
Abstract
The availability of sensitive, accurate and specific analytical methods for the measurement of polymyxins in biological fluids has enabled an understanding of the pharmacokinetics of these important antibiotics in healthy humans and patients. Colistin is administered as its inactive prodrug colistin methanesulfonate (CMS) and has especially complex pharmacokinetics. CMS undergoes conversion in vivo to the active entity colistin, but the rate of conversion varies from brand to brand and possibly from batch to batch. The extent of conversion is generally quite low and depends on the relative magnitudes of the conversion clearance and other clearance pathways for CMS of which renal excretion is a major component. Formed colistin in the systemic circulation undergoes very extensive tubular reabsorption; the same mechanism operates for polymyxin B which is administered in its active form. The extensive renal tubular reabsorption undoubtedly contributes to the propensity for the polymyxins to cause nephrotoxicity. While there are some aspects of pharmacokinetic behaviour that are similar between the two clinically used polymyxins, there are also substantial differences. In this chapter, the pharmacokinetics of colistin, administered as CMS, and polymyxin B are reviewed, and the therapeutic implications are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Colistimethate
- Colistin
- Polymyxin B
- Pharmacokinetics in humans
- Comparison of colistin and polymyxin B pharmacokinetics
- Pharmacodynamics and toxicodynamics
- Clinical implications
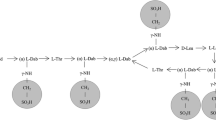
As discussed in Chap. 3, the polymyxins were discovered in the 1940s. While the polymyxin family of antibiotics comprises several members, only colistin (also known as polymyxin E) and polymyxin B were developed for clinical use . These two polymyxins differ from each other by just one amino acid in the heptapeptide ring (Fig. 15.1) and they possess very similar in vitro antibacterial activity [1,2,3]. Since they are products of fermentation, they are multicomponent mixtures with the major respective components being colistin A and B, and polymyxin B1 and B2. Notwithstanding the similarities mentioned above, colistin and polymyxin B differ substantially in regard to the chemical form that is contained within the parenteral and inhalational products that are administered to patients [1, 4]. Polymyxin B is administered parenterally as its sulfate salt; that is, patients directly receive the active antibacterial entity. In contrast, colistin is administered as the sodium salt of colistin methanesulfonate (CMS, also known as colistimethate ) (Fig. 15.1). It is now well established that CMS is an inactive prodrug [5, 6], and it requires conversion in vivo to colistin to unmask antibacterial activity [5, 7,8,9]. However, CMS is an extremely complex mixture of up to ~30 methanesulfonated derivatives for each colistin component, and the composition of CMS pharmaceutical products may vary from brand-to-brand and even from batch-to-batch [10]. The numerous chemical pathways for the de-methanesulfonation process of the multiple CMS components is likely to affect the overall time-course for in vivo generation of colistin. Indeed, in a study conducted in rats the time-course of plasma colistin concentrations differed across four brands of parenteral CMS [10]. As discussed in this chapter, the difference in the chemical forms of colistin and polymyxin B as administered to patients has a major effect on the pharmacokinetic profiles of the active forms of the two polymyxins, with significant clinical pharmacological implications [1].

Structures of colistin A and B, and polymyxin B1 and B2 are shown in the upper panel. In polymyxin B, D-Phe (phenylalanine) replaces the D-Leu (leucine) marked with the asterisk. The lower panel depicts structures of colistin methanesulfonate (CMS) A and B. All five primary amines of colistin are shown as being methanesulfonated, but it should be recognized that CMS is a complex mixture containing numerous partially methanesulfonated derivatives [1]. In addition, recently it has been shown that some primary amines may not be derivatized while others may have two methanesulfonate groups attached [128]. Fatty acid: 6-methyloctanoic acid for colistin A and polymyxin B1; and 6-methylheptanoic acid for colistin B and polymyxin B2. Thr: threonine; Dab: α,γ-diaminobutyric acid. α and γ indicate the respective –NH2 involved in the peptide linkage
The pharmacokinetic studies conducted on the polymyxins during the twentieth century relied on microbiological assays for quantification of the drugs in biological fluids; unfortunately, such methods are still used in some more recent studies. As discussed in Chap. 6, these methods have general limitations in regard to assay performance criteria such as selectivity (interference by coadministered antibiotics), sensitivity (inability to quantify low concentrations), accuracy and reproducibility. Microbiological assays are especially problematic for quantification of ‘colistin’ concentrations in biological fluids following administration of CMS [8]. These assays are incapable of differentiating the colistin present in a biological sample at the time of its collection from a patient and the colistin formed in vitro by hydrolysis of CMS during the microbiological assay. The resultant measured concentration of ‘colistin’ can substantially over-estimate the true concentration present in the patient, especially in the early hours after administration of a dose of CMS when its concentration vastly exceeds that of colistin (see below). Prior to 2015, the pharmacokinetic information provided in all product labels (summary of product characteristics (SPCs)) for CMS parenteral products around the world was still based upon the erroneous plasma concentration versus time profiles that were generated with microbiological assays decades ago [8, 11, 12]. Fortunately, in December 2014 the European Commission accepted recommendations of the European Medicines Agency (EMA) to modernize the SPCs in use across Europe [13]. It is regrettable that such a process has not yet occurred in the USA and many other parts of the world. In the present chapter, in general only pharmacokinetic data from studies in which appropriate sample handling and chromatographic methods [8] (see also Chap. 6) were used will be reviewed. Studies that do not satisfy these criteria will only be mentioned to highlight the importance of appropriate sample handling and bioanalytical procedures.
In the present chapter, the pharmacokinetic, and where possible the pharmacodynamic and toxicodynamic, data for CMS/colistin and polymyxin B will be reviewed in turn. For each of the polymyxins, initially data obtained following intravenous dosing will be considered as this is the most common mode of administration and the route for which most information exists. Where data are available, other routes, such as nebulization delivery to the airways and intrathecal dosing, will also be considered. As discussed in Chap. 7 in relation to disposition in animals and below in regard to humans, there are major and clinically important differences in the pharmacokinetics of CMS and colistin. However, there is no substantial difference in the pharmacokinetics of the major components of the polymyxins (colistin A and B; polymyxin B1 and B2) [14,15,16,17,18]. Therefore the pharmacokinetics of the individual components will not be considered in this review.
15.1 Colistin Methanesulfonate and Formed Colistin
Doses of CMS below are expressed in terms of both milligrams of colistin base activity (CBA) and number of international units (IU). The conversion factor between these two conventions is: 1 million IU is equivalent to ~33 mg CBA [19].
15.1.1 Intravenous Administration
Healthy Volunteers
With a new drug, typically the disposition in healthy volunteers is elucidated early in the clinical development of the drug, and this provides baseline information for comparison with data obtained later in various patients groups. There have been only three published reports in which chromatographic methods were used to define the disposition of CMS and colistin in healthy volunteers [18, 20, 21]. The first two of these studies were reported in 2011 while the third study was reported in 2018, approximately 50–60 years after colistin, administered as its inactive prodrug CMS, began to be used in patients.
In the first of these studies [18], the disposition of CMS and formed colistin was examined in 12 young (mean ± SD; 29.5 ± 5.5 years) healthy male volunteers (creatinine clearance 121 ± 18 mL/min) in France. The intravenous dose of CMS was 1 million IU (~33 mg CBA) and this was administered as a 1-h infusion. CMS was predominantly cleared by excretion into urine; the typical values of the total and renal clearances of CMS were 148 and 103 mL/min, respectively; the latter was similar to the glomerular filtration rate (GFR) estimated from the creatinine clearance values. As had been reported from an earlier study in patients with cystic fibrosis [22], the formation of colistin in vivo was observed in healthy human volunteers [18]. Maximum plasma concentrations of formed colistin were achieved at ~2 to 3 h after commencement of the CMS infusion. The terminal half-life of colistin (~3 h) was longer than that of the prodrug (~2 h) indicating that the disposition of formed colistin was rate-limited by its own elimination, not its conversion from CMS; [18] the same finding had been reported from an earlier study in rats [7]. Population pharmacokinetic analysis (structural model had two compartments for CMS and one compartment for colistin) was performed, but subject factors (e.g. renal function) influencing the disposition of CMS and colistin were not identified. This is not surprising given the small sample size and the fact that all subjects were healthy young males. Both CMS and formed colistin were recovered in urine; however, it was recognized based upon earlier studies in rats [7, 14], that much of the recovered colistin was formed from excreted CMS that underwent spontaneous hydrolysis in tubular and/or bladder urine. After correcting for this post-excretion conversion of CMS to colistin, the renal clearance of the latter was only 1.9 mL/min. While plasma protein binding of colistin was not examined in this study [18], subsequent studies have shown that colistin is approximately 50% bound in the plasma of humans [23]. Thus, the renal clearance value of colistin (1.9 mL/min) [18] is much less than the product of the unbound fraction in plasma and GFR indicating extensive renal tubular reabsorption of colistin that is formed from CMS prior to renal excretion. Very extensive renal tubular reabsorption of colistin following its direct administration to rats had been observed previously [14]. Based upon a urinary recovery of CMS and colistin (the latter mainly formed within the urinary tract) of approximately 70% of the administered dose, the authors speculated that ~30% of the prodrug was converted to colistin within the body prior to the renal excretion events [18]. This assumes that the only clearance pathways for CMS are conversion to colistin within the body and renal excretion. However, this approach almost certainly over-estimates the percentage conversion of the prodrug within the body, prior to renal excretion events. In an earlier study in rats in which the area under the plasma concentration-time curve of formed colistin after intravenous administration of CMS [7] was compared with the dose-normalized area after direct administration of colistin [14] it was found that only ~7% of a dose of CMS was converted to colistin within the body; [7] other similar studies in rats concluded that ~2.5–12% of a CMS dose was converted systemically to colistin [24,25,26]. These figures for percentage systemic conversion (~2.5–12%) [7, 24,25,26] are substantially lower than what would be predicted (~40%) based upon the urinary recovery (as CMS and colistin) of ~60% of the dose in rats intravenously administered CMS [7]. Thus, the percentage of a CMS dose converted systemically to colistin in the study in healthy young human volunteers was very likely lower than the 30% proposed by the authors [18]. Clearly, CMS is an extremely inefficient prodrug in both rats and humans with good kidney function.
In the second study in healthy human volunteers [20], the disposition of CMS and formed colistin was investigated in 15 male Japanese subjects (age 26.3 ± 6.7 years; creatinine clearance 125 ± 29 mL/min/1.73 m2). CMS was administered as a 0.5-h intravenous infusion of a single dose (2.5 mg/kg CBA (~75,000 IU per kg)); after a 7-day washout, 14 of the subjects received the same dose twice daily for 2.5 days. Non-compartmental pharmacokinetic analysis was used for the plasma and urinary data from both the single- and multiple-dose phases of the study [20]. It is not clear why the authors chose to extrapolate the area under the plasma concentration-time curve (AUC) to infinite time after both single and repeat dosing, because for a drug exhibiting linear pharmacokinetics the AUC to infinite time after the first (single) dose should be the same as the AUC across a dosage interval at steady state [27]. It is also not understood why the accumulation (steady-state) ratios for CMS and formed colistin were determined as the ratio of the respective AUC over a 12-h dosing interval on day 3 to the AUC to infinite time after the single dose; [20] both AUCs should have been determined over a 12-h interval following the respective doses [27]. In the Japanese subjects, ~40% of the dose of CMS was excreted in urine as CMS plus colistin [20]. The authors made the same assumption as that discussed for the study conducted in France [18] and therefore concluded that ~60% of each dose of CMS was converted to colistin in the body. As discussed above, that almost certainly over-estimates the actual extent of systemic conversion.
