
Abstract
Mitochondrial disorders are a clinically heterogeneous group of disorders that arise as a result of dysfunction of the mitochondrial energy metabolism, and they represent one of the largest groups of inborn errors of metabolism. Mitochondrial disorders can be caused by mutation of genes encoded by either nuclear DNA or mitochondrial DNA (mtDNA), with more than 300 disease-associated genes identified to date. Among these genes, around 100 have so far been associated with cardiac manifestations. Cardiomyopathy is estimated to occur in 20–40% of children with mitochondrial disorders. Genetic defects can affect a vast range of different mitochondrial functions including electron transport chain complex subunits and their assembly factors, mitochondrial transfer or ribosomal RNAs, factors involved in translation or mtDNA maintenance, and cofactor metabolism such as coenzyme Q10 synthesis. With collectively more than 1000 described cases, the most frequent mitochondrial cardiac diseases include Barth syndrome, Sengers syndrome, ACAD9- or TMEM70-related mitochondrial complex I or V deficiency, and Friedreich ataxia. Hypertrophic cardiomyopathy is the most common type of cardiomyopathy, but mitochondrial cardiomyopathies might also present as dilated, restrictive, left ventricular non-compaction, and histiocytoid cardiomyopathies. Mitochondrial cardiomyopathy can vary in severity from asymptomatic to severe manifestations including heart failure and sudden cardiac death. Congenital arrhythmias and congenital heart defects (CHDs) are also part of the clinical spectrum of mitochondrial disorders. In this chapter, we provide an overview of the constantly growing number of mitochondrial cardiac disorders and comment on the current practice in the diagnosis and treatment of patients with mitochondrial cardiomyopathy, including optimal therapeutic management and long-term monitoring.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
3.1 Introduction
Mitochondrial disorders are a heterogeneous group of inborn errors of metabolism encompassing a wide range of clinical presentations, with > 300 disease-associated genes identified to date [1, 192]. Mitochondria are largely known as the powerhouse of the cell due to their crucial function in the generation of cellular energy. They exploit the energy stored in fats, carbohydrates, and proteins to produce ATP in a process called oxidative phosphorylation (OXPHOS). OXPHOS requires the transport of electrons to molecular oxygen via the mitochondrial respiratory chain, which involves four multi-subunit complexes (complex I–complex IV) and two mobile electron carriers, coenzyme Q10 (CoQ10) and cytochrome c. The respiratory chain generates a transmembrane proton gradient that is used by complex V (also known as ATP synthase) to synthesize ATP.
Major metabolic consequences of OXPHOS impairment include accumulation of intermediary metabolic products, increased generation of reactive oxygen species, and decreased energy production. As a consequence of energy deficiency, high-energy demand tissues such as muscle, brain, and liver can be impaired, with resulting multi-organ disease, often with a progression of symptoms developing over time. The variable and frequently systemic nature of mitochondrial disorders make molecular diagnosis difficult. Many different medical specialties are often involved in patient care (with individual physicians often becoming discouraged from solving such complex phenotypes). On the other hand, its very systemic nature aids in raising suspicion for the diagnosis of mitochondrial disorders. Specific combinations of the clinical features associated with mitochondrial disorders can subsequently be grouped into specific syndromes [2].
With an estimated prevalence of 1 in 5000 live births, mitochondrial disorders represent one of the largest groups of inborn errors of metabolism [3]. The onset of mitochondrial disorders has an apparent peak in early childhood (first 3 years of life) and a second broad peak beginning toward the second and the fourth decade of life (adult-onset diseases) [3]. Childhood-onset mitochondrial disorders are generally more severe, especially when clinical manifestation occurs in infancy (<1 year of age) or if they include cardiac involvement (mortality of 71% vs 26% without cardiac phenotype) [4].
Cardiac involvement, specifically cardiomyopathy and arrhythmias, are common features associated with both early- and late-onset forms of mitochondriopathy. Cardiomyopathy is estimated to occur in 20–40% of children with mitochondrial disorders [4, 5]. However, screening for cardiac involvement is not always performed, and this figure is therefore possibly underestimated. The most common form of cardiomyopathy is hypertrophic; however dilated cardiomyopathy and left ventricular (LV) non-compaction also occur relatively frequently [6, 7]. Conduction system and bradyarrhythmias, in addition to WPW syndrome and tachyarrhythmias, are the most commonly encountered arrhythmias in mitochondrial disorders [8, 9].
On the other hand, when a mitochondrial condition affects at the initial stage selectively the heart, the mitochondrial cardiomyopathy may be clinically indistinguishable from other genetic determined cardiomyopathies [147]. Hence, mitochondrial disorders should be suspected in idiopathic isolated forms of cardiomyopathies. Conventional cardiomyopathy gene panels often do not include all the mitochondrial cardiomyopathy-associated genes, and only the increasing use of whole-exome sequencing (WES) has led to unmasking of such cases. In general, the introduction of next-generation sequencing (NGS) has dramatically improved diagnostic yield for mitochondrial disorders and shifted the focus from mtDNA to the nuclear genome. Indeed, both mtDNA and nuclear genomes can be analyzed for pathogenic variants within a single experiment. Consequently, NGS has enabled the identification of a constantly growing list of new disease genes, thereby setting the foundation to uncover an increasing variety of pathophysiologic disease mechanisms [10,11,12,13].
Defects of the mitochondrial energy metabolism can be caused by mutations in a number of mitochondrial pathways and functions, including the ATP producing chain from pyruvate dehydrogenase via Krebs cycle to OXPHOS by affecting replication of mtDNA or transcription and translation of mitochondrially encoded OXPHOS subunits. Furthermore, there are numerous additional functions involved in the mitochondrial energy metabolism, such as transport processes of proteins and substrates through the mitochondrial membranes, quality control systems, essential cofactors, motility of the organelle, and membrane integrity.
Mitochondrial genetics is complex, as the workhorse of mitochondria, the mitochondrial proteome (around 1500 proteins), is under the control of two genomes, the nuclear genome and the mitochondrial genome (mtDNA). Although only a small fraction of their proteins are encoded by mtDNA, abnormalities in mtDNA affect more than half of the adult cases, whereas nuclear DNA defects account for up to 80% of mitochondrial disorders in children [14]. Therefore, the most common mode of inheritance of mitochondrial disorders in children is autosomal recessive. For mtDNA variants, the number of wild-type copies is a key factor that determines whether a cell expresses a biochemical defect. This is usually determined by the proportion of mutated copies versus wild-type copies of mtDNA. Homoplasmy describes the setting in which all mtDNA copies have the same mutation, while heteroplasmy, on the other hand, occurs if a mixture of different genotypes, e.g., wild-type and mutant mtDNA, coexist. Variants in mtDNA causing mitochondrial disorders can be further classified into three types: (a) mutations in genes encoding structural proteins or in genes involved in protein synthesis, (b) single or multiple mtDNA deletions, and (c) mtDNA depletion. Multiple mtDNA deletions and mtDNA depletion are caused by nuclear gene defects. Screening the literature, we found an cassociated cardiac phenotype in around 100 genes (n = 1855 cases) among the 300 mitochondrial disease-associated genes described to date. However, concerning the number of patients per gene defect, a non-equal long tail distribution was observed. There are disorders like Friedreich ataxia for which more than 1200 patients have a reported cardiac involvement, Barth syndrome with more than 200 patients, and ACAD9 with over 50 patients, while there are around 70 disease entities with less than 10 published cases with cardiac involvement (Table 3.1). Many individuals display a cluster of clinical features which fall into defined mitochondrial syndromes. Among them, MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) and MERRF (myoclonic epilepsy with ragged red fibers) represent the syndromes most frequently associated with cardiac manifestations.
3.2 Cardiac Manifestation in Mitochondrial Disorders
Cardiomyopathies represent a significant clinical manifestation associated with mitochondrial disorders that can result in sudden cardiac death. Neonatal cardiomyopathies are often characterized by hypertrophy of one (mostly left) or both ventricles [147]. Ventricular dysfunction may be progressive in utero and after birth. Neonatal cardiomyopathy is often followed by fatal heart failure or, in other cases, can progress into a dilated form, non-compaction of the left ventricle, improve, or even regress completely [147].
Hypertrophic cardiomyopathy (HCM) is the most frequent manifestation in mitochondrial disorders. HCM is characterized by progressive myocardial thickening; diastolic and systolic ventricular dysfunction; histopathologic changes, such as myocyte disarray and fibrosis; and arrhythmias which may cause sudden cardiac death. In adulthood, hypertrophic cardiomyopathy has a prevalence of 1 in 500 and is typically caused by mutations in sarcomere genes [148]. Pediatric HCM is most frequently associated with Noonan syndrome (1 in 10,000), but it is also frequently found with inborn errors of metabolism, including mitochondrial disorder [149]. HCM was reported to be associated with about 90 mitochondrial disease genes out of 100 with a cardiac phenotype (Table 3.1).
Dilated cardiomyopathy (DCM) is characterized by progressive myocardial dilatation and thinning. Both diastolic and systolic ventricular dysfunctions can occur, and DCM is often associated with the occurrence of cardiac conduction system diseases, arrhythmias, and sudden arrhythmic death. DCM as a sequela of myocarditis is the most common cardiomyopathy in childhood, but it can also be inherited or part of systemic diseases, such as a mitochondrial disorder [150]. In mitochondrial disorders, DCM is often a consequence of a progressed HCM. It has been found in association with 30 disease genes, most frequently with Barth and DCMA syndromes [151].
Mitochondrial disorders can present with cardiac conduction defects typically involving sinus node dysfunction, atrioventricular block, ventricular conduction delay, or WPW syndrome. Progression in intraventricular conduction defect is unpredictable and responsible for sudden cardiac death. Overall, 17 mitochondrial disease genes were found in association with arrhythmias. Prevalence of Wolff-Parkinson-White syndrome among patients with mtDNA mutations is high (up to 15%), specifically in patients with MELAS and MERRF [152]. Also in the clinical presentation of Kearns-Sayre syndrome are cardiac conduction defects, together with progressive external ophthalmoplegia and pigmentary retinopathy [153].
Congenital heart defects (CHDs) are rarely recognized to be linked to mitochondrial disorders. CHDs have been identified in patients with mitochondrial disorders due to 16 different genetic defects; however the pathomechanism remains largely unknown (Table 3.1). The CHDs include patent ductus arteriosus (PDA), ventricular and septal defects (VSD and ASD), or more complex CHD defects (tetralogy of Fallot, transpositions of great arteries). Among them, if we consider three of the most frequent causes of cardiac disorder, we found CHDs to be reported in 30% of TMEM70 cases and 10% of ACAD9 and TAZ cases ([34, 53]; https://www.barthsyndrome.org;). This figure is clearly above the 1% of cases with CHDs identified in the general population. The observation of CHD is present in only a fraction of patients with the clinical presentation of mitochondrial disorders. Usually not all patients with a certain gene defect develop a cardiac phenotype, but the mitochondrial dysfunction predisposes patients to cardiac abnormalities [154]. Sengers syndrome or ELAC2 patients seemed to be an exception, but for most gene defects which were tightly linked to cardiac manifestations, NGS diagnostics extended the clinical spectrum to patients without cardiac phenotypes [59, 79, 80, 125].
Left ventricular non-compaction (LVNC) is a morphological abnormality of excessive trabeculation of the left ventricular myocardium, which is often complicated by ventricular dysfunction, arrhythmias, and cardioembolism. LVNC can be part of a structural heart disease. Additionally, mutations in a variety of genes, such as transcription factors, structural, nuclear, or ion channel genes, may cause LVNC. In general, the etiology of this disease entity is still poorly understood and object of current investigations. In mitochondrial disorders, Barth syndrome is most often associated with LVNC, but it is also typical for DNAJC19 defects and reported in a total of 11 mitochondrial genes [62, 153, 155].
3.3 Genetic Causes of Mitochondrial Cardiac Manifestation
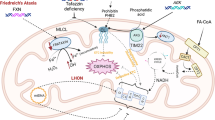
With about 300 gene defects reported in association with human mitochondrial disorders, the spectrum of respiratory chain defects is rather intricate. To break down the complexity, we have grouped the genes here into (1) disorders of oxidative phosphorylation (OXPHOS) subunits and their assembly factors; (2) defects of mitochondrial DNA, RNA, and protein synthesis; (3) defects in the substrate-generating upstream reactions of OXPHOS; (4) defects in cofactor metabolism; (5) defects in mitochondrial homeostasis; and (6) defects in relevant inhibitors (Fig. 3.1). In the following text, we will describe these groups in more details.

