
Abstract
Bacteria are equipped with sophisticated metabolic control systems to adapt to variable nutrient conditions. While metabolic pathways are controlled at enzyme activity levels and at mRNA levels, scarcity in preferred nutrients causes transcriptional induction of genes for alternative nutrient source utilization. Transcriptional regulatory systems controlling metabolisms of carbon, nitrogen, phosphate, and sulfur in Corynebacterium glutamicum have been extensively studied for the last two decades. The knowledge of the regulators, including regulon members, operator sequences, and effectors, has deepened our understanding of the C. glutamicum physiology and has led to develop synthetic biology tools for metabolic engineering, maximizing the C. glutamicum potential as a production host. In this chapter, we review the studies of the transcriptional regulators, especially focus on those with global regulatory roles in the primary metabolism in C. glutamicum.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Transcriptional regulation
- Carbon metabolism
- Nitrogen metabolism
- Phosphorus metabolism
- Sulfur metabolism
1 Introduction
In bacterial genomes, uptake and metabolic systems for versatile nutrient sources, including carbon, nitrogen, phosphorus, and sulfur sources, are encoded to adapt to fluctuating conditions. Elaborate mechanisms for sensing the environmental and cellular nutrient status and those for switching gene expression in response to the status are also encoded to express the corresponding genes at the appropriate degree and timing and to avoid the expression of unnecessary genes. When multiple resources exist, selective and successive utilization can be achieved by global transcriptional regulatory mechanisms. Hierarchical transcriptional regulation mediated by global regulators and pathway-specific local regulators constitute a transcriptional regulatory network. Genome-wide analysis, including transcriptome using microarray and high-throughput sequencer, has revealed the network structure in some organisms. Since the genomic sequence of Corynebacterium glutamicum was reported in 2003 (Ikeda and Nakagawa 2003; Kalinowski et al. 2003), genes for the primary metabolic pathways and transcriptional regulators involved in their regulation have been intensively studied [reviewed in (Schröder and Tauch 2010; Nešvera and Pátek 2011; Pátek and Nešvera 2011; Bott and Brocker 2012; Neshat et al. 2014)]. These studies revealed that the transcriptional regulatory systems different from the paradigms established in model microorganisms, like Escherichia coli and Bacillus subtilis, are working in the suborder Corynebacterineae in the class Actinobacteria. For example, coordinated simultaneous carbon metabolism not subjected to the catabolite control, adenylylation-mediated protein interaction of a transcriptional regulator is involved in nitrogen control and coordinated transcriptional regulation of methionine and cysteine synthesis pathways. In this chapter, we review the current understanding of transcriptional regulators involved in carbon, nitrogen, phosphorus, and sulfur metabolism in C. glutamicum.
2 Carbon Catabolism Control
C. glutamicum is capable of utilizing a variety of carbon sources, including sugars, sugar alcohols, organic acids, alcohols, amino acids, and aromatic compounds. Studies identifying genes involved in the metabolism of these compounds were followed by those identifying transcriptional regulators controlling the genes. All of these carbon compounds are finally catabolized through the central carbon metabolic pathways consisting of glycolysis, the pentose phosphate pathway, the TCA cycle, and the glyoxylate shunt. The genes constituting these pathways are coordinately regulated by multiple transcriptional regulators.
2.1 Sugar Uptake
2.1.1 Phosphotransferase System-Dependent Sugar Uptake
The phosphoenolpyruvate (PEP): carbohydrate phosphotransferase system (PTS) is the major sugar uptake system in most bacteria (Postma et al. 1993). It consists of three components: two common cytoplasmic proteins, so-called general components, enzyme I (EI) and HPr, and sugar-specific enzyme IIs (EIIs). Phosphoryl group of PEP is transferred to EII via EI and HPr, then EIIs couple sugar transport with phosphorylation. C. glutamicum possesses EIIs specific to glucose, fructose, and sucrose, which are encoded by the ptsG, ptsF, and ptsS genes (cg1537, cg2120, and cg2925 in the type strain ATCC 13032, cgR_1425, cgR_1766, and cgR_2547 in strain R; hereafter the locus tag “cg----” indicates a gene in strain ATCC 13032, “cgR_----” indicates that in strain R), respectively, while the general components EI and HPr are encoded by the ptsI and ptsH genes (cg2117 or cgR_1763 and cg2121 or cgR_1767) (Kotrba et al. 2001; Parche et al. 2001). While ptsG and ptsS are dispersed in the genome, the other components are encoded in a cluster; an operon consisting of fruR-pfkB1-ptsF-ptsH is located upstream of ptsI, which is divergently transcribed. The genes, fruR (cg2118 or cgR_1764) and pfkB1 (cg2119 or cgR_1765) encode a DeoR-type transcriptional regulator and fructose-1-phosphate kinase, respectively. Sugar-dependent transcriptional regulation of the pts genes has been first reported in 2008 (Tanaka et al. 2008a). Northern analysis demonstrated that fructose and sucrose induce all pts genes whereas glucose upregulates ptsI, ptsH, and ptsG, the last two of which are generally expressed even in the absence of PTS sugars. The analysis also showed that ptsH is transcribed not only as a part of the operon but from its own promoter.
FruR, encoded by the first gene of the fruR-pfkB1-ptsF-ptsH operon, is not involved in the sugar-dependent upregulation because, in the fruR deletion mutant, the fructose-dependent upregulation of the fruR operon and ptsI was enhanced (Tanaka et al. 2008a), which led to the idea that FruR attenuates the induction of expression of ptsI, ptsH, and its own operon in the presence of fructose. Although FruR was obtained by DNA affinity purification using a DNA fragment encompassing the intergenic region between ptsI and fruR (Tanaka et al. 2008a), its binding motif was not determined.
Another DeoR-type transcriptional regulator, SugR, globally represses the pts genes. SugR is encoded by the sugR gene (cg2115 or cgR_1761), whose genomic locus is close to the ptsI-ptsF cluster, motivating its functional analysis (Engels and Wendisch 2007). Transcriptome analysis using microarray revealed that in the absence of PTS sugars, expression of all pts genes in the sugR deletion mutant was higher than in the wild type (Engels and Wendisch 2007; Gaigalat et al. 2007) (Fig. 1). The recombinant protein of SugR bound to the promoter region of these genes in vitro. The binding of SugR to the ptsG promoter region in vitro was inhibited by fructose 6-phosphate (Engels and Wendisch 2007). However, the binding to the promoter region of ptsI was not inhibited by fructose-6-phosphate, but by some sugar phosphates including fructose 1-phosphate and glucose 6-phosphate, each of which is yielded by fructose and glucose uptake via PtsF and PtsG, respectively, and fructose 1,6-bisphosphate (Gaigalat et al. 2007). Of the sugar phosphates tested, fructose-1-phosphate has the strongest inhibitory effect. This is consistent with the fact that the pts genes were highly induced in the presence of fructose compared with that in the presence of glucose (Tanaka et al. 2008a). The binding sites of SugR have been investigated by different groups, confining the region required for the SugR binding, but the consensus motif presented were different from each other (Engels and Wendisch 2007; Gaigalat et al. 2007; Tanaka et al. 2008b). Chromatin immunoprecipitation with microarray (ChIP-chip) analysis detected the SugR binding in vivo to the promoter regions of the pts genes (Engels et al. 2008). The alignment of the detected promoter regions determined the consensus binding motif as TCRRACA (R: A or G) (Engels et al. 2008).

Transcriptional regulators of genes involved in sugar uptake and metabolism. Metabolic pathways are indicated by thick red arrows. Genes constituting the metabolic pathways are indicated by italic letters. Sugars and metabolites are indicated by bold letters. Transcriptional regulators are indicated by ovals. Arrows from the ovals indicate transcriptional activation, whereas T-bars indicate repression. Lines ended with squares indicate the in vivo or in vitro binding of transcriptional regulators with unknown function. G6P glucose 6-phosphate, F1P fructose 1-phosphate, 6PGL 6-phosphoglucono-1,5-lactone, 6PG 6-phosphogluconate, Ru5P ribulose 5-phosphate, Xu5P xylulose 5-phosphate, R5P ribose 5-phosphate, S7P sedoheptulose 7-phosphate, F6P fructose 6-phosphate, E4P erythrose 4-phosphate, F1,6BP fructose 1,6-bisphosphate, DHAP dihydroxyacetone phosphate, GAP glyceraldehyde 3-phosphate, 1,3-BGP glycerate 1,3-bisphosphate, 3-GP glycerate 3-phosphate, 2-GP glycerate 2-phosphate, PEP phosphoenolpyruvate
Functionally redundant two GntR-type transcriptional regulators GntR1 and GntR2, the latter of which is strain-specific, have been first identified as transcriptional repressors of gluconate utilization genes, encoding gluconate permease, gluconate kinase, and 6-phosphogluconate dehydrogenase in strain ATCC 13032 (Frunzke et al. 2008). Transcriptome analysis of gntR1/R2 (cg2783 or cgR_2434/cg1935) double deletion mutant using microarray revealed the downregulation of ptsG and ptsS as well as upregulation of the pentose phosphate pathway genes and the gluconate metabolic genes. In vitro DNA binding assay and in vivo promoter reporter assay confirmed that GntR1/R2 also function as transcriptional activators of the pts genes. The DNA binding activity of GntR1/R2 was inhibited by gluconate and glucono-δ-lactone (Frunzke et al. 2008). Consistent with this, the glucose uptake rate was reduced by gluconate. The coordinated regulation of gluconate utilization pathway and PTS sugar uptake likely represents the molecular mechanism of simultaneous utilization of different carbon sources of C. glutamicum rather than sequential utilization observed in other model organisms. A ChIP-chip analysis of GntR1 in C. glutamicum strain R, in which GntR2 is not encoded, not only confirmed the previous results but also expanded its regulon and identified consensus binding motif more precisely, 5′-AWWGGTMRYACCWWT-3′ (Tanaka et al. 2014), unveiling the more global physiological function of GntR1 as described later.
The MerR-type transcriptional regulator RamB (cg0444 or cgR_0442) has been first identified as a transcriptional repressor of genes for acetate metabolism, including glyoxylate shunt genes, aceA (cg2560 or cgR_2212) and aceB (cg2559 or cgR_2211), and acetate activation genes, pta (cg3048 or cgR_2656) and ackA (cg3047 or cgR_2655) (Gerstmeir et al. 2004). This study identified the RamB binding motif, 5′-ARAACTTTGCAAA-3′ (R: A or G) and in silico search for the motif in the genome identified a putative binding site in the ptsG promoter region. Transcriptome analysis followed by in vitro DNA binding assay indicated that RamB directly activates ptsG and ptsS expression during growth in minimal medium containing glucose as a sole carbon source (Auchter et al. 2011a). In contrast, promoter reporter assay demonstrated that RamB acts as a transcriptional repressor of ptsG during growth in LB medium with or without glucose and acetate (Subhadra and Lee 2013). The putative binding site is located at position +72 with respect to the transcriptional start site (TSS) of ptsG, indicating that the repressor function is seemingly reasonable. The putative RamB binding site upstream of ptsS is centered at position −47 with respect to the TSS, which is likely consistent with the activator function.
RamA (cg2831 or cgR_2464), a LuxR-type transcriptional regulator, was isolated by DNA affinity purification using DNA fragments containing either the promoter region of the pta-ackA operon or the intergenic region of aceA and aceB, as RamB (Gerstmeir et al. 2004; Cramer and Eikmanns 2007). Thus, the names of these regulators RamA and RamB (regulators of acetate metabolism A and B) represent genes first identified to be regulated by themselves. Transcriptome analysis of the ramA deletion mutant and in vitro DNA binding assay demonstrated that ptsG and ptsH are directly repressed by RamA (Auchter et al. 2011a). There are seven and two putative binding sites (consecutive four to six G or C) of RamA in the ptsG and ptsH promoter regions, respectively, despite no direct evidence that RamA binds to all of these sites. A part of the sites is located downstream of the TSS, implying the repressor function of RamA.
GlxR (cg0350 or cgR_0377) is a homolog of E. coli cyclic AMP (cAMP) receptor protein (CRP). It has been first identified as a transcriptional repressor of the aceB gene encoding malate synthase in the glyoxylate shunt (Kim et al. 2004). In silico search for its binding motif, 5′-TGTGANNNNNNTCACA-3′, which is almost the same as that of E. coli CRP, and in vitro DNA binding assay confirmation detected 77 binding sites in the genome, identifying putative 150 genes belonging to the GlxR regulon (Kohl et al. 2008; Kohl and Tauch 2009). ChIP-chip and ChIP-seq (ChIP with high-throughput sequencing) analyses (Toyoda et al. 2011; Jungwirth et al. 2013) confirmed and extended the GlxR regulon by detecting GlxR binding to DNA in vivo. The GlxR binding sites upstream of ptsG and in the intergenic region between ptsI and the fruR-pfkB1-ptsF operon were detected by all the analyses whereas those upstream of ptsS was detected only by ChIP-chip (Toyoda et al. 2011). Although the function of GlxR in vivo at those binding sites has not been demonstrated, the position of the binding sites with respect to the TSS suggest a repressor function at these sites. Indeed, promoter reporter assay has recently shown that the ptsG promoter activity was upregulated in deletion mutants of glxR (cg0350 or cgR_0377) and cyaB (cg0375 or cgR_0397) (Subhadra and Lee 2013), the latter of which encodes adenylate cyclase producing cAMP. As the intracellular cAMP levels of the glucose-grown cells are higher than those of acetate-grown cells (Kim et al. 2004), GlxR may suppress ptsG expression in the presence of glucose where SugR-mediated repression is alleviated.
