
Abstract
Endoplasmic Reticulum stress (ER stress) is a condition whereby unfolded and misfolded proteins accumulate in the ER lumen as a result of perturbation of the ER homeostasis. Nutrient deprivation, hypoxia, and calcium depletion are some conditions which can compromise the homeostasis of this compartment. To overcome the imbalanced ER protein-folding capacity, cells have evolved an evolutionary conserved signal transduction pathway called unfolded protein response (UPR) which primarily aims to reestablish ER homeostasis. Meanwhile, autophagy is a process responsible for the turnover of unnecessary or dysfunctional organelles and proteins. It facilitates normal cell growth and development and it is also a survival pathway, required during starvation or growth factor deprivation. Massive vacuolation as a result of uncontrolled autophagy leads to cell death. It is now widely acknowledged that various proteins and pathways related to ER stress and autophagy are deregulated during cancer development. This chapter highlights the signaling pathways of ER stress and autophagy, the relevant therapeutic targets in cancer, and summarizes the current state of development of novel therapeutics in various phases of clinical trials. In addition, crosstalks in apoptosis, autophagy, and ER stress signaling pathways and future treatment strategies are reviewed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
18.1 Introduction
The endoplasmic reticulum (ER) is an important organelle responsible for protein folding and modification and disturbances in the ER environment will lead to ER stress and subsequently causes accumulation of unfolded or misfolded proteins. Although ER stress activates the unfolded protein response (UPR) mechanism to reestablish ER homeostasis, unresolved ER stress can lead to cellular processes such as apoptosis or autophagy. In cancer, tumor cells are dependent on these processes to combat and neutralize the chronic stress and harsh conditions of the tumor microenvironment, leading to tumor survival and tumor expansion; hence, the ER stress response is thought to be cytoprotective. It is now known that ER stress, apoptosis, and autophagy share overlapping molecular pathways and can occur in parallel under similar conditions. Fundamental knowledge in these processes has also generated a great deal of insight into the pathophysiological aspects of cancer, and has provided important considerations in strategizing cancer pharmacotherapy. A number of drugs targeting these processes have been developed and were proven to be promising in both preclinical and clinical studies.
18.2 Endoplasmic Reticulum Stress (ER Stress)
The ER is an intracellular organelle that provides crucial biosynthetic, stress-sensing, and signaling functions in eukaryotic cells [1, 2]. It is the main subcellular compartment for the synthesis, folding, modification, and transport of proteins which are destined to be secreted or embedded in the plasma membrane [3, 4]. The ER is also the major site for the biosynthesis of steroid, cholesterol, and lipid. It is the major intracellular calcium (Ca2+) storage organelle in the cell, and thus plays an important role in calcium homeostasis and calcium-mediated signaling pathways [5]. Nascent proteins are folded and modified correctly in the ER before being transported via the Golgi apparatus to the cell surface or other destination. It is an orchestrated process involving folding, assembly, modification, quality control, and recycling of proteins in a highly oxidizing and calcium-rich ER environment. Proteins translocated into the ER lumen are folded into their proper three-dimensional shapes and modified and assisted by ER-resident enzymes, such as chaperones, glycosylating enzymes, and oxidoreductases [6,7,8]. Incomplete or misfolded forms are eliminated by quality control systems, including the ER-associated degradation (ERAD) pathway and autophagy [7, 9, 10].
Physiological and pathological conditions such as hypoxia, nutrient fluctuations, altered ER-calcium levels, oxidative injury, inflammation, and viral infections may disrupt the protein folding environment in the ER, causing the accumulation of unfolded or misfolded proteins in the ER lumen [3]. This cellular condition is known as ER stress. ER stress leads to a complex intracellular signal transduction pathway, known as unfolded protein response (UPR), an adaptive mechanism to reestablish ER homeostasis [5, 11]. The UPR primarily aims at reestablishing ER homeostasis by coordinating temporal shut down in protein translation, upregulating ER chaperone genes to increase protein-folding capacity in the ER, and promoting ERAD pathway to remove misfolded proteins [4, 5]. However, when the initial cellular responses fail to restore ER homeostasis, persistent ER stress will elicit an alternative response called the “terminal UPR,” which actively promotes cell death to eliminate the damaged cells [7, 12, 13]. Activation of the UPR represents the defining criterion of ER stress, although the terms UPR and ER stress are often used interchangeably [8].
18.3 Unfolded Protein Response (UPR)
The UPR in mammalian cells is governed by three transmembrane ER stress sensors, namely PERK (protein kinase RNA-like ER kinase), IRE1α (inositol-requiring enzyme 1α), and ATF6α (activating transcription factor 6α) [3]. In the absence of ER stress, the ER luminal domains of PERK, IRE1α and ATF6α are associated with immunoglobulin heavy chain binding protein known as BIP (also known as GRP78), where this interaction maintains all three transmembrane proteins in their inactive state. BIP, a 78-kDa glucose-regulated protein, is well established as an ER chaperone that participates in protein folding and assembly and has been widely used as a marker for ER stress [14]. During ER stress, the accumulating misfolded or unfolded proteins cause BIP to dissociate from the three transmembrane ER stress sensors, and subsequently bind to these misfolded or unfolded proteins. This is due to higher natural affinity of BIP to unfolded proteins compared with the ER stress sensor luminal domains [4]. The release of BIP causes the homodimerization, trans-auto-phosphorylation, and activation of both IRE1α and PERK and translocation of ATF6α to the Golgi apparatus and subsequent activation [8, 12, 15].
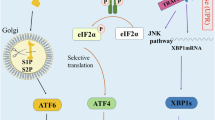
Activated PERK phosphorylates eukaryotic translation initiator factor 2α (eIF2α) and attenuates general protein translation, thereby relieving the protein burden on the stressed ER by reducing new protein synthesis and preventing further accumulation of unfolded proteins. Phosphorylation of eIF2α also regulates translation via inhibition of rRNA synthesis [5, 8]. Paradoxically, eIF2α phosphorylation allows selective translation of activating transcription factor 4 (ATF4), a transcription factor that controls the expression of genes encoding ER chaperones (e.g., BIP and GRP94), autophagy, and apoptosis [16, 17]. ATF4 favors the expression of antioxidant response, amino acid biosynthesis, and transport genes to sustain cell survival [4]. Depending on the severity and duration of stress, PERK activation can lead to either survival or cell death [18, 19]. Figure 18.1 illustrates the UPR pathway upon exposure to moderate ER stress.

The UPR pathway upon exposure to moderate ER stress
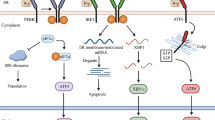
During prolonged ER stress, ATF4 stimulates the transcription of DNA-damage-inducible transcript 3 (DDIT3; also known as CHOP [CCAAT/enhancer binding protein homologous transcription factor] or GADD153 [growth arrest and DNA damage-inducible gene 153]), a transcription factor that is activated by all three arms of the UPR [5]. DDIT3 itself is a transcription factor that is critical in supporting the ER stress-induced apoptotic program [20]. In addition to its prodeath functions, DDIT3 participates in relieving the general block on translation via induction of growth arrest and DNA damage-inducible protein 34 (GADD34). GADD34 activates protein phosphatase 1 alpha (PP1A) to dephosphorylate eIF2α and dephosphorylated eIF2α resumes its function in general translation. If the protein folding capacity of the ER has not been reestablished, a premature restoration of protein synthesis will increase protein load in the stressed ER, thus amplifying the damage [5, 8]. Although eIF2α phosphorylation is downregulated during prolonged ER stress, PERK signaling is sustained, possibly to sensitize cells to cell death via DDIT3 induction [17]. Figure 18.2 illustrates the UPR pathway during severe and prolonged ER stress.

The UPR pathway and ER stress-Ca2+ signaling during severe and prolonged ER stress and antitumor targets
Similar to PERK, the release of BIP allows IRE1α to undergo dimerization and autophosphorylation. IRE1α is a bifunctional molecule with serine/threonine protein kinase and endoribonuclease (RNase) activity in its cytosolic domain [8]. Hence, this process leads to the activation of its cytosolic RNase domain, which removes a 26-nucleotide intron from the mRNA encoding the transcription factor X box-binding protein 1 (XBP1), producing mature spliced XBP1 mRNA. The spliced XBP1 mRNA is subsequently translated into an active and stable transcription factor, termed spliced XBP1 (XBP1s). XBP1s regulates the transcription of several genes involved in protein folding and quality control, ERAD, and phospholipid synthesis [21, 22]. ERAD is a process where misfolded proteins are retro-translocated from ER to the cytosol to be degraded by the 26S proteasome. Meanwhile, phospholipid synthesis is required for ER membrane expansion during ER stress [5, 11]. Through a process known as regulated IRE1-dependent decay (RIDD) of mRNA, IRE1 RNase domain degrades a subset of mRNAs encoding certain proteins of the secretory pathways and proteins located in the ER [11, 16].
Upon severe ER stress, XBP1s upregulates the expression of DDIT3 [5]. On the other hand, prolonged activation of IRE1α recruits the adaptor molecule TNF receptor-associated factor 2 (TRAF2), which further recruits apoptosis signal regulating kinase 1 (ASK1). This leads to a mitogen-activated protein (MAP) kinase activation cascade that activates c-jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) which further activates BIM and cause the inactivation of BCL-2 [3, 5, 23]. However, IRE1α is turned off upon prolonged ER stress, leading to ablation of the prosurvival XBP1s expression. Attenuation of IRE1α signaling is one possible mechanism to explain the transition from the adaptive UPR to prodeath events [11, 17] (Fig. 18.2).
ATF6α, a type II transmembrane protein, translocates to the Golgi apparatus once released from BIP, where it is proteolytically cleaved to generate a transcriptionally active fragment, termed ATF6f transcription factor. ATF6f mediates the adaptive response to ER protein misfolding by increasing the transcription of genes that increase ER capacity and the expression of Xbp1 [24, 25]. The transcription target of ATF6f includes genes involved in ERAD, phospholipid synthesis, and ER chaperones, thereby enhancing cellular folding and degradation capacity [8, 16, 17] (Fig. 18.1). ATF6f also contributes to upregulation of DDIT3 during prolonged ER stress [5] (Fig. 18.2).
Taken together, the three UPR transcription factors, ATF4, XBP1s and ATF6f, regulate a large set of partially overlapping UPR target genes during ER stress which modulates adaptation to stress or the induction of cell death under severe conditions [11]. The mechanisms underlying the switch from adaptive phase to prodeath events are still unclear, although it could be possibly through programs that sense the duration of the ER stress condition [17]. If the UPR is successful to increase the protein folding capacity and reduce the amount of misfolded proteins in the ER, BIP reassociates with PERK, IRE1α, and ATF6α, thereby inactivating these signaling modules. However, in case of excessive or prolonged ER stress, signaling pathways leading to cell death, either as apoptosis or autophagy, would be initiated [5, 8]. In certain situations, UPR may upregulate the autophagy machinery to eliminate damaged ER and abnormal protein aggregates [11]. In this context, autophagy is activated as an adaptive mechanism to reestablish ER homeostasis. However, if autophagy reaches a point of no return, cell death will be triggered. Therefore, just like in the case of UPR, persistent ER stress switches the cytoprotective functions of autophagy to cell death-promoting mechanisms [5, 26].
18.4 ER Stress and Cell Death
Several signaling pathways leading to apoptosis and autophagy would be initiated if ER stress is too severe to be relieved [27]. DDIT3 plays an important role in ER stress-induced cellular death, as this factor is a target gene common to all three apical ER stress sensors/executioners [1]. Duration and/or strength of PERK signaling may determine whether prosurvival or prodeath outcome predominates. Transient PERK signaling protects cells by temporarily reducing protein synthesis and thus reducing misfolded protein levels in the ER, but may be insufficient to induce DDIT3 to threshold level, given DDIT3’s inherent mRNA and protein instability. Since DDIT3 mRNA and protein have short half-lives, a strong and chronic activation of PERK is necessary to increase steady-state level of DDIT3 to promote cell death [28]. Persistent PERK signaling during prolonged ER stress is known to impair cell proliferation and promotes apoptosis via DDIT3 [29]. DDIT3 represses BCL-2 expression, upregulates BCL-2-interacting mediator of cell death (BIM) transcription, and promotes translocation of BAX to mitochondria [30,31,32]. It is also known to bind and induce the promoters of p53 upregulated modulator of apoptosis (PUMA), lipocalin 2 (LCN2), tribbles homologue 3 (TRIB3), and death receptor 5 (DR5) [33,34,35,36,37].
As a mediator of the mitochondrial apoptotic pathway, PUMA is known to result in the displacement and activation of BAX/BAK through its binding to antiapoptotic BCL-2 proteins, leading to mitochondrial dysfunction and caspase activation, hence initiating apoptosis [38, 39]. LCN2 is known to exacerbate hypoxia-induced cytochrome c release from mitochondria and caspase-3 activation [40]. Meanwhile, TRIB3 induces both apoptosis and autophagy. It indirectly activates unc-51-like autophagy-activating kinase1 (ULK1) which augments autophagosome formation and reduces autophagy flux. TRIB3 levels inhibit the activity of the kinase Akt by interacting with it and activating forkhead box O1 (FoxO1), a transcription factor that is negatively regulated by Akt, where it is translocated to the nucleus, and induces the proapoptotic gene, BIM [41]. It is also noted that DDIT3-mediated DR5 induction is responsible for ER stress apoptosis via caspase 8 [42]. PERK-dependent activation of ATF4 and DDIT3 has been demonstrated to upregulate the transcription of a set of autophagy genes, which are implicated in the formation, elongation, and function of the autophagosome [43].
In addition, IRE1α promotes cell death by recruiting a TRAF2-ASK1 complex, leading to the activation of JNK and p38 MAPK cascades upon prolonged ER stress. JNK promotes apoptosis through the phosphorylation-mediated regulation of Bcl-2 family members [5, 31, 44]. JNK exerts its proapoptotic effect by activating proapoptotic BH3-only protein BIM and by suppressing the antiapoptotic BCL-2 [5]. The p38 MAPK also phosphorylates and suppresses the antiapoptotic BCL-2 protein [45]. BCL-2 not only functions as an antiapoptotic protein, but also acts as an antiautophagy protein via its inhibitory interaction with BECN1. Both JNK and p38 MAPK have been proposed to induce autophagy by promoting dissociation of BECN1 from BCL-2. BECN1 is an essential autophagy regulator that participates in autophagosome formation [5, 45, 46]. In addition, p38 MAPK is known to phosphorylate DDIT3 and enhances DDIT3’s ability to function as a transcriptional activator [5, 47] (Fig. 18.2). The apoptosis-inducing activity of the third arm of UPR, ATF6α, has not been widely recognized. This is at least partly due to the fact that ATF6α does not induce apoptosis in cell lines commonly used in research. However, it has been shown that ATF6f mediates apoptosis via suppression of antiapoptotic protein, myeloid cell leukemia sequence 1 (Mcl-1) [48].
The mechanisms underlying the switch from adaptive phase to prodeath events remain elusive, although several hypotheses were suggested. The expression of the transcription factor DDIT3 is thought to be a decisive effector of the switch between adaptive UPR to cell death and the duration and amount of elevated DDIT3 level were hypothesized to be the decisive factor in determining the cell’s fate [26]. Upon severe ER stress, ATF4, XBP1s, and ATF6f transcription factors induce the transcription of DDIT3. On the other hand, PERK/eiF2α/ATF4 branch is essential to upregulate DDIT3 protein expression. The transcriptional activity of DDIT3 is then enhanced through the phosphorylation by p38 MAPK [5, 31]. Prolonged high level of DDIT3 protein expression is considered an indicator of the switch to proapoptotic module [8]. DDIT3 alters the balance between prosurvival and proapoptotic Bcl-2 family members and thus promotes apoptosis through the mitochondrial pathway. In addition, a molecular switch to cell death events could also involve TRIB3, a downstream transcriptional target of DDIT3. TRIB3 binds directly to prosurvival Akt kinase, thereby preventing its phosphorylation and reducing its kinase activity. During severe or persistent ER stress, induction of TRIB3 would be more robust, leading to autophagy and apoptosis through TRIB3-mediated inhibition of Akt/mTOR axis [5, 31, 49, 50] (Fig. 18.2).
In fact, IRE1α activities, namely (1) XBP1 mRNA splicing, (2) regulated IRE1-dependent decay of mRNAs, and (3) JNK/p38 MAPK activation, are also thought to be responsible for the life/death switch under prolonged ER stress conditions [51, 52]. Recently, the role of E2F1 has been described as a potential mechanistic survival/death switch under ER stress conditions [4, 53]. E2F1 is a member of the E2F family of transcription factors involved in several cellular functions such as proliferation, differentiation, and cell death [54, 55]. Upon ER stress induction, E2F7 as one of XBP1 target gene has been demonstrated to be positively regulated and the combined activity of E2F7 and activated ATF6 results in a specific but timely downregulation of E2F1 expression. This results in the removal of E2F1-dependent basal inhibition of both PUMA and NOXA that will induce the apoptotic program [4]. Timely and coordinated expression levels of E2F1 are crucial for determining the survival/death cell fate under ER stress conditions [4].
