
Abstract
It is becoming increasingly evident that stromal cells such as macrophages, mast cells, adipocytes and mesenchymal cells associated with tumors significantly contribute to tumorigenesis. Some types of cancer indeed profoundly rely on extrinsic signals afforded by infiltrating or neighbouring cells for survival, proliferation and dissemination. Tissue disruption that results from tumor growth further activates tissue repair and inflammatory reactions that significantly shape the nature of the developing tumors. Over the past recent years, several studies have revealed that mesenchymal stem cells (MSCs) are recruited to tumors and play a particularly important role in the regulation of both solid and haematological malignancies. The tumor-homing properties of MSCs have further led to studies investigating their therapeutic use as targeted delivery vehicles of gene products. I hereafter discuss the role of MSCs in cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) are non-hematopoietic precursor cells, mainly found in the bone marrow, which contribute to the maintenance and regeneration of a variety of connective tissues, such as bone, cartilage, adipose and muscle. MSCs are identified based on their ability to differentiate into mesenchymal lineage cells and to express specific membrane-bound surface antigens [1]. Purification of SSEA-4 expressing bone marrow cells can significantly enrich for MSCs cultures devoid of hematopoietic cells [2]. SSEA-4 is an antigen of globo-series glycolipids previously thought to be restricted to embryonic stem cells and some neural ganglion cells. Human and mouse MSCs express CD105 (endoglin), CD73 (ecto-5′-nucleotidase) and CD44 (hyaluronate receptor). MSCs are also routinely tested for the absence of CD45 and CD31 expression, which are markers for hematopoietic and endothelial cells, respectively. MSCs are also characterized by their low levels of major histocompatibility complex (MHC) class I molecules expression. Following IFN-γ treatment, MSCs upregulate MHC class I and II molecules, process and present antigenic peptides and can induce IL-2 production of antigen-specific T cells [3, 4]. A unique feature of MSCs is their ability to suppress immune responses, although the exact mechanisms by which MSCs mediate their immunosuppression are not fully understood [1]. An important question that remains unanswered is whether endogenous MSCs behave similarly as ex vivo expanded MSCs. The development of more efficient in vivo tracking methods will be key in defining the physiological functions of MSCs in health and disease.
MSC in Tissue Repair
Several studies have shown that MSCs possess the intrinsic ability to home to injured tissues and actively participate in tissue repair. TGF-β family members (TGF-β1/2/3, activins and bone morphogenetic proteins) and Wnt signaling play important roles in MSCs-mediated tissue repair [5, 6]. Wnt activation is essential, for instance, for the differentiation of MSCs into osteoblasts. Accordingly, the secreted soluble factor Dickkopf-1 (Dkk-1), which inhibits Wnt signalling, also inhibits MSC-mediated osteogenesis [7]. It has been reported that MSCs can repair injured tissue through differentiation, cell fusion or by secreting cytokines and growth factors [8]. In the context of myocardium regeneration for instance, it has been proposed that trophic factors secreted by MSCs are mainly responsible for their therapeutic effects after cardiac infarction [9]. The release of trophic factors by MSCs has also been suggested to be beneficial in experimental models of stroke [10].
MSCs in Solid Tumors
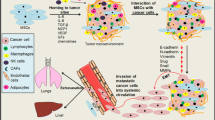
The microenvironment of a solid tumor closely resembles the environment of an injured tissue. As such, tumor growth is often associated with a variety of stromal cells in a manner that closely ressembles wound healing and tissue repair sites. Several studies have now demonstrated that similar to injury sites, developing tumors recruit MSCs through the release of endocrine and paracrine signals. Although molecules such as CXCR4, CXCR12 and CCL2 have been implicated in the tissue-homing ability of MSCs [11], the exact mechanism governing MSC migration—in response to injury or oncogenesis—is still not fully characterized. One of the difficulties in studying the migratory properties of MSCs may come from the fact that ex vivo cultured MSCs often loose expression of chemokine receptors and responsiveness to chemokines. Indeed, early passage MSCs, but not long-term cultured MSCs, have been described to express a broad range of chemokine receptors, including CCR1, CCR7, CCR9, CX3CL1, CXCR4, CXCR5 and CXCR6, and to migrate in response to CXCL12, CXCL13, CXCL16, CCL19, CCL5 and CCL25 [12].