2.1.2 β-Glucoside Uptake and Utilization
Unlike the type strain ATCC 13032, strain R can utilize β-glucosides, including salicin and arbutin (Kotrba et al. 2003). Genes for uptake and utilization of the glucosides form a gene cluster, bglF-bglA-bglG (cgR_2729-2727), each gene of which encodes β-glucoside-specific enzyme II of PTS, phospho-β-glucosidase, and an antiterminator, respectively. The antiterminator is required for substrate-dependent expression of the operon. In the absence of a substrate, transcription of the operon terminates at a terminator present in the 5′-UTR (untranslated region) of mRNA. In the presence of a substrate, the antiterminator is postulated to bind to the ribonucleic antiterminator sequence in the terminator, thereby disrupting the terminator structure and allowing elongation of transcripts. C. glutamicum R carries another orthologous cluster consisting of bglF2-bglA2-bglG2 (cgR_2610-2608) for β-glucosides utilization. Genetic analysis demonstrated that the components are functionally equivalent, but the antiterminators BglG and BglG2 are specific to cognate transcripts with minor cross-talk regulation (Tanaka et al. 2009). In contrast to other PTS components, SugR is not involved in the expression of the gene clusters (Tanaka et al. 2009). However, ChIP-chip analysis identified GlxR binding sites in the promoter region of the operons (unpublished data), suggesting its involvement in the regulation of the gene clusters.
In the type strain ATCC 13032 genome, the bglF-bglA-bglG cluster is present as pseudogenes and bglA2 and bglG2 in the bglF2-bglA2-bglG2 cluster were lost (Kalinowski et al. 2003; Yukawa et al. 2007). This suggests that the clusters were present in the common ancestor of the two strains. This is supported by the fact that, at the end of 2018, NCBI BLAST search revealed that other than strain R, at least eight strains of C. glutamicum, whose genomic sequences are available, possess orthologous gene clusters for β-glucosides utilization.
2.1.3 Non-PTS Sugars
Non-PTS sugars are transported into the cytosol via other transporters but not PTS. The genes, iolT1 (cg0226 or cgR_0261) and iolT2 (cg3387 or cgR_2943), encoding the myo-inositol transporters IolT1 and IolT2, were identified as inositol-responsive genes by transcriptome analysis of cells grown on myo-inositol (Krings et al. 2006). These transporters belong to the major facilitator superfamily. Genetic analysis and sugar uptake study demonstrated that these transporters are functionally complementary inositol transporters (Krings et al. 2006). The gene cluster I and II comprising 13 and 8 genes, respectively, were upregulated in response to inositol. The iolT1 gene is located about 10 kb downstream of the cluster I. Gene deletion experiment showed that the cluster II, containing iolT2, is dispensable for myo-inositol utilization. The iolR gene (cg0196 or cgR_0236), encoding a GntR-type transcriptional regulator, is located upstream of the iol operon in the gene cluster I and transcribed divergently. Transcriptome analysis of the iolR deletion mutant revealed that the iol operon (cg0197-cg207 or cgR_0237-cgR_0246) and iolT1 (cg0223 or cgR_261) were highly upregulated compared with the wild type (Klaffl et al. 2013), demonstrating the transcriptional repressor function of IolR. The DNA-binding activity of IolR is probably controlled by inositol or its catabolite. The consensus binding site KGWCHTRACA (K: G or T, W: A or T, H: not G, R: A or G) was deduced using the MEME tool (Bailey and Elkan 1994) and confirmed by in vitro DNA binding assay (Klaffl et al. 2013). ChIP-chip analysis of GntR1 detected the GntR1 binding site in the iolT1 promoter region, but deletion of gntR1 had little effect on the iolT1 expression (Tanaka et al. 2014). A factor directly involved in inositol-responsive induction of the gene cluster II has not been identified. IpsA, a LacI-type transcriptional regulator, activates expression of ino1, encoding myo-inositol phosphate synthase (cg3323 or cgR_2885), a key enzyme for endogenous inositol synthesis, in the absence of inositol (Baumgart et al. 2013). Transcriptome analysis of the ipsA (cg2910 or cgR_2534) deletion mutant showed that a part of genes (but not including iolT2) in the gene cluster II were upregulated in the mutant, suggesting repression of the gene cluster II by IpsA in the absence of inositol (Baumgart et al. 2013).
The in silico search detected the GlxR binding sites upstream of iolT1 and iolT2 (Kohl et al. 2008; Kohl and Tauch 2009). ChIP-chip and ChIP-seq analyses also confirmed the binding sites and additionally detected the binding site in the intergenic region between iolR and iolC (the first gene of the iol operon) (Toyoda et al. 2011; Jungwirth et al. 2013). These results suggest the GlxR involvement in the regulation of inositol uptake and catabolism, although whether it acts as a repressor or activator is not clear.
The IolT1 and IolT2 transporters are biotechnologically important owing to their broad substrate specificity for glucose (Ikeda et al. 2011; Lindner et al. 2011), fructose (Bäumchen et al. 2009), and xylose (Brüsseler et al. 2018), in addition to myo-inositol. Deletion of iolR, mutations in the iolR promoter, or myo-inositol supplementation, all of which induced iolT1 and iolT2, rescued the growth defect of the deletion mutant of ptsH, encoding the PTS general component HPr, on glucose as a sole carbon source. Substitution of glucose uptake pathway via PTS with that via IolT1 was achieved by inactivation of PTS components and overexpression of IolT1 and glucokinase (Ikeda et al. 2011; Lindner et al. 2011). This enables cells to save PEP consumption during glucose uptake and use it for bioproduction of PEP-derived compounds, including amino acids and aromatic compounds, contributing to the improvement of production yield.
Another example of non-PTS sugar is maltose, a disaccharide composed of glucose. Uptake of maltose is mediated by an ABC transporter system, MusEFGK2I (Henrich et al. 2013), which is essential for maltose uptake and utilization. Deletion of ramA significantly decreased expression of the transporter-encoding gene cluster, musK2 (cg2708 or cgR_2369), musE (cg2705 or cgR_2367), and musFGI (cg2704-2703-2701 or cgR_2365-2364-2363), indicating an activator role of RamA (Auchter et al. 2011a). In addition, as predicted by in silico analysis (Kohl et al. 2008; Kohl and Tauch 2009), the GlxR binding sites were detected in the upstream regions of musK2 and musE by ChIP-chip and ChIP-seq analyses (Toyoda et al. 2011; Jungwirth et al. 2013). Because the binding sites are located downstream of the TSS of musK2 and overlap with the −10 region of the musE promoter, GlxR probably represses the mus genes. Uptake of maltose enhances the expression of ptsG, encoding the glucose-specific EII of PTS. The upregulation of ptsG was abolished in the ramA mutant. As RamA is involved in upregulation of mus genes and ptsG, it is likely that RamA mediates the maltose effect on the ptsG upregulation (Henrich et al. 2013).
The wild-type C. glutamicum is incapable of utilizing the sugar alcohol mannitol. A deletion mutant of cgR_0187 (cg0146) is able to catabolize it (Peng et al. 2011). cgR_0187 encodes a DeoR-type transcriptional regulator, which has been first identified as the repressor SucR of the sucCD operon, encoding succinyl-CoA synthetase (Cho et al. 2010). Later, SucR was also shown to repress its own gene sucR and adhA (cg3107 or cgR_2695), encoding alcohol dehydrogenase (Auchter et al. 2011b). mtlD, encoding mannitol 2-dehydrogenase (MtlD), is encoded upstream of cgR_0187 (sucR) with rbtT, encoding a putative ribitol transporter. These genes are transcribed divergently from sucR. xylB, encoding xylulose kinase, is located downstream of cgR_0187 (sucR) in the same direction. These genes were upregulated in the cgR_0187 (sucR) deletion mutant and shown to be required for mannitol catabolism (Peng et al. 2011), demonstrating the repressor function of the regulator. Mannitol is oxidized to fructose, which is excreted once, then imported again by PtsF, yielding fructose-1-phosphate. Taken together, this gene cgR_0187 was renamed mtlR (Peng et al. 2011). However, mannitol did not induce the genes regulated by MtlR. Finally, arabitol was shown to be a genuine inducer of the regulator (Laslo et al. 2012), leading to the designation of the regulator AtlR. The adjacent genes, mtlD, rbtT, and xylB, were induced by arabitol supplementation, enabling the wild type C. glutamicum to grow on the carbon source. rbtT encodes a probable transporter for arabitol, not mannitol, and xylulokinase encoded by xylB is required for catabolism of xylulose generated by arabitol oxidation (Laslo et al. 2012). The ChIP-chip analysis detected the GlxR binding site overlapping the TSS of rbtT (Toyoda et al. 2011), indicating that GlxR also represses the rbtT-mtlD operon.
2.2 Glycolysis
Sugars transported by the uptake systems described above are metabolized in the glycolytic pathway. C. glutamicum uses the Embden–Meyerhof–Parnas (EMP) pathway comprising nine reactions for conversion of glucose-6-phosphate into pyruvate. For the glycolytic genes, their transcript organization and transition of their expression levels during the different growth phase on different carbon sources have been demonstrated (Han et al. 2007). The gapA-pgk-tpi-ppc gene cluster (cg1791-1790-1789-1787 or cgR_1636-1635-1634-1633) encodes the key enzyme glyceraldehyde-3-phosphate dehydrogenase, which is the first glycolytic enzyme that generates the reducing equivalent NADH, with phosphoglycerate kinase, triosephosphate isomerase, and phosphoenolpyruvate carboxylase, respectively. Of the glycolytic genes, this cluster was specifically and highly upregulated during the growth-arrested bioprocess, where cells were packed to a high density under oxygen deprivation (Inui et al. 2007). To clarify the molecular mechanism underlying the upregulation, transcriptional regulators interacting with the gapA promoter were isolated using DNA affinity purification. GlxR, GntR1, SugR, RamA, and MalR (cg3315 or cgR_2877), the former four of which have been shown to globally regulate gene expression, as described above, have been obtained (Toyoda et al. 2008, 2009a), illustrating the importance of the control of the cluster expression. As none of the regulators had been shown to regulate the glycolytic genes at that time, this study first indicated their global regulatory function. ChIP-chip and ChIP-seq analyses of GlxR and GntR1 detected the gapA promoter region, confirming their binding in vivo (Toyoda et al. 2011; Jungwirth et al. 2013; Tanaka et al. 2014).
SugR represses the gapA expression during growth on a gluconeogenic substrate like acetate or complex medium as it does the pts genes (Toyoda et al. 2008). ChIP-chip analysis detected the SugR binding at the promoter regions of pfk (cg1409 or cgR_1327, phosphofructokinase), eno (cg1111 or cgR_1071, enolase), pyk (cg2291 or cgR_1973, pyruvate kinase), and fba (cg3068 or cgR_2667, fructose-bisphosphate aldolase) of the glycolytic genes as well as those of pts genes (Engels et al. 2008). Although the binding of SugR to these promoter regions was confirmed by in vitro DNA binding assay, transcriptome analysis of the sugR deletion mutant revealed the significant upregulation of only eno owing to derepression. This suggests that complex regulation of the genes by multiple transcriptional regulators as observed for gapA. Even though not detected by ChIP-chip, another pyruvate kinase-encoding gene, pyk2 (cg3218 or cgR_2811), was upregulated in the sugR mutant, but this is probably due to the derepression of its upstream gene, ldhA (cg3219 or cgR_2812), which is also repressed by SugR (Engels et al. 2008; Dietrich et al. 2009; Toyoda et al. 2009c).
RamA activates gapA expression by binding to three sites in the promoter with a different affinity (Toyoda et al. 2009a). In contrast, transcriptome analysis followed by in vitro DNA binding assay demonstrated that RamA negatively regulates pfk (6-phosphofructokinase) and pyk (pyruvate kinase) during growth on glucose (Auchter et al. 2011a). The transcriptome study showed that RamB is not involved in the regulation of the glycolytic genes.
GlxR also activates gapA expression (Toyoda et al. 2011) through its binding to the promoter region at position −244 with respect to the TSS. ChIP-chip analysis detected the GlxR binding sites in the promoter regions of the glycolytic genes pgi, pfk, gapA, pgk, and eno (Toyoda et al. 2011; our unpublished data). The putative binding sites were found at positions −65, −174, −244, −56, and −100 with respective to the TSSs of these genes, suggesting the activator role of GlxR. Indeed, promoter reporter assay demonstrated that GlxR activates the expression of pfk by binding to the putative site (Toyoda et al. 2011). The gapA operon yields four transcripts: gapA, gapA-pgk-tpi, pgk-tpi, and pgk-tpi-ppc (Schwinde et al. 1993). GlxR regulates the promoters upstream of gapA and pgk.
DNA affinity purification and ChIP-chip analysis detected the GntR1 binding to the gapA promoter (Toyoda et al. 2009a; Tanaka et al. 2014). The binding site is located farther upstream of the GlxR binding site. Based on these findings, GntR1 may be involved in activation of gapA along with other transcriptional regulators, although transcriptome analysis showed no discernible change in the gapA expression in the gntR1 mutant. The ChIP-chip analysis also detected the GntR1 binding at the promoter regions of pyk and gapB (also known as gapX), encoding glyceraldehyde-3-phosphate dehydrogenase isozyme with a different cofactor preference (Omumasaba et al. 2004). Combined with the results of transcriptome analysis and promoter reporter assay, GntR1 directly activates gapB expression and represses the pyk expression in the absence of gluconate, the negative effector of GntR1 (Tanaka et al. 2014). Transcriptome analysis and in vitro DNA binding assay showed that gapB expression is also directly activated by RamA (Auchter et al. 2011a).