Known disease genes (n = 309) in different parts of the mitochondrial energy metabolism. Q (coenzyme Q10), IS (iron-sulfur clusters), B (biotin), T (thiamine pyrophosphate), L (lipoic acid), H (heme), A (coenzyme A), F (riboflavin/FMN/FAD), Fe (iron), Cu (copper), M (s-adenosyl methionine), N (NADH/NADPH). Summary of mitochondrial genes with cardiac manifestations: in red are highlighted genes in which the cardiac involvement represents a major symptom among the other clinical features associated with the gene defect; in green are highlighted genes in which the cardiac involvement represents a minor symptom among the other clinical features associated with the gene defect
3.3.1 OXPHOS Subunits and Their Assembling Factors
Complex I deficiency is the most common biochemical phenotype observed in individuals with mitochondrial disorder and is responsible for approximately 30% of childhood-onset cases [156]. Complex I is composed of 44 subunits (7 encoded by mtDNA and 37 by nDNA), with genetic defects identified in 28 structural genes, in 11 assembly factors, and in a number of factors involved in mitochondrial translation (Fig. 3.1) [154].
Complex I deficiency is clinically heterogeneous, with a diverse spectrum of clinical presentations, such as Leigh syndrome, fatal infantile lactic acidosis (FILA), leukoencephalopathy, MELAS, or also hypertrophic cardiomyopathy. HCM, in particular, can be isolated or associated with multi-organ disease. Isolated HCM has been reported with mutations in nuclear-encoded subunits (NDUFS2, NDUFV2) and assembly factors (ACAD9, and less commonly NDUFAF1). Less frequently patients manifest with dilated cardiomyopathy, left ventricle non-compaction, or conduction defects (such as Wolff-Parkinson-White) [157]. Among 95 cases with complex I deficiency and cardiac manifestations described in the literature (Table 3.1), ACAD9 deficiency, with 56 cases, represents the most frequent genetic cause observed [34]. Considering the number of ACAD9 patients and the minor allele frequency of deleterious variants described so far, an incidence of 59 new ACAD9 patients born every year in Europe has been recently estimated [34]. The clinical spectrum of ACAD9 deficiency is characterized by predominant heart involvement. Other clinical recurrent symptoms include lactic acidosis and muscular weakness [158]. Age of onset, severity of the disease, and progression can vary significantly [34]. On a biochemical level, residual ACAD9 enzyme activity, and not complex I activity, seems to correlate with the severity of clinical symptoms in ACAD9-deficient patients [159]. Regarding the clinical presentation, the typical age of onset of patients with ACAD9 deficiency is in the neonatal period or in early childhood, with the majority of the patients presenting in the first year of life. Looking at the survival data, for this subgroup with early onset, the survival was poor (50% not surviving the first 2 years) and the outcome extremely severe compared to patients with a later presentation (more than 90% surviving 10 years) [34]. The patients with severe developmental delay or intellectual disability also showed an early disease onset; however neurological manifestations seem to be a rare presentation of the disease [34]. Furthermore, most patients currently alive are able to perform routine activities of daily living. This aspect could also influence the management of ACAD9 patients, for example, for the decision of performing a cardiac transplant. Four patients with ACAD9 defects underwent a cardiac transplant. Unfortunately, the two patients who presented within the first year died despite all efforts. In contrast, the two patients presenting after the age of 1 year developed normally, and their clinical status remains stable after years of follow-up (currently aged 15 and 35 years, respectively). However, additional longitudinal studies are warranted to better identify which patients with ACAD9 deficiency are appropriate heart transplant candidates [160]. Supplementation with riboflavin showed improvement in complex I activity in the majority of patient-derived fibroblasts, and most patients similarly were reported to have clinical benefit with treatment. Most notably, patients presenting within the first year of life show a significantly better survival when treated with riboflavin [34]. However, detailed data about the starting point of riboflavin treatment, the dosage, etc. in large cohorts are needed [34, 161].
Isolated complex II deficiency is a rare cause of mitochondrial disorder. Complex II, or succinate dehydrogenase, is formed of four subunits, and it is the only respiratory chain complex entirely encoded by nDNA. Genetic defects were identified in four structural genes and two assembly factors. Clinically, neurological symptoms represent the most common presentation (especially Leigh syndrome or leukoencephalopathy), but hypertrophic and dilated cardiomyopathy, as well as left ventricular non-compaction, were identified in six of the reported cases [162]. In particular, early-onset hypertrophic cardiomyopathy with left ventricular non-compaction and severe complex II deficiency has been observed in association with SDHD defects [40]. Isolated and severe neonatal dilated cardiomyopathy has been associated with SDHA mutations [163]. Conduction abnormalities were present in the same patients and thus should be investigated in patients with complex II deficiency [162].
Complex III (coenzyme Q or cytochrome reductase) is a multi-subunit transmembrane protein encoded by both the mitochondrial (cytochrome b) and the nuclear genomes (ten subunits). Gene defects have been identified in five structural genes and five assembly factors [164]. Complex III or combined complex I and III deficiency typically manifests in infancy as a severe, multisystem disorder that includes features such as hypotonia, seizures, lactic acidosis, hypoglycemia, and intellectual disability. Variants in BCS1L can result in Björnstad syndrome and GRACILE syndrome, severe neonatal syndromes with multisystem and neurological manifestations. However, milder cases with survival into adulthood have also been described. In such cases, HCM has been reported in two patients [41]. In addition, different forms of cardiomyopathy (hypertrophic, dilated, and histiocytoid cardiomyopathy), either isolated or accompanied by multisystem mitochondrial disorder, have been described in three patients with complex III deficiency associated with mutations in mitochondrial DNA-encoded cytochrome b (MT-CYB) [42, 165].
Complex IV—also called cytochrome c oxidase—is the terminal enzyme of the respiratory chain and consists of 14 subunits, 3 of which (named COX1, COX2, and COX3) are encoded by mitochondrial DNA. Complex IV deficiency has been associated with mutations in 11 structural genes and 8 assembly factors.
There are four types of complex IV deficiency differentiated by symptoms and age of onset: benign infantile mitochondrial type, French-Canadian type, infantile mitochondrial myopathy type, and Leigh syndrome. The clinical spectrum can vary among affected individuals, even within the same family. In mildly affected individuals can occur muscle weakness and hypotonia. Whereas in more severely affected individuals neurological dysfunction, heart and liver manifestations, lactic acidosis, and/or Leigh syndrome may also be present [166]. Cardiac involvement has been described in 16 published patients with complex IV deficiency (Table 3.1). Isolated dilated cardiomyopathy has been reported in individuals with complex IV deficiency and carrying MT-CO2 and MT-CO3 variants [167]. SURF1, a nuclear gene encoding a complex IV assembly factor, represents the most common cause of Leigh syndrome due to complex IV deficiency and is sometimes accompanied by cardiomyopathy [48]. In addition, there are a number of cofactor deficiencies which result in complex IV deficiency with cardiac manifestations (see Sect. 3.4 and Table 3.1).
Mitochondrial ATP synthase (complex V) synthesizes ATP from ADP and inorganic phosphate using the energy provided by the proton electrochemical gradient (proton-motive force) across the inner mitochondrial membrane. It consists of two functional domains, F1 and Fo. F1 comprises five different subunits, while the Fo region includes three main subunits a, b, and c, and six additional subunits. Gene defects have been identified in six structural genes and two assembly factors.
Most cases present with neonatal-onset hypotonia, lactic acidosis, hyperammonemia, hypertrophic cardiomyopathy, and 3-methylglutaconic aciduria. Among 55 cases with cardiac manifestations and complex V deficiency present in literature, 49 have been associated with variants in TMEM70. TMEM70 deficiency is the most common genetic defect affecting the ATP synthase [51]. Frequent symptoms at onset are poor feeding, hypotonia, lethargy, and respiratory and heart failure, accompanied by lactic acidosis, 3-methylglutaconic aciduria, and hyperammonemia. In children with TMEM70 deficiency, the most common heart problem is non-obstructive concentric HCM with preserved systolic function [52, 54]. With the exception of neonates with heart failure, the prognosis of cardiomyopathy in TMEM70 patients is favorable, because HCM is mostly non-progressive or even regressive during long-term follow-up. Conduction defects (Wolff-Parkinson-White syndrome) have been found in 13% of TMEM70 patients. Cardiomyopathy associated with systemic mitochondrial disorder has also been described with mutations in mitochondrial genes encoding complex V subunits, including MT-ATP6 [191] and MT-ATP8 [57, 168, 169].
3.3.2 Defects of Mitochondrial DNA, RNA, and Protein Synthesis
Mitochondrial protein translation defects typically cause multiple OXPHOS abnormalities and severe mitochondrial disorder. Gene defects have been identified in genes encoding elongation factors, aminoacyl-tRNA synthetases, tRNA-modifying enzymes, a mitochondrial peptide release factor, and an RNase that processes mitochondrial RNA. Mitochondrial disorders linked to protein translation defects manifest with neurological involvement and hypertrophic cardiomyopathy, as well as other multisystem abnormalities. Conditions with cardiac manifestations include defects in mitochondrial ribosomal proteins (MRPS22, MRPL3, MRPL44), mitochondrial tyrosine (YARS2) or alanine (AARS2) tRNA aminoacylation, and other enzymes involved in mitochondrial RNA metabolism [186, 188]. Cardiomyopathy resulting from mutations in ELAC2 are usually associated with early severe forms of hypertrophic cardiomyopathy in the context of a multisystem disorder. However, isolated forms of hypertrophic and dilated cardiomyopathy may also be present.
Posttranscriptional modification of mitochondrial tRNAs is necessary for their stability and function. Abnormalities in these processes are illustrative of recently identified protein translation disorders that lead to cardiomyopathy. Hypertrophic cardiomyopathy and cardiac conduction defects in combination with psychomotor delay, encephalopathy, hypotonia, and lactic acidosis were found in children with variants in MTO1 [86]. Mutations in GTPBP3 have been described in association with neonatal hypertrophic or dilated cardiomyopathy and conduction defects. Again, most of the patients also showed extra-cardiac symptoms, such as encephalopathy, lactic acidemia, hypoglycemia, and hyperammonemia [84]. Another posttranscriptional modification defect of mitochondrial tRNAs was found in tRNA methyltransferase 5 (TRMT5) deficiency. Mutations in TRMT5 have been described in association with hypertrophic cardiomyopathy, exercise intolerance, global developmental delay, hypotonia, peripheral neuropathy, renal tubulopathy, and lactic acidosis [88].
3.3.3 Defects in the Substrate-Generating Upstream Reactions of OXPHOS
Deficiencies of the mitochondrial phosphate carrier (SLC25A3) have been described in cases with hypertrophic cardiomyopathy accompanied by myopathy and lactic acidosis [137]. Lahrouchi et al. have recently reported a loss of function of the carnitine transport SLC22A5 in a family with history of pediatric cardiomyopathy and sudden cardiac death. The patients showed a reduction in the degree of cardiac hypertrophy, and her exercise tolerance improved markedly after l-carnitine supplementation [144]. Biallelic missense variants in the nuclear-encoded mitochondrial inorganic pyrophosphatase (PPA2) were identified in 17 individuals from 7 unrelated pedigrees presenting with seizures, lactic acidosis, cardiac arrhythmia, and exquisite sensitivity to alcohol, leading to sudden cardiac death [146].
3.3.4 Defects in Relevant Cofactors
Many cofactors have a key role in mitochondrial energy metabolism. Among them, some are required for the respiratory chain enzymes like coenzyme Q, iron-sulfur clusters, riboflavin, and heme. Their deficiency typically results in defects of more than one respiratory enzyme. Primary CoQ10 deficiency is a phenotypically and genetically heterogeneous condition, with various clinical presentations including encephalomyopathy, myopathy, cerebellar ataxia, nephrotic syndrome, and severe infantile multisystem mitochondrial disorder. Hypertrophic cardiomyopathy has been reported in cases with mutations in COQ2, COQ4, and COQ9 [118, 119, 170, 187]. Fatal infantile cardioencephalomyopathy due to cytochrome c oxidase (COX) deficiency 1 is caused by biallelic variants in the SCO2 gene. SCO2 is a mitochondrial copper-binding protein involved in the biogenesis of the Cu(A) site in the cytochrome c oxidase (CcO) subunit Cox2 and in the maintenance of cellular copper homeostasis. Mutations in both SCO1 and SCO2 are associated with distinct clinical phenotypes in addition to tissue-specific cytochrome c oxidase deficiency. SCO2 is highly expressed in the muscle, whereas SCO1 is expressed at higher levels in the liver. This reflects the different clinical presentation of SCO1, mostly associated with hepatic liver failure, and SCO2, predominantly associated with severe early-onset cardiac failure. The onset of cardiomyopathy is either in utero or in the first days of life [132]. Copper-histidine supplementation in cell culture, and also the treatment in one patient, was reported to be beneficial for the cardiac phenotype [131].
3.3.5 Defects in Mitochondrial Homeostasis
Mitochondrial homeostasis involves several essential aspects of mitochondrial biogenesis, lipid synthesis, protein import, fission and fusion, quality control, and targeted degradation.
