
Abstract
Cognitive dysfunction is one of the comorbidities of diabetes mellitus, but hippocampus-dependent learning and memory, a component of cognitive function, shows particular decline in type 2 diabetes, suggesting an increased risk for dementia and Alzheimer’s disease. Cognitive function is related to dysregulated glucose metabolism, which is the typical cause of type 2 diabetes; however, hippocampal glycogen and its metabolite lactate are also crucial for hippocampus-dependent memory function. Type 2 diabetes induced hippocampus-dependent learning and memory dysfunction can be improved by chronic exercise and this improvement may possibly mediate through an adaptation of the astrocyte-neuron lactate shuttle (ANLS). This chapter focuses on the dysregulation of hippocampal glycometabolism in type 2 diabetes examining both existing evidence as well as the potential underlying pathophysiological mechanism responsible for memory dysfunction in type 2 diabetes, and showing for the first time that chronic exercise could be an effective therapy for type-2-diabetes-induced hippocampal memory decline.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Type 2 diabetes mellitus
- Hippocampus-dependent learning and memory
- Hippocampal glycometabolism
- Monocarboxylate transporter 2
1 Introduction
Diabetes mellitus is a metabolic disorder that is associated with a number of serious complications. The International Diabetes Federation reported 425 million persons as having diabetes mellitus in 2018, and this number is estimated to increase to 629 million by 2045. The World Health Organization (WHO) ranked diabetes mellitus among the top 10 causes of the 56.9 million deaths worldwide in 2016, indicating that diabetes mellitus is a common and serious disorder worldwide warranting global attention.
A complication of diabetes mellitus that has received considerable attention in recent years is hippocampal cognitive dysfunction, which is a recognized risk factor for dementia and Alzheimer disease (Heijer et al. 2003; Cukierman et al. 2005; Mccrimmon et al. 2012; Umegaki et al. 2013). Learning and memory deficits in cognitive components are more profound in patients with type 2 diabetes than in those with type 1 diabetes (Biessels et al. 2006; Biessels and Despa 2018; Mccrimmon et al. 2012), and this could be associated with hippocampal atrophy (Gold et al. 2007; Mccrimmon et al. 2012). The American College of Sports Medicine (ACSM) and the American Diabetes Association (ADA), based on a number of studies, recommend regular physical activity for improvement of diabetes and indicate that chronic exercise has health benefits that go beyond blood glucose control, weight control, and a reduction of the risk of well-known complications of type 2 diabetes although its effects are yet to be investigated for hippocampal memory dysfunction (Mu et al. 2001; Holloszy 2005; O’Gorman et al. 2006; Sriwijitkamol et al. 2007; Colberg et al. 2010; Lee et al. 2011; Jenkins et al. 2012).
At present, no consensus has been reached regarding the underlying mechanism of memory dysfunction in type 2 diabetes, including hippocampus-based memory decline, but dysregulation of glucose utilization in the brain (Sickmann et al. 2010), brain-derived neurotrophic factor (BDNF) (Stranahan et al. 2009), neuroinflammation (Whitmer 2007), and oxidative stress have all been associated with memory dysfunction in patients with type 2 diabetes (Whitmer 2007; Stranahan 2015). Interestingly, one metabolic adaptation shown in type 2 diabetes is elevated glycogen deposition in the heart (Bhattacharjee et al. 2006; Shearer et al. 2011). This suggests that a metabolic adaptation involving glycogen might also occur in the hippocampus, a crucial site for memory formation, and lead to memory dysfunction in type 2 diabetes as hippocampal glycogen-derived lactate in astrocytes and its transport into neurons plays a crucial role in hippocampus-dependent memory formation (Newman et al. 2011; Suzuki et al. 2011).
Chronic exercise , which positively affects hippocampus-dependent memory function in animals (van Praag et al. 1999; Brown et al. 2003; Liu et al. 2009; Creer et al. 2010), also increases hippocampal glycogen levels in normal rats (Matsui et al. 2012) and lactate transporter levels (monocarboxylate transporters; MCT) in type 1 diabetic rats (Aveseh et al. 2014), suggesting that chronic exercise could potentially ameliorate type-2-diabetes–related memory dysfunction through an adaptation of hippocampal glycometabolism which means glycogen metabolism (metabolism of sugars and other carbohydrate). In this chapter, we summarize the evidence for an association between brain glycometabolism dysregulation and cognitive decline in type 2 diabetes and the effects of chronic exercise on hippocampus-dependent memory dysfunction.
2 Cognitive Decline and Dysregulated Hippocampal Glycometabolism in Type 2 Diabetes
There are two types of diabetes mellitus: type 1 tends to develop in childhood and is characterized by a lack of insulin due to destruction of the βcells of the islet of Langerhans in the pancreas. By contrast, obesity and lack of physical activity are the crucial risk factors for type 2 diabetes, a disease condition with reduced insulin sensitivity and insulin resistance. Both type 1 and type 2 diabetes are associated with high blood glucose levels (hyperglycemia), which is the cause of numerous complications including cognitive dysfunction (Mccrimmon et al. 2012). Furthermore, insulin resistance is also a risk factor for cognitive dysfunction (Geroldi et al. 2005). In human studies, neuroimaging provides evidence that diabetes affects the brain, as adverse effects are evident in the cortical and subcortical regions, such as the medial temporal lobe (hippocampus and amygdala) (Heijer et al. 2003; Manschot et al. 2006), which are related to cognitive impairment associated with attention, executive function, information processing speed, and memory in patients with type 2 diabetes (Manschot et al. 2006). In these patients, the hippocampus may be the first site affected by type 2 diabetes (Gold et al. 2007). The neuropathological changes seen in the hippocampus with type 2 diabetes are also characteristic of Alzheimer’s disease, but the same changes are not seen in patients diagnosed with dementia.
Conversely, in rodents, cognitive function tested using the T-maze and Morris water maze shows a decline in streptozotocin (STZ)-induced type 1 diabetic mice (Flood et al. 1990; Biessels et al. 1996; Kamal et al. 1999). In terms of alterations related to learning and memory at the cellular level, N-methyl-d-aspartate (NMDA)-dependent long-term potentiation (LTP) in the CA1 and NMDA-independent LTP in the CA3 fields of the hippocampus are also impaired in STZ-induced diabetic rats, and this impairment is correlated with the severity of hyperglycemia (Biessels et al. 1996; Chabot et al. 1997; Tekkök and Krnjevic 1999). Based on this research, type 2 diabetes could have a similar type of impact on cognitive function, particularly through its effects on the hippocampus. Much information is available regarding the effects of glucose metabolism on cognitive function, but the role of glycometabolism on cognitive deficits in type 2 diabetes is not yet clearly understood. Recent findings suggest that hippocampal glycogen plays an important role in memory processing (Newman et al. 2011; Suzuki et al. 2011), and that hippocampal glycometabolism could be altered/dysregulated in type 2 diabetes (Fig. 1).