The group 2 sigma factor SigB globally activates the expression of the glycolytic genes, namely, pfk, fba, gapA, pgk, tpi, and eno (Ehira et al. 2008). This finding suggested the SigB-dependent promoter motif, distinct from the primary sigma factor SigA-dependent one. The promoter motifs specific for the sigma factors encoded in the C. glutamicum genome are described in detail in the Chap. 4.
2.3 Pentose Phosphate Pathway and Pentose Utilization
Pentoses, including ribose, xylose, and arabinose, are metabolized in the pentose phosphate pathway. The pathway generates not only NADPH for reducing equivalents, but also important intermediates, including riboflavin, phosphoribosyl pyrophosphate (PRPP), and erythrose-4-phosphate (E4P). PRPP is used for nucleic acids and histidine synthesis whereas E4P enters the shikimate pathway, through which chorismate, a precursor for aromatic amino acids, folate, and menaquinone, is synthesized. Although the wild-type C. glutamicum is incapable of utilizing xylose and arabinose, the metabolic engineering successfully developed strains that are able to utilize the pentoses simultaneously with glucose as carbon and energy source (Kawaguchi et al. 2006, 2008). Xylose and arabinose enter the pentose phosphate pathway as xylulose-5-phosphate.
Genes involved in the pentose phosphate pathway are: tkt (cg1774 or cgR_1624, transketolase), tal (cg1776 or cgR_1625, transaldolase), zwf-opcA (cg1778-1779 or cgR_1626-1627, glucose 6-phosphate dehydrogenase), devB (cg1780 or cgR_1628, 6-phosphogluconolactonase), all of which form an operon in this order, gnd (cg1643 or cgR_1513, 6-phosphogluconate dehydrogenase), rpi (cg2658 or cgR_2318, ribose 5-phosphate isomerase), and rpe (cg1801 or cgR_1646, ribulose 5-phosphate 3-epimerase), three of which are dispersed in the genome. As described above, GntR1 negatively regulates the tkt operon and gnd gene, as well as the gluconate utilization genes (Frunzke et al. 2008).
RamA represses the tkt operon (Auchter et al. 2011a), whereas it activates gnd (Tanaka et al. 2012). As the RamA binding site, upstream of the gnd gene, overlaps with one of the two binding sites of GntR1, RamA-mediated activation of the gnd gene is blocked in the presence of GntR1.
The binding of SugR to the upstream region of the tkt operon was detected by ChIP-chip analysis and further corroborated by the in vitro DNA binding assay, although deletion of sugR had little effect on the expression of the operon (Engels et al. 2008). ChIP-chip analysis also detected the binding of GlxR to the upstream region of the tkt operon (Toyoda et al. 2011), but the role of GlxR in the regulation of the operon has not been demonstrated.
ChIP-chip analysis of the alternative sigma factor SigH identified the SigH-dependent promoters upstream of tal in the tkt-tal-zwf-opcA-devB operon and upstream of ribC in the ribGCAH cluster (cg1800-1799-1798-1797 or cgR_1645-1644-1643-1642), which encodes riboflavin biosynthetic enzymes (Toyoda et al. 2015). SigH is responsible for heat and oxidative stress response and upregulates genes that are involved in protein quality control and oxidative stress protection as well as genes for enzymes carrying oxidative stress labile cofactors (Ehira et al. 2009; Busche et al. 2012; Toyoda et al. 2015). Deletion of rshA, encoding the cognate anti-sigma factor RshA, upregulated expression of tal-zwf-opcA-devB and ribCAH, enhancing the production of NADPH and riboflavin. NADPH is utilized by the thioredoxin system and dehydrogenases under the control of SigH. Riboflavin is the precursor of FMN and FAD, redox cofactors for flavoenzymes, which are also induced by SigH.
An ABC transporter essential for ribose uptake is encoded by the genes rbsACBD, which form the operon rbsRACBD (cg1410-1411-1412-1413-1414 or cgR_1328-1329-1330-1331-1332) with rbsR, encoding a LacI-type transcriptional regulator. Genetic analysis and in vitro DNA binding assay demonstrated that RbsR acts as a transcriptional repressor of its own operon in the absence of ribose (Nentwich et al. 2009). Ribokinases phosphorylating ribose to ribose 5-phosphate, which enters the pentose phosphate pathway, are encoded by rbsK1 in the uriR-rbsK1-uriT-uriH operon (cg1547-1546-1545-1544 or cgR_1432-1431-1430-1429) and rbsK2 (cg2554 or cgR_2206). RbsR binding sites were found upstream of the operon and the rbsK2. The uri operon is required for uridine uptake and metabolism. UriR, a LacI-type transcriptional regulator, represses its own operon (Brinkrolf et al. 2008). The rbs operon was no longer induced by ribose in the rbsK1-rbsK2 double mutant, suggesting that an effector molecule affecting RbsR DNA binding activity is derived from ribose-5-phosphate, although ribose-5-phosphate itself does not affect the activity (Nentwich et al. 2009). The GlxR binding site with the unknown physiological role was identified by ChIP-chip at position −90 with respect to the TSS of rbsR (our unpublished data). In addition, the rbs operon, the uri operon, and rbsK2 were significantly downregulated in the gntR1 deletion mutant (Tanaka et al. 2014). As only a weak ChIP-chip peak was detected at the intragenic region of uriT, GntR1 may be indirectly involved in the regulation of these genes. Transcriptome analysis of gntR1-gntR2 double deletion mutant of strain ATCC 13032 did not detect the downregulation of these genes (Frunzke et al. 2008). The difference in the results may be attributable to the basal medium used.
2.4 The Tricarboxylic Acid (TCA) Cycle and Glyoxylate Shunt
The TCA cycle generates reducing equivalents (in the form of NADH and menaquinol), which are used for ATP synthesis by oxidative phosphorylation. Meanwhile, the intermediates 2-oxoglutarate and oxaloacetate in the cycle are used as precursors of glutamate and aspartate family amino acids. The TCA cycle is essential for organic acid metabolism as well. For the acetate metabolism, the glyoxylate shunt bypasses decarboxylation reactions in the cycle. For the metabolism of propionate, the 2-methyl citrate cycle generates the TCA cycle intermediates for further metabolism (Plassmeier et al. 2012). Genes involved in these metabolic pathways are also controlled by the global regulators, like the genes for sugar uptake and the glycolytic pathway as described above.
Pyruvate generated by glycolysis is first transformed into acetyl-CoA by pyruvate dehydrogenase complex to enter the TCA cycle. Of the genes encoding the three subunits AceE, AceF (SucB), and Lpd, aceE (cg2466 or cgR_2120) is positively regulated by GlxR and RamB and aceF (cg2421 or cgR_2087) is activated by RamA (Blombach et al. 2009; Auchter et al. 2011a; Toyoda et al. 2011) (Fig. 2). The binding of SugR to the lpd (cg0441 or cgR_0439) promoter region was detected in vivo and in vitro, although sugR deletion had no apparent effect on the lpd expression (Engels et al. 2008). Lpd and AceF constitute 2-oxoglutarate dehydrogenase complex with OdhA in place of AceE (Hoffelder et al. 2010).

Transcriptional regulators of genes involved in the TCA cycle, glyoxylate shunt, and acetate activation. Similar to Fig. 1, the metabolic pathways are indicated by thick red arrows, genes constituting the metabolic pathways are indicated by italic letters, and intermediate metabolites are indicated by bold letters. Transcriptional regulators are indicated by ovals. Arrows from the ovals indicate transcriptional activation, whereas T-bars indicate repression. Lines ended with squares indicate the in vivo binding of transcriptional regulators with unknown function. PEP, phosphoenolpyruvate; Acetyl-P, acetyl phosphate
In silico analysis, in vitro DNA binding assay, ChIP-chip and ChIP-seq analyses detected the potential GlxR binding sites upstream of the TCA cycle genes, namely, gltA (cg0949 or cgR_0943, citrate synthase), acn (cg1737 or cgR_1598, aconitase), sucC (cg2837 or cgR_2469, succinyl-CoA synthetase), sdhCD (cg0445 or cgR_0443, succinate dehydrogenase), and fum (cg1145 or cgR_1099, fumarase) (Han et al. 2008; Kohl et al. 2008; Kohl and Tauch 2009; Toyoda et al. 2011; Jungwirth et al. 2013) (Fig. 2). Promoter reporter assay and glxR overexpression study showed that GlxR negatively regulates gltA and sdhCDAB (Bussmann et al. 2009; van Ooyen et al. 2011). The GlxR binding sites are found around the TSS of sucC, indicating the repressor function of GlxR. The GlxR binding sites are located upstream of the fum promoter, but the regulatory role of GlxR in the regulation of fum expression should be confirmed by in vivo experiment.
ChIP-chip analysis and promoter reporter assay demonstrated that GntR1 directly activates the expression of icd (cg0766 or cgR_0784), encoding isocitrate dehydrogenase (Tanaka et al. 2014). This is the first transcriptional regulator directly involved in the icd regulation. GntR1 also positively regulates malE (maeB)(cg3335 or cgR_2895), encoding malic enzyme for NADP+-dependent malate decarboxylation (Gourdon et al. 2000) (Fig. 2). Isocitrate dehydrogenase from C. glutamicum is a monomer and catalyzes the oxidation of isocitrate with NADP+, not NAD+, as an electron acceptor (Eikmanns et al. 1995). Thus, GntR1 activates expression of genes encoding NADPH-generating enzymes. This activation is mitigated in the presence of gluconate, which also alleviates the GntR1-mediated repression of genes for the pentose phosphate pathway, resulting in compensation for NADPH generation reactions (Tanaka et al. 2014).
Transcriptome analysis of the ramA deletion mutant followed by in vitro DNA binding assay indicated the involvement of RamA in the regulation of gltA, acn, aceF, sucC, sdhCD, fum, mdh (cg2613 or cgR_2262), and mqo (cg2192 or cgR_1830) (Auchter et al. 2011a). RamA activates the expression of these genes, except that of mdh, which is repressed by RamA (Fig. 2).
The two anaplerotic enzymes PEP carboxylase and pyruvate carboxylase are encoded by ppc and pyc (cg0791 or cgR_0809). Since ppc is transcribed as a part of the glycolytic genes operon pgk-tpi-ppc, it is positively regulated by GlxR (Toyoda et al. 2011; Jungwirth et al. 2013) and SigB (Ehira et al. 2008), as described above in the glycolysis part 2.2. Expression of pyc is negatively regulated by RamB, although its binding sites are over 150 bp upstream of the TSS (Auchter et al. 2011a). The binding sites of GlxR were located downstream of the TSS of pyc, suggesting the repressor role of GlxR (Kohl et al. 2008; Kohl and Tauch 2009; Toyoda et al. 2011). The in vitro DNA binding assay clearly confirmed RamA binding to the pyc promoter region and identified the putative RamA binding sites. However, ramA deletion had no apparent effect on the pyc expression during growth in glucose minimal medium and complex medium (Auchter et al. 2011a).
The pckA gene (cg3169 or cgR_2751), encoding PEP carboxykinase, which catalyzes the reverse reaction of PEP carboxylase for gluconeogenesis, is also under complex regulation. The binding of GlxR to the pckA promoter was detected in vivo and in vitro studies (Han et al. 2007; Toyoda et al. 2011). The position of the binding site is located at 30 bp upstream of the TSS, which suggests a repressor function of GlxR. RamA, GntR1/R2, and IolR were isolated by DNA affinity purification (Klaffl et al. 2013). Gene deletion analysis combined with in vitro DNA binding assay demonstrated that RamA acts as a repressor and GntR1/2 and IolR act as activators. Deletion of iolR upregulated gluconate permease and gluconate kinase genes as well as the iol gene cluster for inositol uptake and catabolism as described above and downregulated glyoxylate shunt genes and pckA (Klaffl et al. 2013). However, direct interaction was confirmed only for the iol gene cluster and pckA.
DNA affinity purification using the promoter of the malE (maeB) gene, encoding malic enzyme, isolated the MarR-type transcriptional regulator MalR, which is characterized as a transcriptional repressor (Krause et al. 2012). MalR has been first isolated using DNA affinity purification using the gapA promoter as a ligand (Toyoda et al. 2009a), although whether it acts as a repressor or activator was not demonstrated. Thus, the malic enzyme gene malE (maeB) is regulated by two repressors, MalR and RamB, and two activators, RamA and GntR1/R2.
Global transcriptional regulators, not described earlier in this chapter, are involved in the regulation of the TCA cycle enzymes containing iron as a cofactor, namely, aconitase and succinate dehydrogenase (Fig. 2). The global iron regulator DtxR positively regulates sdhCDAB (succinate dehydrogenase) under iron-rich conditions (Brune et al. 2006). Upon iron limitation, DtxR-dependent repression of ripA (cg1120 or cgR_1076), encoding an AraC-type regulator, is relieved, resulting in RipA-dependent repression of sdhCDAB and acn (Wennerhold et al. 2005). acn is also repressed by the TetR-type regulator AcnR, encoded by the immediately downstream gene acnR (cg1738 or cgR_1599) (Krug et al. 2005), although a sensing molecule of AcnR remains elusive.
Isocitrate lyase and malate synthase, encoded by the aceA and aceB genes, respectively, form the glyoxylate shunt. This pathway is required for the metabolism of simple carbon sources, such as acetate. Acetate is activated to acetyl-CoA by phosphotransacetylase and acetate kinase, which are encoded by the pta-ackA operon, then enters the TCA cycle. To prevent the loss of the acetate-derived two carbons, the glyoxylate shunt bypasses the decarboxylation reactions in the cycle. These four genes are specifically induced during growth on acetate when compared with on glucose (Wendisch et al. 1997; Hayashi et al. 2002; Muffler et al. 2002). Due to the evident expression alteration, the promoters of the genes have been utilized to isolate transcriptional regulators, GlxR (Kim et al. 2004), RamA (Cramer et al. 2006), and RamB (Gerstmeir et al. 2004), all of which were later identified as key global regulators in C. glutamicum as described above. GlxR and RamB were isolated as transcriptional repressors of aceB, whereas RamA was as a transcriptional activator.