Barth syndrome is due to mutations in the X-linked TAZ gene, which codes for Tafazzin, a phospholipid transacylase involved in the remodelling of cardiolipin. Barth syndrome is characterized by cardiomyopathy, skeletal myopathy, distinctive facial features, developmental delay, neutropenia, and increased urinary levels of 3-methylglutaconic acid. Cardiomyopathy is the presenting manifestation in more than 70% of affected males and usually appears in infancy. Interestingly, in these patients, left ventricular non-compaction and dilated cardiomyopathies are more frequent cardiological findings than hypertrophic cardiomyopathy [171]. Patients can manifest with supraventricular and ventricular arrhythmias, sometimes related to sudden cardiac death [172]. Recently, a systematic mutation screening of TAZ in a large cohort of pediatric patients with primary cardiomyopathy identified pathogenic variants in 3.5% of the male patients [173]. In a mouse model of Barth syndrome, cardiac-specific loss of succinate dehydrogenase (complex II) activity has been described as a key event in the pathogenesis of cardiomyopathy [174]. Sengers syndrome is caused by the deficiency of the acylglycerol kinase (AGK) which is also involved in the mitochondrial protein import. The clinical spectrum is characterized by the presence of hypertrophic cardiomyopathy, cataracts, myopathy, exercise intolerance, and lactic acidosis [60]. 3-Methylglutaconic aciduria associated with DNAJC19 mutations (DCMA syndrome) is mainly associated with dilated cardiomyopathy or left ventricular non-compaction and non-progressive cerebellar ataxia [63]. Variants in C1QBP, encoding a complement component 1 Q subcomponent-binding protein, have been recently associated with severe forms of neonatal or later-onset cardiomyopathy associated with combined respiratory chain deficiencies [66].
3.3.6 Defects in Relevant Inhibitors
Mitochondrial short-chain enoyl-CoA hydratase-1 deficiency (ECHS1) is an autosomal recessive inborn error of metabolism caused by compound heterozygous mutations in the ECHS1 gene and characterized by severely delayed psychomotor development, neurodegeneration, increased lactic acid, and cardiomyopathy due to the accumulation of toxic metabolites. Usually these patients are affected by hypertrophic cardiomyopathy and, more rarely, by the dilated form [136, 189].
3.3.7 Mitochondrial Syndromes
Mitochondrial syndromes represent a spectrum of symptoms typically associated with specific abnormalities of mitochondrial DNA (Table 3.2). They can be associated with cardiomyopathy, conduction abnormalities, or both. Kearns-Sayre syndrome is characterized by extra-cardiac manifestations such as pigmentary retinopathy, progressive external ophthalmoplegia, increased cerebrospinal fluid protein concentration, and cerebellar ataxia. Cardiac conduction block is one of the cardinal manifestations; however hypertrophic cardiomyopathy may also be observed. Sudden cardiac death occurs in up to 20% of cases [175].
Cardiomyopathy and/or conduction abnormalities may also accompany MELAS, MERRF, and mtDNA-associated Leigh syndrome/NARP (neuropathy, ataxia, and retinitis pigmentosa) syndromes. Cardiomyopathy occurs in 18–30% of individuals with MELAS syndrome [176]. Both dilated and hypertrophic cardiomyopathies have been observed in MELAS syndrome; however, more typical is non-obstructive concentric hypertrophy [177]. Cardiac conduction abnormalities including Wolff-Parkinson-White syndrome has been reported in 13–27% of individuals with MELAS syndrome [176]. Cardiomyopathy has been identified in 30% of MERRF cases and Wolff-Parkinson-White in 22% [178]. In NARP syndrome, the cardiological manifestations described thus far are hypertrophic cardiomyopathy and atrioventricular block [179].
3.4 Characterization of the Cardiac Involvement
The clinical presentation in patients with cardiomyopathy varies from asymptomatic to severe heart failure with asphyxia. In some cases, a sudden cardiac death can even be the first manifestation of the underlying cardiac dysfunction in an apparently healthy individual. Therefore, the assessment of the cardiac status represents a fundamental step for the management, follow-up, and prognosis of these patients. History and physical examination might reveal multisystem involvement, such as failure to thrive, global developmental delay, muscle weakness, respiratory abnormalities, epilepsy, vision problems, and others [180]. Family history might be positive for the occurrence of multisystem disorders in ancestors (Fig. 3.2). If a significant ventricular dysfunction is present, the cardiac examination might show signs of heart failure, such as dyspnea, sweating, failure to thrive, bilateral lung crackles, pitting edema, hepatomegaly, and signs of hypoperfusion. Severe cardiac manifestations including heart failure, ventricular tachyarrhythmias, and sudden cardiac death can occur during a metabolic crisis often precipitated by physiologic stressors such as febrile illness or surgery. The cardiac investigation should include chest X-ray, transthoracic echocardiography (TTE), electrocardiography (ECG), Holter ECG, cardiac magnetic resonance (CMR), and, in older children, cardiopulmonary exercise testing (CPET) (Fig. 3.2). TTE is performed to assess ventricular chamber sizes, myocardial thickness, and systolic and diastolic function [181]. If hypertrophy is present, it is mostly concentric [182] (Fig. 3.3), but asymmetric septal hypertrophy with or without left ventricular outflow obstruction has been described. ECG and Holter recordings are performed to screen for the presence of arrhythmias and cardiac conduction system disorders, such as sinus node dysfunction, atrioventricular block, WPW syndrome, or intraventricular conduction delay [181] (Fig. 3.3e-f). CPET is performed to assess exercise capacity as reflected by the maximum rate of oxygen consumption (peak VO2) [181] and to evaluate the risk for exercise-triggered arrhythmias or conduction system disease. CMR should be performed to fully evaluate cardiac morphology and ventricular function, to screen for storage diseases, and to assess the presence of interstitial or focal fibrosis. The indication for cardiac catheterization is on an individual basis and should be performed if ventricular function is very poor in order to assess hemodynamics for possible heart transplantation or if the underlying diagnosis is not clear, so that endomyocardial biopsy results are made available. The conventional clinical approach of patients with mitochondrial cardiomyopathy includes detailed family and personal history and a clinical work-up to evaluate the cardiac defect, followed by histologic, enzymologic, molecular, and metabolic findings [153] (Fig. 3.2). If the cardiac phenotype is suspected to be based on a mitochondrial disorder, laboratory tests should include basic measurements, as well as detailed metabolic tests including ammonia, serum pyruvate, serum lactate, creatine kinase, quantitative amino acids, plasma acyl carnitine profile, and quantitative urine organic acids [153]. Care should be taken for proper processing of blood samples, as many metabolic tests require specific handling for accurate results. However, in clinical practice can frequently occur a mitochondrial cardiomyopathy without alteration of such classical metabolic findings.

Clinical work-up of patient with suspected mitochondrial cardiomyopathy

Case presentation: cardiac manifestation of mitochondrial disorder. (a, b, c) A 15-year-old girl with complex I and IV deficiency and concentric hypertrophic cardiomyopathy presenting with metabolic crisis and seizures, but preserved cardiac function. Parasternal long axis (a) and parasternal short axis (b) on transthoracic echocardiography show thickening of the left ventricular posterior wall (star) and of the interventricular septum (arrowhead); there is left ventricular hypertrophy on electrocardiography with deep S waves in the right (star) and tall R waves in the left (arrowhead) precordial leads. (d, e, f) A 2-month-old boy with complex I deficiency and severe concentric hypertrophic cardiomyopathy presenting with heart failure. Four-chamber view reveals massive thickening of the left ventricular myocardium (star) and pericardial effusion (arrowhead) on transthoracic echocardiography (d); telemetric monitoring shows brief runs of non-sustained self-limiting polymorphic ventricular tachycardia (star) (e); there is WPW syndrome with short PR interval (star) and delta-waves (arrowhead) on 12-lead surface electrocardiogram
3.5 Diagnosis of Potential Mitochondrial Cardiac Disease
The clinical spectrum of mitochondrial disorders often overlaps with common cardiological and extra-cardiac diseases. The diagnostic approach of these disorders is complex, time-consuming, and expensive. As mentioned above, although a positive biochemical result can substantiate a clinical diagnosis, the results are often inconclusive [11]. In this scenario, today’s use of genetic testing represents a fundamental step in the diagnosis of mitochondrial disorders [12, 13] (Fig. 3.2). Whole-exome sequencing enables rapid, cost-effective, genome-wide screening and dramatically increases the diagnostic yield to greater than 60%, revealing a remarkable heterogeneity of underlying gene defects [11, 192]. With a genome-wide approach, missing genotype-phenotype correlations are revealed, and subsequently the list of new disease genes is ever growing. Indeed, in clinical practice, many mitochondrial cardiomyopathies would have been clinically misdiagnosed without the identification of the causal gene defects by NGS techniques. On the other hand, early-onset forms of hypertrophic cardiomyopathy related to specific disorders, such as Noonan syndrome, could also be erroneously suspected to be due to mitochondrial defects without a genetic confirmation. In case of identification of variants of uncertain significance (VUS), a further valuable diagnostic step to validate the genetic finding is represented by a targeted biochemical analysis in fibroblasts or tissue biopsies (endomyocardial biopsy) (Fig. 3.2). Fibroblasts should be stored in parallel to be available for functional confirmation without further delaying diagnosis [193].
3.6 Management of Patients with Mitochondrial Cardiomyopathy
During the past 6 years, more than 100 novel mitochondrial disorders have been identified, affecting diverse mitochondrial pathways. The application of NGS technologies facilitates our understanding of the pathophysiology of mitochondrial disorders in general and potentially treatable disease subgroups; but the clinical management of affected individuals is challenging, and diagnostic strategies are in flux [102]. Unfortunately, in current clinical practice, therapeutic options for the majority of classical mitochondrial syndromes are limited to supportive care. Nevertheless, there are several mitochondrial defects, especially related to cofactor metabolism, that can be corrected by specific treatment strategies [102]. If the patient is severely and progressively ill (e.g., neonatal lactic acidosis), a more rapid diagnostic procedure is important, and a muscle biopsy with functional investigations is necessitated in parallel with starting genetic testing. This holds true especially if the suspected diagnosis influences the disease management (e.g., PDHc deficiency: initiation of ketogenic diet) or if it can guide the end of life decisions (e.g., Leigh syndrome). In this regard, empirical therapy with thiamine (20 mg/kg/day), biotin (5 mg/kg/day), riboflavin (20 mg/kg/day), and coenzyme Q10 (15 mg/kg/day) might be considered in patients with rapidly progressive or potential life-threatening course of disease. The treatment effect depends on the underlying mitochondrial disorders, for example, l-carnitine supplementation is highly effective in patients who have dilated cardiomyopathy secondary to primary systemic carnitine deficiency and in Barth syndrome. On the other hand, it has low effect on other types of mitochondrial cardiomyopathy. l-Arginine is effective in MELAS syndrome. Riboflavin is highly effective in complex I deficiencies and in particular in ACAD9 patients [34]. A low-fat, high-protein diet is important for long-chain fatty acid metabolism disorders [102]. Patients with mitochondrial disorders should avoid certain medications that interfere with mitochondrial function and can precipitate a crisis state, such as metformin, propofol, statins, valproic acid, macrolide antibiotics, or tetracyclines [153]. Regular clinical checkups of all organs affected are necessitated depending on the severity of multisystem involvement. Cardiac involvement is treated by conventional heart failure therapy which includes diuretics, angiotensin-converting enzyme inhibitors, beta-blockers, and calcium antagonists. Oral anticoagulation may be indicated with poor systolic ventricular function according to the clinical guidelines. Implantation of a pacemaker might be necessary in severe cardiac conduction system disease. An implantable cardioverter-defibrillator device might be indicated if there is a significant risk for sudden arrhythmic death or for secondary prevention after survived sudden death. Prophylactic cardiac pacing device implantation is thus generally proposed to prevent cardiac death. In the absence of consistent data regarding the incidence of ventricular arrhythmia in patients with mitochondrial cardiomyopathy, an implantable cardiac defibrillator should be proposed according to current clinical guidelines. A metabolic crisis with acute or subacute multi-organ failure secondary to physiologic stressors, such as infection, toxic triggers, medications, psychological stress, heat, or dehydration, is an emergency and needs to be managed appropriately. Cardiac complications during a crisis include cardiogenic shock, arrhythmias, and sudden cardiac death. Management should be focused to the underlying cause of the crisis and on treatment that can improve mitochondrial function. Fever needs to be treated and empiric antibiotics administered if an infection is suspected. Mechanical ventilation is required in case of respiratory failure and care needs to be taken with oxygenation, as it worsens the crisis by increasing free radical production. Correction of acid-base and electrolyte disturbances should be gradual. Continuous infusion of 10% arginine and dextrose-containing intravenous fluids should be best administered via central venous access given the risk for phlebitis and necrosis. Hemodialysis might be necessary for treatment-resistant hyperammonemia, hyperkalemia, or lactic acidosis [183, 184].
