
Abstract
Purpose
Sirolimus, an oral mTOR inhibitor, may complement the anti-angiogenic and anti-tumor activity of sunitinib, an oral small molecule inhibitor of multiple receptor tyrosine kinases, by vertical disruption of vascular epithelial growth factor receptor (VEGFR) signaling, by reducing the compensatory production of VEGF in sunitinib-treated patients and also by directly inhibiting tumor cell proliferation. We conducted this phase 1 study to investigate the maximum tolerated dose (MTD) for this combination of sunitinib and sirolimus in humans.
Patients and methods
Sunitinib was given at 50 mg daily × 28 every 6 weeks. The first cohort received sunitinib alone for cycle 1 (50 mg daily for 2 weeks followed by 2 weeks off) and received sunitinib at standard dose 50 mg daily for 4 weeks followed by 2 weeks off in combination with sirolimus 4 mg weekly; this dose and schedule were further investigated in second cohort. The third cohort received decreased dose of sunitinib at 37.5 mg daily for 4 weeks followed by 2 weeks off in combination with sirolimus at 4 mg weekly. Sirolimus dose was escalated to 8 mg weekly in fourth cohort.
Results
Eighteen patients with ECOG PS of 0 or 1 were enrolled, median age 57 years (range 24–76), M:F ratio: 11:7. Median number of prior treatments is 2 (range 0–5); six patients had no prior systemic therapy. Half of patients from the first two cohorts required dose reduction or early discontinuation of treatment; therefore, sunitinib dose was decreased to 37.5 mg daily in third and fourth cohort. In third and fourth cohort, one-third of patients required dose modification during cycle 1 or cycle 2. Multiple patients had significant toxicities including fatigue and hand–foot syndrome. One patient developed interstitial pneumonitis, and one patient died suddenly on day 8 due to progressive disease. There were six patients who tolerated four or more cycles. Among these six patients, two patients with renal cell carcinoma (RCC) achieved partial response; one subsequently underwent surgical resection of residual renal mass and lymph node dissection and achieved complete response afterward. One with metastatic melanoma also achieved complete response after metastatectomy. There was no apparent pharmacokinetic interaction between sunitinib and sirolimus. 4 mg weekly sirolimus did not reduce the sunitinib-induced circulating VEGF production but stimulated more VEGF production through some unknown compensatory mechanism.
Conclusion
Toxicity precluded dose escalation of weekly sirolimus in combination with a standard sunitinib dose/schedule. These results suggest caution when combining targeted agents lacking specificity for tumor signaling or vasculature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sunitinib (Sutent) is a small molecule that selectively targets and blocks the intracellular signaling pathways of multiple receptor tyrosine kinases (RTKs) including: vascular epithelial growth factor receptor (VEGFR), platelet derived growth factor receptor (PDGFR), FLT3, c-KIT, and RET [1]. It is currently FDA-approved for the treatment of advanced RCC, imatinib-resistant or refractory gastrointestinal stromal tumor (GIST), and advanced pancreatic neuroendocrine tumor [2–5]. In addition, sunitinib also showed broad anti-tumor activity in other malignancies such as lung, pancreas, thyroid cancer, sarcoma, and melanoma in preclinical and early phase studies [6–9].
Standard dose of sunitinib is 50 mg oral daily, 4 weeks on, and 2 weeks off [2]. The commonly observed toxicities are associated with sunitinib included fatigue, nausea, diarrhea, stomatitis, myelosuppression, dermatitis. Cardiac toxicities such as asymptomatic decrease in ejection fraction (EF), especially with pre-existing anthracycline treatment or cardiovascular disease.
Sirolimus (rapamycin), a commonly used immunosuppressant for organ transplantation, is an inhibitor of mammalian target of rapamycin (mTOR) signaling pathway. mTOR pathway dysregulation has been shown in a variety of cancer cells, making mTOR pathway an attractive therapeutic target in treating cancers [10–12]. Sirolimus was found to control angiogenic process by reducing tumor cell production of VEGF and inhibit VEGF-stimulated cell proliferation in vitro and in vivo [13].
