
Abstract
The liver has a complex vascular system, with a dual blood supply divided between the hepatic artery and the portal vein, which ultimately mixes within the hepatic sinusoids before draining into the systemic circulation via the hepatic venous system. Each of its components can be damaged by various types of injury. Vascular disorders of the liver are rare conditions, usually affecting young people, with high morbidity and mortality that can occur in the absence of a proper diagnosis and disease-specific management. In the last years, international collaborations provided data-supported approaches, which allowed to increase knowledge and awareness in understanding and management of these conditions. Given the rarity of some vascular disorders of the liver, not all will be discussed in this chapter. Definition, aetiology, diagnosis, treatment and prognosis of the most common vascular disorders of the liver will be considered. Moreover morphological and functional changes relevant to the clinical aspect are explained.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hepatic venous outflow tract obstruction
- Budd-Chiari syndrome
- Portal vein thrombosis
- Portal cavernoma
- Nodular regenerative hyperplasia
- Sinusoidal obstruction syndrome
- Congenital vascular malformations of the liver
- Hereditary haemorrhagic telangiectasia
- Congenital arteriovenous shunt
- Congenital arterioportal shunt
- Congenital portosystemic shunt
-
Vascular disorders of the liver are rare conditions, usually affecting young people, with high morbidity and mortality that can occur in the absence of a proper diagnosis and disease-specific management.
-
An underlying systemic prothrombotic condition is found in patients with Budd-Chiari Syndrome and portal vein thrombosis
-
Anticoagulation should be initiated without waiting in patients with Budd-Chiari Syndrome and acute portal vein thrombosis.
-
Sinusoidal obstruction syndrome occurs as an iatrogenic complication of exposure to toxic agents for sinusoidal endothelium of the liver and hematopoietic bone marrow cells.
-
Vascular malformations in hereditary haemorrhagic telangiectasia affect the liver extensively and evolve continuously from small telangiectasia to large arteriovenous malformations.
1 Introduction
The liver is a very vascular organ and at rest receives up to 25% of total cardiac output [1]. It is divided into eight independent segments, each segment having a separate hepatic artery and portal vein in the centre and hepatic veins in the periphery [2]. The microscopic units of the liver are known as hexagonal hepatic lobules, formed by radiating hepatocytes and many specialized capillaries known as sinusoids [2]. Every component of the hepatic vascular system (hepatic arteries, portal and hepatic veins, sinusoids, and lymphatics) can present a spectrum of variants and pathologic conditions. In the last years, international collaborations provided data-supported approaches, which allowed to increase knowledge and awareness in understanding and management of these conditions. Vascular disorders of the liver affect less than 5/10,000 patients and together comprise a number of rare conditions that can cause non-cirrhotic portal hypertension with high morbidity and mortality [3]. Moreover, they are usually diagnosed in young people, with a contrarily normal life expectancy if these conditions are timely diagnosed and managed properly [3]. Diagnosis is based on a high degree of clinical suspicion and usually confirmed by imaging. Doppler ultrasound, angio-MR, angio-CT provide information of similar accuracy depending of the type of vascular disorder and have the advantage of being non-invasive. Liver biopsy is excessive in most of the case and usually not recommended. Treatment depends on the type of vascular disorder, associated conditions and patient’s clinical status. Given the rarity of some vascular disorders of the liver, not all will be discussed in this chapter. A spectrum of variants and diseases involving the hepatic venous outflow tract will be considered, notably Budd-Chiari Syndrome (BCS). Anomalies, and disease involving the portal vein such as acute portal vein thrombosis (PVT), chronic PVT, and cavernous transformation will be discussed. Sinusoidal obstruction syndrome (SOS) also reviewed. Congenital vascular malformations involving the liver are also explained, with an emphasis on hepatic vascular malformations in hereditary haemorrhagic telangiectasia (HHT) and congenital shunts.
2 Budd-Chiari Syndrome or Hepatic Venous Outflow Tract Obstruction
Budd-Chiari syndrome (BCS) known also as hepatic venous outflow tract obstruction (HVOTO) is define by an obstruction on the hepatic veins, at any level between small intrahepatic veins to the entrance of the inferior vena cava and right atrium, independent of the mechanism of obstruction, leading to an impaired hepatic venous drainage [3]. Cardiac disease, pericardial disease or sinusoidal obstruction syndrome (SOS) are excluded from this definition [3, 4]. BCS can be classified depending on the level of obstacle: small hepatic veins, inferior vena cava, and any combination thereof, having among these categories distinct presentation and geographical distribution [5].
Aetiology: According to the cause, BCS can be classified into primary or secondary. Primary BCS is the consequence of a primarily venous disease (thrombosis or phlebitis), and secondary BCS is the result of an external compression or invasion by a lesion originating outside the vein (benign or malignant tumour or infectious process) [3, 4].
An underlying systemic prothrombotic condition is found in up to 80% in patients with BCS [6]. However, the aetiology is often multifactorial. A combination of such conditions is present in nearly half, particularly in patients with heterozygous factor V Leiden or in patients taken oral contraceptives or pregnant women [7]. Antiphospholipid antibodies are responsible for 30% of BCS cases whereas lupus anticoagulant or anti-beta-2 glycoprotein 1 antibodies for 4–5% [3]. The aetiology of primarily BCS differ greatly among countries. In Europe, BCS is mostly secondary to thrombosis in hepatic veins, whereas in Asia, Behcet disease, membranous obstruction of the inferior vena cava are the most common aetiologies [8]. BCS has been related to myeloproliferative neoplasms in 35–50% cases in western countries, JAK2 mutation, V617F mutation account for 90% of them and CARL mutations in 2–5% [9].
The main primary tumours involving in secondary BCS are carcinoma: hepatocellular, renal and adrenal, primary hepatic hemangiosarcoma, epithelioid hemangioendothelioma, sarcoma of the inferior vena cava, right atrial myxoma, alveolar hydatid disease [10]. Infectious processes account for a small number of cases and most common are: amoebic and pyogenic abscess, polycystic liver disease [7]. Moreover, BCS may occur following trauma [7], hepatic resection or transplantation [11].
The local factor that develops thrombosis of the hepatic venous tract remains unidentified in most patients [4].
The diagnosis of the underlying cause (primary or secondary) of BCS has important implications for treatment. If left untreated, symptomatic BCS is lethal within a few days, to a few years [12].
Morphological changes: hepatic venous outflow tract obstruction induces venous wall inflammation, increased sinusoid pressure and portal hypertension. Centrilobular sinusoidal dilatation and congestion, liver cell loss and fibrosis are considered histopathological features for BCS [4]. However, these features are not specific, being also seen in heart failure, constrictive pericarditis and SOS [12]. Ultimately, a cirrhotic pattern develops, nodular regenerative hyperplasia, macroregenerative nodules being common in advanced cases [13]. These lead to fibrous enlargement of the portal tract, with portovenous and portoportal bridging fibrosis and thrombosis of intrahepatic portal veins [4, 13]. Because of the marked heterogeneous distribution of these lesions, liver biopsy is not recommended, the assessment of fibrosis proved irrelevant for prognosis [12].