Proposed pathophysiological mechanisms in type 2 diabetes. In contrast to type 1 diabetes, which is caused by insulin deficit due to the immune-mediated βcell destruction of pancreatic cells in the islets of Langerhans, type 2 diabetes occurs through insulin resistance and reduced insulin sensitivity that presents as hyperglycemia and is induced by genetic susceptibility, several environmental, lifestyle factors, such as obesity, physical inactivity, and ageing, leading to various comorbidities. The chronicity of these symptoms on the peripheral tissues affects the central nervous system and causes cognitive decline, which has been recently recognized as one of the comorbidities of diabetes. Brain glycometabolism is associated with cognitive function, particularly in the hippocampus, so dysregulated glycometabolism could be the underlying mechanism leading to the dysfunction of memory in type 2 diabetes
2.1 Hippocampal Glycometabolism and Cognitive Function
The energy requirements of the brain are high, and brain energy metabolism changes depending on neuronal activity. Pioneering work on brain energy metabolism using the 2-deoxyglucose (2-DG) autoradiographic technique demonstrated that blood glucose is metabolized in the brain by neuronal activity, suggesting a metabolic coupling between neuronal activity and blood vessels (Newman et al. 2011). Furthermore, activation of glutamatergic neurons causes the release of glutamate into the intercellular space, and glutamate is a trigger that induces glycolysis in astrocytes (Magistretti and Pellerin 1999). The glutamate released from glutamatergic synapses is co-transported with Na+ into astrocytes, which express the glutamate transporters GLAST and GLT1. This co-transport activates the astrocytic Na+/K+-ATPase, and glycolysis is subsequently stimulated in the astrocytes through activation of phosphoglycerate kinase (PGK) (Magistretti and Pellerin 1999). The lactate produced through glycolysis in the astrocytes is transported to the neurons via MCT (astrocyte: MCT1 and MCT4; neuron: MCT2) for use as an energy substrate, and this metabolic coupling between these two cell types has led to the proposal of a hypothetical ‘astrocyte-neuron lactate shuttle’ (ANLS) (Pellerin et al. 1998). In the case of high or long-lasting neuronal activity, the glucose supply from the blood vessels to neurons becomes insufficient, thus, the contribution of glycogen stored exclusively in astrocytes is important for sustaining neuronal activity and maintaining a high rate of glycolysis (Swanson et al. 1992).
Astrocytic glycogen degradation (via glycogenolysis) is stimulated by noradrenaline (NA), vasoactive intestinal peptide (VIP), adenosine, and serotonin resulting in the release of lactate that is transported to neurons in the same manner as lactate derived from glycolysis (Magistretti et al. 1981; Hof et al. 1988; Sorg and Magistretti 1991, 1992; Dringen et al. 1993; Choi et al. 2012; Matsui et al. 2015). Interestingly, the tight coupling between neuronal activity and energy metabolism suggests that ‘metabolic plasticity’ underlies neuronal plasticity (Magistretti 2006; Choi et al. 2012). Indeed, metabolic adaptation occurs via glycogen metabolic processes under in vitro conditions. As mentioned above, neurotransmitters (namely NA, VIP, and adenosine) and neuromodulators induce glycogenolysis in a short time (seconds to minutes); however, NA has long-term effect in that it can stimulate a transcriptionally regulated action that results in glycogen synthesis (Allaman et al. 2000, 2003). The long-term effects of NA include NA-triggered cyclic-AMP production, which increases the expression of the transcriptional factor CCAAT/enhancer-binding protein (C/EBP), glycogen synthase, and protein targeting to glycogen (PTG) (Cardinaux and Magistretti 1996; Ruchti et al. 2016) (Fig. 2).

Simplified illustration of the astrocyte-neuron lactate shuttle: Contribution of glycogen-derived lactate to cognitive function. During high or long-lasting neuronal activity, brain glycogen-derived lactate becomes an important energy source to satisfy neuronal energy demand. Glycogen degradation is induced by noradrenaline (NA), vasoactive intestinal peptide (VIP), and adenosine (ADE), resulting in lactate production. Particularly in the hippocampus, lactate is transported via monocarboxylate transporters (MCT) and serves both as an energy substrate and in memory processing through induction of protein expression related to neural plasticity, such as Arc, phosphorylated-cAMP-response-element-binding protein (pCREB), and phosphorylated-Cofilin (pCofilin). Early growth response protein 1 (EGR1) and brain-derived neurotrophic factor (BDNF) are also induced by lactate in in vitro experiments. These lactate effects are suppressed by blocking MCT2 or inhibiting glycogen degradation, suggesting that glycogen-derived lactate is crucial for memory processing and neuronal plasticity. NA induces both glycogenolysis as a short-term effect and glycogen synthesis as a long-term effect by inducing protein targeting to glycogen (PTG) and glycogen synthase (GS)
Recent studies have demonstrated that l-lactate is used as an energy substrate for neurons (Bélanger et al. 2011), and it also acts as a signaling molecule to induce the expression of neural plasticity related genes (i.e., activity-regulated cytoskeleton-associated protein [Arc], c-Fos, and Zif268) in cultured neurons (Yang et al. 2014). These are immediate early genes (IEGs), which are induced during both long-term memory and long-term plasticity (Alberini 2009; Bramham et al. 2010; Caroni et al. 2012). These effects of l-lactate are mediated by NMDA receptor activity in neurons and its downstream signaling Erk1/2 cascade, which means that l-lactate potentiates glutamatergic neuronal currents by ensuring an increase in intracellular calcium through NMDA receptor activation and changes in the cellular redox state, and that it acts as a neuromodulator to induce plasticity-related gene expression (Yang et al. 2014). Although most of these observations about ANLS were elucidated in vitro experiments using cortical cultured neurons or astrocytes, the most important current work on lactate effects associated with learning and memory in the hippocampus has shown that lactate derived from astrocytes is essential for long-term memory formation (Suzuki et al. 2011). Hippocampus-dependent long-term memory formation (measured using an inhibitory avoidance task) and LTP were inhibited when lactate production from astrocytes was blocked using DAB (1,4-dideoxy-1,4-imino-d-arabinitol). By contrast, co-injection of DAB and l-lactate rescued inhibited memory consolidation and LTP (Suzuki et al. 2011).