More recently, the pharmacokinetics of CMS and formed colistin in healthy Chinese subjects after single and multiple intravenous doses of a new product of CMS developed in China have been reported [21]. A total of 12 subjects (6 female and 6 male; age 25.6 ± 3.2 years; creatinine clearance 129 ± 9.8 mL/min) received nominally 2.5 mg CBA per kg as a single dose infused over 1 h; the same dose was administered twice daily for 7 days to another group of 12 subjects, again with an equal number of women and men (age 25.4 ± 3.2 years; creatinine clearance 133 ± 13.8 mL/min). Because the maximum dose to be administered at any time was 150 mg CBA, the average (range) actual CMS doses in the single- and multiple-dose studies were 2.36 (2.19–2.50) mg CBA per kg and 2.35 (2.01–2.50) mg CBA per kg, respectively. The plasma and urinary data on CMS and colistin were subjected to non-compartmental pharmacokinetic analysis. As for the study in Japanese subjects [20], AUC from zero time to infinity was determined for both the single- and multiple-dose studies in the Chinese subjects [21]. In the latter study, the accumulation ratio was determined as the ratio of the AUC over 12 h on day 7 of the multiple-dose study to the AUC over 12 h after the single dose in the other group of subjects. As occurred in the other two studies in healthy subjects [18, 20], the fraction of CMS converted to colistin in the body in the Chinese subjects was estimated as one minus the fraction of CMS in the body that was excreted in urine. Thus, the resulting estimate of ~37% for the fractional conversion of CMS to colistin in the Chinese subjects [21] very likely is an over-estimate of the actual fraction of each CMS dose converted systemically to colistin. Indeed, in discussing their results, the authors suggested that the fractional conversion of CMS to colistin may increase by three to fivefold in renally impaired patients [21], which is clearly not possible if the fractional conversion in healthy subjects (i.e. with normal kidney function) is actually ~37%.
Across all three studies in healthy subjects [18, 20, 21] there were a number of similarities [21]. In all studies: (a) the maximum plasma concentration of CMS occurred at the end of the infusion of the prodrug while that of formed colistin occurred 1–3 h later; (b) the maximum plasma concentration of CMS was substantially higher than that of colistin; (c) the terminal half-life of formed colistin was longer than that of its prodrug, indicating that the disposition of colistin was rate-limited by its own elimination; (d) the renal clearance of CMS (84–103 mL/min) was substantially greater than that of colistin (1.9–10.5 mL/min); (e) given that colistin is ~50% unbound in human plasma [23] the very low renal clearance of colistin in all three studies was consistent with extensive net renal tubular reabsorption; and, (f) the conversion of CMS to colistin in the body was far from complete, and the fraction converted was very likely over-estimated in all three studies.
The major difference across the three studies in healthy volunteers is the dose-normalized plasma concentration of formed colistin following a single dose. In the studies in Japanese [20] and Chinese [21] subjects, the CMS dose administered was approximately 150 mg CBA, and the average maximum plasma colistin concentration was 0.69 mg/L and 2.55 mg/L, respectively, representing ~3.7-fold difference across these two studies [21]. In the study conducted in France [18], the single dose administered was only 33 mg CBA and the average maximum plasma colistin concentration was 0.83 mg/L [21]. When normalized to a dose of 150 mg CBA, the maximum plasma colistin concentration predicted from the study in France would be ~3.8 mg/L. Thus, across the three studies the range of plasma colistin concentrations (0.69 mg/L to ~3.8 mg/L) differs by ~5.5-fold. The possible reasons for the wide range of plasma colistin concentrations achieved from the same dose of CMS are: (a) brand-to-brand or batch-to-batch variation in the composition of the CMS products administered; [10] (b) differing extents of unrecognized conversion of CMS to colistin during sample collection, processing and analysis; [5, 6, 28] and, (c) inter-ethnic differences.
Patients with Cystic Fibrosis
Reed et al. [29] were the first to report a study of the pharmacokinetics of ‘colistin’ in patients with cystic fibrosis in which an HPLC method was used for quantification of concentrations in biological fluids. The report did not indicate that blood samples had been collected and plasma harvested and stored in such a way as to minimize ex vivo conversion of CMS to colistin [30, 31]. In addition, unfortunately their analytical method involved heating at 54 °C for 2 h during preparation of the samples for HPLC analysis, conditions under which a substantial portion of the CMS contained within samples would undergo conversion to colistin [28]. This likely accounts for the very high plasma concentrations of ‘colistin’ and the very low values of clearance and volume of distribution that they reported, relative to later studies [22, 32]. Thus, the pharmacokinetic parameters reported from the study by Reed et al. [29] may not be reliable. This highlights the importance of ensuring that sample handling procedures and analytical methods do not lead to in vitro conversion of CMS to colistin.
Li et al. conducted a study in which the pharmacokinetics of CMS and formed colistin were determined across a dosage interval at steady state [22]. The study subjects were 12 patients (6 female; age 21.7 ± 6.9 years (mean ± SD); body weight 56 ± 9 kg) with cystic fibrosis, none of whom had renal impairment. According to the clinical protocol at the study site, patients weighing more than 50 kg received 2 million IU of CMS (~66 mg CBA) whereas those less than 50 kg were administered 1 million IU (~33 mg CBA) intravenously every 8 h, with each dose infused over 15–60 min. Plasma concentrations of CMS and formed colistin were quantified by two previously validated HPLC assays [33, 34]. Thus, this study was the first to demonstrate the in vivo conversion of CMS to colistin in humans and report the time-course of both species in plasma [22]. The total body clearance, volume of distribution and half-life of CMS were 2.01 ± 0.46 mL/min/kg, 340 ± 95 mL/kg and 124 ± 52 min, respectively. Colistin had a significantly longer mean half-life of 251 ± 79 min; the half-life of formed colistin was longer than that of CMS in each of the 12 patients. Apparent clearance of formed colistin was not reported in this study. It was noted that the protocol-driven dosage regimens employed resulted in plasma colistin concentrations across a dosage interval (maximum and minimum concentration ranges of 1.2–3.1 mg/L and 0.14–1.3 mg/L, respectively) that were low based upon emerging pharmacodynamic data at the time [35]. On that basis the authors suggested that dose-ranging studies to examine higher daily doses should be considered [22]. The dosage regimens used in this study (3–6 million IU per day, equivalent to ~100–200 mg CBA per day) [22] would now be regarded as low for patients without renal impairment.
More recently, the pharmacokinetics of intravenously administered CMS and the colistin formed from it were determined in cystic fibrosis patients as part of a study by Yapa et al. [32] to elucidate the pulmonary and systemic pharmacokinetics of inhaled and intravenous CMS. One of the study arms involved administration of a single intravenous dose of CMS (150 mg CBA, equivalent to ~4.5 million IU) infused over 45 min. The six study subjects were male patients with cystic fibrosis (age range 20–35 years; body weight 56–85 kg; creatinine clearance 103–148 mL/min/1.73m2). The mean ± SD values for clearance, volume of distribution and terminal half-life of the administered CMS were 5.96 ± 1.07 L/h, 16.9 ± 4.68 L and 2.66 ± 0.60 h, respectively. When clearance and volume of distribution are normalized for body weight they are in good agreement with the values reported several years earlier by Li et al. [22], and the CMS half-life values reported from both studies are also similar. Across the 6 subjects, the plasma concentrations of formed colistin increased slowly to maxima of 0.40–0.77 mg/L within ~5 h after commencement of the CMS infusion. The time of the maximum colistin concentration was later than had been observed in healthy volunteers [18] and the previous study in patients with cystic fibrosis [22]; this may be the result of brand-to-brand variability in the complex composition of CMS products with impact on the rate of in vivo conversion to colistin, as has been observed in rats [10]. In the study of Yapa et al. [32], the post-maximal colistin concentrations declined with a longer half-life than that of CMS in each subject (mean ± SD 7.34 ± 1.41 h). Of the intravenous dose of CMS, 40.0% ± 18.7% was recovered as CMS and colistin in urine collected over 24 h, with approximately half of the recovered CMS dose (19.5% ± 8.79%) in the form of colistin. Based on evidence from other studies [7, 18, 28], most of the colistin recovered in urine would have been formed within the urinary tract. An important finding of the study of Yapa et al. was that negligible concentrations of colistin were measured in sputum samples collected across the 12 h sampling period [32]. While care is needed in the interpretation of this finding from a single-dose study, it may have implications for the ability to achieve colistin concentrations in lung fluids sufficiently high to elicit an adequate antibacterial effect in the many hours after initiation of therapy; any binding of colistin to mucin [36] or other components within the lung must also be considered. This study also investigated the potential targeting advantage that may be achieved by inhalational administration [32], and this will be discussed below.
In summary, the two evaluable studies in patients with cystic fibrosis [22, 32] revealed pharmacokinetics of CMS and formed colistin that were remarkably consistent with each other and also with the overall disposition profile that has been observed in healthy volunteers [18]. Notable features were: a total clearance of CMS similar to creatinine clearance and dominated by renal excretion of CMS with subsequent ongoing formation of colistin within the urinary tract; relatively slow and variable formation of colistin from CMS; a terminal half-life of CMS of approximately 2–3 h while that of formed colistin was 1.5–2.5 times longer. All of these studies had relatively small numbers of subjects with quite homogeneous demographic and clinical presentations. Thus, while the studies provide essential information on the overall disposition profiles of CMS and formed colistin in healthy volunteers and patients with cystic fibrosis, they were not designed to explore the full spectrum of patient characteristics (e.g. renal function) that may influence the exposure to colistin from a given dosage regimen of CMS.
Critically-Ill Patients
Li et al. [37] were the first to report plasma concentrations of CMS and formed colistin in a critically-ill patient. The patient had developed multiple organ failure and was receiving continuous venovenous hemodiafiltration. The product information for CMS provided no guidance on dosage selection for such a patient and so the patient was administered 2.5 mg CBA (~76,000 IU) per kg every 48 h; this regimen had been proposed in a review on antibiotic dosing in patients receiving continuous renal replacement therapy [38], although there was no supporting data for the colistin regimen proposed. The case report of Li et al. [37] demonstrated that both CMS and colistin were cleared by the extracorporeal system and that plasma concentrations of colistin were substantially lower than 1 mg/L (the minimum inhibitory concentration (MIC) of the infecting organism) for ~85% of the 48-h dosage interval. Unfortunately, the patient succumbed. This report highlighted the urgent need for pharmacokinetic information to guide dosage regimens of CMS in various categories of critically-ill patients.
Subsequently, Makou et al. [39] reported plasma concentrations of formed colistin (CMS was not quantified) across a CMS dosage interval in 14 critically-ill patients (1 female, age range 18–84 years; body weight 60–85 kg; creatinine clearance 46–200 mL/min) who were receiving 3 million IU of CMS (~100 mg CBA) every 8 h. The half-life of formed colistin was 7.4 ± 1.7 h (mean ± SD). The maximum plasma colistin concentration within the dose interval ranged from 1.15 to 5.14 mg/L (2.93 ± 1.24 mg/L) and the corresponding range for the minimum concentration was 0.35–1.70 mg/L (1.03 ± 0.44 mg/L); 8 of the 14 patients had minimum plasma colistin concentrations less than 1 mg/L. A similar study was conducted by Imberti et al. [40] in 13 critically-ill patients (3 female, age range 20–70 years; body weight 55–110 kg; creatinine clearance 96–215 mL/min) who were receiving 2 million IU of CMS (~66 mg CBA) every 8 h. Blood samples were collected across a dosage interval at least 2 days after commencement of the regimen, and plasma was analysed for colistin concentration. The terminal half-life of colistin was 5.9 ± 2.6 h (mean ± SD) and maximum and minimum plasma colistin concentrations were 2.21 ± 1.08 mg/L and 1.03 ± 0.69 mg/L, respectively. The authors of both reports expressed concern about the relatively low plasma colistin concentrations achieved in their patients, all of whom had creatinine clearance greater than ~50 mL/min [39, 40]. Because of the small sample sizes in these studies, it was not possible to identify patient factors influencing exposure to colistin. A study of similar size (15 critically-ill patients) was reported by Karnik et al. [41]. The investigators collected blood samples after the first dose and across a dosage interval on the fourth day of therapy with CMS. The resultant plasma colistin concentration versus time profiles were highly unusual. The profiles for the two doses were very similar with little evidence of accumulation that has been observed in other studies [15, 31, 42]. The reported maximal plasma colistin concentrations were as high as 22–23 mg/L and occurred at the end of the 30 min infusion of CMS, while the median half-life of colistin after the first (2.7 h, range 1.1–4.6 h) and day 3–4 dose (3.3 h, 1.2–5.4 h) [41] was very short in comparison with other studies in critically-ill patients [15, 31, 39, 42]. It seems likely that these findings were the result at least in part of ex vivo conversion of CMS to colistin artificially elevating the measured plasma colistin concentrations at early time points after administration of a dose.