Combination of doxorubicin and sirolimus was proved to have effect on T cell acute lymphoblastic leukemia (T-ALL) [14]. Other mTOR inhibitors such as temsirolimus and everolimus were also tested in metastatic renal cell carcinoma [15–18]. In a phase III trial, 626 untreated RCC with poor-prognostic features were randomized to receive either temsirolimus, interferon-α, or both agents [16]. The temsirolimus arm achieved significant survival benefit compared with the other two arms (10.9 vs. 7.3 vs. 8.4 months, respectively). This trial led to the approval of temsirolimus for RCC patients with unfavorable prognosis by Food and Drug Administration (FDA). Everolimus in a similar fashion also obtained FDA approval for treatment of RCC as salvage therapy [17].
Doses of sirolimus range from 2 to 40 mg per day in organ transplantation setting. However, the pharmacokinetic and in vitro data suggest that low-dose sirolimus given once weekly could effectively inhibit mTOR and downstream p70S6k phosphorylation in cancer cells [19].
Inhibition of mTOR pathway may complement the anti-angiogenic and anti-tumor activities of receptor tyrosine kinase inhibitors (RTKi) such as sunitinib, by disrupting VEGFR downstream signals, by reducing production of VEGF (which often increases in response to VEGFR inhibition), by inhibiting cell proliferation. To test the hypothesis, we conducted a phase I study to investigate the combination of sunitinib and sirolimus in humans to determine the MTD/schedule for future use in phase II trials. In addition, we assessed dose effects of this combination on serum levels of VEGF and circulating endothelial cells, and to obtain efficacy of this combination in treating solid malignancies using RECIST criteria.
Patients and methods
Patient selection
Patients of equal or more than 18 years of age, with histologically confirmed malignancy that was metastatic or unresectable and for which standard measures do not exist or no longer effective, with measurable disease by RECIST criteria, adequate hematopoietic with Hgb ≥ 9 g/dl, ANC ≥ 1000/μl, platelet >100,000/μl; liver functions with total bilirubin <1.5 × upper limit normal (ULN), AST/ALT < 3 × ULN; and renal function with serum creatinine ≤2 × ULN, ECOG performance status of 0–1, were enrolled in the study. All subjects had normal cardiac ejection fraction as assessed by echocardiogram (ECHO) or multiple gated acquisition (MUGA) scan prior to treatment.
Sexually active men and women with child-bearing potential used acceptable contraceptive methods. Pregnant and nursing patients were excluded from the study.
Written informed consent was obtained according to federal and institutional guidelines. The protocol was approved by institution’s human investigational committee.
Drug administration
Sunitinib was supplied by Pfizer Global Research and Development (La Jolla, CA) as capsules in strengths of 12.5, 25, or 50 mg. Sirolimus was administered orally once weekly, beginning at 4 mg, and escalating to 8 mg weekly.
The first cohort of this study received sunitinib alone at 50 mg daily for 2 weeks followed by 2 weeks off as cycle 1, then followed by standard sunitinib dose and schedule (50 mg daily for 4 weeks, and 2 weeks off) in combination with sirolimus 4 mg weekly as described above for subsequent cycles (Table 2).
Pretreatment and follow-up studies
Prior to the first course of treatment, histories and physical examinations were performed, and the following evaluations were also obtained: complete blood count, routine chemistries and electrolytes, clotting studies, urinalysis, 24 h urine creatinine clearance, 12-lead EKG, and ECHO or MUGA scan. Tumor stagings appropriate to the disease including CT of chest and abdomen, CT of pelvis, bone scan, PET scan, CT or MRI of the brain, and tumor serum markers were obtained pre-treatment and repeated every 8 weeks. Patients were evaluated weekly for adverse events. The cardiac ejection fraction was measured every 3 months while on study and at the completion of all study treatment, using the same technique (echocardiogram, MUGA) used for the baseline assessment and follow-up monitor.
