
Abstract
Metabolomics is the analysis of endogenous and exogenous low molecular mass metabolites within cells, tissues, or biofluids of an organism in response to an external stressor. In this chapter, we highlight the importance of the subdiscipline of environmental metabolomics, which investigates the interactions of organisms with environmental stressors such as biotic and abiotic factors, xenobiotics, temperature shifts, and chemical contaminants. Over the past decade, there has been increasing scientific interest in environmental metabolomics, most likely attributable to the comprehensive nature of nontargeted metabolomics. Hypotheses have therefore been developed on complex environmental stressor effects, especially those with unknown modes of action. The availability of a wide variety of model organisms such as freshwater organisms of the food chain has promoted the potential of metabolomics to detect stress from an extensive range of external factors. Furthermore, these dynamics may shift from individuals to populations, contemplating the traditional fields of the ecophysiology and ecology from instantaneous effects to those over evolutionary timescales. In this chapter, we provide an overview of analytical instrumentation, extraction methods, general experimental design, and the statistical methods generally used in environmental metabolomics. Despite the difficulty in understanding the consequences of environmental exposure due to inter- and intra-individual variability, we believe that environmental metabolomics may enrich our understanding of the responses of organisms to the numerous types of environmental stressors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Great scientific change in molecular biology and biochemistry approaches and techniques has begun since the sequencing of human genome in the 1990s. Automated micro-array methods to detect changes in gene expression and the ability to assay and identify proteins by mass spectroscopy methods led respectively to two new revolutionary disciplines: transcriptomics and proteomics which are useful for the comprehension of the complex interactions between genetic make-up and environmental factors. It is crucial to underline that the small molecules involved in biochemical processes provide a wide range of information on the status of living systems when studying changes in genes expression due to every variation in life conditions and external perturbations. The process of monitoring and evaluating such changes is termed “metabonomics” or “metabolomics” when mostly model organism or plant system are studied (Lindon et al. 2006).
The use of NMR spectroscopy combined with multivariate statistic investigation of the complex data sets led to the following definition: “the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification” by Jeremy Nicholson and colleagues (1999).
The metabolites, or small molecules, within a cell, tissue, organ, biological fluid, or entire organism constitute the metabolome (Miller 2007). Metabolomic analyses can be categorized as either nontargeted or targeted (Issaq et al. 2008; Verpoorte et al. 2008). Nontargeted metabolomics is a nonbiased quantitative analysis of all—or a large number of—metabolites found in a biological sample (Issaq et al. 2008). By contrast, targeted metabolomics analyzes a specific group of metabolites (Issaq et al. 2008; Verpoorte et al. 2008).
The complementary role of metabolomics in regard to other omic techniques may actually provide a potential solution to the many weak points that are encountered with other omic methods (Griffin and Bollard 2004; Bilello 2005; van Ravenzwaay et al. 2007). Indeed, despite the development of methods to detect changes in genomic, transcriptomic, and proteomic profiles, key information needed to make significant interpretations based on these data are usually not promptly available (Ankley et al. 2006). Changes in gene expressions and protein synthesis due to external stressor exposure of an organism usually cause the activation of homeostatic controls and feedback mechanisms; these changes could be intensified at the metabolome level (Nicholson et al. 2008; Ankley et al. 2006; van Ravenzwaay et al. 2007). As a result, metabolomics could be considered a more sensitive and reliable indicator of the external stress than other omic technologies (Nicholson et al. 2008; Ankley et al. 2006; van Ravenzwaay et al. 2007).
The popularity of metabolomics in many fields of scientific research like nutrition, medicine, clinical pharmacology, and toxicology has grown considerably thanks to the ability to detect subtle molecular changes and the comprehensive nature of metabolite measurements (Lin et al. 2006; Chen et al. 2008; Fialho et al. 2008). As a result, metabolomics is nowadays considered a rapid and sensitive technique that would be able of clear up relationships between metabolite levels and an external stressor, be it contaminant exposure, nutritional deficit, or disease (Miller 2007). For example, cancer cells’ metabolic profiles have been used to monitor and comprehend tumor progression (Griffiths et al. 2002; Griffiths and Stubbs 2003; Griffin and Shockcor 2004). Metabolomic investigations in non-model organisms will be particularly exciting in the characterization of organism metabolic responses to anthropogenic (or manmade) stressors, such as pollutants and climate changes, highlighting another important application of the environmental metabolomics. This approach would utilize “sentinel” (or representative) organisms of a particular ecosystem to reveal the condition of the environment (Viant 2008b).
In the last decades, in order to understand the toxic effects of several kinds of xenobiotic residues (pharmaceuticals, pesticides, nanoparticles, heavy metals) in the environment and the related biological changes, various nontarget organisms of the aquatic food chain have been selected as bioindicators for their suitable characteristics. In fact, invertebrates such as bivalve molluscs Mytilus edulis and Mytilus galloprovincialis (Tuffnail et al. 2009; Fasulo et al. 2012; Bonnefille et al. 2018), crustaceans Daphnia magna and Gammarus pulex (Taylor et al. 2010; Gómez-Canela et al. 2016; Kovacevic et al. 2016; Nagato et al. 2016; Wagner et al. 2017) and vertebrates like fish such as Danio rerio and Oncorhynchus mykiss (Samuelsson et al. 2006; De Sotto et al. 2017) represent a valid tool to study the ecotoxicogenomics and the metabolomics. These bioindicators are chosen for these kinds of studies because: they are important components of the aquatic ecosystem, are easy to recognize, are sensitive to a wide range of stressors, are abundant and widely distributed, and are suitable for laboratory experiments. In addition, these aquatic nontarget organisms have been utilized by different scientific environmental researchers (Brezovšek et al. 2014; Parrella et al. 2014, 2015; Isidori et al. 2016a; Kundi et al. 2016; Russo et al. 2018; Kovács et al. 2015, 2016; Gačić et al. 2014) through specific tests according to international standard guidelines not only for highlighting physiological alterations (mortality, offspring reduction, inhibition of growth, malformations) but also for evaluating genotoxic, mutagenic and teratogenic damages. Since there is not a clear relationship between biochemical mode of action of xenobiotics and defined endpoints such as mortality and reproduction, a detection of the metabolic profile in aquatic sentinel organisms may be of great scientific interest. In fact, significant variations in amino acids, in glucose and other metabolite concentrations in cells, tissues, or biofluids (Kovacevic et al. 2016; Wagner et al. 2017) may occur in organisms after sub-lethal exposure to hazardous and pseudo-persistent contaminants like pharmaceuticals. Indeed, among all contaminants, pharmaceuticals are frequently detected in aquatic ecosystems because of their high consumption and scarce removal efficiencies by wastewater treatment plants (Negreira et al. 2014; Lenz et al. 2007; Isidori et al. 2016b).
Among pharmaceuticals, antineoplastic drugs are suspected to be hazardous for aquatic nontarget species (Parrella et al. 2014, 2015; Isidori et al. 2016a; Kundi et al. 2016; Russo et al. 2018). To the best of our knowledge, there are no studies in literature regarding the overall metabolomic responses in different nontarget organisms exposed to antineoplastic drugs, while only few studies (Table 18.1) have utilized metabolomics as a robust tool to evaluate the biological environmental responses in the aquatic sentinel species exposed to other pharmaceutical classes. On the other hand, in recent studies (Laith et al. 2017; Mumtaz et al. 2017; Ruiz-Torres et al. 2017), metabolomic approach has been considered a good strategy to identify, select, and provide secondary metabolites from natural promising sources, such as plants and marine invertebrates and vertebrates, as new drugs for cancer therapy, rather than using metabolomics for studying environmental biological effects.
In this chapter, we provide an overview of the experimental design, analytical techniques, and statistical methods used in environmental metabolomics as well as an overview of recent studies using aquatic nontarget organisms in metabolomics to demonstrate the potential of this technique to detect and understand the mechanisms of exposure to some pharmaceuticals.
2 Experimental Design and Analytical Methods Used in Environmental Metabolomics
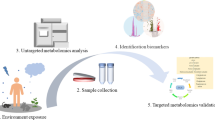
The basic procedure used in an environmental metabolomics study is outlined in Fig. 18.1. Furthermore, an accurate outline of the entire metabolomic experimental scheme from the experimental design, to data mining and biological interpretation is described by Wolfender et al. (2013).

Environmental metabolomics experiment workflow (from Simpson and McKelvie 2009; Lankadurai et al. 2013). NMR, nuclear magnetic resonance spectroscopy; MS, mass spectrometry; PCA, principal component analysis; PLS-regression, partial least-squares regression analysis. In some cases, a further step might be present between the design of experiment and the sampling, consisting in the experimental performance when the experiment has to be performed in laboratory controlled conditions
The first step in any metabolomics experiment is the experimental design, which, in case of environmental metabolomics, involves the selection of a model organism or microorganism, a type of external stressor (e.g., exposure to contaminants, heat/cold, starvation, or disease), and the mode/route of exposure. A good experimental design mainly depends on the starting point and goals of the research. Focusing on the biological question underpinning the research is the most crucial step, as metabolomics usually takes into account a large number of samples (Brown et al. 2008). Taking into consideration the biological variability and choosing a suitable number or replicates from an early stage of the research is also compelling, for organisms grown in either natural or controlled conditions.
When planning and carrying out the experiment, it is extremely important to consider that metabolism is highly dynamic and changes occur at different timescales, depending on the organism, and on the metabolic pathway considered. For example, some changes might occur according to the developmental stage or phenology (Scognamiglio et al. 2014) while others to circadian clock (Eckel-Mahan et al. 2012), with the results that the first changes can only be observed on a longer timescale, while the second ones are responsible for cyclic changes in metabolites’ concentration during the light/darkness alternation. It has to be specified that there are many other physiological reasons for metabolic alterations that also depend on the organism (and on the biological medium) taken into consideration. Therefore, this dynamic feature has to be borne in mind when planning timing for treatment, sampling, and so on, in order to make sure to detect and discriminate the changes caused by the external perturbation from the ones due to physiological changes in the metabolism. Taking into consideration the two parameters previously mentioned (developmental stage and circadian clock), for example, it is crucial to collect samples at the same moment of the day and at the same organism growth stage. Monitoring environmental variations (e.g., photoperiod, relative humidity, temperature) throughout the progress of the experiment is important as well.