An important report by Plachouras et al. [15] identified a major problem that may arise if CMS regimens are commenced without administration of a loading dose. The study involved 18 critically-ill patients (6 female, age range 40–83 years; body weight 65–110 kg; creatinine clearance 41–126 mL/min). The CMS regimens (either 3 million IU (~100 mg CBA) or 2 million IU (~66 mg CBA) every 8 h) for these patients were commenced without administration of a loading dose. The result was a very gradual increase in plasma concentrations of formed colistin over several hours after the initial dose. Indeed, the population estimate of the terminal half-life of formed colistin in the critically-ill patients was ~14 h; thus, in the absence of a loading dose steady-state plasma concentrations of colistin would not be achieved before ~2 days of therapy. This raised concern about the possible negative impact on antibacterial effect in the early hours and days after initiation of a regimen. In a subsequent study from the same group [42], the pharmacokinetics of CMS and formed colistin were evaluated following administration of a loading dose of 6 million IU (~200 mg CBA) in 10 critically-ill patients (4 female, age range 32–88 years; body weight 60–140 kg; creatinine clearance 25–192 mL/min). As expected, the plasma concentrations of formed colistin across the first 8 h after this loading dose were higher than those observed in the earlier study by this group [15]. However, even with a loading dose of 6 million IU, only three of the ten patients had achieved a plasma colistin concentration of 2 mg/L by 8 h. A later study by this group examined the administration of a CMS loading dose of 9 million IU (~300 mg CBA) in 19 critically-ill patients (8 female, age range 18–86 years, body weight 50–120 kg, creatinine clearance 29–220 mL/min) [43]. The average maximum plasma concentration of formed colistin after the loading dose was 2.65 mg/L, with a time to maximum concentration of 8 h. It should be noted, however, that a wide variation was observed in the maximum plasma colistin concentration (0.95–5.1 mg/L) after the loading dose of CMS.
Based on these [15, 42, 43] and other [31, 44] studies CMS regimens should commence with a loading dose [13], but even when this is done there is still a delay of several hours in achievement of plasma concentrations of colistin greater than 2 mg/L. It is evident that the enormously complex chemistry of CMS [Chap. 3, 1] has the potential to lead to variation across brands and even across batches in the composition of partially methanesulfonated derivatives of colistin. This in turn may impact the rate of in vivo conversion of CMS to colistin, as has been observed in rats administered different brands of parenteral CMS [10]. The same situation may apply to CMS in patients, and indeed may account for the wide inter-patient variability in the plasma concentrations of formed colistin observed both after a loading dose and at steady state [31, 43,44,45]. The need for a loading dose in critically-ill patients would be greatest under circumstances where the rate of in vivo conversion to colistin is slow. Unfortunately, there is no a priori mechanism for determining the likely rate of in vivo conversion among different brands and batches of parenteral CMS.
The three aforementioned studies reported by the same group [15, 42, 43], involved collection of plasma samples after the first dose and again after the fourth to eighth dose administered every 8 h. The data from all three studies were pooled and subjected to population pharmacokinetic analysis [43]. The authors needed to invoke a different availability to account for the somewhat higher concentrations of the A and B forms of colistimethate and colistin measured in the third study. Patients with creatinine clearance >80 mL/min had a reduced ability to achieve maximum plasma colistin concentrations above 2 mg/L at steady state despite the administration of a maintenance dose of 9 million IU (300 mg CBA) per day. Only 4 of 12 patients with creatinine clearance >80 mL/min achieved maximum plasma concentrations of 2 mg/L at steady state, and the proportion of patients may have been even lower if an average steady-state plasma concentration (Css,avg) of colistin of 2 mg/L had been considered. This confirmed an earlier report which first indicated the difficulty of achieving such a colistin concentration in patients with good renal function [31].
Up until the end of 2016, the largest population pharmacokinetic study of CMS and formed colistin in critically-ill patients was that reported by Garonzik et al. [31]. The study involved 105 patients (37 female, age range 19–92 years; body weight 30–106 kg) with a wide range of renal function. The report [31] presented an interim analysis while the study proceeded to eventually recruit 215 patients; the population pharmacokinetic analysis on the full cohort of patients was reported in 2017 [46] and the results are discussed below.
Of the 105 patients in the interim population pharmacokinetic analysis [31], 89 were not receiving renal replacement therapy (creatinine clearance range 3–169 mL/min/1.73 m2) while 16 patients were recipients of such support (12 patients receiving intermittent hemodialysis and 4 continuous renal replacement therapy [3 continuous veno-venous hemodialysis, 1 continuous veno-venous hemofiltration]). The CMS dosage regimen for each patient was at the discretion of the respective treating physician (range of maintenance doses 75–410 mg CBA per day, equivalent to ~2.3 to 12.4 million IU per day). Blood samples for quantification of plasma CMS and colistin concentrations were collected across an inter-dosing interval on the third or fourth day of the CMS regimen. The plasma CMS and colistin concentrations achieved varied greatly across the patients (Fig. 15.2); there was an approximate 20-fold range in the Css,avg of the active antibacterial colistin (0.48–9.38 mg/L). Preliminary analysis of the data suggested that, in addition to the daily dose of CMS, renal function was an important contributor to the wide range of plasma colistin concentrations (Fig. 15.3). The data in this figure also indicate that in patients with creatinine clearance greater than ~80 mL/min/1.73 m2, administration of a daily dose of CMS at or in the vicinity of the upper limit of the recommended dose range (300 mg CBA (~9 million IU) per day) [8, 13, 47] was not able to reliably generate a plasma colistin Css,avg of 2 mg/L. This concentration may be regarded as a reasonable target, based upon: [47] translation of current evidence from pharmacokinetic/pharmacodynamic studies of colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in the mouse thigh infection model; [23] and, as discussed in more detail below, on the basis of pharmacokinetic/toxicodynamic analyses in patients which indicate that the risk of nephrotoxicity substantially increases above a plasma colistin Css,avg of ~2–3 mg/L [48, 49]. Unfortunately, in patients with creatinine clearance >80 mL/min it may not be possible to simply increase the daily dose of CMS to compensate for the low conversion to colistin because of the increased risk of nephrotoxicity , which is the major dose-limiting adverse effect [8, 50]. It should be noted that studies in the mouse lung infection model revealed that these infections were more resilient than those in the mouse thigh; [23] accordingly a plasma colistin Css,avg of 2 mg/L may be insufficient for such infections. Thus, combination therapy should be carefully considered in patients with relatively good renal function, especially if the MIC for the infecting organism is towards the upper end of the current breakpoint range and/or the patient is being treated for a respiratory tract infection [31, 47]. As discussed in Chap. 11, uncertainty still surrounds the effectiveness of colistin combination therapy compared with colistin monotherapy.

Steady-state plasma concentration versus time profiles of the prodrug CMS (Panel A) and formed colistin (Panel B) in 105 critically-ill patients (89 not on renal replacement, 12 on intermittent hemodialsys and 4 on continuous renal replacement therapy) [31]. Physician-selected CMS dosage intervals ranged from 8 to 24 h and hence the inter-dosing blood sampling interval spanned the same range. (Reproduced with permission from Garonzik et al. [31] Copyright © American Society for Microbiology, Antimicrobial Agents and Chemotherapy 55 (7):3284–3294. doi:https://doi.org/10.1128/AAC.01733-10)

Relationship of physician-selected daily dose of CMS (expressed as colistin base activity (CBA)) (Panel A) and the resultant steady-state plasma colistin concentration (Panel B) with creatinine clearance in 105 critically-ill patients [31]. (Reproduced with permission from Garonzik et al. [31] Copyright © American Society for Microbiology, Antimicrobial Agents and Chemotherapy 55 (7):3284–3294. doi:https://doi.org/10.1128/AAC.01733-10)
In the study by Garonzik et al. [31], the influence of renal function on the overall disposition of CMS and formed colistin was demonstrated via population pharmacokinetic analysis (structural model with two compartments for CMS and one compartment for colistin). Creatinine clearance was an important covariate on the total clearance of both CMS and colistin. It is easy to understand the dependence of total clearance of CMS on creatinine clearance because in animals [7] and humans [18] with good renal function, the prodrug CMS is mainly cleared by renal excretion; and only a relatively small fraction of a dose is converted to colistin [7, 18]. As a result, it is not unexpected that the total clearance of CMS declines with creatinine clearance. Animal studies involving direct administration of colistin have revealed that it is cleared predominantly by non-renal mechanisms, with only a very small fraction of the administered dose recovered in urine in unchanged form; [14] this is very similar to the disposition of polymyxin B in animals [17] and patients [16, 51]. Thus, it may seem surprising that renal function would influence the plasma Css,avg of formed colistin achieved from a given daily dose of CMS. This relationship occurs because of the relatively complex overall disposition of CMS and formed colistin (Fig. 15.4, left panel). As discussed above, in subjects with good kidney function only a small fraction of each dose of CMS is converted to colistin. Because CMS is cleared predominantly by renal excretion, as renal function declines a progressively larger fraction of each dose of CMS is converted to colistin. Thus, the apparent clearance of colistin (i.e. the actual clearance divided by the fraction of the CMS dose converted to colistin in a given patient) declines with reduction in kidney function [31]. Accordingly, creatinine clearance was the patient factor incorporated into the algorithm developed by the authors to calculate the CMS daily maintenance dose needed to generate a desired target plasma concentration of formed colistin in a patient not receiving renal replacement therapy. It should be noted, however, that at a given creatinine clearance there was a very large degree of inter-patient variability (up to ~tenfold) in the apparent clearance of colistin and consequently in the CMS dosage requirements to achieve a desired steady-state plasma colistin concentration, reflected by the data in Fig. 15.3. The variability probably arises because of the complexity of the chemical composition, and the inefficiency, of CMS as a prodrug (i.e. substantially less than 100% conversion in vivo ). These factors impact the fractional conversion to colistin at a given creatinine clearance [1]. This is consistent with a study in rats which demonstrated that the time-course of plasma concentration of formed colistin varied across four different brands of parenteral CMS [10]. The inter-patient variability in the plasma colistin concentration achieved at a given creatinine clearance and daily dose of CMS complicates the use of CMS, particularly since colistin has a low therapeutic index [1].

Overview of the pharmacokinetic pathways for colistin methanesulfonate (CMS) and colistin (left panel) and polymyxin B (right panel). The thickness of the arrows indicates the relative magnitude of the respective clearance pathways when kidney function is normal. CMS includes fully and all partially methanesulfonated derivatives of colistin. After administration of CMS, extensive renal excretion of the prodrug occurs with some of the excreted CMS converting to colistin within the urinary tract. In addition to renal clearance of CMS and conversion to colistin, one or more ‘other clearance’ pathways must exist for the prodrug, although the mechanisms involved are not known [1]. (Reproduced with permission from Nation et al. [1])
Of the 89 patients not on renal replacement in the above-mentioned report [31], all but two had been prescribed a CMS maintenance dose of 300 mg CBA (~9 million IU) per day or less, and 48% had a rise in serum creatinine of >50% from baseline; this is in keeping with nephrotoxicity rates of up to ~60% in other studies [50, 52,53,54,55]. In population pharmacokinetic/toxicodynamic analyses based upon 153 patients not receiving renal replacement at the start of CMS therapy (from the full cohort of 215 patients mentioned above), the time-course of changes in renal function after commencing CMS have been examined throughout the entire course of treatment [49]. Possible relationships with a range of factors, including pharmacokinetic exposure to CMS and colistin have been sought. As a component of these analyses, risk factors for a colistin-associated decline in kidney function have been identified. A creatinine clearance >80 mL/min and a plasma colistin Css,avg > 1.9–2.3 mg/L were identified as independent risk factors; the presence of both factors was at least additive [49]. In a study involving 102 patients receiving CMS, the pre-dose plasma colistin concentration on day 3 of CMS therapy was a predictor of acute kidney injury on day 7 and at end of treatment (the only two time-points considered). When the plasma colistin concentration on day 3 was evaluated as a categorical variable, the breakpoints that better predicted acute kidney injury were 3.33 mg/L on day 7 and 2.42 mg/L at end of treatment [48]. The end-of-treatment breakpoint of 2.42 mg/L was subsequently validated in a small study [56].
