
Abstract
Our study describes a novel phenotype in a series of nine Saudi patients with lactic acidosis, from four consanguineous families three of which are related. Detailed genetic studies including linkage, homozygosity mapping and targeted sequencing identified a causative mutation in the BCS1L gene. All affected members of the families have an identical mutation in this gene, mutations of which are recognized causes of Björnstad syndrome, GRACILE syndrome and a syndrome of neonatal tubulopathy, encephalopathy, and liver failure (MIM 606104) leading to isolated mitochondrial respiratory chain complex III deficiency. Here we report the appearance of a novel behavioral (five patients) and psychiatric (two patients) phenotype associated with a p.Gly129Arg BCS1L mutation, differing from the phenotype in a previously reported singleton patient with this mutation. The psychiatric symptoms emanated after childhood, initially as hypomania later evolving into intermittent psychosis. Neuroradiological findings included subtle white matter abnormalities, whilst muscle histopathology and respiratory chain studies confirmed respiratory chain dysfunction. The variable neuro-psychiatric manifestations and cortical visual dysfunction are most unusual and not reported associated with other BCS1L mutations. This report emphasizes the clinical heterogeneity associated with the mutation in BCS1L gene, even within the same family and we recommend that defects in this gene should be considered in the differential diagnosis of lactic acidosis with variable involvement of different organs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mitochondrial respiratory chain deficiency is implicated in the pathogenesis of a wide range of neurological disorders affecting both adults and children, and may be caused by mutations in either the mitochondrial (mtDNA) or nuclear genome (McFarland et al 2010). Mitochondrial complex III (ubiquinol:cytochrome c reductase; bc1 complex; CIII; E.C.1.10.2.2) is composed of 11 polypeptide subunits, all except one of which—cytochrome b (encoded by the mitochondrial genome)—are encoded by nuclear genes. Isolated CIII deficiency is reported to cause a heterogeneous group of neuromuscular and non-neuromuscular disorders affecting adults and children (Fernandez-Vizarra et al 2007). The bc1 synthesis like (BCS1L) gene, a member of the AAA (ATPases associated with various cellular activities) family, encode a 419-amino-acid mitochondrial chaperone protein (BCS1L) required for the assembly of the Rieske iron-sulfur subunit of CIII (de Lonlay et al 2001). The spectrum of manifestations of BCS1L mutation is wide ranging from highly restricted disease like the Björnstad syndrome (sensorineural hearing loss and pili torti; MIM 262000) (Hinson et al 2007) to profound multisystem organ failure such as GRACILE syndrome (intrauterine growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, and early death; MIM number 603358) (Visapaa et al 2002). Furthermore, it can cause isolated fatal mitochondrial encephalopathy (Fernandez-Vizarra et al 2007) and a syndrome of neonatal tubulopathy, encephalopathy, and liver failure (MIM 606104) (de Lonlay et al 2001). With the exception of the Björnstad syndrome, BCS1L mutations cause CIII deficiency since they alter the ATP-binding residues (Hinson et al 2007).
The majority of reported cases of lactic acidosis due to BCS1L defect have early-onset disease that result in early death with the exception of one recently-reported adult case (Tuppen et al 2010). Here we report nine patients from four families with an identical BCS1L gene mutation and variable clinical presentations representing multisystem involvement of the brain, vision and hearing, skeletal and cardiac muscles.
Materials and methods
Patient ascertainment
A total of nine patients from four consanguineous families, three of which are related, with suspected mitochondrial disease were identified via the genetics clinic at King Faisal Specialist Hospital and Research Center under an approved project (RAC#2050009). Written informed consents for the study were obtained from participating families.
DNA isolation, PCR, mtDNA, and BCS1L sequencing and mutation detection
Whole blood samples were collected into EDTA-coated vacutainer (BD Vacutainer Systems, Plymouth, UK). DNA isolation was performed using QIAamp DNA Blood Mini Kit (Qiagen Inc.) that was used for PCR. The entire coding region the mitochondrial genome was amplified as described elsewhere (Kaya et al 2008). Intronic primers were designed to amplify the coding exons of BCS1L. PCR and sequencing were performed according to standard protocols.