Dose escalation and modification rules
Dose escalation was proceeded according to standard phase I rules [20]. Both drugs were held for poorly tolerated grade 2 non-hematologic toxicities and any dose-limiting toxicity (DLT) as defined (see Table 3). All patients were observed for at least 6 weeks with no more than 1 DLT before patients may be entered at the next higher dose level. Accrual of new patients continued until two patients in a cohort developed DLT.
Maximum tolerated dose (MTD) was defined as one dose level below that at which two or more patients experience a DLT or the dose level at which one patient experience a DLT.
Missed treatment within a cycle was not made up. Treatment with both drugs was restarted when non-hematologic toxicities resolved to grade 1 and hematologic toxicities resolved to grade 2. All treatments on study were discontinued permanently for patients who experienced grade 4 non-hematologic DLT.
Toxicity assessment
The National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 3.0 was used to grade toxicity.
Evaluation of response
Tumor response was evaluated based on RECIST criteria every two cycles. In addition to a baseline scan, confirmatory scans were obtained 4–8 weeks following initial documentation of an objective response.
Pharmacokinetic (PK) study
Whole blood samples were drawn for PK of sunitinib and metabolite SU012662 on day 15 of cohort 1 cycle 2 at pre-dose, and 1, 4, 6, 8, 24 h post-dose. Whole blood samples (4 ml) were collected into a K-EDTA tube at the specified time points. Following collection, blood sample was gently inverted 15 times to mix whole blood and anticoagulant. Samples were kept into an ice bath at 2–8 °C during harvesting. Whole blood samples were centrifuged at 4 °C for 10 min at 1000×g to separate the plasma from the blood cells. The plasma layer (1.5 ml) was transferred into an amber cryovial using a pipette. Plasma samples were stored at −20 °C prior to shipment to Yale Bioanalytical Laboratory for analysis. Fully validated HPLC–MS/MS assay methods were applied to quantify plasma concentrations of sunitinib [21]. Blood samples were collected from three patients at each time point.
Whole blood samples for PK of sirolimus were collected using aforementioned methods on day 15 of cycle 2 at pre-dose, and 1, 2, 4, 24 h post-dose. Sirolimus levels were determined by routine Yale New Haven Hospital (YNHH) clinical laboratory assay [22]. Blood samples were collected from three patients at each time point.
Determination of VEGF (VEGF-A) and soluble VEGF-R2 (sVEGFR2) in plasma
For the first cohort, blood (10 ml) was drawn into tubes containing sodium heparin within 15 min before the administration of sunitinib on days 1 and 15 of cycles 1 and 2, day 29 of cycle 2, and day 1 of cycle 3.
Plasma VEGF-A was measured using the Quantikine sandwich ELISA (R&D Systems). Samples were diluted 1:1 in assay diluent and added to wells coated with a monoclonal antibody specific for VEGF. After washing away unbound substances, a second horseradish peroxidase-conjugated antibody was added to the wells. Antibody binding was detected by adding tetramethylbenzidine as a substrate, and after development of the color, the reaction was stopped by adding 2 N sulfuric acid. Absorbance was measured using a microplate reader set at 450 nm with wavelength correction set at 570 nm.
Statistical method blood samples were collected from three patients at each time point for PK data. Serum drug concentrations were calculated using mean ± SD; p value was calculated using Student’s t test.
Results
Baseline demographic characteristics
Eighteen patients consented for participating this phase I trial between April 2006 and August 2007. Among eighteen patients, five had kidney cancers including one clear cell renal cell carcinoma (RCC), one papillary RCC, one medullary RCC, one mixed cell type RCC (60 % of sarcomatoid component and 40 % clear cell component), and one metastatic kidney angiomyolipoma (Table 1).
Median age of accrued patients was 57 years old. Eleven patients were male, and seven were female. Majority patients (14 out of 18) had ECOG performance status of 1.
Twelve out of eighteen patients had prior chemotherapies with a median number of regimens of 2.