Sixteen of the critically-ill patients in the report of Garonzik et al. [31] were recipients of renal support at the time of initiating the CMS regimen (12 intermittent hemodialysis and 4 continuous renal replacement). The results from these patients revealed the substantial impact of these modalities on the plasma colistin concentration achieved from a given daily dose of CMS [31], in accord with other reports [37, 57,58,59,60,61,62,63,64,65,66,67]. The reason why renal replacement has such a large impact is easily understood when one considers that the rate of extracorporeal removal of a drug (or prodrug) is equal to the product of the intrinsic dialysis clearance (this is determined by the physicochemical properties of the drug (or prodrug) and the membrane) and the plasma concentration of the compound being delivered to the extracorporeal circuit. Not surprisingly given the similar molecular size of CMS and colistin, the intrinsic dialysis clearances of the two are of similar magnitude [37, 60, 63, 66]. However, the plasma concentration of CMS is substantially higher than that of colistin for a significant portion of a dosage interval (Fig. 15.2). Thus, much of the CMS/colistin that is removed by these renal replacement modalities is in the form of the prodrug, before it has had an opportunity to be converted in vivo to the active form, colistin. An additional reason for the impact of renal replacement relates to the respective handling of formed colistin in a functioning kidney versus that occurring in a renal replacement cartridge. In the kidney, an extremely large fraction of colistin filtered at glomeruli undergoes carrier-mediated tubular reabsorption [14], whereas in an extracorporeal cartridge no such reabsorprtion mechanism is in operation for colistin that diffuses into dialysate. The clinical implication is that dosage regimens for such patients must be carefully tailored to the circumstances. By way of population pharmacokinetic modeling, Garonzik et al. [31] proposed daily doses of CMS to achieve a target plasma colistin concentration in patients receiving intermittent hemodialysis. The dosage regimen algorithm included administration of a supplemental dose of CMS at the completion of dialysis to replace CMS and colistin lost during the session. These authors also provided an algorithm for the daily dose of CMS to achieve a desired plasma colistin concentration in patients receiving continuous renal replacement therapy [31]. In contrast to the efficient extracorporeal clearance of CMS and colistin via intermittent hemodialysis and continuous renal replacement [31], continuous ambulatory peritoneal dialysis has been reported to contribute little to the overall clearance of CMS and colistin; consequently it has been suggested that daily doses of CMS should not be increased to accommodate any drug loss via this renal support modality [68].
Following on from the interim analysis and dosage suggestions reported by Garonzik et al. [31], the results of the population pharmacokinetic analysis on the complete cohort of patients have been recently reported [46]. That analysis included data from a total of 215 patients, 29 of whom were receiving renal replacement therapy on the pharmacokinetic study day. Briefly, the characteristics for the 214 patients whose data were fully evaluable were: age range 19–101 years; body weight 30–122 kg; Appache II score 4–43; creatinine clearance 0–236 mL/min; 29 were receiving renal replacement therapy (16 intermittent hemodialysis, 4 sustained low-efficiency dialysis, 9 continuous renal replacement therapy)). Protein binding was conducted on plasma collected from 66 of the critically-ill patients in the study by Garonzik et al. and Nation et al. [31, 46]. The median and mean unbound fraction in plasma was approximately 0.5. The population pharmacokinetic analysis on the full cohort [46] confirmed the finding from Garonzik et al. [31] that the apparent clearance of formed colistin was related to the renal function of the patient. The impact of that relationship is shown in Fig. 15.5 which portrays the daily dose of CBA needed to achieve each 1 mg/L of plasma colistin Css,avg. The extensive inter-individual variability that had been observed by Garonzik et al. [31] is clearly evident in both panels of Fig. 15.5. The relationship in the right-hand panel of Fig. 15.5 formed the basis of a renal function-based algorithm to determine the daily dose needed to achieve a desired plasma colistin Css,avg. In developing the algorithm, the investigators needed to minimize the proportion of patients who would be likely to achieve plasma colistin concentrations that may increase the risk of colistin-associated nephrotoxicity [46]. Also reported from the population pharmacokinetic analysis were suggested daily doses for patients receiving renal replacement therapy; those suggestions included the need for any supplemental dosing after intermittent forms of dialysis and the timing of CMS dosing relative to the dialysis session [46]. All of the daily dose suggestions updated those that had been reported by Garonzik et al. [31] The ability of the updated daily dose suggestions to achieve a target plasma colistin Css,avg ≥ 2 mg/L were evaluated for various creatinine clearance clusters, for patients not in receipt of renal support. A plasma colistin Css,avg of 2 mg/L was chosen for the reasons discussed above and also because the MIC of the infecting pathogen may not be known at the time CMS treatment is initiated. While it was possible to achieve target attainment rates for a plasma colistin Css,avg of ≥2 mg/L in 80–90% of patients with creatinine clearance <80 mL/min, the attainment rate was only ~40% in patients with higher creatinine clearance. This highlighted the potential importance of considering active combination therapy, especially in patients with good renal function. Target attainment rates for a plasma colistin Css,avg of ≥2 mg/L with the proposed daily dose suggestions for patients receiving renal support were 85–89% [46].

Relationship between the dose of colistin base activity (CBA) per day needed for each 1 mg/L of plasma colistin Css,avg and creatinine clearance. Panels A and B show the relationship in linear-linear and log-linear forms, respectively. (Reproduced with permission from Nation et al. [46])
In summary, the study by Garonzik et al. [31] and Nation et al. [46] lead to the proposing of the first scientifically-based dosage regimens for various categories of critically-ill patients. The authors reported an algorithm for determination of a loading dose of CMS, based upon body weight being a covariate on the volume of the central compartment of CMS. Others have questioned whether it may be more appropriate to use a non-weight based loading dose [69]. Following on from the work of Garonzik et al. [31], Nation et al. [46] proposed daily maintenance doses of CMS for patients receiving or not receiving renal support.
Results from the final population pharmacokinetic analysis on the data from 162 patients not in receipt of renal replacement therapy have been used to evaluate the recently updated daily dose guidelines approved by the European Medicines Agency (EMA) [13] and the US Food and Drug Administration (FDA) [70], as reported [47]. The EMA-approved daily doses had greatly superior attainment rates for plasma colistin Css,avg ≥ 0.5, ≥1, ≥2 and ≥ 4 mg/L than did the dose suggestions approved by the FDA [47]. The EMA-approved dosing suggestions had been informed by a thorough review of recent pharmacological data [19], especially key studies discussed above [31, 42], while those approved by the FDA had not.
Little is known about the pharmacokinetics of CMS and colistin in pediatric patients. An investigation involving three patients aged 1.5 months (this patient was also studied during two other courses of CMS at 2.5 and 5.5 months of age), 5.5 years and 14 years (1 female; body weight range 6.2–40 kg) has been reported [71]. The CMS dosage regimens across the five courses were 0.06, 0.13, 0.2, 0.2 and 0.225 million IU/kg/day, corresponding to ~2.0, 4.3, 6.6, 6.6 and 7.4 mg/kg/day of CBA. The daily dose was divided and administered as 20-min infusions at 8 h intervals. After administration of at least 4 doses, blood samples were collected immediately before and 30 min after the end of a CMS infusion; cerebrospinal fluid samples were collected concomitantly (results discussed in the following paragraph). The authors noted that despite the CMS daily doses being in the range of those currently recommended and even higher, the serum colistin concentrations were greater than 2 mg/L in only the 14-year old patient (mean pre-dose and post-dose concentrations in this patient of 2.29 and 2.20 mg/L). Consequently, a key observation from this study was that CMS daily doses higher than previously recommended may be needed for pediatric patients to treat bloodstream infections caused by Gram-negative bacteria, especially if the causative organism exhibits borderline susceptibility to colistin [71]. The observation of the likely need for higher than traditionally used body-weight-based doses of CMS in neonates and in children is supported by results of other studies [72, 73]. Unfortunately, plasma colistin concentrations reported from another study as occurring in seven pediatric patients [74] were almost certainly spuriously high measurements due to ongoing conversion of CMS to colistin after samples were collected [75, 76]. Clearly, substantially more reliable information on the pharmacokinetics of colistin in pediatric patients is required to guide therapy.
There are very sparse data on the distribution of colistin into extravascular sites where infection may exist. The available data suggest that following intravenous administration of CMS, the concentrations of colistin in cerebrospinal fluid (CSF) [71, 77, 78] are only ~5% of concurrent plasma concentrations; very limited data from the study in pediatric patients discussed above suggest increased penetration in the presence of meningitis [71]. Similarly, the available data indicate that following intravenous administration of CMS, only relatively low concentrations of colistin are achieved in sputum of patients with cystic fibrosis [32] and in bronchoalveolar lavage (BAL) fluid from critically-ill patients [40, 79]. Care is required when interpreting the BAL concentrations from one of the latter studies [40] because the limit of quantification of the assay (0.05 mg/L) was for the analysed matrix (BAL) and there was ~100-fold dilution of the epithelial lining fluid (ELF) in performing the lavage procedure. Microdialysis studies in pigs have revealed ELF concentrations of colistin substantially lower than concomitant unbound concentrations in plasma [80]. Binding of colistin to mucin (and/or other components) in the airways must be considered as this may serve to decrease antibacterial activity [36]. As noted above, mouse lung infections were shown to be substantially more difficult to treat with colistin administered systemically than were thigh infections in the same species [23]. Thus, direct administration to these sites may be advantageous. Pharmacokinetics following intrathecal/intraventricular and inhalation administration of CMS are discussed below.
Burn Patients
Severely burned patients are very vulnerable to nosocomial infections with Gram-negative bacteria. Because burn injury can lead to many changes in physiological status, including blood flows, fluid distribution and glomerular filtration rate [81], it is important to understand the impact of such changes on the disposition of antibiotics. Lee et al. conducted a population pharmacokinetic study in 50 patients who had burns to 4–85% (mean ± SD; 50.5 ± 21.8%) of total body surface area [82]. The patients (11 female; age range 26–80 years; body weight 50–98 kg; creatinine clearance 23–309 mL/min, with 17 patients receiving continuous renal replacement therapy of unspecified type; 18 patients with edema) were administered 150 mg of CBA (~4.5 million IU) infused over 30 min every 12 h. Blood samples were collected before and across the 8 h following a CMS infusion, at least 3 days after the first dose of CMS. The plasma concentration versus time data for colistin (CMS was not quantified) were subjected to population pharmacokinetic analysis. The structural model involved one compartment for colistin; in the absence of plasma concentrations for the prodrug, a single compartment was assumed for CMS. The terminal half-life of colistin for the typical burn patient was 6.6 h, which is somewhat shorter than that observed in critically-ill patients [15, 31]. The identified covariates in the final model for burn patients were the presence of edema and, as in the study by Garonzik et al. in critically-ill patients [31], creatinine clearance. It was estimated that the fractional conversion of CMS to colistin in an anephric burn patient was approximately five times greater than in a patient with a creatinine clearance of ~120 mL/min, consistent with expectations (Fig. 15.4, left panel). The final population pharmacokinetic model was used in simulations for 1000 virtual burn patients to estimate steady-state values for the area under the plasma colistin concentration-time curve across a day at steady state for a dosage regimen of 150 mg CBA as a 30-min infusion every 12 h. Based upon the results of the simulation presented graphically in Figure 5 of the paper [82], this dosage regimen would achieve a plasma colistin Css,avg in a typical burn patient with a creatinine clearance <70 mL/min of ~1.5 mg/L and only ~1 mg/L in a patient with a creatinine clearance of ≥70 mL/min. The authors suggested that these relatively low concentrations may prompt the need to consider escalating doses above the currently approved dose that was used in this study. That patients with burns may require higher doses of CMS was supported by a recent brief report describing data from eight patients (median (IQR) age, weight, body mass index and serum creatinine concentration were 62 years (49–69 years), 69 kg (65–75 kg), 23.5 kg/m2 (21.8–26.4 kg/m2) and 0.75 mg/dL (0.68–1.36 mg/dL)) [83]. The patients received a median CMS dose of 2.25 million IU (2.00–3.00 million IU) (median ~74 mg CBA (IQR ~66–100 mg CBA)) every 12 h, but the plasma colistin concentrations were below the limit of quantification of the assay (~0.1–0.2 mg/L). The possible need for high doses in burn patients was exemplified in a case report describing a patient with burns who required a daily dose substantially higher than the currently approved dose range [84]. It is to be expected that renal function would be an important determinant of CMS dose requirements and that patients with augmented renal function in particular would be candidates for high doses. Data from two burn patients during continuous venovenous hemofiltration suggested efficient extracorporeal clearance of colistin [85], similar to findings in critically-ill patients as discussed above. However, it appears that CMS, rather than colistin (sulfate), was used to establish the calibration curve for colistin in this study.