The underlying mechanisms by which glycogen-derived lactate induces memory formation could be associated with the induction of Arc, phosphorylated-cAMP-response-element-binding protein (pCREB), and phosphorylated-Cofilin (pCofilin), which are related to synaptic plasticity (Suzuki et al. 2011) (Fig. 2). A similar observation relating hippocampal glycogen to working memory has also been made with another behavioral paradigm (closed plus maze) (Newman et al. 2011). Little in vivo evidence supports a role for brain glycogen in memory function, but the blockade of glycogenolysis with DAB impaired a discrimination avoidance learning task in chicks (Gibbs et al. 2006). Similarly, the lack of brain glycogen generated by knockout of glycogen synthase specifically (GYS1) in the nervous system impaired memory in an operant conditioning task and LTP (Boury-Jamot et al. 2015).
2.2 Dysregulated Hippocampal Glycometabolism and Cognitive Decline in Type 2 Diabetes
A number of studies have shown that the central nervous system is affected by diabetes, and that diabetes-related cognitive decline is an independent risk factor for dementia and Alzheimer’s disease (Biessels et al. 2006; Biessels and Despa 2018; Mccrimmon et al. 2012). These diseases are accompanied by morphological changes and metabolic alteration/dysregulation in the hippocampus and cortex, which are regions associated with cognitive function (Heijer et al. 2003; Sickmann et al. 2012; Biessels and Despa 2018). The hippocampus, in particular, shows atrophy in the initial stage of these neuro-degenerative disorders (Heijer et al. 2003; Gold et al. 2007). The energy supply to the brain depends strongly on glucose supply from blood vessels; thus, impairment of glucose metabolism by insulin deficiency, as commonly occurs in diabetes, could be one of the risk factors for impaired cognitive function. Moreover, hippocampal glycogen-derived lactate metabolism associated with cognitive function could also be affected by diabetes. However, little research has examined the relationship between hippocampal glycometabolism and cognitive decline/dysfunction in diabetes.
Previous studies examining the glycogen levels in the hippocampus and/or cortex in diabetic rats have demonstrated that STZ-induced type 1 diabetic rats had increases in glycogen levels in the cortex depending on the STZ treatment duration (Plaschke and Hoyer 1993). Conversely, in type 2 diabetes models, Zucker obese (ZO) rats had hyper-levels of hippocampal and cortical glycogen than did their counterpart Zucker lean (ZL) genetic control rats, whereas Zucker diabetic fatty (ZDF) rats had lower levels of cortical glycogen compared with control ZDF lean rats (Sickmann et al. 2010). One possible explanation for the conflicting results between these two studies is that the methodology used for brain tissue preparation was neither optimal nor standard/validated for glycogen measurement (Swanson et al. 1989; Dringen et al. 1993). Brain glycogenolysis degraded rapidly by ischemia; therefore, microwave irradiation can inactivate glycogenolysis enzymes needed for the accurate measurement of brain glycogen (Kong et al. 2002).
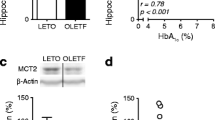
Our laboratory has endeavored to elucidate the glycometabolism pattern in type 2 diabetic rats by measuring glycogen and MCT levels in the hippocampus using microwave irradiation, with consideration to the issues mentioned above. For three reasons we used Otsuka Long-Evans Tokushima Fatty (OLETF) rats as our type 2 diabetes model: (a) OLETF rats exhibit hyperphagia; (b) they have a dysregulated sympathoadrenal response and show a decline in executive function (Suge et al. 2012); and (c) the onset of diabetes takes time, in agreement with human type 2 diabetes (Kawano et al. 1992). Type 2 diabetic rats exhibit hyper-glycogen levels and diminished MCT2 protein levels in the hippocampus when compared with their counterpart genetic control rats Long-Evans Tokushima Otsuka (LETO), and a negative relationship exists between the two components of the glycometabolism system in neurons (Shima et al. 2016a) (Fig. 3). Notably, the negative correlation exhibited in type 2 diabetic rats has been observed only in the hippocampus, and not in the hypothalamus and the cortex, even though these regions exhibit changes similar to those seen in the hippocampus, namely, glycogen (hypothalamus and cortex) and MCT2 (hypothalamus) increases, implying that the hippocampus may be the first region affected by dysregulation of glycometabolism induced by type 2 diabetes (Shima et al. 2016a). The significance of hyper-hippocampal glycogen in type 2 diabetic rats may indicate metabolic compensation for the decreased lactate utilization via MCT2 and this consequence may have linkage with ANSL although concrete evidence demands. Indeed, the same phenomenon was reported in the hearts of patients with type 2 diabetes (Bhattacharjee et al. 2006; Shearer et al. 2011). Notably, MCT1 and MCT4 are expressed in astrocytes and have a similar transport roles in astrocytes in all three brain regions (hippocampus, hypothalamus and cortex) as stated above (Shima et al. 2016a). Thus, collectively the findings observed in the study by Shima et al. at the progressive stage of type 2 diabetic hippocampus in concern to glycometabolism and the associated molecular mechanism warrant future studies to be conducted at pre diabetes or at the early stage of diabetes for the advancement of clinical improvement of diabetic complications.

Dysregulated hippocampal glycometabolism in a rat model of type 2 diabetes. Glycogen levels in the hippocampus, hypothalamus, and cerebellum are higher in the OLETF type 2 diabetes rat model than in control LETO rats. Conversely, MCT2 expression in the hippocampus and hypothalamus is lower in OLETF rats than in LETO rats, even though MCT1 and MCT4 levels show no significant differences between the LETO and OLETF rats. However, a significant correlation exists between MCT2 and glycogen levels, but only in the hippocampus, suggesting that memory dysfunction in OLETF rats could be caused by dysregulated hippocampal glycometabolism. (a) Glycogen levels in the hippocampus, cortex, hypothalamus, cerebellum, and brainstem, (b) MCT1 levels, (c) MCT2 levels, (d) MCT4 levels, (e) Correlation between MCT2 and glycogen levels in the hippocampus, (f) Correlation between MCT2 and glycogen levels in the hypothalamus. (Referenced by Shima et al., J Physiol Sci, 2016)
3 Chronic Exercise Effects on Dysregulated Hippocampal Glycometabolism and Cognitive Decline in Type 2 Diabetes
Chronic exercise can normalize blood glucose level control by enhancing glucose uptake in the peripheral tissues in an insulin-independent manner and is an effective therapy for type 2 diabetes complications such as neuropathy (Winder et al. 1987; Mu et al. 2001; Balducci et al. 2006). Cognitive decline is one of the complications of type 2 diabetes (Gispen and Biessels 2000; Whitmer 2007), with hippocampal dysfunction being a particularly serious disability associated with the development of dementia and Alzheimer’s disease (Pouwer et al. 2003; Witting et al. 2006; Ahtiluoto et al. 2010). Since a number of studies have demonstrated an improvement of cognitive function, such as hippocampus-dependent memory (van Praag et al. 1999; Liu et al. 2009; Creer et al. 2010; Bolz et al. 2015), with chronic exercise, this type of exercise could have the potential to affect hippocampal dysfunction in type 2 diabetes in a beneficial manner. Recently, our laboratory identified a chronic exercise regimen effective for hippocampal dysfunction in type 2 diabetes (Shima et al. 2016b); thus, the effects of acute and chronic exercise on hippocampal glycometabolism in normal and type 2 diabetic rats will be reviewed next.
