
Abstract
Cancer develops in multicellular organisms from cells that ignore the rules of cooperation and escape the mechanisms of anti-cancer surveillance. Tumorigenesis is jointly encountered by the host and microbiota, a vast collection of microorganisms that live on the external and internal epithelial surfaces of the body. The largest community of human microbiota resides in the gastrointestinal tract where commensal, symbiotic and pathogenic microorganisms interact with the intestinal barrier and gut mucosal lymphoid tissue, creating a tumor microenvironment in which cancer cells thrive or perish. Aberrant composition and function of the gut microbiota (dysbiosis) has been associated with tumorigenesis by inducing inflammation, promoting cell growth and proliferation, weakening immunosurveillance, and altering food and drug metabolism or other biochemical functions of the host. However, recent research has also identified several mechanisms through which gut microbiota support the host in the fight against cancer. These mechanisms include the use of antigenic mimicry, biotransformation of chemotherapeutic agents, and other mechanisms to boost anti-cancer immune responses and improve the efficacy of cancer immunotherapy. Further research in this rapidly advancing field is expected to identify additional microbial metabolites with tumor suppressing properties, map the complex interactions of host-microbe ‘transkingdom network’ with cancer cells, and elucidate cellular and molecular pathways underlying the impact of specific intestinal microbial configurations on immune checkpoint inhibitor therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Holobiont
- Immune checkpoint inhibitors
- Dysbiosis
- Biofilm
- Butyrate paradox
- Intestinal barrier
- Transkingdom network
- Fecal microbiota transplantation
1 Introduction
Multicellular life first evolved about 1 billion years ago (Maynard Smith and Szathmáry 1995). Multicellularity requires cooperation among cells to ensure division of labor, allocation of resources and maintenance of replication with effective mechanisms to control cell proliferation and suppression of cheating (Aktipis et al. 2015). Multicellular organisms co-evolved with their microbial environment from the very beginning. Microorganisms living inside and out of the multicellular host are termed microbiota, and the totality of their genetic information is termed microbiome, while the host-microbiota ecosystem is often referred to as ‘super-organism’ or ‘holobiont’ (Huitzil et al. 2018; Schwabe and Jobin 2013). Microbiota are involved in diverse interactions with each other while contributing key functions to host physiology, aptly described within the context of a ‘transkingdom network’ (Greer et al. 2016). The largest community of human microbiota resides in the gastrointestinal tract and contains over 1000 different bacterial species (Human Microbiome Project 2012). Gut microbiota support the host by maximizing dietary energy extraction, generating essential metabolites, assisting the biotransformation of xenobiotics, shaping innate and adaptive immunity, and protecting from the invasion of pathogenic microorganisms (Backhed et al. 2005; Lee and Hase 2014; Nicholson et al. 2012). Similarly, host-to-microbe effects related to nutrition, medications and other lifestyle or health factors are critical for the gut microbiota (Fischbach and Sonnenburg 2011; Foster et al. 2017; Zmora et al. 2019). Perturbations of the host-microbial relationship result in dysbiosis, which is defined as a loss of balance in microbiota composition and function and potentially results in disease phenotypes (Llorente and Schnabl 2015).
Cancer is an inherent feature of multicellular existence that develops when multicellular cooperation breaks down and cells reject cooperation to favor self-autonomy, evasion of growth control and programmed cell death, replicative immortality, and a limitless ability to invade and metastasize (Hanahan and Weinberg 2000, 2011). This process involves abandonment of multicellularity and a reverse evolution of cancer cells back to a unicellular state (Chen et al. 2015). Cancer-like phenomena characterized by abnormal cell proliferation and neoplastic growth have been observed in all seven branches of multicellular life (Aktipis et al. 2015). Cancer cells become increasingly robust during their emergence and induce reciprocal changes in the host and microbiota (Aktipis and Nesse 2013). Uncontrolled cancer growth eventually leads to demise of the host, making it a common existential threat for the entire holobiont. However, because natural selection primarily works at species level (Maynard Smith 1998), each individual microbial strain and the host itself are distinct entities in this conflict with potentially divergent selective pressures and it may be wrong to assume they are acting with common interests (Foster et al. 2017). Thus, while coexistence of host and microbiota may normally serve the entire holobiont’s homeostasis, interactions between cancer and microbiota do not necessarily benefit the host. In fact, the range of host-microbial relationship extends from mutualistic (mutually beneficial) to parasitic (harmful to the host and beneficial to the parasite) (Wasielewski et al. 2016). A better understanding of the interplay between host, cancer and microbiota is therefore critical to take advantage of between-species cooperation and of the potential gains from modulating this relationship (Fig. 5.1). This review will focus on some recent insights into the complex role of gut microbiota as an essential partner of the host in facing the initiation, progression and prognosis of cancer.

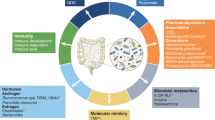
Host—microbiota—cancer interplay
Schematic illustration of key interactions between host, microbiota and cancer, each multicellular community manifesting distinct behavior (shown in parentheses) in their triangular relationship. Genetic and environmental factors (e.g., diet and medications) may determine the success of host-mediated control over microbiota and tumorigenesis. Innate and adaptive immunity is essential in both cases, while additional structural (e.g., intestinal epithelial barrier) and functional elements (e.g., antimicrobial peptides, not shown), regulate the microbiota and sustain host homeostasis. Cancer cells seek defection with an aggressive agenda for limitless growth and proliferation at the host’s expense, including coercion (‘enslavement’) of host cells into metabolic reprogramming (a.k.a. reverse Warburg effect in cancer-associated fibroblasts) and the use of immune checkpoints to stifle anti-cancer immune responses. Gut microbiota and cancer have an opportunistic relationship, since commensal microorganisms as individual species or in consortia respond to various selection pressures that may fortuitously assist cancer cells and act therefore not in the host’s interest. Thus, providing unconventional nutrients to cancer cells, creating biofilms that impair the gut barrier and induce inflammation, or promoting genotoxicity in host cells may tilt the balance toward tumorigenesis in the gastrointestinal tract and beyond. However, eubiotic gut microbiota have also been shown to strengthen anti-cancer immunosurveillance through a variety of mechanisms such as synthesis of tumor suppressor metabolites (e.g., butyrate), cancer cell recognition via antigenic mimicry, or enhancement of anti-cancer chemotherapy and immunotherapy. Please see details in the main text.
2 Tumor Microenvironment: A Collective Affair
Tumor microenvironment includes several types of cells that modulate the growth, proliferation and dissemination of cancer cells (Leong et al. 2018). The holobiont has developed powerful mechanisms in this milieu to suppress tumorigenesis, which may explain why—against the mathematical odds—clinically apparent cancer is relatively rare (Aktipis and Nesse 2013). Cancer cells become members of a ‘social microenvironment’ that includes cellular elements of both the host immunosurveillance and commensal microbiota, embedded in a physicochemical microenvironment and threatened by adversities such as poor nutrient availability, hypoxia, low pH and redox stress (Sun et al. 2018). Local expansion and metastatic spreading of cancer cells occur through a web of key permissive and controlling factors within this microenvironment and has been the topic of several excellent reviews in recent years (Leong et al. 2018; Morgillo et al. 2018; Quail and Joyce 2013; Quante et al. 2013; Swartz et al. 2012).