Once the organism has been exposed to the external stressor, the biological medium to study will be harvested and may include blood, urine, or other biological fluids, and (or) tissue/organ extracts (Simpson and McKelvie 2009). Quenching metabolism immediately after exposure is essential to minimize the influence of puzzling variables in the analysis of the metabolic response (Lin et al. 2006). This is commonly done by flash freezing with liquid nitrogen and storing at −80°C. However, in order to improve precipitation and inactivation of soluble enzymes, acid treatment, or extraction with cold mixtures of organic solvents such as methanol, ethanol, acetone, or acetonitrile might be used (Lin et al. 2006). When suitable, also freeze drying of the samples is a good practice (Kim and Verpoorte 2010).
The further step is the choice and development of a suitable extraction method. Considering the high variability and complexity of matrixes to be tested, sample extraction and preparation methods can vary a lot depending on the matrix to be analyzed and on the analytical platform used (Kim and Verpoorte 2010). As no single extraction method can isolate every metabolite within a sample, a proper extraction procedure will need to be selected and tested, based on the goals of the experiment. In general, an aqueous buffer extraction is sufficient to obtain a polar metabolite profile, but a more rigorous extraction involving a mixture of polar and nonpolar solvents is required to extract both polar and nonpolar metabolites (Wu et al. 2008). Various extraction methods involving a mixture for organic solvents (methanol, chloroform, and acetonitrile) and water in varying ratios have been examined by Lin et al. (2006). The methanol/chloroform/water (final solvent ratio of 2/2/1.8, respectively) extraction method, which was first described by Bligh and Dyer (1959), has been considered one of the most reproducible and with the highest recovery of both polar and nonpolar metabolites. Wu et al. (2008) then went a step further, and examined three different strategies to add the methanol, chloroform, and water to the tissue samples for extraction: (i) stepwise addition—the original Bligh and Dyer (1959) method of adding each solvent one by one; (ii) two-step addition—methanol and water are added in step one and chloroform and water are added in step two; (iii) all-in-one addition—all three solvents are added together. They stated that the two-step addition was the best out of the three: this assessment is based on metabolite yield, extraction reproducibility, and sample throughput. Recently, Liebeke and Bundy (2012) compared four different solvent systems for the extraction of metabolites from the tissue of the earthworm Lumbricus rubellus: (i) chloroform: methanol: water, 2:1:1 (CMW); (ii) 75% hot ethanol (hEt); (iii) acetonitrile:methanol:water, 2:2:1 (AMW); (iv) isopropanol:methanol: water, 2:5:2 (IMW). Extracts were analyzed using NMR, gas chromatography (GC)–MS and Fourier transform ion cyclotron resonance (FT-ICR) MS. They determined that the AMW extraction gave the best results in terms of reproducibility and good yield for metabolite extraction (Liebeke and Bundy 2012). It has furthermore been shown that in case of plant material, a mixture of phosphate buffer and MeOH (1:1) is usually able to extract big part of the metabolome (Kim and Verpoorte 2010).
Also, the presence of proteins binding the typical used NMR internal standards 2, 2 dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) and sodium 3-trimethylsilyltetradeuteriopropionate (TSP) in aqueous buffer extracts has led to large differences in the quantification of metabolite concentrations (Nowick et al. 2003). Consequently, the development of extraction methods that not only capture both polar and nonpolar metabolites but also include the precipitation of proteins is essential. Nevertheless, one of the main problems of metabolomics is still the lack of a standardized and reproducible extraction protocol, so a great effort should be made in this direction.
Once the extractions are completed, the samples need to be prepared for the analytical platform of choice. A list of the available techniques and of the viewpoints on their advantages and disadvantages in metabolomics applications have been extensively discussed in the literature (Table 18.2) (Scognamiglio et al. 2015). It is important to emphasize that the choice of the analytical platform and strategy heavily depends on the object of the research, and it is commonly acknowledged that the better results can be achieved combining different extractions and analytical measurements (Allwood et al. 2008; Kim and Verpoorte 2010). However, nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS) are doubtlessly considered the most powerful tools and the ones that will be here described more in detail. NMR and MS use in metabolomics is so widespread that in the past there was a common misconception that “metabonomics” dealt with NMR-derived metabolic profiling studies, while “metabolomics” dealt with MS-derived metabolic profiling studies (Robertson 2005).
In MS, the analysis is done after the fragmentation of the molecules, which this leads to the generation of ions that are later separated by their mass-to-charge ratio and finally analyzed by a detector. A number of ion sources and of analyzers is available (Xiao et al. 2012). Direct injection MS enables the injection of a crude extract directly into an electrospray mass spectrometer, resulting in one spectrum per sample, but this method is not particularly quantitative and not often used (Lin et al. 2006).
MS use in metabolomics is usually coupled to liquid chromatography (LC; Wu et al. 2005; Bajad and Shulaev 2007; Hughes et al. 2009), gas chromatography (GC; McKelvie et al. 2009; Flores-Valverde et al. 2010; Warren et al. 2012), or capillary electrophoresis (CE; Sato et al. 2004; Ramautar et al. 2012; Yamamotoya et al. 2012). These chromatographic techniques separate the complex sample mixtures so that they can be analyzed by MS, but this can make the overall analysis time consuming (Robertson 2005; Pan and Raftery 2007). Also, GC methods usually entail elaborate derivatization steps that are very lengthy and thus inconvenient for high-throughput analysis (Lin et al. 2006), besides introducing bias in the analysis due to the used chemical reactions. However, the combination of MS with chromatography, when there is availability of pure certified chemical standards, is a useful approach for identification and quantification. Indeed, retention time can be considered as an additional hint of metabolite identity, while the chemical standards are used also to set up proper external calibration curves for compounds quantification (Allwood et al. 2008).
Mass fragment databases are usually employed for easy preliminary identification of compounds (Noctor et al. 2007; Pan and Raftery 2007). Only the use of tandem MS (MS-MS), preferably HR (MS)n instruments allows for structural definition of compound identities. This usually excludes the countless, to date unknown metabolites, which are reported as “unknowns” or as putatively identified metabolites. In this case, isolation and characterization by NMR is fundamental to definite structural elucidation (Sumner et al. 2007).
The newest LC-MS/MS approaches are a useful tool for metabolite identification and quantification (Xiao et al. 2012), although it is always recommended to follow published guidelines and to refer to minimum reporting standards for the level of confidence of chemical structural elucidation (Fernie et al. 2011; Goodacre et al. 2007; Sumner et al. 2007).
The biggest advantage of MS-based techniques is sensitivity (typically picogram level), making the technique very suitable in studies that are targeting novel biomarkers (Robertson 2005; Pan and Raftery 2007) but, on the other hand, the identification of unknown metabolites is problematic. FT-ICR MS in particular provides high resolution and mass accuracy, but the instrument is costly and is thus not widely used (Pan and Raftery 2007).
The principal downside of MS-based approaches is their difficult standardization, as a consequence of a number of combinations of chromatographic systems (the separation step also includes bias in the analysis), ion sources, and different analyzers that highly impact the analysis output.
Beyond the potentially long and not always reproducible chromatographic separations, the difficult standardization, and the structural elucidation power limitation, other drawbacks of MS include: the presence of matrix effects, the destructive nature, its selectivity for certain analytes, ion suppression causing extensive variations of signal intensities, and the lack of more robust methods for chromatographic separations (Robertson 2005; Pan and Raftery 2007).
Often, due to its selectivity, MS has been used in targeted metabolomics studies (Edwards et al. 2006; Lutz et al. 2006; Issaq et al. 2008; Southam et al. 2011; García-Cañaveras et al. 2012) or in the elucidation or confirmation of metabolites first observed by NMR (Bundy et al. 2002a; Crockford et al. 2006).
On the other hand, NMR is nondestructive, nonselective, possesses cross-laboratory reproducibility, and lacks sample bias (Robertson 2005; Pan and Raftery 2007; Viant et al. 2009). As a result, NMR has been used extensively in the nontargeted or comprehensive studies of all or most of all the metabolites in a biological sample (Verpoorte et al. 2008; Simpson and McKelvie 2009; Whitfield Åslund et al. 2011a, b; Li et al. 2012; Ritota et al. 2012).
NMR is an instrumental analytical technique that allows obtaining detailed information on the structure of molecules by observing the behavior of atomic nuclei in a magnetic field. The frequency at which each nucleus resonates depends on its chemical surrounding environment so each compound has a highly specific spectrum, which is an information-rich graph. Rapid identification of all of the compounds present in a mixture can be performed thanks to the combination of one-dimensional (1D) and two-dimensional (2D) NMR techniques. Recent advances in the identification of unknown compounds in the analyzed mixtures allow NMR to obtain important structural information without requiring further purification (Forseth and Schroeder 2011). Indeed, one of the main strengths of this technique is the unique set of structural information furnished that in most cases guarantees the definitive structural elucidation of the compounds, sometimes including stereochemistry.
Besides its power in structural elucidation, 1H NMR is commonly used in metabolomics thanks to several other advantages: easy sample preparation, ease of standardization, and high reproducibility, and the solvent used and the magnetic field strength being the only variables (Verpoorte et al. 2007). Indeed, in NMR-based metabolomics the only bias is introduced by the solvent choice (Allwood et al. 2008) and the reproducibility appears very good and allows a comprehensive identification and quantification of a large number of compounds with short analytical times (including the extraction procedures). The need of a standardization procedure for the metabolome extraction previously mentioned makes NMR-based metabolomics convenient thanks to minimum sample preparation. In fact, extraction can be carried out directly in deuterated solvents, often with a mixture of phosphate buffer in D2O and MeOD (1:1). Furthermore, NMR is fully quantitative (Kim et al. 2011; Wishart 2008). The calculated precision and accuracy of a 500 MHz NMR spectrometer in a quantitative 1H NMR analysis of external standards has been demonstrated to be around 1% (Burton et al. 2005).