3.1 Hippocampal Glycometabolism During Acute Exercise in Normal Rats
Exercise requires mobilization of every system of the body, including muscles and the brain (Vissing et al. 1996; Secher et al. 2008). Glucose molecules stored as glycogen play a crucial role in the immediate energy supply needed by exercising muscles to maintain muscular contraction because depletion of muscle glycogen during exercise causes a decline in endurance performance (Secher et al. 2008). Muscle glycogen decreases in an activity-dependent manner (Gollnick et al. 1974), and depletion of muscle and liver glycogen during hypoglycemia occurs when rats are subjected to prolonged exhaustive exercise (Winder et al. 1987). Exercise also affects the brain and increases neuronal activity (Saito and Soya 2004; Ohiwa et al. 2006, 2007, Soya et al. 2007a, b); furthermore, glucose is utilized as an energy source by neurons during exercise (Vissing et al. 1996). However, whether brain glycogen, and particularly hippocampal glycogen, decreases in a similar manner to that seen for muscle glycogen following prolonged exhaustive exercise remains an unanswered question.
In one previous experiment, glycogen levels in muscles and the liver were depleted after exhaustive exercise (treadmill running), which induced hypoglycemia (Matsui et al. 2011). Hippocampal glycogen also decreased with acute moderate exhaustive exercise (20 m/min, until exhaustion) (Matsui et al. 2011). Furthermore, levels of methoxyhydroxyphenylglycol (MHPG) and 5-Hydroxyindoleacetic acid (5-HIAA), which are the respective metabolites of NA and serotonin (5-TH), were increased in cerebral cortex after exercise, and their levels were negatively correlated with a decrease in cortical glycogen, suggesting that NA and 5-HT could be part of an underlying mechanism of exercise-induced brain glycogen degradation (Matsui et al. 2011).
Based on these results, next we will discuss exercise-induced hippocampal glycogen dynamics, especially in the recovery phase of glycometabolism. Muscle glycogen decreases with exhaustive exercise and then returns to above basal levels (supercompensation) 24 h after exercise (Bergström and Hultman 1966). Surprisingly, brain glycogen, including hippocampal glycogen, also exhibits supercompensation 6 h after exhaustive exercise (Matsui et al. 2012); furthermore, the rate of brain glycogen supercompensation depends on the rate of glycogen decrease during exercise, similar to the situation seen in muscles (Gaesser and Brooks 1980) and consistent with studies demonstrating brain glycogen supercompensation at 4–7 h after insulin-induced hypoglycaemia (Choi et al. 2003; Canada et al. 2011).
An adaptation to chronic exercise occurs, as indicated by glycogen supercompensation following exhaustive exercise (James and Kraegen 1984), as 4 weeks of chronic moderate exercise resulted in an increase of basal hippocampal and cortical glycogen levels in normal rats (Matsui et al. 2012). The underlying mechanisms of hippocampal glycogen synthesis is not clear, but exercise is reported to activate NA neurons (Kitaoka et al. 2010), and NA metabolites, such as MHPG, increase following exhaustive exercise and are negatively correlated with brain glycogen decreases (Matsui et al. 2011), suggesting that NA-induced glycogen synthesis might be involved (Allaman et al. 2000; Crosson et al. 2003; Ruchti et al. 2016) (Fig. 4).

Brain glycometabolism during acute exhaustive exercise. Brain glycogen levels decrease in the hippocampus, cortex, hypothalamus, cerebellum, and brainstem following acute exhaustive treadmill running exercise, while increases in 5-HIAA and MHPG, which are metabolites of serotonin (5-HT) and NA , respectively, occur after exercise. The decrease in glycogen levels in the cortex during exercise was negatively correlated with increased 5-HIAA and MHPG levels in the cortex, suggesting that 5-HT and NA may be involved in glycogenolysis during exercise. (Summarized figure based on Matsui et al., J Physiol, 2011)
3.2 Hippocampal Glycometabolism with Acute Exercise in Type 2 Diabetes Rats
A number of studies have shown that exercise is effective in restoring dysregulated glucose control in type 2 diabetes (Sigal et al. 2007; Ruchti et al. 2016); however, the most effective/beneficial exercise conditions and exercise effects on hippocampal glycometabolism remain unestablished. To address this, we have to use a relatively identical exercise intensity protocol for both non-diabetic rats and diabetic rats. Exercise increases blood lactate level which depends on the exercise duration and intensity; the exercise intensity at which blood lactate level begins to exponentially increase is called lactate threshold. Stress responses (plasma ACTH and adrenaline release) are also taken place at lactate threshold (Soya et al. 2007a), thus it is useful to determine the relatively same exercise intensity protocol based on the lactate threshold for the study rats to be compared. We used an acute moderate exercise protocol similar to a moderate exercise intensity based on the lactate threshold of type 2 diabetic rats (OLETF: 22.6 ± 0.3 m/min) and genetic control rats (LETO: 28.3 ± 1.8 m/min) (Shima et al. 2016b), and we then examined the effect of acute moderate exercise on hippocampal glycometabolism (Shima et al. 2016b). We first noted a decrease in hippocampal glycogen following 30 minutes of acute moderate exercise in both OLETF and LETO rats that was positively correlated with the hippocampal lactate increase (Shima et al. 2016b). Diabetes patients exhibit hyper-glycogen in the heart (Bhattacharjee et al. 2006), and the OLETF rats exhibited hyper-glycogen in the hippocampus, which could be a result of adaptation to declining lactate utilization due to the downregulation of MCT2 (Shima et al. 2016a, b). These results suggest that hippocampal glycometabolism although dysregulated in type 2 diabetic rats is activated with acute moderate exercise and this activation with acute exercise is also observed in normal rats hippocampus.