SNP genotyping, linkage and homozygosity mapping analyses
GeneChip Human Mapping 10 K Array Xba142 (Affymetrix Inc., Santa Clara, CA, USA) was used for genotyping according to manufacturer guidelines and protocols. Pedigree check and genome-wide linkage analysis were done by PedCheck (O’Connell and Weeks 1998) and Allegro (Gudbjartsson et al 2005), respectively, using Easy Linkage software (Lindner and Hoffmann 2005). Parametric multipoint logarithm of the odds (LOD) score analysis was performed with sets of 100 markers and a disease allele frequency of 0.0001 % with minor allele frequencies and no phenocopies. Homozygous blocks were determined by identifying loss of heterozygous regions (LOH) calculated by GT Console (GTC) 3.0 (Affymetrix Inc.) and CNAG version 3.0 applying default settings of both softwares.
Results
Clinical descriptions
Patient 1
This gentleman was evaluated at the age of 14 years because of poor school performance. Bilateral horizontal nystagmus and moderate sensorineural hearing loss (SNHL) was noted, whilst neuropsychological testing revealed significant impairment of dexterity abilities and visual spatial skills. His IQ was 80 with more pronounced weakness in conceptual language use and abstractions, and he had mild attention deficit hyperactivity disorder (ADHD). He also had poor vision (OD 20/300, OS 20/300), but the anterior ocular and retinal examination was normal. The electroretinogram (ERG) was normal whilst visual evoked potentials showed prolonged P100 latencies over the mid-occipital region. Repeat IQ testing at the age of 19 years was 64. He had a notable sense of humor with sarcasm associated with silly smiles. He also had talkativeness, hyperactivity, insomnia, obsessional intrusive thoughts and preferred isolation. At the age of 21 years, he developed auditory and visual hallucination associated with paranoid delusion and depressive symptoms that lasted for 2–4 weeks. Since then, he has exhibited persistent hypomanic symptoms controlled by olanzapine. Echocardiogram and Cardiac MRI evaluations showed non-obstructive hypertrophic cardiomyopathy (HCM) (Fig. 1a and b). Urine GC/MS showed moderate elevation of lactate, pyruvate, 2-hydroxybutyric and 2-hydroxyisovaleric acids. Plasma amino acid analysis showed mild elevation of alanine (770 μmol/L; normal range 70–700 μmol/L). MS/MS analysis was within normal limits. Pyruvate dehydrogenase complex, pyruvate carboxylase, and phosphoenol pyruvate carboxykinase activities were normal. The patient has three siblings with the lactic acidosis (patients 2, 3, and 4) (Table 1). Of note, patient 2 had SNHL and patient 4 developed insulin-dependent diabetes mellitus (IDDM) at the age of 8 years. Families II (patients 5 and 6) and III (patient 7) are related to family I (Table 1).

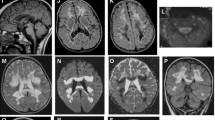
Cardiac MRI findings of patient 1 (upper panel): a MRI four chamber view image shows severe generalized biventricular hypertrophy with impaired contractile and diastolic function. b Late contrast enhancement MRI image shows area of fibrosis (arrow) on anterior LV septum with severe concentric LVH. Neuroradiological findings (lower panel): Fluorine-18 fluoro-deoxy-glucose Positron Emission Tomography (18F-FDG PET) axial images of patient 1 in color c showing symmetrical but disproportionate increased FDG activity at the basal ganglia, mainly at the putamen, in comparison to the rest of grey matter. The MRS image d at the level of the right basal ganglia shows a small doublet lactate peak at 1.3–1.4 PPM (arrow)
Patient 8
This Saudi boy developed hypoglycemia and severe metabolic acidosis on the first day of life. At the age of 5 years, he had an IQ of 78 with problems in the areas of organization, pure problem solving and social skills, whilst by the age of 15 years his IQ had declined to 63 associated with mild attentive ADHD. He was also found to have horizontal nystagmus with visual acuity of 20/200 although the afferent visual system was normal. Anxiety symptoms were also observed with emotional disturbance associated with talkativeness, verbal disinhibition, and yelling. He was described as detached, stubborn and unsociable. He was also noted to have restlessness, polyphagia, frequent hand washing and frequent visits to the bathroom, symptoms consistent with hypomania. Later, he started to have intermittent bouts of psychosis with visual and auditory hallucinations. Investigations were remarkable for the presence of α-ketoglutarate in the urine organic acid analysis. He showed gradual neurological regression, and at the age of 26 years, developed severe rhabdomyolysis (creatine kinase (CK) >21500, normal 24–195) and acute renal failure. He is now in a vegetative state due to recurrent episodes of metabolic acidosis and encephalopathy. The patient has a 13-year-old brother (patient 9) who had early onset of lactic acidosis, mild speech immaturity and attentive ADHD (Table 1), and two previous siblings (a boy and a girl) who died with suspected lactic acidosis.