The main disadvantage of NMR, compared to MS, is its low sensitivity. This is an issue in the analyses of endogenous metabolites and in particular detecting novel biomarkers. Indeed, these metabolites are usually present at very low levels that cannot be reached with NMR. The sensitivity of 1H NMR is also dependent on the number of protons in the molecule, the structure and size of the molecule. Nowadays, the problem of low sensitivity can be attenuated using ultrahigh magnetic field strength NMR spectrometers and probes that are cryogenically cooled to 4.5 K; these probes may result in a four-times raise in sensitivity (Logan et al. 1999; Griffin 2003; Pan and Raftery 2007; Grimes and O’Connell 2011) reaching the stage at which structures can be solved using very small quantities (in the microgram range) (Harvey et al. 2015). The other advantage of microcoil probe use is also the lower sample mass requirements, which is a big benefit for small organisms (Lacey et al. 1999; Pan and Raftery 2007; Grimes and O’Connell 2011; Poynton et al. 2011). The only disadvantage with these methods is the affordability of such high-end instrumentation and although sensitivity has been drastically increased, NMR is still surely less sensitive when compared to mass spectrometry (Forseth and Schroeder 2011; Kim et al. 2011). Nonetheless, compared to MS, the sensitivity of NMR is independent from metabolite pKa or hydrophobicity, making it a very adaptable choice for representative analyses (Pan and Raftery 2007). An additional downside of NMR is that some classes of lipids can only be identified as total groups and not as individual compounds by means of 1D NMR.
A large part of environmental metabolomics studies still use NMR because of the comprehensive nature of nontargeted metabolomics and the ability to generate hypotheses involving complex environmental stressors for which there are no known modes of action (Bundy et al. 2001; Tjeerdema 2008; Viant 2008a; Viant et al. 2009; Brown et al. 2009; Simpson and McKelvie 2009). For all these reasons, NMR is an ideal environmental metabolomics discovery tool. It should be noted that once metabolites of interest are discovered using NMR, targeted MS-based methods can be subsequently developed for the routine monitoring of these metabolites.
The majority of NMR-based metabolomics studies still use one-dimensional (1D) 1H NMR experiments (Bundy et al. 2002b, c; Samuelsson et al. 2006; Brown et al. 2010; McKelvie et al. 2010, 2011). 1D 1H NMR experiments are advantageous for metabolomic studies, which usually have hundreds of samples, because of their short acquisition times (10–15 min per sample), allowing for high-throughput analyses and a high number of sample replicates (Pan and Raftery 2007; Yuk et al. 2010).
Analyzing aqueous samples using 1H NMR requires the application of water suppression techniques (Nicholson and Wilson 1989; McKay 2009). Water concentration in samples is much higher (50 mol/L) compared to the millimolar metabolite concentrations, leading to the suppression of signal intensities in the peaks of other compounds due to saturation of the NMR receiver by the H2O signal (Bothwell and Griffin 2011). While deuterated solvents are mostly used (deuterium resonates at a different frequency than 1H in the NMR), there is always residual H2O present in samples. The best and most used water suppression methods for metabolomics are presaturation, nuclear overhauser effect spectroscopy (NOESY) presaturation, and presaturation utilizing relaxation gradients and echoes (PURGE; Bundy et al. 2002a; Viant et al. 2003; Simpson and Brown 2005; Wishart 2008; McKelvie et al. 2010; Poynton et al. 2011). Several studies have used PURGE water suppression for NMR-based metabolomic analyses (McKelvie et al. 2011, 2013; Yuk et al. 2011, 2013). Among the several water suppression techniques, PURGE provided superior water suppression with the least amount of parameter optimization and the fewest number of spectral regions that need to be excluded because of variations in the suppression of the solvent peak. Furthermore, comparing various 1D and 2D NMR techniques, PURGE 1H NMR has demonstrated to be the most rapid, informative, and economic method for analyzing aqueous metabolomics samples (McKay 2009; Yuk et al. 2010).
The complexity of any biological sample due to the large number of molecules that they possess, results in a large number of peaks within the small chemical shift range of a 1H NMR spectrum (0–14 ppm). This leads to difficulty in identifying compounds that are present at low concentrations, considering the common chance of peak overlapping generated by different metabolites. This usually means that some peaks are masked by larger peaks from compounds present in higher concentrations (Pan and Raftery 2007). Some of the best techniques to alleviate the spectral overlap and improve resolution between peaks are: Carr–Purcell–Meiboom–Gill (CPMG), J-resolved spectroscopy (J-RES), and other various 2D NMR techniques.
Carr–Purcell–Meiboom–Gill (CPMG) is used to remove broad resonances associated with molecules of high molecular mass or molecules with constrained motion and hereby offers better resolution of low molecular mass metabolites (Weckwerth 2007; Wishart 2008).
J-resolved spectroscopy (J-RES) projection may improve spectral resolution. J-RES is a two-dimensional (2D) NMR technique, in which the chemical shift information is on one axis and the spin–spin coupling information is on another. Projecting only the chemical shift axis, an equivalent to a 1D proton decoupled spectrum is obtained, which has less spectral overlap and enables better detection of specific metabolites (Viant et al. 2003; Pan and Raftery 2007; Yuk et al. 2010), although it also results in the loss of some information.
Other 2D NMR techniques have also been used to increase spectral resolution because they have an additional dimension into which the signals can be dispersed. Besides J-RES, some of the other common 2D NMR techniques in metabolomics are 1H correlation spectroscopy (COSY) and 1H-13C heteronuclear single quantum coherence (HSQC) spectroscopy (Xi et al. 2008; Ludwig and Viant 2010; Yuk et al. 2010; Flores-Sanchez et al. 2012). The main benefit of using 2D NMR techniques, such as HSQC, is that the 13C axis has a large chemical shift range (200 ppm), which allows for greater spectral dispersion and enhanced resolution (Xi et al. 2008; Chylla et al. 2011; Hu et al. 2011a, b). However, a drawback of most 2D NMR techniques is the lower sensitivity, which results in longer acquisition times—sometimes many times more than 1D experiments (Jacobsen 2007). In fact, 2D experiments such as HSQC may require long acquisition times for adequate signal-to-noise (S/N) ratios. For this reason, the use of 2D NMR techniques in metabolomic studies is limited to complementing compound identification from 1D 1H NMR experiments (Yuk et al. 2010).
After the samples are analyzed, the data are processed and statistical analyses are performed using multivariate and univariate analyses. These are done in conjunction with the quantification and identification of the metabolites.
The final step then involves biological interpretation of the data to make a connection between the external stressor and the metabolic response of the organism.
3 Statistical Methods Used in Metabolomics
The interest in metabolomics is due to its ability to generate large volumes of data in a high-throughput way, so one of the biggest challenges is to find a way to visually analyze all of the collected data (i.e., NMR or MS data) to identify differences between samples in a reasonable time frame (Robertson 2005). Both, multivariate statistical and pattern recognition methods are employed to smooth the analysis of metabolomics data sets and to obtain meaningful relationships between the external stressor and the metabolic response (Trygg et al. 2007; Coen et al. 2008). Pattern recognition methods are able to reduce the dimensionality of metabolomics data from hundreds of variables into two or three components that are orthogonal to each other (Trygg et al. 2007).
PCA is an unsupervised method, meaning that the model is not provided with any prior information concerning the identity of the samples (Holmes and Antti 2002), and is the most widely used multivariate statistical approach in metabolomics (Bundy et al. 2002a, 2004; Trygg et al. 2007; Wishart 2008; Simpson and McKelvie 2009). The association of the samples in a PCA scores plot is based on the similarities in their metabolic profiles. PCA figures out the comprehensive variability in a data set, which is explained by a set of uncorrelated variables called principal components (PCs); these are linear combinations of the original variables (Trygg et al. 2007). The first PC (PC1) explains most of the variation in the data and PC2, which is orthogonal to PC1, explains the second most variation in the data and so on. PCA allows for dimensional reduction of the data into a low-dimensional plane, such as PC1 versus PC2. The scores plot (e.g., PC1 vs. PC2) allows for a visual examination of the relationship between the samples based on their metabolic profiles.
PLS regression analysis and PLS discriminant analysis (PLS-DA) are also used often as multivariate statistical tools in metabolomics (Barker and Rayens 2003; van Ravenzwaay et al. 2007; Ekman et al. 2008; Jones et al. 2008; Whitfield Åslund et al. 2011b). PLS-regression and PLS-DA are supervised methods. In this case, the classification of the samples as either control or experimental is known to the model. Predictive models are built adding predefined variables to maximize the separation between the sample classes. These variables are measurable quantities such as the contaminant exposure concentration. In PLS-DA these are dummy variables: for example, we can assign all the controls a value of zero and the experimental group a value of one to distinguish the sample classes (Trygg et al. 2007).
In order to reduce models’ components, make it easier to interpret and more relevant, supervised methods such as orthogonal projections to latent structures (OPLS) and OPLS-DA have been increasingly used in metabolomics studies (Trygg and Wold 2002). These are basically extensions of PLS and PLS-DA where the orthogonal variation to the predefined variables is removed from the model (Trygg and Wold 2002; Bylesjö et al. 2006), but could also be analyzed together with the identification of the uncorrelated variables sources.