Future investigations in burn patients should aim to provide additional information, including relationships of bacteriological and clinical outcomes and of nephrotoxicity with plasma colistin concentrations.
15.1.2 Direct Administration to the Lungs and Central Nervous System
One of the most common reasons for administration of CMS/colistin is the treatment of pulmonary infections such as ventilator-associated pneumonia in an intensive care setting. While the need to treat Gram-negative infections within the central nervous system (CNS) is less common, the critical and sensitive nature of this site poses special challenges. The common goal in treating these infections is the delivery of adequate colistin concentrations to the respective infection site, without exposing that site or systemic regions (e.g. kidneys) to concentrations likely to cause toxicity . As noted above, it is evident from the data available that intravenous administration of CMS results in relatively low colistin concentrations in CSF and pulmonary fluids. In contrast, direct administration to these sites leads to a very substantial targeting advantage, with significantly higher concentrations of colistin in the region to which CMS is delivered relative to the colistin concentrations reaching the systemic circulation.
A pulmonary targeting advantage has been demonstrated in six cystic fibrosis patients where relative sputum versus plasma exposures (area under the respective colistin concentration – time curve) were determined after intravenous and nebulization administration of CMS on different occasions [32]. The systemic availability of CMS was low (<10%) following nebulization of 2 and 4 million IU (~66 mg and 130 mg of CBA, respectively) and the plasma colistin concentrations were below the limit of quantification of the assay (0.125 mg/L). It was not possible in the study to perform bronchoalvealoar lavage and it remains to be determined whether the findings for sputum are representative of those for ELF. In this context, the relative concentrations of colistin in ELF and serum have been determined after inhalation of CMS (1 million IU (~33 mg CBA) every 8 h) in 20 mechanically ventilated critically-ill patients [86]. Median (25–75% interquartile range) colistin concentrations in ELF were 6.7 (4.8–10.1) mg/L, 3.9 (2.5–6.0) mg/L and 2.0 (1.0–3.8) mg/L at 1, 4 and 8 h post-nebulization, respectively. These concentrations were ~fivefold higher than those achieved concurrently in serum. The relative concentrations in the two biological fluids were not determined after intravenous administration and therefore it is not possible from this study to determine the targeting advantage of inhalational delivery. A recent study in adult critically-ill patients has demonstrated the ability of nebulized CMS to achieve concentrations of colistin in ELF much higher than concomitant concentrations in plasma, and substantially higher than the ELF colistin concentrations achieved with intravenous administration of CMS [79]. Similarly, a brief report has described six ventilated neonates (median gestational age 39 weeks (range, 32–39 weeks)) with ventilator-associated pneumonia who received a single dose of nebulized CMS (not clear if the dose was 120,000 IU/kg or 120,000 IU) [87]. Tracheal aspirate concentrations of colistin were very much higher than those in plasma. The results from these studies in cystic fibrosis [32] and critically-ill [79, 86,87,88] patients, and in a large animal model [89], suggest that inhalational delivery of CMS (perhaps together with intravenous administration) may be advantageous for the treatment of lung infections. This would be expected to allow attainment of higher colistin concentrations in lung fluid/tissue and at the same time lower systemic exposure to spare the kidneys. Recent clinical studies and systematic reviews and meta analyses reported in Chap. 11 and elsewhere [90,91,92,93,94,95,96] are suggestive of a beneficial effect of nebulized CMS either together with intravenous CMS or alone (including non-inferiority to intravenous CMS but with lower incidence of nephrotoxicity). A recent report of a relatively small retrospective clinical study suggested lack of benefit of inhaled CMS added to intravenous CMS versus intravenous CMS, although the study may have been confounded by a very high proportion of patients in both groups who were co-administered at least one other antibiotic [97]. Larger well-controlled studies are required.
Assuming no or negligible publication bias, there is a growing body of evidence that intrathecal or intraventricular administration of CMS (often in combination with intravenous therapy) is a generally safe and effective treatment for CNS infections (ventriculitis/meningitis) caused by Gram-negative bacteria [98,99,100,101,102,103,104]. From a relatively low dose of CMS administered via these routes, it is possible to achieve CSF colistin concentrations substantially higher than can be achieved by intravenous administration of a very much larger dose [71, 77, 78]. Interestingly, there is little information on the plasma colistin concentrations achieved from the relatively low doses of CMS administered by the intrathecal or intraventricular routes, but it would be expected that the systemic exposure would be low. It is not surprising that colistin-associated nephrotoxicity is an extremely rare occurrence with intrathecal or intraventricular administration [98, 100]. Importantly, administration of CMS via these routes gives rise to concentrations of colistin in CSF that could never be safely achieved with intravenous administration alone.
15.2 Polymyxin B
At the outset, it is important to recognize that there has been only one report of a pharmacokinetic/pharmacodynamic study on systemically administered polymyxin B (against Klebsiella pneumoniae) in an animal infection model [105], and there has not been a prospective pharmacokinetic/toxicodynamic study in patients to identify the association between plasma polymyxin B concentration and risk of nephrotoxicity. However, recently Lakota et al. proposed a tentative plasma polymyxin B concentration range of 2–4 mg/L [106] based upon animal pharmacokinetic/pharmacodynamic data for colistin against P. aeruginosa and A. baumannii [23] and polymyxin B against K. pneumoniae [105] to define the lower end of the range, and a pharmacokinetic/toxicodynamic meta-analysis of retrospective nephrotoxicity data gleaned from the literature to define the upper end of the range [106].
Studies on the disposition of intravenously administered polymyxin B in healthy humans with analysis of concentrations in plasma by chromatographic methods have not been performed. There is also a lack of information on pharmacokinetics of polymyxin B after administration to patients via routes other than intravenous (e.g. inhalation , intrathecal, intraventricular). Thus, the focus of the review below will be the pharmacokinetics of polymyxin B following intravenous administration in patients, most of whom were critically ill. The review will only include studies in which investigators used chromatographic methods for the analysis of drug concentration in biological fluids.
15.2.1 Intravenous Administration
The first reports on the disposition of polymyxin B in the modern era were in 2008 [16, 107]. The study by Kwa et al. [107] was conducted in nine adult patients (one female; age range 16–70 years; body weight 46–80 kg), all of whom were described as being among the general patient population and having normal renal function; patients with cystic fibrosis were excluded. The dosage regimens were at the discretion of the attending physicians and ranged from 0.75 to 2 million units (~75–200 mg of polymyxin B base) per day; each dose was infused over 2–6 h. Sparse blood sampling was performed (19 samples from the 9 patients) and serum was analysed for the concentration of one of the components (polymyxin B1) of the material administered. The resulting data were subjected to population pharmacokinetic analysis (one-compartment structural model) that yielded a typical half-life and clearance of 13.1 h and 2.2 L/h, respectively. The latter value may have over-estimated the clearance because only polymyxin B1 was quantified.
Zavascki et al. [16] studied the pharmacokinetics of polymyxin B in eight critically-ill patients (four female; age range 42–86 years; body weight 50–80 kg; creatinine clearance <10 to 246 mL/min). The physician-selected dosage regimens of polymyxin B ranged from 0.5 mg/kg (~5000 units/kg) every 48 h to 1.25 mg/kg (~12,500 units/kg) every 12 h. At the time, it was the practice at the clinical site to use lower daily doses in patients with poorer kidney function; three patients with the lowest creatinine clearances (<10, <10 and 26 mL/min) were dosed every 48 h and the remainder were dosed every 12 h. Each dose was infused over 60 min, and after at least 2 days of therapy seven blood samples were collected from each patient across a dosage interval including samples at the end of the 1-h infusion and at 0.5, 1 and 2 h thereafter. The two patients with creatinine clearance <10 mL/min were receiving intermittent hemodialysis; they were studied on a non-dialysis day. Importantly, for four patients it was possible to quantitatively collect urine over the dosage interval. The samples of plasma and urine were analysed for the concentration of polymyxin B with the resultant data subjected to non-compartmental pharmacokinetic analysis. Peak plasma concentration at the end of the short-term infusion ranged from 2.38 to 13.9 mg/L. There were several key findings from the pharmacokinetic analysis. First, after the infusion ceased the plasma polymyxin B concentrations declined in a multi-exponential manner indicating that more than one pharmacokinetic compartment was involved in the disposition of polymyxin B. Second, notwithstanding the diverse renal functions in the patients, there was remarkably little inter-individual variability in the total body clearance (range, 0.27–0.81 mL/min/kg) and volume of distribution (71–194 mL/kg) of polymyxin B. Third, urinary recovery of unchanged drug was extremely low, with each of the 4 patients for whom data were available excreting <1% of the administered dose in urine. The same very low urinary recovery (<1%) has been observed in rats for polymyxin B [17] and colistin [14], each after administration of the respective sulfate salt. Fourth, consideration of the relative magnitudes of the calculated glomerular filtration clearance of polymyxin B and the ultimate overall renal clearance indicated that very extensive renal tubular reabsorption must have been occurring in the patients. Again, a similar conclusion was reached for the renal handling of colistin in rats [14]. Fifth, and arguably most important from a clinical pharmacokinetic perspective, even though the renal clearance of polymyxin B varied ~12-fold among patients, consistent with the wide range of creatinine clearance values, there was very little variability (~threefold range) in the total body clearance of polymyxin B. This arose because renal clearance was only a very small component (<1%) of the total body clearance. The study by Zavascki et al. [16] was the first to imply that dose adjustment based upon renal function (as may be appropriate for CMS) may not be required for polymyxin B, a proposition supported by the pharmacokinetic data from a single case report 3 years later involving a patient with renal insufficiency [108].
Subsequently, Sandri and coworkers reported on a larger study on the pharmacokinetics of polymyxin B in critically-ill patients [51]. That study involved 24 patients, including 2 receiving continuous venovenous hemodialysis (CVVHD) for whom greater detail was reported separately [109]. The 24 patients were typical of the intensive care setting (11 female; age range 21–87 years; total body weight 41–250 kg; lean body weight 29–99 kg; creatinine clearance in the 22 non-CVVHD patients 10–143 mL/min). One patient was extremely obese (250 kg); the next heaviest patient was 110 kg. The study design was similar to that described in the earlier report from the same group as discussed above [16]; in the study of Sandri et al. eight blood samples were collected across the dosage interval from each patient, with even more intensive sampling shortly after the end of the polymyxin B infusion [51]. The plasma polymyxin B concentration – time profiles resulting from the physician-selected dosage regimens are shown in Fig. 15.6. These data were well described in a population pharmacokinetic analysis (two-compartment structural model). The concentration-time profiles from all patients (i.e. on and not on renal replacement therapy) were successfully described simultaneously with one set of population pharmacokinetic parameter estimates because only a small amount was removed by CVVHD (12.2% and 5.62% for the two patients). The apparent small effect of extracorporeal clearance on total body clearance and hence dosage requirements of polymyxin B contrasts with the situation for CMS/colistin [31, 37, 57, 59, 62]. This difference may be explained by the fact that in the latter case a substantial amount of the CMS/colistin removed by the cartridge is in the form of the prodrug which circulates in plasma at concentrations that are generally higher than those of colistin across the early stage of a dosage interval. Sandri et al., the authors of the population pharmacokinetic study on polymyxin B, reported that the median unbound fraction in plasma was 0.42 [51]. Urinary excretion data for polymyxin B were available for 17 non-CVVHD patients. The median percentage of the polymyxin B dose excreted in urine was 4.04% (range 0.98–17.4%). Thus, renal clearance was only a very small component of the total body clearance. While renal clearance of polymyxin B is a minor elimination pathway, it is important to understand the renal handling mechanisms because of the very high probability of a link with the propensity to cause nephrotoxicity. Application of physiological concepts to the polymyxin B renal clearance data revealed extensive renal tubular reabsorption [51], consistent with previous findings for polymyxin B in critically-ill patients [16].