2.5 L-Lactate Metabolism
NADH oxidation under oxygen deprivation depends on fermentation. C. glutamicum produces L-lactate as one of the fermentation products from pyruvate using L-lactate dehydrogenase, encoded by the ldhA gene (Inui et al. 2004) (Fig. 1). ChIP-chip analysis of the sugar-responsive regulator SugR identified its binding sites in the ldhA promoter region. Transcriptome analysis of SugR demonstrated that SugR acts as a transcriptional repressor in the absence of sugar (Engels et al. 2008). Thus, SugR coordinates sugar uptake and metabolism with fermentative pyruvate reduction. This was corroborated by DNA affinity purification identifying SugR as a protein binding to the ldhA promoter region, in vitro DNA binding assay, and genetic analysis (Dietrich et al. 2009; Toyoda et al. 2009c). The DNA affinity purification using the ldhA promoter region as a ligand also identified a GntR-type transcriptional regulator, LldR, which has been shown to repress the cg3226 (cgR2818)-lldD (cg3227 or cgR_2819) operon required for L-lactate utilization as a carbon source (Stansen et al. 2005; Georgi et al. 2008). DNA binding activity of LldR is inactivated by L-lactate, leading to derepression of the operon for uptake and oxidation of L-lactate (Georgi et al. 2008). lldR (cg3224 or cgR_2816) deletion had no apparent effect on ldhA expression. However, the finding that the ldhA promoter activity was decreased in the ldhA deletion mutant and that the promoter activity in the mutant was restored upon deletion of lldR indicates that L-lactate-mediated repression of ldhA by LldR is not relieved in the ldhA deletion mutant background in contrast to in the wild type (Toyoda et al. 2009b). The SugR-mediated repression is epistatic to the LldR-mediated one. This is consistent with the position of the binding sites for the regulators with respect to the TSS: around the −10 and −35 regions of the ldhA promoter for SugR and LldR, respectively (Toyoda et al. 2009b). Transcriptome analysis of the lldR mutant revealed that ptsF, encoding the fructose-specific EII of PTS, is also repressed by LldR, indicating its global function (Gao et al. 2008). Because it was isolated by DNA affinity purification using the ldhA promoter region as a ligand, AtlR, a repressor of the arabitol utilization operon, is likely involved in the expression of ldhA (Dietrich et al. 2009; Toyoda et al. 2009c), in addition to that of sucCD and adhA. As none of these genes (sucCD, adhA, and ldhA) were detected by transcriptome analysis of the atlR deletion mutant (Laslo et al. 2012), the role of AtlR in the regulation of these genes may be masked by other regulators. In addition, MalR was also isolated by the DNA affinity purification study, although its role in the regulation of ldhA expression was unclear (Dietrich et al. 2009).
2.6 Carbon Catabolite Control
Carbon catabolite control (repression) enables the preferential consumption of one carbon source over the others, leading to a biphasic (diauxic) growth. CRP in E. coli and catabolite control protein A (CcpA) in B. subtilis represent transcriptional regulators and govern the catabolite repression. The intracellular cAMP levels in E. coli are lowered in the presence of glucose due to the abolishment of the activation of adenylate cyclase by glucose-specific EII of PTS (Reddy and Kamireddi 1998; Takahashi et al. 1998), deactivating catabolic genes for other carbon sources. The B. subtilis CcpA represses catabolic genes for various carbon sources by forming a protein complex with the PTS general component HPr, which is phosphorylated by HPr kinase in the presence of glucose (Deutscher et al. 1995; Jones et al. 1997).
In contrast to E. coli and B. subtilis, C. glutamicum co-metabolizes glucose with a variety of other carbon sources, including fructose, gluconate, lactate, pyruvate, acetate, propionate, vanillate, shikimate, and serine (Teramoto et al. 2009; Cocaign-Bousquet et al. 1993; Dominguez et al. 1997; Wendisch et al. 2000; Claes et al. 2002; Netzer et al. 2004; Merkens et al. 2005; Stansen et al. 2005; Frunzke et al. 2008). Ethanol and glutamate have been exceptionally shown to cause diauxic growth when provided in the mixture of glucose (Kronemeyer et al. 1995; Arndt and Eikmanns 2007; Kotrbova-Kozak et al. 2007). Although the E. coli CRP ortholog GlxR is encoded and involved in multiple carbon metabolic genes in C. glutamicum as described above, the intracellular cAMP levels of glucose-grown cells are higher than those of acetate-grown cells in contrast to E. coli (Kim et al. 2004). The intracellular cAMP levels of C. glutamicum are controlled by the adenylate cyclase CyaB and phosphodiesterase CpdA (Cha et al. 2010; Schulte et al. 2017b). In addition, HPr kinase, a component of the CcpA system, is not encoded in the C. glutamicum genome. Thus, C. glutamicum has been thought to use a molecular mechanism distinct from that functions in the model organisms.
GlxR is involved in the catabolite repression observed for these carbon sources utilization (Park et al. 2010; Subhadra and Lee 2013). The gluABCD operon (cg2136-2139 or cgR_1780-1783) encoding glutamate uptake transporter is upregulated by glutamate, but that upregulation is suppressed in the presence of glucose. The glucose-mediated suppression of the gluA promoter activity was alleviated in the glxR deletion mutant (Park et al. 2010). GlxR was shown to bind to the consensus site found in the gluA promoter region in vitro (Kohl et al. 2008). Ethanol is oxidized to acetate by alcohol dehydrogenase encoded by adhA and aldehyde dehydrogenase encoded by ald (cg3096 or cgR_2686). These genes are positively and negatively regulated by RamA and RamB, respectively (Arndt and Eikmanns 2007; Auchter et al. 2009). However, an additional regulator is suggested to be involved in the glucose-mediated repression of these genes. The activities of these ethanol catabolic enzymes in the glxR deletion mutant in the presence of glucose were higher than those in the wild type (Subhadra and Lee 2013). The two GlxR binding sites are located downstream of the TSS of the adhA and ald genes (Kohl et al. 2008; Kohl and Tauch 2009). Taken together, the CRP ortholog GlxR is involved in the catabolite repression observed during utilization of these carbon sources, which is different from E. coli CRP that functions as a transcriptional activator.
3 Global Nitrogen Control
Nitrogen is an essential component of almost all macromolecules, including nucleic acids, proteins, and cell wall. To supply an appropriate amount of nitrogen and to cope with environmental changes in nitrogen sources, bacteria use global nitrogen control systems optimized to their metabolisms and life cycles. Regarding C. glutamicum, which is used as a host for amino acids production, the nitrogen control system is important for not only homeostatic function but also for the industrial production of amino acids.
3.1 AmtR, a Global Nitrogen Regulator
As ammonium, the most favorable nitrogen source for C. glutamicum, is in equilibrium with ammonia, which diffuses into the cytosol via the cytoplasmic membrane, no specific protein is needed for uptake unless its concentration is limiting. When nitrogen source is scarce, various nitrogen uptake and assimilation genes are induced to utilize diverse nitrogen sources in the environment. A TetR-type transcriptional regulator, AmtR (encoded by cg0986 or cgR_0978), is responsible for the induction in C. glutamicum; it represses these genes under nitrogen-rich conditions. Since AmtR has been first identified as a transcriptional repressor of the ammonium transporter gene amtA under nitrogen surplus conditions (Jakoby et al. 2000), bioinformatic analysis and multi-omics analysis expanded its regulon to comprise over 30 genes for transport and assimilation of ammonium (amtA, amtB, glnA, gltBD; cg1785 or cgR_1631, cg2261 or cgR_1948, cg2429 or cgR_2093, cg0029-0030 or cgR_0263-0264), creatinine (codA, crnT; cg0104 or cgR_0096; cg0103 or cgR_0095) and urea uptake and metabolism (urtABCDE, ureABCEFGD, cg1061-1062-1064-1066 or cgR_1031-1035), signal transduction proteins (glnD, glnK, cg2260-2259 or cgR_1947-1946), meso-diaminopimelate synthesis (dapD, cg1256 or cgR_1189), malic enzyme (malE), and proteins with unknown function (Jakoby et al. 2000; Beckers et al. 2005; Buchinger et al. 2009). The vanABK operon (cg2616-2618 or cgR_2265-2267) encoding vanillate uptake transporter (VanK) and a metabolic enzyme (VanAB) is included as an indirect target due to the lack of the AmtR binding site (AmtR box) upstream of the operon. This operon is directly negatively regulated by VanR, which is encoded by the upstream gene vanR (cg2615 or cgR_2264) (Morabbi Heravi et al. 2015).
The AmtR box represented by the consensus motif, 5′–TTTCTATN6ATAGAWA–3′, with the strong conserved CTAT motif, is found upstream of the AmtR regulon. The similarity of the AmtR box to the consensus motif is likely correlated with the stringency of repression (Hasselt et al. 2011). Whereas transcription of ammonium uptake systems, encoded by amtA and amtB, as well as that of the gltBD operon encoding glutamate synthase are tightly regulated and fully repressed in nitrogen-rich medium, expression of gdh encoding glutamate dehydrogenase and glnA encoding glutamine synthetase is only loosely controlled. The strictly repressed genes possess multiple AmtR boxes with the conserved palindromic motif, CTATN6ATAG, while the AmtR boxes found upstream of the loosely regulated genes have an imperfect motif (Hasselt et al. 2011).
The importance of the conserved motif in the AmtR box for the interaction with AmtR was demonstrated by the structure of AmtR in free and DNA-bound forms (Palanca and Rubio 2016). AmtR exists as a homodimer with N-terminal DNA-binding (DNB) domain containing the helix-turn-helix (HTH) motif and a C-terminal ligand binding and dimerization (LBD) domain as other TetR-type transcriptional regulators. At the C-terminal region, there is an extra helix being a characteristic of corynebacterial AmtR proteins. Crystal structure of the DNA-bound form was obtained by mixing AmtR and self-complementary quasi-palindromic oligonucleotides containing the AmtR box. The recognition helices of the HTH motif of each subunit in the dimer were inserted into the two successive turns of the major groove of the double helix containing the AmtR box. The interacting residues were reasonable for the strongly conserved motif CTAT in the AmtR box. In addition, the N-terminal extension, which is rich in Arg and Pro and characteristic to corynebacterial AmtR, also interacted with DNA via the minor groove flanking to the major groove interacting with the helix of the HTH motif, which explains AT bases flanking the conserved CTAT motif. The mutant AmtR protein lacking this N-terminal 19-residues does not have the DNA binding ability in vitro. This extension may guide AmtR to the canonical box. The structure of the DNA-bound form revealed that the DNB domain of each subunit undergoes movement with the hinge at the junction between the two domains. This movement might be involved in triggering the dissociation of AmtR from DNA.
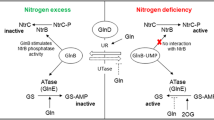
GlnK, a sole PII-type signal transduction protein in this organism, is involved in the derepression of the AmtR regulon expression (Nolden et al. 2001) (Fig. 3). The conserved Tyr residue 51 of GlnK is adenylylated by the adenylyltransferase GlnD during nitrogen starvation. The replacement of this Tyr residue with Phe, or the deletion of glnK or glnD, all of which result in permanent repression of the AmtR regulon (Nolden et al. 2001). These findings led to the assumption that direct interaction between GlnK and AmtR is required for release of AmtR from DNA. Indeed, adenylylated GlnK interacted with AmtR in vitro and in vivo and inhibited the binding of AmtR to DNA in vitro (Beckers et al. 2005). Deadenylylated GlnK is trapped by membrane via interaction with the ammonium transporter AmtB (Strösser et al. 2004), encoded in the operon amtB-glnK-glnD. GlnK is subjected to proteolysis by the membrane-bound proteinase FtsH, followed by the Clp system. Membrane localization is prerequisite for GlnK proteolysis because, in the absence of AmtB, GlnK is stable in the deadenylylated form, which is incapable of interacting with AmtR (Strösser et al. 2004). The structure of the AmtR-GlnK complex will provide a clue for understanding the molecular mechanism of AmtR dissociation from DNA. In this context, the crystal structure of AmtR suggested a loop possibly interacting with the GlnK T-loop, which is subjected to adenylylation (Palanca and Rubio 2016). Switching the interaction partner of the loop from the other subunit of AmtR to the T-loop of adenylylated GlnK may cause structural alteration resulting in dissociation of AmtR from the AmtR box.