2.1 Gut Microbiota
Microbial gene richness is a key feature of healthy gut microbiome, while diminishing bacterial diversity is often associated with disease (Le Chatelier et al. 2013). Metagenomic analysis of the feces by culture-independent methods found that most intestinal bacteria belong to 6 phyla: Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Verrucomicrobia, and Fusobacteria (Human Microbiome Project 2012). Similar to other living systems of multicellular aggregates, gut microbiota members interact through cooperation and competition. Cooperative activities between microbial species include the formation of biofilms, fermentation of complex substrates, and the use of supply chains such as exchange of metabolites and cross-feeding, whereby the metabolic end product of one strain becomes the nutrient for a different strain (Muller et al. 2018; Plichta et al. 2016). At the same time, microbial strains use diverse mechanisms to compete with each other for resources and adhesion sites, or produce antimicrobial substances to gain advantage (Foster et al. 2017). Gut microbial ecosystems analyzed in a large number of individuals fall into three major ‘enterotypes’ distinguished by variable microbial abundance and molecular functions (Arumugam et al. 2011). Interestingly, whereas microbial species composition differs from individual to individual, the overall distribution of expressed gene functions remains relatively constant (Plichta et al. 2016). A remarkable process regulating coexistence of microbial species in the gut is complementary silencing of genes that encode overlapping functions such as anaerobic fermentation, biosynthesis of short-chain fatty acids, and starch degradation (Plichta et al. 2016). Local stability analysis of microbiota networks indicates the importance of limiting positive feedbacks and weakening ecological interactions (Coyte et al. 2015). Cooperation can create dependency and increases the probability that perturbations rapidly spread and destabilize the system. Accordingly, high diversity is key to microbiota stability, characterized by competitiveness and weak interactions (Coyte et al. 2015).
2.2 Intestinal Barrier
Gut microbiota and the host are physically separated by a complex intestinal barrier (Goto and Kiyono 2012). The epithelial component is a single sheet of columnar cells tightly connected by intercellular complexes and covered by a thick mucous layer secreted by goblet cells (Marchiando et al. 2010). The endothelial or gut vascular barrier is another layer of tightly connected endothelial cells surrounded by pericytes and glial cells (Bouziat and Jabri 2015). Host self-defense is further enhanced via anti-microbial peptides secreted by Paneth cells at the crypt base (Dupont et al. 2014). Microbial metabolites or structural components of entire microorganisms residing in the gastrointestinal tract are sampled by the pattern recognition receptors of dendritic cells (Quante et al. 2013). Matured antigen-presenting dendritic cells migrate to mesenteric lymph nodes where they shape adaptive immunity by activating memory and naïve T cells (Steinman 2007). Depending on the context of microbial metabolites or byproducts, dendritic cells may turn naïve T cells into helper, effector (cytotoxic), or regulatory (Treg or suppressor) T cells. TH17 cells, a special subset of CD4+ helper T cells, protect the host from pathogenic microbes by strengthening tight junctions, stimulating the production of anti-microbial peptides, and by recruiting polymorphonuclear neutrophils (Garrett et al. 2010). Systemic immune responses are also affected by gut microbiota when T cells primed by dendritic cells circulate from local lymph nodes to distant sites or if microbial components and viable microorganisms enter the portal venous system. This response escalation is increasingly likely in the setting of dysbiosis characterized by reduced species diversity, enrichment of opportunistic pathogenic bacteria, and impaired gut barrier (Giannelli et al. 2014; Gopalakrishnan et al. 2018a).
2.3 Host Immunosurveillance
Augmented inflammation due to activation of innate and adaptive immune responses may promote cancer initiation and progression. Tumorigenic properties of dendritic cells, polymorphonuclear neutrophils, tumor-associated macrophages and myeloid-derived suppressor cells immature myeloid cells have been linked to the release of cytokines, growth factors, tissue-degrading enzymes and angiogenic mediators (Noonan et al. 2008; Quante et al. 2013). Several subsets of the adaptive immune system such as TH17 cells have also been associated with inflammation and their increased presence of TH17 cells in colorectal cancer predicts poor prognosis (Quante et al. 2013). The impact of Tregs on cancer is similarly controversial as they create a local anti-inflammatory milieu and mitigate tumorigenesis, but may simultaneously weaken anti-tumor immunosurveillance, often making their presence an ambivalent prognostic parameter (Quante et al. 2013). By contrast, cytotoxic CD8+ T cells may specifically identify tumor differentiation antigens via their T cell receptors and destroy cancer cells, indicating that increased infiltration of the tumor tissue with these cells is a favorable prognostic sign (Reticker-Flynn and Engleman 2019).
However, anti-cancer immunosurveillance is not guaranteed by the presence of tumor-infiltrating lymphocytes, which are typically restrained by immune checkpoints consisting of a large and heterogeneous group of ligands and receptors preventing indiscriminate activation of the immune response (Restifo et al. 2012). Ligands that are able to activate immune checkpoints such as programmed cell death protein 1 (PD1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA4) often become overexpressed in cancer cells and other components of the tumor microenvironment, thereby assisting evasion of immune-mediated destruction (Pardoll 2015). Tumor immunotherapy targeting immune checkpoints has been one of the fastest-developing and successful chapters of cancer research and, as discussed later, its clinical efficacy is remarkably determined by the composition and function of gut microbiota.
3 Gut Microbiota and Mechanisms of Tumor Promotion
In general, microbial contribution to tumorigenesis can be classified into three mechanisms: direct impact by modulating host cell proliferation and death, interference with the host innate and adaptive immune system, and altering food and drug metabolism or host biochemistry (Garrett 2015). Altogether, an estimated 20–30% of cancers are associated with chronic microbial infections (Garrett 2015; Yang et al. 2013). So far, however, only 10 microorganisms have been irrefutably designated as carcinogens (i.e., bona fide oncomicrobes), including Helicobacter pylori (gastric adenocarcinoma), hepatitis B and C viruses (liver cancer), Schistosoma haematobium (urinary bladder cancer), Opisthorchis viverrini and Clonorchis sinensis (cholangiocellular neoplasia), Epstein-Barr virus (nasopharyngeal carcinoma and lymphoma), human herpes virus type 8 (Kaposi’s sarcoma), human T cell lymphotropic virus type 1 (lymphoma), and human papilloma viruses (cervical and anogenital cancer) (de Martel et al. 2012).