Functional changes: The hepatic vein obstruction is predominantly due to the occlusion of at least two hepatic veins, but the occlusion is not synchronous, an acute clinical presentation coincides in most of the cases with the ultimate obstruction of an individual hepatic vein overlapping a chronic obstruction [12, 14]. It is associated with an obstruction of the IVC in approximately one third of patients, while isolated IVC obstruction is rare [14]. Hepatic vein occlusion causes elevated sinusoidal pressure, liver congestion and increased lymphatic filtration of interstitial fluid [15]. Increased sinusoidal pressure within proportions of the hepatic parenchyma with blood stasis induce portal hypertension and ascites, which is also increased by the impaired lymphatic drainage capability. Several mechanisms tend to preserve blood perfusion to the liver: increased arterial blood flow, redistribution of portal blood flow to the areas with preserved outflow and the development of venous collateral circulation (intrahepatic and extrahepatic) [13]. Although these mechanisms can prevent the development of clinical manifestations of liver disease, in the absence of treatment, irreversible liver abnormalities progressively develop to centrilobular fibrosis [4].
Diagnosis: Clinical presentation ranges from absence of symptoms to fulminant hepatic failure [16]. Asymptomatic BCS cases accounts for up to 20% of cases, and is often associated with the presence of large hepatic venous collaterals [3, 4]. Classical signs and symptoms include fever, abdominal pain, ascites, hepatomegaly, ankle swelling, gastrointestinal bleeding and hepatic encephalopathy [4, 17]. In a multicentre prospective study, ascites were present in 83% percent of patients with BCS, hepatomegaly in 67%, abdominal pain in 61% and gastrointestinal bleeding in 5% [18]. The course of these manifestations can be progressive or with periods of exacerbations and remissions. BCS can present a long insidious course, or a short period of prodrome followed by an accelerated falling course. In approximately 15% of cases portal venous obstruction is associated, suggesting a more severe form [17, 18].
Diagnosis is establish by confirming the hepatic venous outflow obstruction. Doppler ultrasound, angio-MR and angio-CT provide information of similar accuracy and have the advantages of being non-invasive or minimally invasive [3, 12]. Doppler ultrasound is the first line investigation and has a sensitivity of more than 75% but the awareness and expertise of the examiner are crucial [3].
Imaging findings classify BCS lesions as direct signs—visualization the obstacle and indirect signs (secondary to the venous obstruction)—intrahepatic or extrahepatic collateral circulation, perfusion abnormalities and anatomical changes to the liver, all resulting from portal hypertension [19]. The obstruction can present with several aspects, including short-length, extended narrowing of venous lumen, a complete obstruction simulating a membrane or a fibrous cord, with upstream dilatation [12, 19]. Venography is recommended if the diagnosis is uncertain and it is compulsory for percutaneous interventions [3]. Hepatic nodules can be seen using imaging in more than a half of patients with BCS, resulting from perfusion disturbances and being usually benign [3]. These nodules are usually small (<4 cm in diameter), multiple, hypervascularized and disseminated through the liver [20]. Although hepatocellular carcinoma in BCS account for 4% of the cases, currently there are no clear diagnosis criteria, a biopsy should be performed in selected cases (less or equal to three nodules, nodules >3 cm in diameter, heterogeneity or washed out on venous phase, patients with high levels of alphafetoprotein [20]. Secondary BCS related to an external compression or invasion is ruled out also by using the same radiological approaches [3]. Liver biopsy should only be taken into consideration in selected cases where the imaging has failed to demonstrate obstruction, due to the associated risk of bleeding that may delay the initiation of anticoagulation therapy [4]. To note that the assessment of fibrosis proved irrelevant for prognosis [4].
Treatment: In most cases, the underlying disorder causing BCS in unrecognized at presentation. Patients with BCS presenting with ascites and varices require the same treatment as cirrhotic patients [21].
Anticoagulant therapy should be initiated without waiting, as soon as possible and for an indefinite period of time in order to reduce the clot extensions and new thrombotic episodes [18]. Although ineffective on chronic liver disease, early anticoagulation has improved survival in patients with moderate BCS, probably by a preventive systemic effect in other sites on thrombosis [19]. Currently, there are no data regarding the use of Non-Vitamin K Antagonists, treatment with Warfarin or Acenocumarol should be consider for an indefinite period. Treatment for the underlying prothrombotic condition should be started in the same time.
The experience of thrombolysis is limited, and complications can be fatal [3].
Patients with focal or segmental obstruction of the hepatic venous outflow tract are eligible for percutaneous angioplasty, with or without stenting. Focal or segmental stenosis are present in 60% of patients with IVC obstruction, and 25–60% of those with hepatic vein obstruction [3]. These patients may benefit by percutaneous angioplasty of one HV, of the IVC or both [22]. The rationale for angioplasty is to re-establish the physiological drainage of portal and sinusoidal blood [22]. At present, less than 10% of patients with BCS are eligible for stenting (Fig. 11.1) [6, 23].

(a) Pre-procedure sonography coupled with Doppler ultrasound showing a patent IVC. (b) Segmental narrowing of all hepatic veins in a case of BCS. This patient underwent right hepatic vein angioplasty and stenting and follow-up sonography (c) and Doppler (d) showed a patent stent. (Reproduced with permission from Das CJ, Soneja M, Tayal S, Chahal A, Srivastava S, Kumar A, Baruah U (2018) Role of radiological imaging and interventions in management of Budd-Chiari syndrome. Clin Radiol 73:610–624)
Patients who do not improve with medical and endoscopic treatment are proposed for transjugular intrahepatic portosystemic shunt (TIPS) [12]. The rationale for TIPS is to decompress the liver by transforming the portal vein into an outflow tract [12]. Patients are selected based on the extent of the vascular lesions, particularly patients with multitroncular HV obstructions with mall intrahepatic collaterals [24].
The minority of patients in which TIPS fails or those with fulminant hepatic failure are proposed for liver transplantation, the rationale being a complete correction of the hepatic consequences of vascular obstructions [12].
Prognosis: Without treatment, symptomatic BCS is lethal within a few days to a few years [12]. Current therapeutic strategies permits to achieve 5-year survival rates over 80% [6]. Child-Pugh score and its components have been found to be independent prognostic factors. Moreover, prognostic scores based on a combination of these factors have been developed [4]. These scores are useful for the assessment of transplant-free survival and clinical studies, but not for individual management [25]. At present, long term prognosis is determined by hepatocellular carcinoma or by complications of the underlying blood disease (leukaemia in patients with myeloproliferative disease) [12].
3 Portal Vein Thrombosis
Portal vein thrombosis (PVT): is characterised by an obstruction of the portal veins, and its branches, which include splenic, superior and inferior mesenteric veins [26]. Isolated splenic or superior mesenteric vein obstruction is included in the entity of splanchnic vein obstruction [12]. Obstruction may be complete or partial [3].