Regulation of the AmtR regulon in response to nitrogen availability. The PII protein GlnK is adenylylated by the adenylyltransferase GlnD upon nitrogen limitation. Adenylation is indicated with small circles indicated with A. The adenylylated GlnK captures AmtR, resulting in derepression of the AmtR regulon. The adenylylated GlnK is deadenylylated by GlnD upon nitrogen supplementation. GlnK in the deadenylylated form is bound to the ammonium transporter AmtB and digested by FtsH and the Clp system. AmtR released from GlnK binds to the AmtR box, thereby repressing its regulon. PM, plasma membrane
3.2 Nitrogen Sensing
In E. coli, nitrogen limitation reduces the ratio of glutamine to 2-oxoglutarate, activating the uridylyltransferase GlnD to modify the PII proteins GlnB and GlnK. The uridylylated PII proteins induce positive autoregulation of the two-component NtrBC system, which directly and indirectly activates the expression of genes for nitrogen uptake and metabolism (reviewed in Merrick and Edwards 1995; Ninfa et al. 2000). In B. subtilis, cellular nitrogen availability is sensed by the glutamine synthetase GlnA, controlling the activity of two transcriptional regulators, TnrA and GlnR. Under nitrogen limitation, TnrA activates genes for ammonium transport and utilization of other nitrogen sources and represses those for glutamine and glutamate synthesis (Wray et al. 2001; Yoshida et al. 2003). Under nitrogen-rich conditions, GlnR represses tnrA and glnA encoding glutamine synthetase (Brown and Sonenshein 1996). The DNA binding activity of these regulators is controlled by the interaction with GlnA in the feedback-inhibited form (Wray et al. 2001; Fisher and Wray 2008, 2009; Schumacher et al. 2015). In contrast to the situations in these model organisms, glutamine is unlikely a sensing molecule in C. glutamicum, based on the finding that the AmtR regulon is derepressed in the glutamine-grown cells, in which glutamine concentrations are higher than those in ammonium-grown cells (Rehm et al. 2010). Intracellular ammonium levels, which are reduced upon nitrogen starvation (Nolden et al. 2001), are the possible indicators of the nitrogen status, although a sensor protein is still not identified.
3.3 Cross-Talk with Transcriptional Regulators for Other Metabolisms
The GlxR regulon contains the AmtR regulon including the gltBD operon (cgR_0263-0264), the amtB-glnK-glnD operon, the urtABCDE operon, gdh, the cg2181-cg2184 (cgR_1819-cgR_1822) operon, the gluABCD operon, and glnA1, thus connecting the carbon and nitrogen metabolisms (Kohl and Tauch 2009). Although its binding in vitro and in vivo was confirmed, its physiological role remains elusive, except for the repression of the gluABCD operon (Park et al. 2010). ChIP-chip analysis detected GntR1 binding to the intergenic region between gdh and glxK, although the gdh expression was not affected by gntR1 deletion (Tanaka et al. 2014).
4 Phosphate Control
Another macromolecule essential for cell growth is phosphorus, which is used for nucleic acid synthesis and protein modification. Bacteria are able to utilize inorganic and organic phosphorus, including orthophosphate, pyrophosphate, sugar phosphates, nucleotides, and phospholipids, to adapt to fluctuating phosphorus concentrations in the environment. Molecular mechanism of adaptation to phosphate limitation, namely, upregulation of genes for transporters, extracellular enzymes, and metabolic enzymes for phosphonates and organophosphates utilization, is intensively studied in model organisms and known to be under the control of two-component systems consisting of a sensor kinase and a response regulator (Santos-Beneit 2015).
4.1 The Two-Component System PhoRS-Mediated Phosphate-Limitation Response
C. glutamicum utilizes a variety of phosphorus-containing compounds as a phosphorus source (Wendisch and Bott 2008). Transcriptional response to phosphate limitation in C. glutamicum was investigated by transcriptome analysis using microarray (Ishige et al. 2003). Phosphate limitation by transferring cells from phosphate sufficient (13 mM) medium to phosphate limiting (0.13 mM) medium induced the genes for phosphate uptake and metabolism as observed in other bacteria. These genes include the pstSCAB operon (cg2846-2843 or cgR_2478-2475, high-affinity ABC-type phosphate uptake system), nucH (cg2868 or cgR_2495, extracellular nuclease), phoH1 (cg0085 or cgR_0084, ATPase with unknown function), the ugpAEBC operon (cg1568-1571 or cgR_1446-1449, ABC-type sn-glycerol 3-phosphate uptake system), glpQ1 (cg3215 or cgR_2805, glycerophosphoryl diester phosphodiesterase), phoC (cg3393 or cgR_2949, putative phosphoesterase), the pctABCD operon (cg1652-1649 or cgR_1522-1519, ABC-type transporter for phosphorus-containing compounds), and ushA (cg0397 or cgR_0412, putative 5′-nucleotidase) (Rittmann et al. 2005). Genes encoding proteins unrelated to phosphate metabolism were also upregulated but their roles in adaptation to phosphate limitation are not known.
C. glutamicum intracellularly accumulates polyphosphate synthesized by a polyphosphate kinase encoded by cg3007 (cgR_2616) (Lindner et al. 2007). The polyphosphate is hydrolyzed by exopolyphosphatases encoded by ppx1 (cg0488 or cgR_0480) and ppx2 (cg1115 or cgR_1074) to release orthophosphate (Lindner et al. 2009). Although the polyphosphate can be utilized as a phosphorus source, expression of the genes involved in its synthesis and hydrolysis was not affected by phosphate limitation.
PhoRS, a two-component system comprising a pair of sensor kinase and response regulator encoded by the operon phoR-phoS (cg2888-2887 or cgR_2511-2510), was transiently induced in response to phosphate limitation. Screening of deletion mutants of the 13 two-component systems encoded in the C. glutamicum genome for growth under phosphate limitation confirmed PhoRS as a key system for induction of the genes in response to phosphate limitation (Kočan et al. 2006). Indeed, microarray analysis and primer extension analysis revealed that the upregulation of the phosphate limitation-responsive genes was abolished in the phoRS deletion mutant, except for the pstSCAB operon, which was still induced with lower induction levels than in the wild type. Thus, PhoRS positively regulates the expression of genes induced under phosphate limitation. In vitro analysis demonstrated that the sensor kinase PhoS, in the form lacking its transmembrane region, is capable of autophosphorylation and transferring the phosphoryl group to the response regulator PhoR (Schaaf and Bott 2007). In vitro DNA binding assay revealed that PhoR binds to the promoter regions of the genes that are induced under phosphate limitation. The affinity of PhoR to DNA was enhanced upon phosphorylation by PhoS. The affinity of the promoter regions of the PhoR regulon to PhoR are different; the pstS promoter has the highest affinity and the glpQ1 has the lowest (Schaaf and Bott 2007). In the course of the in vitro DNA binding assay, two additional genes were found to belong to the PhoR regulon: the porB gene (cg1109 or cgR_1069), encoding an anion-specific porin (Costa-Riu et al. 2003), and pitA (cg0545 or cgR_0533), encoding low-affinity phosphate uptake transporter (Schaaf and Bott 2007). Expression of porB is independent of phosphate concentration, but it is activated by PhoR. In contrast to the other PhoR regulon, pitA is negatively regulated by PhoR. Indeed, pitA expression decreased after phosphate limitation shift (Schaaf and Bott 2007). Successive deletion and mutation of the DNA fragments used in in vitro DNA binding assay confined the location of the PhoR binding site and identified important nucleotides in the site. Since an artificial 8-bp direct repeat with a 3-bp spacer, 5′-CCTGTGAANNNCCTGTGAA-3′, had the currently highest affinity to PhoR, the PhoR consensus binding motif is the direct repeat, although there is no identical sequence upstream of any of the PhoR regulon. Promoter reporter assay of the phoR promoter showed that, in contrast to the full-length promoter, the promoter fragment lacking the PhoR binding site lost induction of a reporter gene under phosphate limitation, demonstrating physiological importance of the PhoR binding site (Schaaf and Bott 2007). In the pstS promoter region, two PhoR binding sites were found, but only upstream one, more closely matched with the consensus motif, is required for the PhoR-dependent upregulation of pstS (Schaaf and Bott 2007).
4.2 Involvement of Other Transcriptional Regulators
Although the two-component system has served as a paradigm of the transcriptional response to phosphate limitation in bacteria, interaction with other transcriptional regulators involved in different metabolisms is important to coordinate whole metabolism (reviewed in Santos-Beneit 2015). Of the phosphate-responsive genes in C. glutamicum, the pstSCAB operon encoding ABC-type phosphate uptake system has been shown to be still induced upon phosphate limitation in the absence of PhoRS (Kočan et al. 2006), as described above. DNA affinity purification using the pstS promoter region as a ligand identified the global regulator GlxR (Panhorst et al. 2011). Bioinformatic analysis followed by in vitro DNA binding assay identified the GlxR binding site at position −133 to −117 with respect to the TSS. Mutational inactivation of the site decreased the pstS promoter activity after phosphate limitation, demonstrating the transcriptional activator function of GlxR. Overexpression of GlxR promotes the growth under phosphate-limiting conditions, supporting the GlxR function as an activator. The effect of the binding site mutations was larger in glucose-grown cells compared with the acetate-grown cells. This is likely because intracellular cAMP levels in glucose-grown cells were higher than in acetate-grown cells (Kim et al. 2004).
The pstS promoter lacking the cis elements (binding sites) of PhoR and GlxR is still induced upon phosphate limitation. These findings motivated the search for transcriptional regulators involved in the PhoRS-independent regulation. DNA affinity purification using the DNA fragment containing the pstS promoter region but lacking the binding sites for PhoR and GlxR identified RamB (Sorger-Herrmann et al. 2015). Two RamB binding sites having similarity to the consensus binding motif were identified around the position −80 and −4 with respect to the TSS and demonstrated to be bound by RamB in vitro. These sites are important for phosphate limitation induction of pstS. Thus, RamB functions as a transcriptional activator of the operon.
5 Sulfur Control
Sulfur is essential for the biosynthesis of biomolecules including cysteine, methionine, Coenzyme A, and iron-sulfur clusters. Bacteria utilize sulfate, sulfite, and sulfonate as sulfur sources. These compounds should be degraded and reduced to disulfide for the biosynthesis. Because the reduction of sulfate is a high energy-requiring process and the resulting disulfide is highly toxic, expression of genes encoding enzymes involved in the reduction and the biosynthesis should be coordinately regulated.
C. glutamicum utilizes organic and inorganic sulfur compounds as sulfur sources. Organic sulfur compounds that C. glutamicum utilizes are sulfonates like ethane sulfonate, 3-(N-morpholino)propane sulfonate (MOPS), and taurine, and sulfonate esters like busulfan, ethyl methanesulfonate, and propanesulfonate (Koch et al. 2005b). Combining genomic analysis based on the knowledge obtained from model organisms (Kertesz 2000) with genetic studies identified genes responsible for the utilization of these compounds (Koch et al. 2005b). The ABC transporter encoded by ssuCBA (cg1377-1379-1380 or cgR_1299-1301), which has a similarity to the reported sulfonate transporter SsuABC, is required for uptake of sulfonates and sulfonate esters, although the involvement of additional transporter is suggested by gene deletion analysis. Sulfonates and sulfonate esters are degraded to liberate sulfite for anabolism. Two functionally equivalent sulfonatases encoded by ssuD1 (cg1376 or cgR_1298) and ssuD2 (cg1156) decompose sulfonates, whereas monooxygenases encoded by seuA, seuB, and seuC (cg1151, 1152, and 1153) are also involved in the degradation of sulfonate esters in addition to the sulfonatases. These sulfonatases and monooxygenases are FMNH2-dependent. FMNH2 is provided by the reductase SsuI, which is essential for the utilization of all sulfonates and sulfonate esters. ssuD1 and ssuCBA apparently form the operon ssuD1CBA. Although the gene cluster seuABC (cg1151-1153)-ssuD2 (cg1156) is encoded in the genome of limited strains of C. glutamicum, including ATCC 13032, ssuI (cg1147 or cgR_1100), which is closely located to the cluster, is still present in the same locus of other strain genomes, likely supporting the ability of the strains not carrying the seuABC-ssuD2 cluster to utilize the organic sulfur compounds.
Genomic analysis followed by genetic analysis also identified a gene cluster, fpr2–cysIXHDNYZ (cg3119-3112 or cgR_2704-2703-cgR_6148-cgR_2702-2698), for transporter and enzymes responsible for uptake and reduction of inorganic sulfur compounds, sulfate, and sulfite, to sulfide, which is utilized in the cysteine and methionine biosynthetic pathways (Rückert et al. 2005). Sulfate and sulfite are transported via CysZ. Sulfate is adenylated by CysDN, then reduced by CysH to sulfite. Sulfite, which is also generated from sulfonates and sulfonate esters degradation, is reduced to sulfide by CysI, for which the reducing equivalents are provided by the ferredoxin CysX and the ferredoxin reductases Fpr1 (cg3049 or cgR_2657) and Fpr2.
These sulfur compound utilization genes, together with genes constituting the biosynthetic pathway of sulfur-containing amino acids, cysteine and methionine (Rückert et al. 2003), are under the control of the global regulator McbR (Rey et al. 2005). McbR was first identified using DNA affinity purification as a transcriptional repressor of metY (cg0755 or cgR_0775) encoding O-acetyl-L-homoserine sulfhydrylase (Rey et al. 2003). The McbR regulon identified using transcriptome analysis of the mcbR (cg3253 or cgR_2850) deletion mutant and in vitro DNA binding assay comprises at least 45 genes, including the ssu and seu genes for sulfonates and sulfonate esters utilization, the cys cluster for sulfate uptake and reduction, and genes for cysteine and methionine biosynthesis. Bioinformatic analysis of the upstream region of the genes upregulated in the mcbR mutant identified the McbR binding motif, TAGAC-N6-GTCTA. This analysis also indicated the autoregulation of mcbR. The DNA binding activity of McbR is modulated by S-adenosylmethionine (SAM) and S-adenosylhomocysteine, which are a substrate and a product, respectively, of methyltransferases (Fig. 4) (Rey et al. 2005; Suda et al. 2008).