3.7 Conclusion and Perspectives
Cardiomyopathy and conduction defects are the most frequent manifestations of mitochondrial cardiac disease. Mitochondrial cardiomyopathy is associated with an ever-growing list of genes, currently around 100. This list outmatches the number of non-mitochondrial cardiomyopathy genes. Therefore, screening for cardiomyopathy should be a routine part of the management of individuals with known or suspected mitochondrial disorders. The diagnosis of mitochondrial disorders, however, remains challenging in many cases due to the myriad of different symptoms in overlap with other clinical conditions. NGS techniques of leukocyte-derived DNA are the state-of-the-art tool to diagnose mitochondrial disorders, enabling rapid, cost-effective, genome-wide screening with a diagnostic yield of greater than 60% [12, 13, 191]. During the past 6 years, more than 100 novel mitochondrial disorders have been identified and have facilitated our understanding of the pathophysiology of mitochondrial disorders. For the remaining undiagnosed cases, the role of complementary NGS approaches, such as genome sequencing and RNA sequencing, is gaining importance [185, 193]. These genomic approaches are still used on a research-oriented base, but we predict that in the future, they will become a valuable tool also in a diagnostic setting. Despite the scientific progress discussed thus far, it is important to underline the fact that most of these disorders do not have a treatment. This is with the exception of the group of cofactor metabolism deficiencies for which there are promising treatment options, highlighting the need for molecular diagnostics. There are still more than 70 diseases with less than 10 patients described. With the increasing number of patients, we will better understand the genotype-phenotype correlations which will help in counseling families. The better we understand the pathomechanism, the greater the chance we will identify new treatment options.
References
Mayr JA, Haack TB, Freisinger P, Karall D, Makowski C, Koch J, Feichtinger RG, Zimmermann FA, Rolinski B, Ahting U, Meitinger T, Prokisch H, Sperl W. Spectrum of combined respiratory chain defects. J Inherit Metab Dis. 2015;38:629–40.
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, Turnbull DM. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080.
Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, Taylor RW, Turnbull DM, McFarland R. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–9.
Holmgren D, Wåhlander H, Eriksson BO, Oldfors A, Holme E, Tulinius M. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J. 2003;24:280–8.
Scaglia F, Towbin JA, Craigen WJ, Belmont JW, Smith EO, Neish SR, Ware SM, Hunter JV, Fernbach SD, Vladutiu GD, Wong L-JC, Vogel H. Clinical spectrum, morbidity, and mortality in 113 pediatric patients with mitochondrial disease. Pediatrics. 2004;114:925–31.
Limongelli G, Masarone D, Pacileo G. Mitochondrial disease and the heart. Heart. 2017;103:390–8.
Tang S, Batra A, Zhang Y, Ebenroth ES, Huang T. Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Mitochondrion. 2010;10:350–7.
Bates MGD, Bourke JP, Giordano C, d’Amati G, Turnbull DM, Taylor RW. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33:3023–33.
Finsterer J, Kothari S. Cardiac manifestations of primary mitochondrial disorders. Int J Cardiol. 2014;177:754–63.
Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241:236–50.
Horvath R, Chinnery PF. The effect of neurological genomics and personalized mitochondrial medicine. JAMA Neurol. 2017;74:11.
Wortmann S, Mayr J, Nuoffer J, Prokisch H, Sperl W. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics. 2017a;48:309–14.
Wortmann SB, Timal S, Venselaar H, Wintjes LT, Kopajtich R, Feichtinger RG, Onnekink C, Mühlmeister M, Brandt U, Smeitink JA, Veltman JA, Sperl W, Lefeber D, Pruijn G, Stojanovic V, Freisinger P, v Spronsen F, Derks TG, Veenstra-Knol HE, Mayr JA, Rötig A, Tarnopolsky M, Prokisch H, Rodenburg RJ. Biallelic variants in WARS2 encoding mitochondrial tryptophanyl-tRNA synthase in six individuals with mitochondrial encephalopathy. Hum Mutat. 2017b;38:1786–95.
Goldstein AC, Bhatia P, Vento JM. Mitochondrial disease in childhood: nuclear encoded. Neurotherapeutics. 2013;10:212–26.
Hoefs SJG, Dieteren CEJ, Distelmaier F, Janssen RJRJ, Epplen A, Swarts HGP, Forkink M, Rodenburg RJ, Nijtmans LG, Willems PH, Smeitink JAM, van den Heuvel LP. NDUFA2 complex I mutation leads to Leigh disease. Am J Hum Genet. 2008;82:1306–15.
Hoefs SJG, van Spronsen FJ, Lenssen EWH, Nijtmans LG, Rodenburg RJ, Smeitink JAM, van den Heuvel LP. NDUFA10 mutations cause complex I deficiency in a patient with Leigh disease. Eur J Hum Genet. 2011;19:270–4.
Berger I, Hershkovitz E, Shaag A, Edvardson S, Saada A, Elpeleg O. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol. 2008;63:405–8.
Rea G, Homfray T, Till J, Roses-Noguer F, Buchan RJ, Wilkinson S, Wilk A, Walsh R, John S, McKee S, Stewart FJ, Murday V, Taylor RW, Ashworth M, Baksi AJ, Daubeney P, Prasad S, Barton PJR, Cook SA, Ware JS. Histiocytoid cardiomyopathy and microphthalmia with linear skin defects syndrome: phenotypes linked by truncating variants in NDUFB11. Cold Spring Harb Mol Case Stud. 2017;3:a001271.
Shehata BM, Cundiff CA, Lee K, Sabharwal A, Lalwani MK, Davis AK, Agrawal V, Sivasubbu S, Iannucci GJ, Gibson G. Exome sequencing of patients with histiocytoid cardiomyopathy reveals a de novo NDUFB11 mutation that plays a role in the pathogenesis of histiocytoid cardiomyopathy. Am J Med Genet A. 2015;167A:2114–21.
Torraco A, Bianchi M, Verrigni D, Gelmetti V, Riley L, Niceta M, Martinelli D, Montanari A, Guo Y, Rizza T, Diodato D, Di Nottia M, Lucarelli B, Sorrentino F, Piemonte F, Francisci S, Tartaglia M, Valente EM, Dionisi-Vici C, Christodoulou J, Bertini E, Carrozzo R. A novel mutation in NDUFB11 unveils a new clinical phenotype associated with lactic acidosis and sideroblastic anemia. Clin Genet. 2017;91:441–7.
van Rahden VA, Fernandez-Vizarra E, Alawi M, Brand K, Fellmann F, Horn D, Zeviani M, Kutsche K. Mutations in NDUFB11, encoding a complex I component of the mitochondrial respiratory chain, cause microphthalmia with linear skin defects syndrome. Am J Hum Genet. 2015;96:640–50.
Loeffen J, Elpeleg O, Smeitink J, Smeets R, Stöckler-Ipsiroglu S, Mandel H, Sengers R, Trijbels F, van den Heuvel L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann Neurol. 2001;49:195–201.
Loeffen J, Smeitink J, Triepels R, Smeets R, Schuelke M, Sengers R, Trijbels F, Hamel B, Mullaart R, van den Heuvel L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–608.
Ngu LH, Nijtmans LG, Distelmaier F, Venselaar H, van Emst-de Vries SE, van den Brand MAM, Stoltenborg BJM, Wintjes LT, Willems PH, van den Heuvel LP, Smeitink JA, Rodenburg RJT. A catalytic defect in mitochondrial respiratory chain complex I due to a mutation in NDUFS2 in a patient with Leigh syndrome. Biochim Biophys Acta Mol basis Dis. 2012;1822:168–75.
Haack TB, Madignier F, Herzer M, Lamantea E, Danhauser K, Invernizzi F, Koch J, Freitag M, Drost R, Hillier I, Haberberger B, Mayr JA, Ahting U, Tiranti V, Rötig A, Iuso A, Horvath R, Tesarova M, Baric I, Uziel G, Rolinski B, Sperl W, Meitinger T, Zeviani M, Freisinger P, Prokisch H. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J Med Genet. 2012b;49:83–9.
Haack TB, Haberberger B, Frisch E-M, Wieland T, Iuso A, Gorza M, Strecker V, Graf E, Mayr JA, Herberg U, Hennermann JB, Klopstock T, Kuhn KA, Ahting U, Sperl W, Wilichowski E, Hoffmann GF, Tesarova M, Hansikova H, Zeman J, Plecko B, Zeviani M, Wittig I, Strom TM, Schuelke M, Freisinger P, Meitinger T, Prokisch H. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J Med Genet. 2012a;49:277–83.
Bénit P, Beugnot R, Chretien D, Giurgea I, De Lonlay-Debeney P, Issartel J-P, Corral-Debrinski M, Kerscher S, Rustin P, Rötig A, Munnich A. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003;21:582–6.
Cameron JM, MacKay N, Feigenbaum A, Tarnopolsky M, Blaser S, Robinson BH, Schulze A. Exome sequencing identifies complex I NDUFV2 mutations as a novel cause of Leigh syndrome. Eur J Paediatr Neurol. 2015;19:525–32.
Zifa E, Theotokis P, Kaminari A, Maridaki H, Leze H, Petsiava E, Mamuris Z, Stathopoulos C. A novel G3337A mitochondrial ND1 mutation related to cardiomyopathy co-segregates with tRNALeu(CUN) A12308G and tRNAThr C15946T mutations. Mitochondrion. 2008;8:229–36.
Alila OF, Rebai EM, Tabebi M, Tej A, Chamkha I, Tlili A, Bouguila J, Tilouche S, Soyah N, Boughamoura L, Fakhfakh F. Whole mitochondrial genome analysis in two families with dilated mitochondrial cardiomyopathy: detection of mutations in MT-ND2 and MT-TL1 genes. Mitochondrial DNA A. 2016;27:2873–80.
Dunning CJR, McKenzie M, Sugiana C, Lazarou M, Silke J, Connelly A, Fletcher JM, Kirby DM, Thorburn DR, Ryan MT. Human CIA30 is involved in the early assembly of mitochondrial complex I and mutations in its gene cause disease. EMBO J. 2007;26:3227–37.
Fassone E, Taanman J-W, Hargreaves IP, Sebire NJ, Cleary MA, Burch M, Rahman S. Mutations in the mitochondrial complex I assembly factor NDUFAF1 cause fatal infantile hypertrophic cardiomyopathy. J Med Genet. 2011;48:691–7.
Saada A, Edvardson S, Rapoport M, Shaag A, Amry K, Miller C, Lorberboum-Galski H, Elpeleg O. C6ORF66 is an assembly factor of mitochondrial complex I. Am J Hum Genet. 2008;82:32–8.
Repp BM, Mastantuono E, Alston CL, Schiff M, Haack TB, Rötig A, Ardissone A, Lombes A, Catarino CB, Diodato D, Schottmann G, Poulton J, Burlina A, Jonckheere A, Munnich A, Ghezzi D, Rokicki D, Wellesley D, Martinelli D, Lamantea E, et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: is riboflavin supplementation effective? Orphanet J Rare Dis. 2018;13(1):120.
Fassone E, Duncan AJ, Taanman J-W, Pagnamenta AT, Sadowski MI, Holand T, Qasim W, Rutland P, Calvo SE, Mootha VK, Bitner-Glindzicz M, Rahman S. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum Mol Genet. 2015;24:4183.
Alston CL, Compton AG, Formosa LE, Strecker V, Oláhová M, Haack TB, Smet J, Stouffs K, Diakumis P, Ciara E, Cassiman D, Romain N, Yarham JW, He L, De Paepe B, Vanlander AV, Seneca S, Feichtinger RG, Płoski R, Rokicki D, Pronicka E, Haller RG, Van Hove JLK, Bahlo M, Mayr JA, Van Coster R, Prokisch H, Wittig I, Ryan MT, Thorburn DR, Taylor RW. Biallelic mutations in TMEM126B cause severe complex I deficiency with a variable clinical phenotype. Am J Hum Genet. 2016;99:217–27.
Alston CL, Davison JE, Meloni F, van der Westhuizen FH, He L, Hornig-Do H-T, Peet AC, Gissen P, Goffrini P, Ferrero I, Wassmer E, McFarland R, Taylor RW. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J Med Genet. 2012;49:569–77.
Van Coster R, Seneca S, Smet J, Van Hecke R, Gerlo E, Devreese B, Van Beeumen J, Leroy JG, De Meirleir L, Lissens W. Homozygous Gly555Glu mutation in the nuclear-encoded 70 kDa flavoprotein gene causes instability of the respiratory chain complex II. Am J Med Genet A. 2003;120A:13–8.
Courage C, Jackson CB, Hahn D, Euro L, Nuoffer J-M, Gallati S, Schaller A. SDHA mutation with dominant transmission results in complex II deficiency with ocular, cardiac, and neurologic involvement. Am J Med Genet A. 2017;173:225–30.