The number of enrollment of each cohort and drug dose/schedule is listed (Table 2).
Tolerability
The combination of sunitinib and sirolimus was observed to be quite toxic in all cohorts.
In cohort 1, the first three patients received 17, 5, and 4 cycles, respectively. The patient with metastatic RCC (clear cell) tolerated a total of 17 cycles of treatment but experienced grade 3 toxicities including abdominal pain, dyspnea, fatigue, diarrhea, electrolytes disturbance, and asymptomatic drop of LVEF from 57 to 49 % requiring a dose reduction of sunitinib from 50 to 37.5 mg daily since cycle 5. The patient with medullary RCC experienced grade 3 electrolytes imbalance with hyperglycemia and hypophosphatemia, rash, abdominal pain and headache, venous thromboembolic event (VTE), as well as congestive heart failure and pleural effusion requiring dose reduction of sunitinib to 37.5 mg daily in third cycle and then to 25 mg daily in forth cycle. The third patient with spindle cell carcinoma developed grade 3 anemia, neutropenia, and dyspnea requiring dose reduction of sunitinib to 37.5 mg daily in fourth cycle. The patient did not finish the 4th cycle due to severe fatigue and worsening performance status. Two patients only received two cycles of treatment, one patient opted not to continue due to diffuse side effects, and the other one was found to have progressive disease on scan.
Given dose reduction or discontinuation of sunitinib resulted in resolution of toxicity within 7 days despite continued administration of sirolimus suggested that the adverse events were primarily caused by sunitinib. Therefore, we continued with cohort 2. Three patients were enrolled into this cohort. The first patient tolerated a total of four cycles, but required early discontinuation in cycle 1 and twice dose reduction at cycle 2 and then cycle 3 due to fatigue. Similarly, the second patient required early discontinuation of sunitinib in cycle 1 and dose reduction at cycle 2. However, even with a 25 % reduced dose, this patient only tolerated 15 days in cycle 2. The third patient, a 59-year-old white male with metastatic non-small cell lung cancer (squamous cell carcinoma in histology) underwent laser treatment for right airway debridement during cycle 3 treatment; procedure was complicated with grade 3 pulmonary hemorrhage. Embolization of two bronchial arteries was performed in an attempt of stopping the bleeding; the patient was further complicated with grade 4 motor neuropathy (paralysis) after embolization. The patient expired 14 days after laser treatment. The death was believed not to be related to study drugs.
The overwhelming toxicity we encountered in cohort 1 and 2 precluded further dose escalation, protocol was amended to allow a change to proceed with de-escalation schedule as listed in Table 2. Cohort 3 started with sunitinib 37.5 mg daily in combination with sirolimus 4 mg weekly. Three patients were enrolled. The first patient, a 54-year-old white male with diagnosis of metastatic esophageal cancer only tolerated 15 days in cycle 1 due to grade 3 thrombocytopenia. He was then started cycle 2 with sunitinib 25 mg daily. This cycle was complicated with a grade 4 hemolysis which precluded him from receiving further treatment. The other two patients were able to tolerate two full cycles of treatment; one did require dose reduction from 37.5 mg daily to 25 mg daily at second cycle due to grade 2 mucositis. Both patients subsequently were taken off the study due to evidence of disease progression on following CT scans (Table 3).
Since no more than two patients experienced DLTs in cohort 3, we started cohort 4 with an effort of dose escalating sirolimus only. Seven patients enrolled in cohort 4.
A 65-year-old black female in cohort 4 with remote history of RCC, status post-nephrectomy, and a diagnosis of adenocarcinoma with unknown primary expired suddenly while on this cohort for only 8 days. The cause of death was attributed to tumor compression and invasion of the left carotid bundle, although other causes such as pulmonary embolism or myocardial infarction could not be completely excluded. The death was believed not to be related to study drugs (Table 4).
Other patients in this cohort developed grade 3 hypophosphatemia (2), severe flank/kidney pain (1), limb pain (1), rash/hand–foot syndrome (HFS) (1), and fatigue (1). Overall, the toxicities of each cohort are summarized in Table 5.
Efficacy
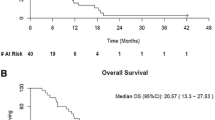
Median cycle of sunitinib/sirolimus treatment were two cycles with a range of 1–17; only six patients tolerated more than four cycles including one medullary RCC (four cycles), one papillary RCC (five cycles), one clear cell RCC (17 cycles), one spindle cell carcinoma (five cycles), one breast cancer (four cycles), and one pancreatic neuroendocrine tumor (PNET) (16 cycles).
RECIST criteria were used to evaluate the tumor response. Two patients (one in cohort 1 and one in cohort 4) only received treatment less than two cycles and therefore were excluded for response assessment due to lack of restaging scans. Response was assessed for patients who tolerated more than two cycles (Table 6). Two patients achieved partial response (PR); both had RCC. The one with papillary RCC eventually achieved no evidence of disease (NED) after surgical resection of residual renal mass and lymph nodes. He remained NED for more than 5 years.
Two patients achieved stable disease (SD). The patient with clear cell RCC subsequently moved onto sorafenib as second-line salvage therapy. The other patient with pancreatic NET had stable disease since the initiation of this trial, tolerated a total of 17 cycles.
Three patients demonstrated transient central necrosis or size reduction in some tumors/metastases, but not qualified for a best response of PR or SD per RECIST. Fourteen patients had progressive disease; 10 of 14 developed new metastatic lesions.
Pharmacokinetic analysis
Plasma was collected and separated from peripheral blood samples harvested at different time points. Three specimens were taken for each time point. Serum concentrations of sunitinib and its main metabolite, SU012662, as well as sirolimus were measured in bioanalytical laboratory and YNHH clinical laboratory, respectively. Sunitinib and SU012662 serum concentrations reached relatively stable level after 15 days of continuous administration (Fig. 1). There is some variation among individual measurement; however, the addition of sirolimus did not significantly affect the concentration of sunitinib. Sirolimus levels were measured and charted (Fig. 1). 4 mg weekly dose of sirolimus reached peak at 2 h post-administration; within 24-h, the level has returned back to pre-administration level.

Pharmacokinetics data of cohort 1 (top sunitinib and its active metabolite SU012662 serum concentration (ng/ml), bottom sirolimus serum concentration (ng/ml) in cohort 1)
Pharmacodynamic analysis
We measured VEGF-A, one of the major VEGFR ligands, by ELISA on days 1 and 15 of cycles 1 and 2, day 29 of cycle 2, and day 1 of cycle 3 (Fig. 2). VEGF-A was expectedly rising after initiation of sunitinib and peaked at day 15 and then dropped down to almost baseline level on day 1 of cycle 2. Interestingly, in cycle 2, the circulating VEGF-A was not suppressed by the addition of sirolimus as we hypothesized but further elevated and peaked at day 29 along with sunitinib administration, subsequently dropped down to almost baseline on day 1 of cycle 3 when off sunitinib. The addition of sirolimus in cycle 2 appears to have induced an efficient compensatory mechanism in tumor cells to produce VEGF.

Serum VEGF-A level (pg/ml) in cohort 1
Discussion
The standard dose of sunitinib is 50 mg daily for 4 weeks followed by 2 weeks of rest. However, this dose and schedule has been universally noted to cause significant toxicities [2–5]. Dose reduction is generally accepted; 37.5 mg dose achieves similar efficacy. Previously, tumor re-growth during the off-treatment time had been reported clinically. Adding another signal transduction pathway blocker to enhance the anti-tumor effect of sunitinib was then considered. mTOR inhibitors were thought to be a reasonable candidate to work synergistically with sunitinib by double blocking both VEGF and PI3k/akt pathways [10–13, 23–26].