Several independent studies have now shown that MSCs are recruited to tumors of breast carcinomas, colon carcinomas, ovarian carcinomas, gliomas, melanomas and Kaposi’s sarcomas [13–18]. In the context of Kaposi’s sarcomas [18], MSCs appear to play an anti-tumoral role via inhibition of the PI-3K/AKT pathway in an E-cadherin-dependent manner. The anti-tumoral effects of endogenous MSCs are still not clear and warrant further investigation. Recently, Qiao L. et al. [19] demonstrated that human MSCs could inhibit the proliferation and colony-forming ability of human cancer cell lines, possibly through the downregulation of anti-apoptotic Bcl-2. In human breast cancer, human MSCs have been shown to migrate up to 11-fold more towards conditioned media from cancer cells than from non cancer cells [20]. Building upon these observations, Karnoub et al. [20] recently demonstrated that human MSCs are particularly important to enhance the metastatic ability of human tumors xenografts. Out of 4 human breast cancer cell lines tested, namely MCF7/Ras, MDA-MB-231, MDA-MB-435 and HMLER cells, all showed an increased metastatic potential to the lungs when co-injected with ex vivo expanded human MSCs. However, only one cell line, MCF7/Ras, showed accelerated primary tumor growth when co-injected with MSCs. Importantly, the co-injection of tumor cells with other types of mesenchymal cells such as BJ fibroblasts did not enhanced tumor growth or metastasis. In a screen for cytokines, chemokines and growth factors differentially expressed in co-cultures of human breast cancer cells and MSCs, CCL5 (RANTES) was revealed to be upregulated up to 60-fold in co-cultures compared to monocultures. Using short hairpin RNA technology, it was demonstrated that MSCs were the source of CCL5 and that physical interactions between the two cell types were required, i.e. MSCs needed to be “educated” by the cancer cells by a mechanism yet to be identify. CCL5 can induce the recruitment of tumor-associated macrophages and endothelial cells. However, the enhanced ability of breast cancer to metastasize in response to CCL5 was rather shown to be due to an enhanced ability of the tumor cells to colonize foreign microenvironments, possibly through enhanced extravasation, although direct evidence of this remains to be shown. Taken together, these studies revealed that solid tumors of various cell origins can recruit ex vivo expanded MSCs and, in the context of breast cancer at least, tumor recruitment of MSCs facilitates metastatic spread through the release of CCL5 by MSCs. However, a better understanding of the biological pathways governing the functions of endogenous MSCs (as opposed to ex vivo expanded MSCs) is crucial if new therapeutic strategies targeting tumor-stroma interactions are to be developed.
Mesenchymal fibroblasts within solid tumors are often referred to as activated or carcinoma-associated fibroblast (CAFs). Although, in most cases, the tissue origin of CAFs is not well defined, the fact that CAFs share common surface antigens and exhibit common functions with MSCs suggest they may origin from bone marrow MSCs [21]. In a model of human prostate cancer, it was recently shown that production of TGF-β by CAFs increased CXCR4 expression on initiated non-tumorigenic prostate epithelial cells. Importantly, suppression of CXCR4 expression abrogated the subsequent tumorigenic potential of prostate epithelial cells [22]. Hence, induced expression of CXCR4 via TGF-β modified the response of prostate epithelial, inducing epithelial cells to respond to stromal-derived factor (SDF)-1—the ligand of CXCR4—produced by MSCs. CAFs have also been described to play an important role in the development of mammary carcinomas. Orimo A. et al. [23] demonstrated that CAFs extracted from human breast tumors promoted the growth of tumor cells through the release of SDF-1, which was also in part responsible for the recruitment of endothelial progenitor cells and angiogenesis.
The tumor-homing properties of MSCs have led to studies investigating their potential as targeted delivery vehicles for therapeutic genes. Michael Andreeff and is team [14, 15] showed that intravenously injected human MSCs gene-modified to secrete IFN-β preferentially engrafted into xenografted tumors, thereby significantly delaying tumor growth. We have shown that MSCs could also be used in a syngeneic setting to deliver immunostimulatory gene products such as interleukin-2 and induce anti-cancer immune responses [24]. The selective engraftment of MSCs to tumors may thus constitute a new therapeutic avenue to target anti-tumor gene products to the tumor microenvironment.
MSCs in Haematological Malignancies
MSCs have also been implicated in the pathology of several forms of haematological malignancies. Myeloma bone disease, which is known to be associated with an increased production of bone-resorbing osteoclast-activating factors, is also associated with an increase production of Dkk-1 that inhibits MSCs differentiation [25]. Dkk-1 also predisposes undifferentiated MSCs to enter cell division, as addition of synthetic recombinant Dkk-1 peptides has been shown to stimulate proliferation of undifferentiated MSCs [7]. In patients with multiple myeloma, inhibition of MSCs differentiation by Dkk-1 produced by myeloma cells is associated with the development of osteolytic lesions. MSCs cocultured with human myeloma cells were shown to produce high levels of IL-6, stimulating the proliferation of Dkk1-secreting myeloma cells, which in turn induced the proliferation of MSCs and inhibited their differentiation into osteogenic cells. Notably, blocking Dkk-1 with a monoclonal antibody can significantly reduce the development of osteolytic lesions and significantly inhibit the growth multiple myeloma in mice [25]. Anti-Dkk-1 antibody therapy may thus constitute a novel therapeutic approach in the treatment of multiple myeloma and associated bone disease.
Recently, Mukherjee et al. [26] reported that the proteasome inhibitor bortezomib, a first-in-class agent for patients with relapsed and refractory myeloma, can induce differentiation of MSCs into osteoblasts thereby preventing bone disease. The studies demonstrated that low doses of bortezomib increased osteoblastogenesis, bone formation and mineralized trabecular bone in mice. The authors suggested that bortezomib prevented proteasomal degradation of the runt-related transcription factor 2 (Runx-2), which is a key transcription factor required for osteoblast differentiation. Indeed, Runx-2 together with the transcriptional modulator TAZ drives MSCs to differentiate into osteoblasts [27]. Of note, in addition to its effect on MSCs, Bortezomib has also been shown to inhibit ostoeclast formation through blockade of NF-κB activity [28]. Thus, recent studies suggested that pharmacologic manipulation of MSCs by administration of anti-Dkk1 antibodies or by administration of the proteasome inhibitor bortezomib could be a useful strategy for conditions associated with loss of bone function.
Through the production of SDF-1, bone marrow MSCs have been suggested to mediated chemotaxis of CD34+ acute myelogenouse leukemia cells and to play an important role in the homing of these cells to the bone marrow microenvironment [29]. The preferential homing of potential cancer stem cells in the bone marrow via production of SDF-1 has been proposed as a mechanism of chemoresistance in different haematological malignancies, including myeloma, acute myelogenous leukemia, acute lymphoblastic leukemia (ALL) and chronic lymphocytic leukemia [30–32]. In the context of chronic myelogenous leukemia (CML), the role of tumor-stroma interactions is generally considered as low since the chimeric fusion protein Bcr-Abl (resulting from the translocation of c-Abl gene on chromosome 9 to the breakpoint region (Bcr) on chromosome 22) can inhibit SDF-1-induced migration of CML cells [33]. Recently, Jin et al. [34] hypothesized that imatinib, a tyrosine kinase inhibitor developed to specifically inhibit Bcr-Abl, may restore SDF-1-induced migration of CML cells and cause resistant CML cells to localize to bone marrow microenvironment. They observed that imatinib treatment significantly increased CXCR4 expression (the receptor for SDF-1) on CML cells and increased their migratory ability towards bone marrow MSCs. Importantly, they demonstrated that CML cells co-cultured with MSCs were significantly more resistant to imatinib, and that addition of the CXCR4 antagonist AMD3465 restored the sensitivity of CML cells to imatinib when co-cultured with MSCs. Thus, therapeutic strategies aimed at blocking the protective effects of bone marrow MSCs on CML cells via the SDF-1/CXCR4 axis could benefit relapsing CML patients.
Recent studies by Iwamoto et al. [35] revealed that ALL resistance to asparaginase therapy may rely on the interaction between ALL cells and bone marrow MSCs. Asparagine depletion in an essential component of ALL treatment because the capacity of ALL cells to produce their own asparagine is extremely low due to their low expression levels of asparaginase synthetase (ASNS) [36]. Since bone marrow MSCs express high levels of ASNS and that ALL cells grow in close association with MSCs, it was hypothesized that MSCs might confer resistance to asparaginase therapy. When ALL cells were co-cultured with human MSCs, they were indeed significantly protected against asparaginase cytotoxicity. Notably, the authors observed patient-dependent variability in the levels of ASNS in MSCs and subsequent resistance to asparaginase. Theses studies suggest that resistance to asparaginase may be mediated by ASNS expression by MSCs rather than endogenous ASNS expression in ALL cells. Thus, the development of ASNS inhibitors to complement asparaginase therapy should target both ALL cells and MSCs.
MSCs in Graft-versus-host Disease
One of the most fascinating properties of MSCs is their ability to suppress immune responses, both in vitro and in vivo. Soluble factors secreted by MSCs play a major role in their immunosuppressive effects, including hepatocyte growth factor, prostaglandin E2, TGF-β1, indoleamine 2,3-dioxygenase, nitric oxide and IL-10. Some studies reported that contact-dependent mechanisms, such as B7H1 interactions, might also be implicated in MSC-mediated immune suppression. Although not fully characterized, the immunosuppressive properties of MSCs affect the function of a broad range of immune cells, including T cells, B cells, NK cells and antigen-presenting cells [1]. Co-transplantation of MSCs has been proposed for the treatment of graft-versus-host disease (GVHD) and other autoimmune diseases. In a landmark case study of severe grade IV GVHD, LeBlanc and colleagues [37] reported that administration of haploidentical human MSCs following allogeneic stem cell transplantation completely reversed the GVHD. One year following treatment, the patient was still free of GVHD and had no minimal residual disease of his leukemia in blood and bone marrow. Recently Ning et al. [38] reported the results of the first randomized study on the use of MSCs for the prevention of GvHD. The study is significant in that it revealed that although MSCs co-transplantation was efficient at preventing GvHD (one out of ten versus eight of ten patients), it was also associated with a greatly higher incidence of leukemia relapse (60% versus 20%). The non-specific immune suppression induced by MSCs may have accounted form simultaneous inhibition of graft-versus-leukemia effect. However, patients injected with MSCs did not display any differences in infectious events, which would have been expected from systemic immune suppression. It is therefore possible that other mechanisms are responsible for the higher incidence of leukemia relapse in the MSC-treated group. A possible explanation could be that MSCs directly enhanced survival of residual malignant cells through the release of exogenous factors as described above.
MSCs as a Source of Cancer Stem Cells
In addition to their ability to incorporate tumors’ stromal environment and to influence tumor growth, MSCs may also constitute an important source of cancer stem cells. Human MSCs have indeed been shown to spontaneously transform following ex vivo culture [39]. In tissue cultures, human and mouse MSCs are susceptible to spontaneous mutations, including loss of p53 function. Recently, Li et al. [40] examined age-related fibrosarcomas developing in control-treated or MSC-injected mice and observed that MSCs spontaneously form fibrosarcomas in mice as a result of aging, possibly involving acquisition of p53 mutation. Interestingly, immunohistopathological analyses of MSC-derived tumors identified them as poorly differentiated carcinomas, suggesting a mesenchymal-epithelial transition [41]. This was corroborated by microarray and protein expression analysis that showed that almost all mesenchymal genes were severely repressed in MSC-derived tumors. In support of a role for endogenous MSCs in the development of spontaneous oncogenesis, Houghton et al. [42] demonstrated that chronic infection with Helicobacter pylori induced the recruitment of bone marrow-derived cells (BMDCs) that developed into intraepithelial cancer. This study revealed that epithelial cancers can arise from BMDCs and suggested that MSCs were likely involved in this process. Recently, Bernardo et al. [43] investigated the susceptibility to transformation of human bone marrow-derived MSCs at different in vitro time points. Using genomic hybridization, karyotyping, subtelomeric fluorescent in situ hybridization and telomerase activity, the authors failed to observe malignant transformation in any of the ten human MSCs preparations tested. This suggests that human MSCs isolated from the bone marrow are not susceptible to cellular transformation when cultured for less than 25 passages.
Conclusion
As I have discussed in this review, MSCs constitute a rare population of adult stem cells mainly found in the bone marrow that play an important role in a number of malignant diseases. Recently, Karnoub et al. [20] demonstrated that human MSCs were particularly potent at enhancing the metastatic ability of human breast tumor xenografts. Future studies focusing on resident MSCs will be of great importance to validate observations made with ex vivo expanded MSCs, which have been the sole basis of our knowledge of MSCs biology. Notwithstanding this, evidence strongly suggests MSC-tumor cells interactions are crucial in the development of solid and haematological malignancies. MSCs may further constitute a source of cancer stem cells, as suggested by the observation that bone marrow-derived cells are directly responsible for the development of epithelial gastric cancers triggered by chronic Helicobacter infections [42]. It will be of great interest to further investigate the role of resident MSCs as a source of cancer stem cells in other types of malignancies. Defining the role and function of resident MSCs will thus be of great importance in our understanding of the biology and function of adult stem cells and in the development of therapeutic strategies aimed at exploiting or targeting adult MSCs.
References
Stagg, J. (2007). Immune regulation by mesenchymal stem cells: Two sides to the coin. Tissue Antigens, 69(1), 1–9.
Gang, E. J., Bosnakovski, D., Figueiredo, C. A., Visser, J. W., & Perlingeiro, R. C. (2007). SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood, 109(4), 1743–1751.
Stagg, J., Pommey, S., Eliopoulos, N., & Galipeau, J. (2006). Interferon-gamma-stimulated marrow stromal cells: A new type of nonhematopoietic antigen-presenting cell. Blood, 107(6), 2570–2577.
Krampera, M., Cosmi, L., Angeli, R., Pasini, A., Liotta, F., Andreini, A., et al. (2006). Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells, 24(2), 386–398.
Massague, J., Blain, S. W., & Lo, R. S. (2000). TGFbeta signaling in growth control, cancer, and heritable disorders. Cell, 103(2), 295–309.
Mishra, L., Derynck, R., & Mishra, B. (2005). Transforming growth factor-beta signaling in stem cells and cancer. Science, 310(5745), 68–71.
Gregory, C. A., Perry, A. S., Reyes, E., Conley, A., Gunn, W. G., & Prockop, D. J. (2005). Dkk-1-derived synthetic peptides and lithium chloride for the control and recovery of adult stem cells from bone marrow. Journal of Biological Chemistry, 280(3), 2309–2323.
Spees, J. L., Olson, S. D., Ylostalo, J., Lynch, P. J., Smith, J., Perry, A., et al. (2003). Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proceedings of the National Academy of Sciences of the United States of America, 100(5), 2397–2402.
Gnecchi, M., He, H., Liang, O. D., Melo, L. G., Morello, F., Mu, H., et al. (2005). Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nature Medicine, 11(4), 367–368.
Li, Y., Chen, J., Chen, X. G., Wang, L., Gautam, S. C., Xu, Y. X., et al. (2002). Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology, 59(4), 514–523.
Dwyer, R. M., Potter-Beirne, S. M., Harrington, K. A., Lowery, A. J., Hennessy, E., Murphy, J. M., et al. (2007). Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clinical Cancer Research, 13(17), 5020–5027.
Honczarenko, M., Le, Y., Swierkowski, M., Ghiran, I., Glodek, A. M., & Silberstein, L. E. (2006). Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells, 24(4), 1030–1041.
Nakamura, K., Ito, Y., Kawano, Y., Kurozumi, K., Kobune, M., Tsuda, H., et al. (2004). Antitumor effect of genetically engineered mesenchymal stem cells in a rat glioma model. Gene Therapy, 11(14), 1155–1164.
Nakamizo, A., Marini, F., Amano, T., Khan, A., Studeny, M., Gumin, J., et al. (2005). Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Research, 65(8), 3307–3318 Apr 15.
Studeny, M., Marini, F. C., Dembinski, J. L., Zompetta, C., Cabreira-Hansen, M., Bekele, B. N., et al. (2004). Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. Journal of the National Cancer Institute, 96(21), 1593–1603.
Hung, S. C., Deng, W. P., Yang, W. K., Liu, R. S., Lee, C. C., Su, T. C., et al. (2005). Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clinical Cancer Research, 11(21), 7749–7756.
Komarova, S., Kawakami, Y., Stoff-Khalili, M. A., Curiel, D. T., & Pereboeva, L. (2006). Mesenchymal progenitor cells as cellular vehicles for delivery of oncolytic adenoviruses. Molecular Cancer Therapeutic, 5(3), 755–766.
Khakoo, A. Y., Pati, S., Anderson, S. A., Reid, W., Elshal, M. F., Rovira, I. I., et al. (2006). Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. Journal of Experimental Medicine, 203(5), 1235–1247.
Qiao, L., Xu, Z., Zhao, T., Zhao, Z., Shi, M., Zhao, R. C., et al. (2008). Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Research, 18(4), 500–507.
Karnoub, A. E., Dash, A. B., Vo, A. P., Sullivan, A., Brooks, M. W., Bell, G. W., et al. (2007). Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature, 449(7162), 557–563.
Haniffa, M. A., Wang, X. N., Holtick, U., Rae, M., Isaacs, J. D., Dickinson, A. M., et al. (2007). Adult human fibroblasts are potent immunoregulatory cells and functionally equivalent to mesenchymal stem cells. Journal of Immunology, 179(3), 1595–1604.
Ao, M., Franco, O. E., Park, D., Raman, D., Williams, K., & Hayward, S. W. (2007). Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer Research, 67(9), 4244–4253.
Orimo, A., Gupta, P. B., Sgroi, D. C., Arenzana-Seisdedos, F., Delaunay, T., Naeem, R., et al. (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell, 121(3), 335–348.
Stagg, J., Lejeune, L., Paquin, A., & Galipeau, J. (2004). Marrow stromal cells for interleukin-2 delivery in cancer immunotherapy. Human Gene Therapy, 15(6), 597–608.
Gunn, W. G., Conley, A., Deininger, L., Olson, S. D., Prockop, D. J., & Gregory, C. A. (2006). A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: A potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells, 24(4), 986–991.
Mukherjee, S., Raje, N., Schoonmaker, J. A., Liu, J. C., Hideshima, T., Wein, M. N., et al. (2008). Pharmacologic targeting of a stem/progenitor population in vivo is associated with enhanced bone regeneration in mice. Journal of Clinical Investigation, 118(2), 491–504.
Hong, J. H., Hwang, E. S., McManus, M. T., Amsterdam, A., Tian, Y., Kalmukova, R., et al. (2005). TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science, 309(5737), 1074–1078.
von Metzler, I., Krebbel, H., Hecht, M., Manz, R. A., Fleissner, C., Mieth, M., et al. (2007). Bortezomib inhibits human osteoclastogenesis. Leukemia, 21(9), 2025–2034.
Kim, C. H., & Broxmeyer, H. E. (1998). In vitro behavior of hematopoietic progenitor cells under the influence of chemoattractants: Stromal cell-derived factor-1, steel factor, and the bone marrow environment. Blood, 91(1), 100–110.
Hideshima, T., Bergsagel, P. L., Kuehl, W. M., & Anderson, K. C. (2004). Advances in biology of multiple myeloma: Clinical applications. Blood, 104(3), 607–618.
Garrido, S. M., Appelbaum, F. R., Willman, C. L., & Banker, D. E. (2001). Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Experimental Hematology, 29(4), 448–457.
Burger, J. A., & Kipps, T. J. (2006). CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood, 107(5), 1761–1767.
Geay, J. F., Buet, D., Zhang, Y., Foudi, A., Jarrier, P., Berthebaud, M., et al. (2005). p210BCR-ABL inhibits SDF-1 chemotactic response via alteration of CXCR4 signaling and down-regulation of CXCR4 expression. Cancer Research, 65(7), 2676–2683.
Jin, L., Tabe, Y., Konoplev, S., Xu, Y., Leysath, C. E., Lu, H., et al. (2008). CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Molecular Cancer Therapeutics, 7(1), 48–58.
Iwamoto, S., Mihara, K., Downing, J. R., Pui, C. H., & Campana, D. (2007). Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. Journal of Clinical Investigation, 117(4), 1049–1057.
Oettgen, H. F., Old, L. J., Boyse, E. A., Campbell, H. A., Philips, F. S., Clarkson, B. D., et al. (1967). Inhibition of leukemias in man by L-asparaginase. Cancer Research, 27(12), 2619–2631.
Le Blanc, K., Rasmusson, I., Sundberg, B., Gotherstrom, C., Hassan, M., Uzunel, M., et al. (2004). Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet, 363(9419), 1439–1441.
Ning, H., Yang, F., Jiang, M., Hu, L., Feng, K., Zhang, J., et al. (2008). The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: Outcome of a pilot clinical study. Leukemia, 22(3), 593–599.
Rubio, D., Garcia-Castro, J., Martin, M. C., de la Fuente, R., Cigudosa, J. C., Lloyd, A. C., et al. (2005). Spontaneous human adult stem cell transformation. Cancer Research, 65(8), 3035–3039.
Li, H., Fan, X., Kovi, R. C., Jo, Y., Moquin, B., Konz, R., et al. (2007). Spontaneous expression of embryonic factors and p53 point mutations in aged mesenchymal stem cells: A model of age-related tumorigenesis in mice. Cancer Research, 67(22), 10889–10898.
Rubio, D., Garcia, S., De la Cueva, T., Paz, M. F., Lloyd, A. C., Bernad, A., et al. (2008). Human mesenchymal stem cell transformation is associated with a mesenchymal-epithelial transition. Experimental Cell Research, 314(4), 691–698.
Houghton, J., Stoicov, C., Nomura, S., Rogers, A. B., Carlson, J., Li, H., et al. (2004). Gastric cancer originating from bone marrow-derived cells. Science, 306(5701), 1568–1571.
Bernardo, M. E., Zaffaroni, N., Novara, F., Cometa, A. M., Avanzini, M. A., Moretta, A., et al. (2007). Human bone marrow derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Research, 67(19), 9142–9149 Oct 1.
Acknowledgments
J.S. is supported by a post-doctoral fellowship from the Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stagg, J. Mesenchymal Stem Cells in Cancer. Stem Cell Rev 4, 119–124 (2008). https://doi.org/10.1007/s12015-008-9030-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-008-9030-4