While only a few microorganisms have been specifically implicated in tumorigenesis, microbiota as a multicellular aggregate appears to have influence on the initiation and progression of cancer (Drewes et al. 2016; Garrett et al. 2010; Morgillo et al. 2018). Current view is that cancer is more likely to develop in dysbiosis that includes a marked decrease in both microbial diversity and community stability (Bhatt et al. 2017). Moreover, dysbiosis modulates the impact of anti-cancer therapy on host responses and adverse events (Gopalakrishnan et al. 2018a). Importantly, the tumorigenic impact of dysbiosis is not confined to colorectal cancer as dysbiosis has also been associated with other forms of cancers including breast, lung, urogenital tract and liver (Pope et al. 2017). In fact, gut microbiota, metabolites, and immune cells may exit the gut via the circulation and influence tumorigenesis at distant sites (Bhatt et al. 2017). Supporting this notion, metastases from patients with colorectal cancer continue to harbor bacteria, in particular Fusobacterium but also Bacteroides, Selenomonas and Prevotella species (Kroemer and Zitvogel 2018).
3.1 Microbiota-Associated Genotoxicity and Tumorigenesis
Many bacteria have developed competitive strategies, which include the ability to damage the genome of competing organisms. Some of these bacteria are part of the commensal human microbiota. Microbial-induced genotoxic mechanisms and the activation of related oncogenic signaling pathways also affect the host, potentially leading to cancer (Garrett 2015). For instance, Salmonella typhi strains possessing avrA, a virulence gene encoding acetyltransferase activity, establish chronic infection and activate epithelial β-catenin signaling, which has been associated with hepatobiliary and colorectal cancers (Dutta et al. 2000; Lu et al. 2017). Colibactin is a bacterial toxin, which is synthesized by the pks genomic island and found in members of the Enterobacteriaceae family such as group B2 Escherichia coli (Fais et al. 2018). Colibactin promotes colorectal tumorigenesis by inducing double-stranded DNA breaks and a senescence-associated secretory phenotype in intestinal epithelial cells (Fulbright et al. 2017; Schwabe and Jobin 2013). Analysis of the colonic mucosa of patients with colorectal cancer found significantly higher abundance of pks-harboring E. coli strains compared to healthy controls (Buc et al. 2013). Enterotoxigenic Bacteroides fragilis (ETBF), a gut commensal and opportunistic pathogen enriched in patients with colorectal cancer, secretes a zinc-dependent metalloprotease that cleaves and degrades E-cadherin (Sears 2009). This toxin enables nuclear translocation of β-catenin and transcription of Myc proto-oncogene, promoting cellular proliferation in the colon epithelium. In the ApcMin/+ mouse model, ETBF facilitates colon tumorigenesis by triggering TH17-mediated colitis and STAT3 activation (Wu et al. 2009).
3.2 Gut Microbiota Metabolism Favoring Tumorigenesis
Robust cancer phenotypes emerge from comprehensive reprogramming of macromolecular biosynthesis and energy metabolism (DeBerardinis et al. 2008). A prominent metabolic feature of cancer cells is the Warburg effect or reallocation of energy production from mitochondrial oxidative phosphorylation to the disproportionate use of glycolysis (Warburg et al. 1924). While the causes and rationale of the Warburg effect remain debated, it has been proposed that glycolytic ATP production can be scaled up with fewer regulatory constraints to match the vast energetic needs of rapidly proliferating cancer cells (Pfeiffer et al. 2001). In addition, diversion of substrates from the electron transport chain may diminish mitochondrial oxidative stress that is already substantial in cancer cells (Brand 1997). Also, glycolysis can be viewed as a versatile production line of precursors for almost all major biosynthetic routes including the pentose phosphate pathway, serine synthesis pathway, and de novo lipogenesis (Pavlova and Thompson 2016; Sun et al. 2018). Finally, excess lactate production gives cancer cells a competitive advantage by sustaining an acidic tumor microenvironment increasingly inhabitable for normal cells (DeBerardinis et al. 2008; Hsu and Sabatini 2008).
Cancer cells exploit all available resources to support limitless growth and proliferation in an increasingly harsh and nutrient-deprived microenvironment. Thus, cancer cells may engulf and digest apoptotic bodies or entire living cells and utilize cellular waste products such as lactate, acetate, branched-chain keto acids, ketone bodies and ammonia (Sun et al. 2018). Currently, it is impossible to tell how many gut microbial species actually provide benefits and how many may ‘collude’ with cancer cells while interacting via diffusible metabolites (Wegiel et al. 2018). Many dietary and digestive components are metabolized by bacteria in the gastrointestinal tract, yielding putative metabolites that have either oncogenic or tumor suppressing properties (Bhatt et al. 2017). Unconventional nutrients potentially made available by the microbiota for the energy metabolism of cancer cells include short-chain fatty acids, bile acids, polyamines, choline metabolites, indole derivatives and vitamins (Sun et al. 2018). This list is almost certainly incomplete since close to half of the metabolites found in human plasma have been estimated to originate in the microbiota (Martin et al. 2007). Moreover, many genes identified in gut microbiomes of participants in the Human Microbiome Project could not be characterized by standard annotation methods (Joice et al. 2014), leaving us with a possibly large number of small molecules that may influence tumorigenesis (Donia and Fischbach 2015; Foster et al. 2017).
Bile acids represent an important connection between microbial and host metabolism. Primary bile acids synthesized in the liver as cholic acid or chenodeoxycholic acid become conjugated with glycine or taurine and excreted into bile (Wahlstrom et al. 2016). Most conjugated bile acids are reabsorbed in the terminal ileum and return to the liver via the enterohepatic circulation, while a small amount is deconjugated by intestinal bacteria into secondary bile acids such as deoxycholic acid and lithocholic acid (Long et al. 2017). Secondary bile acids may contribute to colon tumorigenesis by triggering inflammation and promoting β-catenin and NF-κB signaling. In addition, intestinal deoxycholic acid was found to contribute to the development of liver cancer by inducing senescence-associated secretory phenotype in hepatic stellate cells and stimulating pro-inflammatory and tumor-promoting reactions in a mouse model of obesity-associated hepatocellular carcinoma (Yoshimoto et al. 2013). Consumption of a diet rich in saturated fat leads to dysbiosis with expansion of sulfur-reducing gut bacteria such as Bilophila and Desulfovibrio, which use sulfur as a terminal electron acceptor, primarily obtained from taurine-conjugated bile acids (Devkota et al. 2012; Wang 2012). Microbial-derived hydrogen sulfide (H2S) has been implicated in the development of colitis and colorectal cancer based on its ability to impair the gut barrier and cause genotoxicity (Arkan 2017; Singh and Lin 2015).
Microbial contribution to amino acid metabolism has a significant impact on host immunosurveillance. A key example is tryptophan, an essential amino acid catabolized by both host and microbial enzymes and having derivatives with multiple biological functions. Endogenous tryptophan metabolites include kynurenine, serotonin and melatonin, whereas bacterial breakdown of tryptophan yields indole, indole acetate, indole propionate, skatole, and tryptamine (Gao et al. 2018). Tryptophan catabolism is an important effector system that modulates T cell responses and promotes immune tolerance (Fallarino et al. 2006). Not surprisingly, accelerated tryptophan catabolism has been reported by various tumors of the colon, breast, lung and brain due to higher expression of tryptophan-2, 3-dioxygenase and indoleamine-2, 3-dioxygenase (Sun et al. 2018). Endogenous and microbial derivatives of tryptophan activate the aryl hydrocarbon receptor (AhR), a xenobiotic sensor and regulator of inflammation and immunity (Zelante et al. 2013). AhR mediates activation of group 3 innate lymphoid cells (ILC3) resulting in enhanced secretion of IL-22, which dampens pro-inflammatory signals and protects the intestinal mucous barrier (Hernandez et al. 2018), but AhR activation also limits immunosurveillance by promoting apoptosis of effector T cells and creating therefore a cancer-permissive microenvironment (Grohmann et al. 2003).