In the study reported by Sandri and coworkers, the population estimate of polymyxin B clearance not adjusted for body weight was 1.87 L/h and that scaled by body weight was 0.0276 L/h/kg [51]. In view of the minor contribution of renal clearance to total clearance, it was not surprising that the population pharmacokinetic analysis did not identify creatinine clearance as a covariate of the total body clearance of polymyxin B (Fig. 15.7). The use of unscaled (i.e. absolute) clearance (left panel of the figure) resulted in the obese patient being an outlier. In the population pharmacokinetic analysis, linear scaling of polymyxin B clearance by total body weight reduced the unexplained between subject variability. Allometric scaling of polymyxin B clearance by total body weight, and linear and allometric scaling by lean body weight were also considered, but they resulted in only marginal improvement. To simplify application in the clinic, the results from the model with linear scaling by total body weight were adopted. After linear scaling of clearance by total body weight, the parameter estimates for the two patients receiving CVVHD, including the patient with 250-kg total body weight, were within the range of estimates from the other patients (Fig. 15.7, right panel). It is evident that neither the unscaled nor the scaled polymyxin B clearance values were related to creatinine clearance. Notwithstanding the very wide range of total body weights and creatinine clearance values there was only approximately threefold variation in the total body weight scaled clearance values of polymyxin B (Fig. 15.7, right panel). Accordingly, the population pharmacokinetic model yielded a between subject variability in clearance (coefficient of variation 32.4%) that was remarkably low in this diverse group of critically-ill patients [51]. The authors went on to perform Monte Carlo simulations for a number of clinically relevant dosage regimens scaled by total body weight.

Individual polymyxin B clearance estimates versus creatinine clearance. Polymyxin B clearance was either unscaled (L/h, left panel) or scaled by total body weight (L/h/kg, right panel). Open circles represent patients not receiving continuous venovenous hemodialysis (CVVHD), the filled diamond represents the CVVHD patient who weighed 250 kg, and the filled triangle the lean CVVHD patient [51]. (Reproduced with permission from Sandri et al. [51])
In 2017, Thamlikitkul et al. reported pharmacokinetic data on polymyxin B in 19 patients (creatinine clearance range 15–110 mL/min) [110]. At least 48 h after initiating polymyxin B, four blood samples were collected across a dosage interval. The resulting plasma concentration-time data were subjected to two forms of analysis: a standard two-stage approach which involved a one-compartment model being used to describe the data; and a maximum a posteriori Bayesian approach (which used the results of Sandri et al. [51] as Bayes priors) resulting in a two-compartment model providing the best fit to the data. The primary subsequent analysis involved comparison of the dose-normalized plasma exposure to polymyxin B in 5 patients with creatinine clearance ≥80 mL/min with that of the 14 patients with creatinine clearance <80 mL/min; there was no difference in exposure (P > 0.4) [110], consistent with results reported previously [16, 51, 108].
In a paper published in 2018 Manchandani et al. reported a population pharmacokinetic study in 35 patients (12 female; age range 25–89 years; body weight 36–112 kg; creatinine clearance 15–175 mL/min) [111]. Each polymyxin B dose was infused over 1–3 h, and four blood samples were collected over one dosage interval at steady state (prior to the dose, at 1–2 h and 8–12 h after the end of the infusion, and prior to the next dose). Population pharmacokinetic analysis was conducted and a one-compartment model was chosen as the best-fit model. The population estimate of polymyxin B clearance was 2.5 L/h, a value similar to those reported from earlier studies [16, 51, 107, 110], and the inter-patient variability was similar to that reported by Sandri et al. [51]. In the study reported by Manchandani and coworkers, aside from one patient who presented as an outlier, the polymyxin B clearance estimates for the remaining 34 patients were within an approximate four to fivefold range across the creatinine clearance spectrum represented in this patient population. While creatinine clearance was found to be a statistically significant covariate of polymyxin B clearance, the magnitude was regarded as clinically insignificant [111]. Volume of distribution of polymyxin B was poorly predicted by actual body weight, but given the small number of samples collected from each patient, the timing of the samples and the resultant application of a one-compartment model it is possible that the volume estimates were subject to bias.
Kubin et al. [112] and Miglis et al. [113] published companion papers in 2018 reporting the population pharmacokinetics of polymyxin B in patients, other than those with cystic fibrosis. Both studies involved sparse blood sample collection (samples collected primarily for therapeutic drug monitoring (TDM)) with an average of three blood samples per patient. The study by Kubin and coworkers included 43 patients while that of Miglis et al. included data for 52 patients, but 43 of those patients were from the study of Kubin et al. (i.e. 83% of the patients reported in the second study were also included in the first study). Interestingly, even though the data from the 43 patients in the study of Kubin et al. were used to develop the base model described in Miglis et al., the former study found that a one-compartment model satisfactorily described the data while a two-compartment model proved superior in the study by Miglis et al. [112, 113]. A finding in common across both reports was that actual body weight was not a covariate of polymyxin B clearance; the commonality of findings is not surprising given the very large degree of overlap in the patient populations across the two studies. The mean population estimates of polymyxin B clearance reported by Kubin et al. (2.37 L/h) and Miglis et al. (2.63 L/h) were similar to those reported previously [51, 110, 111]. However, it was noticeable that the inter-patient variability in polymyxin B clearance was substantially greater than in the earlier studies; even after disregarding an outlier, the report by Miglis et al. suggests there was >tenfold inter-patient variability in the clearance of polymyxin B [113]. It is possible that this difference across studies arose because of the sparse sampling used by Miglis et al.; the resultant inability to accurately define area under the plasma concentration-time curve very likely resulted in biased estimates of clearance [112]. The sparse blood sampling approach was also used by the same group to undertake a pilot pharmacokinetic study in nine adult patients with cystic fibrosis [114]. In that study, patients had a median of two blood samples collected during polymyxin B therapy for the purpose of TDM. The authors commented that while the pharmacokinetic parameters in the patients with cystic fibrosis were similar to those without this condition, substantial care is required in regard to interpreting this and other findings because of the small number of patients and the sparse blood sampling [114].
It is instructive to consider the relationship between polymyxin B clearance and creatinine clearance using data pooled from studies reviewed above (Fig. 15.8) [16, 51, 108, 109]. It is evident from these studies and more recent investigations [110, 111, 113] that polymyxin B total body clearance is not influenced by renal function to a clinically important extent. It is also evident from Fig. 15.8 that polymyxin B clearance is subject to a remarkably low degree of inter-individual variability across the wide range of creatinine clearance values examined. These two characteristics contrast sharply with those for the apparent clearance of formed colistin after administration of CMS, as discussed above. The clinically insignificant effect of kidney function on the total clearance of polymyxin B is behaviour expected of a drug that is excreted in urine to only a minor extent; see Fig. 15.4 (right panel) for a diagrammatic representation of the overall disposition of polymyxin B. Therefore, based on currently available data, in contrast to information provided in package inserts for polymyxin B, daily doses should not be based on renal function. A reduction in daily dose in a renally-impaired patient will potentially have negative impact on microbiological and clinical responses [51, 108, 115].

15.3 Commentary and Conclusions
From the foregoing, it is clear that while very significant progress has been made in understanding key features of the clinical pharmacology of colistin and polymyxin B, there is still much to be learned. Large clinical population pharmacokinetic, pharmacodynamic and toxicodynamic studies have been performed on colistin, but there are much sparser data for polymyxin B and large-scale studies are urgently needed. There is a dearth of pharmacokinetic information for both polymyxins in important groups, including obese patients, those with burns, and neonates and children. There is also need to delineate the role of inhaled polymyxin in treatment of pneumonia, and of when and how to implement polymyxin combination regimens [12].
Even at this stage, it is very evident that while both clinically used polymyxins share similar characteristics in some areas (e.g. similar in vitro antibacterial activity against important species of Gram-negative bacteria (Chap. 3) [2, 3], propensity to cause damage to renal tubular cells (Chap. 18) [52, 116]), they differ substantially in some important aspects of their clinical pharmacology . The difference in in vivo behaviour arises because colistin is administered as an inactive prodrug (CMS) that is predominantly renally cleared before significant conversion can occur, while polymyxin B is administered directly as its sulfate salt (Fig. 15.4).
The potential clinical pharmacological consequences of the different disposition profiles have been discussed previously [1] and therefore they will be presented only briefly here. Firstly, the generally slow, incomplete and variable (due to batch-to-batch variability in CMS) in vivo conversion to colistin often impedes the ability to achieve desired plasma concentrations of colistin in the first several hours of therapy, even with a loading dose [10, 31, 42, 43]. This may have a negative impact on prognosis given the link between delayed initiation of appropriate antibiotic therapy and patient outcome [117, 118]. As noted earlier, the need for a loading dose would be greater if administering a parenteral product of CMS that is subject to very slow in vivo conversion to colistin; unfortunately, the rate of conversion of the product available in a given hospital is generally not known by the clinicians caring for a patient. Because polymyxin B is not administered as a prodrug it is possible to more rapidly attain plasma concentrations that are likely to be effective [51]. Second, in patients with creatinine clearance greater than ~80 mL/min, the avid renal clearance of CMS competes very successfully with the pathway for conversion to colistin (Fig. 15.4), and therefore it is not possible to reliably achieve plasma colistin concentrations of ≥2 mg/L in such patients, even with daily doses at the upper limit of the current prescribing information (Fig. 15.3) [31, 43, 46, 47]. This is not the case with polymyxin B [16, 51]. Third, because the apparent clearance of colistin is related to creatinine clearance, the daily maintenance dose of CMS may require adjustment based upon renal function [31, 43, 46, 47]. Based upon current evidence, this is not appropriate for polymyxin B because its total body clearance is not dependent on creatinine clearance to a clinically significant degree [16, 51, 108, 110, 111]. Fourth, based upon studies with intensive blood sampling across a dosage interval to define the area under the plasma concentration versus time curve, there is substantially greater inter-individual variability in the apparent clearance of colistin at a given creatinine clearance (up to ~tenfold variability) [31, 46] than there is in the clearance of polymyxin B across a very wide range of creatinine clearance values (only ~threefold variability in the clearance of polymyxin B) [51]. This point together with that immediately above means that it is substantially easier to predict a daily maintenance dose to achieve a desired steady-state plasma concentration for polymyxin B than it is for colistin. Fifth, both colistin and polymyxin B have low therapeutic indices and as such TDM is likely to be beneficial [12]. Some laboratories are already providing a TDM service to assist in optimization of dosage regimens [37, 84, 119, 120]. There is an even stronger case for TDM (and ideally adaptive feedback control (AFC) [121]) with CMS/colistin because of the very substantial variability in the overall pharmacokinetic profile. However, that variability will render difficult the successful implementation of AFC for colistin. In addition, TDM is inherently more difficult for CMS/colistin, because of the critical need to ensure that all sample collection, handling and assay procedures do not lead to ongoing in vitro conversion of CMS to colistin [30]. Thus, application of TDM and AFC is expected to be more straightforward for polymyxin B than for CMS/colistin; indeed, early work towards the application of AFC for polymyxin B has already occurred [106]. Sixth, CMS/colistin and polymyxin B differ in the concentrations of the active antibacterial that can be achieved in urine. This occurs because CMS is extensively excreted into urine and then it undergoes partial conversion to colistin within the urinary tract as a result of instability of CMS in an aqueous environment; [7, 18, 122] in contrast, polymyxin B undergoes minimal excretion into urine [16, 51] (Fig. 15.4).
Thus, polymyxin B would appear to have superior pharmacokinetic characteristics for infections where it is important to rapidly and reliably achieve and then maintain a desired concentration in plasma. Interestingly, a meta-analysis of five recent clinical studies [52, 53, 123,124,125] which compared relative rates of nephrotoxicity associated with CMS/colistin versus polymyxin B revealed that the risk was lower for polymyxin B [126], probably related to the administration of colistin as a prodrug [127]. CMS may be the preferred polymyxin for the treatment of urinary tract infections [1]. The relative safety and effectiveness of CMS and polymyxin B delivered directly to sites such as the lungs and CSF needs to be evaluated. CMS may be less irritating to membranes lining the lungs and to the meninges, and there may be ongoing conversion to colistin from the prodrug ‘trapped’ in these regions due to its slow absorption into the systemic circulation and eventual delivery to the kidneys. It would be advantageous for clinicians in all parts of the world to have access to parenteral products of both CMS and polymyxin B, so that they can choose between the two in particular circumstances [12]. Head-to-head comparisons of the two polymyxins against various types of infections are needed, and such studies should evaluate both efficacy and toxicity endpoints. A compilation of high priority aspects requiring attention to optimize the clinical use of the polymyxins has been formulated [12]. It is hoped that progress on addressing these areas will be made with good speed. In the meantime, it is pleasing that international consensus guidelines for the optimal use of the polymyxins have been published [129]. The information used to formulate the dosing regimens of colistin and polymyxin B recommended in those guidelines was from key studies reviewed in the present chapter.