The ssu and seu genes are also transcriptionally regulated by the ROK (Repressor ORF kinase) family transcriptional regulator SsuR (Koch et al. 2005a) (Fig. 4), which belongs to the McbR regulon (Rey et al. 2005). The deletion of the ssuR gene (cg0012 or cgR_0010) lost the capability of sulfonate utilization as a sulfur source and abolished the expression of the ssu and seu genes, demonstrating that SsuR acts as a transcriptional activator. Competitive EMSA (electrophoretic mobility shift assay) using non-labeled oligonucleotides, followed by alignment analysis, revealed that the SsuR binding motif consisting of a T-, a GC- and an A-rich domain with a length of six, nine and six nucleotides, respectively. The motif was found around or upstream of the −35 region of the promoter of the target genes, consistent with the SsuR activator function. The genome-wide search for the motif revealed that no gene other than the ssu and seu genes is directly regulated by SsuR. Although the DNA binding activity of SsuR is inhibited by sulfate, APS, sulfite, and sulfide, it is not affected by the sulfonates including ethanesulfonate and methyl-ethanesulfonate, which are degraded by the Ssu and Seu systems. The induction of the ssu and seu genes during growth with sulfonates as a sole sulfur source was abolished in the presence of sulfate. Taken together, the absence of more preferred inorganic sulfur source is a signal for the SsuR regulon (Koch et al. 2005a).

The McbR regulon. Transcriptional regulators are indicated with ovals. Uptake systems and metabolic pathways under the control of the regulators are shown by rounded rectangles, whereas molecules and metabolites involved in the pathways are indicated with rectangles. Arrows and T-bars from the transcriptional regulators indicate transcriptional activation and repression, respectively. Those from the molecules and metabolites indicate activation and inactivation of the transcriptional regulators, except for arrows from SAM, indicating the methylation reaction catalyzed by O-methyltransferase (not indicated). SAH, S-adenosyl homocysteine; SAM, S-adenosyl methionine; O-AcHS, O-acetylhomoserine; O-AcSer, O-acetylserine; Sox, sulfate and sulfite; APS, adenosine-5′-phosphosulfate
The fpr2–cysIXHDNYZ gene cluster, required for sulfate uptake and reduction, is under the control of another ROK family transcriptional regulator CysR (Rückert et al. 2008), which also belongs to the McbR regulon. The deletion mutant of the cysR gene (cg0156 or cgR_0197) loses the ability to utilize sulfate and sulfonates as a sulfur source and has no expression of the ssu and seu genes as well as that of the cys gene cluster. Transcriptional analysis of the cysR deletion mutant revealed that CysR activates the ssuR gene encoding the transcriptional activator of the ssu and seu genes, indicating a hierarchical transcriptional regulation (Fig. 4). Comparison of the transcriptome profiles of the triple mutant of mcbR, ssuR, and cysR, and the mutant with constitutive expression of cysR distinguish the direct and indirect effect of the cysR deletion. While the cys gene cluster is directly activated by CysR, the ssu and seu genes are indirectly activated via activation of ssuR by CysR. In addition to the cys gene cluster, CysR positively regulates two transcript units, cg3372-3375 (cgR_2929-2932) and cg1514-4005 (cgR_6095-6094), encoding proteins with unknown function, and negatively regulates other two transcript units cg2810 (cgR_2451, dicarboxylate symporter family protein) and cg3138-3139 (cgR_2721-2722, proteins with unknown function), indicating its dual transcriptional regulatory function (Rückert et al. 2008). Alignment of the promoter regions of the genes activated by CysR revealed the CysR binding motif consisting of two 10-bp motifs forming an inverted repeat with a variable spacer of 6–8 bp. The 3′ region of the CysR binding site resembles that of the SsuR binding site. The DNA binding activity of CysR requires O-acetyl-L-serine or O-acetyl-L-homoserine, each of which serves as a sulfide acceptor for cysteine and methionine synthesis, respectively. Although sequence-specific binding in vitro was not confirmed, the importance of the binding site was confirmed by promoter reporter assay using DNA fragments with or without the binding site. Thus, CysR controls the expression of genes for sulfur uptake and reduction in response to the availability of sulfide acceptor to avoid an accumulation of toxic sulfide (Rückert et al. 2008). Since McbR also regulates the expression of the ssuR gene, the ssu and seu genes are only activated when additional sulfur is required and the concentration of inorganic sulfur sources is low. Because the genes with unknown function in the CysR regulon are co-regulated by McbR, proteins encoded are probably involved in the sulfur metabolic pathway. In contrast to the regulons of AmtR and PhoR, the involvement of other global transcriptional regulators has not been reported.
6 Concluding Remarks and Perspectives
The studies summarized in this chapter provide the overview of transcriptional regulatory systems for the metabolism of primary elements, i.e., carbon, nitrogen, sulfur, and phosphorus, in C. glutamicum. These studies demonstrated that the key global regulators were first identified as a transcriptional repressor or activator of a single gene in the metabolism. Such regulators have been isolated by either screening of the genomic library using reporter expression, which identified GlxR and AmtR, or by purification of proteins bound to the promoter region of the target gene, which identified RamA, RamB, IolR, AtlR, and McbR. Moreover, the genomic locus and orthologous regulators function also provided important clues to the identification of the global regulators, such as SugR, GntR1/2, and PhoRS. Accurate identification of the regulon members supports the understanding of the transcriptional regulators’ functions in three contexts. Firstly, a common feature among the regulon genes regulated in the same way gives a hint to understand the function of the regulator. SugR has been first identified as a transcriptional repressor of one of the PTS components, but it was demonstrated to repress genes involved in sugar uptake and metabolism in response to sugar availability. Secondly, the alignment of the upstream regions of the regulon genes gives a clue to determine the binding motif of the regulator as a matrix. Finally, the establishment of the regulon list also helps identify effector molecules controlling the DNA binding activity of the regulators as described for the regulators involved in sulfur control. In this context, ChIP-chip or ChIP-seq analysis directly detects the binding of the transcriptional regulator to the chromosomal DNA in vivo, enabling identification of the regulon and the binding motif.
The knowledge of the global transcriptional regulators in C. glutamicum has been accumulated for the last decade. It has not only deepened the understanding of the C. glutamicum physiology at the molecular level but also has been recently applied to metabolic engineering. For example, deletion of sugR was applied to enhance sugar metabolic pathways (Bartek et al. 2010; Xu et al. 2016; Pérez-García et al. 2018) and that of mcbR has increased the productivity of methionine and SAM (Rey et al. 2003; Han et al. 2016). Moreover, the knowledge has been used to construct biosensors to detect changes in metabolites (Mustafi et al. 2012; Schulte et al. 2017a). The sensors coupled with fluorescence-activated cell sorting were used together with random mutagenesis to screen for beneficial mutations that improve productivity (Mahr et al. 2015; Zhang et al. 2018). There are over 50 transcriptional regulators, including response regulators of the two-component systems, that have not been characterized yet in C. glutamicum. Investigation of these regulators will provide further insights into the physiology and transcriptional regulatory network, and contribute to the development of synthetic biological tools.
References
Arndt A, Eikmanns BJ (2007) The alcohol dehydrogenase gene adhA in Corynebacterium glutamicum is subject to carbon catabolite repression. J Bacteriol 189:7408–7416
Auchter M, Arndt A, Eikmanns BJ (2009) Dual transcriptional control of the acetaldehyde dehydrogenase gene ald of Corynebacterium glutamicum by RamA and RamB. J Biotechnol 140:84–91
Auchter M et al (2011a) RamA and RamB are global transcriptional regulators in Corynebacterium glutamicum and control genes for enzymes of the central metabolism. J Biotechnol 154:126–139
Auchter M et al (2011b) Control of adhA and sucR expression by the SucR regulator in Corynebacterium glutamicum. J Biotechnol 152:77–86
Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36
Bartek T et al (2010) Studies on substrate utilisation in L: -valine-producing Corynebacterium glutamicum strains deficient in pyruvate dehydrogenase complex. Bioprocess Biosyst Eng 33:873–8883
Bäumchen C, Krings E, Bringer S, Eggeling L, Sahm H (2009) Myo-inositol facilitators IolT1 and IolT2 enhance D-mannitol formation from D-fructose in Corynebacterium glutamicum. FEMS Microbiol Lett 290:227–235
Baumgart M, Luder K, Grover S, Gätgens C, Besra GS, Frunzke J (2013) IpsA, a novel LacI-type regulator, is required for inositol-derived lipid formation in Corynebacteria and Mycobacteria. BMC Biol 11:122. https://doi.org/10.1186/1741-7007-11-122
Beckers G et al (2005) Regulation of AmtR-controlled gene expression in Corynebacterium glutamicum: mechanism and characterization of the AmtR regulon. Mol Microbiol 58:580–595
Blombach B, Cramer A, Eikmanns BJ, Schreiner M (2009) RamB is an activator of the pyruvate dehydrogenase complex subunit E1p gene in Corynebacterium glutamicum. J Mol Microbiol Biotechnol 16:236–239
Bott M, Brocker M (2012) Two-component signal transduction in Corynebacterium glutamicum and other corynebacteria: on the way towards stimuli and targets. Appl Microbiol Biotechnol 94:1131–1150. https://doi.org/10.1007/s00253-012-4060-x
Brinkrolf K et al (2008) The LacI/GalR family transcriptional regulator UriR negatively controls uridine utilization of Corynebacterium glutamicum by binding to catabolite-responsive element (cre)-like sequences. Microbiology 154:1068–1081
Brown SW, Sonenshein AL (1996) Autogenous regulation of the Bacillus subtilis glnRA operon. J Bacteriol 178:2450–2454
Brune I, Werner H, Hüser AT, Kalinowski J, Pühler A, Tauch A (2006) The DtxR protein acting as dual transcriptional regulator directs a global regulatory network involved in iron metabolism of Corynebacterium glutamicum. BMC Genomics 7:21
Brüsseler C, Radek A, Tenhaef N, Krumbach K, Noack S, Marienhagen J (2018) The myo-inositol/proton symporter IolT1 contributes to d-xylose uptake in Corynebacterium glutamicum. Bioresour Technol 249:953–961. https://doi.org/10.1016/j.biortech.2017.10.098
Buchinger S et al (2009) A combination of metabolome and transcriptome analyses reveals new targets of the Corynebacterium glutamicum nitrogen regulator AmtR. J Biotechnol 140:68–74
Busche T, Šilar R, Pičmanová M, Pátek M, Kalinowski J (2012) Transcriptional regulation of the operon encoding stress-responsive ECF sigma factor SigH and its anti-sigma factor RshA, and control of its regulatory network in Corynebacterium glutamicum. BMC Genomics 13:445. https://doi.org/10.1186/1471-2164-13-445
Bussmann M, Emer D, Hasenbein S, Degraf S, Eikmanns BJ, Bott M (2009) Transcriptional control of the succinate dehydrogenase operon sdhCAB of Corynebacterium glutamicum by the cAMP-dependent regulator GlxR and the LuxR-type regulator RamA. J Biotechnol 143:173–182
Cha PH et al (2010) Characterization of an adenylate cyclase gene (cyaB) deletion mutant of Corynebacterium glutamicum ATCC 13032. Appl Microbiol Biotechnol 85:1061–1068
Cho HY, Lee SG, Hyeon JE, Han SO (2010) Identification and characterization of a transcriptional regulator, SucR, that influences sucCD transcription in Corynebacterium glutamicum. Biochem Biophys Res Commun 401:300–305
Claes WA, Pühler A, Kalinowski J (2002) Identification of two prpDBC gene clusters in Corynebacterium glutamicum and their involvement in propionate degradation via the 2-methylcitrate cycle. J Bacteriol 184:2728–2739
Cocaign-Bousquet M, Monnet C, Lindley ND (1993) Batch kinetics of Corynebacterium glutamicum during growth on various carbon substrates: use of substrate mixtures to localise metabolic bottlenecks. Appl Microbiol Biotechnol 40:526–530
Costa-Riu N, Maier E, Burkovski A, Krämer R, Lottspeich F, Benz R (2003) Identification of an anion-specific channel in the cell wall of the Gram-positive bacterium Corynebacterium glutamicum. Mol Microbiol 50:1295–1308
Cramer A, Eikmanns BJ (2007) RamA, the transcriptional regulator of acetate metabolism in Corynebacterium glutamicum, is subject to negative autoregulation. J Mol Microbiol Biotechnol 12:51–59