PVT is classified into acute or chronic [4]. Acute thrombosis refers to recent and symptomatic, while chronic thrombosis refers to long standing process. They represent successive stages of the same disease and share similar causes, differing in their management [4]. After acute thrombosis, in the absence of recanalization, portal lumen obliterates and a set of collateral portoportal veins develops to replace the portal vein [4, 12]. This term is called portal cavernoma or cavernomatous transformation of the portal vein and corresponds to a long-standing process [4, 12]. In children, cavernoma might result from a malformation [27]. PVT is responsible for up to 30% and 75% of cases of portal hypertension in adults and children, in developing countries [4].
Aetiology: Except from childhood portal cavernoma, a thrombus is the cause of the disease. PVT is caused by a combination of local and systemic factors. Local factors include cancer (any abdominal organ), cirrhosis (these two are the leading local risk factors) and intraabdominal infections (such as secondary peritonitis) [4]. PVT is common in patients with cirrhosis, more that 30% of liver transplant recipients have PVT at the time of transplant [28]. The risk of developing PVT associated with cirrhosis is correlated to the severity of liver disease and the presence of the inherited prothrombotic disorders [29]. Systemic factors refer to an inherited or acquired prothrombotic condition [3]. Usually, one or several systemic factors are identified, the most common being myeloproliferative disease (25–30%) and factor II Leiden (in 15%) [30]. The simultaneous presence of several prothrombotic causes in patients with PVT is more frequent than in general population [31]. Identification of a local risk factor does not exclude the possibility that a general risk factor is present [31]. In this section, we will focus on the non-cirrhotic, non-malignant portal thrombosis, cirrhotic PVT and malignant being discussed separately elsewhere.
Morphological changes: In patients with non-cirrhotic portal vein thrombosis, alterations in portal venous flow result in a spectrum of altered hepatic histology, ranging from large regenerative nodules to nodular regenerative hyperplasia without bridging fibrosis [32]. Unlike the central atrophy that is characteristic for cirrhosis, the central portion of the liver is relatively spared due to collateral portal venous flow developing over time [33]. However, peripheral liver cells apoptosis may occur, because collateral blood flow to subcapsular regions is insufficient [33]. Abnormal liver circulation result in a distorted architecture of the liver with micro- and macroscopic nodules of hyperplastic hepatocytes that are not surrounded by fibrous septae [34]. Nodular regenerative hyperplasia, as in BCS, must be differentiated from hepatocellular carcinoma [3].
Functional changes: Despite acute complete portal thrombosis, there is limited evidence for liver ischemia, because the immediate development of porto-portal collaterals, involving the porta hepatis and because the compensated increase in hepatic arterial blood flow [35]. Above the obstacle, ischemia does not develop because of the small tributaries to superior or inferior mesenteric veins [36]. Spontaneous recanalization is exceptional, and in a matter of weeks a portal cavernoma is formed by collateral veins which contribute to maintain the perfusion of portal blood to the liver [37]. Because they cannot reduce portal pressure, spleen enlargement and portosystemic collaterals develop [12]. Within a year gastroesophageal varices will be formed [12, 37]. Synchronous, liver architecture is compromised, with preserved blood perfusion to the central of the liver and hardship in the periphery, with increase in size of segments I and IV of the liver and atrophy of left liver lobe and peripheral parts of the right lobe [12]. However, frank liver dysfunction is absent, subtle signs being common, such as a decrease in coagulation factors levels and subclinical hepatic encephalopathy [38].
Diagnosis: Diagnosis of PVO is presently made in 50–70% of the cases in the acute setting [39]. Common symptoms of acute complete PVT include acute abdominal or lumbar pain, with moderated distended abdomen by ileus, without any other features of intestinal occlusion [37]. Partial thrombosis is associated with fewer symptoms, PVT being recognised only at the stage of cavernomatous transformation [3]. In patients with chronic PVT, the severity of portal hypertension typically contrasts with a mild or absent liver dysfunction (with normal levels of transaminases, alkaline phosphatase and gamma-glutamyl transferase) [4]. 50% of the patients present with ascites, but ascites emerge after a triggering event like bleeding or infection, and it is usually reversible [30]. Features of hypersplenism may be marked and bleeding related to portal hypertension is massive though better tolerated than patients with cirrhosis [12].
Acute PVT is rapidly diagnosed using noninvasive imaging. It shows thrombus occupying the lumen of the portal vein or its branches, with a poorly development of porto-portal collaterals [40], Doppler ultrasound, CT scan and MRI are almost equivalent, depending on the expertise of the operator [3, 4]. Standard abdominal echography reveals a hyperechogenic material in the lumen with distensions of the portal veins [4]. Doppler imaging allows to prove the absence of flow in part of the lumen [3, 4]. Because the mesenteric veins are difficult to visualize at echography, CT or MRI are more sensitive for assessment of thrombus extension [3, 4]. Thrombus is revealed by CT scan as a hyperattenuating material in the portal vein (Fig. 11.2a) [41, 42]. After contrast injection is revealed as a lack of luminal enhancement, with increased hepatic enhancement in the arterial phase and decreased hepatic enhancement in the portal phase [41]. If the thrombus is less than a week old, it appears as a hyperintense material on MRI T1-weighted sequences [40]. Portal cavernoma is seen as a lattice of serpiginous structures that enhance the portal phase of vascular contrast while the normal portal vein is not visible [12].

(a, b) Contrast-enhanced CT scans show severe thrombosis (arrowhead) in the intrahepatic portal branches (a) and the main portal vein (b). (c) Direct portal venogram shows extensive thrombosis in the portal venous system, multiple collaterals (arrows), and hepatofugal flow. (d) One covered stent was deployed after the competing collaterals were embolized. Fifteen months after TIPS placement (e, f) CT scans show complete recanalization of the portal vein. (Reproduced with permission from Luo X et al. (2015) Advanced Cirrhosis Combined with Portal Vein Thrombosis: A Randomized Trial of TIPS versus Endoscopic Band Ligation Plus Propranolol for the Prevention of Recurrent Esophageal Variceal Bleeding. Radiology 276:286–293)
Treatment: Since the aim of the treatment between acute and chronic portal vein thrombosis differs, we will discuss them separately. The aim of the treatment of acute PVT is the recanalization of the obstructed veins and prevention of the extension of thrombosis to mesenteric veins followed by intestinal infarction and portal hypertension [3, 4].
For acute PVT, immediate initiation of anticoagulation prevents thrombus extension [30]. There have been no controlled studies of anticoagulation therapy in patients with acute PVT [4]. In a recent prospective study, intestinal infarction was a rare complication (2/95 patients) which require only limited intestinal resection, even if in 60% of the patients superior mesenteric vein was involved [30]. In the setting of intestinal infarction, emergency laparotomy should be performed [4]. Full recanalization of the portal vein was achieved in 40% of patients by 6 months of treatment, and did not occur in any of the patients beyond 6 months of treatment [30]. Also, high recanalization rates were observed after anticoagulation in post splenectomy PVT patients or for acute thrombosis involving the superior mesenteric vein [43]. Splenic vein thrombosis and ascites suggest failure in recanalization [30]. Spontaneous recanalization is infrequent in patients not receiving anticoagulation therapy [30]. The optimal duration of anticoagulation therapy has not been determined, however, according to a panel of international experts [3, 4], at least 3-month period should be considered, while permanent anticoagulation for patients with permanent prothrombotic conditions should be taken into consideration [43]. In most of the studies, anticoagulation was based on unfractionated heparin, LMWH or VKA targeting an INR between 2 and 3 [3].