Neuro-radiological evaluation
Brain MRI showed subtle findings (Table 1). Abnormalities include ill-defined T2 periventricular white matter abnormalities mainly involving the occipital regions and mild atrophic changes. The basal ganglia appeared normal. The FDG PET (patient 1) demonstrated increased metabolic activity involving the putamen bilaterally, and the caudate nuclei to a lesser extent (Fig. 1c). Small lactate peak was found on MRS in three patients (Fig. 1d).
Muscle histopathology and respiratory chain studies
Patient 1
There was minimal variation in myofiber size but Gomori trichrome revealed frequent fibers with increased subsarcolemmal mitochondria suggestive of “early” ragged-red fibers (RRF) (Fig. 2a). Oxidative enzyme reactions, nicotinamide dehydrogenase (NADH) and succinic dehydrogenase (SDH) reactions revealed correspondingly accentuated activities (Fig. 2b and c). Lipid accumulation as evident by the oil red O stain was moderate, especially in type 1 myofibers.

Panels a–c depict muscle biopsy findings in patient 1. Mitochondrial proliferation (asterisks) is noted in the modified Gomori trichrome stain (a) as ragged red fibers. With the oxidative enzyme NADH-TR reaction (b) and SDH (c) reaction they appear “ragged blue”. In all these stains the mitochondria accumulate in the subsarcolemmal zone in most fibers in a crescentic pattern. Panels D-I depict muscle biopsy findings in patient 8. Ragged red fibers (asterisks) are seen with the modified Gomori trichrome (d), appear “ragged blue” in the SDH reaction (f) and are COX-positive (g). Fibers with prominent lipid accumulation (open arrows) are seen with oil red O stain (e). Ultrastructurely, accumulation of mitochondria (m) is noted at low magnification (h) along with glycogen granules (g) in a myofiber. At high magnification (i), the mitochondria (m) appear round and swollen with altered internal architecture
Patient 8
There was mild variation in myofiber size with occasional necrotic and regenerative fibers, and scattered, elongated mildly atrophic fibers. Gomori Trichrome revealed increased subsarcolemmal staining and RRF (Fig. 2d). Oil red O showed moderate increase in lipid content in many fibers (Fig. 2e). NADH, SDH and cytochrome c oxidase (COX) reactions revealed frequent fibers with accentuated subsarcolemmal activity (Fig. 2f and g) but no COX-deficient fibers. The ATPase reactions showed no selective myofiber type atrophy or fiber type grouping. Transmission electron microscopy revealed moderate accumulation of mitochondria and glycogen; mitochondria were spherical and had abnormal cristae (Fig. 2h and i).
Mitochondrial respiratory chain activities were determined in the muscle biopsy of patient 8, showing an increased citrate synthetase (CS) activity concomitant with the observed mitochondrial proliferation. After correction for CS activities, we observed a clear deficiency in the activity of complexes II + III but with normal CII activity, indicating a mitochondrial respiratory chain disorder involving complex III in isolation (Table 2).
Genetic and genomic analyses
The entire mitochondrial genome was sequenced in patients from families 1 and 2, but no pathogenic mutations were identified. Genotypes of the four affected children and other healthy members in family 1 were generated using SNP chip arrays, and whole-genome linkage analysis was performed in family 1. The multipoint parametric analysis identified a single significant peak on chromosome 2q35-q36.3 with a LOD score of 3.2597 (Fig. 3a). The region of interest (13.5 megabases in size) was localized between SNP probes SNP_A-1516727 (chr2:216,595,534 bp) and SNP_A-1518963 (chr2:230,092,889 bp) and contained 128 genes based on NCBI MapViewer 36.3 Build including BCS1L. Haplotype construction visualized by Haplopainter (Fig. 3b) suggested homozygosity by descent for the alleles in the critical region, which was confirmed by the presence of a large LOH block extending on the same region based on SNP calls. Microsatellite markers narrowed down the region further and BCS1L was screened, identifying a previously reported mutation (c.385 G > A; p.Gly129Arg) (Tuppen et al 2010) in patients 1–7 with parents confirmed as heterozygous carriers (Fig. 3c). Due to clinical similarities, we sequenced the DNA samples from the two patients in family 4, identifying the same BCS1L gene mutation.