This phase I trial was designed to test this hypothesis in humans. The starting dose of sunitinib and sirolimus were chosen based on previously known data in the literature; unfortunately, we encountered overwhelmingly significant toxicities in cohort 1 and 2. More detailed assessment of toxicities revealed prominent toxicities related to sunitinib administration. Thus, dose de-escalation was proposed in cohort 3 with a 25 % reduced dose of sunitinib. Among the three patients in this cohort, toxicities seemed to be milder compared to prior cohorts. Cohort 4 with an effort of escalating sirolimus dose was attempted. In this cohort, the toxicity profile was not very overwhelming; however, the efficacy was not encouraging either. Our overall impression of this combination was very toxic. A few patients were able to tolerate full courses of treatment with planned dose and schedule; however, majority of patients in each cohort required dose modification.
This was also observed in other sunitinib combination therapies. Rini et al. reported patients receiving sunitinib at 50 mg daily in combination with VEGF monoclonal antibody bevacizumab at 10 mg/kg experienced significant toxicities including hypertension, fatigue, and cytopenias. Dose reduction or delay in continued therapy was required, although patients in that study tolerated relatively better at lower dose levels [27].
Sunitinib seems to be the one causing the majority of adverse events although whether there is synergistic effect between sunitinib and sirolimus is not clear yet.
Pharmacokinetics study did not reveal apparent drug–drug interaction between sunitinib and sirolimus; this was also proved in other study [28]. Although the dose and schedule of sirolimus seem to be low and infrequent, the effect on sunitinib-induced compensatory VEGF production was strikingly interesting. The addition of sirolimus was thought to be able to decrease the sunitinib-induced VEGF production. However, in contrast to our original hypothesis, our data demonstrated a totally unexpected phenomenon—VEGF went further up along with sunitinib and sirolimus administration, only came down during the sunitinib off weeks. This observation disappointed us in the thinking that current dose/schedule of sirolimus had no effect on interrupting the compensatory VEGF production although we could not prove whether sirolimus has direct effect on tumor cell proliferation with these data.
This interesting observation made us think whether the true molecular mechanisms of sirolimus and sunitinib are fully understood. In our opinion, we should be very cautious when combining two anti-angiogenic agents lacking specificity for tumor signaling.
In summary, data from this trial suggested that a combination of sunitinib and sirolimus at current tested doses/schedules is not acceptable due to overwhelming toxicities. Dose escalation was not achieved as originally planned; dose reduction with sunitinib and dose escalation of sirolimus strategy were not able to produce enough data to warrant a phase II trial.
References
Mendel DB, Laird AD, Xin X et al (2003) In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9(1):327–337
Motzer RJ, Hutson TE, Tomczak P et al (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356(2):115–124
Motzer RJ, Hutson TE, Tomczak P et al (2009) Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 27(22):3584–3590
Demetri GD et al (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368(9544):1329–1338
Raymond E, Dahan L, Raoul J-L et al (2011) Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. NEJM 364(6):501–513
Kim S, Ding W, Zhang L et al (2014) Clinical response to sunitinib as a multitargeted tyrosine-kinase inhibitor (TKI) in solid cancers: a review of clinical trials. Onco Targets Ther 7:719–728
Reni M, Cereda S, Milella M et al (2013) Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: a phase II randomized trial. Eur J Cancer 49(17):3609–3615
Ready N, Pang HH, Gu L et al (2015) Chemotherapy with or without maintenance sunitinib for untreated extensive-stage small-cell lung cancer: a randomized, double-blind, placebo-controlled phase II study—CALGB 30504 (alliance). J Clin Oncol 33(15):1660–1665
Ravaud A, Fouchardiere C, Asselineau J et al (2010) Efficacy of sunitinib in advanced medullary thyroid carcinoma: intermediate results of phase II THYSU. Oncologist 15(2):212–213
Sun SY, Rosenberg LM, Wang X et al (2005) Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 65(16):7052–7058
Wendel HG, De Stanchina E, Fridman JS et al (2004) Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428(6980):332–337
Moschetta M, Reale A, Marasco C et al (2014) Therapeutic targeting of the mTOR-signalling pathway in cancer: benefits and limitations. Br J Pharmacol 171(16):3801–3813
Lane HA, Wood JM, McSheehy PM et al (2009) mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin Cancer Res 15(5):1612–1622
Batista A, Barata JT, Raderschall E et al (2011) Targeting of active mTOR inhibits primary leukemia T cells and synergizes with cytotoxic drugs and signaling inhibitors. Exp Hematol 39(4):457–472
Vogelzang NJ, Bhor M, Liu Z et al (2013) Everolimus vs. temsirolimus for advanced renal cell carcinoma: use and use of resources in the US oncology network. Clin Genitourin Cancer 11(2):115–120
Hudes G, Carducci M, Tomczak P et al (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281
Motzer RJ, Escudier B, Oudard S et al (2010) Phase 3 trial of everolimus for metastatic renal cell carcinoma : final results and analysis of prognostic factors. Cancer 116(18):4256–4265
Flaherty KT, Manoma JB, Pins M et al (2015) BEST: a randomized phase II study of vascular endothelial growth factor, RAF kinase, and mammalian target of rapamycin combination targeted therapy with bevacizumab, sorafenib, and temsirolimus in advanced renal cell carcinoma—a trial of the ECOG-ACRIN Cancer Research Group (E2804). J Clin Oncol 60:9727
Molhoek KR, Brautigan DL, Slingluff CL et al (2005) Synergistic inhibition of human melanoma proliferation by combination treatment with B-raf inhibitor BAY43-9006 and mTOR inhibitor rapamycin. J Transl Med 3:39–45
Storer BE (1989) Design and analysis of phase I clinical trials. Biometrics 45(3):925–937
Lankheet NA, Steeghs N, Rosing H et al (2013) Quantification of sunitinib and N-desethyl sunitinib in human EDTA plasma by liquid chromatography coupled with electrospray ionization tandem mass spectrometry: validation and application in routine therapeutic drug monitoring. Ther Drug Monit 35(2):168–176
Zimmerman JJ, Kahan BD (1997) Pharmacokinetics of sirolimus in stable renal transplant patients after multiple oral dose administration. J Clin Pharmacol 37:405–415
Pawaskar DK, Straubinger RM, Fetterly GJ et al (2013) Synergistic interactions between sorafenib and everolimus in pancreatic cancer xenografts in mice. Cancer Chemother Pharmacol 71(5):1231–1240
Pignochino Y, Dell’Aglio C, Inghilleri S et al (2015) The combination of sorafenib and everolimus shows antitumor activity in preclinical models of malignant pleural mesothelioma. BMC Cancer 15:374
Altomare I, Bendell JC, Bullock KE et al (2011) A phase II trial of bevacizumab plus everolimus for patients with refractory metastatic colorectal cancer. Oncologist 16(8):1131–1137
Slingluff CL, Petroni GR, Molhoek KR et al (2013) Clinical activity and safety of combination therapy with temsirolimus and bevacizumab for advanced melanoma: a phase II trial (CTEP 7190/Mel47). Clin Cancer Res 19(13):3611–3620
Rini BI, Garcia JA, Cooney MM et al (2009) A phase I study of sunitinib plus bevacizumab in advanced solid tumors. Clin Cancer Res 15:6277–6283
Gangadhar TC, Cohen EW, Wu K et al (2011) Two drug interaction studies of sirolimus in combination with sorafenib or sunitinib in patients with advanced malignancies. Clin Cancer Res 17(7):1956–1963
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, J., Kluger, H., Devine, L. et al. Phase I study of safety and tolerability of sunitinib in combination with sirolimus in patients with refractory solid malignancies and determination of VEGF (VEGF-A) and soluble VEGF-R2 (sVEGFR2) in plasma. Cancer Chemother Pharmacol 77, 1193–1200 (2016). https://doi.org/10.1007/s00280-016-3033-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-016-3033-7