The reported experience with thrombolytic therapy, systemic or in situ, is extremely limited [3, 4]. The reported recanalization rates have been similar to those achieved with anticoagulation alone, but with major procedure-related complications. A higher mortality rate was noted with approaches using transhepatic route [44]. Surgical thrombectomy has proved a benefit in 30% of the patients, but with a high recurrence rate when performed more than 30 days after the onset [45].
Current studies report that balloon angioplasty with or without stenting without thrombolysis or thrombectomy could be an alternative and save treatment for post-operative main portal vein and superior mesenteric vein thrombosis [46].
Data on TIPS are limited, beyond the technical challenge of the procedure, medium-term efficacy require further evaluation (Fig. 11.2b) [4, 42].
The aim of the treatment for chronic PVT is to prevent recurrent thrombosis, and the prevention and treatment of the associated complications, gastrointestinal bleeding and portal cholangiopathy [4]. At present there are no controlled studies regarding treatment of gastroesophageal varices in patient with chronic PVT. Some retrospective multivariate studies found that screening for gastroesophageal varices, beta-adrenergic blockers or endoscopic therapy reduces the risk of bleeding and by thus, improves survival [4]. In a number of uncontrolled surveys, endoscopic sclerotherapy has achieved eradication of varices and a reduction in bleeding rate [4].
The experience in splenectomy, devascularisation and TIPS insertion in patients with portal cavernoma is limited [47]. The prevention of recurrent thrombosis in chronic PVT requires also anticoagulation, with the same mentions as in acute PVT [3, 4]. To note that a recent retrospective study showed that warfarin had independently improved the survival of patients with chronic PVT, most of them having a risk factor for thrombosis [4]. Another retrospective analysis found a decreased incidence of gastrointestinal bleeding after starting anticoagulation therapy [4]. Patients with portal cholangiopathy usually present with jaundice and biliary symptoms. Insertion of a biliary prosthesis after endoscopic extraction of stones is a proved therapy and the lack of the recurrence after prosthesis removal was noted in almost half of patients [48]. Other techniques involve portosystemic shunting, bilio-enteric anastomosis, biliary surgery without portal decompression, but current data are limited [3, 4].
Prognosis: With the effective prevention and control of bleeding and thrombosis, the outcome is given by age and the course of the underlying disease [12].
The mortality of acute PVT is high due to late recognition of intestinal infarction, and portal hypertension developed with associated complications (variceal bleeding) [4]. Above half of the patients do not achieve recanalization and will develop gastroesophageal varices with a 2 year high probability of bleeding [37].
The outcome for treated patients with chronic PVT is currently good [4]. Mortality is related to the recurrent bleeding from portal hypertension, followed by recurrent thrombosis at splanchnic or extrasplanchnic sites. In a 5-year followed up period, less than 5% of patients with PVT died from classical complications [4].
4 Sinusoidal Obstruction Syndrome
Sinusoidal obstruction syndrome (SOS) is characterized by a loss of sinusoidal endothelium integrity with a consequent sinusoidal obstruction by outflow block [3]. Damaged sinusoids can be associated with a partial or complete occlusion of small hepatic veins, therefore being previously known as hepatic veno-occlusive disease (VOD) [49]. SOS is a primary circulatory disorder that can occur in the absence of central vein occlusion, the involvement of central vein being related to more severe disease [4]. Therefore the alternative term of SOS was considered in replacement of VOD.
Aetiology: SOS occurs as an iatrogenic complication of exposure to toxic agents for sinusoidal endothelium of the liver and hematopoietic bone marrow cells [3]. A large number of drugs and toxins have been associated with SOS: plant pyrrolizidine alkaloids, myeloablative regimens used in the setting of haematopoietic stem cell transplantation, chemotherapy for liver metastasis of colorectal cancer, thiopurine derivatives [50]. Other reported conditions are liver irradiation and platelet transfusion containing ABO-incompatible plasma [3]. Lately, an inherited condition combining VOD and immunodeficiency associated with mutations in Sp110, has been described. The acronym for this condition is VODI [51]. Although its mechanism is unknown, one possible explanation is an accompanying opportunistic viral infection affecting the endothelium of sinusoids or central vein [51].
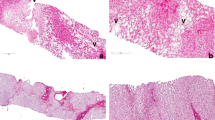
Morphological changes: Despite of its multiple causes, patients with SOS present similar morphological changes [50]. Circulatory obstruction precedes liver dysfunction. According to the level of obstruction, various degrees of centrilobular hepatocellular necrosis may occur [3]. As a result, SOS lesions appear to have a patchy distribution [3]. It may also associate one or more other lesions such as centrilobular perisinusoidal and endovenular fibrosis, peliosis and nodular regenerative hyperplasia (Fig. 11.3) [3, 49, 50]. All of these changes seem to be related to SOS severity or represent late lesions [50].

Acute sinusoidal obstruction syndrome related to gemtuzumab use (Mylotarg) after haematopoietic stem cell transplantation (a, b) Masson trichrome, (c hematein eosin-safran; d: argentation stain): dilatation and congestion of sinusoids are limited to centrilobular zones around the terminal hepatic vein ∗; endothelial cells of veins and sinusoids are damaged, leading to a huge hematic deposition in Disse space and to hepatocyte necrosis around the central veins. (Reproduced from Valla D-C, Cazals-Hatem D. (2016) MINI REVIEW Sinusoidal obstruction syndrome, Clin Res Hepatol Gastroenterol 40:378–385)
Functional changes: SOS is a clinical diagnosis based on several common liver disease signs and symptoms: weight gain with or without ascites, hepatomegaly and jaundice [52]. However, patients can be asymptomatic or can present with features of portal hypertension or multiple organ dysfunction syndrome.
Diagnosis: Starting from the definition, the diagnosis of SOS expects a histologic examination of the liver [12]. Percutaneous liver biopsy is usually contraindicated by thrombocytopenia, coagulopathy or ascites [4]. Transjugular liver biopsy with hepatic venous pressure gradient is of major help in confirming SOS [3]. In patients receiving myeloablative regimens used in the context of haematopoietic stem cell transplantation, a hepatic venous pressure gradient >10 mmHg proved to have a specificity of 91% and a sensitivity of 52% for the diagnosis of SOS [3].
After excluding confiding situations, the diagnosis can be made based on a high index of clinical suspicion in a patient who has signs and symptoms of SOS and had received a therapy known to cause liver injury [3, 4]. Increased serum level of bilirubin is a sensitive but not specific marker [4]. The American Association for Study of Liver Disease (ASSLD) introduced clinical criteria for SOS diagnosis [4]. These criteria, known as Seattle or Baltimore criteria, present clinical features for diagnosis used for defining populations for research [3]. For example, the Seattle criteria were developed for patients receiving myeloablative regimens containing cyclophosphamide, clinical findings must occur within 20 days of transplantation [3]. These criteria does not apply in patients receiving regimens to cause late onset disease [3]. Their sensitivity and specificity are currently not well established, and their use in different settings of SOS aetiology have not been evaluated [3].