3.3 Effects of Chronic Exercise on Type-2-Diabetes-Induced Dysregulation of Glycometabolism Accompanied with Cognitive Decline
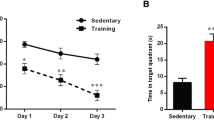
Regular exercise is an effective therapy for type 2 diabetes because it leads to improvement of glycometabolism in peripheral organs (Mu et al. 2001; Holloszy 2005; O’Gorman et al. 2006; Sigal et al. 2007). In addition, chronic exercise not only enhances hippocampus-dependent memory but it also increases hippocampal glycogen levels in normal rats and MCT expression in both peripheral organs and brain tissues (hippocampus and cortex) in diabetes (van Praag et al. 1999; Juel et al. 2004; Liu et al. 2009; Creer et al. 2010; Nikooie et al. 2013; Aveseh et al. 2014; Bolz et al. 2015), suggesting that chronic exercise could be an appropriate exercise therapy for ameliorating hippocampal memory function. OLETF rats exhibited significant hippocampal-dependent memory dysfunction when compared to LETO rats, but, surprisingly, after four weeks of chronic moderate exercise, which is reported to increase hippocampal glycogen levels, memory function was also ameliorated in the OLETF rats (Shima et al. 2016b) (Fig. 5). Chronic exercise induced hyper-glycogen levels in the hippocampus of normal rats (Matsui et al. 2012) as well as in OLETF rats with concomitant normalization of downregulated MCT2 levels in neurons, increased glycogen synthase (GS) levels, and also ameliorated glucose transporter 1 (GLUT1) expression in astrocytes in the hippocampus without any significant alteration in the expression of GLUT3, MCT1 and MCT4 (Shima et al. 2016b). Interestingly, chronic exercise increased BDNF levels in the hippocampus of LETO rats, whereas no change in BDNF expression levels occurred in OLETF rats; furthermore, expression of other proteins associated with the downstream BDNF signaling, such as tyrosine receptor kinase B (TrkB), pCREB, and phosphorylated-GS kinase-3β(GSK-3β), were not changed by chronic exercise in either LETO or OLETF rats, implying that normalization of MCT2 levels and further increases in glycogen levels in the hippocampus could be an adaptation in response to chronic exercise that could contribute to the amelioration of hippocampus-dependent memory dysfunction in OLETF rats (Fig. 6).

The effects of chronic exercise on hippocampus-dependent memory and dysregulated hippocampal glycometabolism in type 2 diabetes. The escape latency during learning test of OLETF rats was shortened by 4 weeks of moderate exercise, the time spent in platform area during probe test was increased after 4 weeks of exercise. Furthermore, hippocampal glycogen levels were increased in exercised group of both LETO and OLETF rats, and MCT2 levels in the hippocampus were normalized in exercised OLETF rats group, suggesting that4 weeks of moderate exercise ameliorated hippocampus-dependent memory via improvement of MCT2-mediated lactate uptake into the neurons in OLETF rats (a) Escape latency, (b) Time spent in platform area during probe test, (c) Hippocampal glycogen levels, (d) Hippocampal MCT2 levels. Circles, LETO rats; squares, OLETF rats; white symbols and bars, sedentary rats; black symbols and bars, exercised rats, n = 6–8 rats per group. ∗p < 0.05 and ∗∗∗p < 0.001 vs LETO rats; †p < 0.05, ††p < 0.01 and †††p < 0.001 vs sedentary rats (Referenced by Shima et al., Diabetologia, 2016)

Conceptual diagram of the chronic exercise effect on ameliorating hippocampus-dependent memory in type 2 diabetes. A 4-week regimen of moderate exercise improves memory dysfunction in type 2 diabetes via amelioration of hippocampal glycometabolism dysregulation. The dysregulated hippocampal glycometabolism and its relationship to lactate transport might be one of the possible etiologies of memory dysfunction in type 2 diabetes. (Summarized figure based on the Shima et al., Diabetologia, 2016)
A dysregulation of MCT2-mediated lactate uptake in neurons may be a novel etiology of memory dysfunction in type 2 diabetes, especially in the context of the hippocampus (Shima et al. 2016b). However, four weeks of chronic exercise did not improve well-known peripheral symptoms/abnormalities of type 2 diabetes, such as alterations in circulatory glucose, insulin and HbA1c levels, which means that more long-term exercise is needed to simultaneously ameliorate the pathology concerned not only with cognitive dysfunction but also with dysregulated glucose control at the peripheral level. Nevertheless, the amelioration of memory dysfunction induced with chronic (four weeks) exercise was exhibited earlier than other pathologies in the peripheral organs. Based on our preliminary findings, abnormal hippocampal glycometabolism and memory dysfunction are already evident at the pre-type2diabetes stage, so the nervous system may require more rapid adaptation to type 2 diabetes than do other organs. These findings suggest that conventional clinical diagnostic indices or criteria for type 2 diabetes, such as glucose and HbA1c levels, may not reflect the earliest organ involvement, which would be the brain with functional hippocampal impairment, as is evident in our unpublished observations and in other studies during the early phase of pre-type-2 diabetes (Soares et al. 2013). Thus, new diagnostic criteria might be necessary to prevent or delay the onset of type 2 diabetes with an urgent focus on the initiation and progression of brain memory dysfunction. Further, it is important to innovate an appropriate exercise regimen either from pre-diabetes or early diabetes, and even starting at advanced stages, which would clinically improve peripheral diabetic or pre-diabetic symptoms with exerting a beneficial and effective positive role on memory dysfunction through in depth comprehensive future studies.
4 Concluding Remarks and Future Direction
The rapid increase in the number of patients with type 2 diabetes worldwide is a great global problem; hence, a strategy is needed apart from pharmacotherapy such as insulin treatment or other established anti-diabetic drugs, to treat or solve this problem. Cognitive decline is a particularly devastating complication of type 2 diabetes, as hippocampal dysfunction is a serious disability that is associated with dementia, Alzheimer disease, and depression (Pouwer et al. 2003; Ahtiluoto et al. 2010) ultimately causing a remarkable morbidity in social life. Although there are to date still very few studies focusing on this area, in this chapter we largely reviewed the ameliorating effects of chronic exercise on memory dysfunction in type 2 diabetes at the advanced stage observed in our recent findings. The dysregulation of hippocampal MCT2-mediated lactate uptake into neurons could be the etiology of hippocampus-dependent memory dysfunction in type 2 diabetes, and this is a completely novel addition to this field. Although we do not have any clear and concrete evidence linking our results to ANLS, we assume that the exercise induced amelioration of dysregulated glycometabolism in hippocampus together with cognitive impairment may take place through the adaptation of ANLS although future studies should clarify this speculation through extensive in vivo and in vitro investigations. The current findings have potential implications for translation to human subjects, especially in the context of exercise intensity and duration, as lactate threshold has been a crucial determinant for the exercise models in our diabetes studies. Further investigation is needed to determine the optimal exercise condition to ameliorate type-2-diabetes-induced memory dysfunction together with beneficial effects on peripheral diabetic symptoms and complications, even with advanced diabetes, from the perspective of lifestyle-based therapeutic strategies and intervention. Indeed, we are currently on the way to explore the best exercise regimen for diabetes and diabetes-induced organ complications both in terms of prevention and therapeutics through a multi-disciplinary and multi-faceted translational research approach .
Abbreviations
- ANLS:
-
Astrocyte-neuron lactate shuttle
- BDNF:
-
Brain-derived neurotrophic factor
- DAB:
-
1,4-Dideoxy-1,4-imino-D-arabinitol
- GLUT:
-
Glucose transporter
- GSK-3β:
-
Phosphorylated-GS kinase-3β
- LETO:
-
Long-Evans Tokushima Otsuka
- LTP:
-
Long-term potentiation
- MCT:
-
Monocarboxylate transporters
- MHPG:
-
NA metabolites
- NA:
-
Noradrenaline
- NMDA:
-
N-methyl-d-aspartate
- OLETF:
-
Otsuka Long-Evans Tokushima Fatty
- pCofilin:
-
Phosphorylated-Cofilin
- pCREB:
-
Phosphorylated-cAMP-response-element-binding protein
- PGK:
-
Phosphoglycerate kinase
- PTG:
-
Protein targeting to glycogen
- STZ:
-
Streptozotocin
- VIP:
-
Vasoactive intestinal peptide
- ZDF:
-
Zucker diabetic fatty
- ZL:
-
Zucker lean
- ZO:
-
Zucker obese
References
Ahtiluoto S, Polvikoski T, Peltonen M, Solomon A, Tuomilehto J, Winblad B, Sulkava R, Kivipelto M (2010) Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology 75:1195–1202
Alberini CM (2009) Transcription factors in long-term memory and synaptic plasticity. Physiol Rev 89:121–145
Allaman I, Pellerin L, Magistretti PJ (2000) Protein targeting to glycogen mRNA expression is stimulated by noradrenaline in mouse cortical astrocytes. Glia 30:382–391
Allaman I, Pellerin L, Magistretti PJ (2003) Glucocorticoids modulate neurotransmitter-induced glycogen metabolism in cultured cortical astrocytes. J Neurochem 88:900–908
Aveseh M, Nikooie R, Sheibani V, Esmaeili-Mahani S (2014) Endurance training increases brain lactate uptake during hypoglycemia by up regulation of brain lactate transporters. Mol Cell Endocrinol 394:29–36
Balducci S, Iacobellis G, Parisi L, Di Biase N, Calandriello E, Leonetti F, Fallucca F (2006) Exercise training can modify the natural history of diabetic peripheral neuropathy. J Diabetes Complications 20:216–223
Bélanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–738
Bergström J, Hultman E (1966) Muscle glycogen synthesis after exercise: an enhancing factor localized to the muscle cells in man. Nature 210:309–310
Bhattacharjee M, Venugopal B, Wong K, Goto Y, Bhattacharjee M (2006) Mitochondrial disorder, diabetes mellitus, and findings in three muscles, including the heart. Ultrastruct Pathol 30:481–487
Biessels GJ, Despa F (2018) Cognitive decline and dementia in diabetes mellitus: mechanisms and clinical implications. Nat Rev Endocrinol 14:591–604
Biessels G-J, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH (1996) Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes 45:1259–1266
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P (2006) Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5:64–74
Bolz L, Heigele S, Bischofberger J (2015) Running improves pattern separation during novel object recognition. Brain Plast 1:129–141
Boury-Jamot B, Carrad A, Martin J, Halfon O, Magistretti P, Boutrel B (2015) Disrupting astrocyte–neuron lactate transfer persistently reduces conditioned responses to cocaine. Mol Psychiatry 21:1–7
Bramham CR, Alme MN, Bittins M, Kuipers SD, Nair RR, Pai B, Panja D, Schubert M, Soule J, Tiron A, Wibrand K (2010) The arc of synaptic memory. Exp Brain Res 200:125–140
Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG (2003) Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci 17:2042–2046
Canada SE, Weaver SA, Sharpe SN, Pederson BA (2011) Brain glycogen supercompensation in the mouse after recovery from insulin-induced hypoglycemia. J Neurosci Res 89:585–591
Cardinaux JR, Magistretti PJ (1996) Vasoactive intestinal peptide, pituitary adenylate cyclase-activating peptide, and noradrenaline induce the transcription factors CCAAT/enhancer binding protein (C/EBP)-beta and C/EBP delta in mouse cortical astrocytes: involvement in cAMP-regulated glyco. J Neurosci 16:919–929
Caroni P, Donato F, Muller D (2012) Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci 13:478–490
Chabot C, Massicotte G, Milot M, Trudeau F, Gagné J (1997) Impaired modulation of AMPA receptors by calcium-dependent processes in streptozotocin-induced diabetic rats. Brain Res 768:249–256
Choi IY, Seaquist ER, Gruetter R (2003) Effect of hypoglycemia on brain glycogen metabolism in vivo. J Neurosci Res 72:25–32
Choi HB, Gordon GRJ, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, Levin LR, Buck J, MacVicar BA (2012) Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl Cyclase. Neuron 75:1094–1104
Colberg SR, Sigal RJ, Fernhall B, Regensteiner JG, Blissmer BJ, Rubin RR, Chasan-Taber L, Albright AL, Braun B (2010) Exercise and type 2 diabetes: the American College of Sports Medicine and the American Diabetes Association: joint position statement. Diabetes Care 33:e147–e167
Creer DJ, Romberg C, Saksida LM, van Praag H, Bussey TJ (2010) Running enhances spatial pattern separation in mice. Proc Natl Acad Sci 107:2367–2372
Crosson SM, Khan A, Printen J, Pessin JE, Saltiel AR (2003) PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance. J Clin Invest 111:1423–1432
Cukierman T, Gerstein HC, Williamson JD (2005) Cognitive decline and dementia in diabetes - systematic overview of prospective observational studies. Diabetologia 48:2460–2469
Dringen R, Gebhardt R, Hamprecht B (1993) Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res 623:208–214
Flood JF, Mooradian AD, Morley JE (1990) Characteristics of learning and memory in streptozocin-induced diabetic mice. Diabetes 39:1391–1398
Gaesser GA, Brooks GA (1980) Glycogen repletion following continuous and intermittent exercise to exhaustion. J Appl Physiol Resp Env Ex Physiol 49:722–728
Geroldi C, Frisoni GB, Paolisso G, Bandinelli S, Lamponi M, Abbatecola AM, Zanetti O, Guralnik JM, Ferrucci L (2005) Insulin resistance in cognitive impairment. Arch Neurol 62:1067–1072
Gibbs ME, O’Dowd BS, Hertz E, Hertz L (2006) Astrocytic energy metabolism consolidates memory in young chicks. Neuroscience 141:9–13