Alston CL, Ceccatelli Berti C, Blakely EL, Oláhová M, He L, McMahon CJ, Olpin SE, Hargreaves IP, Nolli C, McFarland R, Goffrini P, O’Sullivan MJ, Taylor RW. A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Hum Genet. 2015;134:869–79.
Al-Owain M, Colak D, Albakheet A, Al-Younes B, Al-Humaidi Z, Al-Sayed M, Al-Hindi H, Al-Sugair A, Al-Muhaideb A, Rahbeeni Z, Al-Sehli A, Al-Fadhli F, Ozand PT, Taylor RW, Kaya N. Clinical and biochemical features associated with BCS1L mutation. J Inherit Metab Dis. 2013;36:813–20.
Hagen CM, Aidt FH, Havndrup O, Hedley PL, Jespersgaard C, Jensen M, Kanters JK, Moolman-Smook JC, Møller DV, Bundgaard H, Christiansen M. MT-CYB mutations in hypertrophic cardiomyopathy. Mol Genet Genomic Med. 2013;1:54–65.
Abdulhag UN, Soiferman D, Schueler-Furman O, Miller C, Shaag A, Elpeleg O, Edvardson S, Saada A. Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur J Hum Genet. 2015;23:159–64.
Campos Y, García-Redondo A, Fernández-Moreno MA, Martínez-Pardo M, Goda G, Rubio JC, Martín MA, del Hoyo P, Cabello A, Bornstein B, Garesse R, Arenas J. Early-onset multisystem mitochondrial disorder caused by a nonsense mutation in the mitochondrial DNA cytochrome C oxidase II gene. Ann Neurol. 2001;50:409–13.
Arbustini E, Favalli V, Narula N, Serio A, Grasso M. Left ventricular noncompaction: a distinct genetic cardiomyopathy? J Am Coll Cardiol. 2016;68:949–66.
Indrieri A, van Rahden VA, Tiranti V, Morleo M, Iaconis D, Tammaro R, D’Amato I, Conte I, Maystadt I, Demuth S, Zvulunov A, Kutsche K, Zeviani M, Franco B. Mutations in COX7B cause microphthalmia with linear skin lesions, an unconventional mitochondrial disease. Am J Hum Genet. 2012;91:942–9.
Piekutowska-Abramczuk D, Magner M, Popowska E, Pronicki M, Karczmarewicz E, Sykut-Cegielska J, Kmiec T, Jurkiewicz E, Szymanska-Debinska T, Bielecka L, Krajewska-Walasek M, Vesela K, Zeman J, Pronicka E. SURF1 missense mutations promote a mild Leigh phenotype. Clin Genet. 2009;76:195–204.
Wedatilake Y, Brown RM, McFarland R, Yaplito-Lee J, Morris AAM, Champion M, Jardine PE, Clarke A, Thorburn DR, Taylor RW, Land JM, Forrest K, Dobbie A, Simmons L, Aasheim ET, Ketteridge D, Hanrahan D, Chakrapani A, Brown GK, Rahman S. SURF1 deficiency: a multi-centre natural history study. Orphanet J Rare Dis. 2013;8:96.
Weraarpachai W, Sasarman F, Nishimura T, Antonicka H, Auré K, Rötig A, Lombès A, Shoubridge EA. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am J Hum Genet. 2012;90:142–51.
Huigsloot M, Nijtmans LG, Szklarczyk R, Baars MJH, van den Brand MAM, Hendriksfranssen MGM, van den Heuvel LP, Smeitink JAM, Huynen MA, Rodenburg RJT. A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:488–93.
Čížková A, Stránecký V, Mayr JA, Tesařová M, Havlíčková V, Paul J, Ivánek R, Kuss AW, Hansíková H, Kaplanová V, Vrbacký M, Hartmannová H, Nosková L, Honzík T, Drahota Z, Magner M, Hejzlarová K, Sperl W, Zeman J, Houštěk J, Kmoch S. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat Genet. 2008;40:1288–90.
Honzík T, Tesarová M, Mayr JA, Hansíková H, Jesina P, Bodamer O, Koch J, Magner M, Freisinger P, Huemer M, Kostková O, van Coster R, Kmoch S, Houstêk J, Sperl W, Zeman J. Mitochondrial encephalocardio-myopathy with early neonatal onset due to TMEM70 mutation. Arch Dis Child. 2010;95:296–301.
Magner M, Dvorakova V, Tesarova M, Mazurova S, Hansikova H, Zahorec M, Brennerova K, Bzduch V, Spiegel R, Horovitz Y, Mandel H, Eminoğlu FT, Mayr JA, Koch J, Martinelli D, Bertini E, Konstantopoulou V, Smet J, Rahman S, Broomfield A, Stojanović V, Dionisi-Vici C, van Coster R, Morava-Kozicz E, Sperl W, Zeman J, Honzik T, Honzik T. TMEM70 deficiency: long-term outcome of 48 patients. J Inherit Metab Dis. 2015;38:417–26.
Spiegel R, Khayat M, Shalev SA, Horovitz Y, Mandel H, Hershkovitz E, Barghuti F, Shaag A, Saada A, Korman SH, Elpeleg O, Yatsiv I. TMEM70 mutations are a common cause of nuclear encoded ATP synthase assembly defect: further delineation of a new syndrome. J Med Genet. 2011;48:177–82.
Imai A, Fujita S, Kishita Y, Kohda M, Tokuzawa Y, Hirata T, Mizuno Y, Harashima H, Nakaya A, Sakata Y, Takeda A, Mori M, Murayama K, Ohtake A, Okazaki Y. Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein. Int J Cardiol. 2016;207:203–5.
Jonckheere AI, Hogeveen M, Nijtmans LGJ, van den Brand MAM, Janssen AJM, Diepstra JHS, van den Brandt FCA, van den Heuvel LP, Hol FA, Hofste TGJ, Kapusta L, Dillmann U, Shamdeen MG, Smeitink JAM, Rodenburg RJT. A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy. J Med Genet. 2007;45:129–33.
Ware SM, El-Hassan N, Kahler SG, Zhang Q, Ma Y-W, Miller E, Wong B, Spicer RL, Craigen WJ, Kozel BA, Grange DK, Wong L-J. Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes. J Med Genet. 2009;46:308–14.
Barth Syndrome Foundation: Home (n.d.).
Haghighi A, Haack TB, Atiq M, Mottaghi H, Haghighi-Kakhki H, Bashir RA, Ahting U, Feichtinger RG, Mayr JA, Rötig A, Lebre A-S, Klopstock T, Dworschak A, Pulido N, Saeed MA, Saleh-Gohari N, Holzerova E, Chinnery PF, Taylor RW, Prokisch H. Sengers syndrome: six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J Rare Dis. 2014;9:119.
Mayr JA, Haack TB, Graf E, Zimmermann FA, Wieland T, Haberberger B, Superti-Furga A, Kirschner J, Steinmann B, Baumgartner MR, Moroni I, Lamantea E, Zeviani M, Rodenburg RJ, Smeitink J, Strom TM, Meitinger T, Sperl W, Prokisch H. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am J Hum Genet. 2012;90:314–20.
Vukotic M, Nolte H, König T, Saita S, Ananjew M, Krüger M, Tatsuta T, Langer T. Acylglycerol kinase mutated in sengers syndrome is a subunit of the TIM22 protein translocase in mitochondria. Mol Cell. 2017;67:471–483.e7.
Davey KM, Parboosingh JS, McLeod DR, Chan A, Casey R, Ferreira P, Snyder FF, Bridge PJ, Bernier FP. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J Med Genet. 2006;43:385–93.
Ojala T, Polinati P, Manninen T, Hiippala A, Rajantie J, Karikoski R, Suomalainen A, Tyni T. New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr Res. 2012;72:432–7.
Al Teneiji A, Siriwardena K, George K, Mital S, Mercimek-Mahmutoglu S. Progressive cerebellar atrophy and a novel homozygous pathogenic DNAJC19 variant as a cause of dilated cardiomyopathy ataxia syndrome. Pediatr Neurol. 2016;62:58–61.
Ucar SK, Mayr JA, Feichtinger RG, Canda E, Çoker M, Wortmann SB. Previously unreported biallelic mutation in DNAJC19: are sensorineural hearing loss and basal ganglia lesions additional features of dilated cardiomyopathy and ataxia (DCMA) syndrome? JIMD Rep. 2017;35:39–45.
Feichtinger RG, Oláhová M, Kishita Y, Garone C, Kremer LS, Yagi M, Uchiumi T, Jourdain AA, Thompson K, D’Souza AR, Kopajtich R, Alston CL, Koch J, Sperl W, Mastantuono E, Strom TM, Wortmann SB, Meitinger T, Pierre G, Chinnery PF, Chrzanowska-Lightowlers ZM, Lightowlers RN, DiMauro S, Calvo SE, Mootha VK, Moggio M, Sciacco M, Comi GP, Ronchi D, Murayama K, Ohtake A, Rebelo-Guiomar P, Kohda M, Kang D, Mayr JA, Taylor RW, Okazaki Y, Minczuk M, Prokisch H. Biallelic C1QBP mutations cause severe neonatal-, childhood-, or later-onset cardiomyopathy associated with combined respiratory-chain deficiencies. Am J Hum Genet. 2017b;101:525–38.
Heimer G, Eyal E, Zhu X, Ruzzo EK, Marek-Yagel D, Sagiv D, Anikster Y, Reznik-Wolf H, Pras E, Oz Levi D, Lancet D, Ben-Zeev B, Nissenkorn A. Mutations in AIFM1 cause an X-linked childhood cerebellar ataxia partially responsive to riboflavin. Eur J Paediatr Neurol. 2018;22:93–101.
O’Toole JF, Liu Y, Davis EE, Westlake CJ, Attanasio M, Otto EA, Seelow D, Nurnberg G, Becker C, Nuutinen M, Kärppä M, Ignatius J, Uusimaa J, Pakanen S, Jaakkola E, van den Heuvel LP, Fehrenbach H, Wiggins R, Goyal M, Zhou W, Wolf MTF, Wise E, Helou J, Allen SJ, Murga-Zamalloa CA, Ashraf S, Chaki M, Heeringa S, Chernin G, Hoskins BE, Chaib H, Gleeson J, Kusakabe T, Suzuki T, Isaac RE, Quarmby LM, Tennant B, Fujioka H, Tuominen H, Hassinen I, Lohi H, van Houten JL, Rotig A, Sayer JA, Rolinski B, Freisinger P, Madhavan SM, Herzer M, Madignier F, Prokisch H, Nurnberg P, Jackson P, Khanna H, Katsanis N, Hildebrandt F, Hildebrandt F. Individuals with mutations in XPNPEP3, which encodes a mitochondrial protein, develop a nephronophthisis-like nephropathy. J Clin Invest. 2010;120:791–802.
Bonnen PE, Yarham JW, Besse A, Wu P, Faqeih EA, Al-Asmari AM, Saleh MAM, Eyaid W, Hadeel A, He L, Smith F, Yau S, Simcox EM, Miwa S, Donti T, Abu-Amero KK, Wong L-J, Craigen WJ, Graham BH, Scott KL, McFarland R, Taylor RW. Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet. 2013;93:471–81.
Van Goethem G, Luoma P, Rantamäki M, Al Memar A, Kaakkola S, Hackman P, Krahe R, Löfgren A, Martin JJ, De Jonghe P, Suomalainen A, Udd B, Van Broeckhoven C. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–7.
Horvath R, Hudson G, Ferrari G, Fütterer N, Ahola S, Lamantea E, Prokisch H, Lochmüller H, Mcfarland R, Ramesh V, Klopstock T, Freisinger P, Salvi F, Mayr JA, Taylor RW, Turnbull D, Hanna M, Fialho D, Suomalainen A, Zeviani M, Chinnery PF. Phenotypic spectrum associated with mutations of the mitochondrial polymerase g gene. Brain. 2006;129:1674–84.
Verhoeven WM, Egger JI, Kremer BP, de Pont BJ, Marcelis CL. Recurrent major depression, ataxia, and cardiomyopathy: association with a novel POLG mutation? Neuropsychiatr Dis Treat. 2011;7:293–6.
Van Hove JLK, Cunningham V, Rice C, Ringel SP, Zhang Q, Chou P-C, Truong CK, Wong L-JC. Finding twinkle in the eyes of a 71-year-old lady: a case report and review of the genotypic and phenotypic spectrum of TWINKLE-related dominant disease. Am J Med Genet A. 2009;149A:861–7.
Fratter C, Gorman GS, Stewart JD, Buddles M, Smith C, Evans J, Seller A, Poulton J, Roberts M, Hanna MG, Rahman S, Omer SE, Klopstock T, Schoser B, Kornblum C, Czermin B, Lecky B, Blakely EL, Craig K, Chinnery PF, Turnbull DM, Horvath R, Taylor RW. The clinical, histochemical, and molecular spectrum of PEO1 (Twinkle)-linked adPEO. Neurology. 2010;74:1619–26.