The diagnosis may be supported by imaging techniques, Doppler echocardiography showing signs of portal hypertension, liver and spleen enlargement; none of these findings are specific for SOS [53]. Reversal of flow in portal vein and monophasic flow in hepatic vein have been used to diagnose SOS, but lacks sensitivity [54]. Magnetic Resonance imaging may show patchy signal enhancement compatible with histologically severe SOS. Because of its associated toxicity, Computer Tomography is not recommended [4].
Treatment: Identifying patients at risk is useful in preventing SOS. In patients with pre-existing liver disease, previous history of SOS, recent treatment with gemtuzumab ozagamicin or myelofibrosis with extramedullary haematopoiesis, both European and American Guidelines recommend the use of chemotherapy regimens with lower liver toxicity, without cyclophosphamide [55]. At present, the only proved strategy to prevent or reduce the severity of sinusoidal changes and their clinical expression is reducing the intensity of chemotherapeutic regimen [49]. Prophylactic pharmacological strategies have not proved a reduction in overall risk of SOS or the risk of fatal SOS in randomized controlled trials [4]. The routine use of intravenous heparin or subcutaneous low-molecular weight heparin as prophylaxis for SOS, the use of ursodeoxycholate, prostaglandin E1, pentoxifylline or N-acetylcysteine are unproved prophylactic measures [4]. The prophylactic use of ursodeoxycholate reduces the frequency of jaundice and alanine aminotransferase levels, without any benefit in SOS [55].
Treatment of SOS depends on its clinical severity and is based mostly on supportive care with therapy of fluid overload, sepsis and organ failure [3]. Fluid overload should be managed with diuretics, paracentesis, hemofiltration, and haemodialysis [3].
Defibrotide, a mixture of the single-stranded oligodeoxyribonucleotides derived from depolymerisation of porcine intestinal mucosa DNA, proved a benefit for treatment of severe SOS both in adult and children patients [4]. Moreover, it has also demonstrated benefit for SOS prophylaxis in paediatric hematopoietic cell transplantation patients [4].
Heparin and thrombolytic therapy proved no positive effect [56].
TIPS and surgical shunting have been used in selective cases for symptoms relief, but proved no benefit in survival [57].
SOS is usually an iatrogenic complication caused by the therapy used for patients with malignancy [49]. Liver transplantation is limited by the underlying malignancy itself. However, it may be considered in selected cases with a favourable prognosis [4].
Prognosis: The outcome of SOS lies upon the context and the magnitude of exposure to toxic agents [12]. Predictors of poor prognosis are the slope of bilirubin serum levels, weight gain, higher levels of alanine aminotransferase, higher hepatic venous pressure gradient and multiple organ failure [4].
5 Congenital Vascular Malformations Affecting the Liver
Congenital vascular malformations. Vascular malformations of the liver determine an abnormal intra or extrahepatic shunting of blood [4]. They comprise several entities based on the functional shunting. Shunting can develop between the hepatic artery to the portal vein (arterioportal shunt) or to the hepatic vein (arteriovenous shunt) or/and between the portal vein to the systemic circulation (portosystemic or portohepatic shunt) [4]. These types of congenital shunting can be isolated, although rare, and diagnosed in infants or children or may coexist in hereditary haemorrhagic telangiectasia with liver involvement [4]. Although in this chapter we will discuss only the congenital shunts, these shunts can also be acquired, associated with hepatocellular carcinoma and/or cirrhosis, or after trauma (including liver biopsy, transhepatic cholangiography, or biliary surgery).
Hepatic vascular malformations in hereditary haemorrhagic telangiectasia (HHT).
HHT, or Rendu-Osler-Weber disease is a rare, genetic disorder with an autosomal dominant inheritance pattern, characterized by widespread cutaneous, mucosal, and visceral arteriovenous malformations involving the lung, brain and/or liver [3]. Most of patients present a mutation in one of two genes disease related: endoglin and activin A receptor type II-like 1, gene involved in transforming pathway of growth factor b (TGFb). Those dysfunctional gene are expressed predominantly on vascular endothelium [4].
Morphological changes: Vascular malformations in HHT affect the liver extensively, and evolve continuously from tiny telangiectasias to large arteriovenous malformations [58]. Due to a heterogeneous liver blood perfusion, nodular regenerative hyperplasia or focal nodular hyperplasia emerge, the latter having a 100-fold greater prevalence in HHT patients than in general population [59].
Functional changes: Three types of intrahepatic shunting may coexist (arterioportal, arteriovenous, and portosystemic) leading to different, concomitant or successively functional features: high output heart failure, portal hypertension, biliary disease, hepatic encephalopathy, or mesenteric ischemia [60]. High output heart failure is characterized by a hyperdynamic circulation developed through arteriohepatic and/or portohepatic shunting [61]. Portal hypertension emerge from arterioportal shunting and secondary portal fibrosis and/or regenerative hyperplasia [61]. Shunting can cause biliary ischemia which can result in bile duct necrosis and the extreme process of liver necrosis [61].
Diagnostic: Although HHT is a congenital disease, symptoms of liver vascular malformations appear predominantly in females around 30 years of age [4]. Only 8% of patients with liver vascular malformations on imaging are symptomatic [62]. High output heart failure represents the predominant clinical presentation, with exertional dyspnoea, ascites, oedema [4] and atrial fibrillation [63]. The next most common presentation is portal hypertension and the clinical picture includes ascites, varices and variceal bleeding due more often to gastrointestinal telangiectasias than to variceal bleeding [4, 63]. Patients can also present with anicteric cholestasis with or without cholangitis, encephalopathy, or mesenteric angina [3, 4, 63]. Biochemical changes are not specific, with a slight elevation of alkaline phosphatase and gamma glutamyl-transpeptidase, without any changes in the live synthetic function [60].
Currently, according to EASL Clinical Practice Guidelines, the diagnosis of HHT requires several criteria known as Curaçao criteria [3]. Diagnosis of HHT is suspected in a symptomatic patients with clinical features suggesting HHT and requires laboratory assessment and imaging methods such as abdominal Doppler Ultrasound and/or abdominal CT [62]. According to the Curaçao criteria, Doppler ultrasound can give a severity of grading (from 0+ to 4) which correlates with clinical outcome, and enables management and follow-up. Intrahepatic hypervascularization and enlarged hepatic artery seen on Doppler ultrasound or CT have the highest diagnostic accuracy [4, 64]. Moreover, because of the presence of nodular regenerative hyperplasia, the liver may appear nodular on imaging studies, and should be differentiated from cirrhosis. Liver biopsy in the diagnosis of liver vascular malformations in HHT is unnecessary [3, 4]. Genetic testing can be performed to establish the diagnosis in patients with diffuse liver vascular malformations who do not meet clinical diagnostic criteria for HHT [65]. Echocardiography can be performed to evaluate the hemodynamic impact [3]. Further tests (endoscopy, MR, angiography) may be performed in special cases, depending on the severity of liver vascular malformations [4].