a Linkage analysis of the patients in the consanguineous Saudi families. The analysis is done by Allegro/Easy Linkage and produced a peak of LOD score ∼3.25 (y-Axis) on chromosome 2 (x-axis). b Haplotype analysis (Haplopainter); patient haplotypes (labeled as number 1 on the figure) are indicated by SNP Markers on chromosome 2 shared among all the affected individuals. c shows sequencing chromatograms from father, mother and an affected individual. d The location of the p.Gly129Arg mutation (yellow arrow) is indicated on the hypothetical 3D structure of BCS1L
Discussion
The mitochondrial proteome is estimated to be around 1500 proteins in total (Calvo and Mootha 2010) although pathogenic mutations have been identified in only about 150 nuclear genes (Wong 2010). We believe that the autosomally recessive inherited (nuclear) group of mitochondrial disorders is more prevalent in Saudi Arabia than many parts of the world because of the high rate of consanguineous marriages, the tribal structure, and the large family size. However, this entity is undoubtedly underreported in our country.
BSC1L and TTC19 are the only genes involved in CIII assembly that are known in humans (Rotig 2010; Ghezzi et al 2011). All mutations of BCS1L disrupt the assembly of CIII while reducing the activity of the respirasome increases the generation of reactive oxygen species. This is particularly pronounced when the number of mitochondria is increased as in the case of CIII deficiency (Hinson et al 2007). During the formation of neural tube, the expression of BCS1L as well as its distribution, differ from other mitochondrial proteins including the Rieske Fe-S proteins. This suggests another unknown role of BCS1L in the formation, development and function of the CNS, other than its role in assembling CIII (Kotarsky et al 2007).
Dysfunction of the central nervous system is a prominent feature of many mitochondrial disorders (Kisler et al 2010). Beside the variable cognitive impairment, an attentive form of ADHD was seen in the majority of our patients. Relentless neurological regression was noted in the two adults (patients 1 and 8) in our group, associated with a late onset of psychiatric symptoms. Despite being from two different families, both adults exhibited similar findings of hypomania and psychosis and also developed HCM. Additionally, patient 8 developed rhabdomyolysis resulting in acute renal failure. The ophthalmological symptoms seen in patients (1, 2, 3 and 8) are quite unique as it started at the age of 12–15 years with severe reduction in visual acuity causing very poor vision focus. The overall course of disease was generally slowly progressive in all patients, although may be considered static in patients 5 and 9. Neuroradiologically, there were faint white matter abnormalities in the peritrigonal areas seen in several patients (Table 1). In addition, only a small lactate peak was noted on MRS in some patients examined, and the CSF lactate concentration was borderline in patient 3 (2 mmol/L) indicating that these diagnostic modalities may not be of high yield in this disease. The muscle biopsy showed some interesting structural findings (ragged red fibers, mitochondrial accumulation, and glycogen granules). Rhabdomyolysis was noted in one patient. Progressive exercise intolerance and myoglobinuria associated with CIII deficiency due to mutations in the cytochrome b (MTCYB) gene were previously reported (Andreu et al 1999a, b).
A disturbed mitochondrial function has been suggested to underlie the symptoms of common psychiatric disorders like bipolar disorder, schizophrenia and depressive disorders although no clear and direct evidence has been previously found (Rezin et al 2009). The unique finding in the two adults in the present group was an insidiously progressive psychological deterioration and progressive loss of IQ. In fact the appearance of various abnormal psychological signs appear to follow a predestined pattern as ADHD in childhood years, cognitive delay in early adolescence, slow onset dementia during late teenage years and finally hypomania and psychosis in the twenties. The previously reported adult patient with the same p.Gly129Arg BSC1L mutation presented with seizures, optic atrophy, limited exercise tolerance, and isolated CIII deficiency, but with normal intellect, blood lactate, and muscle histology and histochemistry (Tuppen et al 2010). No psychiatric manifestations were reported. In their study, the authors showed pathogenicity of the mutation using yeast complementation studies (Tuppen et al 2010). Therefore, it is apparent that the p.Gly129Arg mutation does not cause early lethality like the other reported mutations although similar to other mutations causing CIII deficient, its location within the BCS1L protein (Fig. 3d) suggests it might alter β-chain structure leading to defective interaction with other proteins involved in the formation of complex III assembly (Tuppen et al 2010). Moreover, this location (Fig. 3d) is highly conserved among different species and predicted to be probably damaging by Polyphen algorithm (Adzhubei et al 2010), and located in the stability domain of N-terminal of BCS1L sitting between mitochondrial import and AAA-ATPase domains (Hinson et al 2007; Moran et al 2010) it is highly likely that BCS1L activity and stability were impaired. We present evidence of a wider spectrum of clinical and histological manifestations related to this mutation including neuropsychiatric involvement related to abnormal mitochondrial proliferation. It is unclear if the complications are age-related, and as such might have been evident in the previously described young patients.