3.3 Biofilm Formation and Tumorigenesis
Colon biofilms are dense consortia of approx. 100 bacterial strains embedded in a complex matrix in close proximity with, and partially invading, the intestinal mucosa (Dejea et al. 2014). Formation of biofilms has also been observed in conditions involving chronic mucosal inflammation beyond the colon (e.g., tonsillitis, otitis media, sinusitis, urethritis and vaginitis) (Costerton et al. 1999). Biofilms have been identified in about 15% of apparently healthy patients (Swidsinski et al. 2007), but they are almost universally present in patients with right-sided, surgically resected colorectal neoplasms and in paired biopsies of tumor-free mucosa (Dejea et al. 2014). Biofilms allow direct bacterial contact with colon epithelial cells, which is believed to trigger chronic inflammation conducive to tumorigenesis, characterized by diminished levels of E-cadherin, enhanced IL-6 and STAT3 activation, and increased crypt epithelial cell proliferation rates (Dejea et al. 2014). Notably, biofilm-positive tumor tissue indicated higher levels of acetylated polyamines compared to biofilm-negative tumors, which may explain how microbial biofilms contribute to colon cancer (Johnson et al. 2015).
Fusobacterium, a gram-negative anaerobic bacterial genus, is a main component of biofilms in various mucosal locations (Zhou et al. 2018). F. nucleatum produces several factors of fusobacterial virulence and associated functions (Wu et al. 2018). Thus, Fap2 engages the inhibitory receptor T cell immunoglobulin and ITIM domain (TIGIT) to silence anti-cancer cytotoxic activity of T cells and natural killer cells (Gur et al. 2015). In addition, FadA is a fusobacterial adhesin that binds to lectins and E-cadherin on the surface of host epithelial cells and activates β-catenin signaling, thus promoting cell proliferation (Rubinstein et al. 2013). F. nucleatum also binds to Toll-like receptor 4 on epithelial cells and activates the Myd88/NF-κB signaling pathway, promoting chemoresistance to colorectal cancer (Yu et al. 2017). Also, abundance of F. nucleatum has been correlated with enrichment of myeloid-derived suppressor cells and tumor-associated macrophages, both of which are known to weaken anti-cancer immunosurveillance (Wu et al. 2018).
4 Gut Microbiota and Mechanisms of Tumor Suppression
Recent research has identified a number of pathways through which commensal microbes support the host against cancer (Perez-Chanona and Trinchieri 2016; Pope et al. 2017). Thus, microbiota may beneficially influence a range of host immune functions including innate and adaptive immune responses, all related to anti-cancer surveillance (Pope et al. 2017). Also, accumulating evidence for the involvement of gut microbiota in cancer pathogenesis has opened new opportunities for preventive or therapeutic interventions. These include the administration of antibiotics, probiotics, prebiotics, and postbiotics (Bultman 2016; Zitvogel et al. 2015), but there are additional efforts to use specific intestinal microbial configurations for selectively manipulating the composition and function of gut microbiota (Comstock and Coyne 2003; Everard et al. 2013; Tanoue et al. 2019). However, caution is advised and some experts warn about the indiscriminate use of probiotics, particularly lactate-producing bacteria such as Lactobacillus, Turicibacter and Streptococcus and anaerobic Lachnospiraceae and Ruminococcaceae that ferment complex carbohydrates and aromatic compounds, in cancer patients (Arkan 2017). It remains to be seen when and which probiotic to use to maximize benefits and minimize potential harm to those with increased susceptibility to cancer.
4.1 Tumor Suppressive Microbial Metabolites and Biotransformation
Carbohydrate fermentation by the gut microbiota yields large amounts of short-chain fatty acids, such as acetate, propionate and butyrate (Riscuta et al. 2018). Short-chain fatty acids are metabolized via β-oxidation and the tricarboxylic acid (TCA) cycle in the mitochondrial matrix, representing an energy source for normal and cancerous colonocytes. Importantly, breakdown of butyrate into acetyl-CoA also stimulates histone acetylase activity, allowing transcriptional activation of genes involved in cell growth and proliferation, therefore making the role of butyrate controversial in tumorigenesis (Donohoe et al. 2012). Microbial-derived short-chain fatty acids may also serve as carbon source for enhanced biosynthetic activity in cancer cells and as ligands for G protein coupled receptors such as GPR41 and GPR43 with an ambiguous role in intestinal inflammation (Ang and Ding 2016). Paradoxically, however, butyrate has mostly been found to inhibit cancer cell growth, referred to as the ‘butyrate paradox’, and the explanation appears to be related to metabolic reprogramming of cancer cells (Donohoe et al. 2012). Thus, mitochondrial breakdown of butyrate may become insufficient due to the Warburg effect with surplus butyrate accumulating in the nucleus where it functions as an inhibitor of histone deacetylase; an effect that can be experimentally reversed by preventing aerobic glycolysis in cancer cells (Donohoe et al. 2012). By contrast, cancer cells that retain the ability to metabolize butyrate are positively selected by the microenvironment and develop a more aggressive and invasive phenotype (Serpa et al. 2010).
There is accumulating evidence that intact commensal microbiota are required for optimal responses to cancer chemotherapy (Kroemer and Zitvogel 2018). Certain health conditions or repeated use of antibiotics may significantly, even if only temporarily, alter the composition and function of gut microbiota and loss of microbial diversity and dysbiosis have been linked to altered pharmacodynamics of anti-cancer agents with unfavorable clinical outcomes (Gopalakrishnan et al. 2018b). Disruption of gut microbiota by antibiotics in a variety of murine tumor models results in profoundly impaired responsiveness of mice to CpG-oligonucleotide immunotherapy and platinum-based chemotherapy (Iida et al. 2013). Platinum compounds such as oxaliplatin and cisplatin require the generation of intratumoral oxidative stress in order to exert DNA damage and apoptosis, an effect that was hampered in mice receiving antibiotics (Iida et al. 2013). Furthermore, oral gavage of bacterial endotoxin to antibiotic-treated mice restored responsiveness to CpG-oligonucleotide immunotherapy evidenced by immunogenic cell death also drives antitumor T cell responses (Iida et al. 2013). The same work also demonstrated that antibiotics reduce the therapeutic efficacy of platinum compounds against subcutaneously transplanted tumors and suggested that the efficacy of oxaliplatin depends on microbiota-based priming of myeloid cells for the release of reactive oxygen species that contribute to genotoxicity and tumor reduction (Iida et al. 2013). This problem is further compounded by chemotherapy-induced dysbiosis and breakdown of the intestinal epithelial barrier, compromising clinical outcomes in cancer patients (Galloway-Pena et al. 2017).