Treatment: In asymptomatic HHT cases with or without liver involvement no treatment is recommended [3, 4]. In symptomatic HHT patients with liver involvement treatment is given by the type of clinical presentation [66]. Patients with high output heart failure should be managed according to heart failure guidelines, with salt retention, diuretics, beta blockers, angiotensin-converting enzyme inhibitors [3, 4]. Complications given by portal hypertension and encephalopathy should be treated as recommended in cirrhotic patients [3, 4]. Supportive care is also important, with blood transfusions or iron administration for anaemia, and treatment of the variceal bleeding [3].
In non-responders to initial medical treatment, peripheral embolization of liver vascular malformations is the most effective and repeatable trans-arterial treatment [65].
Liver transplantation is the only definitive curative option for liver vascular malformations in HHT [3, 4]. It is reserved for selected cases such as ischemic biliary necrosis, complicated portal hypertension and refractory heart failure [65].
Bevacizumab, an antibody to vascular endothelial growth factor (VEGF) has been shown to reduce the liver volume and ameliorate cardiac output after 3-month courses in patients with severe liver vascular malformations and high cardiac output [67]. However, further studies are needed before this therapy should be recommended.
Prognostic: Clinical outcome of liver vascular malformations in HHT correlates with their severity. In a recent cohort study with a median follow-up of 44 months, mortality related to hepatic vascular malformations occurred in 5% of patients, with incidence rates of complications and death 3.6 and 1.1 per person-years, respectively [63].
5.1 Isolated Congenital Liver Shunts
Congenital Arteriovenous (Hepatic artery to hepatic vein) malformations consists of discrete abnormalities with stable evolution and without change in dimensions [68]. These changes are very rare and usually present as a high-output heart failure in a neonate [4]. The diagnosis is based on MRI [68]. Initial treatment is pharmacological and the aim is reducing the symptoms of heart failure [4]. In non-responders to medical treatment, embolization and surgical resection should be considered [4].
Congenital Arterioportal (Hepatic artery to portal vein) malformations are very rare and cause portal hypertension manifested within the first year of life [4]. Clinical features include signs of portal hypertension, splenomegaly, and or variceal bleeding [4]. The diagnosis is based on Doppler ultrasound [68]. Treatment consists of embolization of the feeding artery with or without resection [4]. Liver transplantation should be considered in selected cases [4].
Congenital Portosystemic (Portal vein to systemic circulation) malformations are rare developmental anomalies secondary to abnormal development of the portal venous system [4]. They may be associated with other congenital anomalies [69]. They are divided into intrahepatic and extrahepatic shunts with common clinical features but different treatment [69]. Through the malformations the intestinal blood reaches the systemic circulation bypassing the liver. Due to the lack of metabolization of plasma ammonia, its serum increased level determines cognitive changes [4]. Symptoms include fatigue and mental retardation, with recurrent episodes of portosystemic encephalopathy [70]. Ascites and portal hypertension are not usually seen [4]. Diagnosis and classification is based on MRI [69]. Preoperative evaluation of portal vein by angiography is important, in order to determine portal vein patency, portal pressure and the type of portosystemic shunt [71]. Every shunt that persists after one year of life should be closed before complications emerge [69]. Symptomatic cases are immediately treated either by open surgery or laparoscopy with the intention of shunt ligaturation [69]. Endovascular embolization using periphery metal coils of the shunt is performed in selected centers [4]. The choice of surgical or endovascular approach is based upon patient’s clinical condition, shunt anatomy and size and local expertise [69]. Liver transplantation may be the only treatment of extrahepatic or large intrahepatic multifocal shunts not suitable for embolization, or in cases of previous failed endovascular interventions [69]. To a standard therapeutic approach is not established.
6 Conclusions
Vascular disorders of the liver consist of multiple entities with different pathophysiology background, different clinical picture and different prognosis. As a general rule, therapy implies anticoagulant therapy, endovascular manoeuvres and surgical option. Thrombolysis shows no benefit, while increasing the risk of bleeding complications. TIPS is seldom recommended, while liver transplantation remains a final option for most of the patients with vascular disorders.
Abbreviations
- BCS:
-
Budd-Chiari syndrome
- HVOTO:
-
Hepatic venous outflow tract
- PVT:
-
Portal vein thrombosis
- SOS:
-
Sinusoidal obstruction syndrome
- VOD:
-
Veno-occlusive disease
- HHT:
-
Hereditary haemorrhagic telangiectasia
- Angio-MR:
-
Magnetic resonance angiography
- Angio-CT:
-
Computed tomography angiography
- TIPS:
-
Transjugular intrahepatic portosystemic shunt
References
Abdel-Misih SRZ, Bloomston M. Liver anatomy. Surg Clin North Am. 2010;90:643–53.
Elsayes KM, Shaaban AM, Rothan SM, Javadi S, Madrazo BL, Castillo RP, Casillas VJ, Menias CO. A comprehensive approach to hepatic vascular disease. Radiographics. 2017;37:813–36.
Clinical Practice Guidelines EASL. Vascular diseases of the liver. J Hepatol. 2016;64:179–202.
DeLeve LD, Valla D-C, Garcia-Tsao G. Vascular disorders of the liver. Hepatology. 2009;49:1729–64.
Ludwig j H e, db M, Jona v H. Classification of hepatic venous outflow obstruction: ambiguous terminology of the Budd-Chiari syndrome. Mayo Clin Proc. 1990;65:51–5.
Seijo S, Plessier A, Hoekstra J, et al. Good long-term outcome of Budd-Chiari syndrome with a step-wise management. Hepatology. 2013;57:1962–8.
Denninger M-H, Chaït Y, Casadevall N, Hillaire S, Guillin M-C, Bezeaud A, Erlinger S, Briere J, Valla D. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology. 2003;31:587–91.
Uskudar O, Akdogan M, Sasmaz N, Yilmaz S, Tola M, Sahin B. Etiology and portal vein thrombosis in Budd-Chiari syndrome. World J Gastroenterol. 2008;14:2858–62.
Poisson J, Plessier A, Kiladjian J-J, et al. Selective testing for calreticulin gene mutations in patients with splanchnic vein thrombosis: a prospective cohort study. J Hepatol. 2017;67:501–7.
Valla D-C. Hepatic venous outflow tract obstruction etiopathogenesis: Asia versus the West. J Gastroenterol Hepatol. 2004;19:S204–11.
Kubo T, Shibata T, Itoh K, Maetani Y, Isoda H, Hiraoka M, Egawa H, Tanaka K, Togashi K. Outcome of percutaneous transhepatic venoplasty for hepatic venous outflow obstruction after living donor liver transplantation. Radiology. 2006;239:285–90.
Valla D-C, Cazals-Hatem D. Vascular liver diseases on the clinical side: definitions and diagnosis, new concepts. Virchows Arch. 2018;473:3–13.
Cazals-Hatem D, Vilgrain V, Genin P, Denninger M-H, Durand F, Belghiti J, Valla D, Degott C. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology. 2003;37:510–9.