Cross-validation methods such as the leave-one-out cross validation (LOOCV) are required to evaluate the robustness of the supervised methods such as PLS-regression, PLSDA, OPLS, and OPLS-DA (Westerhuis et al. 2008; Varmuza and Filzmoser 2009; Whitfield Åslund et al. 2011b). LOOCV is performed by first randomly eliminating one of the samples from the original data set, which is called the test set, then the model (PLS/PLS-DA or OPLS/OPLS-DA) is built on the remaining samples (the training set). This process is repeated until all of the samples have been left out of the model at least once. The training set models created are eventually used to predict the test set. The Q2Y, which is known as the goodness of prediction (Westerhuis et al. 2008), represents the ability of the model to predict the test set. This value can be used to evaluate the robustness of a model: typically, a Q2Y > 0.4 is considered a strong model (Jones et al. 2008; Westerhuis et al. 2008). The significance of PLS/OPLS models needs to be evaluated, and this can be done using permutation testing (Eriksson et al. 2006; Alam et al. 2010; Whitfield Åslund et al. 2011b). Permutation testing consists of maintaining the data set constant, while randomly permuting the order of the predefined variables a set number of times. For each permutation a new PLS/OPLS model is fitted, and the Q2Y is calculated providing a reference distribution of the Q2Y statistic. The significance of the original PLS/OPLS model and the confidence in its validity is increased if its Q2Y value is higher than the values obtained for all the PLS/OPLS models built during the permutation tests (Eriksson et al. 2006).
Although, metabolomics studies mostly use multivariate statistics, complementary univariate statistical analyses are also attended to further increase the amount of information obtained from the research. T tests are commonly used to assess the significance of the separation between the controls and stressed organisms in PCA and PLS-DA scores plots, and to define which metabolites in the 1H NMR spectra of the treatment class increased or decreased significantly relative to the controls. A T test filtered difference 1H NMR spectra can also be created by subtracting the buckets of the average controls from that of each average exposure class. Not statistically significant (= 0.05) bucket values metabolite peaks can be replaced with a zero, resulting in a T test filtered 1H NMR difference spectrum (Ekman et al. 2008, 2009). T test filtered difference 1H NMR spectra and loading plots can be used together to determine which metabolites are potential biomarkers of exposure to a particular contaminant.
4 Metabolomic Responses Observed in Aquatic Nontarget Organisms Exposed to Pharmaceuticals
Despite the immense potential of metabolomic research for assessing environmental pollutants, only a small number of studies have been conducted till now to evaluate the metabolomic responses observed in various aquatic nontarget organisms exposed to pharmaceuticals. In fact, Bonefille et al. in 2018, evaluated the effects of the nonselective, nonsteroidal anti-inflammatory drug diclofenac against the marine Mytilus galloprovincialis, chosen for its ease in being handled, for its capability in accumulating toxins, and for its sedentary nature. In this mussel, these authors studied metabolomic perturbations caused by 100 μg/L diclofenac, concentration not affecting organisms’ viability; then, the metabolomic analysis was performed by liquid chromatography-hyphenated to high-resolution mass spectrometry (LC-HRMS) in extracts of digestive gland, and alterations in the tyrosine and tryptophan metabolisms were observed at concentrations only few orders of magnitude higher than those found in seawater (1 μg/L, Gaw et al. 2014). In particular, after a 7-day exposure, tyrosine pathways were down-modulated, while steroid hormone biosynthesis and tryptophan pathways were positively modulated.
In addition to mussels, other nontarget invertebrates such as crustaceans were suitable tools for metabolomic analysis. In fact, Taylor et al. (2010), Kovacevic et al. (2016), and Wagner et al. (2017) studied metabolomic responses in the cladoceran crustacean Daphnia magna after exposure to various pharmaceuticals. In particular, Taylor and coauthors, in 2010, explored D. magna metabolic changings after 24 h exposure to 1.4 mg/L of the nonselective β-adrenergic receptor blocker propranolol by direct infusion Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS). High-quality metabolite profiles were detected both in hemolymph and in the whole-organism extracts from 14-day-old daphnids and metabolic perturbations were found in the multiple fatty acid and oxylipid metabolites. Wagner et al. (2017) performed a similar study testing 0.67 mg/L of propranolol in both neonates (<24 h old) and adult daphnids (18 days old) by nuclear magnet resonance spectroscopy 1H-NMR observing in both populations an increase in amino acid metabolites and a reduction in glucose levels when compared to control. Furthermore, Kovacevic et al. (2016) studied the metabolic profile in the same freshwater crustacean testing three different pharmaceuticals: triclosan (μg/L), carbamazepine, and ibuprofen (mg/L), at sublethal concentrations after 48 h exposure. Triclosan is used for impeding bacterial growth by inhibiting enzymes involved in fatty acid synthesis, carbamazepine is a sodium channel blocker used for the treatment of epilepsy and neuropathic pain for its effects on serotonin systems, while ibuprofen is a nonsteroidal anti-inflammatory drug inhibiting cyclooxygenase enzymes involved in prostaglandins synthesis. Kovacevic and coauthors analyzed adult daphnid metabolites by 1H-NMR, and alterations in amino acid content as well as in sugar glucose levels were observed according to a concentration-dependent relationship between daphnids’ metabolic responses and drug exposure concentrations, reflecting changes at organ, organismal, and population levels. In light of the foregoing, the freshwater consumer D. magna seems to be a very sensitive aquatic bioindicator for the evaluation of the metabolomic responses to many environmental pollutants also considering the advances used in analytical metabolomic techniques.
Other nontarget sentinel species belonging to a higher level of the food aquatic chain are represented by marine and freshwater fish. These organisms were among the earliest organisms used in environmental metabolomics thanks to their similar biochemical mechanisms in comparison to humans, in response to pharmaceuticals. In fact, in 2006, Samuelsson et al., using 1H-NMR, studied the effects of the synthetic contraceptive estrogen ethinylestradiol on the rainbow trout Oncorhynchus mykiss (11 months old) and observed alterations in the plasma metabolite profile at 10 ng/L with a strong induction of the lipoprotein vitellogenin synthesis. Furthermore, Mills et al. (2016) explored physiological responses to the endocrine-active pharmaceutical Tamoxifen in adult fish Tautogolabrus adspersus using the gas chromatograph coupled to a time-of-flight mass spectrometer (GC/ToF-MS). Thus, Mills et al. observed high levels of proline, threonine, alanine, lysine, tyrosine, and tryptophan, and found down-regulated metabolites involved in amino acid synthesis and metabolism, phospholipid synthesis, glucoronidation, and glycolysis, proving that T. adspersus could represent a sensitive nontarget organism, useful for studying metabolomics perturbations after exposure to pharmaceuticals. As reported in scientific literature, fish have been used not only to observe the metabolomics responses of estrogen-like molecules, but also to study metabolomics alterations caused by the exposure to antibiotics. In fact, De Sotto et al. (2017) studied environmental effects of 0.1 mg/L of clarithromycin, florfenicol and sulfamethazine, individually and in mixtures, on adults of Danio rerio after 72 h exposure using high-performance liquid chromatography with quadrupole time-of-flight (QTOF) mass spectrometer. When clarithromycin and florfenicol were tested individually, they were able to yield more metabolites than those found for sulfamethazine, and the most affected pathway was the metabolism of purines, especially guanosine involved in protecting neurons against excitotoxic damages. The similarity between clarithromycin and florfenicol could be explained thanks to the same mode of action of these two antibiotics, which inhibit protein biosynthesis interacting with 50 S subunit in nontarget organisms. Surprisingly, when De Sotto et al. (2017) tested antibiotics in mixtures, a small amount of metabolites was observed, probably due to antagonistic interactions. In line with the scientific literature taken into account here, Danio rerio is a good model in environmental metabolomics to identify the effects of pharmaceuticals, due to its similarity to human metabolism and its ease in absorption of small molecules through skin and gills.
In conclusion, scientific interest is constantly increasing in the wide field of environmental metabolomics, a very useful approach to understand the impact of various environmental xenobiotics in nontarget organisms, through different analytical platforms. In the last years, it has been applied to evaluate metabolic changes in different aquatic organisms of the trophic chain after pharmaceutical exposure (only few scientific papers to date); to the best of our knowledge, no studies on anticancer drugs exist at the moment. Since the consumption and the administration of this class of pseudo-persistent pharmaceuticals are increasing as also their occurrence in the aquatic systems, it would be advisable to use metabolomic strategies to understand anticancer drugs’ environmental toxic effects.