Biotransformation of pharmaceutical agents by the gut microbiota is not necessarily beneficial. One such example involves irinotecan (CPT-11), which is a chemotherapeutic drug often used in the treatment of metastatic colon cancer. Irinotecan is a prodrug, metabolized into the active topoisomerase I inhibitor SN38 and subsequently glucuronidated in the liver to form the inactive SN38-G, which is excreted with the bile into the GI tract. In the colon, SN38-G is converted back to the active form by commensal gut bacteria such as Streptococcus agalactiae, Clostridium perfringens or Bacteroides fragilis that also possess β-glucuronidase activity. Reactivated SN38 causes severe colitis and diarrhea in susceptible patients, and this adverse event may necessitate dose reduction or discontinuation of irinotecan therapy. Small-molecule inhibitors specific to bacterial β-glucuronidases have been developed to avoid this complication (Wallace et al. 2010). Subsequently, inhibitors of E. coli β-glucuronidase were shown to protect the host against gastrointestinal toxicity induced by irinotecan in mice (Pope et al. 2017).
4.2 Gut Microbiota and Anti-cancer Immunosurveillance
Host survival critically depends on timely recognition of tumor-associated antigens and the destruction of cells committed to tumorigenesis. There is increasing evidence that gut microbiota play an important role in this process through their ability to modulate anti-cancer immune responses and immunotherapy (Pope et al. 2017). Neoantigens related to malignant transformation often show sufficient similarity with microbial epitopes, and antigenic mimicry may therefore enhance the recognition of cancer cells (Zitvogel et al. 2016). Related to this concept, the ‘cancer hygiene hypothesis’ suggests that limited exposure to microbial antigens in highly industrialized societies may account for the increased incidence of certain cancers (Thorburn et al. 2014). Moreover, microbe-associated molecular patterns as danger signals may increase the overall ‘vigor’ of innate immune system through the summative impact of extraneous microbial products on pathogen recognition receptors and the generation of soluble mediators such as interferons and cytokines (Zitvogel et al. 2016). Also, activation of T cell subsets implicated in anti-cancer immunosurveillance is impaired in germ-free mice and may be restored upon colonization with various intestinal microbial strains (Sommer and Backhed 2013).
These mechanisms are not necessarily restricted to gut-associated lymphoid tissue as translocation of microbial metabolites or entire microorganisms through a leaky gut-vascular barrier may allow systemic exposure and affects tumorigenesis at distal sites (Bhatt et al. 2017; Kroemer and Zitvogel 2018). In addition, the concept of a ‘common mucosal immune system’ based on animal models postulates that immune cells primed locally in the gut mucosa may travel to other mucosal or lymphoid sites, extending the impact of gut microbiota to the entire host (Wilson and Obradovic 2015).
As recently reported, long-term survivors of pancreatic ductal adenocarcinoma are characterized by high numbers of tumor-infiltrating CD8+ T cells and tumor neoantigens cross-reacting with microbial-derived epitopes, suggesting that enhanced immune response due to antigenic mimicry may account for a more favorable prognosis in this cohort (Balachandran et al. 2017). Cross-reactive clones in these patients show selective loss on metastatic progression, further supporting the favorable impact of microbial homology while precise mechanisms of T cell clone selection and survival remain incompletely understood (Balachandran et al. 2017). The role of gut microbiota in eliciting anti-cancer immune responses was further evidenced by a recent work identifying a consortium of 11 bacterial strains derived from healthy human donor feces and able to induce accumulation of interferon-γ-producing CD8+ T cells in the intestinal lamina propria of germ-free mice (Tanoue et al. 2019). Colonization with the 11-strain mixture protected these animals from dissemination of Listeria monocytogenes and suppressed the growth of syngeneic grafts of colon adenocarcinoma and melanoma, illustrating the profound potential of gut microbiota-based anti-cancer interventions (Tanoue et al. 2019).
4.3 Enhancement of Anti-cancer Immunotherapy by Gut Microbiota
A number of immune checkpoint inhibitor molecules have been developed and marketed in recent years, including monoclonal antibodies against CTLA4 (ipilimumab), PD1, (nivolumab), and PD1 ligand 1 or PDL1 (pembrolizumab) that proved highly efficient in several difficult-to-treat cancers (Fan et al. 2018; Kudo 2018; Robert et al. 2015). However, a truly remarkable impact of immune checkpoint inhibitors on these cancers is only observed in about 25% of patients while the remaining cases show limited or no response (Gopalakrishnan et al. 2018a; Sun et al. 2018). Intriguingly, abnormal composition of gut microbiota profoundly alters the efficacy of immune checkpoint inhibitor therapy and contributes to primary resistance (Bhatt et al. 2017). Anti-CTLA4 antibodies were ineffective when administered to subcutaneous tumors in germ-free or antibiotics-treated mice while CTLA4 blockade was restored by re-colonization with Bacteriodes and Burkholderia, bacterial strains that have markedly reduced abundance in response to anti-CTLA4 treatment in conventional mice (Vetizou et al. 2015). Moreover, fecal microbial transplantation (FMT) from melanoma patients treated with anti-CTLA4 and featuring abundance of Bacteroides fragilis indicated stronger anti-cancer properties when administered to the murine tumor model (Vetizou et al. 2015). In addition, repletion of gut microbiota with B. fragilis and Burkholderia cepacia ameliorated the mucosal toxicity of anti-CTLA4, indicating that microbiota composition may also improve therapeutic effectiveness by preventing adverse reactions (Vetizou et al. 2015).
The efficacy of PDL1 blockade was analyzed by using experimental tumor models in C57BL/6 mice obtained from different facilities and featuring distinct gut microbiota (Sivan et al. 2015). Metagenomic analysis revealed that abundance of Bifidobacterium spp. was associated with increased responsiveness to anti-cancer therapy and the response benefit was transferable between mouse strains by oral Bifidobacterium (Sivan et al. 2015). While Bifidobacterium-primed dendritic cells improved the function of tumor-specific CD8+ T cells, authors postulated a more generic and antigen-independent effect based on the changes in innate immune functions observed in this experimental model (Sivan et al. 2015).
Another recent work has provided additional insights into the molecular and cellular mechanisms by which gut microbiota may influence the tumor microenvironment and enhance immunotherapies (Gopalakrishnan et al. 2018b). Patients with metastatic melanoma under treatment with anti-PD1 therapy were found to harbor significant differences in the composition of gut microbiota according to their response status. Thus, non-responders to immune checkpoint inhibition had increased abundance of Bacteroidales in the gut microbiota, while patients with prolonged progression-free survival in response to anti-PD1 therapy had a significantly higher diversity of bacteria in their gut microbiota as well as a higher relative abundance of Clostridiales, Faecalibacterium and Ruminococcaceae (Gopalakrishnan et al. 2018a). Moreover, tumor tissue infiltration with CD8+ T cells was significantly more prominent among patients with abundance of Faecalibacterium prausnitzii and other Firmicutes in the gut microbiota (Gopalakrishnan et al. 2018a).