References
Nation RL, Velkov T, Li J (2014) Colistin and polymyxin B: peas in a pod, or chalk and cheese? Clin Infect Dis 59:88–94. https://doi.org/10.1093/cid/ciu213
Gales AC, Jones RN, Sader HS (2011) Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006–09). J Antimicrob Chemother 66(9):2070–2074. https://doi.org/10.1093/jac/dkr239
Sader HS, Rhomberg PR, Farrell DJ, Jones RN (2015) Differences in potency and categorical agreement between colistin and polymyxin B when testing 15,377 clinical strains collected worldwide. Diagn Microbiol Infect Dis 83(4):379–381. https://doi.org/10.1016/j.diagmicrobio.2015.08.013
Kwa A, Kasiakou SK, Tam VH, Falagas ME (2007) Polymyxin B: similarities to and differences from colistin (polymyxin E). Expert Rev Anti-Infect Ther 5(5):811–821. https://doi.org/10.1586/14787210.5.5.811
Barnett M, Bushby SR, Wilkinson S (1964) Sodium sulphomethyl derivatives of polymyxins. Br J Pharmacol Chemother 23:552–574
Bergen PJ, Li J, Rayner CR, Nation RL (2006) Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob Agents Chemother 50(6):1953–1958. https://doi.org/10.1128/AAC.00035-06
Li J, Milne RW, Nation RL, Turnidge JD, Smeaton TC, Coulthard K (2004) Pharmacokinetics of colistin methanesulphonate and colistin in rats following an intravenous dose of colistin methanesulphonate. J Antimicrob Chemother 53(5):837–840. https://doi.org/10.1093/jac/dkh167
Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL (2006) Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis 6(9):589–601. https://doi.org/10.1016/S1473-3099(06)70580-1
Gregoire N, Aranzana-Climent V, Magreault S, Marchand S, Couet W (2017) Clinical pharmacokinetics and pharmacodynamics of colistin. Clin Pharmacokinet 56:1441–1460. https://doi.org/10.1007/s40262-017-0561-1
He H, Li JC, Nation RL, Jacob J, Chen G, Lee HJ, Tsuji BT, Thompson PE, Roberts K, Velkov T, Li J (2013) Pharmacokinetics of four different brands of colistimethate and formed colistin in rats. J Antimicrob Chemother 68(10):2311–2317. https://doi.org/10.1093/jac/dkt207
Theuretzbacher U (2014) Product information for parenteral colistin varies substantially across Europe. J Antimicrob Chemother 69:1987–1992. https://doi.org/10.1093/jac/dku064
Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, Mouton JW, Paterson DL, Tam VH, Theuretzbacher U, Tsuji BT, Turnidge JD (2015) Framework for optimisation of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect Dis 15(2):225–234. https://doi.org/10.1016/s1473-3099(14)70850-3
European Medicines Agency (2014) Polymyxin-containing medicines. Polymyxin Article 31 referral – annex III amendments to relevant sections of the summary of product characteristics and the package leaflets. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Polymyxin_31/WC500176332.pdf. Last accessed 16 Jun 2019
Li J, Milne RW, Nation RL, Turnidge JD, Smeaton TC, Coulthard K (2003) Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob Agents Chemother 47(5):1766–1770
Plachouras D, Karvanen M, Friberg LE, Papadomichelakis E, Antoniadou A, Tsangaris I, Karaiskos I, Poulakou G, Kontopidou F, Armaganidis A, Cars O, Giamarellou H (2009) Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by Gram-negative bacteria. Antimicrob Agents Chemother 53(8):3430–3436. https://doi.org/10.1128/AAC.01361-08
Zavascki AP, Goldani LZ, Cao G, Superti SV, Lutz L, Barth AL, Ramos F, Boniatti MM, Nation RL, Li J (2008) Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis 47(10):1298–1304. https://doi.org/10.1086/592577
Abdelraouf K, He J, Ledesma KR, Hu M, Tam VH (2012) Pharmacokinetics and renal disposition of polymyxin B in an animal model. Antimicrob Agents Chemother 56(11):5724–5727. https://doi.org/10.1128/AAC.01333-12
Couet W, Gregoire N, Gobin P, Saulnier PJ, Frasca D, Marchand S, Mimoz O (2011) Pharmacokinetics of colistin and colistimethate sodium after a single 80-mg intravenous dose of CMS in young healthy volunteers. Clin Pharmacol Ther 89(6):875–879. https://doi.org/10.1038/clpt.2011.48
European Medicines Agency (2014) Assessment report on polymyxin-based products. Referral under Article 31 of Directive 2001/83/EC. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Polymyxin_31/WC500179664.pdf. Last accessed 16 Jun 2019
Mizuyachi K, Hara K, Wakamatsu A, Nohda S, Hirama T (2011) Safety and pharmacokinetic evaluation of intravenous colistin methanesulfonate sodium in Japanese healthy male subjects. Curr Med Res Opin 27(12):2261–2270. https://doi.org/10.1185/03007995.2011.626557
Zhao M, Wu XJ, Fan YX, Zhang YY, Guo BN, Yu JC, Cao GY, Chen YC, Wu JF, Shi YG, Li J, Zhang J (2018) Pharmacokinetics of colistin methanesulfonate (CMS) in healthy Chinese subjects after single and multiple intravenous doses. Int J Antimicrob Agents 51(5):714–720. https://doi.org/10.1016/j.ijantimicag.2017.12.025
Li J, Coulthard K, Milne R, Nation RL, Conway S, Peckham D, Etherington C, Turnidge J (2003) Steady-state pharmacokinetics of intravenous colistin methanesulphonate in patients with cystic fibrosis. J Antimicrob Chemother 52(6):987–992. https://doi.org/10.1093/jac/dkg468
Cheah SE, Wang J, Nguyen VT, Turnidge JD, Li J, Nation RL (2015) New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother 70(12):3291–3297. https://doi.org/10.1093/jac/dkv267
Marchand S, Gobin P, Brillault J, Baptista S, Adier C, Olivier JC, Mimoz O, Couet W (2010) Aerosol therapy with colistin methanesulfonate: a biopharmaceutical issue illustrated in rats. Antimicrob Agents Chemother 54(9):3702–3707. https://doi.org/10.1128/AAC.00411-10
Marchand S, Lamarche I, Gobin P, Couet W (2010) Dose-ranging pharmacokinetics of colistin methanesulphonate (CMS) and colistin in rats following single intravenous CMS doses. J Antimicrob Chemother 65(8):1753–1758. https://doi.org/10.1093/jac/dkq183
Yapa SWS, Li J, Porter CJ, Nation RL, Patel K, McIntosh MP (2013) Population pharmacokinetics of colistin methanesulfonate in rats: achieving sustained lung concentrations of colistin for targeting respiratory infections. Antimicrob Agents Chemother 57(10):5087–5095. https://doi.org/10.1128/AAC.01127-13
Gibaldi M, Perrier D (1982) Pharmacokinetics, 2nd edn. Marcel Dekker, New York
Li J, Milne RW, Nation RL, Turnidge JD, Coulthard K (2003) Stability of colistin and colistin methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob Agents Chemother 47(4):1364–1370
Reed MD, Stern RC, O’Riordan MA, Blumer JL (2001) The pharmacokinetics of colistin in patients with cystic fibrosis. J Clin Pharmacol 41(6):645–654
Dudhani RV, Nation RL, Li J (2010) Evaluating the stability of colistin and colistin methanesulphonate in human plasma under different conditions of storage. J Antimicrob Chemother 65(7):1412–1415. https://doi.org/10.1093/jac/dkq134
Garonzik SM, Li J, Thamlikitkul V, Paterson DL, Shoham S, Jacob J, Silveira FP, Forrest A, Nation RL (2011) Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 55(7):3284–3294. https://doi.org/10.1128/AAC.01733-10
Yapa SWS, Li J, Patel K, Wilson JW, Dooley MJ, George J, Clark D, Poole S, Williams E, Porter CJ, Nation RL, McIntosh MP (2014) Pulmonary and systemic pharmacokinetics of inhaled and intravenous colistin methanesulfonate in cystic fibrosis patients: targeting advantage of inhalational administration. Antimicrob Agents Chemother 58(5):2570–2579. https://doi.org/10.1128/AAC.01705-13
Li J, Milne RW, Nation RL, Turnidge JD, Coulthard K, Johnson DW (2001) A simple method for the assay of colistin in human plasma, using pre-column derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and reversed-phase high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 761(2):167–175
Li J, Milne RW, Nation RL, Turnidge JD, Coulthard K, Valentine J (2002) Simple method for assaying colistin methanesulfonate in plasma and urine using high-performance liquid chromatography. Antimicrob Agents Chemother 46(10):3304–3307
Li J, Turnidge J, Milne R, Nation RL, Coulthard K (2001) In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother 45(3):781–785. https://doi.org/10.1128/AAC.45.3.781-785.2001
Huang JX, Blaskovich MA, Pelingon R, Ramu S, Kavanagh A, Elliott AG, Butler MS, Montgomery AB, Cooper MA (2015) Mucin binding reduces colistin antimicrobial activity. Antimicrob Agents Chemother 59(10):5925–5931. https://doi.org/10.1128/aac.00808-15
Li J, Rayner CR, Nation RL, Deans R, Boots R, Widdecombe N, Douglas A, Lipman J (2005) Pharmacokinetics of colistin methanesulfonate and colistin in a critically ill patient receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother 49(11):4814–4815. https://doi.org/10.1128/AAC.49.11.4814-4815.2005
Trotman RL, Williamson JC, Shoemaker DM, Salzer WL (2005) Antibiotic dosing in critically ill adult patients receiving continuous renal replacement therapy. Clin Infect Dis 41(8):1159–1166. https://doi.org/10.1086/444500
Markou N, Markantonis SL, Dimitrakis E, Panidis D, Boutzouka E, Karatzas S, Rafailidis P, Apostolakos H, Baltopoulos G (2008) Colistin serum concentrations after intravenous administration in critically ill patients with serious multidrug-resistant, Gram-negative bacilli infections: a prospective, open-label, uncontrolled study. Clin Ther 30(1):143–151. https://doi.org/10.1016/j.clinthera.2008.01.015
Imberti R, Cusato M, Villani P, Carnevale L, Iotti GA, Langer M, Regazzi M (2010) Steady-state pharmacokinetics and BAL concentration of colistin in critically ill patients after IV colistin methanesulfonate administration. Chest 138(6):1333–1339. https://doi.org/10.1378/chest.10-0463
Karnik ND, Sridharan K, Jadhav SP, Kadam PP, Naidu RK, Namjoshi RD, Gupta V, Gore MS, Surase PV, Mehta PR, Gogtay JA, Thatte UM, Gogtay NJ (2013) Pharmacokinetics of colistin in critically ill patients with multidrug-resistant Gram-negative bacilli infection. Eur J Clin Pharmacol 69(7):1429–1436. https://doi.org/10.1007/s00228-013-1493-9
Mohamed AF, Karaiskos I, Plachouras D, Karvanen M, Pontikis K, Jansson B, Papadomichelakis E, Antoniadou A, Giamarellou H, Armaganidis A, Cars O, Friberg LE (2012) Application of a loading dose of colistin methanesulfonate in critically ill patients: population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob Agents Chemother 56(8):4241–4249. https://doi.org/10.1128/AAC.06426-11
Karaiskos I, Friberg LE, Pontikis K, Ioannidis K, Tsagkari V, Galani L, Kostakou E, Baziaka F, Paskalis C, Koutsoukou A, Giamarellou H (2015) Colistin population pharmacokinetics after application of a loading dose of 9 MU colistin methanesulfonate in critically ill patients. Antimicrob Agents Chemother 59(12):7240–7248. https://doi.org/10.1128/aac.00554-15
Gregoire N, Mimoz O, Megarbane B, Comets E, Chatelier D, Lasocki S, Gauzit R, Balayn D, Gobin P, Marchand S, Couet W (2014) New colistin population pharmacokinetic data in critically ill patients suggesting an alternative loading dose rational. Antimicrob Agents Chemother 58:7324–7330. https://doi.org/10.1128/aac.03508-14