Faraoun SA, Boudjella MEA, Debzi N, Afredj N, Guerrache Y, Benidir N, Bouzid C, Bentabak K, Soyer P, Bendib SE. Budd–Chiari syndrome: a prospective analysis of hepatic vein obstruction on ultrasonography, multidetector-row computed tomography and MR imaging. Abdom Imaging. 2015;40:1500–9.
Simonetto DA, Yang H, Yin M, et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology. 2014;61:648–59.
Janssen HLA, Garcia-Pagan J-C, Elias E, Mentha G, Hadengue A, Valla D-C. Budd-Chiari syndrome: a review by an expert panel. J Hepatol. 2003;38:364–71.
Murad SD, Valla D-C, de Groen PC, Zeitoun G, Hopmans JAM, Haagsma EB, van Hoek B, Hansen BE, Rosendaal FR, Janssen HLA. Determinants of survival and the effect of portosystemic shunting in patients with Budd-Chiari syndrome. Hepatology. 2004;39:500–8.
Murad S, Plessier A, Hernandez-Guerra M, et al. Etiology, management, and outcome of the Budd-Chiari syndrome. Ann Intern Med. 2009;151:167–75.
Faraoun SA, Boudjella MEA, Debzi N, Benidir N, Afredj N, Guerrache Y, Bentabak K, Soyer P, Bendib SE. Budd-Chiari syndrome: an update on imaging features. Clin Imaging. 2016;40:637–46.
Moucari R, Rautou P-E, Cazals-Hatem D, et al. Hepatocellular carcinoma in Budd–Chiari syndrome: characteristics and risk factors. Gut. 2008;57:828 LP–835.
Valla D-C. The diagnosis and management of the Budd-Chiari syndrome: consensus and controversies. Hepatology. 2007;38:793–803.
Klein AS. Management of Budd-Chiari syndrome. Liver Transplant. 2006;12:S23–8.
Das CJ, Soneja M, Tayal S, Chahal A, Srivastava S, Kumar A, Baruah U. Role of radiological imaging and interventions in the management of Budd-Chiari syndrome. Clin Radiol. 2018;73:610–24.
Fu Y-F, Li Y, Cui Y-F, Wei N, Li D-C, Xu H. Percutaneous recanalization for combined-type Budd-Chiari syndrome: strategy and long-term outcome. Abdom Imaging. 2015;40:3240–7.
Rautou P-E, Moucari R, Escolano S, et al. Prognostic indices for Budd–Chiari syndrome: valid for clinical studies but insufficient for individual management. Am J Gastroenterol. 2009;104:1140.
Kinjo N, Kawanaka H, Akahoshi T, et al. Portal vein thrombosis in liver cirrhosis. World J Hepatol. 2014;6:64–71.
de Franchis R. Expanding consensus in portal hypertension. J Hepatol. 2015;63:743–52.
Stine JG, Shah PM, Cornella SL, Rudnick SR, Ghabril MS, Stukenborg GJ, Northup PG. Portal vein thrombosis, mortality and hepatic decompensation in patients with cirrhosis: a meta-analysis. World J Hepatol. 2015;7:2774–80.
Amitrano L, Anna Guardascione M, Brancaccio V, Margaglione M, Manguso F, Iannaccone L, Grandone E, Balzano A. Risk factors and clinical presentation of portal vein thrombosis in patients with liver cirrhosis. J Hepatol. 2004;40:736–41.
Plessier A, Darwish-Murad S, Hernandez-Guerra M, et al. Acute portal vein thrombosis unrelated to cirrhosis: a prospective multicenter follow-up study. Hepatology. 2009;51:210–8.
Janssen HLA, Meinardi JR, Vleggaar FP, et al. Factor V Leiden mutation, prothrombin gene mutation, and deficiencies in coagulation inhibitors associated with Budd-Chiari syndrome and portal vein thrombosis: results of a case-control study. Blood. 2000;96:2364–8.
Kobayashi S, Ng CS, Kazama T, Madoff DC, Faria SC, Vauthey JN, Charnsangavej C. Hemodynamic and morphologic changes after portal vein embolization: differential effects in central and peripheral zones in the liver on multiphasic computed tomography. J Comput Assist Tomogr. 2004;28
Tublin ME, Towbin AJ, Federle MP, Nalesnik MA. Altered liver morphology after portal vein thrombosis: not always cirrhosis. Dig Dis Sci. 2008;53:2784–8.
Nakanuma Y, Hoso M, Sasaki M, Terada T, Katayanagi K, Nonomura A, Kurumaya H, Harada A, Obata H. Histopathology of the liver in non-cirrhotic portal hypertension of unknown aetiology. Histopathology. 2018;28:195–204.
Valla D-C, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol. 2000;32:865–71.
Elkrief L, Corcos O, Bruno O, et al. Type 2 diabetes mellitus as a risk factor for intestinal resection in patients with superior mesenteric vein thrombosis. Liver Int. 2013;34:1314–21.
Turnes J, García–Pagán JC, González M, et al. Portal hypertension and related complications after acute portal vein thrombosis: impact of early anticoagulation. Clin Gastroenterol Hepatol. 2008;6:1412–7.
Sharma P, Sharma BC, Puri V, Sarin SK. Natural history of minimal hepatic encephalopathy in patients with extrahepatic portal vein obstruction. Am J Gastroenterol. 2009;104:885.
Rajani R, Björnsson E, Bergquist A, Danielsson Å, Gustavsson A, Grip O, Melin T, Sangfelt P, Wallerstedt S, Almer S. The epidemiology and clinical features of portal vein thrombosis: a multicentre study. Aliment Pharmacol Ther. 2010;32:1154–62.
Berzigotti A, García-Criado Á, Darnell A, García-Pagán J-C. Imaging in clinical decision-making for portal vein thrombosis. Nat Rev Gastroenterol Hepatol. 2014;11:308.
Bach AM, Hann LE, Brown KT, Getrajdman GI, Herman SK, Fong Y, Blumgart LH. Portal vein evaluation with US: comparison to angiography combined with CT arterial portography. Radiology. 1996;201:149–54.
Seedial SM, Mouli SK, Desai KR. Acute portal vein thrombosis: current trends in medical and endovascular management. Semin Interv Radiol. 2018;35:198–202.
Condat B, Pessione F, Helene Denninger M, Hillaire S, Valla D. Recent portal or mesenteric venous thrombosis: increased recognition and frequent recanalization on anticoagulant therapy. Hepatology. 2003;32:466–70.
Ferro C, Rossi UG, Bovio G, Dahamane M, Centanaro M. Transjugular intrahepatic portosystemic shunt, mechanical aspiration thrombectomy, and direct thrombolysis in the treatment of acute portal and superior mesenteric vein thrombosis. Cardiovasc Intervent Radiol. 2007;30:1070–4.
Sogaard KK, Astrup LB, Vilstrup H, Gronbaek H. Portal vein thrombosis; risk factors, clinical presentation and treatment. BMC Gastroenterol. 2007;7:34.