Another recent study aimed to assess the impact of dysbiosis associated with malignant disease or concomitant antibiotic use on primary resistance to PD1 blockade in patients with non-small cell lung cancer, renal cell carcinoma and urothelial carcinoma (Routy et al. 2018). Clinical review indicated that patients who received antibiotics had shorter progression-free survival, relapsed sooner, and responded poorly to immune checkpoint inhibitor therapy. Metagenomic sequencing indicated major differences in the composition of gut microbiota based on responsiveness to PD1 blockade. Improved clinical outcomes were similarly associated with increased abundance of Akkermansia and Alistipes species in this report (Routy et al. 2018). To establish causality between gut microbiota composition and responsiveness to anti-PD1 therapy, antibiotic-treated mice were given FMT from responder and non-responder cancer patients and then inoculated with tumor cells to assess the efficacy of immune checkpoint blockade in the xenografts. Importantly, tumor growth was delayed in mice that received FMT from responding patients, whereas FMT from non-responding patients had no such effect (Routy et al. 2018). Response to PD1 blockade was also restored by specific re-colonization of mice with Akkermansia muciniphila and Enterococcus hirae, bacterial strains associated with clinical benefits and shown to induce dendritic cells to secrete IL-12, which is involved in the immunogenicity of PD1 blockade in eubiosis (Routy et al. 2018).
Finally, strong correlation between commensal microbial composition and clinical response to immune checkpoint inhibitors in patients diagnosed with metastatic melanoma was recently demonstrated (Matson et al. 2018). Thus, integrated metagenomic analysis identified 10 microbial species differentially enriched in the intestines of responders vs. non-responders to anti-PDL1 or anti-CTLA4 therapy. Bacterial species more abundant in responders included Bifidobacterium longum, Collinsella aerofaciens, Enterococcus faecium, Lactobacillus spp. and Veillonella parvula. FMT from patients to germ-free mice recapitulated the patient phenotypes and animals reconstituted with responder microbiota had increased numbers of tumor antigen-specific CD8+ T cells in their tumor microenvironments (Matson et al. 2018). Anti-PDL1 was highly effective in mice colonized with responder microbiota while it remained completely ineffective in mice receiving FMT from non-responder patients (Matson et al. 2018).
5 Conclusions
Tumorigenesis, the process of host cells opting for cheating over cooperation and breaking the rules of multicellular life, has been the source of much suffering. Advances in preventing cancer that may ultimately destroy the host are eagerly awaited. There is now hope that increasing knowledge about the human commensal microbial community, and in particular the gut microbiota, will give valuable insights into the evolution and ecological interactions of host-cancer-microbiota relationship and identify new molecular targets for preventing and treating cancer. There is already evidence for the importance of eubiosis, which supports the intestinal barrier and gut-associated lymphoid tissue by enhancing innate and adaptive immunity and creating a tumor microenvironment that becomes unwelcoming to cancer cells in the gastrointestinal tract and beyond. Additional research is needed to identify microbial metabolites and specific molecular mechanisms by which individual strains or well-defined consortia of gut microbiota can be utilized in the prevention and treatment of cancer.
References
Aktipis CA, Nesse RM (2013) Evolutionary foundations for cancer biology. Evol Appl 6:144–159. https://doi.org/10.1111/eva.12034
Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, Wilkinson GS (2015) Cancer across the tree of life: cooperation and cheating in multicellularity. Philos Trans R Soc Lond B Biol Sci 370:pii: 20140219. https://doi.org/10.1098/rstb.2014.0219
Ang Z, Ding JL (2016) GPR41 and GPR43 in obesity and inflammation – protective or causative? Front Immunol 7:28. https://doi.org/10.3389/fimmu.2016.00028
Arkan MC (2017) The intricate connection between diet, microbiota, and cancer: a jigsaw puzzle. Semin Immunol 32:35–42. https://doi.org/10.1016/j.smim.2017.08.009
Arumugam M et al (2011) Enterotypes of the human gut microbiome. Nature 473:174–180. https://doi.org/10.1038/nature09944
Backhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI (2005) Host-bacterial mutualism in the human intestine. Science 307:1915–1920. https://doi.org/10.1126/science.1104816
Balachandran VP et al (2017) Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551:512–516. https://doi.org/10.1038/nature24462
Bhatt AP, Redinbo MR, Bultman SJ (2017) The role of the microbiome in cancer development and therapy. CA Cancer J Clin 67:326–344. https://doi.org/10.3322/caac.21398
Bouziat R, Jabri B (2015) Breaching the gut-vascular barrier. Science 350:742–743. https://doi.org/10.1126/science.aad6768
Brand K (1997) Aerobic glycolysis by proliferating cells: protection against oxidative stress at the expense of energy yield. J Bioenerg Biomembr 29:355–364
Buc E et al (2013) High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One 8:e56964. https://doi.org/10.1371/journal.pone.0056964
Bultman SJ (2016) The microbiome and its potential as a cancer preventive intervention. Semin Oncol 43:97–106. https://doi.org/10.1053/j.seminoncol.2015.09.001
Chen H, Lin F, Xing K, He X (2015) The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat Commun 6:6367. https://doi.org/10.1038/ncomms7367
Comstock LE, Coyne MJ (2003) Bacteroides thetaiotaomicron: a dynamic, niche-adapted human symbiont. Bioessays 25:926–929. https://doi.org/10.1002/bies.10350
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322
Coyte KZ, Schluter J, Foster KR (2015) The ecology of the microbiome: networks, competition, and stability. Science 350:663–666. https://doi.org/10.1126/science.aad2602
de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, Plummer M (2012) Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 13:607–615. https://doi.org/10.1016/S1470-2045(12)70137-7
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11–20. https://doi.org/10.1016/j.cmet.2007.10.002
Dejea CM et al (2014) Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A 111:18321–18326. https://doi.org/10.1073/pnas.1406199111
Devkota S et al (2012) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487:104–108. https://doi.org/10.1038/nature11225
Donia MS, Fischbach MA (2015) Small molecules from the human microbiota. Science 349:1254766. https://doi.org/10.1126/science.1254766
Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ (2012) The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell 48:612–626. https://doi.org/10.1016/j.molcel.2012.08.033
Drewes JL, Housseau F, Sears CL (2016) Sporadic colorectal cancer: microbial contributors to disease prevention, development and therapy. Br J Cancer 115:273–280. https://doi.org/10.1038/bjc.2016.189
Dupont A, Heinbockel L, Brandenburg K, Hornef MW (2014) Antimicrobial peptides and the enteric mucus layer act in concert to protect the intestinal mucosa. Gut Microbes 5:761–765. https://doi.org/10.4161/19490976.2014.972238
Dutta U, Garg PK, Kumar R, Tandon RK (2000) Typhoid carriers among patients with gallstones are at increased risk for carcinoma of the gallbladder. Am J Gastroenterol 95:784–787. https://doi.org/10.1111/j.1572-0241.2000.01860.x
Everard A et al (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A 110:9066–9071. https://doi.org/10.1073/pnas.1219451110
Fais T, Delmas J, Barnich N, Bonnet R, Dalmasso G (2018) Colibactin: more than a new bacterial toxin. Toxins (Basel) 10:pii: E151. https://doi.org/10.3390/toxins10040151
Fallarino F et al (2006) The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol 176:6752–6761
Fan CA, Reader J, Roque DM (2018) Review of immune therapies targeting ovarian cancer. Curr Treat Options Oncol 19:74. https://doi.org/10.1007/s11864-018-0584-3
Fischbach MA, Sonnenburg JL (2011) Eating for two: how metabolism establishes interspecies interactions in the gut. Cell Host Microbe 10:336–347. https://doi.org/10.1016/j.chom.2011.10.002
Foster KR, Schluter J, Coyte KZ, Rakoff-Nahoum S (2017) The evolution of the host microbiome as an ecosystem on a leash. Nature 548:43–51. https://doi.org/10.1038/nature23292
Fulbright LE, Ellermann M, Arthur JC (2017) The microbiome and the hallmarks of cancer. PLoS Pathog 13:e1006480. https://doi.org/10.1371/journal.ppat.1006480
Galloway-Pena JR, Jenq RR, Shelburne SA (2017) Can consideration of the microbiome improve antimicrobial utilization and treatment outcomes in the oncology patient? Clin Cancer Res 23:3263–3268. https://doi.org/10.1158/1078-0432.CCR-16-3173
Gao J et al (2018) Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front Cell Infect Microbiol 8:13. https://doi.org/10.3389/fcimb.2018.00013
Garrett WS (2015) Cancer and the microbiota. Science 348:80–86. https://doi.org/10.1126/science.aaa4972
Garrett WS, Gordon JI, Glimcher LH (2010) Homeostasis and inflammation in the intestine. Cell 140:859–870. https://doi.org/10.1016/j.cell.2010.01.023
Giannelli V, Di Gregorio V, Iebba V, Giusto M, Schippa S, Merli M, Thalheimer U (2014) Microbiota and the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World J Gastroenterol 20:16795–16810. https://doi.org/10.3748/wjg.v20.i45.16795
Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA (2018a) The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell 33:570–580. https://doi.org/10.1016/j.ccell.2018.03.015
Gopalakrishnan V et al (2018b) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359:97–103. https://doi.org/10.1126/science.aan4236
Goto Y, Kiyono H (2012) Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev 245:147–163. https://doi.org/10.1111/j.1600-065X.2011.01078.x
Greer R, Dong X, Morgun A, Shulzhenko N (2016) Investigating a holobiont: microbiota perturbations and transkingdom networks. Gut Microbes 7:126–135. https://doi.org/10.1080/19490976.2015.1128625
Grohmann U, Fallarino F, Puccetti P (2003) Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol 24:242–248
Gur C et al (2015) Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42:344–355. https://doi.org/10.1016/j.immuni.2015.01.010
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Hernandez P, Gronke K, Diefenbach A (2018) A catch-22: Interleukin-22 and cancer. Eur J Immunol 48:15–31. https://doi.org/10.1002/eji.201747183
Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134:703–707. https://doi.org/10.1016/j.cell.2008.08.021
Huitzil S, Sandoval-Motta S, Frank A, Aldana M (2018) Modeling the role of the microbiome in evolution. Front Physiol 9:1836. https://doi.org/10.3389/fphys.2018.01836
Human Microbiome Project C (2012) Structure, function and diversity of the healthy human microbiome. Nature 486:207–214. https://doi.org/10.1038/nature11234
Iida N et al (2013) Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342:967–970. https://doi.org/10.1126/science.1240527
Johnson CH et al (2015) Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab 21:891–897. https://doi.org/10.1016/j.cmet.2015.04.011
Joice R, Yasuda K, Shafquat A, Morgan XC, Huttenhower C (2014) Determining microbial products and identifying molecular targets in the human microbiome. Cell Metab 20:731–741. https://doi.org/10.1016/j.cmet.2014.10.003
Kroemer G, Zitvogel L (2018) Cancer immunotherapy in 2017: the breakthrough of the microbiota. Nat Rev Immunol 18:87–88. https://doi.org/10.1038/nri.2018.4
Kudo M (2018) Systemic therapy for hepatocellular carcinoma: latest advances. Cancers (Basel) 10. https://doi.org/10.3390/cancers10110412
Le Chatelier E et al (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546. https://doi.org/10.1038/nature12506
Lee WJ, Hase K (2014) Gut microbiota-generated metabolites in animal health and disease. Nat Chem Biol 10:416–424. https://doi.org/10.1038/nchembio.1535
Leong SP, Aktipis A, Maley C (2018) Cancer initiation and progression within the cancer microenvironment. Clin Exp Metastasis 35:361–367. https://doi.org/10.1007/s10585-018-9921-y
Llorente C, Schnabl B (2015) The gut microbiota and liver disease. Cell Mol Gastroenterol Hepatol 1:275–284. https://doi.org/10.1016/j.jcmgh.2015.04.003
Long SL, Gahan CGM, Joyce SA (2017) Interactions between gut bacteria and bile in health and disease. Mol Aspects Med 56:54–65. https://doi.org/10.1016/j.mam.2017.06.002
Lu R, Bosland M, Xia Y, Zhang YG, Kato I, Sun J (2017) Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget 8:55104–55115. https://doi.org/10.18632/oncotarget.19052
Marchiando AM, Graham WV, Turner JR (2010) Epithelial barriers in homeostasis and disease. Annu Rev Pathol 5:119–144. https://doi.org/10.1146/annurev.pathol.4.110807.092135
Martin FP et al (2007) A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol Syst Biol 3:112. https://doi.org/10.1038/msb4100153
Matson V et al (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359:104–108. https://doi.org/10.1126/science.aao3290
Maynard Smith J (1998) The units of selection. Novartis Found Symp 213:203–211. discussion 211–207
Maynard Smith J, Szathmáry E (1995) The major transitions in evolution. W.H. Freeman Spektrum, Oxford/New York
Morgillo F et al (2018) Carcinogenesis as a result of multiple inflammatory and oxidative hits: a comprehensive review from tumor microenvironment to gut microbiota. Neoplasia 20:721–733. https://doi.org/10.1016/j.neo.2018.05.002
Muller EEL, Faust K, Widder S, Herold M, Martinez Abbas S, Wilmes P (2018) Using metabolic networks to resolve ecological properties of microbiomes. Curr Opin Syst Biol 8:73–80
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S (2012) Host-gut microbiota metabolic interactions. Science 336:1262–1267. https://doi.org/10.1126/science.1223813
Noonan DM, De Lerma Barbaro A, Vannini N, Mortara L, Albini A (2008) Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Rev 27:31–40. https://doi.org/10.1007/s10555-007-9108-5