Gautam V, Shafiq N, Mouton JW, Malhotra S, Kaur S, Ray P (2018) Pharmacokinetics of colistin in patients with multidrug-resistant Gram-negative infections: a pilot study. Indian J Med Res 147(4):407–412. https://doi.org/10.4103/ijmr.IJMR_1464_16
Nation RL, Garonzik SM, Thamlikitkul V, Giamarellos-Bourboulis EJ, Forrest A, Paterson DL, Li J, Silveira FP (2017) Dosing guidance for intravenous colistin in critically ill patients. Clin Infect Dis 64(5):565–571. https://doi.org/10.1093/cid/ciw839
Nation RL, Garonzik SM, Li J, Thamlikitkul V, Giamarellos-Bourboulis EJ, Paterson DL, Turnidge JD, Forrest A, Silveira FP (2016) Updated US and European dose recommendations for intravenous colistin: how do they perform? Clin Infect Dis 62(5):552–558. https://doi.org/10.1093/cid/civ964
Sorli L, Luque S, Grau S, Berenguer N, Segura C, Montero MM, Alvarez-Lerma F, Knobel H, Benito N, Horcajada JP (2013) Trough colistin plasma level is an independent risk factor for nephrotoxicity: a prospective observational cohort study. BMC Infect Dis 13:380. https://doi.org/10.1186/1471-2334-13-380
Forrest A, Garonzik SM, Thamlikitkul V, Giamarellos-Bourboulis E, Paterson DL, Li J, Silveira FP, Nation RL (2017) Pharmacokinetic/toxicodynamic analysis of colistin-associated acute kidney injury in critically ill patients. Antimicrob Agents Chemother 61:e01367–e01317
Pogue JM, Lee J, Marchaim D, Yee V, Zhao JJ, Chopra T, Lephart P, Kaye KS (2011) Incidence of and risk factors for colistin-associated nephrotoxicity in a large academic health system. Clin Infect Dis 53(9):879–884. https://doi.org/10.1093/cid/cir611
Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Bordinhao RC, Wang J, Forrest A, Nation RL, Li J, Zavascki AP (2013) Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis 57(4):524–531. https://doi.org/10.1093/cid/cit334
Phe K, Lee Y, McDaneld PM, Prasad N, Yin T, Figueroa DA, Musick WL, Cottreau JM, Hu M, Tam VH (2014) In vitro assessment and multicenter cohort study of comparative nephrotoxicity rates associated with colistimethate versus polymyxin B therapy. Antimicrob Agents Chemother 58(5):2740–2746. https://doi.org/10.1128/AAC.02476-13
Akajagbor DS, Wilson SL, Shere-Wolfe KD, Dakum P, Charurat ME, Gilliam BL (2013) Higher incidence of acute kidney injury with intravenous colistimethate sodium compared with polymyxin B in critically ill patients at a tertiary care medical center. Clin Infect Dis 57(9):1300–1303. https://doi.org/10.1093/cid/cit453
Hartzell JD, Neff R, Ake J, Howard R, Olson S, Paolino K, Vishnepolsky M, Weintrob A, Wortmann G (2009) Nephrotoxicity associated with intravenous colistin (colistimethate sodium) treatment at a tertiary care medical center. Clin Infect Dis 48(12):1724–1728. https://doi.org/10.1086/599225
Deryke CA, Crawford AJ, Uddin N, Wallace MR (2010) Colistin dosing and nephrotoxicity in a large community teaching hospital. Antimicrob Agents Chemother 54(10):4503–4505. https://doi.org/10.1128/AAC.01707-09
Horcajada JP, Sorli L, Luque S, Benito N, Segura C, Campillo N, Montero M, Esteve E, Mirelis B, Pomar V, Cuquet J, Marti C, Garro P, Grau S (2016) Validation of a colistin plasma concentration breakpoint as a predictor of nephrotoxicity in patients treated with colistin methanesulfonate. Int J Antimicrob Agents 48(6):725–727. https://doi.org/10.1016/j.ijantimicag.2016.08.020
Karvanen M, Plachouras D, Friberg LE, Paramythiotou E, Papadomichelakis E, Karaiskos I, Tsangaris I, Armaganidis A, Cars O, Giamarellou H (2013) Colistin methanesulfonate and colistin pharmacokinetics in critically ill patients receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother 57(1):668–671. https://doi.org/10.1128/AAC.00985-12
Luque S, Sorli L, Li J, Collado S, Barbosa F, Berenguer N, Horcajada JP, Grau S (2014) Effective removal of colistin methanesulphonate and formed colistin during intermittent haemodialysis in a patient infected by polymyxin-only-susceptible Pseudomonas aeruginosa. J Chemother 26(2):122–124. https://doi.org/10.1179/1973947813Y.0000000104
Markou N, Fousteri M, Markantonis SL, Zidianakis B, Hroni D, Boutzouka E, Baltopoulos G (2012) Colistin pharmacokinetics in intensive care unit patients on continuous venovenous haemodiafiltration: an observational study. J Antimicrob Chemother 67(10):2459–2462. https://doi.org/10.1093/jac/dks257
Marchand S, Frat JP, Petitpas F, Lemaitre F, Gobin P, Robert R, Mimoz O, Couet W (2010) Removal of colistin during intermittent haemodialysis in two critically ill patients. J Antimicrob Chemother 65(8):1836–1837. https://doi.org/10.1093/jac/dkq185
Strunk AK, Schmidt JJ, Baroke E, Bode-Boger SM, Martens-Lobenhoffer J, Welte T, Kielstein JT (2014) Single- and multiple-dose pharmacokinetics and total removal of colistin in a patient with acute kidney injury undergoing extended daily dialysis. J Antimicrob Chemother 69:2008–2010. https://doi.org/10.1093/jac/dku075
Leporati M, Bua RO, Mariano F, Carignano P, Stella M, Biancone L, Vincenti M (2014) Determination by LC-MS/MS of colistins A and B in plasma and ultrafiltrate from critically ill patients undergoing continuous venovenous hemodiafiltration. Ther Drug Monit 36(2):182–191. https://doi.org/10.1097/FTD.0b013e3182a8997c
Jacobs M, Gregoire N, Megarbane B, Gobin P, Balayn D, Marchand S, Mimoz O, Couet W (2016) Population pharmacokinetics of colistin methanesulphonate (CMS) and colistin in critically ill patients with acute renal failure requiring intermittent haemodialysis. Antimicrob Agents Chemother 60:1788–1793. https://doi.org/10.1128/aac.01868-15
Karaiskos I, Friberg LE, Galani L, Ioannidis K, Katsouda E, Athanassa Z, Paskalis H, Giamarellou H (2016) Challenge for higher colistin dosage in critically ill patients receiving continuous venovenous haemodiafiltration. Int J Antimicrob Agents 48(3):337–341. https://doi.org/10.1016/j.ijantimicag.2016.06.008
Mariano F, Leporati M, Carignano P, Stella M, Vincenti M, Biancone L (2015) Efficient removal of colistin A and B in critically ill patients undergoing CVVHDF and sorbent technologies. J Nephrol 28(5):623–631. https://doi.org/10.1007/s40620-014-0143-3
Jitmuang A, Nation RL, Koomanachai P, Chen G, Lee HJ, Wasuwattakul S, Sritippayawan S, Li J, Thamlikitkul V, Landersdorfer CB (2015) Extracorporeal clearance of colistin methanesulphonate and formed colistin in end-stage renal disease patients receiving intermittent haemodialysis: implications for dosing. J Antimicrob Chemother 70(6):1804–1811. https://doi.org/10.1093/jac/dkv031
Menna P, Salvatorelli E, Mattei A, Cappiello D, Minotti G, Carassiti M (2018) Modified colistin regimen for critically ill patients with acute renal impairment and continuous renal replacement therapy. Chemotherapy 63(1):35–38. https://doi.org/10.1159/000484974
Koomanachai P, Landersdorfer CB, Chen G, Lee HJ, Jitmuang A, Wasuwattakul S, Sritippayawan S, Li J, Nation RL, Thamlikitkul V (2014) Pharmacokinetics of colistin methanesulfonate and formed colistin in end-stage renal disease patients receiving continuous ambulatory peritoneal dialysis. Antimicrob Agents Chemother 58(1):440–446. https://doi.org/10.1128/AAC.01741-13
Mohamed AF, Cars O, Friberg LE (2014) A pharmacokinetic/pharmacodynamic model developed for the effect of colistin on Pseudomonas aeruginosa in vitro with evaluation of population pharmacokinetic variability on simulated bacterial killing. J Antimicrob Chemother 69(5):1350–1361. https://doi.org/10.1093/jac/dkt520
Food and Drug Administration (2013) Approved drug products. Label and approval history for Coly-Mycin M, NDA 050108. http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/050108s030lbl.pdf. Last accessed 16 Jun 2019
Antachopoulos C, Karvanen M, Iosifidis E, Jansson B, Plachouras D, Cars O, Roilides E (2010) Serum and cerebrospinal fluid levels of colistin in pediatric patients. Antimicrob Agents Chemother 54(9):3985–3987. https://doi.org/10.1128/AAC.01799-09
Nakwan N, Usaha S, Chokephaibulkit K, Villani P, Regazzi M, Imberti R (2016) Pharmacokinetics of colistin following a single dose of intravenous colistimethate sodium in critically ill neonates. Pediatr Infect Dis J 35(11):1211–1214. https://doi.org/10.1097/inf.0000000000001263
Magreault S, Petyt C, Senneville E, Fron D, Nectoux E, Loiez C, Marchand S, Gregoire N, Couet W (2018) Pharmacokinetics of colistin in a 8-year-old child with acute bone infection. Clin Microbiol Infect 24(9):1025–1026. https://doi.org/10.1016/j.cmi.2018.04.021
Mesini A, Loy A, Gattorno M, Moscatelli A, Bandettini R, Faraci M, Cangemi G, Castagnola E (2018) Colistin area under the time-concentration in children treated with intravenous loading dose and maintenance therapy. Clin Infect Dis 66(5):808–809. https://doi.org/10.1093/cid/cix757
Magreault S, Gregoire N, Marchand S, Couet W (2018) Colistin pharmacokinetics in pediatrics. Clin Infect Dis 66(5):809. https://doi.org/10.1093/cid/cix1046
Nation RL (2018) Dose suggestions for intravenous colistin in pediatric patients: caution required. Clin Infect Dis 66(5):810–811. https://doi.org/10.1093/cid/cix1048
Markantonis SL, Markou N, Fousteri M, Sakellaridis N, Karatzas S, Alamanos I, Dimopoulou E, Baltopoulos G (2009) Penetration of colistin into cerebrospinal fluid. Antimicrob Agents Chemother 53(11):4907–4910. https://doi.org/10.1128/AAC.00345-09
Ziaka M, Markantonis SL, Fousteri M, Zygoulis P, Panidis D, Karvouniaris M, Makris D, Zakynthinos E (2013) Combined intravenous and intraventricular administration of colistin methanesulfonate in critically ill patients with central nervous system infection. Antimicrob Agents Chemother 57(4):1938–1940. https://doi.org/10.1128/AAC.01461-12
Boisson M, Jacobs M, Gregoire N, Gobin P, Marchand S, Couet W, Mimoz O (2014) Comparison of intra-pulmonary and systemic pharmacokinetics of colistinmethansulphonate (CMS) and colistin after aerosol delivery and intravenous administration of CMS in critically ill patients. Antimicrob Agents Chemother 58:7331–7339. https://doi.org/10.1128/aac.03510-14
Rottboll LA, Friis C (2016) Penetration of antimicrobials to pulmonary epithelial lining fluid and muscle and impact of drug physicochemical properties determined by microdialysis. J Pharmacol Toxicol Methods 78:58–65. https://doi.org/10.1016/j.vascn.2015.11.007
Weinbren MJ (1999) Pharmacokinetics of antibiotics in burn patients. J Antimicrob Chemother 44(3):319–327
Lee J, Han S, Jeon S, Hong T, Song W, Woo H, Yim DS (2013) Population pharmacokinetic analysis of colistin in burn patients. Antimicrob Agents Chemother 57(5):2141–2146. https://doi.org/10.1128/AAC.00271-13
Corcione S, Baietto L, Malvasio V, Stella M, Di Perri G, D’Avolio A, De Rosa FG (2017) Pharmacokinetics of colistin methanesulfonate (CMS) in burn patients. J Antimicrob Chemother 72(1):319–321. https://doi.org/10.1093/jac/dkw361
Bode-Boger SM, Schopp B, Troger U, Martens-Lobenhoffer J, Kalousis K, