulin signaling in the hallmarks of cancer, including cancer initiation and development, metastasis, as well as therapeutic resistance. Moreover, role of neuregulin signaling in the regulation of tumor microenvironment and targeting of neuregulin signaling in cancer from the therapeutic perspective are also briefly discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keyword
- Neuregulins (NRGs)/heregulins (HRGs)
- Signaling pathway
- Paracrine
- Tumor microenvironment (TME)
- Receptor tyrosine kinases (RTKs)
- ErbB family
- Tumor biology
- Tumorigenesis
- Development of cancer
- Epithelial–mesenchymal transition (EMT)
- Metastasis
- Genetics
- Epigenetics
- Therapeutic resistance
- Target therapy
1.1 Introduction
Over the past two decades, tumor has increasingly been recognized as organ that results from the co-evolution of malignant cells and their direct environment [1, 2]. The tumor microenvironment (TME) encompasses extracellular matrix (ECM) and various non-transformed cells including fibroblasts, immune infiltrates, and vascular vessels recruited from nearby local or distant tissues [3]. Through providing matrices, cytokines, growth factors, as well as vascular networks for nutrient and waste exchange, the TME plays an essential role in tumor initiation, progression, invasion, metastasis, and resistance to therapy [4].
Cumulative studies have demonstrated that the cross talk between cancer cells and their TME involves reciprocal juxtacrine and paracrine signaling pathways. Among them, neuregulin (NRG) signaling has long been recognized as an important player in regulating tumor progression [5]. Neuregulins (NRGs) are members of the largest subclass of polypeptide factors that bind to receptor tyrosine kinases of the ErbB family and mediate a myriad of cellular functions including survival, proliferation, and differentiation in normal tissues [6]. However, there is mounting evidence that dysregulated neuregulin signaling plays important role in initiation and development of a variety of human cancers via regulating cancer cells and/or the TME [7, 8]. Thus, the neuregulin signaling is emerging as therapeutic target in exploring novel strategy against cancer [9, 10]. The aim of this chapter is to discuss how neuregulin signaling takes an important part in regulating the tumor biology. We also address the targeting of neuregulin signaling in cancer from the therapeutic perspective.
1.2 Overview of Neuregulin Signaling
NRGs are a family of structurally related signaling proteins that mediate cell–cell interactions in a broad spectrum of tissues including breast, heart, nervous system, and others [11, 12]. Identified independently over two decades ago by several different research groups, these peptide growth factors were originally described as neu differentiation factor (NDF) [13], heregulins (HRGs), acetylcholine receptor-inducing activity (ARIA) [14], glial growth factors (GGFs) [15], and sensory and motor neuron-derived factor (SMDF) [16]. NRG was first characterized in rat and human as a putative ligand for the ErbB2 [13, 17, 18]; however, it was later showed that the actual receptors of NRG were ErbB3 and ErbB4 [19, 20]. Currently, 4 NRGgenes , NRG-1, NRG-2, NRG-3, and NRG-4, encoding more than 30 different isoforms through multiple promoter usage and alternative splicing, have been described [7, 21,22,23,24]. Moreover, studies are continually revealing that the family of NRGs may be larger than currently known [25,26,27].
The structures and distribution of different isoforms of NRG had been well reviewed [8, 11]. All NRGs share a typical EGF-like domain, an important structure which it alone is sufficient for receptor binding and the activation of intracellular signaling pathways. This extracellular domain contains about 50 amino acids and is characterized by 3 pairs of cysteines that are important for its tertiary structure and biological function. The differences in EGF-like domain define the α and β isoforms of NRG. Among the four members of NRG gene, NRG-1 is the best characterized one whose biological functions had been extensively studied. Transcribed from the same NRG-1 gene located on human chromosome 8p12, the isoforms of NRG-1 mRNA differ in their coding segment composition due to initiation of transcription from different NRG-1 gene promoters and alternative splicing which give rise to six types of NRG-1 (types I, II, III, IV, V, and VI). These different types of NRG-1 are also divided based on the differences in their extracellular amino-terminal domains with types I, II, IV, and V sharing an immunoglobulin-like (IgG-like) domain followed by a glycosylation-rich region (type I) or a GGF-specific (kringle) domain (type II) while type III of NRG-1 containing a cysteine-rich domain (CRD) that loops back intracellularly along with its N-terminal sequence. In addition, whether the isoform is initially synthesized as a transmembrane or nonmembrane protein adds more dimensionality to different isoforms of NRG. The human NRG-2 gene located on chromosome 5q31.2 encodes six isoforms of NRG-2 (α, β, αν, βν, α*1, and α*2) [21, 28,29,30]. The human NRG-3 and NRG-4 genes were mapped to chromosome 10q23.1 and 15q24.2, respectively [31, 32]. While the NRG-1 transcripts were evidenced to be broadly expressed in multiple tissues, both NRG-2 and NRG-3 were found to be mainly expressed in the nervous system. Although the expression of NRG-4 mRNA has only been detected in the pancreas and to a lesser extent in muscle [24], NRG-4 protein expression was found in bladder as well as prostate cancer [32, 33]. Structurally, NRG-4 is distinct from other NRGs, which contain no recognizable motifs other than the EGF-like domains. Most NRGs are synthesized as large transmembrane precursor proteins with the EGF-like domain connected to the transmembrane domain by a juxtamembrane linker. These NRGs can be activated and released from the membrane through proteolytic processing, which has also been extensively studied and reviewed [34,35,36,37].
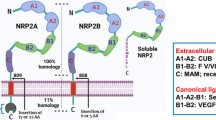
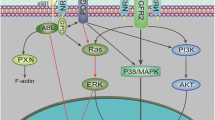
In the current body of knowledge, NRGs mainly elicit their biological functions via binding of ErbB3 or ErbB4 in an autocrine, paracrine, or juxtacrine way (Fig. 1.1) [5, 38, 39]. The ErbB subfamily of transmembrane receptor tyrosine kinases (RTKs) consists of four closely related transmembrane receptors: ErbB1 (also known as EGFR or HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4) [40]. Among all four ErbB subfamily members, NRGs can directly bind to the ErbB3 and ErbB4 receptors. All products of four NRG genes can bind ErbB4, whereas only NRG-1 and NRG-2 proteins can bind ErbB3 [41, 42]. Binding of ligands to the extracellular domain of ErbB3 or ErbB4 induces the formation of kinase active homo- or hetero-oligomers, which further induces transphosphorylation of the ErbB dimer partner and stimulates downstream intracellular pathways including RAS/RAF/MEK/ERK, PI3K/Akt/mTOR, Src kinases, and JAK/STAT [43, 44]. To date, there is no known ligand for ErbB2; however, ErbB2 is constantly in a conformation that resembles a ligand-activated state and favors dimerization [45, 46]. Unlike other ErbB receptors, ErbB3 can bind ATP and catalyze autophosphorylation, whereas it has a weak kinase activity [47]. Interestingly, ErbB3 possesses most tyrosine residues in its intracellular domain ready to be phosphorylated. It must interact with other RTKs to exhibit its biological functions [48]. Among many interactive partners of ErbB3, ErbB2 is the most important one [49]. Upon ligand binding, ErbB3 triggers the formation of ErbB2/ErbB3 heterodimer, in which ErbB3 benefits from ErbB2’s strong kinase activity for phosphorylation on its intracellular tyrosine residues. Thus, the “dumb” ErbB3 (weak kinase activity, but has ligands) and the “deaf” ErbB2 (no known ligand, but has kinase activity) make a perfect sense to form a potent ErbB2/ErbB3 heterodimer leading to activation of the downstream signaling pathways (Fig. 1.2) [42]. Accordingly, co-expression of ErbB3 and ErbB2 in normal or cancer tissues is frequently observed [50, 51], and ErbB3 has been increasingly evidenced to play a critical role in ErbB2-mediated cancer development as well as therapeutic resistance [52,53,54,55,56].

Schematic diagram of acting models of NRGs

Schematic diagram of NRG signalings and their biological consequences
1.3 Role of Neuregulin Signaling in Hallmarks of Cancer
NRG is initially identified as an important regulator in the development of nervous system [57]; however, NRG-induced signaling has also been shown to play a pivotal role in a wide variety of tissues, including breast [58, 59], heart [60], lung [61], muscle [62], and stomach [63]. Given the importance of ErbB receptors in hallmarks of cancer [64, 65], it has been evidenced that aberrant activation of the NRG signaling acts as a key player in the development and progression of various human cancers, which will be discussed in detail in the following section of this chapter (Table 1.1) [5, 7, 8].
1.3.1 Role of Neuregulin Signaling in Cancer Initiation and Development
Cancer is a genetic disease. Several lines of evidence indicate that tumorigenesis is a multistep process and that these steps reflect genetic alterations that drive the progressive transformation of normal cells into highly malignant derivatives [66]. Among the eight distinctive and complementary capabilities—hallmarks of cancer—enabling tumor growth and metastatic dissemination proposed by Hanahan et al. [2, 66], sustaining proliferative signaling is fundamental in cancer initiation and development.
In normal tissues, the production and release of growth-promoting signals governing cell growth and proliferation is strictly controlled, thereby ensuring a homeostasis of cell number and thus maintenance of normal tissue architecture and function. In contrast, malignant cells acquire the ability to sustain proliferative signaling via accumulating genetic as well as epigenetic alterations. The mitogenic signals are largely conveyed by growth factors that bind cell surface receptors such as RTKs, typically containing intracellular tyrosine kinase domains. The RTKs proceed to elicit signals via branched intracellular signaling pathways that regulate progression through the cell cycle as well as cell growth [67, 68].
Over the past several decades, it has become evident the ErbB subfamily members of RTKs have a prominent role in the initiation and development of several types of cancer. The evidence for a role of ErbB receptorsin cancer was first inferred from a study on ErbB2, a human ortholog of rat Neu, as forced overexpression of human ErbB2 was shown to transform diploid cells [69]. Almost at the same time, the EGFR was initially identified as an oncogene owing to its homology to v-erb-B [70]. Overexpression of wild-type Neu or ErbB2 under the control of a mammary-specific promoter was latterly shown to lead to metastatic mammary tumors in transgenic mice [71, 72]. Consistently, the amplification and/or overexpression of ErbB2 was found to be significantly and independently associated with a worse prognosis for breast cancer patients [73, 74]. Less frequent activating mutations of ErbB2 in several cancer types without gene amplification were further unveiled [75]. Comparably, the causal role of EGFR in tumorigenesis of a relatively broad spectrum of cancers was well recognized thereafter its first identification as oncogene [76,77,78]. Although mutations in both ErbB3 and ErbB4 were identified in human cancers [79,80,81], the involvement of ErbB3 and ErbB4 in cancer was mainly revealed as partners in promoting signaling from EGFR and ErbB2 currently [44].
As aforementioned, most of the time, homo- or hetero-dimerization of ErbB receptors triggered by binding of ligand is prerequisite for fulfilling their biological function. Ever since NRGs were identified as the ligands of ErbB3 and/or ErbB4, the role of NRGs in cancer initiation and development has attracted much research interest. While forced overexpression of NRGs in the mammary gland of transgenic mice was shown to provoke the development of breast adenocarcinomas [82], NRGs were also evidenced to function as potent stimulators of proliferation of both malignant human breast and ovarian epithelial cells in vitro [83]. Moreover, development of an autocrine NRG signaling loop results in transformation of breast epithelial cells into malignant derivates [84]. Consistently, NRG is overexpressed in about 30% of breast cancer biopsies that do not overexpress ErbB2, and this overexpression is sufficient to promote tumorigenesis and metastasis of breast cancer cells [85, 86]. In addition to growth factors, the development of breast cancer is also regulated by a plethora of signals mediated by steroid receptors such as estrogen receptor (ER) and progestogen receptor (PR) [87, 88]. Importantly, although NRG was showed to induce an estrogen-independent phenotype of breast cancer cells [85, 89], the interactions between progestins and NRG signaling pathways were unveiled in the development of mouse mammary adenocarcinomas [90]. NRG not only induces transcriptional activation of the progesterone receptor via an ErbB2-dependent manner in breast cancer cells [91] but also drives breast cancer growth through the co-option of progesterone receptor signaling [92]. Mechanistically, while the extracellular region of NRG was shown to promote mammary gland proliferation and tumorigenesis [93], a recent study demonstrated that the Ig-like region of NRGs may exert an important role in their capability to activate ErbB receptors and mitogenic responses [94]. Consequently, circulating NRG1 has been demonstrated to be additional biomarkers indicative of prognosis and outcomes for breast cancer patients [95].
Lung cancer remains the most commonly diagnosed cancer and the leading cause of cancer-related deaths worldwide [96]. Of all pathological types, non-small-cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers [97]. Extensive genomic characterization of NSCLC has led to the identification of molecular subtypes of NSCLC that are oncogene addicted, including overexpression and/or activating mutations in epidermal growth factor receptor (EGFR) [98]. However, there is mounting evidence that substantial molecular and clinical heterogeneity exists within oncogenic driver-defined subgroups of NSCLC [99, 100]. In comparison with a higher prevalence of EGFR mutations in NSCLC [78], the aberrations of other ErbB subfamily members have also been observed in NSCLC but with a relative low frequency [101]. The mutations of ErbB2, ErbB3, and ErbB4 were reported to occur in about 4% [102], 1% [79], and 5.4% [103] of human primary lung tumors, respectively. Nevertheless, in addition to EGFR, the overexpression of other ErbB subfamily members was frequently detected in NSCLC [102, 104]. While ErbB2, ErbB3, ErbB4, and NRG were found to be differentially expressed in normal bronchial epithelial and NSCLC cell lines [105], an autocrine activation of ErbB2/ErbB3 receptor complex by NRG-1 was further unraveled in NSCLC cells [106]. These studies raised the potential role of NRG signaling in initiation and development of NSCLC. More importantly, through transcriptome sequencing of 25 lung adenocarcinomas of never smokers, a novel somatic gene fusion, CD74–NRG1, was identified [107]. The CD74–NRG1 was demonstrated to give rise to the extracellular expression of the EGF-like domain of NRG1 III-β3 which acts as the ligand for ErbB2/EebB3 receptor complexes. Thereafter, NRG1 fusions were reported in different populations with different frequencies in NSCLC [108,109,110,111]. In addition to canonical NRG signaling, a recent study demonstrated that EGF and NRG may induce phosphorylation of ErbB3 by EGFR using distinct oligomeric mechanisms which even broadens our understanding of NRG signaling in cancer development [112]. The increasing recognition of the role of NRG signaling in lung cancer has raised the possibility that specific inactivation of NRG signaling may have therapeutic potential in this malignant disease [113]. More detailed information regarding the NRG signaling-targeted therapy for cancer treatment will be discussed in the rear part of this chapter.
Dysregulated ErbB signaling has also been reported to be involved in development of neoplasms of gastrointestinal tract, which mainly include esophageal carcinoma, gastric cancer, liver cancer, colorectal cancer (CRC), and pancreatic cancer accounting for more than 26% and 35% of all new cancer cases and deaths worldwide [44, 96]. Overexpression of ErbB2 in gastric cancer was firstly reported in 1986 [114]. Thereafter, accumulated data indicates the association of ErbB2 and/or ErbB3 overexpression with poor prognosis of patients with this disease [115,116,117]. While HRG-α was showed to affect epithelial cell proliferation through mesenchymal–epithelial interaction in the gastric mucosa [63], an interesting study demonstrated that overexpression of NRG1 could promote progression of gastric cancer by regulating the self-renewal of cancer stem cells [118]. In contrast to EGFR and ErbB2, which have been widely studied in CRC [119], the role of ErbB3 and ErbB4 in CRC is largely underestimated previously. Although the expression of ErbB3 in CRC was reported to range from 36 to 89% [120, 121], the information regarding its prognostic role in CRC was ever limited. This situation changed when two groups reported that ErbB3 overexpression was significantly associated with decreased time to CRC progression [122, 123]. Actually, autocrine heregulin has been demonstrated to generate growth factor independence and block apoptosis in CRC cells as early as in 2002 [124]. Interestingly, bone marrow-derived mesenchymal stem cells (MSCs) in TME were evidenced to promote CRC progression through paracrine NRG1/ErbB3 signaling [125]. In addition to acting on cancer cells themselves, NRG signaling was shown to induce expression of vascular endothelial growth factor (VEGF) in colon cancer cells, which might further affect the growth of colon cancer in vivo [126]. ErbB4 is the least recognized ErbB subfamily member. However, phosphorylated ErbB4 and NRG have also been demonstrated to contribute to poorer patient prognosis in CRC [127, 128].
The worldwide mortality of pancreatic cancer ranks the seventh among all cancers in both sexes accounting for about 4.5% of all cancer deaths [96]. Extensive genetic studies have revealed that during the progression of three broad stages of pancreatic cancer, acquired somatic mutations in oncogenes and tumor suppressor genes accumulate and account for initiation and aggressive development of this malignant disease. These mutations occur most frequently in KRAS, CDKN2A, TP53, and SMAD4 [129,130,131]. Activating mutations of the KRAS oncogene, which encodes a member of the RAS family of GTP-binding proteins, are the most common genetic abnormality presenting in approximately 95% of pancreatic tumors analyzed [132, 133]. In addition, wild-type KRAS is also normally activated in response to the binding of extracellular signals such as growth factors to RTKs [134]. Accumulating evidence shows that the ErbB receptors are overexpressed in approximately 60% of pancreatic cancers [135]. Recently, several studies have uncovered that recurrent gene rearrangements such as NRG1 fusions are prevalent in patients with KRAS wild-type pancreatic cancer [136, 137]. Thus, in a subset of KRAS wild-type pancreatic cancer cases, NRG signaling may act as targetable oncogenic driver, providing a potential treatment strategy in this disease [138].
Given the role of NRG in the development of nervous system, it is thus not surprising that aberrant NRG signaling could result in development of glioma and schwannoma [139,140,141,142]. Gliomas are the leading cause of death among adults with primary brain malignancies due to lack of effective remedy [143, 144]. In an attempt to search for novel target therapy against glioma, an RNAi-based inhibition of presenilin 2 was demonstrated to inhibit glioma cell growth and invasion via regulation of NRG1/ErbB signaling [145]. MicroRNA is emerging as important player in tumor biology [146]. It was revealed that the expression of miR-125a-3p was significantly decreased in malignant glioma and lower expression of miR-125a-3p was associated with a poor prognosis of patients with glioblastoma. Further mechanistic study suggested that miR-125a-3p might perform an important role in development of glioma via direct targeting of NRG-1 [147]. In addition to aforementioned cancer types, the aberrant NRG signaling was also involved in development of many other cancers derived from different tissues as listed in Table 1.1 which will not be discussed in detail [148,149,150,151,152,153,154,155,156,157,158,159].
1.3.2 Role of Neuregulin Signaling in Cancer Metastasis
Currently, metastatic disease is largely incurable and remains the main cause of cancer-related deaths worldwide [160]. Metastasis is the end result of a multistage process that includes acquisition of invasive phenotype of primary tumor cells, local invasion, intravasation into the blood or lymphatic system, survival in circulation, arrest at a distant organ, extravasation, survival in a new environment, and adaptation and proliferation to form metastases [161]. Each of these steps in the complex metastatic cascade depends on the genetic and epigenetic alterations acquired by primary tumor cells, as well as interactions with the host microenvironment and the immune system [162, 163].
Change in cell phenotype between epithelial and mesenchymal states, defined as epithelial–mesenchymal transition (EMT), has been increasingly recognized as an initial step of tumor metastasis [164]. The EMT program is generally induced in epithelial cells by heterotypical signals, among which the transforming growth factor-β (TGFβ) family of cytokines are the best characterized inducers of EMTs [165]. In addition, RTK signaling has also been shown to play crucial roles in the induction of EMT [166]. NRG-1 was able to enhance motility and migration of human glioma cells via not only activation of focal adhesion kinase (FAK) [167] but also induction of expression of cell adhesion molecule L1 [168]. While the constitutive activation of ERBB3-dependent signaling driven by an NRG-1/ERBB3 autocrine mechanism was strongly associated with microscopic vascular invasion and poor prognosis of hepatocellular carcinoma (HCC) [149], a recent study further demonstrated that miR-296-5p suppressed EMT of HCC via targeting NRG1/ErbB2/ErbB3 signaling [169].
Metastatic lesions develop from disseminated cancer cells (DCCs) that can remain dormant [170]. Accumulating data suggest that metastatic dissemination often occurs early during tumor formation [171, 172]. Progesterone-induced signaling was shown to trigger migration of cancer cells from early lesions shortly after HER2 activation in a HER2-driven mouse mammary tumor model which exhibited capability for early metastatic dissemination [173]. Using the same mammary tumor model, a subpopulation of Her2+p-p38lop-Atf2loTwist1hiE-cadlo early cancer cells with invasive ability to spread to target organs was identified in the early lesions of the transgenic mice [174]. These studies strongly suggested that aberrant ErbB2 signaling played a pivotal role in early dissemination of breast cancer cells. Furthermore, while the NRG expression in breast tumors was evidenced to be associated with lymph node invasion and poor patient outcome, it was demonstrated that NRG expression favored in situ tumor growth, local spreading, and metastatic dissemination via an ERK1/2 kinase-dependent upregulation of collagenase-3 [175].
Tumor-associated macrophages (TAMs), derived from recruited circulating monocytes by a wide variety of growth factors such as colony-stimulating factor 1 (CSF1), are critical for regulating processes of tumor including various steps in the metastatic cascade [176,177,178,179]. The first direct evidence for a synergistic interaction between macrophages and tumor cells during cell migration in vivo was provided in 2004 [180]. Macrophages are conventionally subdivided into antitumor pro-inflammatory M1 or pro-tumor immune-suppressive M2 phenotypes; however, the diversity of macrophage types in different tissues and cancers indicates that this classification is an oversimplification [181]. Accumulating data have revealed different TAM behaviors linked to their locations, including migration-associated streaming and perivascular populations [182,183,184]. Perivascular macrophages are an essential component of structures termed TMEM (tumor microenvironment of metastasis) that consist of a TAM in direct contact with a Mena-overexpressing tumor cell and endothelial cell [185]. While the EGF–CSF1 paracrine loop between the tumor cells and macrophages was shown to mediate pairing and stream formation [186], it was further demonstrated that the hepatocyte growth factor (HGF) supplied by endothelial cells is required for sustained directional migration of both tumor cells and macrophages toward blood vessels [184]. Moreover, the temporal aspects of macrophage subtype specification within primary tumors and the possibility of interconversion among subtypes had been verified by Arwert and colleagues whose study indicate that a unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation [187]. Notably, the EGF/CSF-1 paracrine invasion loop has been reported to be triggered by HRG-β1 and CXCL12 [188]. In addition, heregulin/ErbB3 signaling has also been shown to enhance breast cancer cell motility via hypoxia-inducible factor 1α (HIF-1α)-dependent upregulation of CXCR4, the receptor of SDF-1/CXCL12 [189].
The Notch signaling pathway regulates many aspects of cancer biology, including metastasis [190]. Upon ligand binding, the transmembrane Notch receptor is cleaved sequentially, leading to the release of the Notch intracellular domain (NICD). The NICD translocates to the nucleus where it orchestrates the transcription of specific genes. Ligands of Notch receptor expressed by the signal-sending cell are transmembrane proteins of the Delta/Serrate/LAG-2 (DSL) family which comprises three delta-like ligands (Dll1, Dll3, and Dll4) and two jagged ligands (Jagged 1 and Jagged 2) in mammals. It has been demonstrated that tumor-derived Jagged 1 (JAG1) could promote osteolytic bone metastasis of breast cancer by engaging Notch signaling in bone cells [191]. Interestingly, stimulation of macrophages by tumor cell-derived NRG-1 results in upregulation of JAG1, which in turn enhances transendothelial migration and intravasation of breast cancer cells [192]. The cross talk between different signaling pathways may play a synergistic role in tumor biology. Through a transcriptome meta-analysis, higher number of gene fusions affecting core members of the Hippo pathway, Neurofibromatosis 1 (NF1), and NRG1 genes was shown to be an independent prognostic factor for poor survival in lung cancer [193]. A direct evidence of cross talk between Hippo and NRG signaling in regulating aggressive behavior of tumor cells was provided by Haskins and colleagues, whose study demonstrated that NRG 1-activated ErbB4 interacted with YAP to induce Hippo pathway target genes and promoted cell migration [194].
The dysregulated NRG signaling in development of lung cancer has been described in the front part of this chapter. In respect to metastasis, some significant differences in ErbB family receptor-related abnormalities have been shown in NSCLC brain metastases in comparison with primary lung tumors [195]. As aforementioned, oncogene fusions have increasingly been identified as driver mutations in lung adenocarcinoma. In an attempt to identify druggable oncogenic fusions in invasive mucinous adenocarcinoma (IMA) of the lung, Nakaoku and colleagues found that two oncogenic fusions involving NRG1 (CD74–NRG1 and SLC3A2–NRG1) occurred mutually exclusive with KRAS mutations [196]. The SLC3A2–NRG1 fusion was further demonstrated to increase cell migration via promoting ErbB2–ErbB3 phosphorylation and heteroduplex formation and activating the downstream PI3K/Akt/mTOR pathway through paracrine signaling [197].
1.3.3 Role of NRG Signaling in Therapeutic Resistance in Cancer
Rapid progress in our understanding of cancer biology has dramatically improved our therapeutic strategies against cancer. In addition to the conventional operations, such as chemotherapy and radiotherapy, molecularly targeted therapies and immunotherapy recently emerge as the principal modes of cancer treatment. However, resistance to these therapies frequently occurs and currently represents a major clinical problem. Therapeutic resistance can be divided into two broad categories: primary (de novo) or acquired. Primary resistance is usually caused by resistance-mediating factors pre-existing in the bulk of tumor cells, which make the therapy ineffective. Acquired resistance can develop during the treatments of cancers that are initially sensitive. Both primary and acquired resistances can occur at many levels, including increased drug efflux and decreased drug influx, drug inactivation, alterations in drug target, processing of drug-induced damage, and evasion of apoptosis [198].
Acquired resistance can arise through therapy-induced selection of a resistant subpopulation of cells that is present in the original tumors with a high degree of heterogeneity [199]. It can also be caused by mutations arising during the treatments, by increased expression of the therapeutic target, as well as through various other adaptive responses such as activation of the compensatory signaling pathways [200].
1.3.3.1 Resistance to Chemotherapy
Chemotherapy , as an important conventional treatment for human cancers, usually induces cancer cell death by cytotoxicity, thereby reducing the tumor bulk [201]. The mechanism of antitumor activity of chemotherapeutic drugs is complex and involves various biological pathways, including apoptosis, autophagy, necrosis, and mitotic catastrophe [202]. Consequently, the mechanisms of chemoresistance are intricate and not fully understood. Taking into consideration that many clinically used chemotherapeutic drugs mainly exert antitumor activity via induction of apoptosis, it is understandable that cancer cells with enhanced survival signaling and/or defects in apoptotic pathways may escape from those therapies [203]. Since the PI3K/Akt pathway is an important survival signaling and readily activated by RTKs, including ErbB3, it is rational that aberrant NRG signaling may result in chemoresistance in cancer treatment [204]. In an early study, the co-expression of ErbB2 and ErbB3 in human breast cancer cell lines was shown to be associated with an increased resistance to multiple chemotherapeutic agents , such as paclitaxel, doxorubicin, 5-fluorouracil, etoposide, and camptothecin, via activation of PI3K/Akt signaling [205]. In our own attempt to identify the key downstream mediator of ErbB3 signaling that contributed to chemoresistance, we discovered that elevated expression of ErbB3 conferred paclitaxel resistance in ErbB2-positive breast cancer cells via PI3K/Akt-dependent upregulation of Survivin [56]. MSCs are connective tissue progenitor cells that contribute to fibrotic reactions during tissue remodeling and repair in places of wounding and inflammation. In response to chemokines from tumor cells, MSCs are continuously recruited to and become integral components of the tumor microenvironment [206, 207]. MSCs in tumor microenvironment have been shown to influence multiple hallmarks of cancer, including resistance to chemotherapy [208, 209]. More recently, we demonstrated that the aforementioned ErbB2/ErbB3 → PI3K/Akt → Survivin signaling axis underlying paclitaxel resistance in breast cancer cells could also be activated via the paracrine stimulation by MSCs-derived NRG-1 [210]. Doxorubicin is another widely used drug in chemotherapy against multiple types of cancer. It was reported that treatment with doxorubicin resulted in activation of the ErbB3–PI3K–Akt signaling cascade in ovarian cancer cells through transcription-dependent upregulation of NRG1 and specific blockade of ErbB3 enhanced the doxorubicin-induced apoptosis [211]. These data suggest that an activated NRG1/ErbB3 autocrine loop may account for doxorubicin resistance in ovarian cancer cells.
1.3.3.2 Resistance to Targeted Therapy
Multiple processes of cancer initiation and development involve the progressive acquisition of genetic mutations and epigenetic abnormalities in the expression of various genes with highly diverse functions. Nonetheless, the observation that some cancer cells can seemingly exhibit dependence on a single oncogenic pathway or protein for their sustained proliferation and/or survival has led to a new concept referred to as oncogene addiction [212]. The notion of oncogene addiction reveals a possible “Achilles’ heel” within the cancer cells that can be therapeutically targeted [213, 214].
Owing to the improved understanding of the oncogenic driver mutations in NSCLC , targeted therapy has been extensively exploited while combating this malignant disease. Systematic analysis has revealed that almost two-thirds of patients with NSCLC harbor an oncogenic driver mutation, approximately half of whom have a therapeutically targetable lesion [215]. Although treatment with a targeted therapy improves outcomes in patients with NSCLC, responses to the therapeutic agents are generally incomplete and temporary followed by resistance [216]. Cancer cells develop several mechanisms of resistance to targeted therapy, which can be classified as “on-target” or “off-target” [217]. Alterations of the primary target of the drug typically result in on-target resistance [218]. In the circumstance of off-target resistance, activation of collateral signaling events that are parallel to, or downstream of, signaling by the driver oncoprotein bypass the requirement of the driver oncoprotein for cell survival and growth. Among the expanding spectrum of oncogenic driver mutations identified in NSCLC, somatic activating mutations in EGFR are the most common ones occurring in ~16% of patients with advanced lung adenocarcinoma [219]. Currently, two strategies including blocking antibody such as cetuximab and tyrosine kinase inhibitors (TKIs, such as gefitinib, lapatinib, and erlotinib) are mainly used in EGFR-targeted therapy in clinic [220]. Cumulative data have indicated that activation of NRG/ErbB3 signaling is one of the major mechanisms contributing to the resistance to EGFR-targeted therapy [221,222,223,224]. It has been reported that MET amplification leads to gefitinib resistance in lung cancer treatment by activating ErbB3 signaling [225]. Besides, NRG/ErbB3 signaling was also shown to induce resistance to lapatinib-mediated growth inhibition of ErbB2-amplified gastric cancer cells and cetuximab-based therapy in colorectal cancer, respectively [226, 227]. The EML4–ALK fusion gene was detected in 3% to 7% of patients with NSCLC [228, 229]. It has been demonstrated that crizotinib, an ALK inhibitor, shows remarkable antitumor effect in EML4–ALK-positive lung cancer [230]. However, patients receiving treatment with crizotinib eventually acquire resistance to this drug. While paracrine receptor activation by ligands from the microenvironment was demonstrated to trigger resistance to ALK inhibitors in EML4–ALK lung cancer cells [231], an analysis of patient-derived cancer cell further revealed that activation of neuregulin/ErbB3 signaling accounted for crizotinib resistance in EML4–ALK lung cancer [232]. In addition, paracrine effect of NRG1 was also evidenced to drive resistance to MEK inhibitors in metastatic uveal melanoma [233].
The finding that amplification and/or overexpression of ErbB2 occurs in approximately 25% of invasive breast cancer and is significantly associated with a worse prognosis for breast cancer patients has made ErbB2 an attractive therapeutic target [73, 74, 234]. ErB2-targeted therapy in breast cancer is thus another paradigm in cancer research. As the first ErbB2-targeted agent approved by the US Food and Drug Administration (FDA), trastuzumab (also known as Herceptin, a humanized monoclonal antibody against ErbB2) has demonstrated significant activity in the treatment of both early-stage and metastatic ErbB2-overexpressing (ErbB2+) breast cancer [235,236,237,238]. Subsequently, lapatinib, an orally bioavailable small molecule TKI dual targeting ErbB2 and EGFR, was also approved by FDA for the treatment of patients with advanced ErbB2+ breast cancer in combination with capecitabine [239]. Unfortunately, resistance to both trastuzumab and lapatinib has greatly limited the efficacy of ErbB2-targeted therapy [240]. Numerous mechanisms including loss of phosphatase and tensin homolog (PTEN) and activating mutations in genes coding for components of the PI3K/Akt/mTOR pathway have been proposed that may mediate de novo and acquired resistance to trastuzumab and lapatinib [241, 242]. Moreover, our studies and others have demonstrated that increased activation of ErbB3 signaling plays a critical role in the resistance to ErbB2-targeted therapy in breast cancer [243,244,245]. These findings are further confirmed by a study indicating that ADAM10-mediated release of heregulin confers resistance to trastuzumab by activating ErbB3 [246]. Recently, to explore the influence of microenvironment signals on HER2-targeted TKI response , Watson et al. assessed the effects of >2500 different combinations of 56 soluble and 46 matrix microenvironmental proteins on outgrowth of lapatinib-treated HER2+ breast cancer cells using an emerging technology termed microenvironment microarrays (MEMA) [247]. It was demonstrated that NRG1β and hepatocyte growth factor (HGF) are the most significant factors to enhance outgrowth of HER2+ breast cancer cells in the presence of lapatinib. Specifically, NRG1β attenuated the response of the luminal-like HER2+ (L-HER2+) cells to lapatinib, whereas HGF attenuated the lapatinib sensitivity in the basal-like (HER2E) cells. This elegant work suggests that different mechanisms underlying resistance to HER2-TKI lapatinib may be due to the complexity of microenvironment as well as differences in signaling network wiring and architecture in different subtype cells. These findings reinforce the notion that tissue microenvironments are remarkably complex and intertwined in cancer biology [248].
1.3.3.3 Resistance to Other Therapy
Endocrine therapy such as tamoxifen, an antiestrogen agent, is commonly used in the treatment of patients with estrogen receptor-positive (ER+) breast cancer, a subtype accounting for about 80% of all breast cancers [249]. Although patients benefit a lot from tamoxifen treatment, resistance to this agent is a serious problem in clinic [250]. In the current body of knowledge, multiple mechanisms responsible for endocrine resistance have been proposed and include deregulation of various components of the ER pathway itself [251], alterations in cell cycle regulators such as MYC or cyclin D1, and the activation of alternative signaling pathways [252]. Among these, increased expression or aberrant activation of ErbB signaling has been extensively investigated and associated with both experimental and clinical endocrine therapy resistance [204, 253]. ER+ breast cancer patients with co-expression of ErbB2 and ErbB3 were significantly more likely to relapse on tamoxifen [254]. Direct evidence of the involvement of neuregulin/ErbB3 in tamoxifen resistance was provided by Liu et al. whose studies showed that downregulation of ErbB3 by a siRNA abrogated ErbB2-mediated tamoxifen resistance in breast cancer cells [55]. Furthermore, overexpression of HRG-β2 was reported to induce hormone-independent phenotype of ER+ breast cancer cells and resistant to tamoxifen via constitutive activation of ErbB signaling [89].
1.4 Role of Neuregulin Signaling in Regulation of Tumor Microenvironment
As an important part of immune infiltrates in TME , tumor-educated macrophages at the primary site promote tumor initiation and malignant progression via supporting tumor-associated angiogenesis; enhancing tumor cell invasion, migration, and intravasation; as well as suppression of antitumor immune responses [255, 256]. As earlier mentioned, a paracrine interaction involving reciprocal signaling between carcinoma cells and macrophages involving EGF and CSF-1 has been demonstrated to be required for tumor cell migration in mammary tumors [180, 257]. The finding that this EGF/CSF-1 paracrine invasion loop can be triggered by HRG-β1 [188], along with the fact that tumor cell-derived NRG-1 induces upregulation of JAG1 in macrophages [192], indicates that macrophages are subjected to a fine tune regulated by NRG signaling.
Nerves in the TME also play roles in tumor progression [258, 259]. It has been shown that, in addition to vascular and lymphatic systems, nerve system may serve as an alternative route for dissemination of cancer cells [260]. Cancer cells can grow around existing nerves and eventually invade them, in a process defined as perineural invasion (PNI) [261]. To date, PNI has been observed in a variety of cancers, including pancreatic, prostate, colorectal, and others. PNI is generally associated with a poor prognosis and can cause severe pain in patients [262]. In a landmark publication, Magnon and colleagues provided experimental evidence showing that autonomic nerve sprouting in prostate tumors was essential for the progression of prostate cancers [263]. Later on, vagal innervation was demonstrated to contribute to gastric tumorigenesis via M3 receptor-mediated Wnt signaling in the stem cells [264]. Further investigation revealed that Dclk1+ tuft cells and nerves were the main sources of acetylcholine (ACh) within the gastric mucosa [265]. While cholinergic stimulation was evidenced to induce nerve growth factor (NGF) expression in the gastric epithelium, NGF/Trk signaling in turn was demonstrated to regulate mucosal innervation and promote carcinogenesis [265]. Considering the importance of NRG signaling in the development of nervous system [12, 266], these findings have shed a new light on the possible nerve-dependent mechanism of NRG signaling in regulating tumorigenesis [267].
Inducing angiogenesis is one of the key hallmarks of both primary and metastatic cancer [2]. When the primary tumor or metastases grow to a certain extent, oxygen and nutrient supply and the discharge of metabolic wastes and carbon dioxide are insufficient. Thus, the diameter of cancer can rarely exceed 2–3 mm without neovascularization. Hypoxia in TME is a key driver of transition from pre-vascular hyperplasia to highly vascularized and progressively outgrowing tumors termed “angiogenic switch,” a process being fine-tuned by factors that either induce or inhibit angiogenesis [268, 269]. Among many reported pro-angiogenic proteins, vascular endothelial growth factor (VEGF) mainly secreted by cancer cells is now known to be central to this process [270]. Moreover, a large body of evidence indicates that angiogenic programming of tumor tissue is regulated by TME in concert with cancer cells [271]. Once the blood vessels grow into the tumor, the way of supplying nutrients and oxygen to the tumor tissue changes from peripheral diffusion to blood perfusion, and its metabolic wastes can be eliminated in time. In addition, tumor blood vessels can also determine the pathophysiology, growth, invasion, and metastasis of cancer, as well as its response to various treatments [272]. In normal condition, the neuregulin signaling has been demonstrated to play an important role in development of cardiovascular system [273, 274]. In a recent study, Nrg4 was also showed to be an angiogenic factor involved in maintaining adipose tissue vasculature [275]. Given the findings that the expression of pro-angiogenic proteins can be induced by several activated oncogenes including ErbB2 [276, 277], the role of neuregulin signaling in the process of tumor angiogenesis has attracted much research interest as early as in the very beginning of this century. It was revealed by Yen et al. for the first time that HRG β1 can selectively upregulate the expression of VEGF in ErbB2-overexpressing breast cancer cells [278]. The induced expression of VEGF by HRG β1 was further confirmed by two independent research groups thereafter [279, 280]. Furthermore, HRG signaling-induced VEGF expression was also found in colon cancer [281].
Upon activation, normally quiescent vasculature sprouts new vessels to sustain the expanding growth of tumor. During this process, the recruitment of pericytes by platelet-derived growth factor-B (PDGFB) in TME was demonstrated to play an important role in maintaining appropriate vascular morphogenesis [282,283,284]. Pericytes vary not only in their morphology but also in protein markers they expressed as well as their origin [285]. Importantly, only type-2 pericytes were demonstrated to participate in normal and tumoral angiogenesis [286]. Taking into consideration the role of HIF signaling in regulating the secretion of PDGFB by endothelial cells [287], as well as the reported cross talk between HER2 and HIF signalings [288, 289], it is of great interest to explore the more exact role of NRG signaling in tumor angiogenesis.
1.5 Targeting of Neuregulin Signaling in Cancer Therapy
Given the pivotal oncogenic role of aberrant NRG signaling in a wide variety of human cancers, therapeutic targeting of NRG signaling in cancer treatment has long been proposed and extensively studied. Strategies developed for NRG signaling-targeted therapy are different owing to the broad spectrum of casual mutations/aberrations unraveled. Some of them have been successfully approved for clinic application in cancer therapy and achieved favorable outcome. Since both EGFR- and ErbB2-targeted therapies have been extensively reviewed [290,291,292], our discussion mainly focuses on, but is not restricted to, NRG/ErbB3-targeted therapy in the following section.
Unlike its close relatives EGFR and ErbB2, less attention has been paid to the oncogenic functions of ErbB3 due to its weak kinase activity. However, during the last two decades, ErbB3 has been shown to be a direct therapeutic target in cancer treatment because of its role in tumor biology [293]. Currently, strategies developed for ErbB3-targeted therapy are mainly focused on blocking its activation through antibodies. The various ErbB3-targeted antibodies, including seribantumab (also known as MM-121/SAR256212) [294], LJM716 [295], and patritumab (or U3–1287) [296], have been demonstrated to inhibit ErbB3 pathway activation via different ways. MM-121, a fully humanized monoclonal antibody that interferes in binding of HRG to ErbB3, has been shown to effectively block ligand-dependent activation of ErbB3 induced by either EGFR, HER2, or MET [297]. In our own studies, we found that targeting of ErbB3 with MM-121 not only potentiated antitumor activity of paclitaxel against ErbB2+ breast cancer [298] but also had a potential of reversing trastuzumab resistance [299]. Recently, patritumab was evidenced to overcome ligand-mediated resistance to trastuzumab in ErbB2+ breast cancer via synergistically targeting of ErbB2/ ErbB3 signaling axis [300]. With the significant advances in antibody engineering technologies, strategies that simultaneously target multiple receptors with bispecific or multi-specific antibodies have been developed and demonstrated to circumvent the limitations of conventional mono-specific therapies and achieved enhanced therapeutic efficacy [301]. MEHD7945A, a monoclonal antibody that dually targets EGFR and ErbB3 [302], has been shown to be more effective than a combination of cetuximab and anti-HER3 antibody at inhibiting both EGFR/HER3 signaling and tumor growth [303]. Although multiple phase I and II trials have been opened for various ErbB3-targeted antibodies [304, 305], clinical benefits of the antibodies as single agents have not been reported. In a randomized phase II trial of paclitaxel with or without seribantumab in patients with advanced platinum-resistant or platinum-refractory ovarian cancer, although no difference in progression-free survival (PFS) between the two arms was reported, the authors’ exploratory analyses suggested that high HRG and low ErbB2 might be predictive of seribantumab benefit [306]. More recently, it was reported that the combination of SAR256212 and PI3K inhibitor SAR245408 for patients with metastatic or locally advanced solid tumors resulted in stable disease as the best response without effect on the pharmacokinetics of either drug, and the side effects seen in combination were similar to the profiles of each individual drug [307]. These results of clinical trials indicate that deliberate selection of patient and combinatory application with other treatments will be critical for the clinical practice of ErbB3-targeted therapy in the future [308].
In addition to blocking antibody, novel approaches targeting ErbB3 have also been proposed [309]. EZN-3920, a locked nucleic acid (LNA)-based ErbB3 antisense oligonucleotide, has been demonstrated to inhibit growth of xenograft models of breast and lung cancer cell lines via specific downregulation of ErbB3 [310]. As alterations in chromatin structure by histone modification and/or DNA methylation have been extensively correlated with cancer development, progression, and resistance to therapy [311, 312], epigenetic targeting is emerging as a promising therapeutic strategy for cancer treatment [313]. Histone deacetylases (HDACs), whose deregulation is evidenced to play an important role in aberrant gene expression in tumorigenesis, have long been recognized as druggable targets [314]. We previously reported that the class I HDAC inhibitor entinostat (also known as MS-275 or SNDX-275) specifically enhanced expression of miR-125a, miR-125b, and miR-205, which acted in concert to downregulate ErbB2 and ErbB3 and selectively induced apoptosis in ErbB2-overexpressing breast cancer cells [315,316,317]. Recently, we found that valproic acid (VPA), a safely used anticonvulsant drug with reported HDACi capability, held an antitumor activity selectively against EGFR/ErbB2/ErbB3-coexpressing pancreatic cancer via induction of ErbB family members-targeting microRNAs [318]. Thus, miRNA-mediated epigenetic regulation may represent a new mechanism inactivating ErbB2/ErbB3 [319].
Since ligand binding is indispensable for successful eliciting of NRG/ErbB signaling, it is rational that specific downregulation of NRG and/or blocking its binding to ErbB3 should be an attractive strategy for interrupting this signaling. Actually, in the very beginning of this century, it was discovered that blockade of HRG expression using an antisense nucleotide technology resulted in inhibition of cell proliferation and anchorage-independent growth, as well as suppression of invasive potential of breast cancer cells in vitro [320]. Targeting of ADAM17, a major ErbB ligand sheddase, was also reported to inhibit ErbB3 and EGFR pathways through preventing the processing and activation of multiple ErbB ligands [38]. Neutralization antibodies have been extensively explored in targeting excess ligands in cancer therapeutics. By using two self-developed blocking antibodies of NRG1, Hegde et al. reported a successful inhibition of NRG1 signaling in NSCLC, leading to an enhanced duration of the response to chemotherapy [321]. Moreover, a NRG1-specific antibody 7E3 has been demonstrated as a promising antitumor agent against pancreatic cancer recently. 7E3 not only promotes antibody-dependent cellular cytotoxicity (ADCC) in NRG1-positive pancreatic cancer cells and cancer-associated fibroblasts (CAFs) and inhibits NRG1-associated signaling pathway induction in vitro; it also suppresses migration and growth of pancreatic cancer cells co-cultured with CAFs, both in vitro and in vivo using orthotopic pancreatic cancer xenografts [322]. Large body of functional studies has confirmed that miRNA dysregulation plays a causal role in many cases of cancer. Insights into the roles of miRNAs in cancer have made them attractive tools and targets for novel therapeutic approaches [323]. MiRNAs may act as tumor suppressors or oncogenes (oncomiRs), and miRNA mimics and molecules targeted at miRNAs (antimiRs) have shown promise in preclinical development. In a recent study, miR-296-5p was demonstrated to be significantly downregulated in HCC tissues, and introduced miR-296-5p was able to suppress EMT of HCC via direct targeting of NRG1 [169]. Thus, these findings have raised the possibility of miRNA therapeutics in NRG1-targeted therapy, which deserves further investigation. In addition, with the landmark finding of CD74–NRG1 gene fusions in lung adenocarcinoma in 2014 [107], much effort has been made in exploring NRG1 fusion-targeted therapy in cancer management [324]. Although strategies proposed for NRG1 fusion-targeted therapy are still focused on interruption of downstream ErbB signaling [325,326,327], whether NRG1 fusion itself may act as a direct druggable target is of great interest.
1.6 Conclusions and Future Perspectives
Although extensive effort has concentrated on elucidating the role of NRG signaling in the tumor microenvironment, several critical questions have been raised owing to the progress in some rapidly evolving fields of cancer research. Firstly, with the increasing recognition of link between the microbiota and cancer [328], the identification of intratumor microbiota has broadened the dimension of complexity of TME [329]. Microbiota has been demonstrated to play a key role in carcinogenesis and regulation of the response to therapy through a variety of mechanisms such as bacterial dysbiosis, production of genotoxins, pathobionts, and disruption of the host metabolism [330]. In a cross-sectional study, Tsay et al. reported that enrichment of the lower airway microbiota with oral commensals was associated with upregulation of the PI3K signaling pathway in lung cancer [331]. Since the underlying mechanism of microbiota-induced upregulation of PI3K pathway remains undiscovered, whether NRG signaling is involved in this biological process deserves further exploration. Secondly, although anticancer immunotherapies involving the use of immune checkpoint inhibitors or adoptive cellular transfer have achieved promising clinical outcome, resistance to immunotherapy is still a major obstacle to be overcome [332, 333]. As has been revealed by two independent groups, loss of PTEN results in resistance to T-cell-mediated immunotherapy [334, 335]. These findings may hint a causal role of aberrant NRG signaling in resistance to immunotherapy. Thus, it will be of special interest to figure out whether simultaneous targeting of NRG signaling may enhance the efficacy of immunotherapy. Finally, exosome-mediated communication within TME has been recognized as an important player in regulating tumor progression [336]. An improved understanding of whether NRGs may be present in exosome and act locally and/or distantly on eliciting downstream signaling will be of great help in the guidance of developing novel NRG-targeted therapy, such as eliminating oncogenic exosomes with aptamer-functionalized nanoparticles [337]. In conclusion, we believe that the answer of these questions will offer a clearer understanding of the role of NRG signaling in the TME and point out the future direction of mechanism-based NRG signaling-targeted therapy.
References
Bissell MJ, Radisky D (2001) Putting tumours in context. Nat Rev Cancer 1(1):46–54
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–774
Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21(3):309–322
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437
Montero JC, Rodriguez-Barrueco R, Ocana A, Diaz-Rodriguez E, Esparis-Ogando A, Pandiella A (2008) Neuregulins and cancer. Clin Cancer Res 14(11):3237–3241
Seroogy KB, Dickerson JW, Cassella SN, Zhang-Auberson L (2013) Neuregulins. In: Handbook of biologically active peptides. Elsevier Academic Press, Amsterdam, pp 1633–1638
Stove C, Bracke M (2004) Roles for neuregulins in human cancer. Clin Exp Metastasis 21(8):665–684
Breuleux M (2007) Role of heregulin in human cancer. Cell Mol Life Sci 64(18):2358–2377
Kawakami H, Yonesaka K (2016) HER3 and its ligand, heregulin, as targets for cancer therapy. Recent Pat Anticancer Drug Discov 11(3):267–274
Jones MR, Lim H, Shen Y, Pleasance E, Ch’ng C, Reisle C, Leelakumari S, Zhao C, Yip S, Ho J, Zhong E, Ng T, Ionescu D, Schaeffer DF, Mungall AJ, Mungall KL, Zhao Y, Moore RA, Ma Y, Chia S, Ho C, Renouf DJ, Gelmon K, Jones SJM, Marra MA, Laskin J (2017) Successful targeting of the NRG1 pathway indicates novel treatment strategy for metastatic cancer. Ann Oncol 28(12):3092–3097
Falls DL (2003) Neuregulins: functions, forms, and signaling strategies. Exp Cell Res 284(1):14–30
Esper RM, Pankonin MS, Loeb JA (2006) Neuregulins: versatile growth and differentiation factors in nervous system development and human disease. Brain Res Rev 51(2):161–175
Wen D, Peles E, Cupples R, Suggs SV, Bacus SS, Luo Y, Trail G, Hu S, Silbiger SM, Levy RB (1992) Neu differentiation factor: a transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 69(3):559–572
Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD (1993) ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell 72(5):801–815
Marchionni MA, Goodearl AD, Chen MS, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy F, Misumi D, Sudhalter J, Kobayashi K, Wroblewski D, Lynch C, Baldassare M, Hiles I, Davis JB, Hsuan JJ, Totty NF, Otsu M, McBurney RN, Waterfield MD, Stroobant P, Gwynne D (1993) Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature 362(2618):312–318
Ho WH, Armanini MP, Nuijens A, Phillips HS, Osheroff PL (1995) Sensory and motor neuron-derived factor. A novel heregulin variant highly expressed in sensory and motor neurons. J Biol Chem 270(24):14523–14532
Peles E, Bacus SS, Koski RA, Lu HS, Wen D, Ogden SG, Levy RB, Yarden Y (1992) Isolation of the neu/HER-2 stimulatory ligand: a 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell 69(1):205–216
Holmes WE, Sliwkowski MX, Akita RW, Henzel WJ, Lee J, Park JW, Yansura D, Abadi N, Raab H, Lewis GD (1992) Identification of heregulin, a specific activator of p185erbB2. Science 256(5060):1205–1210
Carraway KL, Sliwkowski MX, Akita R, Platko JV, Guy PM, Nuijens A, Diamonti AJ, Vandlen RL, Cantley LC, Cerione RA (1994) The erbB3 gene product is a receptor for heregulin. J Biol Chem 269(19):14303–14306
Tzahar E, Levkowitz G, Karunagaran D, Yi L, Peles E, Lavi S, Chang D, Liu N, Yayon A, Wen D (1994) ErbB-3 and ErbB-4 function as the respective low and high affinity receptors of all Neu differentiation factor/heregulin isoforms. J Biol Chem 269(40):25226–25233
Carraway KL, Weber JL, Unger MJ, Ledesma J, Yu N, Gassmann M, Lai C (1997) Neuregulin-2, a new ligand of ErbB3/ErbB4-receptor tyrosine kinases. Nature 387(6632):512–516
Chang H, Riese DJ, Gilbert W, Stern DF, McMahan UJ (1997) Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature 387(6632):509–512
Zhang D, Sliwkowski MX, Mark M, Frantz G, Akita R, Sun Y, Hillan K, Crowley C, Brush J, Godowski PJ (1997) Neuregulin-3 (NRG3): a novel neural tissue-enriched protein that binds and activates ErbB4. Proc Natl Acad Sci U S A 94(18):9562–9567
Harari D, Tzahar E, Romano J, Shelly M, Pierce JH, Andrews GC, Yarden Y (1999) Neuregulin-4: a novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene 18(17):2681–2689
Watanabe E, Maeda N, Matsui F, Kushima Y, Noda M, Oohira A (1995) Neuroglycan C, a novel membrane-spanning chondroitin sulfate proteoglycan that is restricted to the brain. J Biol Chem 270(45):26876–26882
Kinugasa Y, Ishiguro H, Tokita Y, Oohira A, Ohmoto H, Higashiyama S (2004) Neuroglycan C, a novel member of the neuregulin family. Biochem Biophys Res Commun 321(4):1045–1049
Uchida T, Wada K, Akamatsu T, Yonezawa M, Noguchi H, Mizoguchi A, Kasuga M, Sakamoto C (1999) A novel epidermal growth factor-like molecule containing two follistatin modules stimulates tyrosine phosphorylation of erbB-4 in MKN28. Biochem Biophys Res Commun 266(2):593–602
Ring HZ, Chang H, Guilbot A, Brice A, LeGuern E, Francke U (1999) The human neuregulin-2 (NRG2) gene: cloning, mapping and evaluation as a candidate for the autosomal recessive form of Charcot-Marie-tooth disease linked to 5q. Hum Genet 104(4):326–332
Busfield SJ, Michnick DA, Chickering TW, Revett TL, Ma J, Woolf EA, Comrack CA, Dussault BJ, Woolf J, Goodearl AD, Gearing DP (1997) Characterization of a neuregulin-related gene, Don-1, that is highly expressed in restricted regions of the cerebellum and hippocampus. Mol Cell Biol 17(7):4007–4014
Higashiyama S, Horikawa M, Yamada K, Ichino N, Nakano N, Nakagawa T, Miyagawa J, Matsushita N, Nagatsu T, Taniguchi N, Ishiguro H (1997) A novel brain-derived member of the epidermal growth factor family that interacts with ErbB3 and ErbB4. J Biochem 122(3):675–680
Gizatullin RZ, Muravenko OV, Al-Amin AN, Wang F, Protopopov AI, Kashuba VI, Zelenin AV, Zabarovsky ER (2000) Human NRG3 gene Map position 10q22-q23. Chromosom Res 8(6):560
Memon AA, Sorensen BS, Melgard P, Fokdal L, Thykjaer T, Nexo E (2004) Expression of HER3, HER4 and their ligand heregulin-4 is associated with better survival in bladder cancer patients. Br J Cancer 91(12):2034–2041
Hayes NV, Blackburn E, Smart LV, Boyle MM, Russell GA, Frost TM, Morgan BJ, Baines AJ, Gullick WJ (2007) Identification and characterization of novel spliced variants of neuregulin 4 in prostate cancer. Clin Cancer Res 13(11):3147–3155
Montero JC, Yuste L, Diaz-Rodriguez E, Esparis-Ogando A, Pandiella A (2000) Differential shedding of transmembrane neuregulin isoforms by the tumor necrosis factor-alpha-converting enzyme. Mol Cell Neurosci 16(5):631–648
Shirakabe K, Wakatsuki S, Kurisaki T, Fujisawa-Sehara A (2001) Roles of Meltrin beta/ADAM19 in the processing of neuregulin. J Biol Chem 276(12):9352–9358
Fleck D, Voss M, Brankatschk B, Giudici C, Hampel H, Schwenk B, Edbauer D, Fukumori A, Steiner H, Kremmer E, Haug-Kröper M, Rossner MJ, Fluhrer R, Willem M, Haass C (2016) Proteolytic processing of Neuregulin 1 type III by three intramembrane-cleaving proteases. J Biol Chem 291(1):318–333
Willem M (2016) Proteolytic processing of Neuregulin-1. Brain Res Bull 126(Pt 2):178–182
Zhou BB, Peyton M, He B, Liu C, Girard L, Caudler E, Lo Y, Baribaud F, Mikami I, Reguart N, Yang G, Li Y, Yao W, Vaddi K, Gazdar AF, Friedman SM, Jablons DM, Newton RC, Fridman JS, Minna JD, Scherle PA (2006) Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 10(1):39–50
Montero JC, Rodriguez-Barrueco R, Yuste L, Juanes PP, Borges J, Esparis-Ogando A, Pandiella A (2007) The extracellular linker of pro-Neuregulin-alpha2c is required for efficient sorting and juxtacrine function. Mol Biol Cell 18(2):380–393
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signaling network. Nat Rev Mol Cell Biol 2(2):127–137
Carpenter G (2003) ErbB-4: mechanism of action and biology. Exp Cell Res 284(1):66–77
Citri A, Skaria KB, Yarden Y (2003) The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res 284(1):54–65
Yarden Y, Pines G (2012) The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 12(8):553–563
Arteaga CL, Engelman JA (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25(3):282–303
Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, Leahy DJ (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421(6924):756–760
Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Kofler M, Jorissen RN, Nice EC, Burgess AW, Ward CW (2003) The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell 11(2):495–505
Shi F, Telesco SE, Liu Y, Radhakrishnan R, Lemmon MA (2010) ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc Natl Acad Sci U S A 107(17):7692–7697
Schulze WX, Deng L, Mann M (2005) Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol 1:2005.0008
Olayioye MA, Neve RM, Lane HA, Hynes NE (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J 19(13):3159–3167
DeFazio A, Chiew YE, Sini RL, Janes PW, Sutherland RL (2000) Expression of c-erbB receptors, heregulin and oestrogen receptor in human breast cell lines. Int J Cancer 87(4):487–498
Bieche I, Onody P, Tozlu S, Driouch K, Vidaud M, Lidereau R (2003) Prognostic value of ERBB family mRNA expression in breast carcinomas. Int J Cancer 106(5):758–765
Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE (2003) The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A 100(15):8933–8938
Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM (2008) A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res 68(14):5878–5887
Liu B, Ordonez-Ercan D, Fan Z, Huang X, Edgerton SM, Yang X, Thor AD (2009) Estrogenic promotion of ErbB2 tyrosine kinase activity in mammary tumor cells requires activation of ErbB3 signaling. Mol Cancer Res 7(11):1882–1892
Liu B, Ordonez-Ercan D, Fan Z, Edgerton SM, Yang X, Thor AD (2007) Down-regulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int J Cancer 120(9):1874–1882
Wang S, Huang X, Lee C-K, Liu B (2010) Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene 29(29):4225–4236
Buonanno A, Fischbach GD (2001) Neuregulin and ErbB receptor signaling pathways in the nervous system. Curr Opin Neurobiol 11(3):287–296
Yang Y, Spitzer E, Meyer D, Sachs M, Niemann C, Hartmann G, Weidner KM, Birchmeier C, Birchmeier W (1995) Sequential requirement of hepatocyte growth factor and neuregulin in the morphogenesis and differentiation of the mammary gland. J Cell Biol 131(1):215–226
Kogata N, Zvelebil M, Howard BA (2013) Neuregulin 3 and erbb signalling networks in embryonic mammary gland development. J Mammary Gland Biol Neoplasia 18(2):149–154
Kramer R, Bucay N, Kane DJ, Martin LE, Tarpley JE, Theill LE (1996) Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc Natl Acad Sci U S A 93(10):4833–4838
Patel NV, Acarregui MJ, Snyder JM, Klein JM, Sliwkowski MX, Kern JA (2000) Neuregulin-1 and human epidermal growth factor receptors 2 and 3 play a role in human lung development in vitro. Am J Respir Cell Mol Biol 22(4):432–440
Kim D, Chi S, Lee KH, Rhee S, Kwon YK, Chung CH, Kwon H, Kang MS (1999) Neuregulin stimulates myogenic differentiation in an autocrine manner. J Biol Chem 274(22):15395–15400
Noguchi H, Sakamoto C, Wada K, Akamatsu T, Uchida T, Tatsuguchi A, Matsui H, Fukui H, Fujimori T, Kasuga M (1999) Expression of heregulin alpha, erbB2, and erbB3 and their influences on proliferation of gastric epithelial cells. Gastroenterology 117(5):1119–1127
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5(5):341–354
Hynes NE, MacDonald G (2009) ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 21(2):177–184
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):67–70
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134
Witsch E, Sela M, Yarden Y (2010) Roles for growth factors in cancer progression. Physiology (Bethesda) 25(2):85–101
Schechter AL, Stern DF, Vaidyanathan L, Decker SJ, Drebin JA, Greene MI, Weinberg RA (1984) The neu oncogene: an erb-B-related gene encoding a 185,000-Mr tumour antigen. Nature 312(5994):513–516
Downward J, Yarden Y, Mayes E, Scrace G, Totty N, Stockwell P, Ullrich A, Schlessinger J, Waterfield MD (1984) Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 307(5951):521–527
Andrechek ER, Hardy WR, Siegel PM, Rudnicki MA, Cardiff RD, Muller WJ (2000) Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc Natl Acad Sci U S A 97(7):3444–3449
Finkle D, Quan ZR, Asghari V, Kloss J, Ghaboosi N, Mai E, Wong WL, Hollingshead P, Schwall R, Koeppen H, Erickson S (2004) HER2-targeted therapy reduces incidence and progression of midlife mammary tumors in female murine mammary tumor virus huHER2-transgenic mice. Clin Cancer Res 10(7):2499–2511
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235(4785):177–182
Thor AD, Schwartz LH, Koerner FC, Edgerton SM, Skates SJ, Yin S, McKenzie SJ, Panicali DL, Marks PJ, Fingert HJ, Wood WC (1989) Analysis of c-erbB-2 expression in breast carcinomas with clinical follow-up. Cancer Res 49(24 Pt 1):7147–7152
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Sugawa N, Ekstrand AJ, James CD, Collins VP (1990) Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A 87(21):8602–8606
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350(21):2129–2139
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304(5676):1497–1500
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz RP, Bowman KK, Lorenzo M, Li H, Wu J, Yuan W, Peters BA, Kan Z, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, de Sauvage FJ, Seshagiri S (2013) Oncogenic ERBB3 mutations in human cancers. Cancer Cell 23(5):603–617
Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC, Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel SB, Lander ES, Gibbs RA, Meyerson M, Wilson RK (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455(7216):1069–1075
Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y (2009) Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet 41(10):1127–1132
Krane IM, Leder P (1996) NDF/heregulin induces persistence of terminal end buds and adenocarcinomas in the mammary glands of transgenic mice. Oncogene 12(8):1781–1788
Aguilar Z, Akita RW, Finn RS, Ramos BL, Pegram MD, Kabbinavar FF, Pietras RJ, Pisacane P, Sliwkowski MX, Slamon DJ (1999) Biologic effects of heregulin/neu differentiation factor on normal and malignant human breast and ovarian epithelial cells. Oncogene 18(44):6050–6062
Li Q, Ahmed S, Loeb JA (2004) Development of an autocrine neuregulin signaling loop with malignant transformation of human breast epithelial cells. Cancer Res 64(19):7078–7085
Atlas E, Cardillo M, Mehmi I, Zahedkargaran H, Tang C, Lupu R (2003) Heregulin is sufficient for the promotion of tumorigenicity and metastasis of breast cancer cells in vivo. Mol Cancer Res 1(3):165–175
Schmitt M, Walker MP, Richards RG, Bocchinfuso WP, Fukuda T, Medina D, Kittrell FS, Korach KS, DiAugustine RP (2006) Expression of heregulin by mouse mammary tumor cells: role in activation of ErbB receptors. Mol Carcinog 45(7):490–505
Richman J, Dowsett M (2019) Beyond 5 years: enduring risk of recurrence in oestrogen receptor-positive breast cancer. Nat Rev Clin Oncol 16(5):296–311
Carroll JS, Hickey TE, Tarulli GA, Williams M, Tilley WD (2017) Deciphering the divergent roles of progestogens in breast cancer. Nat Rev Cancer 17(1):54–64
Tang CK, Perez C, Grunt T, Waibel C, Cho C, Lupu R (1996) Involvement of heregulin-beta2 in the acquisition of the hormone-independent phenotype of breast cancer cells. Cancer Res 56(14):3350–3358
Balañá ME, Lupu R, Labriola L, Charreau EH, Elizalde PV (1999) Interactions between progestins and heregulin (HRG) signaling pathways: HRG acts as mediator of progestins proliferative effects in mouse mammary adenocarcinomas. Oncogene 18(46):6370–6379
Labriola L, Salatino M, Proietti CJ, Pecci A, Coso OA, Kornblihtt AR, Charreau EH, Elizalde PV (2003) Heregulin induces transcriptional activation of the progesterone receptor by a mechanism that requires functional ErbB-2 and mitogen-activated protein kinase activation in breast cancer cells. Mol Cell Biol 23(3):1095–1111
Proietti CJ, Rosemblit C, Beguelin W, Rivas MA, Díaz Flaqué MC, Charreau EH, Schillaci R, Elizalde PV (2009) Activation of Stat3 by heregulin/ErbB-2 through the co-option of progesterone receptor signaling drives breast cancer growth. Mol Cell Biol 29(5):1249–1265
Weinstein EJ, Leder P (2000) The extracellular region of heregulin is sufficient to promote mammary gland proliferation and tumorigenesis but not apoptosis. Cancer Res 60(14):3856–3861
Centa A, Rodríguez-Barrueco R, Montero JC, Pandiella A (2018) The immunoglobulin-like domain of neuregulins potentiates ErbB3/HER3 activation and cellular proliferation. Mol Oncol 12(7):1061–1076
De Iuliis F, Salerno G, Taglieri L, Lanza R, Cardelli P, Scarpa S (2017) Circulating neuregulin-1 and galectin-3 can be prognostic markers in breast cancer. Int J Biol Markers 32(3):e333–e336
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424
Morgensztern D, Ng SH, Gao F, Govindan R (2010) Trends in stage distribution for patients with non-small cell lung cancer: a National Cancer Database survey. J Thorac Oncol 5(1):29–33
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK (2014) Non- small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer 14(8):535–546
Skoulidis F, Heymach JV (2019) Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer 19(9):495–509
Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, McGraw T, Mittal V (2019) The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer 19(1):9–31
Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511(7511):543–550
Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J (2004) Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431(7008):525–526
Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E et al (2012) Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150(6):1107–1120
Sithanandam G, Anderson LM (2008) The ERBB3 receptor in cancer and cancer gene therapy. Cancer Gene Ther 15(7):413–448
al Moustafa AE, Alaoui-Jamali M, Paterson J, O’Connor-McCourt M (1999) Expression of P185erbB-2, P160erbB-3, P180erbB-4, and heregulin alpha in human normal bronchial epithelial and lung cancer cell lines. Anticancer Res 19(1A):481–486
Gollamudi M, Nethery D, Liu J, Kern JA (2004) Autocrine activation of ErbB2/ErbB3 receptor complex by NRG-1 in non-small cell lung cancer cell lines. Lung Cancer 43(2):135–143
Fernandez-Cuesta L, Plenker D, Osada H, Sun R, Menon R, Leenders F et al (2014) CD74-NRG1 fusions in lung adenocarcinoma. Cancer Discov 4(4):415–422
Duruisseaux M, McLeer-Florin A, Antoine M, Alavizadeh S, Poulot V, Lacave R, Rabbe N, Cadranel J, Wislez M (2016) NRG1 fusion in a French cohort of invasive mucinous lung adenocarcinoma. Cancer Med 5(12):3579–3585
Trombetta D, Graziano P, Scarpa A, Sparaneo A, Rossi G, Rossi A et al (2018) Frequent NRG1 fusions in Caucasian pulmonary mucinous adenocarcinoma predicted by Phospho-ErbB3 expression. Oncotarget 9(11):9661–9671
Jonna S, Feldman RA, Swensen J, Gatalica Z, Korn WM, Borghaei H, Ma PC, Nieva JJ, Spira AI, Vanderwalde AM, Wozniak AJ, Kim ES, Liu SV (2019) Detection of NRG1 gene fusions in solid tumors. Clin Cancer Res 25(16):4966–4972
Pan Y, Zhang Y, Ye T, Zhao Y, Gao Z, Yuan H, Zheng D, Zheng S, Li H, Li Y, Jin Y, Sun Y, Chen H (2019) Detection of novel NRG1, EGFR and MET fusions in lung adenocarcinomas in the Chinese population. J Thorac Oncol 14(11):2003–2008
van Lengerich B, Agnew C, Puchner EM, Huang B, Jura N (2017) EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc Natl Acad Sci U S A 114(14):E2836–E2845
Trombetta D, Rossi A, Fabrizio FP, Sparaneo A, Graziano P, Fazio VM, Muscarella LA (2017) NRG1-ErbB lost in translation: a new paradigm for lung cancer? Curr Med Chem 24(38):4213–4228
Sakai K, Mori S, Kawamoto T, Taniguchi S, Kobori O, Morioka Y, Kuroki T, Kano K (1986) Expression of epidermal growth factor receptors on normal human gastric epithelia and gastric carcinomas. J Natl Cancer Inst 77(5):1047–1052
De Vita F, Giuliani F, Silvestris N, Catalano G, Ciardiello F, Orditura M (2010) Human epidermal growth factor receptor 2 (HER2) in gastric cancer: a new therapeutic target. Cancer Treat Rev 36(Suppl 3):S11–S15
Yun S, Koh J, Nam SK, Park JO, Lee SM, Lee K, Lee KS, Ahn SH, Park DJ, Kim HH, Choe G, Kim WH, Lee HS (2018) Clinical significance of overexpression of NRG1 and its receptors, HER3 and HER4, in gastric cancer patients. Gastric Cancer 21(2):225–236
Hayashi M, Inokuchi M, Takagi Y, Yamada H, Kojima K, Kumagai J, Kawano T, Sugihara K (2008) High expression of HER3 is associated with a decreased survival in gastric cancer. Clin Cancer Res 14(23):7843–7849
Han ME, Kim HJ, Shin DH, Hwang SH, Kang CD, Oh SO (2015) Overexpression of NRG1 promotes progression of gastric cancer by regulating the self-renewal of cancer stem cells. J Gastroenterol 50(6):645–656
Khelwatty SA, Essapen S, Seddon AM, Modjtahedi H (2013) Prognostic significance and targeting of HER family in colorectal cancer. Front Biosci (Landmark Ed) 18:394–421
Maurer CA, Friess H, Kretschmann B, Zimmermann A, Stauffer A, Baer HU, Korc M, Büchler MW (1998) Increased expression of erbB3 in colorectal cancer is associated with concomitant increase in the level of erbB2. Hum Pathol 29(8):771–777
Lee JC, Wang ST, Chow NH, Yang HB (2002) Investigation of the prognostic value of coexpressed erbB family members for the survival of colorectal cancer patients after curative surgery. Eur J Cancer 38(8):1065–1071
Grivas PD, Antonacopoulou A, Tzelepi V, Sotiropoulou-Bonikou G, Kefalopoulou Z, Papavassiliou AG, Kalofonos H (2007) HER-3 in colorectal tumourigenesis: from mRNA levels through protein status to clinicopathologic relationships. Eur J Cancer 43(17):2602–2611
Beji A, Horst D, Engel J, Kirchner T, Ullrich A (2012) Towards the prognostic significance and therapeutic potential of HER3 receptor tyrosine kinase in human colon cancer. Clin Cancer Res 18(4):956–968
Venkateswarlu S, Dawson DM, St Clair P, Gupta A, Willson JK, Brattain MG (2002) Autocrine heregulin generates growth factor independence and blocks apoptosis in colon cancer cells. Oncogene 21(1):78–86
De Boeck A, Pauwels P, Hensen K, Rummens JL, Westbroek W, Hendrix A, Maynard D, Denys H, Lambein K, Braems G, Gespach C, Bracke M, De Wever O (2013) Bone marrow-derived mesenchymal stem cells promote colorectal cancer progression through paracrine neuregulin 1/HER3 signalling. Gut 62(4):550–560
Yonezawa M, Wada K, Tatsuguchi A, Akamatsu T, Gudis K, Seo T, Mitsui K, Nagata K, Tanaka S, Fujimori S, Sakamoto C (2009) Heregulin-induced VEGF expression via the ErbB3 signaling pathway in colon cancer. Digestion 80(4):215–225
Mitsui K, Yonezawa M, Tatsuguchi A, Shinji S, Gudis K, Tanaka S, Fujimori S, Sakamoto C (2014) Localization of phosphorylated ErbB1-4 and heregulin in colorectal cancer. BMC Cancer 14:863
Guo Y, Duan Z, Jia Y, Ren C, Lv J, Guo P, Zhao W, Wang B, Zhang S, Li Y, Li Z (2018) HER4 isoform CYT2 and its ligand NRG1III are expressed at high levels in human colorectal cancer. Oncol Lett 15(5):6629–6635
Makohon-Moore A, Iacobuzio-Donahue CA (2016) Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer 16(9):553–565
Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK et al (2013) Recent progress in pancreatic cancer. CA Cancer J Clin 63(5):318–348
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P et al (2015) Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518(7540):495–501
Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL et al (2012) Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491(7424):399–405
Yachida S, White CM, Naito Y, Zhong Y, Brosnan JA, Macgregor-Das AM et al (2012) Clinical significance of the genetic landscape of pancreatic cancer and implications for identification of potential long-term survivors. Clin Cancer Res 18(22):6339–6347
Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D (2011) RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 11(11):761–774
Friess H, Wang L, Zhu Z, Gerber R, Schroder M, Fukuda A et al (1999) Growth factor receptors are differentially expressed in cancers of the papilla of vater and pancreas. Ann Surg 230(6):767–774
Heining C, Horak P, Uhrig S, Codo PL, Klink B, Hutter B et al (2018) NRG1 fusions in KRAS wild-type pancreatic cancer. Cancer Discov 8(9):1087–1095
Jones MR, Williamson LM, Topham JT, Lee MKC, Goytain A, Ho J et al (2019) NRG1 gene fusions are recurrent, clinically actionable gene rearrangements in KRAS wild-type pancreatic ductal adenocarcinoma. Clin Cancer Res 25(15):4674–4681
Aguirre AJ (2019) Oncogenic NRG1 fusions: a new Hope for targeted therapy in pancreatic cancer. Clin Cancer Res 25(15):4589–4591
Westphal M, Meima L, Szonyi E, Lofgren J, Meissner H, Hamel W, Nikolics K, Sliwkowski MX (1997) Heregulins and the ErbB-2/3/4 receptors in gliomas. J Neuro-Oncol 35(3):335–346
von Achenbach C, Weller M, Szabo E (2018) Epidermal growth factor receptor and ligand family expression and activity in glioblastoma. J Neurochem 147(1):99–109
Hansen MR, Linthicum FH (2004) Expression of neuregulin and activation of erbB receptors in vestibular schwannomas: possible autocrine loop stimulation. Otol Neurotol 25(2):155–159
Stonecypher MS, Chaudhury AR, Byer SJ, Carroll SL (2006) Neuregulin growth factors and their ErbB receptors form a potential signaling network for schwannoma tumorigenesis. J Neuropathol Exp Neurol 65(2):162–175
Lapointe S, Perry A, Butowski NA (2018) Primary brain tumours in adults. Lancet 392(10145):432–446
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359(5):492–507
Liu B, Wang L, Shen LL, Shen MZ, Guo XD, Wang T, Liang QC, Wang C, Zheng J, Li Y, Jia LT, Zhang H, Gao GD (2012) RNAi-mediated inhibition of presenilin 2 inhibits glioma cell growth and invasion and is involved in the regulation of Nrg1/ErbB signaling. Neuro-Oncology 14(8):994–1006
Esquela-Kerscher A, Slack FJ (2006) Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer 6(4):259–269
Yin F, Zhang JN, Wang SW, Zhou CH, Zhao MM, Fan WH, Fan M, Liu S (2015) MiR-125a-3p regulates glioma apoptosis and invasion by regulating Nrg1. PLoS One 10(1):e0116759
Srinivasan R, Benton E, McCormick F, Thomas H, Gullick WJ (1999) Expression of the c-erbB-3/HER-3 and cerbB-4/HER-4 growth factor receptors and their ligands, neuregulin-1 a, neuregulin-1 h, and betacellulin, in normal endometrium and endometrial cancer. Clin Cancer Res 5(10):2877–2883
Hsieh SY, He JR, Hsu CY, Chen WJ, Bera R, Lin KY, Shih TC, Yu MC, Lin YJ, Chang CJ, Weng WH, Huang SF (2011) Neuregulin/erythroblastic leukemia viral oncogene homolog 3 autocrine loop contributes to invasion and early recurrence of human hepatoma. Hepatology 53(2):504–516
Gilbertson RJ, Clifford SC, MacMeekin W, Meekin W, Wright C, Perry RH, Kelly P, Pearson AD, Lunec J (1998) Expression of the ErbB-neuregulin signaling network during human cerebellar development: implications for the biology of medulloblastoma. Cancer Res 58(17):3932–3941
Buac K, Xu M, Cronin J, Weeraratna AT, Hewitt SM, Pavan WJ (2009) NRG1/ERBB3 signaling in melanocyte development and melanoma: inhibition of differentiation and promotion of proliferation. Pigment Cell Melanoma Res 22(6):773–784
Gilmour LM, Macleod KG, McCaig A, Sewell JM, Gullick WJ, Smyth JF, Langdon SP (2002) Neuregulin expression, function, and signaling in human ovarian cancer cells. Clin Cancer Res 8(12):3933–3942
Sheng Q, Liu X, Fleming E, Yuan K, Piao H, Chen J, Moustafa Z, Thomas RK, Greulich H, Schinzel A, Zaghlul S, Batt D, Ettenberg S, Meyerson M, Schoeberl B, Kung AL, Hahn WC, Drapkin R, Livingston DM, Liu JF (2010) An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell 17(3):298–310
Kolb A, Kleeff J, Arnold N, Giese NA, Giese T, Korc M, Friess H (2007) Expression and differential signaling of heregulins in pancreatic cancer cells. Int J Cancer 120(3):514–523
Fluge O, Akslen LA, Haugen DR, Varhaug JE, Lillehaug JR (2000) Expression of heregulins and associations with the ErbB family of tyrosine kinase receptors in papillary thyroid carcinomas. Int J Cancer 87(6):763–770
He H, Li W, Liyanarachchi S, Wang Y, Yu L, Genutis LK, Maharry S, Phay JE, Shen R, Brock P, de la Chapelle A (2018) The role of NRG1 in the predisposition to papillary thyroid carcinoma. J Clin Endocrinol Metab 103(4):1369–1379
Leung HY, Weston J, Gullick WJ, Williams G (1997) A potential autocrine loop between heregulin-a and erbB-3 receptor in human prostatic adenocarcinoma. Br J Urol 79(2):212–216
Shames DS, Carbon J, Walter K, Jubb AM, Kozlowski C, Januario T, Do A, Fu L, Xiao Y, Raja R, Jiang B, Malekafzali A, Stern H, Settleman J, Wilson TR, Hampton GM, Yauch RL, Pirzkall A, Amler LC (2013) High heregulin expression is associated with activated HER3 and may define an actionable biomarker in patients with squamous cell carcinomas of the head and neck. PLoS One 8(2):e56765
Hijazi MM, Young PE, Dougherty MK, Bressette DS, Cao TT, Pierce JH, Wong LM, Alimandi M, King CR (1998) NRG-3 in human breast cancers: activation of multiple erbB family proteins. Int J Omcol 13(5):1061–1067
Turajlic S, Swanton C (2016) Metastasis as an evolutionary process. Science 352(6282):169–175
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331(6024):1559–1564
Valastyan S, Weinberg RA (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147(2):275–292
Sethi N, Kang Y (2011) Unravelling the complexity of metastasis-molecular understanding and targeted therapies. Nat Rev Cancer 11(10):735–748
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9(4):265–273
Massagué J (2008) TGFβ in cancer. Cell 134(2):215–230
Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7(2):131–142
Ritch PA, Carroll SL, Sontheimer H (2003) Neuregulin-1 enhances motility and migration of human astrocytic glioma cells. J Biol Chem 278(23):20971–20978
Zhao WJ, Schachner M (2013) Neuregulin 1 enhances cell adhesion molecule l1 expression in human glioma cells and promotes their migration as a function of malignancy. J Neuropathol Exp Neurol 72(3):244–255
Shi DM, Li LX, Bian XY, Shi XJ, Lu LL, Zhou HX, Pan TJ, Zhou J, Fan J, Wu WZ (2018) miR-296-5p suppresses EMT of hepatocellular carcinoma via attenuating NRG1/ERBB2/ERBB3 signaling. J Exp Clin Cancer Res 37(1):294
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14(9):611–622
Hüsemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmüller G, Klein CA (2008) Systemic spread is an early step in breast cancer. Cancer Cell 13(1):58–68
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ (2012) EMT and dissemination precede pancreatic tumor formation. Cell 148(1–2):349–361
Hosseini H, Obradović MMS, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M et al (2016) Early dissemination seeds metastasis in breast cancer. Nature 540(7634):552–558
Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R et al (2016) Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature 540(7634):588–592
Seoane S, Montero JC, Ocaña A, Pandiella A (2016) Breast cancer dissemination promoted by a neuregulin-collagenase 3 signalling node. Oncogene 35(21):2756–2765
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4(1):71–78
Murdoch C, Muthana M, Coffelt SB, Lewis CE (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 8(8):618–631
Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, Harper K, Tardio E, Reyes Torres I, Jones J, Condeelis J, Merad M, Aguirre-Ghiso JA (2018) Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun 9(1):21
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475(7355):222–225
Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64(19):7022–7029
Lewis CE, Harney AS, Pollard JW (2016) The multifaceted role of perivascular macrophages in tumors. Cancer Cell 30(1):18–25
Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, Krummel MF (2014) Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26(5):638–652
Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, Oktay MH, Pollard JW, Jones JG, Condeelis JS (2015) Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA. Cancer Discov 5(9):932–943
Leung E, Xue A, Wang Y, Rougerie P, Sharma VP, Eddy R, Cox D, Condeelis J (2017) Blood vessel endothelium-directed tumor cell streaming in breast tumors requires the HGF/C-Met signaling pathway. Oncogene 36(19):2680–2692
Robinson BD, Sica GL, Liu YF, Rohan TE, Gertler FB, Condeelis JS, Jones JG (2009) Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res 15(7):2433–2441
Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS (2009) Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res 69(24):9498–9506
Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, Condeelis JS (2018) A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep 23(5):1239–1248
Hernandez L, Smirnova T, Kedrin D, Wyckoff J, Zhu L, Stanley ER, Cox D, Muller WJ, Pollard JW, Van Rooijen N, Segall JE (2009) The EGF/CSF-1 paracrine invasion loop can be triggered by heregulin beta1 and CXCL12. Cancer Res 69(7):3221–3227
Lopez-Haber C, Barrio-Real L, Casado-Medrano V, Kazanietz MG (2016) Heregulin/ErbB3 signaling enhances CXCR4-driven Rac1 activation and breast cancer cell motility via hypoxia-inducible factor 1α. Mol Cell Biol 36(15):2011–2026
Meurette O, Mehlen P (2018) Notch signaling in the tumor microenvironment. Cancer Cell 34(4):536–548
Sethi N, Dai X, Winter CG, Kang Y (2011) Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19(2):192–205
Cabrera RM, Mao SPH, Surve CR, Condeelis JS, Segall JE (2018) A novel neuregulin-jagged1 paracrine loop in breast cancer transendothelial migration. Breast Cancer Res 20(1):24
Dhanasekaran SM, Balbin OA, Chen G, Nadal E, Kalyana-Sundaram S, Pan J et al (2014) Transcriptome metaanalysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes. Nat Commun 5:5893
Haskins JW, Nguyen DX, Stern DF (2014) Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci Signal 7(355):ra116
Sun M, Behrens C, Feng L, Ozburn N, Tang X, Yin G, Komaki R, Varella-Garcia M, Hong WK, Aldape KD, Wistuba II (2009) HER family receptor abnormalities in lung cancer brain metastases and corresponding primary tumors. Clin Cancer Res 15(15):4829–4837
Nakaoku T, Tsuta K, Ichikawa H, Shiraishi K, Sakamoto H, Enari M et al (2014) Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin Cancer Res 20(12):3087–3093
Shin DH, Lee D, Hong DW, Hong SH, Hwang JA, Lee BI, You HJ, Lee GK, Kim IH, Lee YS, Han JY (2016) Oncogenic function and clinical implications of SLC3A2-NRG1 fusion in invasive mucinous adenocarcinoma of the lung. Oncotarget 7(43):69450–69465
Longley DB, Johnston PG (2005) Molecular mechanisms of drug resistance. J Pathol 205(2):275–292
Swanton C (2012) Intratumor heterogeneity: evolution through space and time. Cancer Res 72(19):4875–4882
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13(10):714–726
Colvin ME, Sasaki JC, Tran NL (1999) Chemical factors in the action of phosphoramidic mustard alkylating anticancer drugs: roles for computational chemistry. Curr Pharm Des 5(8):645–663
Okada H, Mak TW (2004) Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 4(8):592–603
Kartal-Yandim M, Adan-Gokbulut A, Baran Y (2016) Molecular mechanisms of drug resistance and its reversal in cancer. Crit Rev Biotechnol 36(4):716–726
Lee Y, Ma J, Lyu H, Huang J, Kim A, Liu B (2014) Role of erbB3 receptors in cancer therapeutic resistance. Acta Biochim Biophys Sin Shanghai 46(3):190–198
Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, Schmidt M, Mills GB, Mendelsohn J, Fan Z (2003) HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene 22(21):3205–3212
Stagg J (2008) Mesenchymal stem cells in cancer. Stem Cell Rev 4(2):119–124
Ridge SM, Sullivan FJ, Glynn SA (2017) Mesenchymal stem cells: key players in cancer progression. Mol Cancer 16(1):–31
McMillin DW, Negri JM, Mitsiades CS (2013) The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Disc 12(3):217–228
Shi Y, Du L, Lin L, Wang Y (2017) Tumour-associated mesenchymal stem/stromal cells: emerging therapeutic targets. Nat Rev Drug Disc 16(1):35–52
Chen J, Ren Q, Cai Y, Lin T, Zuo W, Wang J, Lin R, Zhu L, Wang P, Dong H, Zhao H, Huang L, Fu Y, Yang S, Tan J, Lan X, Wang S (2018) Mesenchymal stem cells drive paclitaxel resistance in ErbB2/ErbB3-coexpressing breast cancer cells via paracrine of neuregulin 1. Biochem Biophys Res Commun 501(1):212–219
Bezler M, Hengstler JG, Ullrich A (2012) Inhibition of doxorubicin-induced HER3-PI3K-AKT signalling enhances apoptosis of ovarian cancer cells. Mol Oncol 6(5):516–529
Weinstein IB (2000) Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis 21(5):857–864
Weinstein IB (2002) Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science 297(5578):63–64
Sharma SV, Settleman J (2007) Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 21(24):3214–3231
Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II et al (2014) Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 311(19):1998–2006
Bivona TG, Doebele RC (2016) A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med 22(5):472–478
Rotow J, Bivona TG (2017) Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer 17(11):637–658
Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P et al (2011) Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3(75):75ra26
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C et al (2009) Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 361(10):958–967
de Bono JS, Ashworth A (2010) Translating cancer research into targeted therapeutics. Nature 467(7315):543–549
Vlacich G, Coffey RJ (2011) Resistance to EGFR-targeted therapy: a family affair. Cancer Cell 20(4):423–425
Nguyen KS, Kobayashi S, Costa DB (2009) Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer 10(4):281–289
Kruser TJ, Wheeler DL (2010) Mechanisms of resistance to HER family targeting antibodies. Exp Cell Res 316(7):1083–1100
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, Sutherlin DP, Koeppen H, Merchant M, Neve R, Settleman J (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487(7408):505–509
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316(5827):1039–1043
Sato Y, Yashiro M, Takakura N (2013) Heregulin induces resistance to lapatinib-mediated growth inhibition of HER2-amplified cancer cells. Cancer Sci 104(12):1618–1625
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K, Fukuoka M, Varella-Garcia M, Nakagawa K, Jänne PA (2011) Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 3(99):99ra86
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S et al (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448(7153):561–566
Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ et al (2008) EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 14(13):4275–4283
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B et al (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363(18):1693–1703
Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, Nakamura T, Matsumoto K, Soda M, Mano H, Uenaka T, Yano S (2012) Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res 18(13):3592–3602
Kato K, Imamura F, Inoue M (2015) Analysis of ERBB ligand-induced resistance mechanism to crizotinib by primary culture of lung adenocarcinoma with EML4-ALK fusion gene. J Thorac Oncol 10(3):527–530
Cheng H, Terai M, Kageyama K, Ozaki S, McCue PA, Sato T, Aplin AE (2015) Paracrine effect of NRG1 and HGF drives resistance to MEK inhibitors in metastatic uveal melanoma. Cancer Res 75(13):2737–2748
Rimawi MF, Schiff R, Osborne CK (2015) Targeting HER2 for the treatment of breast cancer. Annu Rev Med 66:111–128
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344(11):783–792
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M (2002) Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 20(3):719–726
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Davidson NE et al (2005) Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 353(16):1673–1684
Hudis CA (2007) Trastuzumab-mechanism of action and use in clinical practice. N Engl J Med 357(1):39–51
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T et al (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355(26):2733–2743
Hurvitz SA, Hu Y, O’Brien N, Finn RS (2013) Current approaches and future directions in the treatment of HER2-positive breast cancer. Cancer Treat Rev 39(3):219–229
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, Hortobagyi GN, Hung MC, Yu D (2004) PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 6(2):117–127
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J (2008) Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 68(22):9221–9230
Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, Esteva FJ, Liu B (2010) Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-I receptor in breast cancer cells resistant to herceptin. Cancer Res 70(3):1204–1214
Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, Arteaga CL (2007) Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res 13(16):4909–4919
Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX (2009) Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 15(5):429–440
Ebbing EA, Medema JP, Damhofer H, Meijer SL, Krishnadath KK, van Berge Henegouwen MI, Bijlsma MF, van Laarhoven HW (2016) ADAM10-mediated release of heregulin confers resistance to trastuzumab by activating HER3. Oncotarget 7(9):10243–10254
Watson SS, Dane M, Chin K, Tatarova Z, Liu M, Liby T, Thompson W, Smith R, Nederlof M, Bucher E, Kilburn D, Whitman M, Sudar D, Mills GB, Heiser LM, Jonas O, Gray JW, Korkola JE (2018) Microenvironment-mediated mechanisms of resistance to HER2 inhibitors differ between HER2+ breast cancer subtypes. Cell Syst 6(3):329–342
Crist SB, Ghajar CM (2018) Friends with benefits: microenvironmental NRG1β and HGF mediate HER2-targeted resistance in L-HER2+ and HER2E breast cancer. Cell Syst 6(3):68–270
Hart CD, Migliaccio I, Malorni L, Guarducci C, Biganzoli L, Di Leo A (2015) Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat Rev Clin Oncol 12(9):541–552
Reinert T, de Paula B, Shafaee MN, Souza PH, Ellis MJ, Bines J (2018) Endocrine therapy for ER-positive/HER2-negative metastatic breast cancer. Chin Clin Oncol 7(3):25
Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R (2015) ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12(10):573–583
Osborne CK, Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62:233–247
Newby JC, Johnston SR, Smith IE, Dowsett M (1997) Expression of epidermal growth factor receptor and c-erbB2 during the development of tamoxifen resistance in human breast cancer. Clin Cancer Res 3(9):1643–1651
Tovey S, Dunne B, Witton CJ, Forsyth A, Cooke TG, Bartlett JM (2005) Can molecular markers predict when to implement treatment with aromatase inhibitors in invasive breast cancer? Clin Cancer Res 11(13):4835–4842
Qian BZ, Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141(1):39–51
Noy R, Pollard JW (2014) Tumor-associated macrophages: from mechanisms to therapy. Immunity 41(1):49–61
Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, Stanley ER, Segall JE, Condeelis JS (2005) Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res 65(12):5278–5283
Jobling P, Pundavela J, Oliveira SM, Roselli S, Walker MM, Hondermarck H (2015) Nerve-cancer cell cross-talk: a novel promoter of tumor progression. Cancer Res 75(9):1777–1781
Marchesi F, Piemonti L, Mantovani A, Allavena P (2010) Molecular mechanisms of perineural invasion, a forgotten pathway of dissemination and metastasis. Cytokine Growth Factor Rev 21(1):77–82
Liebig C, Ayala G, Wilks JA, Berger DH, Albo D (2009) Perineural invasion in cancer: a review of the literature. Cancer 115(15):3379–3391
Bapat AA, Hostetter G, Von Hoff DD, Han H (2011) Perineural invasion and associated pain in pancreatic cancer. Nat Rev Cancer 11(10):695–707
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS (2013) Autonomic nerve development contributes to prostate cancer progression. Science 341(6142):1236361
Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT et al (2014) Denervation suppresses gastric tumorigenesis. Sci Transl Med 6(250):250ra115
Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H et al (2017) Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 31(1):21–34
Boilly B, Faulkner S, Jobling P, Hondermarck H (2017) Nerve dependence: from regeneration to cancer. Cancer Cell 31(3):342–354
Mei L, Xiong WC (2008) Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat Rev Neurosci 9(6):437–452
Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK (2015) Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 15(9):563–572
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3(6):401–410
Baeriswyl V, Christofori G (2009) The angiogenic switch in carcinogenesis. Semin Cancer Biol 19(5):329–337
Ferrara N (2002) VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer 2(10):795–803
De Palma M, Biziato D, Petrova TV (2017) Microenvironmental regulation of tumour angiogenesis. Nat RevCancer 17(8):457–474
Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146(6):873–887
Odiete O, Hill MF, Sawyer DB (2012) Neuregulin in cardiovascular development and disease. Circ Res 111(10):1376–1385
Cote GM, Miller TA, Lebrasseur NK, Kuramochi Y, Sawyer DB (2005) Neuregulin-1alpha and beta isoform expression in cardiac microvascular endothelial cells and function in cardiac myocytes in vitro. Exp Cell Res 311(1):135–146
Nugroho DB, Ikeda K, Barinda AJ, Wardhana DA, Yagi K, Miyata K, Oike Y, Hirata KI, Emoto N (2018) Neuregulin-4 is an angiogenic factor that is critically involved in the maintenance of adipose tissue vasculature. Biochem Biophys Res Commun 503(1):378–384
Rak J, Yu JL, Klement G, Kerbel RS (2000) Oncogenes and angiogenesis: signaling three-dimensional tumor growth. J Invest Dermatol Symp Proc 5:24–33
Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, Kerbelet RS (1997) Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol 151(6):1523–1530
Yen L, You X, Al Moustafa A, Batist G, Hynes NE, Mader S et al (2000) Heregulin selectively upregulates vascular endothelial growth factor secretion in cancer cells and stimulates angiogenesis. Oncogene 19:3460–3469
Xiong S, Grijalva R, Zhang L, Nguyen NT, Pisters PW, Pollock RE, Yu D (2001) Up-regulation of vascular endothelial growth factor in breast cancer cells by the heregulin-beta1-activated p38 signaling pathway enhances endothelial cell migration. Cancer Res 61(4):1727–1732
Tsai P, Shiah S, Lin L, Wu C, Kuo M (2003) Up-regulation of vascular endothelial growth factor C in breast cancer cells by Heregulin-Beta 1: a critical role of p38/nuclear factor-kappa B signaling pathway. J Biol Chem 278(8):5750–5759
Yonezawa M, Wada K, Tatsuguchi A, Akamatsu T, Gudis K, Seo T et al (2009) Heregulin-induced VEGF expression via the ErbB3 signaling pathway in colon cancer. Digestion 80(4):215–225
Chen Z, Xu XH, Hu J (2016) Role of pericytes in angiogenesis: focus on cancer angiogenesis and anti-angiogenic therapy. Neoplasma 63(2):173–182
Teichert M, Milde L, Holm A, Stanicek L, Gengenbacher N, Savant S et al (2017) Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nat Commun 8:16106
Bjarnegård M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A et al (2004) Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development 131(8):1847–1857
Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T (2016) What is a pericyte? J Cereb Blood Flow Metab 36(2):451–455
Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O (2014) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307(1):C25–C38
LaGory E, Giaccia A (2016) The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol 18(4):356–365
Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL (2001) HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol 21(12):3995–4004
Li YM, Zhou BP, Deng J, Pan Y, Hay N, Hung MC (2005) A hypoxia-independent hypoxia-inducible factor-1 activation pathway induced by phosphatidylinositol-3 kinase/Akt in HER2 overexpressing cells. Cancer Res 65(8):3257–3263
Xu MJ, Johnson DE, Grandis JR (2017) EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev 36(3):463–473
Kümler I, Tuxen MK, Nielsen DL (2014) A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev 40(2):259–270
Tebbutt N, Pedersen MW, Johns TG (2013) Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer 13(9):663–673
Gala K, Chandarlapaty S (2014) Molecular pathways: HER3 targeted therapy. Clin Cancer Res 20(6):1410–1416
Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L et al (2009) Therapeutically targeting ErbB3: a key node in ligand-induced activation of the ErbB receptor-PI3K axis. Sci Signal 2(77):ra31
Garner AP, Bialucha CU, Sprague ER, Garrett JT, Sheng Q, Li S et al (2013) An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res 73(19):6024–6035
Garrett JT, Sutton CR, Kuba MG, Cook RS, Arteagam CL (2013) Dual blockade of HER2 in HER2-overexpressing tumor cells does not completely eliminate HER3 function. Clin Cancer Res 19(3):610–619
Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, Fulgham A, Song Y, Nielsen UB, Engelman JA, Wong KK (2010) An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res 70(6):2485–2494
Wang S, Huang J, Lyu H, Cai B, Yang X, Li F, Tan J, Edgerton SM, Thor AD, Lee CK, Liu B (2013) Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res 15(5):R101
Huang J, Wang S, Lyu H, Cai B, Yang X, Wang J, Liu B (2013) The anti-erbB3 antibody MM-121/SAR256212 in combination with trastuzumab exerts potent antitumor activity against trastuzumab-resistant breast cancer cells. Mol Cancer 12(1):134
Watanabe S, Yonesaka K, Tanizaki J, Nonagase Y, Takegawa N, Haratani K, Kawakami H, Hayashi H, Takeda M, Tsurutani J, Nakagawa K (2019) Targeting of the HER2/HER3 signaling axis overcomes ligand-mediated resistance to trastuzumab in HER2-positive breast cancer. Cancer Med 8(3):1258–1268
Zhu Y, Choi SH, Shah K (2015) Multifunctional receptor-targeting antibodies for cancer therapy. Lancet Oncol 16(15):e543–e554
Schaefer G, Haber L, Crocker LM, Shia S, Shao L, Dowbenko D et al (2011) A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer Cell 20(4):472–486
Huang S, Li C, Armstrong EA, Peet CR, Saker J, Amler LC, Sliwkowski MX, Harari PM (2013) Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res 73(2):824–833
Forster MD, Dillon MT, Kocsis J, Remenár É, Pajkos G, Rolland F, Greenberg J, Harrington KJ (2019) Patritumab or placebo, with cetuximab plus platinum therapy in recurrent or metastatic squamous cell carcinoma of the head and neck: a randomised phase II study. Eur J Cancer 123:36–47
Dillon MT, Grove L, Newbold KL, Shaw H, Brown NF, Mendell J, Chen S, Beckman RA, Jennings A, Ricamara M, Greenberg J, Forster M, Harrington KJ (2019) Patritumab with cetuximab plus platinum-containing therapy in recurrent or metastatic squamous cell carcinoma of the head and neck: an open-label, phase Ib study. Clin Cancer Res 25(2):487–495
Liu JF, Ray-Coquard I, Selle F, Poveda AM, Cibula D, Hirte H et al (2016) Randomized phase II trial of seribantumab in combination with paclitaxel in patients with advanced platinum-resistant or -refractory ovarian cancer. J Clin Oncol 34(36):4345–4353
Abramson VG, Supko JG, Ballinger T, Cleary JM, Hilton JF, Tolaney SM et al (2017) Phase Ib study of safety and pharmacokinetics of the PI3K inhibitor SAR245408 with the HER3-neutralizing human antibody SAR256212 in patients with solid tumors. Clin Cancer Res 23(14):3520–3528
Schram AM, Iasonos A, Hyman DM (2016) Picking the right patient for human epidermal growth factor receptor 3-targeted therapy in platinum-resistant ovarian cancer. J Clin Oncol 34(36):4312–4314
Ma J, Lyu H, Huang J, Liu B (2014) Targeting of erbB3 receptor to overcome resistance in cancer treatment. Mol Cancer 13:105
Wu Y, Zhang Y, Wang M, Li Q, Qu Z, Shi V, Kraft P, Kim S, Gao Y, Pak J, Youngster S, Horak ID, Greenberger LM (2013) Downregulation of HER3 by a novel antisense oligonucleotide, EZN-3920, improves the antitumor activity of EGFR and HER2 tyrosine kinase inhibitors in animal models. Mol Cancer Ther 12(4):427–437
Jones PA, Issa JP, Baylin S (2016) Targeting the cancer epigenome for therapy. Nat Rev Genet 17(10):630–641
Morel D, Almouzni G, Soria JC, Postel-Vinay S (2017) Targeting chromatin defects in selected solid tumors based on oncogene addiction, synthetic lethality and epigenetic antagonism. Ann Oncol 28(2):254–269
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C (2019) Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics 14(12):1164–1176
Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6(1):38–51
Huang X, Gao L, Wang S, Lee CK, Ordentlich P, Liu B (2009) HDAC inhibitor SNDX-275 induces apoptosis in erbB2-overexpressing breast cancer cells via down-regulation of erbB3 expression. Cancer Res 69(21):8403–8411
Huang X, Wang S, Lee CK, Yang X, Liu B (2011) HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett 307(1):72–79
Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J, Liu B (2013) Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis 4(3):e556
Lin T, Ren Q, Zuo W, Jia R, Xie L, Lin R, Zhao H, Chen J, Lei Y, Wang P, Dong H, Huang L, Cai J, Peng Y, Yu Z, Tan J, Wang S (2019) Valproic acid exhibits anti-tumor activity selectively against EGFR/ErbB2/ ErbB3-coexpressing pancreatic cancer via induction of ErbB family members-targeting microRNAs. J Exp Clin Cancer Res 38(1):150
Wahdan-Alaswad R, Liu B (2013) “Sister” miRNAs in cancers. Cell Cycle 12(24):3703–3704
Tsai MS, Shamon-Taylor LA, Mehmi I, Tang CK, Lupu R (2003) Blockage of heregulin expression inhibits tumorigenicity and metastasis of breast cancer. Oncogene 22(5):761–768
Hegde GV, de la Cruz CC, Chiu C, Alag N, Schaefer G, Crocker L, Ross S, Goldenberg D, Merchant M, Tien J, Shao L, Roth L, Tsai SP, Stawicki S, Jin Z, Wyatt SK, Carano RA, Zheng Y, Sweet-Cordero EA, Wu Y, Jackson EL (2013) Blocking NRG1 and other ligand-mediated Her4 signaling enhances the magnitude and duration of the chemotherapeutic response of non-small cell lung cancer. Sci Transl Med 5(171):171ra18
Ogier C, Colombo PE, Bousquet C, Canterel-Thouennon L, Sicard P, Garambois V, Thomas G, Gaborit N, Jarlier M, Pirot N, Pugnière M, Vie N, Gongora C, Martineau P, Robert B, Pèlegrin A, Chardès T, Larbouret C (2018) Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett 432:227–236
Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16(3):203–222
Fernandez-Cuesta L, Thomas RK (2015) Molecular pathways: targeting NRG1 fusions in lung cancer. Clin Cancer Res 21(9):1989–1994
Wilson FH, Politi K (2018) ERBB signaling interrupted: targeting ligand-induced pathway activation. Cancer Discov 8(6):676–678
Drilon A, Somwar R, Mangatt BP, Edgren H, Desmeules P, Ruusulehto A et al (2018) Response to ERBB3-directed targeted therapy in NRG1-rearranged cancers. Cancer Discov 8(6):686–695
Shin DH, Jo JY, Han JY (2018) Dual targeting of ERBB2/ERBB3 for the treatment of SLC3A2-NRG1-mediated lung cancer. Mol Cancer Ther 17(9):2024–2033
Schwabe RF, Jobin C (2013) The microbiome and cancer. Nat Rev Cancer 13(11):800–812
Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D et al (2017) Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357(6356):1156–1160
Raza MH, Gul K, Arshad A, Riaz N, Waheed U, Rauf A, Aldakheel F, Alduraywish S, Rehman MU, Abdullah M, Arshad M (2019) Microbiota in cancer development and treatment. J Cancer Res Clin Oncol 145(1):49–63
Tsay JJ, Wu BG, Badri MH, Clemente JC, Shen N, Meyn P et al (2018) Airway microbiota is associated with upregulation of the PI3K pathway in lung cancer. Am J Respir Crit Care Med 198(9):1188–1198
Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A (2017) Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168(4):707–723
O’Donnell JS, Teng MWL, Smyth MJ (2019) Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol 16(3):151–167
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13(1):84–88
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT et al (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6(2):202–216
Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM (2015) Exosome mediated communication within the tumor microenvironment. J Control Release 219:278–294
Xie X, Nie H, Zhou Y, Lian S, Mei H, Lu Y, Dong H, Li F, Li T, Li B, Wang J, Lin M, Wang C, Shao J, Gao Y, Chen J, Xie F, Jia L (2019) Eliminating blood oncogenic exosomes into the small intestine with aptamer-functionalized nanoparticles. Nat Commun 10(1):5476
Acknowledgments
We thank Dr. Bolin Liu at Department of Genetics, Stanley S. Scott Cancer Center, Louisiana State University (LSU) Health Sciences Center, for his critical comments on the manuscript. Work of Shuiliang Wang was supported in part by the National Natural Science Foundation of China No. 81772848 and Joint Funds for the Innovation of Science and Technology from Fujian Province No.2017Y9127.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jia, R., Zhao, H., Wang, S. (2021). Neuregulin Signaling in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1270. Springer, Cham. https://doi.org/10.1007/978-3-030-47189-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-47189-7_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-47188-0
Online ISBN: 978-3-030-47189-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)