Cramer A, Gerstmeir R, Schaffer S, Bott M, Eikmanns BJ (2006) Identification of RamA, a novel LuxR-type transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J Bacteriol 188:2554–2567
Deutscher J, Küster E, Bergstedt U, Charrier V, Hillen W (1995) Protein kinase-dependent HPr/CcpA interaction links glycolytic activity to carbon catabolite repression in gram-positive bacteria. Mol Microbiol 15:1049–1053
Dietrich C, Nato A, Bost B, Le Maréchal P, Guyonvarch A (2009) Regulation of ldh expression during biotin-limited growth of Corynebacterium glutamicum. Microbiology 155:1360–1375
Dominguez H, Cocaign-Bousquet M, Lindley ND (1997) Simultaneous consumption of glucose and fructose from sugar mixtures during batch growth of Corynebacterium glultamicum. Appl Microbiol Biotechnol 47:600–603
Ehira S, Shirai T, Teramoto H, Inui M, Yukawa H (2008) Group 2 sigma factor SigB of Corynebacterium glutamicum positively regulates glucose metabolism under conditions of oxygen deprivation. Appl Environ Microbiol 74:5146–5152
Ehira S, Teramoto H, Inui M, Yukawa H (2009) Regulation of Corynebacterium glutamicum heat shock response by the extracytoplasmic-function sigma factor SigH and transcriptional regulators HspR and HrcA. J Bacteriol 191:2964–2972
Eikmanns BJ, Rittmann D, Sahm H (1995) Cloning, sequence analysis, expression, and inactivation of the Corynebacterium glutamicum icd gene encoding isocitrate dehydrogenase and biochemical characterization of the enzyme. J Bacteriol 177:774–782
Engels V, Wendisch VF (2007) The DeoR-Type regulator SugR represses expression of ptsG in Corynebacterium glutamicum. J Bacteriol 189:2955–2966. https://doi.org/10.1128/jb.01596-06
Engels V, Lindner SN, Wendisch VF (2008) The global repressor SugR controls expression of genes of glycolysis and of the L-lactate dehydrogenase LdhA in Corynebacterium glutamicum. J Bacteriol 190:8033–8044
Fisher SH, Wray LV Jr (2008) Bacillus subtilis glutamine synthetase regulates its own synthesis by acting as a chaperone to stabilize GlnR-DNA complexes. Proc Natl Acad Sci U S A 105:1014–1019. https://doi.org/10.1073/pnas.0709949105
Fisher SH, Wray LV Jr (2009) Novel trans-acting Bacillus subtilis glnA mutations that derepress glnRA expression. J Bacteriol 191:2485–2492. https://doi.org/10.1128/JB.01734-08
Frunzke J, Engels V, Hasenbein S, Gatgens C, Bott M (2008) Co-ordinated regulation of gluconate catabolism and glucose uptake in Corynebacterium glutamicum by two functionally equivalent transcriptional regulators, GntR1 and GntR2. Mol Microbiol 67:305–322
Gaigalat L et al (2007) The DeoR-type transcriptional regulator SugR acts as a repressor for genes encoding the phosphoenolpyruvate:sugar phosphotransferase system (PTS) in Corynebacterium glutamicum. BMC Mol Biol 8:104
Gao Y-G et al (2008) Structural and functional characterization of the LldR from Corynebacterium glutamicum: a transcriptional repressor involved in L-lactate and sugar utilization. Nucleic Acids Res 36:7110–7123. https://doi.org/10.1093/nar/gkn827
Georgi T, Engels V, Wendisch VF (2008) Regulation of L-lactate utilization by the FadR-type regulator LldR of Corynebacterium glutamicum. J Bacteriol 190:963–971
Gerstmeir R, Cramer A, Dangel P, Schaffer S, Eikmanns BJ (2004) RamB, a novel transcriptional regulator of genes involved in acetate metabolism of Corynebacterium glutamicum. J Bacteriol 186:2798–2809
Gourdon P, Baucher MF, Lindley ND, Guyonvarch A (2000) Cloning of the malic enzyme gene from Corynebacterium glutamicum and role of the enzyme in lactate metabolism. Appl Environ Microbiol 66:2981–2987
Han SO, Inui M, Yukawa H (2007) Expression of Corynebacterium glutamicum glycolytic genes varies with carbon source and growth phase. Microbiology 153:2190–2202
Han SO, Inui M, Yukawa H (2008) Effect of carbon source availability and growth phase on expression of Corynebacterium glutamicum genes involved in the tricarboxylic acid cycle and glyoxylate bypass. Microbiology 154:3073–3083
Han G, Hu X, Qin T, Li Y, Wang X (2016) Metabolic engineering of Corynebacterium glutamicum ATCC13032 to produce S-adenosyl-L-methionine. Enzym Microb Technol 83:14–21. https://doi.org/10.1016/j.enzmictec.2015.11.001
Hasselt K, Rankl S, Worsch S, Burkovski A (2011) Adaptation of AmtR-controlled gene expression by modulation of AmtR binding activity in Corynebacterium glutamicum. J Biotechnol 154:156–162. https://doi.org/10.1016/j.jbiotec.2010.09.930
Hayashi M et al (2002) Transcriptome analysis of acetate metabolism in Corynebacterium glutamicum using a newly developed metabolic array. Biosci Biotechnol Biochem 66:1337–1344
Henrich A, Kuhlmann N, Eck AW, Krämer R, Seibold GM (2013) Maltose uptake by the novel ABC transport system MusEFGK2I causes increased expression of ptsG in Corynebacterium glutamicum. J Bacteriol 195:2573–2584. https://doi.org/10.1128/JB.01629-12
Hoffelder M, Raasch K, van Ooyen J, Eggeling L (2010) The E2 domain of OdhA of Corynebacterium glutamicum has succinyltransferase activity dependent on lipoyl residues of the acetyltransferase AceF. J Bacteriol 192:5203–5211. https://doi.org/10.1128/JB.00597-10
Ikeda M, Nakagawa S (2003) The Corynebacterium glutamicum genome: features and impacts on biotechnological processes. Appl Microbiol Biotechnol 62:99–109
Ikeda M, Mizuno Y, Awane S, Hayashi M, Mitsuhashi S, Takeno S (2011) Identification and application of a different glucose uptake system that functions as an alternative to the phosphotransferase system in Corynebacterium glutamicum. Appl Microbiol Biotechnol 90:1443–1451. https://doi.org/10.1007/s00253-011-3210-x
Inui M, Murakami S, Okino S, Kawaguchi H, Vertès AA, Yukawa H (2004) Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J Mol Microbiol Biotechnol 7:182–196
Inui M et al (2007) Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology 153:2491–2504. https://doi.org/10.1099/mic.0.2006/005587-0
Ishige T, Krause M, Bott M, Wendisch VF, Sahm H (2003) The phosphate starvation stimulon of Corynebacterium glutamicum determined by DNA microarray analyses. J Bacteriol 185:4519–4529
Jakoby M, Nolden L, Meier-Wagner J, Krämer R, Burkovski A (2000) AmtR, a global repressor in the nitrogen regulation system of Corynebacterium glutamicum. Mol Microbiol 37:964–977
Jones BE, Dossonnet V, Küster E, Hillen W, Deutscher J, Klevit RE (1997) Binding of the catabolite repressor protein CcpA to its DNA target is regulated by phosphorylation of its corepressor HPr. J Biol Chem 272:26530–26535. https://doi.org/10.1074/jbc.272.42.26530
Jungwirth B et al (2013) High-resolution detection of DNA binding sites of the global transcriptional regulator GlxR in Corynebacterium glutamicum. Microbiology 159:12–22. https://doi.org/10.1099/mic.0.062059-0
Kalinowski J et al (2003) The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol 104:5–25
Kawaguchi H, Vèrtes AA, Okino S, Inui M, Yukawa H (2006) Engineering of a xylose metabolic pathway in Corynebacterium glutamicum. Appl Environ Microbiol 72:3418–3428
Kawaguchi H, Sasaki M, Vertès AA, Inui M, Yukawa H (2008) Engineering of an L: -arabinose metabolic pathway in Corynebacterium glutamicum. Appl Microbiol Biotechnol 77:1053–1062
Kertesz MA (2000) Riding the sulfur cycle–metabolism of sulfonates and sulfate esters in Gram-negative bacteria. FEMS Microbiol Rev 24:135–175. https://doi.org/10.1016/S0168-6445(99)00033-9
Kim HJ, Kim TH, Kim Y, Lee HS (2004) Identification and characterization of glxR, a gene involved in regulation of glyoxylate bypass in Corynebacterium glutamicum. J Bacteriol 186:3453–3460
Klaffl S, Brocker M, Kalinowski J, Eikmanns BJ, Bott M (2013) Complex regulation of the phosphoenolpyruvate carboxykinase gene pck and characterization of its GntR-type regulator IolR as a repressor of myo-inositol utilization genes in Corynebacterium glutamicum. J Bacteriol 195:4283–4296. https://doi.org/10.1128/JB.00265-13
Kočan M, Schaffer S, Ishige T, Sorger-Herrmann U, Wendisch VF, Bott M (2006) Two-component systems of Corynebacterium glutamicum: deletion analysis and involvement of the PhoS-PhoR system in the phosphate starvation response. J Bacteriol 188:724–732
Koch DJ et al (2005a) The transcriptional regulator SsuR activates expression of the Corynebacterium glutamicum sulphonate utilization genes in the absence of sulphate. Mol Microbiol 58:480–494
Koch DJ, Rückert C, Rey DA, Mix A, Pühler A, Kalinowski J (2005b) Role of the ssu and seu genes of Corynebacterium glutamicum ATCC 13032 in utilization of sulfonates and sulfonate esters as sulfur sources. Appl Environ Microbiol 71:6104–6114
Kohl TA, Tauch A (2009) The GlxR regulon of the amino acid producer Corynebacterium glutamicum: detection of the corynebacterial core regulon and integration into the transcriptional regulatory network model. J Biotechnol 143:239–246
Kohl TA, Baumbach J, Jungwirth B, Pühler A, Tauch A (2008) The GlxR regulon of the amino acid producer Corynebacterium glutamicum: in silico and in vitro detection of DNA binding sites of a global transcription regulator. J Biotechnol 135:340–350
Kotrba P, Inui M, Yukawa H (2001) Bacterial phosphotransferase system (PTS) in carbohydrate uptake and control of carbon metabolism. J Biosci Bioeng 92:502–517
Kotrba P, Inui M, Yukawa H (2003) A single V317A or V317M substitution in Enzyme II of a newly identified beta-glucoside phosphotransferase and utilization system of Corynebacterium glutamicum R extends its specificity towards cellobiose. Microbiology 149:1569–1580
Kotrbova-Kozak A, Kotrba P, Inui M, Sajdok J, Yukawa H (2007) Transcriptionally regulated adhA gene encodes alcohol dehydrogenase required for ethanol and n-propanol utilization in Corynebacterium glutamicum R. Appl Microbiol Biotechnol 76:1347–1356
Krause JP, Polen T, Youn JW, Emer D, Eikmanns BJ, Wendisch VF (2012) Regulation of the malic enzyme gene malE by the transcriptional regulator MalR in Corynebacterium glutamicum. J Biotechnol 159:204–215
Krings E et al (2006) Characterization of myo-inositol utilization by Corynebacterium glutamicum: the stimulon, identification of transporters, and influence on L-lysine formation. J Bacteriol 188:8054–8061
Kronemeyer W, Peekhaus N, Krämer R, Sahm H, Eggeling L (1995) Structure of the gluABCD cluster encoding the glutamate uptake system of Corynebacterium glutamicum. J Bacteriol 177:1152–1158
Krug A, Wendisch VF, Bott M (2005) Identification of AcnR, a TetR-type repressor of the aconitase gene acn in Corynebacterium glutamicum. J Biol Chem 280:585–595
Laslo T et al (2012) Arabitol metabolism of Corynebacterium glutamicum and its regulation by AtlR. J Bacteriol 194:941–955. https://doi.org/10.1128/JB.06064-11
Lindner SN, Vidaurre D, Willbold S, Schoberth SM, Wendisch VF (2007) NCgl2620 encodes a class II polyphosphate kinase in Corynebacterium glutamicum. Appl Environ Microbiol 73:5026–5033
Lindner SN, Knebel S, Wesseling H, Schoberth SM, Wendisch VF (2009) Exopolyphosphatases PPX1 and PPX2 from Corynebacterium glutamicum. Appl Environ Microbiol 75:3161–3170
Lindner SN, Seibold GM, Henrich A, Krämer R, Wendisch VF (2011) Phosphotransferase system-independent glucose utilization in Corynebacterium glutamicum by inositol permeases and glucokinases. Appl Environ Microbiol 77:3571–3581. https://doi.org/10.1128/AEM.02713-10
Mahr R, Gätgens C, Gätgens J, Polen T, Kalinowski J, Frunzke J (2015) Biosensor-driven adaptive laboratory evolution of l-valine production in Corynebacterium glutamicum. Metab Eng 32:184–194. https://doi.org/10.1016/j.ymben.2015.09.017
Merkens H, Beckers G, Wirtz A, Burkovski A (2005) Vanillate metabolism in Corynebacterium glutamicum. Curr Microbiol 51:59–65
Merrick MJ, Edwards RA (1995) Nitrogen control in bacteria. Microbiol Rev 59:604–622
Morabbi Heravi K, Lange J, Watzlawick H, Kalinowski J, Altenbuchner J (2015) Transcriptional regulation of the vanillate utilization genes (vanABK operon) of Corynebacterium glutamicum by VanR, a PadR-like repressor. J Bacteriol 197:959–972. https://doi.org/10.1128/JB.02431-14
Muffler A et al (2002) Genome-wide transcription profiling of Corynebacterium glutamicum after heat shock and during growth on acetate and glucose. J Biotechnol 98:255–268
Mustafi N, Grünberger A, Kohlheyer D, Bott M, Frunzke J (2012) The development and application of a single-cell biosensor for the detection of L-methionine and branched-chain amino acids. Metab Eng 14:449–457. https://doi.org/10.1016/j.ymben.2012.02.002
Nentwich SS et al (2009) Characterization of the LacI-type transcriptional repressor RbsR controlling ribose transport in Corynebacterium glutamicum ATCC 13032. Microbiology 155:150–164
Neshat A, Mentz A, Rückert C, Kalinowski J (2014) Transcriptome sequencing revealed the transcriptional organization at ribosome-mediated attenuation sites in Corynebacterium glutamicum and identified a novel attenuator involved in aromatic amino acid biosynthesis. J Biotechnol 190:55–63. https://doi.org/10.1016/j.jbiotec.2014.05.033