Cao G, Ko G-Y, Sung K-B, Yoon H-K, Il GD, Kim J-H. Treatment of postoperative main portal vein and superior mesenteric vein thrombosis with balloon angioplasty and/or stent placement. Acta Radiol. 2013;54:526–32.
Senzolo M, Tibbals J, Cholongitas E, Triantos CK, Burroughs AK, Patch D. Transjugular intrahepatic portosystemic shunt for portal vein thrombosis with and without cavernous transformation. Aliment Pharmacol Ther. 2006;23:767–75.
Harmanci O, Bayraktar Y. How can portal vein cavernous transformation cause chronic incomplete biliary obstruction? World J Gastroenterol. 2012;18:3375–8.
Valla D-C, Cazals-Hatem D. Sinusoidal obstruction syndrome. Clin Res Hepatol Gastroenterol. 2016;40:378–85.
Rubbia-Brandt L, Lauwers GY, Wang H, et al. Sinusoidal obstruction syndrome and nodular regenerative hyperplasia are frequent oxaliplatin-associated liver lesions and partially prevented by bevacizumab in patients with hepatic colorectal metastasis. Histopathology. 2010;56:430–9.
Marquardsen FA, Baldin F, Wunderer F, et al. Detection of Sp110 by flow cytometry and application to screening patients for veno-occlusive disease with immunodeficiency. J Clin Immunol. 2017;37:707–14.
Angliviel B, Benoist S, Penna C, El Hajjam M, Chagnon S, Julié C, Beauchet A, Rougier P, Nordlinger B. Impact of chemotherapy on the accuracy of computed tomography scan for the evaluation of colorectal liver metastases. Ann Surg Oncol. 2009;16:1247–53.
Slade JH, Alattar ML, Fogelman DR, et al. Portal hypertension associated with oxaliplatin administration: clinical manifestations of hepatic sinusoidal injury. Clin Colorectal Cancer. 2009;8:225–30.
Mahgerefteh SY, Sosna J, Bogot N, Shapira MY, Pappo O, Bloom AI. Radiologic imaging and intervention for gastrointestinal and hepatic complications of hematopoietic stem cell transplantation. Radiology. 2011;258:660–71.
Ruutu T, Eriksson B, Remes K, Juvonen E, Volin L, Remberger M, Parkkali T, Hägglund H, Ringdén O. Ursodeoxycholic acid for the prevention of hepatic complications in allogeneic stem cell transplantation. Blood. 2002;100:1977–83.
Imran H, Tleyjeh IM, Zirakzadeh A, Rodriguez V, Khan SP. Use of prophylactic anticoagulation and the risk of hepatic veno-occlusive disease in patients undergoing hematopoietic stem cell transplantation: a systematic review and meta-analysis. Bone Marrow Transplant. 2006;37:677.
Azoulay D, Castaing D, Lemoine A, Hargreaves GM, Bismuth H. Transjugular intrahepatic portosystemic shunt (TIPS) for severe veno-occlusive disease of the liver following bone marrow transplantation. Bone Marrow Transplant. 2000;25:987.
Song W, Zhao D, Li H, Ding J, He N, Chen Y. Liver findings in patients with hereditary hemorrhagic telangiectasia. Iran J Radiol. 2016;13:e31116.
Faughnan ME, Palda VA, Garcia-Tsao G, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48:73–87.
Garcia-Tsao G. Liver involvement in hereditary hemorrhagic telangiectasia (HHT). J Hepatol. 2007;46:499–507.
Blewitt RW, Brown CM, Wyatt JI. The pathology of acute hepatic disintegration in hereditary haemorrhagic telangiectasia. Histopathology. 2003;42:265–9.
Schelker RC, Barreiros AP, Hart C, Herr W, Jung E-M. Macro- and microcirculation patterns of intrahepatic blood flow changes in patients with hereditary hemorrhagic telangiectasia. World J Gastroenterol. 2017;23:486–95.
Buscarini E, Leandro G, Conte D, et al. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic telangiectasia. Dig Dis Sci. 2011;56:2166–78.
Wu JS, Saluja S, Garcia-Tsao G, Chong A, Henderson KJ, White RI. Liver involvement in hereditary hemorrhagic telangiectasia: CT and clinical findings do not correlate in symptomatic patients. Am J Roentgenol. 2006;187:W399–405.
Buscarini E, Plauchu H, Garcia Tsao G, et al. Liver involvement in hereditary hemorrhagic telangiectasia: consensus recommendations. Liver Int. 2006;26:1040–6.
Khalid SK, Garcia-Tsao G. Hepatic vascular malformations in hereditary hemorrhagic telangiectasia. Semin Liver Dis. 2008;28:247–58.
Dupuis-Girod S, Ginon I, Saurin J, et al. Bevacizumab in patients with hereditary hemorrhagic telangiectasia and severe hepatic vascular malformations and high cardiac output. JAMA. 2012;307:948–55.
Gallego C, Miralles M, Marín C, Muyor P, González G, García-Hidalgo E. Congenital hepatic shunts. Radiographics. 2004;24:755–72.
Papamichail M, Pizanias M, Heaton N. Congenital portosystemic venous shunt. Eur J Pediatr. 2018;177:285–94.
Kim MJ, Ko JS, Seo JK, Yang HR, Chang JY, Kim GB, Cheon J-E, Kim WS. Clinical features of congenital portosystemic shunt in children. Eur J Pediatr. 2012;171:395–400.
Murray CP, Yoo S-J, Babyn PS. Congenital extrahepatic portosystemic shunts. Pediatr Radiol. 2003;33:614–20.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Self Study
Self Study
1.1 Questions
-
1.
Which of the following diseases are not treated with anticoagulants?
-
A.
Budd-Chiari Syndrome
-
B.
Acute Portal Vein Thrombosis
-
C.
Sinusoidal Obstruction Syndrome
-
D.
Chronic Portal Vein Thrombosis
-
E.
Splanchnic Vein Thrombosis
-
A.
-
2.
Which statement is true?
-
A.
In Haemorrhagic Hereditary Telangiectasia angio-MRI is the only imaging technique which can give a severity grading of liver vascular malformations which correlates with clinical outcome.
-
B.
The diagnosis of Sinusoidal Obstruction Syndrome is based on CT or liver biopsy.
-
C.
Genetic testing can be made to establish the diagnosis in patients with Budd-Chiari Syndrome.
-
D.
The diagnosis of Chronic Portal Vein Thrombosis requires angiography.
-
E.
Liver transplantation is the only definitive curative option for liver vascular malformations in Haemorrhagic Hereditary Telangiectasia
-
A.
1.2 Answers
-
1.
Which of the following diseases are not treated with anticoagulants?
-
Answer: C
-
-
2.
Which statement is true?
-
Answer: E.
-
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vintilă, V.D., Chitroceanu, A.M., Vintilă, AM. (2020). Vascular Disorders of the Liver. In: Radu-Ionita, F., Pyrsopoulos, N., Jinga, M., Tintoiu, I., Sun, Z., Bontas, E. (eds) Liver Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-24432-3_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-24432-3_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-24431-6
Online ISBN: 978-3-030-24432-3
eBook Packages: MedicineMedicine (R0)