In conclusion, nine patients from four families with a mitochondrial disease caused by a nuclear mitochondrial gene, BCS1L, mutation are described. This report emphasizes the clinical heterogeneity of the mutation of BCS1L gene even within the same family. A breadth of complications (brain, eye, heart, skeletal muscle, endocrine glands) was variably observed in our patients.
References
Adzhubei IA, Schmidt S, Peshkin L et al (2010) A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249
Andreu AL, Bruno C, Dunne TC et al (1999a) A nonsense mutation (G15059A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann Neurol 45(1):127–130
Andreu AL, Hanna MG, Reichmann H et al (1999b) Exercise intolerance due to mutations in the cytochrome b gene of mitochondrial DNA. N Engl J Med 341(14):1037–1044
Calvo SE, Mootha VK (2010) The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 11:25–44
de Lonlay P, Valnot I, Barrientos A et al (2001) A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat Genet 29(1):57–60
Fernandez-Vizarra E, Bugiani M, Goffrini P et al (2007) Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum Mol Genet 16(10):1241–1252
Ghezzi D, Arzuffi P, Zordan M et al (2011) Mutations in TTC19 cause 1212mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat Genet 43(3):259–263
Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A (2005) Allegro version 2. Nat Genet 37(10):1015–1016
Hinson JT, Fantin VR, Schonberger J et al (2007) Missense mutations in the BCS1L gene as a cause of the Bjornstad syndrome. N Engl J Med 356(8):809–819
Kaya N, Imtiaz F, Colak D et al (2008) Genome-wide gene expression profiling and mutation analysis of Saudi patients with Canavan disease. Genet Med 10(9):675–684
Kisler JE, Whittaker RG, McFarland R (2010) Mitochondrial diseases 1212in childhood: a clinical approach to investigation and management. Dev Med Child Neurol 52(5):422–433
Kotarsky H, Tabasum I, Mannisto S, Heikinheimo M, Hansson S, Fellman V (2007) BCS1L is expressed in critical regions for neural development during ontogenesis in mice. Gene Expr Patterns 7(3):266–273
Lindner TH, Hoffmann K (2005) easyLINKAGE: a PERL script for easy and automated two-/multi-point linkage analyses. Bioinformatics 21(3):405–407
McFarland R, Taylor RW, Turnbull DM (2010) A neurological perspective on mitochondrial disease. Lancet Neurol 9(8):829–840
Moran M, Marin-Buera L, Gil-Borlado MC et al (2010) Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum Mutat 31(8):930–941
O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63(1):259–266
Rezin GT, Amboni G, Zugno AI, Quevedo J, Streck EL (2009) Mitochondrial dysfunction and psychiatric disorders. Neurochem Res 34(6):1021–1029
Rotig A (2010) Genetic bases of mitochondrial respiratory chain disorders. Diabetes Metab 36(2):97–107
Tuppen HA, Fehmi J, Czermin B et al (2010) Long-term survival of neonatal mitochondrial complex III deficiency associated with a novel BCS1L gene mutation. Mol Genet Metab 100(4):345–348
Visapaa I, Fellman V, Vesa J et al (2002) GRACILE syndrome, a lethal metabolic disorder with iron overload, is caused by a point mutation in BCS1L. Am J Hum Genet 71(4):863–876
Wong LJ (2010) Molecular genetics of mitochondrial disorders. Dev Disabil Res Rev 16(2):154–162
Acknowledgments
We are very grateful to the patients and their families for their enthusiasm and participation in this study. We also would like to acknowledge generous support of King Faisal Specialist Hospital and Research Center for our research activities. We have no financial interest for disclosure. This work was supported by a King Faisal Specialist Hospital and Research Center’s seed grant for Dr. Kaya’s lab and was approved by KFSHRC’s research advisory council and ethical review board.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Shamima Rahman
Mohammed Al-Owain and Dilek Colak have equal contribution.
Rights and permissions
About this article
Cite this article
Al-Owain, M., Colak, D., Albakheet, A. et al. Clinical and biochemical features associated with BCS1L mutation. J Inherit Metab Dis 36, 813–820 (2013). https://doi.org/10.1007/s10545-012-9536-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-012-9536-4