Pardoll D (2015) Cancer and the immune system: basic concepts and targets for intervention. Semin Oncol 42:523–538. https://doi.org/10.1053/j.seminoncol.2015.05.003
Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23:27–47. https://doi.org/10.1016/j.cmet.2015.12.006
Perez-Chanona E, Trinchieri G (2016) The role of microbiota in cancer therapy. Curr Opin Immunol 39:75–81. https://doi.org/10.1016/j.coi.2016.01.003
Pfeiffer T, Schuster S, Bonhoeffer S (2001) Cooperation and competition in the evolution of ATP-producing pathways. Science 292:504–507. https://doi.org/10.1126/science.1058079
Plichta DR et al (2016) Transcriptional interactions suggest niche segregation among microorganisms in the human gut. Nat Microbiol 1:16152. https://doi.org/10.1038/nmicrobiol.2016.152
Pope JL, Tomkovich S, Yang Y, Jobin C (2017) Microbiota as a mediator of cancer progression and therapy. Transl Res 179:139–154. https://doi.org/10.1016/j.trsl.2016.07.021
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19:1423–1437. https://doi.org/10.1038/nm.3394
Quante M, Varga J, Wang TC, Greten FR (2013) The gastrointestinal tumor microenvironment. Gastroenterology 145:63–78. https://doi.org/10.1053/j.gastro.2013.03.052
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281. https://doi.org/10.1038/nri3191
Reticker-Flynn NE, Engleman EG (2019) A gut punch fights cancer and infection. Nature 565:573–574. https://doi.org/10.1038/d41586-019-00133-w
Riscuta G, Xi D, Pierre-Victor D, Starke-Reed P, Khalsa J, Duffy L (2018) Diet, microbiome, and epigenetic changes in cancer. In: Dumitrescu RG, Verma M (eds) Cancer epigenetics for precision medicine. Methods in molecular biology. Humana Press, Clifton, pp 141–156
Robert C et al (2015) Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372:2521–2532. https://doi.org/10.1056/NEJMoa1503093
Routy B et al (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359:91–97. https://doi.org/10.1126/science.aan3706
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206. https://doi.org/10.1016/j.chom.2013.07.012
Schwabe RF, Jobin C (2013) The microbiome and cancer. Nat Rev Cancer 13:800–812. https://doi.org/10.1038/nrc3610
Sears CL (2009) Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 22:349–369. https://doi.org/10.1128/CMR.00053-08
Serpa J et al (2010) Butyrate-rich colonic microenvironment is a relevant selection factor for metabolically adapted tumor cells. J Biol Chem 285:39211–39223. https://doi.org/10.1074/jbc.M110.156026
Singh SB, Lin HC (2015) Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganisms 3:866–889. https://doi.org/10.3390/microorganisms3040866
Sivan A et al (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350:1084–1089. https://doi.org/10.1126/science.aac4255
Sommer F, Backhed F (2013) The gut microbiota – masters of host development and physiology. Nat Rev Microbiol 11:227–238. https://doi.org/10.1038/nrmicro2974
Steinman RM (2007) Lasker basic medical research award. Dendritic cells: versatile controllers of the immune system. Nat Med 13:1155–1159. https://doi.org/10.1038/nm1643
Sun L, Suo C, Li ST, Zhang H, Gao P (2018) Metabolic reprogramming for cancer cells and their microenvironment: beyond the Warburg effect. Biochim Biophys Acta Rev Cancer 1870:51–66. https://doi.org/10.1016/j.bbcan.2018.06.005
Swartz MA et al (2012) Tumor microenvironment complexity: emerging roles in cancer therapy. Cancer Res 72:2473–2480. https://doi.org/10.1158/0008-5472.CAN-12-0122
Swidsinski A et al (2007) Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut 56:343–350. https://doi.org/10.1136/gut.2006.098160
Tanoue T et al (2019) A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565:600–605. https://doi.org/10.1038/s41586-019-0878-z
Thorburn AN, Macia L, Mackay CR (2014) Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 40:833–842. https://doi.org/10.1016/j.immuni.2014.05.014
Vetizou M et al (2015) Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 350:1079–1084. https://doi.org/10.1126/science.aad1329
Wahlstrom A, Sayin SI, Marschall HU, Backhed F (2016) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24:41–50. https://doi.org/10.1016/j.cmet.2016.05.005
Wallace BD et al (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330:831–835. https://doi.org/10.1126/science.1191175
Wang R (2012) Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92:791–896. https://doi.org/10.1152/physrev.00017.2011
Warburg O, Poesener K, Negelein E (1924) Über den Stoffwechsel der Carcinomzelle. Biochem Z 152:309–344
Wasielewski H, Alcock J, Aktipis A (2016) Resource conflict and cooperation between human host and gut microbiota: implications for nutrition and health. Ann N Y Acad Sci 1372:20–28. https://doi.org/10.1111/nyas.13118
Wegiel B, Vuerich M, Daneshmandi S, Seth P (2018) Metabolic switch in the tumor microenvironment determines immune responses to anti-cancer therapy. Front Oncol 8:284. https://doi.org/10.3389/fonc.2018.00284
Wilson HL, Obradovic MR (2015) Evidence for a common mucosal immune system in the pig. Mol Immunol 66:22–34. https://doi.org/10.1016/j.molimm.2014.09.004
Wu S et al (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15:1016–1022. https://doi.org/10.1038/nm.2015
Wu C et al (2018) Forward genetic dissection of biofilm development by fusobacterium nucleatum: novel functions of cell division proteins FtsX and EnvC. MBio 9. https://doi.org/10.1128/mBio.00360-18
Yang T, Owen JL, Lightfoot YL, Kladde MP, Mohamadzadeh M (2013) Microbiota impact on the epigenetic regulation of colorectal cancer. Trends Mol Med 19:714–725. https://doi.org/10.1016/j.molmed.2013.08.005
Yoshimoto S et al (2013) Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499:97–101. https://doi.org/10.1038/nature12347
Yu T et al (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170:548–563.e516. https://doi.org/10.1016/j.cell.2017.07.008
Zelante T et al (2013) Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39:372–385. https://doi.org/10.1016/j.immuni.2013.08.003
Zhou Z, Chen J, Yao H, Hu H (2018) Fusobacterium and colorectal cancer. Front Oncol 8:371. https://doi.org/10.3389/fonc.2018.00371
Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, Kroemer G (2015) Cancer and the gut microbiota: an unexpected link. Sci Transl Med 7:271ps271. https://doi.org/10.1126/scitranslmed.3010473
Zitvogel L, Ayyoub M, Routy B, Kroemer G (2016) Microbiome and anticancer immunosurveillance. Cell 165:276–287. https://doi.org/10.1016/j.cell.2016.03.001
Zmora N, Suez J, Elinav E (2019) You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol 16:35–56. https://doi.org/10.1038/s41575-018-0061-2
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Baffy, G. (2020). Gut Microbiota and Cancer of the Host: Colliding Interests. In: Serpa, J. (eds) Tumor Microenvironment . Advances in Experimental Medicine and Biology, vol 1219. Springer, Cham. https://doi.org/10.1007/978-3-030-34025-4_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-34025-4_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-34024-7
Online ISBN: 978-3-030-34025-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)