In addition to the three UPR branches, ER stress-Ca2+ signaling also leads to cell death during severe and prolonged ER stress. As ER is the major intracellular calcium storage organelle in the cell, ER stress activation is frequently accompanied by calcium release into the cytosol, causing an increase in cytosolic free calcium ions. Increases in cytosolic calcium concentration upon treatment with different ER stress inducers lead to calcium/calmodulin-dependent kinase kinase-β (CaMKKβ)-dependent activation of AMPK, that ultimately leads to inhibition of mTOR and stimulation of autophagy [5, 56]. In addition, mitochondrial intake of calcium ions following its release into the cytosol from the ER causes a collapse in the inner mitochondrial transmembrane potential (ΔΨm). A long-lasting or permanent ΔΨm dissipation is often associated with cell death [57, 58].
18.5 ER Stress in Cancer and Therapeutic Strategies
Tumor cells are often present within a hostile microenvironment and are confronted with chronic metabolic stress conditions. Following initiation of malignancy, poor vascularization of the tumor mass leads to stressful conditions in the tumor microenvironment, including low oxygen supply, nutrient deprivation, and pH changes. Therefore, many tumor types are thought to be dependent on an adaptive UPR to combat and neutralize the chronic stress and harsh conditions of the tumor microenvironment [5, 26, 44]. On the other hand, most normal cells are not subjected to stress and their UPR pathways are in an inactive state [44].
Both UPR activation and upregulation of BIP represent hallmark of several human cancers. UPR activation enables cancer cells to survive, adapts to adverse environmental conditions, and leads to growth arrest driving dormancy, which promotes resistance to conventional chemotherapy [59,60,61,62]. In addition, there are emerging evidences that linked mutations in three sensor genes such as ATF6α, IRE1α, and PERK in tumorigenesis [63,64,65,66]. The presence of missense, nonsense, and silent mutations in these genes seems to have tumor- or tissue-specific significance.
While BIP is generally too low to be detected in normal cells, many tumor cell lines display permanently elevated levels of BIP, which reflects the cancer cells’ ongoing effort to neutralize the chronic stress within the cells [26]. Elevated BIP is among the critical prosurvival mechanisms of tumor cells to withstand and thrive under detrimental microenvironmental conditions [8]. Similar to BIP, IRE1α/XBP1 signaling pathway is important for tumor growth and survival under stress conditions. An increase in XBP1 expression and splicing has been demonstrated in various human cancers, including breast cancer. Moreover, sustained IRE1α signaling was shown to enhance cell survival and proliferation [44, 67]. PERK/eif2α/ATF4 pathway also plays a role in cancer progression during stress condition. Hypoxia induces activation of the PERK pathway in tumor cells as an adaptive response to promote survival under hypoxic conditions. ATF4 is overexpressed in many solid tumors and is involved in promoting proliferation and survival during nutrient deprivation and severe hypoxia [44, 67].
In addition, several ER stress-associated markers are specifically upregulated in both neuroblastoma and melanoma cells under ER stress conditions [68]. DDIT3 and four other genes associated with ER stress were induced greater than twofold, namely ERdj5 (PDIA19; an ER-resident protein containing DnaJ and thioredoxin domains), ERp57 (GRP58; PDIA3; an ER-resident protein disulfide isomerase), calreticulin, and calnexin (both ER-resident chaperones) [68]. Protein disulfide isomerase (PDI) family members such as ERdj5 and ERp57 are consistently upregulated in neuroectodermal tumors and a generalized inhibition of PDI activity revealed a significant sensitization of tumor cells to ER-stress apoptosis. PDIs are endoplasmic reticulum chaperone proteins, catalyze disulfide bond breakage, formation, and rearrangement, and are required for protein folding in the endoplasmic reticulum (ER). The observation that knockdown of ERdj5 or ERp57 enhanced the extent of cell death induced by chemotherapeutic drugs suggests that downregulating ER stress responses may be therapeutically valuable; the ER resident proteins ERdj5 and ERp57 may thus be anticancer targets and PDI inhibition in general appears to be a novel therapeutic strategy [68,69,70]. Recently, there are a few synthetic small molecule PDI inhibitors such as PACMA31, 16F16, and CCF642 which have proven efficacy in cancer models, but have yet to progress to clinical studies [69, 71,72,73].
Since tumor cells engage adaptive UPR, only a small margin is left for the tumor cells to accommodate additional ER stress. Drugs that aggravate the preexisting ER stress condition in tumor cells may cause a shift from adaptive UPR to severe ER stress, leading to cell death. At the same time, exposure to ER stress-inducing agents causes activation of adaptive UPR in normal cells. Thus, moderate intensity ER stress inducers would be required to sufficiently aggravate ER stress in tumor cells, but at the same time, only modestly trigger ER stress in normal cells, in order to produce tumor-selective cytotoxic outcome. It was hypothesized that exceptionally potent pharmacologic triggers of ER stress might not be ideal in this situation [26].
A variety of distinct pharmacologic agents have been identified to trigger ER stress by different mechanisms. These agents include proteasome inhibitors and sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA) inhibitors, among others [26]. Although these compounds affect the UPR pathway, UPR may not be the primary mechanism of action of these drugs [44]. In the context of cancer research, thapsigargin (an inhibitor of SERCA), tunicamycin (an inhibitor of protein glycosylation), and brefeldin A (an inhibitor of protein transport from ER to Golgi) are frequently used in experiments as ER stress inducers to investigate the details of ER stress response [8].
The degradation of the majority of misfolded proteins is mediated by the 26S proteasome through the ERAD pathway [44]. Inactivation of the proteasome by proteasome inhibitors causes accumulation of misfolded proteins bound for the ERAD pathway, thereby triggering the UPR [26]. Bortezomib is a proteasome inhibitor and was approved by the US FDA in 2003 to treat multiple myeloma and mantle cell lymphoma [8]. Treatment of multiple myeloma cells with bortezomib causes rapid upregulation of the components in the UPR, including PERK, ATF4, and DDIT3, resulting in cell death. On the other hand, bortezomib sensitized pancreatic cancer cells to ER stress-induced apoptosis by induction of DDIT3, GADD34 and JNK, while PERK activation and eIF2α phosphorylation were not detected [44]. Several mechanisms have been proposed to explain the cytotoxicity of bortezomib, including effects on NF-kB, cell cycle proteins, apoptosis-regulatory proteins and caspases, as well as ER stress. Although ER stress represents only one of several processes associated with bortezomib-induced cell death, it is conceivable that it might indeed represent the key component, whereas other observed events might be orchestrated secondary to the aggravation of ER stress [8]. Bortezomib is further discussed in Chap. 17.
Inhibitors of human immunodeficiency virus (HIV) protease are known to inhibit the proteasome [26]. Two widely prescribed HIV protease inhibitors, namely nelfinavir and atazanavir, cause the accumulation of polyubiquitinated proteins, aggresome formation, and an increase in BIP and DDIT3 expression [74, 75]. In addition, nelfinavir has been shown to induce ER stress, autophagy, and apoptosis in vitro and in vivo in nonsmall-cell lung carcinoma [76]. Nelfinavir is currently in clinical trials for repositioning as an anticancer agent [26]. A Phase II trial of nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer (LAPC) revealed that nelfinavir showed acceptable toxicity and promising survival in pancreatic cancer [77]. The study reports the clinical outcome in 23 patients with LAPC treated with chemoradiotherapy plus nelfinavir which shows moderate median and 1-year overall survival at 17.4 months and 73.4%, respectively [77].
In another Phase II trial of nelfinavir in combination with the proteasome inhibitor bortezomib in 12 patients with advanced hematologic malignancies, promising activity in advanced, bortezomib-refractory multiple myeloma was noted [78]. Nelfinavir alone significantly upregulated the expression of proteins related to UPR in peripheral blood mononuclear cells and inhibited proteasome activity. Of ten evaluable patients in the dose escalation cohort, three achieved a partial response, four stable disease for two cycles or more, while three had progressive disease as best response [78]. Of nine patients given oral nelfinavir before and during radiation therapy for advanced rectal cancer, five patients exhibited good tumor regression on MRI assessed by tumor regression grade (mrTRG) [79]. Unfortunately, nelfinavir monotherapy does not result in a meaningful improvement in clinical outcomes among patients with recurrent adenoid cystic carcinoma [80]. Nelfinavir is currently in clinical trials for various cancers such as cervical intraepithelial neoplasia and advanced renal cancers (Table 18.1
). However, atazanavir is not on any clinical trials involving cancer at this moment.
The SERCA is a transmembrane protein that actively imports calcium ions from the cytosol into the ER lumen, thereby establishing a steep calcium gradient between the ER lumen and cytosol. Inhibition of SERCA results in massive leakage of calcium ions from ER to the cytosol and thus efficiently triggers ER stress. Thapsigargin, a naturally occurring sesquiterpene lactone, is an exceptionally potent inhibitor of SERCA. However, its clinical usage is fraught with several challenges; it is quite toxic and not well tolerated by experimental animals. A prodrug of thapsigargin, also known as mipsagargin or G202, has been synthesized and was found to produce substantial tumor regression against a panel of human cancer xenografts in vivo at doses that were minimally toxic to the host [148]. Interestingly, mipsagargin demonstrated an acceptable tolerability and favorable pharmacokinetic profile in a phase I clinical trial in patients with refractory, advanced, or metastatic solid tumors [81].
Certain diaryl-substituted pyrazoles, for example, celecoxib, are another class of compound that has emerged as SERCA inhibitors [26]. Nevertheless, celecoxib might not attain sufficient level of ER stress in tumor tissues because it was initially developed as COX-2 inhibitor. However, celecoxib analogues with minimized COX-2 inhibitory function, but significantly increased ER stress-inducing ability have been developed [8]. AR-12/OSU-03012 is an antitumor celecoxib-derivative that has progressed to Phase I clinical trial as an anticancer agent and has activity against a number of infectious agents including fungi, bacteria, and viruses [149]. It has been shown to suppress tumor cell viability through multiple mechanisms including activation of endoplasmic reticulum stress, inhibition of PDK-1/Akt signaling and the induction of autophagy [150,151,152]. Although a Phase I clinical trial of AR-12 in adult patients with advanced or recurrent solid tumors or lymphoma has been completed, its overall outcome remain unpublished.
In both oncogenic BRAF melanoma cell lines and in patients who failed clinical treatment for skin melanomas, the presence of oncogenic BRAF was responsible for ER stress induction and cell survival [153, 154]. In particular, human skin melanoma is characterized by oncogenic BRAF mutations, such as BRAFV600E. In addition, approximately 8–14% of colorectal cancers (CRC) in early and advanced stages exhibit the BRAFV600E mutation [155,156,157,158]. The BRAF serine/threonine protein kinase is a downstream signaling protein in the epidermal growth factor receptor-mediated MAP kinase pathway, which activates MEK through its phosphorylation. BRAFV600E mutation leads to constitutive BRAF kinase activity, which sustains the MAP kinase signaling pathway. BRAFV600E-mediated p38 MAPK activation stimulates both the IRE1α/ASK1/JNK and TRIB3 pathways. BCL-XL/BCL-2 phosphorylation by active JNK releases BECN1, whereas TRIB3 inhibits the Akt/mTOR axes, resulting in an increase in basal autophagy [154].
Vemurafenib and dabrafenib are BRAF inhibitors which have been approved by the USA FDA and EMA for the treatment of BRAF-mutated metastatic melanoma. In an open-label, multicenter 2-year follow-up of vemurafenib in 3219 patients with BRAFV600 mutation-positive metastatic melanoma, data suggest that long-term vemurafenib treatment is effective and tolerable [83]. Although vemurafenib and dabrafenib demonstrated impressive antitumor activity in advanced melanoma with objective response rates around 50% [85, 159], disappointing results were seen for patients with BRAFV600E-mutated colorectal cancer. In the Phase II study evaluating vemurafenib in patients with metastatic BRAFV600E-mutated colorectal cancer, of 21 patients, only one patient had confirmed partial response (5%) and the median progression-free survival (PFS) was 2.1 months [82]. Dabrafenib monotherapy did not show meaningful clinical activity with only one confirmed partial response among the 11 patients with BRAFV600E-mutated colorectal cancer included in the Phase I trial [84]. Encorafenib, another potent and selective oral BRAF inhibitor, showed signs of efficacy in patients with BRAF-mutant advanced melanoma but lack of objective response in patients with colorectal cancer [160]. All three drugs are currently in several clinical trials for other tumors (Table 18.1).
Treatment of tumor cells with drugs that trigger further ER stress might result in two desirable anticancer outcomes. First, the drugs by themselves might result in increased antitumor effects. Second, the overload and subsequent breakdown of the UPR adaptive system might increase the tumor cells’ sensitivity toward conventional chemotherapeutic agents [26]. Targeting of alternative pathways is an attractive strategy to improve antitumor therapy in apoptosis-resistant cancer. In view of the fact that ER stress is basally activated in many cancers, aggravation of the preexisting ER stress condition and the subsequent activation of autophagy represent an alternative therapeutic target to improve cancer therapy [27].
18.6 Autophagy
The ubiquitin-proteasome system (UPS) and lysosomes are two primary intracellular protein degradation pathways recognized in eukaryotic cells. Differences between these two major protein degradation systems depend on their functional significance and the type of substrates taken in for degradation [161]. The UPS catalyzes the rapid degradation of abnormal proteins and short-lived regulatory proteins, leading to a control of a diversity of essential cellular processes [162]. In the lysosomal protein degradation pathway, degradation of extracellular materials is mediated by endocytosis, whereas degradation of intracellular long-lived cytoplasmic proteins and damaged organelles is mediated by three types of autophagy, macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), which are classified based on their transport of cytoplasmic materials into the lysosome for degradation [163, 164].
Autophagy literally means self-digestion in Greek [165]. Macroautophagy, usually refers to autophagy, is responsible for the turnover of unnecessary or dysfunctional organelles and proteins, such as damaged mitochondria [166]. These processes are important to maintain a well-controlled balance between anabolism and catabolism to facilitate normal cell growth and development. It is also a survival pathway, required during starvation or growth factor deprivation, as it provides an alternative energy source [167, 168]. Autophagy process provides catabolic intermediates for intracellular production of ATP when energy supplies are limited. It plays an essential role during starvation, cellular differentiation, cell death, cell survival, aging, and tumor prevention [164, 166, 169].
Autophagy pathway is a multistep process characterized by induction, vesicle nucleation, extension, and completion of an isolation membrane to form an organelle called autophagosome [170]. Briefly, the autophagy process begins with the formation of a preautophagosomal structure known as isolation membrane or phagophore [171]. The isolation membrane engulfs and elongates to form the autophagosome, surrounding the components destined to be recycled. The autophagosome, which is a double membrane-bounded structure, undergoes maturation, and fuses with both endosomal and lysosomal vesicles to form autolysosome [171,172,173]. The sequestered contents are subsequently degraded by lysosomal hydrolases and are recycled. Based on morphological features, the term “autophagic cell death” has been described in instances of cell death that are accompanied by massive cytoplasmic vacuolization.
The core autophagy machinery is composed of four major functional groups: (1) the unc-51-like kinases (ULKs) (ATG1-ATG13-ATG17 kinase complex), (2) the Class III phosphatidylinositol-3-kinase catalytic subunit type 3 (PI3KC3) complexes, including Class III PI3K (the mammalian orthologue of vascular protein sorting 34; VPS34), p150/VPS15 (the mammalian orthologue of Vps15), BECN-1 (the mammalian orthologue of ATG6/Vps30) and ATG14L (ATG14), (3) two ubiquitin-like conjugation systems: ATG12 and ATG8, and (4) ATG9 and its cycling system [174]. The ULKs (the mammalian orthologues of ATG1, which exist in a large complex with mammalian ATG13), focal adhesion kinase family interacting protein of 200 kDa (FIP200; the mammalian homologue of ATG17), and the recently identified ATG101 play a crucial role in autophagy induction [175,176,177,178,179]. ULK1 is part of a family of kinases in humans (ULK1–4). Isoform ULK1 is the most important component in autophagy and in some cells lines, blocking both ULK1 and ULK2 is necessary to completely shut down autophagy [180].
The ULK1 kinase regulates proautophagic signals by phosphorylating many substrate proteins [181]. The numerous substrates of ULK1 include itself and other subunits of the ULK1 complex; other elements of the core autophagy machinery, including PI3KC3–C1 subunits such as BECN1 and ATG9; and other autophagy-related proteins such as AMBRA1 [180, 181]. Autophosphorylation of the kinase domain’s activation loop at Thr180 of ULK1 is essential for activation upon autophagy induction [182, 183]. Subsequently, phosphorylation of these downstream molecules by ULK1 is an important step in the initiation of autophagy.
The early stages of the phagophore membrane nucleation are dependent on the Class III PI3KC3 complex which consists of the Class III PI3KC3 protein, its regulatory protein kinase p150/VPS15, and BECN1 [184]. BECN1 is a 60-kDa tumor suppressor protein and is identified from a yeast two-hybrid screen as a BCL-2 interacting protein [185]. Several studies have demonstrated that several binding molecules positively regulate BECN1 activity and autophagosome formation and maturation. For example, ultraviolet radiation resistance-associated gene (UVRAG), ATG14L, and activated molecule in BECN1 regulated autophagy protein (AMBRA1) associate with BECN1 to activate autophagy [186,187,188,189,190].
The Class III PI3KC3 phosphorylates phosphatidylinositol to generate PI(3)P which is an essential early event in autophagy initiation, downstream of ULK1 [187, 191, 192]. PI3KC3 forms two distinct complexes, known as complexes I and II (PI3KC3–C1 and PI3KC3–C2) which contain the catalytic subunit VPS34/Vps34, the putative protein kinase VPS15/Vps15 and BECN1/ATG6 [187, 192]. PI3KC3–C1 contains ATG14L/ATG14, which directs the complex to phagophore initiation sites [186, 187, 193,194,195,196]. PI3KC3–C1 facilitates elongation meanwhile PI3KC3–C2, which contains UVRAG, directs endosome and autophagosome maturation [180].
The next stage of phagophore membrane elongation (expansion and closure of the autophagosome) requires two ubiquitin-like systems [197]. The ubiquitin-like protein ATG12 conjugates with ATG5 in an ATG7- and ATG10-dependent manner [161]. The ATG5–ATG12 complex interacts with ATG16 to form a stable and large multimeric complex called the ATG16L complex, which localizes on the outer surface of the extending autophagosomal membrane [170]. This complex is important in the stimulation and localization of the microtubule-associated protein 1 light chain 3 (LC3) conjugation reactions. LC3 is first cleaved by ATG4 to expose a C-terminal glycine residue required for subsequent activation and conjugation reactions [198]. It is then conjugated to the lipid phosphatidylethanolamine (PE), also via ATG7 and E2-like ATG3, and is subsequently recruited to both outer and inner surfaces of the autophagosomal membrane [197, 199]. Actually, two forms of LC3 are produced posttranslationally in various cells; the unconjugated form (LC3-I) is in the cytosol, while the conjugated form (LC3-II) targets the autophagosomal membrane with the assistance of the ATG16L complex [199, 200]. ATG16L complex is a ubiquitin-protein ligase (E3)-like enzyme that functions as a scaffold for LC3-II lipidation by localizing to the source membranes during autophagosome formation [200, 201]. The association of LC3-II to the autophagosome is crucial for membrane elongation of the autophagosome and the final limitation of the membrane to form the vacuoles [161]. The ATG5–ATG12–ATG16 complex is recycled, while the LC3 complex stays on the membrane until it is degraded by the lysosome [161]. In mammalian autophagy, LC3-II protein is used as an index of autophagosome formation or as an autophagosomal marker [202]. These conjugation systems are considered to be uniquely important to the autophagosome formation and have been identified as possible drug targets in cancer [203].
ATG9 system is required for phagophore expansion. It is the only transmembrane protein in the autophagy core machinery and has been proposed to play a key role in directing membrane from donor organelles for autophagosome formation [204]. ATG9 trafficking from the plasma membrane and trans-Golgi network involves two conserved sorting signals for proper function in autophagy, namely ATG9 interaction with the AP1/2 clathrin adaptor complex and phosphorylation of ATG9 at Tyr8 by SRC kinase and at Ser14 by ULK1. SRC kinase directly phosphorylates Tyr8 of ATG9 and promotes the interaction of ATG9 with the AP1/2 complex and leads to the movement of ATG9 away from the juxtanuclear region [205]. As with Tyr8, phosphorylation at Ser14 enhances the binding of ATG9 with the AP2 complex and promotes ATG9-AP1 interaction. Zhou and co-workers showed that phosphorylation of ATG9 at both the Tyr8 and Ser14 sites is required for maintaining proper autophagy under both basal conditions and in response to starvation-induced stress [205]. Finally, ATG9 binds the small Rab GTPases (RABGAP) protein TBC1D5, and both TBC1D5 and the AP2 complex contribute to the correct sorting of ATG9-containing vesicles during the initiation of autophagy [206].
The completed autophagosome membrane subsequently fuses with lysosome via the actions of the lysosomal proteins including the lysosomal-associated membrane protein 1 (LAMP1), LAMP2, member of RAS oncogene family (Rab7), and UVRAG [207]. The eventual autolysosome is a single membrane-bound acidic vesicle where the contents are digested and recycled by lysosomal hydrolases such as cathepsins (CTS), and its nutrient and energy are recycled [208]. These single membrane autolysosomes filled with degraded cytoplasmic materials can be easily observed using transmission electron microscopy (TEM) [170]. In addition, the adapter protein sequestosome 1 (SQSTM1/p62), which targets specific substrates to autophagosomes and LC3II are degraded along with other cargo proteins and are used as a measure of autophagy flux [209]. The autophagy cargo receptor p62/SQSTM1 binds ubiquitin on cargo to deliver cargo proteins to autophagosomes by docking onto LC3 on autophagosomes. P62 itself is an autophagy substrate that accumulates when autophagy is inhibited [210].
The Nomenclature Committee on Cell Death (NCCD) recommends that the term “autophagic cell death” be used based on some biochemical and functional considerations, before indicating that a cell death is mediated by autophagy. Some of the considerations include making sure that the investigated cell death can be suppressed by the inhibition of the autophagic pathway using chemicals and/or genetic means (e.g., gene knock-out or RNAi silencing of essential autophagy modulators such as AMBRA1, ATG5, ATG12, or BECN1) [211].
One of the most-studied and important pathways involved in autophagy regulation is the PI3K-Akt-mTOR signaling pathway. The mammalian target of rapamycin, commonly known as mTOR, is a serine/threonine kinase which belongs to the family of phosphatidylinositol 3-kinase-related kinases. It regulates translation and cell growth by its ability to phosphorylate both binding protein of eukaryotic translation inhibition factor eIF4E (4E-BP1) and p70 ribosomal S6 kinase (p70S6k). Upon stimulation by a variety of signals including cytokines, growth factors, cellular stress such as heat shock, hypoxia, and oxidative stress, PI3K is recruited to the inner cell membrane via phosphorylated receptor tyrosine kinases and catalyzes the phosphorylation of phosphatidylinositol-3,4-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3). The recruitment of inactive Akt from the cytosol to the plasma membrane requires that the pleckstrin homology (PH) domain of Akt binds to PIP3 synthesized at the plasma membrane by PI3K. Akt is then phosphorylated at Thr308 by phosphatidylinositol-dependent kinase 1 (PDK1) [212, 213]. PTEN phosphatase antagonizes PI3K-Akt signaling by converting PIP3 back to PIP2 [212]. (Fig. 18.3).

Autophagy signaling pathway and antitumor targets
Upstream PI3K and Akt activation by growth factors leads to the activation of mTOR and subsequently phosphorylation of downstream substrates. Phosphorylation of p70S6k promotes ribosome biogenesis, and increases the capacity of the translational machinery for protein synthesis [214]. Phosphorylation of 4E-BP1 initiates the transcription of a subset of mRNAs important for cell growth and proliferation [214,215,216]. The mTOR kinase is a key regulatory component that controls the induction of autophagy [217]. Inhibition of mTOR (by nutrient depletion, starvation, or rapamycin) leads to cell cycle arrest, inhibition of cell proliferation, immunosuppression, and induction of autophagy. Increased levels of the mTOR kinase are found to inhibit the autophagy process, resulting in an increased in cell growth and tumor development [173]. Rapamycin, a specific mTOR inhibitor, complexes with the cytosolic receptor FK506-binding protein (FKBP12), and subsequently binds to a distinct region of mTOR upstream of the catalytic domain [218]. It induces autophagy and inhibits the proliferation of a variety of cells [219].
In eukaryotic cells, mTOR exists in two different complexes: mTORC1; a rapamycin-sensitive complex defined by its interaction with the supplementary protein Raptor (regulatory-associated protein of mTOR) and mTORC2; a rapamycin-insensitive complex defined by its interaction with Rictor (rapamycin-insensitive companion of mTOR) [220,221,222]. mTORC1 and mTORC2 accessorial complexes consist of mTOR, mammalian lethal with SEC13 protein 8 (mLST8) (also known as GßL) and DEP domain-containing mTOR-interacting protein (Deptor) [223]. mLST8 binds to the kinase domain of mTOR, and stabilizes the interaction of Raptor with mTOR in a rapamycin-sensitive pathway [224]. Raptor is the first protein shown to bind directly to mTOR that is required to mediate mTOR regulation of p70S6k and 4E-BP1 activities [221, 225]. On the other hand, PRAS40 and Deptor play roles as distinct negative regulators of mTORC1 [226, 227].
In a rapamycin-sensitive mTOR signaling pathway, much of the knowledge about mTORC1 function comes from the use of rapamycin, a bacterial macrolide antibiotic [228]. Upon entering the cell, rapamycin binds FK506-binding protein (FKBP12), its intracellular receptor, which subsequently binds to the FKBP12-rapamycin binding domain (FRB) of mTOR, thus inhibiting the mTORC1 functions [229, 230]. Rapamycin weakens the interaction between mTOR and Raptor [231]. However, the exact mechanism of how rapamycin and several rapamycin derivatives bind to FKBP12 to inhibit mTORC1 signaling is not completely understood [232]. Various conditions including starvation or lack of nutrients such as amino acids and/or glucose mimic rapamycin treatment, hence inhibit mTOR function in cultured cells, as indicated by rapid inactivation of p70S6k and hypophosphorylation of the 4E-BP1 [233].
Studies have shown that mTORC1 controls autophagy through the regulation of a protein complex consisting of ULK1, mAtg13, and FIP200 [176, 178, 234]. ULK complex is directly controlled by mTOR, leading to maintenance of the mAtg13 hyperphosphorylation state and suppression of autophagy induction [235]. A study has demonstrated that inhibition of mTOR by rapamycin leads to dephosphorylation of ULK1, ULK2, and mATG13, and activates ULKs to phosphorylate FIP200. These results suggested that the ULK-ATG13-FIP200 complexes are direct targets of mTOR and important regulators of autophagy in response to mTOR signaling [178]. One of the most important proteins involved in the regulation of mTORC1 activity is the tuberous sclerosis complex (TSC), which is a heterodimer of two proteins, TSC1 (also known as hamartin) and TSC2 (also known as tuberin) [230]. TSC1 and TSC2 function as a GAP (GTPase-activating protein) that negatively regulates a small GTPase called Rheb (Ras homologue-enriched in brain). TSC1 and TSC2 inhibit mTORC1 signaling by transforming Rheb into its inactive GDP-bound state [236, 237].
On the other hand, mTORC2 consists of mTOR, mLST8, Rictor, Deptor, mammalian stress-activated map kinase-interacting protein 1 (mSIN1; also known as MAPKAP1), and the recently identified protein observed with Rictor (PROTOR) [223, 238]. Rictor is defined as a novel mTOR-interacting protein defining a second raptor-independent mTOR complex [220, 239]. Unlike mTOR-Raptor, the mTOR-Rictor complex does not bind to FKBP12-rapamycin, and is insensitive to rapamycin treatment [220, 222]. Therefore, rapamycin treatment does not represent a complete inhibition of mTOR function [240]. mTORC2 stimulates cell signaling through activation and phosphorylation of the proproliferative and prosurvival kinase Akt [241]. Akt regulates cellular processes such as metabolism, survival, apoptosis, growth, and proliferation by phosphorylating various effectors. mTORC2 activates Akt directly by phosphorylation at Ser473, which is a site needed for its maximal activation [242, 243].
In addition, mTORC2 controls various members of the AGC subfamily of kinases which includes serum and glucocorticoid-induced protein kinase 1 (SGK1) and several members of PKC family including PKCα [220], PKCε [244], PKCδ [245], and PKCζ [246]. mTORC2 is also known to phosphorylate mammalian Ste20-like kinases 1 (MST1) which is a core component kinase in the Hippo signaling pathway [247]. The Hippo pathway is composed of a group of evolutionarily conserved protein kinases that inhibit cellular growth and promote apoptosis [248, 249]. MST1 phosphorylates and activates large tumor suppressor (LATS) kinases, which in turn phosphorylate and inhibit Yes-associated protein 1 (YAP1), a co-transcription factor that promotes proliferation and survival [250]. mTORC2 is reported to be involved in the regulation of cytoskeletal organization through Rho GTPases and PKCα [220, 239]. Inhibitors of mTOR kinase domain have been developed to suppress the activity of both mTOR complexes (mTORC1 and mTORC2) [251, 252]. Figure 18.3 illustrates the simplified autophagy signaling pathways.
18.7 Autophagy and Cancer
The role of autophagy in cancer is rather perplexing. It is widely known that the autophagic pathway is deregulated in tumor cells. Several proteins and pathways related to autophagy signaling are deregulated during cancer development [189, 253]. Cell lines derived from hepatic, pancreatic, and breast carcinoma exhibit low autophagic activity, as compared with normal cells from the same origin [189, 254]. Autophagic capacity is known to increase during premalignant stages of pancreatic carcinogenesis, and then decreases during the transition of pancreatic adenoma into adenocarcinoma, suggesting that a decreased autophagic activity possibly contributes to the malignancy of pancreatic cancer [255, 256]. A decrease in autophagic capacity is also observed during animal experimental carcinogenesis, where cells from preneoplastic liver nodules or primary hepatocellular carcinomas induced by chemical carcinogens showed a decreased autophagic capacity as compared to normal liver cells [256, 257]. In addition, BECN1 is found to be mono-allelically deleted in a high percentage of ovarian, breast, and prostate cancers (based on the 17q21 and gene mapping studies). However, BECN1 is adjacent to the known tumor suppressor gene breast cancer 1 (BRCA1) on chromosome 17. Genomic analysis of BECN1 in The Cancer Genome Atlas (TCGA) demonstrated that allelic loss of BECN1 does not occur independently of codeletion with BRCA1, suggesting instead that BRCA1 loss is the driver mutation in hereditary and sporadic breast cancer [258,259,260].
There is a direct link between tumorigenesis and the disruption of the autophagy signaling pathways. PTEN deletions as well as the amplifications of both Class III PI3K and Akt are found in several cancers [261, 262]. The mTOR signaling pathway is constitutively activated in many tumor types. For example, the mTOR pathway is frequently found to be hyperactive in cancers such as breast cancer, suggesting that mTOR is an attractive target for cancer drug development and therapy [263,264,265]. The mTOR signaling network contains a number of tumor suppressor genes which includes PTEN, LKB1 (liver kinase B1), TSC1/2, and a number of proto-oncogenes such as PI3K, Akt, and eIF4E genes [266]. Several alterations in genes such as KRAS, EGFR, LKB1, PTEN, PIK3CA (encoding the p110 catalytic subunit of PI3K), as well as Akt1 mutations, EGFR and PIK3CA amplification, and PTEN deletion have been described in NSCLC, which lead to uncontrolled mTOR pathway signaling [267]. In addition, dysregulation of the mTOR pathway appears to be more common in squamous lung carcinoma than adenocarcinoma [267, 268].
Cancer-related changes in pathways at the downstream of mTOR such as p70S6k and eIF4E are reported in breast carcinoma [269, 270]. In addition, malignant cell types undergo massive autophagosomes and eventually cell death when responding to anticancer agents and traditional herbs indicate the potential utility of autophagic cell death induction in cancer therapy [173, 271, 272]. Autophagic cell death characterized by an increase in the number of autophagic vacuoles in the cytoplasm, followed by cell demise has been observed in various diseases such as Alzheimer’s disease [273], Huntington’s disease [274,275,276,277], and Parkinson’s disease [278]. Thus, manipulation of autophagy is considered an attractive strategy to increase the efficacy of cancer treatments, prevent cancer development, and limit tumor progression.
However, autophagy is divergent in nature in both tumor suppression and tumor progression [279]. Although the argument supports that if cells cannot activate autophagy, protein synthesis will predominate over protein degradation and cellular growth continues (typical characteristic of tumor cells), that was not the case for most. For example, a study in human epidermoid lung carcinoma cells revealed that the autophagic pathway in response to nutrient deprivation is not downregulated when compared to their normal counterparts [280]. Human colon cancer cells which are able to survive for long period of time in the absence of nutrients have a high rate of autophagic activity [281]. Studies in colorectal cancer cells revealed that these cancerous cells harbor functional autophagic machinery to prolong cell survival during shortages of nutrients [282]. A study by Fuji and coworkers has also shown that strong LC3 expression in the peripheral area of pancreatic cancer tissue is correlated with poor outcome and short disease-free period [283]. Activated autophagy observed in pancreatic cancer cells is thought to be a response to factors in the cancer microenvironment, such as hypoxia and poor nutrient supply. In addition, autophagy was found to be upregulated in RAS-transformed cancer cells to promote cancer cells growth, survival, tumorigenesis, invasion, and metastases [284,285,286]. Upregulation of autophagy in cancer cells is caused by direct activation of the transcription factors of the microphthalmia-associated transcription factor (MiTF)/TFE family that control autophagy and lysosomal biogenesis or by removal of a repressive phosphorylation on the autophagy initiation machinery [286,287,288].
In lung cancer, deletion of Atg7 dramatically alters tumor pathology from carcinomas to that of benign oncocytomas [289, 290]. ATG7-deficient tumors accumulate dysfunctional mitochondria and prematurely induce p53 and proliferative arrest. As defective mitochondria is a major autophagy substrate, this indicates that benign human tumors manifest a phenotype of defective autophagy, perhaps explaining their benign status [286]. Autophagy has been identified as the key mechanism of cell survival in estrogen receptor-positive (ER+) breast cancer cells undergoing treatment with 4-hydroxytamoxifen (4-OHT) [291]. Antiestrogen therapy is the standard treatment for ER+ breast cancers which improves overall survival and provides chemoprevention [292, 293]. Unfortunately, approximately half of the women treated with antiestrogen therapy either do not respond or their breast cancer ultimately acquires resistance during treatment [294, 295]. Studies have shown that autophagic activity reduces the efficacy of chemotherapy and tamoxifen therapy in ER+ breast cancer cells [291, 296, 297], supporting the thesis that blocking autophagy signaling pathways may provide a new mechanism of anticancer therapy for resistant tumors.
In another example, electron microscopy examination of autophagic vesicles in melanoma tumors from 12 patients enrolled in a Phase II clinical trial of temozolomide and sorafenib therapy revealed that autophagic index (mean number of autophagic vacuoles per cell) is significantly higher in patients who derived little or no clinical benefit from the combination of temozolomide and sorafenib treatment. Patients who had stable disease or responded to therapy had low levels of autophagy in their tumors. These findings further validate the preclinical evidence that autophagy plays a critical role in resistance to chemotherapy. Results of this study indicate that pretreatment levels of autophagy can predict resistance to therapy. Patients with aggressive melanoma are more likely to have higher levels of autophagy in their tumor and therefore may respond to autophagy inhibition as a therapeutic strategy [298]. Hence, the divergent nature of autophagy has resulted in strategies for using proautophagics or autophagy inhibitors depending on the inherent nature of the cancer involved.
18.8 Autophagy Signaling Pathways and Therapeutic Strategies in Cancer
18.8.1 mTOR Signaling Pathway Inhibitors
Rapamycin (Sirolimus) as the first prototype of an mTOR inhibitor has poor aqueous solubility and strong immunosuppressive properties. Therefore, its utilization at doses capable of exerting anticancer effects is rather limited [299]. Nevertheless, trials utilizing rapamycin as a single agent or combination therapy are still being carried out. In a Phase I study of rapamycin and sunitinib in patients with advanced NSCLC, combination of rapamycin and sunitinib is reported to be well tolerated and has warranted further investigation in Phase II trials [300]. However, the same was not observed in another recent study. Combination of sunitinib and rapamycin was observed to be quite toxic in all cohorts of patients with refractory solid malignancies [93]. The addition of rapamycin was thought to be able to decrease the sunitinib-induced VEGF production, but on the contrary, VEGF levels went further up along with sunitinib and rapamycin administration; it only came down during the sunitinib-off weeks [93]. However, in another recent Phase I trial, combination of oral rapamycin, topotecan, and cyclophosphamide was well tolerated in patients with relapsed/refractory solid tumors. Biomarker studies demonstrated modulation of angiogenic pathways with reduction of thrombospondin-1 and soluble vascular endothelial growth factor receptor-2 levels, respectively [92]. Several Phase II trials with rapamycin in combination therapy are currently recruiting patients with bladder, thyroid, prostate, and central nervous system (CNS) tumors (Table 18.1).
Various rapamycin analogues have since been developed. Temsirolimus (CCI-779) is the first mTOR inhibitor approved by the US FDA for cancer treatment, and is considered a first-line treatment for patients with advanced renal cell carcinoma (RCC) with poor prognostic features [301]. A number of clinical trials were carried out for this drug, mainly as combination therapy with other chemotherapy drugs. Moderate clinical activity was observed in patients with bone and soft-tissue sarcoma given a combination of temsirolimus and cixutumumab in a Phase II trial [302] and in patients with metastatic adrenocortical carcinoma, the same combination therapy resulted in 40% of patients achieving prolonged stable disease [303]. Similarly, in a recent Phase I study of temsirolimus in combination with cetuximab in patients with advanced solid tumours, both the median PFS and overall survival (OS) were <1 year and less than half of the patients had stable disease at the end of the trial, indicating modest clinical activity [94].
In another recent Phase I study combining perifosine (an Akt inhibitor) and temsirolimus, although stable disease was seen in 9 of 11 subjects with high-grade gliomas, no partial or complete responses were achieved [95]. However, the combination of these Akt and mTOR inhibitors was considered safe and feasible in patients with recurrent/refractory pediatric solid tumors [95]. When temsirolimus was tested as a single therapy in patients with relapsed or refractory primary CNS lymphoma in a Phase II trial, complete response was seen in five patients (13.5%), partial response in 12 patients (32.4%), and an overall response rate of just 54% [98]. In platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma, although temsirolimus treatment was well tolerated, it did not meet the predefined efficacy criteria [99]. Phase I and Phase II clinical trials with temsirolimus and sorafenib carried out in patients with metastatic melanoma did not produce sufficient activity to justify further use [304, 305]. Similarly, in a Phase II trial for metastatic colorectal cancer, temsirolimus had limited efficacy in chemotherapy-resistant KRAS mutant disease [306].
Everolimus is another rapamycin analogue which was already approved as an anticancer agent. Everolimus (RAD001; rapamycin derivative 001) is a hydroxyethyl ether derivative of rapamycin that has been developed for oral administration [307]. This drug was approved by FDA for use in a variety of cancers, including advanced renal cell carcinoma, advanced pancreatic neuroendocrine tumors, renal angiomyolipoma, and HER2-negative breast cancer. Everolimus is structurally similar to temsirolimus, binds to an intracellular protein, FKBP12, forming a complex that inhibits the mTOR kinase. In a recent Phase I trial to assess safety and efficacy of everolimus in combination with liposomal doxorubicin and bevacizumab in patients with advanced metaplastic triple negative breast cancer, only patients with the presence of PI3K pathway aberration were associated with a significant improvement in objective response rate, but not the clinical benefit rate [101]. A randomized Phase II study indicated that combination therapy of everolimus with tamoxifen increased the clinical benefit rate (defined as the percentage of all patients with complete or partial response or stable disease at 6 months), time to progression (TTP), and overall survival compared with tamoxifen alone in postmenopausal women with aromatase inhibitor-resistant metastatic breast cancer [308]. Further Phase III trials in combination therapy with aromatase inhibitors and adjuvant hormone therapy in hormone receptor positive metastatic cancer are currently underway.
Everolimus given for 14 days in combination with R-CHOP-21 (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone delivered in a 21-day cycle) in patients with diffuse large B-cell lymphoma was proven to be safe. A total 23 of 24 patients achieved an overall response, and all 23 attained a complete metabolic response by PET, suggesting that drugs that target the PI3K-mTORC pathway added benefit when combined with standard R-CHOP [102]. The combination of everolimus plus CHOP was also effective in patients who are newly diagnosed with peripheral T-cell lymphomas, with objective response rate up to 90% [110]. The combination of mFOLFOX6 and everolimus in patients with metastatic gastroesophageal adenocarcinoma was also considered to be an active regimen with 83% of the patients experiencing a partial response [103]. Everolimus as a single therapy has demonstrated clinically relevant antitumor activity in patients with advanced differentiated thyroid cancer; median PFS and OS were 9 and 18 months, respectively [108].
Ridaforolimus (deforolimus or AP23573) has been tested in Phase I and Phase II clinical trials, and has shown promising results in several tumor types including sarcoma [299, 309]. Ridaforolimus received fast track and orphan drug status from the US FDA, as well as orphan status from the European Medicines Agency. Latest Phase I trials indicate that ridaforolimus as single therapy or in combination with other chemotherapy drugs was safe and well-tolerated [113, 114]. However, in a previous Phase II trial study on the efficacy and safety of single-agent ridaforolimus in patients with relapsed or refractory hematologic malignancies, results were unremarkable. Of the 52 patients evaluated, partial responses were noted in five subjects, while hematologic improvement and stable disease were observed in less than half of the patients [310]. In addition, the combination of ridaforolimus and dalotuzumab was no more effective than exemestane in patients with advanced ER-positive breast cancer, and the incidence of adverse events was higher [116]. Thus, the combination was not further pursued.
PI3K/Akt/mTOR pathway is often constitutively activated in human tumor cells and thus has been considered as a promising drug target. BEZ235 is a potent imidazo (4,5-c) quinoline derivative that inhibits PI3K and mTOR kinase activities by binding to the ATP-binding cleft of these enzymes, and induces G1 arrest [311]. Preclinical studies have suggested that BEZ235 is a potent dual PI3K/mTOR modulator with favorable pharmaceutical properties. For example, it inhibits VEGF-induced HUVEC cell proliferation and survival in vitro and VEGF-induced angiogenesis in vivo [312]. The compound also inhibits microvessel permeability in BN472 mammary carcinoma grown orthotopically in syngeneic rats, suggesting that this compound is potentially antiangiogenic [312]. Deregulated angiogenesis and high tumor vasculature permeability are known VEGF-mediated characteristics of human tumors. In addition, BEZ235 is found to produce significant tumor growth inhibition in xenograft models of pancreatic cancers and breast cancer cells [313, 314]. However, in a Phase II trial of the BEZ235 in patients with everolimus-resistant pancreatic neuroendocrine tumours, BEZ235 was poorly tolerated by patients. Although evidence of disease stability was observed, the study did not proceed to stage two [117]. Similarly, BEZ235 showed modest clinical activity and an unfavorable toxicity profile in patients with advanced and pretreated transitional cell carcinoma, with just a minority of patients experienced a clinical benefit [118]. Several Phase I/II clinical trials of BEZ235 in patients with advanced solid malignancies such as prostate and breast cancer were completed, but reports on the safety and efficacy of this drug have yet to be published.
18.8.2 Proautophagics
Temozolomide is the first proautophagic cytotoxic drug used to overcome apoptosis resistance in cancer cells, and was approved for use in glioblastoma multiforme (GBM) [119]. It has demonstrated therapeutic benefits in patients with glioblastoma, and has been evaluated for several types of apoptosis-resistant cancers [315]. Temozolomide is a prodrug, a monofunctional alkylating agent, and is chemically related to dacarbazine. It is the 3-methyl derivative of the experimental anticancer drug, mitozolomide. The ability of temozolomide in inducing autophagic cell death was reported in various preclinical studies [316,317,318,319]. In addition, temozolomide has demonstrated proapoptotic activities in malignant melanoma cells [320]. In a systematic assessment of three randomized controlled trials addressing whether temozolomide holds any advantage over conventional therapy for high-grade gliomas, it was shown that temozolomide is an effective therapy for GBM. The drug prolongs survival, delays disease progression, and has a low incidence of early adverse events [321]. Similar outcomes were observed in a Phase II study involving erlotinib in combination with radiation therapy and temozolomide to treat GBM and gliosarcoma. Patients treated with the combination of erlotinib and temozolomide during and following radiotherapy had better survival than historical controls [322].
In a later Phase II trial, patients with unresectable or multifocal glioblastoma, an upfront regimen of temozolomide and bevacizumab was well tolerated, and provided a significant level of disease stabilization [323]. In patients with recurrent glioblastoma, either used as a single agent in a dose-intense schedule or in combination with other chemotherapeutic agents, temozolomide was proven to be well tolerated and safe [324,325,326]. In pediatric patients with recurrent solid tumors or brain tumors, low-dose temozolomide improved tolerability and was convenient as outpatient therapy [327]. However, in a recent Phase II trial, bevacizumab plus irinotecan combination resulted in a superior PFS-6 rate and median PFS compared with temozolomide in patients with glioblastoma that harbors a nonmethylated O(6)-methylguanine DNA methyltransferase promoter [124]. Patients with an O(6)-methylguanine–DNA methyltransferase (MGMT) nonmethylated (nmMGMT) glioblastoma (GBM) have a particularly short median survival of 12.6 months and do not substantially benefit from temozolomide chemotherapy [124, 328].
The combination of adjuvant temozolomide and lomustine, an alkylating agent, was associated with a significant improvement in OS and event-free survival (EFS) compared with adjuvant temozolomide alone in the Children’s Oncology Group ACNS0126 study [125]. This effect was most apparent in patients whose tumors had MGMT overexpression, as well as those who did not undergo gross-total resection and in those with glioblastomas. In a current Phase II study, neoadjuvant temozolomide was associated with an encouraging favorable long-term survival with acceptable toxicity in patients with glioblastoma [131]. Temozolomide in combination with vorinostat was also well tolerated in children with recurrent CNS malignancies with myelosuppression [329]. Vorinostat is a broad inhibitor of histone deacetylase (HDAC) activity which induces apoptosis, inhibits angiogenesis, and downregulates immunosuppressive interleukins. Several Phase III trials using temozolomide in combination with targeted monoclonal antibodies or interferon-alpha in glioblastomas and high-grade gliomas are currently recruiting patients (Table 18.1).
However, poor therapeutic effects were observed in patients with NSCLC. In a current efficacy and safety study of temozolomide in a total of 31 pretreated patients with NSCLC, only two patients achieved partial response and three had stable disease [330]. Moreover, the researchers pointed out that prolonged low daily doses of temozolomide produce minimal activity in patients with advanced NSCLC. In a recent Phase II study, combination therapy of pemetrexed and temozolomide group achieved the same efficacy in PFS and OS as the pemetrexed and cisplatin group, but with less toxicity. High-dose pemetrexed plus temozolomide may be a better regimen for treating NSCLC with brain metastasis due to its better safety profile [126]. A further Phase III study in patients with extensive small-cell lung cancer is currently underway.
Arsenic trioxide (ATO) has recently been introduced as part of a regimen in the therapy and management of acute promyelocytic leukemia (APL) [331]. It is now considered to be “the most biologically active single drug in APL” by a panel of International Leukemia Experts for the European Leukemia Net. The North American Intergroup Study Cancer and Leukemia Group B (CALGB) 9710 demonstrated that adults with APL receiving two cycles of ATO consolidation had significantly improved OS and decreased relapse risk (RR) [332]. It also achieves great success as a single agent and in combination with all-trans retinoic acid (ATRA) in the treatment of APL.
Arsenic trioxide (ATO) is known to induce both autophagy and apoptosis depending on cell types; therefore, its role as an autophagy inducer remains largely uncertain. In some preclinical trials, ATO induces the autophagy pathway in ovarian carcinoma cells, and synergizes with everolimus to induce the cytotoxicity of ovarian cancer cells. The enhanced cytotoxicity is accompanied by the upregulation of ATG5-ATG12 conjugate and LC3-II, a hallmark of autophagy [333]. In another recent study, ATO induces the autophagic degradation of the BCR-ABL1 oncoprotein, known to cause chronic myeloid leukemia (CML) and Ph+ acute lymphoblastic leukemia (ALL) [334]. However in other studies, in the presence or absence of ionizing radiation and in specific low concentrations, ATO induces apoptosis in MTLn3 cells, known to be highly malignant and resistant to both radio- and chemotherapy [335]. Interestingly, in human glioma cells, ATO induces both autophagy and apoptosis in vitro and in vivo, mediated by the inhibition of PI3K/Akt and activation of MAPK signaling pathway [336].
In a Phase I clinical study, ATO given concomitantly with radiation therapy in children with newly diagnosed anaplastic astrocytoma, glioblastoma, or diffuse intrinsic pontine glioma, was safe and well tolerated by patients throughout the entire dose escalation [337]. ATO was also reported to be well tolerated when used in combination with temozolomide and radiotherapy in malignant gliomas [338], or when used in combination with bortezomib, high-dose melphalan, and ascorbic acid in multiple myeloma (MM) patients [339]. A Phase II study to evaluate the efficacy and feasibility of a sequential treatment consisting of induction and consolidation with ATO followed by autologous hematopoietic cell transplantation for relapsed APL revealed that ATO demonstrates outstanding efficacy. Of the 23 patients who underwent autologous hematopoietic cell transplantation with PML-RARα-negative PBSC graft, posttransplant relapse occurred only in three patients, and there was no transplant-related mortality. The 5-year event-free and overall survival rates were 65% and 77%, respectively [340].
A recent study showed that the combination of ATO and ATRA exerts at least equal and probably superior antileukemic efficacy compared with ATRA and standard chemotherapy in low- and intermediate-risk APL [136]. In a Phase III study in which a chemotherapy-free ATRA and ATO treatment regimen was compared with the standard chemotherapy-based regimen (ATRA and idarubicin) in both high-risk and low-risk patients with APL, ATRA and ATO have a high cure rate and less relapse and a lower incidence of liver toxicity [138]. Similarly, a recent Phase III trial showed that ATRA-ATO had an edge over ATRA-chemotherapy over time and that there was significantly greater and more sustained antileukemic efficacy in low- and intermediate-risk APL [139]. ATO consolidation cycles are well tolerated in pediatric patients with APL and allow significant reduction in cumulative anthracycline doses while maintaining excellent survival and a low relapse risk for both standard and high-risk patients with APL [140]. Other Phase III clinical trials using ATO as combination therapy with other chemotherapy drugs and/or tretinoin are currently ongoing for APL.
18.8.3 Autophagy Inhibitors
The knowledge that autophagy plays a role as a cell survival pathway in response to therapeutic and cellular stresses in the tumor microenvironment (which is highly acidic and hypoxic) implies that autophagy may work in favor of cancer cells. Therefore, inhibition of protective autophagy may break the resistance mechanism for survival of the harsh tumor microenvironment and lead to cell death [341]. Since autophagy activities are known to differ according to stages of cancer, modulation of autophagy is postulated to enhance efficacy of anticancer therapy. In a preclinical study, effects of imatinib, with or without different types of autophagy inhibitors, on human malignant glioma cells were investigated [342]. It was demonstrated that suppression of imatinib-induced autophagy by 3-methyladenine (3-MA) or siRNA against ATG5 (which inhibits autophagy at an early stage) attenuates the imatinib-induced cytotoxicity. On the other hand, inhibition of autophagy at a late stage by bafilomycin A1 or RTA 203 enhances imatinib-induced cytotoxicity through the induction of apoptosis [342]. The therapeutic efficiency of imatinib may be augmented by inhibition of autophagy at a late stage, which could help sensitize the glioma cells to anticancer therapy [342].
The current autophagy inhibitors used in trials for human cancer are chloroquine (CQ) and hydroxychloroquine (HCQ). Both drugs are widely used as antimalarial agents and have gained much attention as potential chemosensitizers in treating tumors when used in combination with cytotoxic chemotherapeutic agents [343,344,345]. CQ inhibits lysosomal acidification and prevents autophagy by blocking autophagosome fusion and degradation [344, 346, 347]. CQ also sensitizes cancer cells to chemotherapeutic agents through autophagy-independent mechanisms and has other anticancer effects that are independent of its effects on autophagy [348].
A number of clinical trials have revealed the promising role of CQ, an autophagy inhibitor, as a novel antitumor drug. In an early glioblastoma study, where patients were treated with CQ in conjunction with radiation and temozolomide, the results showed a significantly prolonged median survival compared with controls [349]. Addition of CQ to conventional treatment for GBM also improves mid-term survival of patients [350]. Gemcitabine–CQ combination as a first- or late-line treatment in patients with metastatic or unresectable pancreatic cancer is well tolerated and shows promising effects on the clinical response [141]. A number of Phase I/II trials in solid tumors such as breast cancer are currently recruiting patients.
Although initial glioblastoma studies that used CQ in combination with chemotherapy and radiation therapy revealed median survival greater than control, there was no significant improvement in survival of patients with glioblastoma treated with HCQ [145]. A Phase II trial of HCQ as a single agent in patients with previously treated metastatic pancreatic cancer demonstrated no clinical benefit and provided inconsistent evidence of autophagy inhibition [147]. Since this study was carried out in patients with advance disease, thus, there was a limitation for HCQ to improve end-stage disease outcome [348]. The results appear to be similar to an earlier Phase I study involving patients with advanced NSCLC. Although HCQ, with or without erlotinib, was found to be safe and well tolerated, the overall response rate was as low as 5% [351]. In other Phase I/II trials, HCQ in combination therapy with other drugs such as temozolomide, vorinostat, bortezomib, and gemcitabine are proven to be safe and tolerable among patients with advanced solid tumors and myeloma [142,143,144, 146]. So far, clinical trials of CQ and HCQ as autophagy inhibitors have demonstrated the safety of targeting autophagy for cancer therapy. More potent and autophagy-specific inhibitors such as Lys05 and drugs that target ULK1, VPS34, and ATG4B are in development and early preclinical stage [348]. Table 18.1 summarizes the various drugs targeting the autophagy pathways and clinical trial stages based on published reports as well as other trials listed in the NIH ClinicalTrials.gov website.
18.9 Crosstalk in ER Stress, Autophagy, and Apoptosis
Many cellular processes including apoptosis, autophagy, translation, and energy metabolism are controlled by the ER stress and mTOR signaling pathway. However, the crosstalk among these three signaling pathways has been identified only recently. It has been shown that Akt inactivation mediates ER stress-induced cell death. Long-term exposure to ER stress dephosphorylates Akt, induces DDIT3 expression, and causes cell death. Treatment with PI3K inhibitor alone also decreases phosphorylation of Akt, upregulates DDIT3 expression, and causes cell death, suggesting that PI3K/Akt inhibition specifically induces DDIT3 expression. Thus, Akt inactivation is important in ER stress-induced DDIT3 expression and cell death [352]. In addition, ER stress-induced apoptosis has been reported to be partly mediated by reduced insulin signaling through reduced Akt phosphorylation and increased glycogen synthase kinase 3β (GSK3β) activity. GSK3β is a proapoptotic Akt substrate whose activity is inhibited by Akt phosphorylation [353]. Prolonged ER stress has been shown to inhibit Akt/TSC/mTOR pathway, induce DDIT3 expression, and trigger apoptosis cell death [354]. On the other hand, ER stress negatively regulates Akt/TSC/mTOR pathway to enhance autophagy-mediated cell death [355].
It has been suggested that ER stress promotes autophagy and/or apoptosis via TRIB3-dependent inhibition of Akt/mTOR pathway [49, 50]. It was also proposed that ATF4 negatively regulates mTOR via DNA damage inducible transcript 4 (DDIT4, also known as Redd1) expression in response to ER stress. DDIT4 is a cellular stress responsive gene that has been shown to inhibit mTOR activity [356, 357]. ER stress also leads to CaMKKβ-dependent activation of AMPK, which ultimately leads to inhibition of mTOR and stimulation of autophagy [358]. In addition, it has been demonstrated that ER stress induces BIP expression and promotes an interaction between BIP and Akt. The physical interaction between BIP and Akt at the plasma membrane of cells following induction of ER stress prevents Akt phosphorylation [359]. To sum up, these observations suggest that ER stress may negatively regulate Akt and/or mTOR activity via various pathways, and ultimately leads to cell death.
It is also widely accepted that reactive oxygen species (ROS) generation precedes downstream cellular cascades, including those that determine cell fate either survival (autophagy) or death (apoptosis). Excessive ROS production disrupts the electron transport chain and produces reactive oxygen molecules, leading to depolarization of the mitochondrial membrane and initiation of mitochondria-induced apoptosis. However, ROS generation has also been shown to occur downstream after apoptotic stimulation (TRAIL-induced), or autophagy inhibition [360,361,362]. However, cell fate outcomes are largely dependent on the amount of ROS generated and the cell’s antioxidant response. During starvation, reactive oxygen molecules are produced as a result of Class III PI3K activation that stimulates autophagy through oxidation of ATG4, ultimately increasing the formation of lipidated LC3-rich autophagosomes [363]. Both O2− and H2O2− induce autophagy through AMPK activation and subsequent mTOR inhibition, and by transcriptional regulation of autophagy genes such as SQSTM1 (p62) and BECN1 [364,365,366]. A number of studies have similarly demonstrated that exogenously applied ROS leads to autophagy induction or apoptosis.
Functional relationships between apoptosis and autophagy are gaining much interest, as both cell deaths are not mutually exclusive. Perturbations in the apoptotic machinery, such as caspase inhibition, have been reported to induce both autophagic cell death and necroptosis [367, 368]. Inhibition of autophagy in cancer cells results in an accelerated cell death that manifests the hallmarks of apoptosis including chromatin condensation, MOMP, and activation of caspases [369]. In some cases, mixed phenotypes of both autophagy and apoptosis are detected in response to common stimuli [346, 369]. Studies in a variety of experimental systems indicate that autophagic cell death is likely to be context- and cell type-dependent. Autophagy can delay the onset of apoptosis following starvation, DNA damage, and hemodynamic stress [173]. For example, 1-day fasting causes liver autophagy in rats, but when starvation is prolonged for a few days, hepatocytes succumb to apoptosis [370]. Similarly, hematopoietic cell lines withdrawn from growth factor first activate autophagy, and eventually apoptosis [167]. Studies have also demonstrated that certain compounds have the ability to trigger both apoptosis and autophagic cell deaths simultaneously in cancer cells [371, 372]. Blocking of one pathway will trigger the activation of another [373]. Researchers have also hypothesized that there are factors (either external or internal) that may affect the preferential shunting into either biochemical cascades that will ultimately result in either apoptosis or autophagic cell death [374].
Crosstalks between autophagy and apoptosis exist at multiple levels because both pathways share mediators and pathway regulators. Several signals and pathways involved in autophagy are in common with apoptosis. Starvation and oxidative stress can trigger both apoptosis and autophagy. BCL-2 proteins function to inhibit both apoptosis and autophagy, providing another clue to the interplay between both processes. BECN1, the essential autophagy protein and haplo-insufficient tumor suppressor, interacts with several cofactors such as AMBRA1, BIF-1, and UVRAG to activate the lipid kinase Class III PI3K, and induce autophagy [375]. In normal conditions, BECN1 is bound to and inhibited by BCL-2 or the BCL-2 homologue BCL-XL, well-characterized apoptosis regulators, which involve an interaction between the BH3 domain in BECN1 and the BH3 binding groove of BCL-2/BCL-XL. BH3-only proteins can competitively disrupt the interaction between BECN1 and BCL-2/BCL-XL to induce autophagy. Nutrient starvation can stimulate the dissociation of BECN1 from its inhibitors, either by activating BH3-only proteins (such as BAD) or by posttranslational modifications of BCL-2 (such as phosphorylation) that may reduce its affinity for BECN1 and BH3-only proteins [375]. Antiapoptotic BCL-2 family members participate in the inhibition of autophagy, whereas the proapoptotic BH3-only proteins participate in the induction of autophagy.
A recent finding suggests a link between autophagy and the extrinsic apoptotic pathway mediated by p62/SQSTM1. Autophagy is recently known to be responsible in selective degradation of polyubiquitinated proteins via SQSTM1, which encodes for p62 protein. P62 interacts with LC3 via its LC3 interacting region (LIR). Recent studies indicate that p62 is recruited to damaged mitochondria via binding to ubiquitinated outer mitochondrial membrane proteins, suggesting that p62 may serve as an autophagy receptor for ubiquitinated proteins and damaged mitochondria [376,377,378]. In addition to its role in autophagy, p62 mediates a cell’s decision to undergo apoptosis or survival through its organization of signaling complexes in the cytoplasm [377, 379, 380]. Upon cytokine stimulation, p62 activates the NF-κB pathway, which subsequently induces the prosurvival genes, such as antiapoptotic and cell proliferation genes, and induces the expression of inflammatory genes such as cytokines, chemokines, and adhesion molecules [380,381,382,383]. However, p62 is also found to activate caspase-8 in the extrinsic apoptosis pathway, resulting in the initiation of apoptosis and cell death [379].
The expression of Patched (Ptc) induces apoptosis, but this activity is suppressed by its ligand, sonic hedgehog (SHH). Interestingly, hedgehog inhibition is found to induce autophagy through upregulation of BNIP3, and is also found to increase apoptosis in hepatocellular carcinoma cells at the same time [384]. In a recent study, apoptosis suppressed by the knocking down of PP2A can be reversed by the administration of 3-MA, a known autophagy inhibitor. The elevated accumulation of LC3-II and the decline of the autophagy substrate p62 are also observed in PP2Ac-small interfering RNA transfected cells. However, overexpression of PP2Ac suppresses the accumulation of LC3-II and restores p62 [385]. Interestingly, 3-MA increases cell death induced by diamindichloridoplatin (DDP), which suggests the protective function of autophagy in DDP-induced cell death [385].
18.10 Future Directions
There are increasing evidences that three major processes, i.e., apoptosis, ER stress, and autophagy, share overlapping molecular pathways and can occur in parallel under similar conditions. Fundamental knowledge in apoptosis, ER stress, and autophagy has also generated a great deal of insight into the pathogenesis of cancer, and has provided important considerations in strategizing cancer pharmacotherapy. Much effort and investment have been devoted to experimental drugs modulating apoptosis, ER stress, and autophagy. A number of drugs have proven to be promising during preclinical and clinical studies, but these drugs appear to be effective in one type of cancer and not in other. The percentage of patients who totally responded or partially responded to these treatments, either as single agent or in combination therapies, is relatively low, even though the outcome of these trials suggests some potential. These unforeseen effects are probably due to the specific-targeted nature of the therapy, in addition to the interconnected relationships between these cell death pathways. The contradictory role of autophagy and the status of autophagy in the human tumors concerned remain speculative, and further complicate the response to conventional anticancer treatment.
Currently, modulating apoptosis, ER stress, and autophagy by various means may be an important strategy to fight against the disease. Cancers which are resistant to the apoptotic effects of certain chemotherapy drugs may be sensitive to drugs that evoke ER stress or autophagic cell deaths. An intact autophagy pathway has a role in promoting carcinogenesis as well as in suppressing it. It also has a role in the development of resistance to treatment. Therefore, if autophagy response and activity are normal in tumors, combining standard chemotherapy drugs with autophagy inhibitors may sensitize tumor cells to anticancer agents. Cancer cells which present defects in the autophagy pathway may be managed by replacement of autophagy-inducing signals, e.g., proautophagics, or by inhibiting mTOR kinase. In some other cases, utilizing both autophagy and apoptosis inducers may present a deadly strategy against highly resistant tumors. Thus, devising personalized pharmacotherapeutic strategy based on the autophagy status of the tumors has become an attractive option and offers significant potential to be translated into the clinic.
Combination of anticancer drugs of many different classes with autophagy inhibitors and inducers is underway but with little rationale for deciding or selecting patients who are most likely to benefit from these therapies. So far, targeted drugs like oblimersen, bortezomib, and mTOR inhibitors such as everolimus and ridaforolimus have shown to be useful in some clinical trials. These novel classes of drugs appear to work synergistically in combination with other chemotherapeutics, and have also shown specific activities against certain cancers. Clinical trials of CQ or HCQ as autophagy inhibitors have also demonstrated the safety of targeting autophagy for cancer therapy. Since these drugs are specifically targeted against certain molecules or receptors in the pathway, further unveiling of the tumor’s characteristics such as receptor or protein status may be critical in assessing patient’s response and clinical trial success. Furthermore, a number of known genes that play a role in these cell death pathways are either activated or inactivated in several cancers. This will certainly affect not only the promotion and progression of cancer, but also their response to treatment. Therefore, to optimize and personalize treatment strategies, the genetic profile of the tumors is important.
For example, RAS- and BRAF-mutant tumours are often associated with high levels of autophagy and exhibit autophagy dependency. These would be good markers to select patients in which autophagy can be inhibited therapeutically [285, 348, 386,387,388]. Other markers include signal transducer and activator of transcription 3 (STAT3) and IL6 in breast cancer cells [389], JNK1 in colon cancer [390], and EGFR-mutated or amplified tumors [391]. Some clinical trials have already used these markers to evaluate efficacy and for validation. This may provide information on the optimal point in the pathway to be targeted, and can also be identified as prognostic markers. At the same time, the development of both robust tissue markers and relevant techniques that can be used in the clinical context needs to occur along with novel treatments, which will be another challenge.
Although recent studies have incorporated some predictive biomarkers by examining tumor status, the utility of such practice remains nonconclusive. For example, the expression of peptidyl O-glycosyltransferase GaLNT14 has been proposed to be a potential marker of dulanermin or Apo2L/TRAIL activity in NSCLC as high GaLNT14 mRNA and protein expression in tumor cell lines are associated with Apo2L/TRAIL sensitivity [392]. An increase in PFS and OS was observed in GaLNT14-positive patients with advanced NSCLC in the dulanermin arm, indicating the potential predictive response biomarker for Apo2L/TRAIL-based cancer therapy [393]. On the other hand, in a Phase Ib/II trial on mapatumumab, a humanized mAb against TRAIL-R1, strong expression of TRAIL-R1 (indicated by immunohistochemical staining) did not appear to be a prerequisite for the effectiveness of mapatumumab in patients with relapsed or refractory follicular lymphoma [394]. Noteworthy, in the two patients who experienced a partial or complete response, the TRAIL-R1 staining was either undetected or weak [394]. However, this could be an isolated case, and trials with bigger sample size should be carried out. Tumor profiling would remain as a good strategy to identify patients who may respond to the relevant treatment.
Fundamental knowledge of cell death pathways remains an area of major interest among scientists in the field of cancer. More studies to characterize these pathways and identify potential targets, and further evaluation of the efficacy of the current drugs in various cancers are certainly warranted.
References
Bhat TA, Chaudhary AK, Kumar S, O’Malley J, Inigo JR, Kumar R, et al. Endoplasmic reticulum-mediated unfolded protein response and mitochondrial apoptosis in cancer. Biochim Biophys Acta. 2017;1867(1):58–66.
Sasaki K, Yoshida H. Organelle autoregulation-stress responses in the ER, Golgi, mitochondria and lysosome. J Biochem. 2015;157(4):185–95.
Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14(9):581–97.
Corazzari M, Gagliardi M, Fimia GM, Piacentini M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front Oncol. 2017;7:78.
Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking ER stress to autophagy: potential implications for cancer therapy. Int J Cell Biol. 2010;2010:930509.
Tu BP, Weissman JS. Oxidative protein folding in eukaryotes. J Cell Biol. 2004;164(3):341.
Oakes SA. Endoplasmic reticulum proteostasis: a key checkpoint in cancer. Am J Physiol Cell Physiol. 2017;312(2):C93–C102.
Schonthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem Pharmacol. 2013;85(5):653–66.
McCracken AA, Brodsky JL. Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD). BioEssays. 2003;25(9):868–77.
Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7(8):766–72.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102.
Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–94.
Shore GC, Papa FR, Oakes SA. Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol. 2011;23(2):143–9.
Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434(2):181–8.
Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem. 2002;277(15):13045–52.
Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12(9):703–19.
Woehlbier U, Hetz C. Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci. 2011;36(6):329–37.
Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110(10):1383–8.
Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153(5):1011–22.
Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J Biochem. 2012;151(3):217–9.
Ron D, Hubbard SR. How IRE1 reacts to ER stress. Cell. 2008;132(1):24–6.
Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, Lee AH, Qian SB, Zhao H, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93.
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–6.
Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, et al. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13(3):351–64.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–91.
Schonthal AH. Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Lett. 2009;275(2):163–9.
Schleicher SM, Moretti L, Varki V, Lu B. Progress in the unraveling of the endoplasmic reticulum stress/autophagy pathway and cancer: implications for future therapeutic approaches. Drug Resist Updat. 2010;13(3):79–86.
Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4(11):e374.
Lin JH, Li H, Zhang Y, Ron D, Walter P. Divergent effects of PERK and IRE1 signaling on cell viability. PLoS One. 2009;4(1):e4170.
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–59.
Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7(9):880–5.
Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–49.
Zou W, Yue P, Khuri FR, Sun SY. Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res. 2008;68(18):7484–92.
Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279(44):45495–502.
Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30(50):16938–48.
Hsin IL, Hsiao YC, Wu MF, Jan MS, Tang SC, Lin YW, et al. Lipocalin 2, a new GADD153 target gene, as an apoptosis inducer of endoplasmic reticulum stress in lung cancer cells. Toxicol Appl Pharmacol. 2012;263(3):330–7.
Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24(6):1243–55.
Bouchet BP. Caron de Fromentel C, Puisieux A, Galmarini CM. p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol. 2006;58(3):190–207.
Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008;27(Suppl 1):S71–83.
Sung HK, Chan YK, Han M, Jahng JWS, Song E, Danielson E, et al. Lipocalin-2 (NGAL) attenuates autophagy to exacerbate cardiac apoptosis induced by myocardial ischemia. J Cell Physiol. 2017;232(8):2125–34.
Saleem S, Biswas SC. Tribbles pseudokinase 3 induces both apoptosis and autophagy in amyloid-beta-induced neuronal death. J Biol Chem. 2017;292(7):2571–85.
Lu M, Lawrence DA, Marsters S, Acosta-Alvear D, Kimmig P, Mendez AS, et al. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science. 2014;345(6192):98–101.
B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–99.
Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol. 2009;625(1–3):234–46.
De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, et al. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem. 2006;281(30):21353–61.
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–39.
Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science. 1996;272(5266):1347–9.
Morishima N, Nakanishi K, Nakano A. Activating transcription factor-6 (ATF6) mediates apoptosis with reduction of myeloid cell leukemia sequence 1 (Mcl-1) protein via induction of WW domain binding protein 1. J Biol Chem. 2011;286(40):35227–35.
Salazar M, Carracedo A, Salanueva ÍJ, Hernández-Tiedra S, Lorente M, Egia A, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119(5):1359–72.
Zou CG, Cao XZ, Zhao YS, Gao SY, Li SD, Liu XY, et al. The molecular mechanism of endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal cells: the protective effect of insulin-like growth factor I. Endocrinology. 2009;150(1):277–85.
Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013;23(11):547–55.
Tam AB, Koong AC, Niwa M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014;9(3):850–8.
Pagliarini V, Giglio P, Bernardoni P, De Zio D, Fimia GM, Piacentini M, et al. Downregulation of E2F1 during ER stress is required to induce apoptosis. J Cell Sci. 2015;128(6):1166–79.
Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12(15):2245–62.
DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med. 2006;6(7):739–48.
Hoyer-Hansen M, Jaattela M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14(9):1576–82.
Szegezdi E, Macdonald DC, Ni Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am J Physiol Cell Physiol. 2009;296(5):C941–53.
Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163.
Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5(7):723–8.
Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol. 2003;38(5):605–14.
Ranganathan AC, Adam AP, Zhang L, Aguirre-Ghiso JA. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: an adaptive advantage for metastatic cells? Cancer Biol Ther. 2006;5(7):729–35.
Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64(17):5943–7.
Wu X, Xin Z, Zhang W, Zheng S, Wu J, Chen K, et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. Int J Cancer. 2014;135(1):61–8.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8.
Jabouille A, Delugin M, Pineau R, Dubrac A, Soulet F, Lhomond S, et al. Glioblastoma invasion and cooption depend on IRE1alpha endoribonuclease activity. Oncotarget. 2015;6(28):24922–34.
Tang ZH, Su MX, Guo X, Jiang XM, Jia L, Chen X, et al. Increased expression of IRE1alpha associates with the resistant mechanism of osimertinib (AZD9291)-resistant non-small cell lung cancer HCC827/OSIR cells. Anti-Cancer Agents Med Chem. 2018;18(4):550–5.
Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta. 2014;1843(10):2143–9.
Corazzari M, Lovat PE, Armstrong JL, Fimia GM, Hill DS, Birch-Machin M, et al. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: the role of stress proteins ERdj5 and ERp57. Br J Cancer. 2007;96(7):1062–71.
Vatolin S, Phillips JG, Jha BK, Govindgari S, Hu J, Grabowski D, et al. Novel protein disulfide isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Res. 2016;76(11):3340–50.
Yu SJ, Yoon JH, Yang JI, Cho EJ, Kwak MS, Jang ES, et al. Enhancement of hexokinase II inhibitor-induced apoptosis in hepatocellular carcinoma cells via augmenting ER stress and anti-angiogenesis by protein disulfide isomerase inhibition. J Bioenerg Biomembr. 2012;44(1):101–15.
Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, et al. Discovery of an orally active small-molecule irreversible inhibitor of protein disulfide isomerase for ovarian cancer treatment. Proc Natl Acad Sci U S A. 2012;109(40):16348–53.
Won JK, Yu SJ, Hwang CY, Cho SH, Park SM, Kim K, et al. Protein disulfide isomerase inhibition synergistically enhances the efficacy of sorafenib for hepatocellular carcinoma. Hepatology. 2017;66(3):855–68.
Foster CK, Thorpe C. Challenges in the evaluation of thiol-reactive inhibitors of human protein disulfide Isomerase. Free Radic Biol Med. 2017;108:741–9.
Bruning A, Burger P, Vogel M, Rahmeh M, Gingelmaiers A, Friese K, et al. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol Ther. 2009;8(3):226–32.
Pyrko P, Kardosh A, Wang W, Xiong W, Schonthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res. 2007;67(22):10920–8.
Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13(17):5183–94.
Wilson JM, Fokas E, Dutton SJ, Patel N, Hawkins MA, Eccles C, et al. ARCII: a phase II trial of the HIV protease inhibitor Nelfinavir in combination with chemoradiation for locally advanced inoperable pancreatic cancer. Radiother Oncol. 2016;119(2):306–11.
Driessen C, Kraus M, Joerger M, Rosing H, Bader J, Hitz F, et al. Treatment with the HIV protease inhibitor nelfinavir triggers the unfolded protein response and may overcome proteasome inhibitor resistance of multiple myeloma in combination with bortezomib: a phase I trial (SAKK 65/08). Haematologica. 2016;101(3):346–55.
Hill EJ, Roberts C, Franklin JM, Enescu M, West N, MacGregor TP, et al. Clinical trial of oral nelfinavir before and during radiation therapy for advanced rectal cancer. Clin Cancer Res. 2016;22(8):1922–31.
Hoover AC, Milhem MM, Anderson CM, Sun W, Smith BJ, Hoffman HT, et al. Efficacy of nelfinavir as monotherapy in refractory adenoid cystic carcinoma: results of a phase II clinical trial. Head Neck. 2015;37(5):722–6.
Mahalingam D, Wilding G, Denmeade S, Sarantopoulas J, Cosgrove D, Cetnar J, et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br J Cancer. 2016;114(9):986–94.
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Maru D, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–8.
Blank CU, Larkin J, Arance AM, Hauschild A, Queirolo P, Del Vecchio M, et al. Open-label, multicentre safety study of vemurafenib in 3219 patients with BRAFV600 mutation-positive metastatic melanoma: 2-year follow-up data and long-term responders’ analysis. Eur J Cancer. 2017;79:176–84.
Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–901.
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland A, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
Davies MA, Saiag P, Robert C, Grob JJ, Flaherty KT, Arance A, et al. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): a multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017;18(7):863–73.
Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol. 2017;28(7):1631–9.
Long GV, Eroglu Z, Infante J, Patel S, Daud A, Johnson DB, et al. Long-term outcomes in patients with BRAF v600-mutant metastatic melanoma who received dabrafenib combined with trametinib. J Clin Oncol. 2018;36(7):667–73.
Delord JP, Robert C, Nyakas M, McArthur GA, Kudchakar R, Mahipal A, et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin Cancer Res. 2017;23(18):5339–48.
van Geel R, Tabernero J, Elez E, Bendell JC, Spreafico A, Schuler M, et al. A phase ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017;7(6):610–9.
Vo KT, Karski EE, Nasholm NM, Allen S, Hollinger F, Gustafson WC, et al. Phase 1 study of sirolimus in combination with oral cyclophosphamide and topotecan in children and young adults with relapsed and refractory solid tumors. Oncotarget. 2017;8(14):23851–61.
Li J, Kluger H, Devine L, Lee JJ, Kelly WK, Rink L, et al. Phase I study of safety and tolerability of sunitinib in combination with sirolimus in patients with refractory solid malignancies and determination of VEGF (VEGF-A) and soluble VEGF-R2 (sVEGFR2) in plasma. Cancer Chemother Pharmacol. 2016;77(6):1193–200.
Hollebecque A, Bahleda R, Faivre L, Adam J, Poinsignon V, Paci A, et al. Phase I study of temsirolimus in combination with cetuximab in patients with advanced solid tumours. Eur J Cancer. 2017;81:81–9.
Becher OJ, Gilheeney SW, Khakoo Y, Lyden DC, Haque S, De Braganca KC, et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr Blood Cancer. 2017;64(7):e26409.
Waqar SN, Baggstrom MQ, Morgensztern D, Williams K, Rigden C, Govindan R. A phase I trial of temsirolimus and pemetrexed in patients with advanced non-small cell lung cancer. Chemotherapy. 2016;61(3):144–7.
Myers AP, Filiaci VL, Zhang Y, Pearl M, Behbakht K, Makker V, et al. Tumor mutational analysis of GOG248, a phase II study of temsirolimus or temsirolimus and alternating megestrol acetate and tamoxifen for advanced endometrial cancer (EC): an NRG oncology/gynecologic oncology group study. Gynecol Oncol. 2016;141(1):43–8.
Korfel A, Schlegel U, Herrlinger U, Dreyling M, Schmidt C, von Baumgarten L, et al. Phase II trial of temsirolimus for relapsed/refractory primary CNS lymphoma. J Clin Oncol. 2016;34(15):1757–63.
Emons G, Kurzeder C, Schmalfeldt B, Neuser P, de Gregorio N, Pfisterer J, et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol Oncol. 2016;140(3):450–6.
Narayan V, Vapiwala N, Mick R, Subramanian P, Christodouleas JP, Bekelman JE, et al. Phase 1 trial of everolimus and radiation therapy for salvage treatment of biochemical recurrence in prostate cancer patients following prostatectomy. Int J Radiat Oncol Biol Phys. 2017;97(2):355–61.
Basho RK, Gilcrease M, Murthy RK, Helgason T, Karp DD, Meric-Bernstam F, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol. 2017;3(4):509–15.
Johnston PB, LaPlant B, McPhail E, Habermann TM, Inwards DJ, Micallef IN, et al. Everolimus combined with R-CHOP-21 for new, untreated, diffuse large B-cell lymphoma (NCCTG 1085 [Alliance]): safety and efficacy results of a phase 1 and feasibility trial. Lancet Haematol. 2016;3(7):e309–16.
Chung V, Frankel P, Lim D, Yeon C, Leong L, Chao J, et al. Phase Ib trial of mFOLFOX6 and everolimus (NSC-733504) in patients with metastatic gastroesophageal adenocarcinoma. Oncology. 2016;90(6):307–12.
Colon-Otero G, Weroha SJ, Foster NR, Haluska P, Hou X, Wahner-Hendrickson AE, et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol Oncol. 2017;146(1):64–8.
Rugo HS, Seneviratne L, Beck JT, Glaspy JA, Peguero JA, Pluard TJ, et al. Prevention of everolimus-related stomatitis in women with hormone receptor-positive, HER2-negative metastatic breast cancer using dexamethasone mouthwash (SWISH): a single-arm, phase 2 trial. Lancet Oncol. 2017;18(5):654–62.
Bennani NN, LaPlant BR, Ansell SM, Habermann TM, Inwards DJ, Micallef IN, et al. Efficacy of the oral mTORC1 inhibitor everolimus in relapsed or refractory indolent lymphoma. Am J Hematol. 2017;92(5):448–53.
Cirkel GA, Hamberg P, Sleijfer S, Loosveld OJL, Dercksen MW, Los M, et al. Alternating treatment with pazopanib and everolimus vs continuous pazopanib to delay disease progression in patients with metastatic clear cell renal cell cancer: the ROPETAR randomized clinical trial. JAMA Oncol. 2017;3(4):501–8.
Schneider TC, de Wit D, Links TP, van Erp NP, van der Hoeven JJ, Gelderblom H, et al. Everolimus in patients with advanced follicular-derived thyroid cancer: results of a phase II clinical trial. J Clin Endocrinol Metab. 2017;102(2):698–707.
Chow H, Ghosh PM, deVere White R, Evans CP, Dall’Era MA, Yap SA, et al. A phase 2 clinical trial of everolimus plus bicalutamide for castration-resistant prostate cancer. Cancer. 2016;122(12):1897–904.
Kim SJ, Shin DY, Kim JS, Yoon DH, Lee WS, Lee H, et al. A phase II study of everolimus (RAD001), an mTOR inhibitor plus CHOP for newly diagnosed peripheral T-cell lymphomas. Ann Oncol. 2016;27(4):712–8.
Armstrong AJ, Halabi S, Eisen T, Broderick S, Stadler WM, Jones RJ, et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (ASPEN): a multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016;17(3):378–88.
Niikura N, Ota Y, Hayashi N, Naito M, Kashiwabara K, Watanabe K, et al. Evaluation of oral care to prevent oral mucositis in estrogen receptor-positive metastatic breast cancer patients treated with everolimus (oral care-BC): randomized controlled phase III trial. Jpn J Clin Oncol. 2016;46(9):879–82.
Chon HS, Kang S, Lee JK, Apte SM, Shahzad MM, Williams-Elson I, et al. Phase I study of oral ridaforolimus in combination with paclitaxel and carboplatin in patients with solid tumor cancers. BMC Cancer. 2017;17(1):407.
Pearson AD, Federico SM, Aerts I, Hargrave DR, DuBois SG, Iannone R, et al. A phase 1 study of oral ridaforolimus in pediatric patients with advanced solid tumors. Oncotarget. 2016;7(51):84736–47.
Rugo HS, Tredan O, Ro J, Morales SM, Campone M, Musolino A, et al. A randomized phase II trial of ridaforolimus, dalotuzumab, and exemestane compared with ridaforolimus and exemestane in patients with advanced breast cancer. Breast Cancer Res Treat. 2017;165(3):601–9.
Baselga J, Morales SM, Awada A, Blum JL, Tan AR, Ewertz M, et al. A phase II study of combined ridaforolimus and dalotuzumab compared with exemestane in patients with estrogen receptor-positive breast cancer. Breast Cancer Res Treat. 2017;163(3):535–44.
Fazio N, Buzzoni R, Baudin E, Antonuzzo L, Hubner RA, Lahner H, et al. A phase II study of BEZ235 in patients with everolimus-resistant, advanced pancreatic neuroendocrine tumours. Anticancer Res. 2016;36(2):713–9.
Seront E, Rottey S, Filleul B, Glorieux P, Goeminne JC, Verschaeve V, et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. BJU Int. 2016;118(3):408–15.
Cohen MH, Johnson JR, Pazdur R. Food and Drug Administration drug approval summary: temozolomide plus radiation therapy for the treatment of newly diagnosed glioblastoma multiforme. Clin Cancer Res. 2005;11(19 Pt 1):6767–71.
Wong ET, Timmons J, Callahan A, O’Loughlin L, Giarusso B, Alsop DC. Phase I study of low-dose metronomic temozolomide for recurrent malignant gliomas. BMC Cancer. 2016;16(1):914.
Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, et al. Phase I/II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother. 2016;65(12):1499–509.
Aoki T, Arakawa Y, Ueba T, Oda M, Nishida N, Akiyama Y, et al. Phase I/II study of temozolomide plus nimustine chemotherapy for recurrent malignant gliomas: Kyoto neuro-oncology group. Neurol Med Chir (Tokyo). 2017;57(1):17–27.
Glass J, Won M, Schultz CJ, Brat D, Bartlett NL, Suh JH, et al. Phase I and II study of induction chemotherapy with methotrexate, rituximab, and temozolomide, followed by whole-brain radiotherapy and postirradiation temozolomide for primary CNS lymphoma: NRG oncology RTOG 0227. J Clin Oncol. 2016;34(14):1620–5.
Herrlinger U, Schafer N, Steinbach JP, Weyerbrock A, Hau P, Goldbrunner R, et al. Bevacizumab plus irinotecan versus temozolomide in newly diagnosed o6-methylguanine-DNA methyltransferase nonmethylated glioblastoma: the randomized GLARIUS trial. J Clin Oncol. 2016;34(14):1611–9.
Jakacki RI, Cohen KJ, Buxton A, Krailo MD, Burger PC, Rosenblum MK, et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: a report of the Children’s Oncology Group ACNS0423 study. Neuro-Oncology. 2016;18(10):1442–50.
He Q, Bi X, Ren C, Wang Y, Zou P, Zhang H, et al. Phase II study of the efficacy and safety of high-dose pemetrexed in combination with cisplatin versus temozolomide for the treatment of non-small cell lung cancer with brain metastases. Anticancer Res. 2017;37(8):4711–6.
Patel SP, Kim DW, Bassett RL, Cain S, Washington E, Hwu WJ, et al. A phase II study of ipilimumab plus temozolomide in patients with metastatic melanoma. Cancer Immunol Immunother. 2017;66(10):1359–66.
Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. 2017;18(7):946–57.
Calegari MA, Inno A, Monterisi S, Orlandi A, Santini D, Basso M, et al. A phase 2 study of temozolomide in pretreated metastatic colorectal cancer with MGMT promoter methylation. Br J Cancer. 2017;116(10):1279–86.
Modak S, Kushner BH, Basu E, Roberts SS, Cheung NK. Combination of bevacizumab, irinotecan, and temozolomide for refractory or relapsed neuroblastoma: results of a phase II study. Pediatr Blood Cancer. 2017;64(8):e26448.
Shenouda G, Souhami L, Petrecca K, Owen S, Panet-Raymond V, Guiot MC, et al. A phase 2 trial of neoadjuvant temozolomide followed by hypofractionated accelerated radiation therapy with concurrent and adjuvant temozolomide for patients with glioblastoma. Int J Radiat Oncol Biol Phys. 2017;97(3):487–94.
Baumert BG, Hegi ME, van den Bent MJ, von Deimling A, Gorlia T, Hoang-Xuan K, et al. Temozolomide chemotherapy versus radiotherapy in high-risk low-grade glioma (EORTC 22033-26033): a randomised, open-label, phase 3 intergroup study. Lancet Oncol. 2016;17(11):1521–32.
Zhao H, Sun G, Kong D, Zhang Y, Shi W, Zhao M, et al. A phase II study of arsenic trioxide in patients with relapsed or refractory malignant lymphoma. Med Oncol. 2015;32(3):79.
Wang H, Liu Y, Wang X, Liu D, Sun Z, Wang C, et al. Randomized clinical control study of locoregional therapy combined with arsenic trioxide for the treatment of hepatocellular carcinoma. Cancer. 2015;121(17):2917–25.
Modak S, Zanzonico P, Carrasquillo JA, Kushner BH, Kramer K, Cheung NK, et al. Arsenic trioxide as a radiation sensitizer for 131i-metaiodobenzylguanidine therapy: results of a phase II study. J Nucl Med. 2016;57(2):231–7.
Cicconi L, Divona M, Ciardi C, Ottone T, Ferrantini A, Lavorgna S, et al. PML-RARalpha kinetics and impact of FLT3-ITD mutations in newly diagnosed acute promyelocytic leukaemia treated with ATRA and ATO or ATRA and chemotherapy. Leukemia. 2016;30(10):1987–92.
Owonikoko TK, Zhang G, Kim HS, Stinson RM, Bechara R, Zhang C, et al. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J Transl Med. 2016;14(1):111.
Burnett AK, Russell NH, Hills RK, Bowen D, Kell J, Knapper S, et al. Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2015;16(13):1295–305.
Platzbecker U, Avvisati G, Cicconi L, Thiede C, Paoloni F, Vignetti M, et al. Improved outcomes with retinoic acid and arsenic trioxide compared with retinoic acid and chemotherapy in non-high-risk acute promyelocytic leukemia: final results of the randomized Italian-German APL0406 trial. J Clin Oncol. 2017;35(6):605–12.
Kutny MA, Alonzo TA, Gerbing RB, Wang YC, Raimondi SC, Hirsch BA, et al. Arsenic trioxide consolidation allows anthracycline dose reduction for pediatric patients with acute promyelocytic leukemia: report from the children’s oncology group phase III historically controlled trial AAML0631. J Clin Oncol. 2017;35(26):3021–9.
Samaras P, Tusup M, Nguyen-Kim TDL, Seifert B, Bachmann H, von Moos R, et al. Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol. 2017;80(5):1005–12.
Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy. 2014;10(8):1369–79.
Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy. 2014;10(8):1403–14.
Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014;10(8):1380–90.
Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10(8):1359–68.
Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, et al. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 2015;22(13):4402–10.
Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014;19(6):637–8.
Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med. 2012;4(140):140ra86.
Koselny K, Green J, Favazzo L, Glazier VE, DiDone L, Ransford S, et al. Antitumor/antifungal celecoxib derivative ar-12 is a non-nucleoside inhibitor of the ANL-family adenylating enzyme acetyl CoA synthetase. ACS Infect Dis. 2016;2(4):268–80.
Gao M, Yeh PY, Lu YS, Hsu CH, Chen KF, Lee WC, et al. OSU-03012, a novel celecoxib derivative, induces reactive oxygen species-related autophagy in hepatocellular carcinoma. Cancer Res. 2008;68(22):9348–57.
Park MA, Yacoub A, Rahmani M, Zhang G, Hart L, Hagan MP, et al. OSU-03012 stimulates PKR-like endoplasmic reticulum-dependent increases in 70-kDa heat shock protein expression, attenuating its lethal actions in transformed cells. Mol Pharmacol. 2008;73(4):1168–84.
Kucab JE, Lee C, Chen CS, Zhu J, Gilks CB, Cheang M, et al. Celecoxib analogues disrupt Akt signaling, which is commonly activated in primary breast tumours. Breast Cancer Res. 2005;7(5):R796–807.
Giglio P, Fimia GM, Lovat PE, Piacentini M, Corazzari M. Fateful music from a talented orchestra with a wicked conductor: connection between oncogenic BRAF, ER stress, and autophagy in human melanoma. Mol Cell Oncol. 2015;2(3):e995016.
Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015;22(6):946–58.
Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28(3):466–74.
Andre T, de Gramont A, Vernerey D, Chibaudel B, Bonnetain F, Tijeras-Raballand A, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. J Clin Oncol. 2015;33(35):4176–87.
Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20(20):5322–30.
Gavin PG, Colangelo LH, Fumagalli D, Tanaka N, Remillard MY, Yothers G, et al. Mutation profiling and microsatellite instability in stage II and III colon cancer: an assessment of their prognostic and oxaliplatin predictive value. Clin Cancer Res. 2012;18(23):6531–41.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–16.
Gomez-Roca CA, Delord J, Robert C, Hidalgo M, von Moos R, Arance A, et al. 535pencorafenib (lgx818), an oral BRAF inhibitor, in patients (pts) with braf v600e metastatic colorectal cancer (mCRC): results of dose expansion in an open-label, phase 1 study. Ann Oncol. 2014;25(suppl_4):iv182–93.
Gao W, Kang J-H, Liao Y, Li M, Yin X-M. Autophagy and cell death. In: Yin X-M, Dong Z, editors. Essential of apoptosis. Pittsburgh: Humana Press; 2009. p. 671–88.
Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17(24):7151–60.
de Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92.
Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36(12):2445–62.
Lleo A, Invernizzi P, Selmi C, Coppel RL, Alpini G, Podda M, et al. Autophagy: highlighting a novel player in the autoimmunity scenario. J Autoimmun. 2007;29(2–3):61–8.
Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–77.
Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–48.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–6.
Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–7.
Roy S, Debnath J. Autophagy and tumorigenesis. Semin Immunopathol. 2010;32(4):383–96.
Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 2007;581(11):2156–61.
Ferraro E, Cecconi F. Autophagic and apoptotic response to stress signals in mammalian cells. Arch Biochem Biophys. 2007;462(2):210–9.
Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–34.
Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–22.
Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29(1):157–71.
Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–305.
Parthasarathy S, Bin Azizi J, Ramanathan S, Ismail S, Sasidharan S, Said MI, et al. Evaluation of antioxidant and antibacterial activities of aqueous, methanolic and alkaloid extracts from Mitragyna speciosa (Rubiaceae family) leaves. Molecules. 2009;14(10):3964–74.
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003.
Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5(5):649–62.
Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;86:225–44.
Papinski D, Kraft C. Regulation of autophagy by signaling through the Atg1/ULK1 complex. J Mol Biol. 2016;428(9 Pt A):1725–41.
Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440(2):283–91.
Lazarus MB, Novotny CJ, Shokat KM. Structure of the human autophagy initiating kinase ULK1 in complex with potent inhibitors. ACS Chem Biol. 2015;10(1):257–61.
Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186(6):773–82.
Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586–96.
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–96.
Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–72.
Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–5.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–6.
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9(10):1142–51.
Backer JM. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J. 2016;473(15):2251–71.
Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–30.
Kametaka S, Okano T, Ohsumi M, Ohsumi Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J Biol Chem. 1998;273(35):22284–91.
Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes--Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17(4):1527–39.
Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2008;105(49):19211–6.
Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–76.
Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol. 2004;15(2):231–6.
Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29(11):1792–802.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–8.
Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19(5):2092–100.
Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–302.
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–12.
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–12.
Feng Y, Klionsky DJ. Autophagic membrane delivery through ATG9. Cell Res. 2017;27(2):161–2.
Zhou C, Ma K, Gao R, Mu C, Chen L, Liu Q, et al. Regulation of mATG9 trafficking by Src- and ULK1-mediated phosphorylation in basal and starvation-induced autophagy. Cell Res. 2017;27(2):184–201.
Popovic D, Dikic I. TBC1D5 and the AP2 complex regulate ATG9 trafficking and initiation of autophagy. EMBO Rep. 2014;15(4):392–401.
Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10(7):776–87.
Liu B, Cheng Y, Liu Q, Bao JK, Yang JM. Autophagic pathways as new targets for cancer drug development. Acta Pharmacol Sin. 2010;31(9):1154–64.
Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222.
Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282(24):4672–8.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19(1):107–20.
Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22.
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224.
Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4(2):117–26.
Martin KA, Blenis J. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv Cancer Res. 2002;86:1–39.
Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9(3–4):65–76.
Boulay A, Lane HA. The mammalian target of rapamycin kinase and tumor growth inhibition. Recent Results Cancer Res. 2007;172:99–124.
Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–22.
Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–46.
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302.
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–89.
Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–68.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35.
Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904.
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–75.
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–86.
Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–15.
Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009;122(Pt 20):3589–94.
Guertin DA, Kim D-H, Sabatini DM. Growth control through the mTOR network. In: Hall MN, et al., editors. Cell growth: control of cell size. New York: Cold Spring Harbor Laboratory Press; 2004. p. 193–234.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45.
Kirken RA, Wang YL. Molecular actions of sirolimus: sirolimus and mTor. Transplant Proc. 2003;35(3 Suppl):227S–30S.
Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804(3):433–9.
Proud CG. Regulation of mammalian translation factors by nutrients. Eur J Biochem. 2002;269(22):5338–49.
Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5(7):973–9.
Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. 2008;8(2):78–91.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90.
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activation protein complex toward Rheb. Curr Biol. 2003;13:1259–68.
Rosner M, Siegel N, Valli A, Fuchs C, Hengstschlager M. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids. 2010;38(1):223–8.
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–8.
Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11(8):353–61.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–68.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101.
Kaur A, Sharma S. Mammalian target of rapamycin (mTOR) as a potential therapeutic target in various diseases. Inflammopharmacology. 2017;25(3):293–312.
Ikenoue T, Inoki K, Yang Q, Zhou X, Guan KL. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008;27(14):1919–31.
Gan X, Wang J, Wang C, Sommer E, Kozasa T, Srinivasula S, et al. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat Cell Biol. 2012;14(7):686–96.
Li X, Gao T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 2014;15(2):191–8.
Sciarretta S, Zhai P, Maejima Y, Del Re DP, Nagarajan N, Yee D, et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015;11(1):125–36.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505.
Yu FX, Guan KL. The hippo pathway: regulators and regulations. Genes Dev. 2013;27(4):355–71.
Meng Z, Moroishi T, Guan KL. Mechanisms of hippo pathway regulation. Genes Dev. 2016;30(1):1–17.
Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38.
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–32.
Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–5.
Kisen GO, Tessitore L, Costelli P, Gordon PB, Schwarze PE, Baccino FM, et al. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis. 1993;14(12):2501–5.
Toth S, Nagy K, Palfia Z, Rez G. Changes in cellular autophagic capacity during azaserine-initiated pancreatic carcinogenesis. Acta Biol Hung. 2001;52(4):393–401.
Toth S, Nagy K, Palfia Z, Rez G. Cellular autophagic capacity changes during azaserine-induced tumour progression in the rat pancreas. Up-regulation in all premalignant stages and down-regulation with loss of cycloheximide sensitivity of segregation along with malignant transformation. Cell Tissue Res. 2002;309(3):409–16.
Schwarze PE, Seglen PO. Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen-treated rats. Exp Cell Res. 1985;157(1):15–28.
Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–30.
Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol Cancer Res. 2014;12(4):485–90.
White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. 2015;21(22):5037–46.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501.
Thompson JE, Thompson CB. Putting the rap on Akt. J Clin Oncol. 2004;22(20):4217–26.
Yu K, Toral-Barza L, Discafani C, Zhang WG, Skotnicki J, Frost P, et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer. 2001;8(3):249–58.
Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8(23):3831–7.
Chan S. Targeting the mammalian target of rapamycin (mTOR): a new approach to treating cancer. Br J Cancer. 2004;91(8):1420–4.
Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol. 2005;56(1):47–60.
Liu G, Pei F, Yang F, Li L, Amin AD, Liu S, et al. Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci. 2017;18(2):E367.
Fumarola C, Bonelli MA, Petronini PG, Alfieri RR. Targeting PI3K/AKT/mTOR pathway in non-small cell lung cancer. Biochem Pharmacol. 2014;90(3):197–207.
Sorrells DL, Meschonat C, Black D, Li BD. Pattern of amplification and overexpression of the eukaryotic initiation factor 4E gene in solid tumor. J Surg Res. 1999;85(1):37–42.
Easton JB, Houghton PJ. The mTOR pathway and its inhibitors. In: LaRochelle WJ, Shimkets RA, editors. Cancer drug discovery and development: the oncogenomics handbook. Totowa: Humana Press; 2005. p. 553–70.
Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23(16):2891–906.
Tan ML, Muhammad TS, Najimudin N, Sulaiman SF. Growth arrest and non-apoptotic programmed cell death associated with the up-regulation of c-myc mRNA expression in T-47D breast tumor cells following exposure to Epipremnum pinnatum (L.) Engl. hexane extract. J Ethnopharmacol. 2005;96(3):375–83.
Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120(Pt 23):4081–91.
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, et al. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20(19):7268–78.
Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, et al. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12(24):3231–44.
Petersen A, Brundin P. Huntington’s disease: the mystery unfolds? Int Rev Neurobiol. 2002;53:315–39.
Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, et al. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet. 2001;10(12):1243–54.
Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12(1):25–31.
Bialik S, Kimchi A. Autophagy and tumor suppression: recent advances in understanding the link between autophagic cell death pathways and tumor development. Adv Exp Med Biol. 2008;615:177–200.
Lee HK, Jones RT, Myers RA, Marzella L. Regulation of protein degradation in normal and transformed human bronchial epithelial cells in culture. Arch Biochem Biophys. 1992;296(1):271–8.
Houri JJ, Ogier-Denis E, De Stefanis D, Bauvy C, Baccino FM, Isidoro C, et al. Differentiation-dependent autophagy controls the fate of newly synthesized N-linked glycoproteins in the colon adenocarcinoma HT-29 cell line. Biochem J. 1995;309(Pt 2):521–7.
Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res. 2007;67(20):9677–84.
Fujii S, Mitsunaga S, Yamazaki M, Hasebe T, Ishii G, Kojima M, et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008;99(9):1813–9.
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4(4):466–79.
Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22(2):165–78.
Guo JY, White E. Autophagy, metabolism, and cancer. Cold Spring Harb Symp Quant Biol. 2016;81:73–8.
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524(7565):361–5.
Wong PM, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun. 2015;6:8048.
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 2013;27(13):1447–61.
Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3(11):1272–85.
Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther. 2008;7(9):2977–87.
Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90(18):1371–88.
Veronesi U, Maisonneuve P, Rotmensz N, Costa A, Sacchini V, Travaglini R, et al. Italian randomized trial among women with hysterectomy: tamoxifen and hormone-dependent breast cancer in high-risk women. J Natl Cancer Inst. 2003;95(2):160–5.
Clarke R, Leonessa F, Welch JN, Skaar TC. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol Rev. 2001;53(1):25–71.
Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6(3):181–202.
Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14(3):500–10.
Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, et al. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112(3):389–403.
Ma XH, Piao S, Wang D, McAfee QW, Nathanson KL, Lum JJ, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res. 2011;17(10):3478–89.
Mita M, Sankhala K, Abdel-Karim I, Mita A, Giles F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Investig Drugs. 2008;17(12):1947–54.
Waqar SN, Gopalan PK, Williams K, Devarakonda S, Govindan R. A phase I trial of sunitinib and rapamycin in patients with advanced non-small cell lung cancer. Chemotherapy. 2013;59(1):8–13.
Malizzia LJ, Hsu A. Temsirolimus, an mTOR inhibitor for treatment of patients with advanced renal cell carcinoma. Clin J Oncol Nurs. 2008;12(4):639–46.
Schwartz GK, Tap WD, Qin LX, Livingston MB, Undevia SD, Chmielowski B, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14(4):371–82.
Naing A, Lorusso P, Fu S, Hong D, Chen HX, Doyle LA, et al. Insulin growth factor receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with metastatic adrenocortical carcinoma. Br J Cancer. 2013;108(4):826–30.
Margolin KA, Moon J, Flaherty LE, Lao CD, Akerley WL 3rd, Othus M, et al. Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438). Clin Cancer Res. 2012;18(4):1129–37.
Davies MA, Fox PS, Papadopoulos NE, Bedikian AY, Hwu WJ, Lazar AJ, et al. Phase I study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin Cancer Res. 2012;18(4):1120–8.
Spindler KL, Sorensen MM, Pallisgaard N, Andersen RF, Havelund BM, Ploen J, et al. Phase II trial of temsirolimus alone and in combination with irinotecan for KRAS mutant metastatic colorectal cancer: outcome and results of KRAS mutational analysis in plasma. Acta Oncol. 2013;52(5):963–70.
Mita MM, Mita A, Rowinsky EK. Mammalian target of rapamycin: a new molecular target for breast cancer. Clin Breast Cancer. 2003;4(2):126–37.
Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30(22):2718–24.
Mita MM, Mita AC, Chu QS, Rowinsky EK, Fetterly GJ, Goldston M, et al. Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol. 2008;26(3):361–7.
Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, Strair R, et al. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2008;14(9):2756–62.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–63.
Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 2008;68(16):6598–607.
Cao P, Maira SM, Garcia-Echeverria C, Hedley DW. Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. Br J Cancer. 2009;100(8):1267.
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68(19):8022–30.
Lefranc F, Facchini V, Kiss R. Proautophagic drugs: a novel means to combat apoptosis-resistant cancers, with a special emphasis on glioblastomas. Oncologist. 2007;12(12):1395–403.
Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007;14(3):548–58.
Yokoyama T, Iwado E, Kondo Y, Aoki H, Hayashi Y, Georgescu MM, et al. Autophagy-inducing agents augment the antitumor effect of telerase-selve oncolytic adenovirus OBP-405 on glioblastoma cells. Gene Ther. 2008;15(17):1233–9.
Milano V, Piao Y, LaFortune T, de Groot J. Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther. 2009;8(2):394–406.
Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–57.
Naumann SC, Roos WP, Jost E, Belohlavek C, Lennerz V, Schmidt CW, et al. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br J Cancer. 2009;100(2):322–33.
Hart MG, Grant R, Garside R, Rogers G, Somerville M, Stein K. Temozolomide for high grade glioma. Cochrane Database Syst Rev. 2008;4:CD007415.
Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol. 2009;27(4):579–84.
Lou E, Peters KB, Sumrall AL, Desjardins A, Reardon DA, Lipp ES, et al. Phase II trial of upfront bevacizumab and temozolomide for unresectable or multifocal glioblastoma. Cancer Med. 2013;2(2):185–95.
Norden AD, Lesser GJ, Drappatz J, Ligon KL, Hammond SN, Lee EQ, et al. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro-Oncology. 2013;15(7):930–5.
Yust-Katz S, Liu D, Yuan Y, Liu V, Kang S, Groves M, et al. Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma. Cancer. 2013;119(15):2747–53.
Nghiemphu PL, Wen PY, Lamborn KR, Drappatz J, Robins HI, Fink K, et al. A phase I trial of tipifarnib with radiation therapy, with and without temozolomide, for patients with newly diagnosed glioblastoma. Int J Radiat Oncol Biol Phys. 2011;81(5):1422–7.
Wagner L, Turpin B, Nagarajan R, Weiss B, Cripe T, Geller J. Pilot study of vincristine, oral irinotecan, and temozolomide (VOIT regimen) combined with bevacizumab in pediatric patients with recurrent solid tumors or brain tumors. Pediatr Blood Cancer. 2013;60(9):1447–51.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003.
Hummel TR, Wagner L, Ahern C, Fouladi M, Reid JM, McGovern RM, et al. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: a children’s oncology group phase 1 consortium study. Pediatr Blood Cancer. 2013;60(9):1452–7.
Kouroussis C, Vamvakas L, Vardakis N, Kotsakis A, Kalykaki A, Kalbakis K, et al. Continuous administration of daily low-dose temozolomide in pretreated patients with advanced non-small cell lung cancer: a phase II study. Oncology. 2009;76(2):112–7.
Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113(9):1875–91.
Powell BL, Moser B, Stock W, Gallagher RE, Willman CL, Stone RM, et al. Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood. 2010;116(19):3751–7.
Liu N, Tai S, Ding B, Thor RK, Bhuta S, Sun Y, et al. Arsenic trioxide synergizes with everolimus (Rad001) to induce cytotoxicity of ovarian cancer cells through increased autophagy and apoptosis. Endocr Relat Cancer. 2012;19(5):711–23.
Goussetis DJ, Gounaris E, Platanias LC. BCR-ABL1-induced leukemogenesis and autophagic targeting by arsenic trioxide. Autophagy. 2013;9(1):93–4.
Raja WK, Satti J, Liu G, Castracane J. Dose response of MTLn3 cells to serial dilutions of arsenic trioxide and ionizing radiation. Dose Response. 2013;11(1):29–40.
Chiu HW, Ho YS, Wang YJ. Arsenic trioxide induces autophagy and apoptosis in human glioma cells in vitro and in vivo through downregulation of survivin. J Mol Med (Berl). 2011;89(9):927–41.
Cohen KJ, Gibbs IC, Fisher PG, Hayashi RJ, Macy ME, Gore L. A phase I trial of arsenic trioxide chemoradiotherapy for infiltrating astrocytomas of childhood. Neuro-Oncology. 2013;15(6):783–7.
Grimm SA, Marymont M, Chandler JP, Muro K, Newman SB, Levy RM, et al. Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. J Neuro-Oncol. 2012;110(2):237–43.
Sharma M, Khan H, Thall PF, Orlowski RZ, Bassett RL Jr, Shah N, et al. A randomized phase 2 trial of a preparative regimen of bortezomib, high-dose melphalan, arsenic trioxide, and ascorbic acid. Cancer. 2012;118(9):2507–15.
Yanada M, Tsuzuki M, Fujita H, Fujimaki K, Fujisawa S, Sunami K, et al. Phase 2 study of arsenic trioxide followed by autologous hematopoietic cell transplantation for relapsed acute promyelocytic leukemia. Blood. 2013;121(16):3095–102.
Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6(3):322–9.
Shingu T, Fujiwara K, Bogler O, Akiyama Y, Moritake K, Shinojima N, et al. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int J Cancer. 2009;124(5):1060–71.
Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370.
Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmacol. 2009;625(1–3):220–33.
Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res. 2013;73(1):3–7.
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25(3):1025–40.
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17(4):654–66.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–42.
Briceno E, Reyes S, Sotelo J. Therapy of glioblastoma multiforme improved by the antimutagenic chloroquine. Neurosurg Focus. 2003;14(2):e3.
Sotelo J, Briceno E, Lopez-Gonzalez MA. Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 2006;144(5):337–43.
Goldberg SB, Supko JG, Neal JW, Muzikansky A, Digumarthy S, Fidias P, et al. A phase I study of erlotinib and hydroxychloroquine in advanced non-small-cell lung cancer. J Thorac Oncol. 2012;7(10):1602–8.
Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochem Biophys Res Commun. 2006;340(1):286–90.
Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3beta in mouse insulinoma cells. Diabetes. 2005;54(4):968–75.
Di Nardo A, Kramvis I, Cho N, Sadowski A, Meikle L, Kwiatkowski DJ, et al. Tuberous sclerosis complex activity is required to control neuronal stress responses in an mTOR-dependent manner. J Neurosci. 2009;29(18):5926–37.
Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy. 2010;6(2):239–47.
Jin H-O, Seo S-K, Woo S-H, Kim E-S, Lee H-C, Yoo D-H, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-β negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med. 2009;46(8):1158–67.
Corradetti MN, Inoki K, Guan KL. The stress-induced proteins RTP801 and RTP801L are negative regulators of the mammalian target of rapamycin pathway. J Biol Chem. 2005;280(11):9769–72.
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25(2):193–205.
Yung HW, Charnock-Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One. 2011;6(3):e17894.
Lu Y, Zhang R, Liu S, Zhao Y, Gao J, Zhu L. ZT-25, a new vacuolar H+-ATPase inhibitor, induces apoptosis and protective autophagy through ROS generation in HepG2 cells. Eur J Pharmacol. 2016;771(Supplement C):130–8.
Chen J-J, Chou C-W, Chang Y-F, Chen C-C. Proteasome inhibitors enhance TRAIL-induced apoptosis through the intronic regulation of DR5: involvement of NF-κB and reactive oxygen species-mediated p53 activation. J Immunol. 2008;180(12):8030.
Hambright HG, Ghosh R. Autophagy: in the cROSshairs of cancer. Biochem Pharmacol. 2017;126:13–22.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749.
Son Y-O, Pratheeshkumar P, Roy RV, Hitron JA, Wang L, Divya SP, et al. Antioncogenic and oncogenic properties of nrf2 in arsenic-induced carcinogenesis. J Biol Chem. 2015;290(45):27090–100.
Park JS, Kang DH, Bae SH. p62 prevents carbonyl cyanide m-chlorophenyl hydrazine (CCCP)-induced apoptotic cell death by activating Nrf2. Biochem Biophys Res Commun. 2015;464(4):1139–44.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen H-Y, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75.
Madden DT, Egger L, Bredesen DE. A calpain-like protease inhibits autophagic cell death. Autophagy. 2007;3(5):519–22.
Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281(28):19179–87.
Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118(Pt 14):3091–102.
Tessitore L, Tomasi C, Greco M. Fasting-induced apoptosis in rat liver is blocked by cycloheximide. Eur J Cell Biol. 1999;78(8):573–9.
McLean K, Vandeven NA, Sorenson DR, Daudi S, Liu JR. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol Oncol. 2009;112(3):623–30.
Liu B, Cheng Y, Bian HJ, Bao JK. Molecular mechanisms of Polygonatum cyrtonema lectin-induced apoptosis and autophagy in cancer cells. Autophagy. 2009;5(2):253–5.
Moad AI, Tengku Muhammad TS, Oon CE, Tan ML. Rapamycin induces apoptosis when autophagy is inhibited in t-47d mammary cells and both processes are regulated by Phlda1. Cell Biochem Biophys. 2013;66(3):567–87.
Moretti L, Cha YI, Niermann KJ, Lu B. Switch between apoptosis and autophagy: radiation-induced endoplasmic reticulum stress? Cell Cycle. 2007;6(7):793–8.
Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–6.
Williams JA, Thomas AM, Li G, Kong B, Zhan L, Inaba Y, et al. Tissue specific induction of p62/Sqstm1 by farnesoid X receptor. PLoS One. 2012;7(8):e43961.
Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32(2):95–100.
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy. 2010;6(8):1090–106.
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137(4):721–35.
Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19(7):1576–86.
Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–54.
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18(11):3044–53.
Perkins ND. The Rel/NF-kappa B family: friend and foe. Trends Biochem Sci. 2000;25(9):434–40.
Wang Y, Han C, Lu L, Magliato S, Wu T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology. 2013;58(3):995–1010.
Yin X, Zhang N, Di W. Regulation of LC3-dependent protective autophagy in ovarian cancer cells by protein phosphatase 2A. Int J Gynecol Cancer. 2013;23(4):630–41.
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–29.
Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 2014;4(7):773–80.
Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy supports breast cancer stem cell maintenance by regulating IL6 secretion. Mol Cancer Res. 2015;13(4):651–8.
Vasilevskaya IA, Selvakumaran M, Roberts D, O’Dwyer PJ. JNK1 inhibition attenuates hypoxia-induced autophagy and sensitizes to chemotherapy. Mol Cancer Res. 2016;14(8):753–63.
Jutten B, Rouschop KM. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle. 2014;13(1):42–51.
Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13(9):1070–7.
Soria JC, Mark Z, Zatloukal P, Szima B, Albert I, Juhasz E, et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29(33):4442–51.
Younes A, Vose JM, Zelenetz AD, Smith MR, Burris HA, Ansell SM, et al. A phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin’s lymphoma. Br J Cancer. 2010;103(12):1783–7.
Acknowledgments
The authors would like to acknowledge the Ministry of Energy, Science, Technology, Environment and Climate Change (MESTECC) for the Smartfund grant and USM for the Bridging Grant.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Tan, M.L., Tan, H.K., Muhammad, T.S.T. (2020). Endoplasmic Reticulum Stress and Autophagy in Cancer. In: Rezaei, N. (eds) Cancer Immunology. Springer, Cham. https://doi.org/10.1007/978-3-030-30845-2_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-30845-2_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-30844-5
Online ISBN: 978-3-030-30845-2
eBook Packages: MedicineMedicine (R0)