Gispen WH, Biessels GJ (2000) Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci 23:542–549
Gold SM, Dziobek I, Sweat V, Tirsi A, Rogers K, Bruehl H, Tsui W, Richardson S, Javier E, Convit A (2007) Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 50:711–719
Gollnick PD, Piehl K, Saltin B (1974) Selective glycogen depletion pattern in human muscle fibres after exercise of varying intensity and at varying pedalling rates. J Physiol 241:45–57
den Heijer T, Vermeer SE, van Dijk EJ, Prins ND, Koudstaal PJ, Hofman A, Breteler MMB (2003) Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 46:1604–1610
Hof PR, Pascale E, Magistretti PJ (1988) K+ at concentrations reached in the extracellular space during neuronal activity promotes a Ca2+−dependent glycogen hydrolysis in mouse cerebral cortex. J Neurosci 8:1922–1928
Holloszy JO (2005) Exercise-induced increase in muscle insulin sensitivity. J Appl Physiol 63110:338–343
James DE, Kraegen EW (1984) The effect of exercise training on glycogen, glycogen synthase and phosphorylase in muscle and liver. Eur J Appl Physiol Occup Physiol 52:276–281
Jenkins NT, Padilla J, Arce-Esquivel AA, Bayless DS, Martin JS, Leidy HJ, Booth FW, Rector RS, Laughlin MH (2012) Effects of endurance exercise training, metformin, and their combination on adipose tissue leptin and IL-10 secretion in OLETF rats. J Appl Physiol 113:1873–1883
Juel C, Holten MK, Dela F (2004) Effects of strength training on muscle lactate release and MCT1 and MCT4 content in healthy and type 2 diabetic humans. J Physiol 556:297–304
Kamal A, Biessels GJ, Urban IJGW (1999) Hippocampal synaptic plasticity in streptozotocin-diabetic rats: impairment of long-term potentiation and facilitation of long-term depression. Neuroscience 90:737–745
Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T (1992) Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka long-Evans Tokushima fatty (OLETF) strain. Diabetes 41:1422–1428
Kitaoka R, Fujikawa T, Miyaki T, Matsumura S, Fushiki T, Inoue K (2010) Increased noradrenergic activity in the ventromedial hypothalamus during treadmill running in rats. J Nutr Sci Vitaminol (Tokyo) 56:185–190
Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD (2002) Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci 22:5581–5587
Lee H, Chang H, Park JY, Kim SY, Choi KM, Song W (2011) Exercise training improves basal blood glucose metabolism with no changes of cytosolic inhibitor κB kinase or c-Jun N-terminal kinase activation in skeletal muscle of Otsuka long-Evans Tokushima fatty rats. Exp Physiol 96:689–698
Liu YF, Chen HI, Wu CL, Kuo YM, Yu L, Huang AM, Sen WF, Chuang JI, Jen CJ (2009) Differential effects of treadmill running and wheel running on spatial or aversive learning and memory: roles of amygdalar brain-derived neurotrophic factor and synaptotagmin I. J Physiol 587:3221–3231
Magistretti PJ (2006) Neuron-glia metabolic coupling and plasticity. J Exp Biol 209:2304–2311
Magistretti PJ, Pellerin L (1999) Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci 354:1155–1163
Magistretti PJ, Morrison JH, Shoemaker WJ, Sapin V, Bloom FE (1981) Vasoactive intestinal polypeptide induces glycogenolysis in mouse cortical slices: a possible regulatory mechanism for the local control of energy metabolism. Proc Natl Acad Sci U S A 78:6535–6539
Manschot SM, Brands AMA, van der Grond J, Kessels RPC, Algra A, Kappelle LJ, Biessels GJ (2006) Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes 55:1106–1113
Matsui T, Soya S, Okamoto M, Ichitani Y, Kawanaka K, Soya H (2011) Brain glycogen decreases during prolonged exercise. J Physiol 589:3383–3393
Matsui T, Ishikawa T, Ito H, Okamoto M, Inoue K, Lee MC, Fujikawa T, Ichitani Y, Kawanaka K, Soya H (2012) Brain glycogen supercompensation following exhaustive exercise. J Physiol 590:607–616
Matsui T, Soya S, Kawanaka K, Soya H (2015) Brain glycogen decreases during intense exercise without hypoglycemia: the possible involvement of serotonin. Neurochem Res 40:1333–1340
Mccrimmon RJ, Phd R, Mccrimmon RJ, Ryan CM, Frier BM (2012) Diabetes and cognitive dysfunction. Lancet 379:2291–2299
Mu J, Brozinick JT, Valladares O, Bucan M, Birnbaum MJ (2001) A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell 7:1085–1094
Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427
Nikooie R, Rajabi H, Gharakhanlu R, Atabi F, Omidfar K, Aveseh M, Larijani B (2013) Exercise-induced changes of MCT1 in cardiac and skeletal muscles of diabetic rats induced by high-fat diet and STZ. J Physiol Biochem 69:865–877
O’Gorman DJ, Karlsson HKR, McQuaid S, Yousif O, Rahman Y, Gasparro D, Glund S, Chibalin AV, Zierath JR, Nolan JJ (2006) Exercise training increases insulin-stimulated glucose disposal and GLUT4 (SLC2A4) protein content in patients with type 2 diabetes. Diabetologia 49:2983–2992
Ohiwa N, Saito T, Chang H, Omori T, Fujikawa T, Asada T, Soya H (2006) Activation of A1 and A2 noradrenergic neurons in response to running in the rat. Neurosci Lett 395:46–50
Ohiwa N, Chang H, Saito T, Onaka T, Fujikawa T, Soya H (2007) Possible inhibitory role of prolactin-releasing peptide for ACTH release associated with running stress. Am J Physiol Regul Integr Comp Physiol 292:R497–R504
Pellerin L, Pellegri G, Bittar P, Charnay Y, Bouras C, Martin J, Stella N, Magistretti PJ (1998) Evidence supporting the existence of an activity dependent astrocyte-neuron lactate shuttle. Dev Neurosci 20:291–299
Plaschke K, Hoyer S (1993) Action of the diabetogenic drug streptozotocin on glycolytic and glycogenolytic metabolism in adult rat brain cortex and hippocampus. Int J Dev Neurosci 11:477–483
Pouwer F, Beekman ATF, Nijpels G, Dekker JM, Snoek FJ, Kostense PJ, Heine RJ, Deeg DJH (2003) Rates and risks for co-morbid depression in patients with type 2 diabetes mellitus: results from a community-based study. Diabetologia 46:892–898
Ruchti E, Roach PJ, DePaoli-Roach AA, Magistretti PJ, Allaman I (2016) Protein targeting to glycogen is a master regulator of glycogen synthesis in astrocytes. IBRO Rep 1:46–53
Saito T, Soya H (2004) Delineation of responsive AVP-containing neurons to running stress in the hypothalamus. Am J Physiol Regul Integr Comp Physiol 286:R484–R490
Secher NH, Seifert T, Van Lieshout JJ (2008) Cerebral blood flow and metabolism during exercise: implications for fatigue. J Appl Physiol 104:306–314
Shearer J, Ross KD, Hughey CC, Johnsen VL, Hittel DS, Severson DL (2011) Exercise training does not correct abnormal cardiac glycogen accumulation in the db/db mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab 301:E31–E39
Shima T, Jesmin S, Matsui T, Soya M, Soya H (2016a) Differential effects of type 2 diabetes on brain glycometabolism in rats: focus on glycogen and monocarboxylate transporter 2. J Physiol Sci 68:69–75
Shima T, Takashi M, Jesmin S, Okamoto M, Soya M, Inoue K, Liu Y-F, Torres-Aleman I, McEwen BS, Soya H (2016b) Moderate exercise ameliorates dysregulated hippocampal glycometabolism and memory function in a rat model of type 2 diabetes. Diabetologia 60:597–606
Sickmann HM, Waagepetersen HS, Schousboe A, Benie AJ, Bouman SD (2010) Obesity and type 2 diabetes in rats are associated with altered brain glycogen and amino-acid homeostasis. J Cereb Blood Flow Metab 30:1527–1537
Sickmann HM, Waagepetersen HS, Schousboe A, Benie AJ, Bouman SD (2012) Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem Int 60:267–275
Sigal RJ, Kenny GP, Boule NG, Wells GA, Prud D, Fortier M, Reid RD, Tulloch H, Coyle D, Phillips P, Jennings A, Jaffey J (2007) Effects of aerobic training, resistance training, or both on glycemic control in type 2 diabetes. Ann Intern Med 147:357–369
Soares E, Prediger RD, Nunes S, Castro AA, Viana SD, Lemos C, De Souza CM, Agostinho P, Cunha RA, Carvalho E, Fontes Ribeiro CA, Reis F, Pereira FC (2013) Spatial memory impairments in a prediabetic rat model. Neuroscience 250:565–577
Sorg O, Magistretti PJ (1991) Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res 563:227–233
Sorg O, Magistretti PJ (1992) Vasoactive intestinal peptide and noradrenaline exert long-term control on glycogen levels in astrocytes: blockade by protein synthesis inhibition. J Neurosci 12:4923–4931
Soya H, Mukai A, Deocaris CC, Ohiwa N, Chang H, Nishijima T, Fujikawa T, Togashi K, Saito T (2007a) Threshold-like pattern of neuronal activation in the hypothalamus during treadmill running: establishment of a minimum running stress (MRS) rat model. Neurosci Res 58:341–348
Soya H, Nakamura T, Deocaris CC, Kimpara A, Iimura M, Fujikawa T, Chang H, McEwen BS, Nishijima T (2007b) BDNF induction with mild exercise in the rat hippocampus. Biochem Biophys Res Commun 358:961–967
Sriwijitkamol A, Coletta DK, Wajcberg E, Balbontin GB, Reyna SM, Barrientes J, Eagan PA, Jenkinson CP, Cersosimo E, DeFronzo RA, Sakamoto K, Musi N (2007) Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes 56:836–848
Stranahan AM (2015) Models and mechanisms for hippocampal dysfunction in obesity and diabetes. Neuroscience 309:125–139
Stranahan AM, Lee K, Martin B, Maudsley S, Golden E, Cutler G, Mattson MP (2009) Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus 19:951–961
Suge R, Shimazu T, Hasegawa H, Inoue I, Hayashibe H, Nagasaka H, Araki N, Katayama S, Nomura M, Watanabe SI (2012) Cerebral antioxidant enzyme increase associated with learning deficit in type 2 diabetes rats. Brain Res 1481:97–106
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823
Swanson R, Sagar S, Sharp F (1989) Regional brain glycogen stores and metabolism during complete global ischaemia. Neurol Res 11:24–28
Swanson RA, Morton MM, Sagar SM, Sharp FR (1992) Sensory stimulation induces local cerebral glycogenolysis: Demonstration by autoradiography. Neuroscience 51:451–461
Tekkök S, Krnjevic K (1999) Diabetes mellitus preserves synaptic plasticity in hippocampal slices from middle-age rats. Neuroscience 91:185–191
Umegaki H, Hayashi T, Nomura H, Yanagawa M, Nonogaki Z, Nakshima H, Kuzuya M (2013) Cognitive dysfunction: an emerging concept of a new diabetic complication in the elderly. Geriatr Gerontol Int 13:28–34
van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A 96:13427–13431
Vissing J, Andersen M, Diemer NH (1996) Exercise-induced changes in local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab 16:729–736
Whitmer RA (2007) Type 2 diabetes and risk of cognitive impairment and dementia. Curr Neurol Neurosci Rep 7:373–380
Winder WW, Yang HT, Jaussi AW, Hopkins CR (1987) Epinephrine, glucose, and lactate infusion in exercising adrenodemedullated rats. J Appl Physiol 62:1442–1447
Witting A, Chen L, Cudaback E, Straiker A, Walter L, Rickman B, Moller T, Brosnan C, Stella N (2006) Experimental autoimmune encephalomyelitis disrupts endocannabinoid-mediated neuroprotection. Proc Natl Acad Sci 103:6362–6367
Yang J, Ruchti E, Petit J-M, Jourdain P, Grenningloh G, Allaman I, Magistretti PJ (2014) Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc Natl Acad Sci U S A 111:12228–12233
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Soya, M., Jesmin, S., Shima, T., Matsui, T., Soya, H. (2019). Dysregulation of Glycogen Metabolism with Concomitant Spatial Memory Dysfunction in Type 2 Diabetes: Potential Beneficial Effects of Chronic Exercise. In: DiNuzzo, M., Schousboe, A. (eds) Brain Glycogen Metabolism. Advances in Neurobiology, vol 23. Springer, Cham. https://doi.org/10.1007/978-3-030-27480-1_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-27480-1_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27479-5
Online ISBN: 978-3-030-27480-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)