Abbreviations
- CE-MS:
-
Capillary electrophoresis mass spectrometry
- DIMS:
-
Desorption ionization mass spectrometry
- FT-ICR-MS:
-
Fourier transform ion cyclotron resonance mass spectrometry
- FT-IR:
-
Fourier transform infrared spectroscopy
- GC/ToF-MS:
-
Gas chromatography–time-of-flight mass spectrometry
- GC-MS:
-
Gas chromatography mass spectroscopy
- H-NMR:
-
Proton nuclear magnetic resonance
- LC-HRMS:
-
Liquid chromatography high resolution mass spectrometry
- LC-MS:
-
Liquid chromatography mass spectrometry
- MAS-NMR:
-
Magic angle spinning nuclear magnetic resonance
- NMR:
-
Nuclear magnetic resonance
- Q-TOF LC/MS:
-
Quadrupole time-of-flight liquid chromatography mass spectrometry
References
Alam TM, Alam MK, Neerathilingam M, Volk DE, Sarkar S, Ansari GAS, Luxon BA (2010) 1H NMR metabonomic study of rat response to triphenyl phosphate and tri-butyl phosphate exposure. Metabolomics 6(3):386–394. https://doi.org/10.1007/s11306-010-0205-z
Allwood JW, Ellis DI, Goodacre R (2008) Metabolomic technologies and their application to the study of plants and plant–host interactions. Physiol Plant 132:117–135. https://doi.org/10.1111/j.1399-3054.2007.01001.x
Alonso A, Marsal S, Julià A (2015) Analytical methods in untargeted metabolomics: state of the art in 2015. Front Bioeng Biotechnol 5(3):23
Ankley GT, Daston GP, Degitz SJ, Denslow ND, Hoke RA, Kennedy SW, Miracle AL, Perkins EJ, Snape J, Tillitt DE, Tyler CR, Versteeg D (2006) Toxicogenomics in regulatory ecotoxicology. Environ Sci Technol 40(13):4055–4065
Bajad S, Shulaev V (2007) Highly-parallel metabolomics approaches using LC–MS2 for pharmaceutical and environmental analysis. Trends Anal Chem 26(6):625–636. https://doi.org/10.1016/j.trac.2007.02.009. PMID:19756204
Barker M, Rayens W (2003) Partial least squares for discrimination. J Chemom 17(3):166–173. https://doi.org/10.1002/cem.785
Bilello JA (2005) The agony and ecstasy of “OMIC” technologies in drug development. Curr Mol Med 5(1):39–52. https://doi.org/10.2174/1566524053152898. PMID:15720269
Bird SS, Marur VR, Sniatynski MJ, Greenberg HK, Kristal BS (2011) Serum Lipidomics profiling using LC–MS and high-energy collisional dissociation fragmentation: focus on triglyceride detection and characterization. Anal Chem 83(17):6648–6657
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37(8):911–917
Bonnefille B, Gomez E, Alali M, Rosain D, Fenet H, Courant F (2018) Metabolomics assessment of the effects of diclofenac exposure on Mytilus galloprovincialis: potential effects on osmoregulation and reproduction. Sci Total Environ 613–614:611–618
Bothwell JHF, Griffin JL (2011) An introduction to biological nuclear magnetic resonance spectroscopy. Biol Rev 86(2):493–510. https://doi.org/10.1111/j.1469-185X.2010.00157.x. PMID:20969720
Brezovšek P, Eleršek T, Filipič M (2014) Toxicities of four anti-neoplastic drugs and their binary mixtures tested on the green alga Pseudokirchneriella subcapitata and thecyanobacterium Synechococcus leopoliensis. Water Res 52:168–177
Brown SAE, Simpson AJ, Simpson MJ (2008) Evaluation of sample preparation methods for nuclear magnetic resonance metabolic profiling studies with Eisenia Fetida. Environ Toxicol Chem 27(4):828
Brown SAE, Simpson AJ, Simpson MJ (2009) 1H NMR metabolomics of earthworm responses to sub-lethal PAH exposure. Environ Chem 6:432–440. https://doi.org/10.1071/EN09054
Brown SAE, McKelvie JR, Simpson AJ, Simpson MJ (2010) 1H NMR metabolomics of earthworm exposure to sub-lethal concentrations of phenanthrene in soil. Environ Pollut 158(6):2117–2123. https://doi.org/10.1016/j.envpol.2010.02.023. PMID:20338676
Bundy JG, Osborn D, Weeks JM, Lindon JC, Nicholson JK (2001) An NMR-based metabonomic approach to the investigation of coelomic fluid biochemistry in earthworms under toxic stress. FEBS Lett 500(1–2):31–35. https://doi.org/10.1016/S0014-5793(01)02582-0. PMID:11434921
Bundy JG, Lenz EM, Bailey NJ, Gavaghan CL, Svendsen C, Spurgeon D, Hankard PK, Osborn D, Weeks JA, Trauger SA, Speir P, Sanders I, Lindon JC, Nicholson JK, Tang H (2002a) Metabonomic assessment of toxicity of 4-fluoroaniline, 3,5-difluoroaniline and 2 fluoro-4-methylaniline to the earthworm Eisenia veneta (Rosa): identification of new endogenous biomarkers. Environ Toxicol Chem 21(9):1966–1972. https://doi.org/10.1002/etc.5620210926. PMID:12206438
Bundy JG, Lenz EM, Osborn D, Weeks JM, Lindon JC, Nicholson JK (2002b) Metabolism of 4-fluoroaniline and 4-fluorobiphenyl in the earthworm Eisenia veneta characterized by high-resolution NMR spectroscopy with directly coupled HPLC–NMR and HPLC–MS. Xenobiotica 32(6):479–490. https://doi.org/10.1080/00498250210124156. PMID:12160481
Bundy JG, Spurgeon DJ, Svendsen C, Hankard PK, Osborn D, Lindon JC, Nicholson JK (2002c) Earthworm species of the genus Eisenia can be phenotypically differentiated by metabolic profiling. FEBS Lett 521(1–3):115–120. https://doi.org/10.1016/S0014-5793(02)02854-5. PMID:12067738
Bundy JG, Spurgeon DJ, Svendsen C, Hankard PK, Weeks JM, Osborn D, Lindon JC, Nicholson JK (2004) Environmental Metabonomics: applying combination biomarker analysis in earthworms at a metal contaminated site. Ecotoxicology 13(8):797–806
Burton IW, Quilliam MA, Walter JA (2005) Quantitative 1H NMR with external standards: use in preparation of calibration solutions for algal toxins and other natural products. Anal Chem 77(10):3123–3131. https://doi.org/10.1021/ac048385h. PMID:15889900
Bylesjö M, Rantalainen M, Cloarec O, Nicholson JK, Holmes E, Trygg J (2006) OPLS discriminant analysis: combining the strengths of PLS-DA and SIMCA classification. J Chemom 20(8–10):341–351. https://doi.org/10.1002/cem.1006
Cappello T, Pereira P, Maisano M, Mauceri A, Pacheco M, Fasulo S (2016) Advances in understanding the mechanisms of mercury toxicity in wild golden grey mullet (Liza aurata) by 1H NMR-based metabolomics. Environ Pollut 219:139–148
Cappello T, Maisano M, Mauceri A, Fasulo S (2017) 1 H NMR-based metabolomics investigation on the effects of petrochemical contamination in posterior adductor muscles of caged mussel Mytilus galloprovincialis. Ecotoxicol Environ Saf 142:417–422
Chen C, Gonzalez FJ, Idle JR (2008) LC-MS-based metabolomics in drug metabolism. Drug Metab Rev 39(2–3):581–5971
Chen L, Zhou L, Chan ECY, Neo J, Beuerman RW (2011) Characterization of the human tear metabolome by LC–MS/MS. J Proteome Res 10(10):4876–4882
Chiu KH, Dong CD, Chen CF, Tsai ML, Ju YR, Chen TM, Chen CW (2017) NMR-based metabolomics for the environmental assessment of Kaohsiung Harbor sediments exemplified by a marine amphipod (Hyalella azteca). Mar Pollut Bull 124(2):714–724
Chylla RA, Hu K, Ellinger JJ, Markley JL (2011) Deconvolution of twodimensional NMR spectra by fast maximum likelihood reconstruction: application to quantitative metabolomics. Anal Chem 83(12):4871–4880. https://doi.org/10.1021/ac200536b. PMID:21526800
Coen M, Holmes E, Lindon JC, Nicholson JK (2008) NMR-based metabolic profiling and metabonomic approaches to problems in molecular toxicology. Chem Res Toxicol 21(1):9–27. https://doi.org/10.1021/tx700335d. PMID:18171018
Creek DJ, Anderson J, McConville MJ, Barrett MP (2012) Metabolomic analysis of trypanosomatid protozoa. Mol Biochem Parasitol 181(2):73–84
Crockford DJ, Holmes E, Lindon JC, Plumb RS, Zirah S, Bruce SJ, Rainville P, Stumpf CL, Nicholson JK (2006) Statistical heterospectroscopy, an approach to the integrated analysis of NMR and UPLC–MS data sets: application in metabonomic toxicology studies. Anal Chem 78(2):363–371. https://doi.org/10.1021/ac051444m. PMID:16408915
De Sotto RB, Medriano CD, Choc Y, Kimd H, Chungd IC, Seokd SKG, Honge SW, Park Y, Kima S (2017) Sub-lethal pharmaceutical hazard tracking in adult zebrafish using untargeted LC–MS environmental metabolomics. J Hazard Mater 339:63–72
Dunn WB, Ellis DI (2005) Metabolomics: current analytical platforms and methodologies. TrAC Trends Anal Chem 24(4):285–294
Dunn WB, Wilson ID, Nicholls AW, Broadhurst D (2012) The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 4(18):2249–2264
Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, Sassone-Corsi P (2012) Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci 109(14):5541–5546
Edwards JL, Chisolm CN, Shackman JG, Kennedy RT (2006) Negative mode sheathless capillary electrophoresis electrospray ionization-mass spectrometry for metabolite analysis of prokaryotes. J Chromatogr A 1106(1–2):80–88. https://doi.org/10.1016/j.chroma.2005.08.082
Ekman DR, Teng Q, Villeneuve DL, Kahl MD, Jensen KM, Durhan EJ, Ankley GT, Collette TW (2008) Investigating compensation and recovery of fathead minnow (Pimephales promelas) exposed to 17-ethynylestradiol with metabolite profiling. Environ Sci Technol 42(11):4188–4194. https://doi.org/10.1021/es8000618. PMID:18589986
Ekman DR, Teng Q, Villeneuve DL, Kahl MD, Jensen KM, Durhan EJ, Ankley GT, Collette TW (2009) Profiling lipid metabolites yields unique information on sex- and time-dependent responses of fathead minnows (Pimephales promelas) exposed to 17-ethynylestradiol. Metabolomics 5(1):22–32. https://doi.org/10.1007/s11306-008-0138-y
Eriksson L, Johansson E, Kettaneh-Wold N, Trygg J, Wikstrom C, Wold S (2006) Multi- and megavariate data analysis. Part 1: Basic principles and applications, 2nd edn. Umea, Umetrics, AB
Fasulo S, Iacono F, Cappello T, Corsaro C, Maisano M, D’Agata A, Giannetto A, De Domenico E, Parrino V, Lo Paro G, Mauceri A (2012) Metabolomic investigation of Mytilus galloprovincialis (Lamarck1819) caged in aquatic environments. Ecotoxicol Environ Saf 84:139–146
Fernie AR, Aharoni A, Willmitzer L, Stitt M, Tohge T, Kopka J, Carroll AJ, Saito K, Fraser PD, DeLuca V (2011) Recommendations for reporting metabolite data. Plant Cell 23(7):2477–2482
Fialho E, Moreno FS, Ong TP (2008) Nutrição no pós-genoma: fundamentos e aplicações de ferramentas ômicas. Rev Nutr 21(6):757–766
Flores-Sanchez IJ, Choi YH, Verpoorte R (2012) Metabolite analysis of Cannabis sativa L. by NMR spectroscopy. Methods Mol Biol 815:363–375. https://doi.org/10.1007/978-1-61779-424-7
Flores-Valverde AM, Horwood J, Hill EM (2010) Disruption of the steroid metabolome in fish caused by exposure to the environmental estrogen 17 - ethinylestradiol. Environ Sci Technol 44(9):3552–3558. https://doi.org/10.1021/es9039049. PMID:20356033
Forseth RR, Schroeder FC (2011) NMR-spectroscopic analysis of mixtures: from structure to function. Curr Opin Chem Biol 15(1):38–47
Fukusaki E, Kobayashi A (2005) Plant metabolomics: potential for practical operation. J Biosci Bioeng 100(4):347–354
Gačić Z, Kolarević S, Sunjog K, Kračun-Kolarević M, Paunović M, Knežević-Vukčević J, Vuković-Gačić B (2014) The impact of in vivo and in vitro exposure to base analogue 5-FU on the level of DNA damage in haemocytes of freshwater mussels Unio pictorum and Unio tumidus. Environ Pollut 191:145–150
García-Cañaveras JC, Donato MT, Castell JV, Lahoz A (2012) Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J Lipid Res 53(10):2231–2241. https://doi.org/10.1194/jlr.D028803. PMID:22822028
Garreta-Lara E, Campos B, Barata C, Lacorte S, Tauler R (2016) Metabolic profiling of Daphnia magna exposed to environmental stressors by GC–MS and chemometric tools. Metabolomics 12:86. https://doi.org/10.1007/s11306-016-1021-x
Gaw S, Thomas KV, Hutchinson TH (2014) Sources, impacts and trends of pharmaceuticals in the marine and coastal environment. Philos Trans R Soc B 369:20130572. https://doi.org/10.1098/rstb.2013.0572
Ghaste M, Mistrik R, Shulaev V (2016) Applications of Fourier transform ion cyclotron resonance (FT-ICR) and Orbitrap based high resolution mass spectrometry in metabolomics and Lipidomics. Int J Mol Sci 17(6):816
Gil-Solsona R, Nácher-Mestre J, Lacalle-Bergeron L, Sancho JV, Alvar Calduch-Giner J, Hernández F, Pérez-Sánchez J (2017) Untargeted metabolomics approach for unraveling robust biomarkers of nutritional status in fasted gilthead sea bream (Sparus aurata). Peer J 5:e2920
Gillis JD, Price GW, Prasher S (2017) Lethal and sub-lethal effects of triclosan toxicity to the earthworm Eisenia fetida assessed through GC–MS metabolomics. J Hazard Mater 323:203–211
Gómez-Canela C, Miller TH, Bury NR, Tauler R, Barron LP (2016) Targeted metabolomics of Gammarus pulex following controlled exposures to selected pharmaceuticals in water. Sci Total Environ 562:777–788
Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB (2004) Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol 22(5):245–252
Goodacre R, Broadhurst D, Smilde AK, Kristal BS, Baker JD, Beger R, Bessant C, Connor S, Capuani G, Craig A, Ebbels T, Kell DB, Manetti C, Newton J, Paternostro G, Somorjai R, Sjöström M, Trygg J, Wulfert F (2007) Proposed minimum reporting standards for data analysis in metabolomics. Metabolomics 3(3):231–241
Griffin JL (2003) Metabonomics: NMR spectroscopy and pattern recognition analysis of body fluids and tissues for characterisation of xenobiotic toxicity and disease diagnosis. Curr Opin Chem Biol 7(5):648–654. https://doi.org/10.1016/j.cbpa.2003.08.008. PMID:14580571
Griffin JL, Bollard ME (2004) Metabonomics: its potential as a tool in toxicology for safety assessment and data integration. Curr Drug Metab 5(5):389–398. https://doi.org/10.2174/1389200043335432. PMID:15544433
Griffin JL, Shockcor JP (2004) Metabolic profiles of cancer cells. Nat Rev Cancer 4(7):551–561. https://doi.org/10.1038/Nrc1390
Griffiths JR, Stubbs M (2003) Opportunities for studying cancer by metabolomics: preliminary observations on tumors deficient in hypoxia-inducible factor 1. Adv Enzym Regul 43(1):67–76
Griffiths JR, McSheehy PMJ, Robinson SP, Troy H, Chung YL, Leek RD, Williams KJ, Stratford IJ, Harris AL, Stubbs M (2002) Metabolic changes detected by in vivo magnetic resonance studies of HEPA-1 wild-type tumors and tumors deficient in hypoxia-inducible factor-1β (HIF-1β): evidence of an anabolic role for the HIF-1 pathway. Cancer Res 62(3):688–695. PMID:11830521
Grimes JH, O’Connell TM (2011) The application of micro-coil NMR probe technology to metabolomics of urine and serum. J Biomol NMR 49(3–4):297–305. https://doi.org/10.1007/s10858-011-9488-2
Hall RD (2006) Plant metabolomics: from holistic hope, to hype, to hot topic. New Phytol 169(3):453–468
Harvey AL, Edrada-Ebel R, Quinn RJ (2015) The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov 14(2):111
Holmes E, Antti H (2002) Chemometric contributions to the evolution of metabonomics: mathematical solutions to characterising and interpreting complex biological NMR spectra. Analyst 127(12):1549–1557. https://doi.org/10.1039/b208254n. PMID:12537357
Hu K, Ellinger JJ, Chylla RA, Markley JL (2011a) Measurement of absolute concentrations of individual compounds in metabolite mixtures by gradient-selective time-zero 1H-13C HSQC with two concentration references and fast maximum likelihood reconstruction analysis. Anal Chem 83(24):9352–9360. https://doi.org/10.1021/ac201948f. PMID:22029275
Hu K, Westler WM, Markley JL (2011b) Simultaneous quantification and identification of individual chemicals in metabolite mixtures by twodimensional extrapolated time-zero 1H-13C HSQC (HSQC0). J Am Chem Soc 133(6):1662–1665. https://doi.org/10.1021/ja1095304. PMID:21247157
Hughes SL, Bundy JG, Want EJ, Kille P, Sturzenbaum SR (2009) The metabolomic responses of Caenorhabditis elegans to cadmium are largely independent of metallothionein status, but dominated by changes in cystathionine and phytochelatins. J Proteome Res 8(7):3512–3519. https://doi.org/10.1021/pr9001806. PMID:19466807
Isidori M, Piscitelli C, Russo C, Smutná M, Bláha L (2016a) Teratogenic effects of five anticancer drugs on Xenopus laevis embryos. Ecotoxicol Environ Saf 133:90–96
Isidori M, Lavorgna M, Russo C, Kundi M, Zegura B, Novak M, Filipic M, Misík M, Knasmueller S, Lopez de Alda M, Barcelo D, Zonja B, Cesen M, Scancar J, Kosjek T, Heath E (2016b) Chemical and toxicological characterisation of anticancer drugs in hospital and municipal wastewaters from Slovenia and Spain. Environ Pollut 219:275–287
Issaq HJ, Abbott E, Veenstra TD (2008) Utility of separation science in metabolomicstudies. J Sep Sci 31(11):1936–1947. https://doi.org/10.1002/jssc.200700601. PMID:18348322
Jacobsen NE (2007) NMR spectroscopy explained: simplified theory, applications and examples for organic chemistry and structural biology. Wiley, Hoboken, NJ
Jones OAH, Spurgeon DJ, Svendsen C, Griffin JL (2008) A metabolomics based approach to assessing the toxicity of the polyaromatic hydrocarbon pyrene to the earthworm Lumbricus rubellus. Chemosphere 71(3):601–609. https://doi.org/10.1016/j.chemosphere.2007.08.056. PMID:17928029
Kim HK, Verpoorte R (2010) Sample preparation for plant metabolomics. Phytochem Anal 21(1):4–13
Kim HK, Choi YH, Verpoorte R (2011) NMR-based metabolomics: where do we stand, where do we go? Trends Biotechnol 29:267–275
Kokushi E, Shintoyo A, Koyama J, Uno S (2017) Evaluation of 2,4-dichlorophenol exposure of Japanese medaka, Oryzias latipes, using a metabolomics approach. Environ Sci Pollut Res 24(36):27678–27686
Kovacevic V, Simpson AJ, Simpson MJ (2016) 1H NMR-based metabolomics of Daphnia magna responses after sub-lethal exposure to triclosan, carbamazepine and ibuprofen. Comp Biochem Physiol Part D 19:199–210
Kovács R, Csenki Z, Bakos K, Urbányi B, Horváth Á, Garaj-Vrhovac V, Gajski G, Gerić M, Negreira N, López de Alda M, Barceló D, Heath E, Kosjek T, Žegura B, Novak M, Zajc I, Baebler Š, Rotter A, Ramšak Ž, Filipič M (2015) Assessment of toxicity and genotoxicity of low doses of 5-fluorouracil in zebrafish (Danio rerio) two-generation study. Water Res 77:201–212
Kovács R, Bakos K, Urbányi B, Kövesi J, Gazsi G, Csepeli A, Appl ÁJ, Bencsik D, Csenki Z, Horváth Á (2016) Acute and sub-chronic toxicity of four cytostatic drugs in zebrafish. Environ Sci Pollut Res Int 23(15):14718–14729
Kundi M, Parrella A, Lavorgna M, Criscuolo E, Russo C, Isidori M (2016) Prediction and assessment of ecogenotoxicity of antineoplastic drugs in binary mixtures. Environ Sci Pollut Res 23:14771–14779
Lacey ME, Subramanian R, Olson DL, Webb AG, Sweedler JV (1999) High-resolution NMR spectroscopy of sample volumes from 1 nL to 10 _L. Chem Rev 99(10):3133–3152. https://doi.org/10.1021/cr980140f. PMID:11749512
Laith AA, Ambaka M, Abol-Munafi AB, Nurhafizah WWI, Najiaha M (2017) Metabolomic analysis of marine and mud crabs based on antibacterial activity. Aquac Rep 7:7–15
Lankadurai BP, Nagato EG, Simpson MJ (2013) Environmental metabolomics: an emerging approach to study organism responses to environmental stressors. Environ Rev 21(3):180–205
Lenz K, Mahnik SN, Weissenbacher N, Mader RM, Krenn P, Hann S, Koellensperger G, Uhl M, Knasmueller S, Ferk F, Bursh W, Fuerhacker M (2007) Monitoring, removal and risk assessment of cytostatic drugs in hospital wastewater. Water Sci Technol 56(12):141–149
Li W, Ruan CJ, Teixeira da Silva JA, Guo H, Zhao CE (2012) NMR metabolomics of berry quality in sea buckthorn (Hippophae L.). Mol Breed 31:57–67. https://doi.org/10.1007/s11032-012-9768-x
Liebeke M, Bundy JG (2012) Tissue disruption and extraction methods for metabolic profiling of an invertebrate sentinel species. Metabolomics 8(5):819–830. https://doi.org/10.1007/s11306-011-0377-1
Lin CY, Viant MR, Tjeerdema RS (2006) Metabolomics: methodologies and applications in the environmental sciences. J Pestic Sci 31(3):245–251. https://doi.org/10.1584/jpestics.31.245
Lindon JC, Nicholson JK, Holmes E (2006) The handbook of metabonomics and metabolomics. Elsevier Science, Amsterdam. ISBN 9780444528414.
Lloyd AJ, Beckmann M, Wilson T, Tailliart K, Allaway D, Draper J (2017) Ultra high performance liquid chromatography–high resolution mass spectrometry plasma lipidomics can distinguish between canine breeds despite uncontrolled environmental variability and non-standardized diets. Metabolomics 13(2):1–15
Logan TM, Murali M, Wang GS, Jolivet C (1999) Application of a highresolution superconducting NMR probe in natural product structure determination. Magn Reson Chem 37(7):512–515. https://doi.org/10.1002/(SICI)1097-458X(199907)37:7<512::AID-MRC502>3.0.CO;2-B
Ludwig C, Viant MR (2010) Two-dimensional J-resolved NMR spectroscopy: review of a key methodology in the metabolomics toolbox. Phytochem Anal 21(1):22–32. https://doi.org/10.1002/pca.1186. PMID:19904730
Lutz U, Lutz RW, Lutz WK (2006) Metabolic profiling of glucuronides in human urine by LC–MS/MS and partial least-squares discriminant analysis for classification and prediction of gender. Anal Chem 78(13):4564–4571. https://doi.org/10.1021/ac0522299. PMID:16808466
McGhie TK, Rowan DD (2012) Metabolomics for measuring phytochemicals, and assessing human and animal responses to phytochemicals, in food science. Mol Nutr Food Res 56(1):147–158
McKay RT (2009) Chapter 2 Recent advances in solvent suppression for solution NMR: a practical reference. Annu Rep NMR Spectrosc 66:33–76. https://doi.org/10.1016/S0066-4103(08)00402-X
McKelvie JR, Yuk J, Xu YP, Simpson AJ, Simpson MJ (2009) 1H NMR and GC/MS metabolomics of earthworm responses to sub-lethal DDT and endosulfan exposure. Metabolomics 5(1):84–94. https://doi.org/10.1007/s11306-008-0122-6
McKelvie JR, Wolfe DM, Celejewski M, Simpson AJ, Simpson MJ (2010) Correlations of Eisenia fetida metabolic responses to extractable phenanthrene concentrations through time. Environ Pollut 158(6):2150–2157. https://doi.org/10.1016/j.envpol.2010.02.022. PMID:20338675
McKelvie JR, Wolfe DM, Celejewski MA, Alaee M, Simpson AJ, Simpson MJ (2011) Metabolic responses of Eisenia fetida after sub-lethal exposure to organic contaminants with different toxic modes of action. Environ Pollut 159(12):3620–3626. https://doi.org/10.1016/j.envpol.2011.08.002. PMID:21856054
McKelvie JR, Whitfield Åslund M, Celejewski M, Simpson AJ, Simpson MJ (2013) Reduction in the earthworm metabolomic response after phenanthrene exposure in soils with high soil organic carbon content. Environ Pollut 175:75–81. https://doi.org/10.1016/j.envpol.2012.12.018. PMID:23337355
Miller MG (2007) Environmental metabolomics: a SWOT analysis (strengths, weaknesses, opportunities, and threats). J Proteome Res 6(2):540–545. https://doi.org/10.1021/pr060623x. PMID:17269710
Mills LJ, Henderson WM, Jayaraman S, Gutjahr-Gobell RE, Zaroogian GE, Horowitz DB, Laws SC (2016) Approaches for predicting effects of unintended environmental exposure to an endocrine active pharmaceutical, tamoxifen. Environ Toxicol 31(12):1834–1850
Mumtaz MW, Hamid AA, Akhtar MT, Anwar F, Rashid U, AL-Zuaidy MH (2017) An overview of recent developments in metabolomics and proteomics – phytotherapic research perspectives. Front Life Sci 10(1):1–37
Nagato EG, Simpson AJ, Simpson MJ (2016) Metabolomics reveals energetic impairments in Daphnia magna exposed to diazinon, malathion and bisphenol-A. Aquat Toxicol 170:175–186
Negreira N, Lopez de Alda M, Barcelo D (2014) Cytostatic drugs and metabolites in municipal and hospital wastewaters in Spain: filtration, occurrence, and environmental risk. Sci Total Environ 497-498:68–77
Nicholson JK, Wilson ID (1989) High resolution proton magnetic resonance spectroscopy of biological fluids. Prog Nucl Magn Reson Spectrosc 21:449–501. https://doi.org/10.1016/0079-6565(89)80008-1
Nicholson JK, Lindon JC, Holmes E (2008) ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 29(11):1181–1189
Noctor G, Bergot GL, Mauve C, Thominet D, Lelarge-Trouverie C, Prioul JL (2007) A comparative study of amino acid measurement in leaf extracts by gas chromatography-time of flight-mass spectrometry and high performance liquid chromatography with fluorescence detection. Metabolomics 3(2):161–174. https://doi.org/10.1007/s11306-007-0057-3
Nowick JS, Khakshoor O, Hashemzadeh M, Brower JO (2003) DSA: a new internal standard for NMR studies in aqueous solution. Org Lett 5(19):3511–3513. https://doi.org/10.1021/ol035347w. PMID:12967312
Pan ZZ, Raftery D (2007) Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal Bioanal Chem 387(2):525–527. https://doi.org/10.1007/s00216-006-0687-8. PMID:16955259
Parrella A, Lavorgna M, Criscuolo E, Russo C, Fiumano V, Isidori M (2014) Acute and chronic toxicity of six anticancer drugs on rotifers and crustaceans. Chemosphere 115:59–66
Parrella A, Lavorgna M, Criscuolo E, Russo C, Isidori M (2015) Eco-genotoxicity of six anticancer drugs using comet assayin daphnids. J Hazard Mater 286:573–580
Poynton HC, Taylor NS, Hicks J, Colson K, Chan SR, Clark C, Scanlan L, Loguinov AV, Vulpe C, Viant MR (2011) Metabolomics of microliter hemolymph samples enables an improved understanding of the combined metabolic and transcriptional responses of Daphnia magna to cadmium. Environ Sci Technol 45(8):3710–3717. https://doi.org/10.1021/es1037222. PMID:21417318
Ramautar R, Busnel JM, Deelder AM, Mayboroda OA (2012) Enhancing the coverage of the urinary metabolome by sheathless capillary electrophoresis–mass spectrometry. Anal Chem 84(2):885–892. https://doi.org/10.1021/ac202407v. PMID:22148170
Ritota M, Casciani L, Han BZ, Cozzolino S, Leita L, Sequi P, Valentini M (2012) Traceability of Italian garlic (Allium sativum L.) by means of HRMAS–NMR spectroscopy and multivariate data analysis. Food Chem 135(2):684–693. https://doi.org/10.1016/j.foodchem.2012.05.032. PMID:22868146
Robertson DG (2005) Metabonomics in toxicology: a review. Toxicol Sci 85(2):809–822. https://doi.org/10.1093/toxsci/kfi102. PMID:15689416
Ruiz-Torres V, Encinar JA, Herranz-López M, Pérez-Sánchez A, Galiano V, Barrajón-Catalán E, Micol V (2017) An updated review on marine anticancer compounds: the use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 22:1037. https://doi.org/10.3390/molecules22071037
Russo C, Lavorgna M, Cesen M, Kosjek T, Heath T, Isidori M (2018) Evaluation of acute and chronic ecotoxicity of cyclophosphamide, ifosfamide, their metabolites/transformation products and UV treated samples. Environ Pollut 233:356–363
Samuelsson LM, Forlin L, Karlsson G, Adolfsson-Erici M, Larsson DGJ (2006) Using NMR metabolomics to identify responses of an environmental estrogen in blood plasma of fish. Aquat Toxicol 78:341–349
Sato S, Soga T, Nishioka T, Tomita M (2004) Simultaneous determination of the main metabolites in rice leaves using capillary electrophoresis mass spectrometry and capillary electrophoresis diode array detection. Plant J 40(1):151–163. https://doi.org/10.1111/j.1365-313X.2004.02187.x. PMID:15361149
Schripsema J (2009) Application on NMR in plant metabolomics: techniques, problems and prospects. Phytochem Anal 21:14–21
Scognamiglio M, D’Abrosca B, Fiumano V, Golino M, Esposito A, Fiorentino A (2014) Seasonal phytochemical changes in Phillyrea angustifolia L.: metabolomic analysis and phytotoxicity assessment. Phytochem Lett 8:163–170
Scognamiglio M, D’Abrosca B, Esposito A, Fiorentino A (2015) Chemical Composition and Seasonality of Aromatic Mediterranean Plant Species by NMR-Based Metabolomics. J Anal Methods Chem 2015(2015):258570. https://doi.org/10.1155/2015/258570
Simpson AJ, Brown SA (2005) Purge NMR: effective and easy solvent suppression. J Magn Reson 175(2):340–346. https://doi.org/10.1016/j.jmr.2005.05.008. PMID:15964227