Kornblum C, Nicholls TJ, Haack TB, Schöler S, Peeva V, Danhauser K, Hallmann K, Zsurka G, Rorbach J, Iuso A, Wieland T, Sciacco M, Ronchi D, Comi GP, Moggio M, Quinzii CM, DiMauro S, Calvo SE, Mootha VK, Klopstock T, Strom TM, Meitinger T, Minczuk M, Kunz WS, Prokisch H. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet. 2013;45:214–9.
Thompson K, Majd H, Dallabona C, Reinson K, King MS, Alston CL, He L, Lodi T, Jones SA, Fattal-Valevski A, Fraenkel ND, Saada A, Haham A, Isohanni P, Vara R, Barbosa IA, Simpson MA, Deshpande C, Puusepp S, Bonnen PE, Rodenburg RJ, Suomalainen A, Õunap K, Elpeleg O, Ferrero I, McFarland R, Kunji ERS, Taylor RW. Recurrent de novo dominant mutations in SLC25A4 cause severe early-onset mitochondrial disease and loss of mitochondrial DNA copy number. Am J Hum Genet. 2016;99:860–76.
Körver-Keularts IMLW, de Visser M, Bakker HD, Wanders RJA, Vansenne F, Scholte HR, Dorland L, Nicolaes GAF, Spaapen LMJ, Smeets HJM, Hendrickx ATM, van den Bosch BJC. Two novel mutations in the SLC25A4 gene in a patient with mitochondrial myopathy. JIMD Rep. 2015;22:39–45.
Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, Santoro A, Scarcia P, Fontanesi F, Lamantea E, Ferrero I, Zeviani M. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet. 2005;14:3079–88.
Haack TB, Kopajtich R, Freisinger P, Wieland T, Rorbach J, Nicholls TJ, Baruffini E, Walther A, Danhauser K, Zimmermann FA, Husain RA, Schum J, Mundy H, Ferrero I, Strom TM, Meitinger T, Taylor RW, Minczuk M, Mayr JA, Prokisch H. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2013a;93:211–23.
Akawi NA, Ben-Salem S, Hertecant J, John A, Pramathan T, Kizhakkedath P, Ali BR, Al-Gazali L. A homozygous splicing mutation in ELAC2 suggests phenotypic variability including intellectual disability with minimal cardiac involvement. Orphanet J Rare Dis. 2016;11:139.
Taylor RW, Pyle A, Griffin H, Blakely EL, Duff J, He L, Smertenko T, Alston CL, Neeve VC, Best A, Yarham JW, Kirschner J, Schara U, Talim B, Topaloglu H, Baric I, Holinski-Feder E, Abicht A, Czermin B, Kleinle S, Morris AAM, Vassallo G, Gorman GS, Ramesh V, Turnbull DM, Santibanez-Koref M, McFarland R, Horvath R, Chinnery PF. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA. 2014;312:68.
Rauschenberger K, Schöler K, Sass JO, Sauer S, Djuric Z, Rumig C, Wolf NI, Okun JG, Kölker S, Schwarz H, Fischer C, Grziwa B, Runz H, Nümann A, Shafqat N, Kavanagh KL, Hämmerling G, Wanders RJA, Shield JPH, Wendel U, Stern D, Nawroth P, Hoffmann GF, Bartram CR, Arnold B, Bierhaus A, Oppermann U, Steinbeisser H, Zschocke J. A non-enzymatic function of 17β-hydroxysteroid dehydrogenase type 10 is required for mitochondrial integrity and cell survival. EMBO Mol Med. 2010;2:51–62.
Oláhová M, Hardy SA, Hall J, Yarham JW, Haack TB, Wilson WC, Alston CL, He L, Aznauryan E, Brown RM, Brown GK, Morris AAM, Mundy H, Broomfield A, Barbosa IA, Simpson MA, Deshpande C, Moeslinger D, Koch J, Stettner GM, Bonnen PE, Prokisch H, Lightowlers RN, McFarland R, Chrzanowska-Lightowlers ZMA, Taylor RW. LRPPRC mutations cause early-onset multisystem mitochondrial disease outside of the French-Canadian population. Brain. 2015;138:3503–19.
Kopajtich R, Nicholls TJ, Rorbach J, Metodiev MD, Freisinger P, Mandel H, Vanlander A, Ghezzi D, Carrozzo R, Taylor RW, Marquard K, Murayama K, Wieland T, Schwarzmayr T, Mayr JA, Pearce SF, Powell CA, Saada A, Ohtake A, Invernizzi F, Lamantea E, Sommerville EW, Pyle A, Chinnery PF, Crushell E, Okazaki Y, Kohda M, Kishita Y, Tokuzawa Y, Assouline Z, Rio M, Feillet F, de Camaret BM, Chretien D, Munnich A, Menten B, Sante T, Smet J, Régal L, Lorber A, Khoury A, Zeviani M, Strom TM, Meitinger T, Bertini ES, Van Coster R, Klopstock T, Rötig A, Haack TB, Minczuk M, Prokisch H. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am J Hum Genet. 2014;95:708–20.
Baruffini E, Dallabona C, Invernizzi F, Yarham JW, Melchionda L, Blakely EL, Lamantea E, Donnini C, Santra S, Vijayaraghavan S, Roper HP, Burlina A, Kopajtich R, Walther A, Strom TM, Haack TB, Prokisch H, Taylor RW, Ferrero I, Zeviani M, Ghezzi D. MTO1 mutations are associated with hypertrophic cardiomyopathy and lactic acidosis and cause respiratory chain deficiency in humans and yeast. Hum Mutat. 2013;34:1501–9.
Ghezzi D, Baruffini E, Haack TB, Invernizzi F, Melchionda L, Dallabona C, Strom TM, Parini R, Burlina AB, Meitinger T, Prokisch H, Ferrero I, Zeviani M. Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am J Hum Genet. 2012;90:1079–87.
Charif M, Titah SMC, Roubertie A, Desquiret-Dumas V, Gueguen N, Meunier I, Leid J, Massal F, Zanlonghi X, Mercier J, Raynaud de Mauverger E, Procaccio V, de Camaret BM, Lenaers G, Hamel CP. Optic neuropathy, cardiomyopathy, cognitive disability in patients with a homozygous mutation in the nuclear MTO1 and a mitochondrial MT-TF variant. Am J Med Genet A. 2015;167:2366–74.
Powell CA, Kopajtich R, D’Souza AR, Rorbach J, Kremer LS, Husain RA, Dallabona C, Donnini C, Alston CL, Griffin H, Pyle A, Chinnery PF, Strom TM, Meitinger T, Rodenburg RJ, Schottmann G, Schuelke M, Romain N, Haller RG, Ferrero I, Haack TB, Taylor RW, Prokisch H, Minczuk M. TRMT5 mutations cause a defect in post-transcriptional modification of mitochondrial tRNA associated with multiple respiratory-chain deficiencies. Am J Hum Genet. 2015;97:319–28.
Metodiev MD, Thompson K, Alston CL, Morris AAM, He L, Assouline Z, Rio M, Bahi-Buisson N, Pyle A, Griffin H, Siira S, Filipovska A, Munnich A, Chinnery PF, McFarland R, Rötig A, Taylor RW. Recessive mutations in TRMT10C cause defects in mitochondrial RNA processing and multiple respiratory chain deficiencies. Am J Hum Genet. 2016;98:993–1000.
Simon MT, Ng BG, Friederich MW, Wang RY, Boyer M, Kircher M, Collard R, Buckingham KJ, Chang R, Shendure J, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian Genomics, Van Hove JLK, Freeze HH, Abdenur JE. Activation of a cryptic splice site in the mitochondrial elongation factor GFM1 causes combined OXPHOS deficiency. Mitochondrion. 2017;34:84–90.
Smits P, Antonicka H, van Hasselt PM, Weraarpachai W, Haller W, Schreurs M, Venselaar H, Rodenburg RJ, Smeitink JA, van den Heuvel LP. Mutation in subdomain G’ of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur J Hum Genet. 2011a;19:275–9.
Ahola S, Isohanni P, Euro L, Brilhante V, Palotie A, Pihko H, Lönnqvist T, Lehtonen T, Laine J, Tyynismaa H, Suomalainen A. Mitochondrial EFTs defects in juvenile-onset Leigh disease, ataxia, neuropathy, and optic atrophy. Neurology. 2014;83:743–51.
Emperador S, Bayona-Bafaluy MP, Fernández-Marmiesse A, Pineda M, Felgueroso B, López-Gallardo E, Artuch R, Roca I, Ruiz-Pesini E, Couce ML, Montoya J. Molecular-genetic characterization and rescue of a TSFM mutation causing childhood-onset ataxia and nonobstructive cardiomyopathy. Eur J Hum Genet. 2016;25:153–6.
Ng YS, Alston CL, Diodato D, Morris AA, Ulrick N, Kmoch S, Houštěk J, Martinelli D, Haghighi A, Atiq M, Gamero MA, Garcia-Martinez E, Kratochvílová H, Santra S, Brown RM, Brown GK, Ragge N, Monavari A, Pysden K, Ravn K, Casey JP, Khan A, Chakrapani A, Vassallo G, Simons C, McKeever K, O’Sullivan S, Childs A-M, Østergaard E, Vanderver A, Goldstein A, Vogt J, Taylor RW, McFarland R. The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease. J Med Genet. 2016;53(11):768–75. https://doi.org/10.1136/jmedgenet-2016-103910.
Yin X, Tang B, Mao X, Peng J, Zeng S, Wang Y, Jiang H, Li N. The genotypic and phenotypic spectrum of PARS2-related infantile-onset encephalopathy. J Hum Genet. 2018;63:971–80.
Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A, Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784–6.
Smits P, Saada A, Wortmann SB, Heister AJ, Brink M, Pfundt R, Miller C, Haas D, Hantschmann R, Rodenburg RJT, Smeitink JAM, van den Heuvel LP. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur J Hum Genet. 2011b;19:394–9.
Baertling F, Haack TB, Rodenburg RJ, Schaper J, Seibt A, Strom TM, Meitinger T, Mayatepek E, Hadzik B, Selcan G, Prokisch H, Distelmaier F. MRPS22 mutation causes fatal neonatal lactic acidosis with brain and heart abnormalities. Neurogenetics. 2015b;16:237–40.
Galmiche L, Serre V, Beinat M, Assouline Z, Lebre A-S, Chretien D, Nietschke P, Benes V, Boddaert N, Sidi D, Brunelle F, Rio M, Munnich A, Rötig A. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum Mutat. 2011;32:1225–31.
Bursle C, Narendra A, Chuk R, Cardinal J, Justo R, Lewis B, Coman D. COXPD9 an evolving multisystem disease; congenital lactic acidosis, sensorineural hearing loss, hypertrophic cardiomyopathy, cirrhosis and interstitial nephritis. JIMD Rep. 2017;34:105–9.
Carroll CJ, Isohanni P, Pöyhönen R, Euro L, Richter U, Brilhante V, Götz A, Lahtinen T, Paetau A, Pihko H, Battersby BJ, Tyynismaa H, Suomalainen A. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet. 2013;50:151–9.
Distelmaier F, Haack TB, Wortmann SB, Mayr JA, Prokisch H. Treatable mitochondrial diseases: cofactor metabolism and beyond. Brain. 2017;140:e11.
Haack TB, Gorza M, Danhauser K, Mayr JA, Haberberger B, Wieland T, Kremer L, Strecker V, Graf E, Memari Y, Ahting U, Kopajtich R, Wortmann SB, Rodenburg RJ, Kotzaeridou U, Hoffmann GF, Sperl W, Wittig I, Wilichowski E, Schottmann G, Schuelke M, Plecko B, Stephani U, Strom TM, Meitinger T, Prokisch H, Freisinger P. Phenotypic spectrum of eleven patients and five novel MTFMT mutations identified by exome sequencing and candidate gene screening. Mol Genet Metab. 2014;111:342–52.
Tucker EJ, Hershman SG, Köhrer C, Belcher-Timme CA, Patel J, Goldberger OA, Christodoulou J, Silberstein JM, McKenzie M, Ryan MT, Compton AG, Jaffe JD, Carr SA, Calvo SE, RajBhandary UL, Thorburn DR, Mootha VK. Mutations in MTFMT underlie a human disorder of formylation causing impaired mitochondrial translation. Cell Metab. 2011;14:428–34.
Götz A, Tyynismaa H, Euro L, Ellonen P, Hyötyläinen T, Ojala T, Hämäläinen RH, Tommiska J, Raivio T, Oresic M, Karikoski R, Tammela O, Simola KOJ, Paetau A, Tyni T, Suomalainen A. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88:635–42.
McMillan HJ, Schwartzentruber J, Smith A, Lee S, Chakraborty P, Bulman DE, Beaulieu CL, Majewski J, Boycott KM, Geraghty MT. Compound heterozygous mutations in glycyl-tRNA synthetase are a proposed cause of systemic mitochondrial disease. BMC Med Genet. 2014;15:36.
Kohda M, Tokuzawa Y, Kishita Y, Nyuzuki H, Moriyama Y, Mizuno Y, Hirata T, Yatsuka Y, Yamashita-Sugahara Y, Nakachi Y, Kato H, Okuda A, Tamaru S, Borna NN, Banshoya K, Aigaki T, Sato-Miyata Y, Ohnuma K, Suzuki T, Nagao A, Maehata H, Matsuda F, Higasa K, Nagasaki M, Yasuda J, Yamamoto M, Fushimi T, Shimura M, Kaiho-Ichimoto K, Harashima H, Yamazaki T, Mori M, Murayama K, Ohtake A, Okazaki Y. A comprehensive genomic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;12:e1005679.
Verrigni D, Diodato D, Di Nottia M, Torraco A, Bellacchio E, Rizza T, Tozzi G, Verardo M, Piemonte F, Tasca G, D’Amico A, Bertini E, Carrozzo R. Novel mutations in KARS cause hypertrophic cardiomyopathy and combined mitochondrial respiratory chain defect. Clin Genet. 2017;91:918–23.
Riley LG, Rudinger-Thirion J, Schmitz-Abe K, Thorburn DR, Davis RL, Teo J, Arbuckle S, Cooper ST, Campagna DR, Frugier M, Markianos K, Sue CM, Fleming MD, Christodoulou J. LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure. JIMD Rep. 2016;28:49–57.
Ardissone A, Lamantea E, Quartararo J, Dallabona C, Carrara F, Moroni I, Donnini C, Garavaglia B, Zeviani M, Uziel G. A novel homozygous YARS2 mutation in two Italian siblings and a review of literature. JIMD Rep. 2015;20:95–101.
Pitceathly RDS, Smith C, Fratter C, Alston CL, He L, Craig K, Blakely EL, Evans JC, Taylor J, Shabbir Z, Deschauer M, Pohl U, Roberts ME, Jackson MC, Halfpenny CA, Turnpenny PD, Lunt PW, Hanna MG, Schaefer AM, McFarland R, Horvath R, Chinnery PF, Turnbull DM, Poulton J, Taylor RW, Gorman GS. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain. 2012;135:3392–403.
Bruni F, Di Meo I, Bellacchio E, Webb BD, Mcfarland R, Chrzanowska-Lightowlers ZMA, He L, Skorupa E, Moroni I, Ardissone A, Walczak A, Tyynismaa H, Isohanni P, Mandel H, Prokisch H, Haack T, Bonnen PE, Enrico B, Pronicka E, Ghezzi D, Taylor RW, Diodato D. Clinical, biochemical, and genetic features associated with VARS2-related mitochondrial disease. Hum Mutat. 2018;39(4):563–78.
Riley LG, Menezes MJ, Rudinger-Thirion J, Duff R, de Lonlay P, Rotig A, Tchan MC, Davis M, Cooper ST, Christodoulou J. Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J Rare Dis. 2013;8:193.
Carrozzo R, Verrigni D, Rasmussen M, de Coo R, Amartino H, Bianchi M, Buhas D, Mesli S, Naess K, Born AP, Woldseth B, Prontera P, Batbayli M, Ravn K, Joensen F, Cordelli DM, Santorelli FM, Tulinius M, Darin N, Duno M, Jouvencel P, Burlina A, Stangoni G, Bertini E, Redonnet-Vernhet I, Wibrand F, Dionisi-Vici C, Uusimaa J, Vieira P, Osorio AN, McFarland R, Taylor RW, Holme E, Ostergaard E. Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: phenotype and genotype correlations in 71 patients. J Inherit Metab Dis. 2016;39:243–52.
WebHome < MITOMAP < Foswiki, (n.d.).
Bannwarth S, Procaccio V, Lebre AS, Jardel C, Chaussenot A, Hoarau C, Maoulida H, Charrier N, Gai X, Xie HM, Ferre M, Fragaki K, Hardy G, de Camaret BM, Marlin S, Dhaenens CM, Slama A, Rocher C, Bonnefont JP, Rötig A, Aoutil N, Gilleron M, Desquiret-Dumas V, Reynier P, Ceresuela J, Jonard L, Devos A, Espil-Taris C, Martinez D, Gaignard P, Sang K-HLQ, Amati-Bonneau P, Falk MJ, Florentz C, Chabrol B, Durand-Zaleski I, Paquis-Flucklinger V. Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet. 2013;50:704–14.
Scalais E, Chafai R, Van Coster R, Bindl L, Nuttin C, Panagiotaraki C, Seneca S, Lissens W, Ribes A, Geers C, Smet J, De Meirleir L. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2). Eur J Paediatr Neurol. 2013;17:625–30.
Desbats MA, Lunardi G, Doimo M, Trevisson E, Salviati L. Genetic bases and clinical manifestations of coenzyme Q10 (CoQ10) deficiency. J Inherit Metab Dis. 2015a;38:145–56.
Brea-Calvo G, Haack TB, Karall D, Ohtake A, Invernizzi F, Carrozzo R, Kremer L, Dusi S, Fauth C, Scholl-Bürgi S, Graf E, Ahting U, Resta N, Laforgia N, Verrigni D, Okazaki Y, Kohda M, Martinelli D, Freisinger P, Strom TM, Meitinger T, Lamperti C, Lacson A, Navas P, Mayr JA, Bertini E, Murayama K, Zeviani M, Prokisch H, Ghezzi D. COQ4 mutations cause a broad spectrum of mitochondrial disorders associated with CoQ10 deficiency. Am J Hum Genet. 2015;96:309–17.
Chung WK, Martin K, Jalas C, Braddock SR, Juusola J, Monaghan KG, Warner B, Franks S, Yudkoff M, Lulis L, Rhodes RH, Prasad V, Torti E, Cho MT, Shinawi M. Mutations in COQ4, an essential component of coenzyme Q biosynthesis, cause lethal neonatal mitochondrial encephalomyopathy. J Med Genet. 2015;52:627–35.
Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong W-H, Ogilvie I, Shoubridge EA, Robinson BH. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–95.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won H-H, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG. Exome aggregation consortium, analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Mollet J, Giurgea I, Schlemmer D, Dallner G, Chretien D, Delahodde A, Bacq D, de Lonlay P, Munnich A, Rötig A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J Clin Invest. 2007;117:765–72.
Kollberg G, Tulinius M, Melberg A, Darin N, Andersen O, Holmgren D, Oldfors A, Holme E. Clinical manifestation and a new ISCU mutation in iron–sulphur cluster deficiency myopathy. Brain. 2009;132:2170–9.
Haack TB, Rolinski B, Haberberger B, Zimmermann F, Schum J, Strecker V, Graf E, Athing U, Hoppen T, Wittig I, Sperl W, Freisinger P, Mayr JA, Strom TM, Meitinger T, Prokisch H. Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis. 2013b;36:55–62.
Mayr JA, Feichtinger RG, Tort F, Ribes A, Sperl W. Lipoic acid biosynthesis defects. J Inherit Metab Dis. 2014;37:553–63.
Antonicka H, Leary SC, Guercin G-H, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12:2693–702.
Vondrackova A, Vesela K, Hansikova H, Docekalova DZ, Rozsypalova E, Zeman J, Tesarova M. High-resolution melting analysis of 15 genes in 60 patients with cytochrome-c oxidase deficiency. J Hum Genet. 2012;57:442–8.
Alfadhel M, Lillquist YP, Waters PJ, Sinclair G, Struys E, McFadden D, Hendson G, Hyams L, Shoffner J, Vallance HD. Infantile cardioencephalopathy due to a COX15 gene defect: Report and review. Am J Med Genet A. 2011;155:840–4.
Stiburek L, Vesela K, Hansikova H, Hulkova H, Zeman J. Loss of function of Sco1 and its interaction with cytochrome c oxidase. AJP Cell Physiol. 2009;296:C1218–26.
Freisinger P, Horvath R, Macmillan C, Peters J, Jaksch M. Reversion of hypertrophic cardiomyopathy in a patient with deficiency of the mitochondrial copper binding protein Sco2: is there a potential effect of copper? J Inherit Metab Dis. 2004;27:67–79.
Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, Coster RV, Lyon G, Scalais E, Lebel R, Kaplan P, Shanske S, De Vivo DC, Bonilla E, Hirano M, DiMauro S, Schon EA. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23:333–7.
Tran-Viet K-N, Powell C, Barathi VA, Klemm T, Maurer-Stroh S, Limviphuvadh V, Soler V, Ho C, Yanovitch T, Schneider G, Li Y-J, Nading E, Metlapally R, Saw S-M, Goh L, Rozen S, Young TL. Mutations in SCO2 are associated with autosomal-dominant high-grade myopia. Am J Hum Genet. 2013;92:820–6.
Baertling F, van den Brand MAM, Hertecant JL, Al-Shamsi A, van den Heuvel LP, Distelmaier F, Mayatepek E, Smeitink JA, Nijtmans LGJ, Rodenburg RJT. Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum Mutat. 2015a;36:34–8.
Kishita Y, Pajak A, Bolar NA, Marobbio CMT, Maffezzini C, Miniero DV, Monné M, Kohda M, Stranneheim H, Murayama K, Naess K, Lesko N, Bruhn H, Mourier A, Wibom R, Nennesmo I, Jespers A, Govaert P, Ohtake A, Van Laer L, Loeys BL, Freyer C, Palmieri F, Wredenberg A, Okazaki Y, Wedell A. Intra-mitochondrial methylation deficiency due to mutations in SLC25A26. Am J Hum Genet. 2015;97:761–8.
Haack TB, Jackson CB, Murayama K, Kremer LS, Schaller A, Kotzaeridou U, de Vries MC, Schottmann G, Santra S, Büchner B, Wieland T, Graf E, Freisinger P, Eggimann S, Ohtake A, Okazaki Y, Kohda M, Kishita Y, Tokuzawa Y, Sauer S, Memari Y, Kolb-Kokocinski A, Durbin R, Hasselmann O, Cremer K, Albrecht B, Wieczorek D, Engels H, Hahn D, Zink AM, Alston CL, Taylor RW, Rodenburg RJ, Trollmann R, Sperl W, Strom TM, Hoffmann GF, Mayr JA, Meitinger T, Bolognini R, Schuelke M, Nuoffer J-M, Kölker S, Prokisch H, Klopstock T. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann Clin Transl Neurol. 2015;2:492–509.
Bhoj EJ, Li M, Ahrens-Nicklas R, Pyle LC, Wang J, Zhang VW, Clarke C, Wong LJ, Sondheimer N, Ficicioglu C, Yudkoff M. Pathologic variants of the mitochondrial phosphate carrier SLC25A3: two new patients and expansion of the cardiomyopathy/skeletal myopathy phenotype with and without lactic acidosis. JIMD Rep. 2015;19:59–66.
Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, Hashimoto T, Korson MS. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr. 1998;133:247–53.
Mathur A, Sims HF, Gopalakrishnan D, Gibson B, Rinaldo P, Vockley J, Hug G, Strauss AW. Molecular heterogeneity in very-long-chain acyl-CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation. 1999;99:1337–43.
Longo N, di San Filippo CA, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006;142C:77–85.
Orngreen MC, Madsen KL, Preisler N, Andersen G, Vissing J, Laforet P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: a randomized clinical trial. Neurology. 2014;82:607–13.
Martins E, Cardoso ML, Rodrigues E, Barbot C, Ramos A, Bennett MJ, Teles EL, Vilarinho L. Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: the clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis. 2011;34:835–42.
Choi J-H, Yoon H-R, Kim G-H, Park S-J, Shin Y-L, Yoo H-W. Identification of novel mutations of the HADHA and HADHB genes in patients with mitochondrial trifunctional protein deficiency. Int J Mol Med. 2007;19(1):81–7.
Lahrouchi N, Lodder EM, Mansouri M, Tadros R, Zniber L, Adadi N, Clur S-AB, van Spaendonck-Zwarts KY, Postma AV, Sefiani A, Ratbi I, Bezzina CR. Exome sequencing identifies primary carnitine deficiency in a family with cardiomyopathy and sudden death. Eur J Hum Genet. 2017;25:783–7.
Iacobazzi V, Invernizzi F, Baratta S, Pons R, Chung W, Garavaglia B, Dionisi-Vici C, Ribes A, Parini R, Huertas MD, Roldan S, Lauria G, Palmieri F, Taroni F. Molecular and functional analysis of SLC25A20 mutations causing carnitine-acylcarnitine translocase deficiency. Hum Mutat. 2004;24:312–20.