Nešvera J, Pátek M (2011) Tools for genetic manipulations in Corynebacterium glutamicum and their applications. Appl Microbiol Biotechnol 90:1641–1654
Netzer R, Peters-Wendisch P, Eggeling L, Sahm H (2004) Cometabolism of a nongrowth substrate: L-serine utilization by Corynebacterium glutamicum. Appl Environ Microbiol 70:7148–7155
Ninfa AJ, Jiang P, Atkinson MR, Peliska JA (2000) Integration of antagonistic signals in the regulation of nitrogen assimilation in Escherichia coli. Curr Top Cell Regul 36:31–75
Nolden L, Ngouoto-Nkili CE, Bendt AK, Krämer R, Burkovski A (2001) Sensing nitrogen limitation in Corynebacterium glutamicum: the role of glnK and glnD. Mol Microbiol 42:1281–1295
Omumasaba CA, Okai N, Inui M, Yukawa H (2004) Corynebacterium glutamicum glyceraldehyde-3-phosphate dehydrogenase isoforms with opposite, ATP-dependent regulation. J Mol Microbiol Biotechnol 8:91–103
Palanca C, Rubio V (2016) Structure of AmtR, the global nitrogen regulator of Corynebacterium glutamicum, in free and DNA-bound forms. FEBS J 283:1039–1059. https://doi.org/10.1111/febs.13643
Panhorst M, Sorger-Herrmann U, Wendisch VF (2011) The pstSCAB operon for phosphate uptake is regulated by the global regulator GlxR in Corynebacterium glutamicum. J Biotechnol 154:149–155. https://doi.org/10.1016/j.jbiotec.2010.07.015
Parche S, Burkovski A, Sprenger GA, Weil B, Krämer R, Titgemeyer F (2001) Corynebacterium glutamicum: a dissection of the PTS. J Mol Microbiol Biotechnol 3:423–428
Park SY, Moon MW, Subhadra B, Lee JK (2010) Functional characterization of the glxR deletion mutant of Corynebacterium glutamicum ATCC 13032: involvement of GlxR in acetate metabolism and carbon catabolite repression. FEMS Microbiol Lett 304:107–115
Pátek M, Nešvera J (2011) Sigma factors and promoters in Corynebacterium glutamicum. J Biotechnol 154:101–113
Peng X, Okai N, Vertès A, Inatomi K, Inui M, Yukawa H (2011) Characterization of the mannitol catabolic operon of Corynebacterium glutamicum. Appl Microbiol Biotechnol 91:1375–1387. https://doi.org/10.1007/s00253-011-3352-x
Pérez-García F, Jorge JMP, Dreyszas A, Risse JM, Wendisch VF (2018) Efficient production of the dicarboxylic acid glutarate by Corynebacterium glutamicum via a novel synthetic pathway. Front Microbiol 9:2589. https://doi.org/10.3389/fmicb.2018.02589
Plassmeier P, Persicke M, Pühler A, Sterthoff C, Rückert C, Kalinowski J (2012) Molecular characterization of PrpR, the transcriptional activator of propionate catabolism in Corynebacterium glutamicum. J Biotechnol 159:1–11
Postma PW, Lengeler JW, Jacobson GR (1993) Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Mol Biol Rev 57:543–594
Reddy P, Kamireddi M (1998) Modulation of Escherichia coli adenylyl cyclase activity by catalytic-site mutants of protein IIAGlc of the phosphoenolpyruvate:sugar phosphotransferase system. J Bacteriol 180:732–736
Rehm N et al (2010) L-Glutamine as a nitrogen source for Corynebacterium glutamicum: derepression of the AmtR regulon and implications for nitrogen sensing. Microbiology 156:3180–3193. https://doi.org/10.1099/mic.0.040667-0
Rey DA, Pühler A, Kalinowski J (2003) The putative transcriptional repressor McbR, member of the TetR-family, is involved in the regulation of the metabolic network directing the synthesis of sulfur containing amino acids in Corynebacterium glutamicum. J Biotechnol 103:51–65
Rey DA et al (2005) The McbR repressor modulated by the effector substance S-adenosylhomocysteine controls directly the transcription of a regulon involved in sulphur metabolism of Corynebacterium glutamicum ATCC 13032. Mol Microbiol 56:871–887
Rittmann D, Sorger-Herrmann U, Wendisch VF (2005) Phosphate starvation-inducible gene ushA encodes a 5' nucleotidase required for growth of Corynebacterium glutamicum on media with nucleotides as the phosphorus source. Appl Environ Microbiol 71:4339–4344
Rückert C, Pühler A, Kalinowski J (2003) Genome-wide analysis of the L-methionine biosynthetic pathway in Corynebacterium glutamicum by targeted gene deletion and homologous complementation. J Biotechnol 104:213–228
Rückert C et al (2005) Functional genomics and expression analysis of the Corynebacterium glutamicum fpr2-cysIXHDNYZ gene cluster involved in assimilatory sulphate reduction. BMC Genomics 6:121
Rückert C, Milse J, Albersmeier A, Koch DJ, Pühler A, Kalinowski J (2008) The dual transcriptional regulator CysR in Corynebacterium glutamicum ATCC 13032 controls a subset of genes of the McbR regulon in response to the availability of sulphide acceptor molecules. BMC Genomics 9:483
Santos-Beneit F (2015) The Pho regulon: a huge regulatory network in bacteria. Front Microbiol 6:402. https://doi.org/10.3389/fmicb.2015.00402
Schaaf S, Bott M (2007) Target genes and DNA-binding sites of the response regulator PhoR from Corynebacterium glutamicum. J Bacteriol 189:5002–5011
Schröder J, Tauch A (2010) Transcriptional regulation of gene expression in Corynebacterium glutamicum: the role of global, master and local regulators in the modular and hierarchical gene regulatory network. FEMS Microbiol Rev 34:685–737
Schulte J, Baumgart M, Bott M (2017a) Development of a single-cell GlxR-based cAMP biosensor for Corynebacterium glutamicum. J Biotechnol 258:33–40. https://doi.org/10.1016/j.jbiotec.2017.07.004
Schulte J, Baumgart M, Bott M (2017b) Identification of the cAMP phosphodiesterase CpdA as novel key player in cAMP-dependent regulation in Corynebacterium glutamicum. Mol Microbiol 103:534–552. https://doi.org/10.1111/mmi.13574
Schumacher MA, Chinnam NB, Cuthbert B, Tonthat NK, Whitfill T (2015) Structures of regulatory machinery reveal novel molecular mechanisms controlling B. subtilis nitrogen homeostasis. Genes Dev 29:451–464. https://doi.org/10.1101/gad.254714.114
Schwinde JW, Thum-Schmitz N, Eikmanns BJ, Sahm H (1993) Transcriptional analysis of the gap-pgk-tpi-ppc gene cluster of Corynebacterium glutamicum. J Bacteriol 175:3905–3908
Sorger-Herrmann U, Taniguchi H, Wendisch VF (2015) Regulation of the pstSCAB operon in Corynebacterium glutamicum by the regulator of acetate metabolism RamB. BMC Microbiol 15:113. https://doi.org/10.1186/s12866-015-0437-1
Stansen C, Uy D, Delaunay S, Eggeling L, Goergen JL, Wendisch VF (2005) Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol 71:5920–5928
Strösser J, Ludke A, Schaffer S, Kramer R, Burkovski A (2004) Regulation of GlnK activity: modification, membrane sequestration and proteolysis as regulatory principles in the network of nitrogen control in Corynebacterium glutamicum. Mol Microbiol 54:132–147
Subhadra B, Lee JK (2013) Elucidation of the regulation of ethanol catabolic genes and ptsG using a glxR and adenylate cyclase gene (cyaB) deletion mutants of Corynebacterium glutamicum ATCC 13032. J Microbiol Biotechnol 23:1683–1690
Suda M, Teramoto H, Imamiya T, Inui M, Yukawa H (2008) Transcriptional regulation of Corynebacterium glutamicum methionine biosynthesis genes in response to methionine supplementation under oxygen deprivation. Appl Microbiol Biotechnol 81:505–513
Takahashi H, Inada T, Postma P, Aiba H (1998) CRP down-regulates adenylate cyclase activity by reducing the level of phosphorylated IIAGlc, the glucose-specific phosphotransferase protein, in Escherichia coli. Mol Gen Genet 259:317–326
Tanaka Y, Okai N, Teramoto H, Inui M, Yukawa H (2008a) Regulation of the expression of phosphoenolpyruvate: carbohydrate phosphotransferase system (PTS) genes in Corynebacterium glutamicum R. Microbiology 154:264–274
Tanaka Y, Teramoto H, Inui M, Yukawa H (2008b) Regulation of expression of general components of the phosphoenolpyruvate: carbohydrate phosphotransferase system (PTS) by the global regulator SugR in Corynebacterium glutamicum. Appl Microbiol Biotechnol 78:309–318
Tanaka Y, Teramoto H, Inui M, Yukawa H (2009) Identification of a second beta-glucoside phosphoenolpyruvate: carbohydrate phosphotransferase system in Corynebacterium glutamicum R. Microbiology 155:3652–3660
Tanaka Y, Ehira S, Teramoto H, Inui M, Yukawa H (2012) Coordinated regulation of gnd, which encodes 6-phosphogluconate dehydrogenase, by the two transcriptional regulators GntR1 and RamA in Corynebacterium glutamicum. J Bacteriol 194:6527–6536. https://doi.org/10.1128/JB.01635-12
Tanaka Y, Takemoto N, Ito T, Teramoto H, Yukawa H, Inui M (2014) Genome-wide analysis of the role of global transcriptional regulator GntR1 in Corynebacterium glutamicum. J Bacteriol 196:3249–3258. https://doi.org/10.1128/JB.01860-14
Teramoto H, Inui M, Yukawa H (2009) Regulation of expression of genes involved in quinate and shikimate utilization in Corynebacterium glutamicum. Appl Environ Microbiol 75:3461–3468
Toyoda K, Teramoto H, Inui M, Yukawa H (2008) Expression of the gapA gene encoding glyceraldehyde-3-phosphate dehydrogenase of Corynebacterium glutamicum is regulated by the global regulator SugR. Appl Microbiol Biotechnol 81:291–301
Toyoda K, Teramoto H, Inui M, Yukawa H (2009a) Involvement of the LuxR-type transcriptional regulator, RamA, in regulation of expression of the gapA gene encoding glyceraldehyde-3-phosphate dehydrogenase of Corynebacterium glutamicum. J Bacteriol 191:968–977
Toyoda K, Teramoto H, Inui M, Yukawa H (2009b) The ldhA gene, encoding fermentative L-lactate dehydrogenase of Corynebacterium glutamicum, is under the control of positive feedback regulation mediated by LldR. J Bacteriol 191:4251–4258. https://doi.org/10.1128/jb.00303-09
Toyoda K, Teramoto H, Inui M, Yukawa H (2009c) Molecular mechanism of SugR-mediated sugar-dependent expression of the ldhA gene encoding L-lactate dehydrogenase in Corynebacterium glutamicum. Appl Microbiol Biotechnol 83:315–327
Toyoda K, Teramoto H, Inui M, Yukawa H (2011) Genome-wide identification of in vivo binding sites of GlxR, a cyclic AMP receptor protein-type regulator in Corynebacterium glutamicum. J Bacteriol 193:4123–4133. https://doi.org/10.1128/JB.00384-11
Toyoda K, Teramoto H, Yukawa H, Inui M (2015) Expanding the regulatory network governed by the extracytoplasmic function sigma factor σH in Corynebacterium glutamicum. J Bacteriol 197:483–496. https://doi.org/10.1128/JB.02248-14
van Ooyen J, Emer D, Bussmann M, Bott M, Eikmanns BJ, Eggeling L (2011) Citrate synthase in Corynebacterium glutamicum is encoded by two gltA transcripts which are controlled by RamA, RamB, and GlxR. J Biotechnol 154:140–148
Wendisch VF, Bott M (2008) Phosphorus metabolism and its regulation. In: Burkovski A (ed) Corynebacteria: genomics and molecular biology. Caister Academic Press, pp 203–216
Wendisch VF, Spies M, Reinscheid DJ, Schnicke S, Sahm H, Eikmanns BJ (1997) Regulation of acetate metabolism in Corynebacterium glutamicum: transcriptional control of the isocitrate lyase and malate synthase genes. Arch Microbiol 168:262–269
Wendisch VF, de Graaf AA, Sahm H, Eikmanns BJ (2000) Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J Bacteriol 182:3088–3096
Wennerhold J, Krug A, Bott M (2005) The AraC-type regulator RipA represses aconitase and other iron proteins from Corynebacterium under iron limitation and is itself repressed by DtxR. J Biol Chem 280:40500–40508
Wray LV Jr, Zalieckas JM, Fisher SH (2001) Bacillus subtilis glutamine synthetase controls gene expression through a protein-protein interaction with transcription factor TnrA. Cell 107:427–435
Xu J, Zhang J, Liu D, Zhang W (2016) Increased glucose utilization and cell growth of Corynebacterium glutamicum by modifying the glucose-specific phosphotransferase system (PTS(Glc)) genes. Can J Microbiol 62:983–992. https://doi.org/10.1139/cjm-2016-0027
Yoshida K, Yamaguchi H, Kinehara M, Ohki YH, Nakaura Y, Fujita Y (2003) Identification of additional TnrA-regulated genes of Bacillus subtilis associated with a TnrA box. Mol Microbiol 49:157–165
Yukawa H et al (2007) Comparative analysis of the Corynebacterium glutamicum group and complete genome sequence of strain R. Microbiology 153:1042–1058
Zhang X, Xu G, Shi J, Xu Z (2018) Integration of ARTP mutagenesis with biosensor-mediated high-throughput screening to improve L-serine yield in Corynebacterium glutamicum. Appl Microbiol Biotechnol 102:5939–5951. https://doi.org/10.1007/s00253-018-9025-2
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Toyoda, K., Inui, M. (2020). Global Transcriptional Regulators Involved in Carbon, Nitrogen, Phosphorus, and Sulfur Metabolisms in Corynebacterium glutamicum . In: Inui, M., Toyoda, K. (eds) Corynebacterium glutamicum. Microbiology Monographs, vol 23. Springer, Cham. https://doi.org/10.1007/978-3-030-39267-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-39267-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-39266-6
Online ISBN: 978-3-030-39267-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)