Simpson MJ, McKelvie JR (2009) Environmental metabolomics: new insights into earthworm ecotoxicity and contaminant bioavailability in soil. Anal Bioanal Chem 394(1):137–149
Southam AD, Lange A, Hines A, Hill EM, Katsu Y, Iguchi T, Tyler CR, Viant MR (2011) Metabolomics reveals target and off-target toxicities of a model organophosphate pesticide to roach (Rutilus rutilus): implications for biomonitoring. Environ Sci Technol 45(8):3759–3767. https://doi.org/10.1021/es103814d. PMID:21410251
Sumner LW, Mendes P, Dixon RA (2003) Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry 62(6):817–836
Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR (2007) Proposed minimum reporting standards for chemical analysis. Metabolomics 3(3):211–221
Taylor NS, Weber RJM, White TA, Viant MR (2010) Discriminating between different acute chemical toxicities via changes in the Daphnid Metabolome. Toxicol Sci 118(1):307–317
Tjeerdema RS (2008) Application of NMR-based techniques in aquatic toxicology: brief examples. Mar Pollut Bull 57(6–12):275–279. https://doi.org/10.1016/j.marpolbul.2008.01.002. PMID:18282584
Trygg J, Wold S (2002) Orthogonal projections to latent structures (O-PLS). J Chemom 16(3):119–128. https://doi.org/10.1002/cem.695
Trygg J, Holmes E, Lundstedt T (2007) Chemometrics in metabonomics. J Proteome Res 6(2):469–479. https://doi.org/10.1021/pr060594q. PMID:17269704
Tuffnail W, Mills GA, Cary P, Greenwood R (2009) An environmental 1H NMR metabolomic study of the exposure of the marine mussel Mytilus edulis to atrazine, lindane, hypoxia and starvation. Metabolomics 5:33–43
van der Hooft JJJ, Padmanabhan S, Burgess KEV, Barrett MP (2016) Urinary antihypertensive drug metabolite screening using molecular networking coupled to high-resolution mass spectrometry fragmentation. Metabolomics 12(7):1–15
van Ravenzwaay B, Cunha GCP, Leibold E, Looser R, Mellert W, Prokoudine A, Walk T, Wiemer J (2007) The use of metabolomics for the discovery of new biomarkers of effect. Toxicol Lett 172(1–2):21–28. https://doi.org/10.1016/j.toxlet.2007.05.021. PMID:17614222
Varmuza K, Filzmoser P (2009) Introduction to multivariate statistical analysis in chemometrics. CRC Press, Boca Raton, FL
Verpoorte R, Choi YH, Kim HK (2007) NMR-based metabolomics at work in phytochemistry. Phytochem Rev 6(1):3–14
Verpoorte R, Choi YH, Mustafa NR, Kim HK (2008) Metabolomics: back to basics. Phytochem Rev 7(3):525–537. https://doi.org/10.1007/s11101-008-9091-7
Viant MR (2008a) Environmental metabolomics using 1H-NMR spectroscopy. Methods Mol Biol 410:137–150. PMID:18642599
Viant MR (2008b) Recent developments in environmental metabolomics. Mol Bio Syst 4(10):980–986. https://doi.org/10.1039/b805354e
Viant MR, Rosenblum ES, Tjeerdema RS (2003) NMR-based metabolomics: a powerful approach for characterizing the effects of environmental stressors on organism health. Environ Sci Technol 37(21):4982–4989. https://doi.org/10.1021/es034281x. PMID:14620827
Viant MR, Bearden DW, Bundy JG, Burton IW, Collette TW, Ekman DR, Ezernieks V, Karakach TK, Lin CY, Rochfort S, de Ropp JS, Teng Q, Tjeerdema RS, Walter JA, Wu H (2009) International NMR-based environmental metabolomics intercomparison exercise. Environ Sci Technol 43(1):219–225. https://doi.org/10.1021/es802198z. PMID:19209610
Wagner ND, Simpson AJ, Simpson MJ (2017) Metabolomic responses to sublethal contaminant exposure in neonate and adult daphnia magna. Environ Toxicol Chem 36:938–946
Wang M, Chen L, Liu D, Chen H, Tang D-D, Zhao Y-Y (2017) Metabolomics highlights pharmacological bioactivity and biochemical mechanism of traditional Chinese medicine. Chem Biol Interact 273:133–141
Warren CR, Aranda I, Cano FJ (2012) Metabolomics demonstrates divergent responses of two Eucalyptus species to water stress. Metabolomics 8(2):186–200. https://doi.org/10.1007/s11306-011-0299-y
Weckwerth W (2007) Metabolomics: methods and protocols. Humana Press Inc., Totowa, NJ
Westerhuis JA, Hoefsloot HCJ, Smit S, Vis DJ, Smilde AK, van Velzen EJJ, van Duijnhoven JPM, van Dorsten FA (2008) Assessment of PLSDA cross validation. Metabolomics 4(1):81–89. https://doi.org/10.1007/s11306-007-0099-6
Whitfield Åslund M, Celejewski M, Lankadurai BP, Simpson AJ, Simpson MJ (2011a) Natural variability and correlations in the metabolic profile of healthy Eisenia fetida earthworms observed using 1H NMR metabolomics. Chemosphere 83(8):1096–1101. https://doi.org/10.1016/j.chemosphere.2011.01.036. PMID:21316730
Whitfield Åslund M, Simpson AJ, Simpson MJ (2011b) 1H NMR metabolomics of earthworm responses to polychlorinated biphenyl (PCB) exposure in soil. Ecotoxicology 20(4):836–846. https://doi.org/10.1007/s10646-011-0638-9. PMID:21424327
Wishart DS (2008) Quantitative metabolomics using NMR. TrAC Trends Anal Chem 27(3):228–237
Wolfender J-L, Rudaz S, Hae Choi Y, Kyong Kim H (2013) Plant metabolomics: from holistic data to relevant biomarkers. Curr Med Chem 20(8):1056–1090
Wu B, Zhu L, Le XC (2017) Metabolomics analysis of TiO 2 nanoparticles induced toxicological effects on rice (Oryza sativa L.). Environ Pollut 230:302–310
Wu L, Mashego MR, van Dam JC, Proell AM, Vinke JL, Ras C, van Winden WA, van Gulik WM, Heijnen JJ (2005) Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal Biochem 336(2):164–171. https://doi.org/10.1016/j.ab.2004.09.001. PMID:15620880
Wu H, Southam AD, Hines A, Viant MR (2008) High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal Biochem 372(2):204–212. https://doi.org/10.1016/j.ab.2007.10.002. PMID:17963684
Xi Y, de Ropp JS, Viant MR, Woodruff DL, Yu P (2008) Improved identification of metabolites in complex mixtures using HSQC NMR spectroscopy. Anal Chim Acta 614(2):127–133. https://doi.org/10.1016/J.Aca.2008.03.024
Xiao JF, Zhou B, Ressom HW (2012) Metabolite identification and quantitation in LC-MS/MS-based metabolomics. TrAC Trends Anal Chem 32:1–14
Yamamotoya T, Dose H, Tian Z, Faure A, Toya Y, Honma M, Igarashi K, Nakahigashi K, Soga T, Mori H, Matsuno H (2012) Glycogen is the primary source of glucose during the lag phase of E. coli proliferation. Biochim Biophys Acta 1824(12):1442–1448. https://doi.org/10.1016/j.bbapap.2012.06.010
Yingrong Chen, Zhihong Ma, Lishan Min, Hongwei Li, Bin Wang, Jing Zhong, Licheng Dai, (2015) Biomarker identification and pathway analysis by serum metabolomics of lung Cancer. Biomed Res Int 1–9
Yuk J, McKelvie JR, Simpson MJ, Spraul M, Simpson AJ (2010) Comparison of 1-D and 2-D NMR techniques for screening earthworm responses to sub-lethal endosulfan exposure. Environ Chem 7(6):524–536. https://doi.org/10.1071/EN10084
Yuk J, Simpson AJ, Simpson MJ (2011) 1-D and 2-D NMR metabolomics of earthworm responses to sub-lethal trifluralin and endosulfan exposure. Environ Chem 8:281–294. https://doi.org/10.1071/EN11033
Yuk J, Simpson MJ, Simpson AJ (2013) 1-D and 2-D NMR-based metabolomics of earthworms exposed to endosulfan and endosulfan sulfate in soil. Environ Pollut 175:35–44. https://doi.org/10.1016/j.envpol.2012.12.007. PMID:23333485
Zhao L, Huang Y, Hu J, Zhou H, Adeleye AS, Keller AA (2016) Environ Sci Technol 50(4):2000–2010
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mastroianni, G., Scognamiglio, M., Russo, C., Fiorentino, A., Lavorgna, M. (2020). Environmental Metabolomics: A Powerful Tool to Investigate Biochemical Responses to Drugs in Nontarget Organisms. In: Heath, E., Isidori, M., Kosjek, T., Filipič, M. (eds) Fate and Effects of Anticancer Drugs in the Environment. Springer, Cham. https://doi.org/10.1007/978-3-030-21048-9_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-21048-9_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-21047-2
Online ISBN: 978-3-030-21048-9
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)