Kennedy H, Haack TB, Hartill V, Mataković L, Baumgartner ER, Potter H, Mackay R, Alston CL, O’Sullivan S, McFarland R, Connolly G, Gannon C, King R, Mead S, Crozier I, Chan W, Florkowski CM, Sage M, Höfken T, Alhaddad B, Kremer LS, Kopajtich R, Feichtinger RG, Sperl W, Rodenburg RJ, Minet JC, Dobbie A, Strom TM, Meitinger T, George PM, Johnson CA, Taylor RW, Prokisch H, Doudney K, Mayr JA. Sudden cardiac death due to deficiency of the mitochondrial inorganic pyrophosphatase PPA2. Am J Hum Genet. 2016;99:674–82.
El-Hattab AW, Scaglia F. Mitochondrial cardiomyopathies. Front Cardiovasc Med. 2016;3:25.
Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785–9.
Elliott P, Charron P, Blanes JRG, Tavazzi L, Tendera M, Konté M, Laroche C, Maggioni AP, Anastasakis A, Arbustini E, Asselbergs FW, Axelsson A, Brito D, Caforio ALP, Carr-White G, Czekaj A, Damy T, Devoto E, Favalli V, Findlay I, Garcia-Pavia P, Hagège A, Heliö T, Iliceto S, Isnard R, Jansweijer JA, Limongelli G, Linhart A, Cuenca DL, Mansencal N, McKeown P, Mogensen J, Mohiddin SA, Monserrat L, Olivotto I, Rapezzi C, Rigopoulos AG, Rosmini S, Pfeiffer B, Wicks E, Podzimkova J, Kuchynka P, Palecek T, Bundgaard H, Thune JJ, Kumme A, Vestergaard LD, Hey T, Ollila L, Kaartinen M, Dubourg O, Arslan M, Tsieu MS, Guellich A, Tissot CM, Guendouz S, Thevenin S, Khelifa RC, Gandjbakhch E, Komajda M, Neugebauer A, Pfeiffer B, Steriotis A, Ritsatos K, Vlagkouli V, Biagini E, Gentile N, Longhi S, Arretini A, Fornaro A, Cecchi F, Spirito P, Formisano F, Masarone D, Valente F, Pacileo G, Schiavo A, Testolina M, Serio A, Grasso M, Wilde A, Pinto Y, Klöpping C, Van Der Heijden JF, De Jonge N, Sikora-Puz A, Wybraniec M, Czekaj A, Francisco AR, Brito D, Madeira H, Ortiz-Genga M, Barriales-Villa R, Fernandez X, Lopez-Cuenca D, Gomez-Milanes I, Lopez-Ayala JM, Guzzo-Merello G, Gallego-Delgado M, Muir A, McOsker J, Jardine T, Iqbal H, Sekhri N, Rajani R, Bueser T, Watkinson O. European cardiomyopathy pilot registry: EURObservational research programme of the European society of cardiology. Eur Heart J. 2016;37(2):164–73.
Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children. Circulation. 2007;115:773–81.
Nugent AW, Daubeney PEF, Chondros P, Carlin JB, Colan SD, Cheung M, Davis AM, Chow CW, Weintraub RG. National Australian childhood cardiomyopathy study, clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation. 2005;112:1332–8.
Montaigne D, Pentiah AD. Mitochondrial cardiomyopathy and related arrhythmias. Card Electrophysiol Clin. 2015;7:293–301.
Meyers DE, Basha HI, Koenig MK. Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Texas Hear Inst J. 2013;40:385–94.
Enns GM. Pediatric mitochondrial diseases and the heart. Curr Opin Pediatr. 2017;29(5):541–51.
Spencer CT, Bryant RM, Day J, Gonzalez IL, Colan SD, Thompson WR, Berthy J, Redfearn SP, Byrne BJ. Cardiac and clinical phenotype in Barth syndrome. Pediatrics. 2006;118:e337–46.
Brunel-Guitton C, Levtova A, Sasarman F. Mitochondrial diseases and cardiomyopathies. Can J Cardiol. 2015;31:1360–76.
Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49:578–90.
Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, Uziel G, Lamantea E, Invernizzi F, Poulton J, Rolinski B, Iuso A, Biskup S, Schmidt T, Mewes H-W, Wittig I, Meitinger T, Zeviani M, Prokisch H. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–4.
Schiff M, Haberberger B, Xia C, Mohsen A-W, Goetzman ES, Wang Y, Uppala R, Zhang Y, Karunanidhi A, Prabhu D, Alharbi H, Prochownik EV, Haack T, Häberle J, Munnich A, Rötig A, Taylor RW, Nicholls RD, Kim J-J, Prokisch H, Vockley J. Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum Mol Genet. 2015;24:3238–47.
Collet M, Assouline Z, Bonnet D, Rio M, Iserin F, Sidi D, Goldenberg A, Lardennois C, Metodiev MD, Haberberger B, Haack T, Munnich A, Prokisch H, Rötig A. High incidence and variable clinical outcome of cardiac hypertrophy due to ACAD9 mutations in childhood. Eur J Hum Genet. 2016;24:1112–6.
Gerards M, van den Bosch BJC, Danhauser K, Serre V, van Weeghel M, Wanders RJA, Nicolaes GAF, Sluiter W, Schoonderwoerd K, Scholte HR, Prokisch H, Rötig A, de Coo IFM, Smeets HJM. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain. 2011;134:210–9.
Jain-Ghai S, Cameron JM, Al Maawali A, Blaser S, MacKay N, Robinson B, Raiman J. Complex II deficiency – a case report and review of the literature. Am J Med Genet A. 2013;161A:285–94.
Levitas A, Muhammad E, Harel G, Saada A, Caspi VC, Manor E, Beck JC, Sheffield V, Parvari R. Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur J Hum Genet. 2010;18:1160–5.
Feichtinger RG, Brunner-Krainz M, Alhaddad B, Wortmann SB, Kovacs-Nagy R, Stojakovic T, Erwa W, Resch B, Windischhofer W, Verheyen S, Uhrig S, Windpassinger C, Locker F, Makowski C, Strom TM, Meitinger T, Prokisch H, Sperl W, Haack TB, Mayr JA. Combined respiratory chain deficiency and UQCC2 mutations in neonatal encephalomyopathy: defective supercomplex assembly in complex III deficiencies. Oxidative Med Cell Longev. 2017a;2017:1–11.
Andreu AL, Checcarelli N, Iwata S, Shanske S, Dimauro S. A missense mutation in the mitochondrial cytochrome b gene in a revisited case with histiocytoid cardiomyopathy. Pediatr Res. 2000;48:311–4.
Rak M, Bénit P, Chrétien D, Bouchereau J, Schiff M, El-Khoury R, Tzagoloff A, Rustin P. Mitochondrial cytochrome c oxidase deficiency. Clin Sci (Lond). 2016;130:393–407.
Marin-Garcia J, Goldenthal MJ, Ananthakrishnan R, Pierpont ME. The complete sequence of mtDNA genes in idiopathic dilated cardiomyopathy shows novel missense and tRNA mutations. J Card Fail. 2000;6:321–9.
Finsterer J, Stöllberger C, Schubert B. Acquired left ventricular hypertrabeculation/noncompaction in mitochondriopathy. Cardiology. 2004;102:228–30.
Mayr JA, Havlícková V, Zimmermann F, Magler I, Kaplanová V, Jesina P, Pecinová A, Nusková H, Koch J, Sperl W, Houstek J. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum Mol Genet. 2010;19:3430–9.
Desbats MA, Vetro A, Limongelli I, Lunardi G, Casarin A, Doimo M, Spinazzi M, Angelini C, Cenacchi G, Burlina A, Rodriguez Hernandez MA, Chiandetti L, Clementi M, Trevisson E, Navas P, Zuffardi O, Salviati L. Primary coenzyme Q10 deficiency presenting as fatal neonatal multiorgan failure. Eur J Hum Genet. 2015b;23:1254–8.
Vernon HJ, Sandlers Y, McClellan R, Kelley RI. Clinical laboratory studies in Barth syndrome. Mol Genet Metab. 2014;112:143–7.
Jefferies JL. Barth syndrome. Am J Med Genet C Semin Med Genet. 2013;163C:198–205.
Wang J, Guo Y, Huang M, Zhang Z, Zhu J, Liu T, Shi L, Li F, Huang H, Fu L. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J Rare Dis. 2017;12:26.
Dudek J, Cheng I-F, Chowdhury A, Wozny K, Balleininger M, Reinhold R, Grunau S, Callegari S, Toischer K, Wanders RJ, Hasenfuß G, Brügger B, Guan K, Rehling P. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol Med. 2016;8:139–54.
Kabunga P, Lau AK, Phan K, Puranik R, Liang C, Davis RL, Sue CM, Sy RW. Systematic review of cardiac electrical disease in Kearns-Sayre syndrome and mitochondrial cytopathy. Int J Cardiol. 2015;181:303–10.
El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015;116:4–12.
Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. 2004;27:349–62.
DiMauro S, Hirano M. MERRF. Seattle: University of Washington; 1993.
Santorelli FM, Tanji K, Shanske S, DiMauro S. Heterogeneous clinical presentation of the mtDNA NARP/T8993G mutation. Neurology. 1997;49:270–3.
Rapezzi C, Arbustini E, Caforio ALP, Charron P, Gimeno-Blanes J, Helio T, Linhart A, Mogensen J, Pinto Y, Ristic A, Seggewiss H, Sinagra G, Tavazzi L, Elliott PM. Diagnostic work-up in cardiomyopathies: bridging the gap between clinical phenotypes and final diagnosis. A position statement from the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2013;34:1448–58.
Limongelli G, Tome-Esteban M, Dejthevaporn C, Rahman S, Hanna MG, Elliott PM. Prevalence and natural history of heart disease in adults with primary mitochondrial respiratory chain disease. Eur J Heart Fail. 2010;12:114–21.
Guenthard J, Wyler F, Fowler B, Baumgartner R. Cardiomyopathy in respiratory chain disorders. Arch Dis Child. 1995;72:223–6.
Golden AS, Law YM, Shurtleff H, Warner M, Saneto RP. Mitochondrial electron transport chain deficiency, cardiomyopathy, and long-term cardiac transplant outcome. Pediatr Transplant. 2012;16:265–8.
Koga Y, Akita Y, Nishioka J, Yatsuga S, Povalko N, Tanabe Y, Fujimoto S, Matsuishi T. L-Arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64:710–2.
Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, Haack TB, Graf E, Schwarzmayr T, Terrile C, Koňaříková E, Repp B, Kastenmüller G, Adamski J, Lichtner P, Leonhardt C, Funalot B, Donati A, Tiranti V, Lombes A, Jardel C, Gläser D, Taylor RW, Ghezzi D, Mayr JA, Rötig A, Freisinger P, Distelmaier F, Strom TM, Meitinger T, Gagneur J, Prokisch H. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824.
Distelmaier F, Haack TB, Catarino CB, Gallenmüller C, Rodenburg RJ, Strom TM, Baertling F, Meitinger T, Mayatepek E, Prokisch H, Klopstock T. MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics. 2015;16:319–23.
Duncan AJ, Bitner-Glindzicz M, Meunier B, Costello H, Hargreaves IP, López LC, Hirano M, Quinzii CM, Sadowski MI, Hardy J, Singleton A, Clayton PT, Rahman S. A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease. Am J Hum Genet. 2009;84:558–66.
Van Haute L, Pearce SF, Powell CA, D’Souza AR, Nicholls TJ, Minczuk M. Mitochondrial transcript maturation and its disorders. J Inherit Metab Dis. 2015;38:655–80.
Peters H, Buck N, Wanders R, Ruiter J, Waterham H, Koster J, Yaplito-Lee J, Ferdinandusse S, Pitt J. ECHS1 mutations in Leigh disease: a new inborn error of metabolism affecting valine metabolism. Brain. 2014;137:2903–8.
Weidemann F, Rummey C, Bijnens B, Störk S, Jasaityte R, Dhooge J, Baltabaeva A, Sutherland G, Schulz JB, Meier T. Mitochondrial protection with idebenone in cardiac or neurological outcome (MICONOS) study group, the heart in Friedreich ataxia: definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation. 2012;125:1626–34.
Ganetzky RD, Stendel C, McCormick EM, Zolkipli-Cunningham Z, Goldstein AC, Klopstock T, Falk MJ. MT-ATP6 mitochondrial disease variants: phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum Mutat. 2019;40(5):499–515.
Stenton SL, Prokisch H. Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018;62(3):399–408.
Stenton SL, Kremer LS, Kopajtich R, Ludwig C, Prokisch H. The diagnosis of inborn errors of metabolism by an integrative “multi-omics” approach: a perspective encompassing genomics, transcriptomics, and proteomics. J Inherit Metab Dis. 2019.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mastantuono, E., Wolf, C.M., Prokisch, H. (2019). Genetic Basis of Mitochondrial Cardiomyopathy. In: Erdmann, J., Moretti, A. (eds) Genetic Causes of Cardiac Disease. Cardiac and Vascular Biology, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-030-27371-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-27371-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27370-5
Online ISBN: 978-3-030-27371-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)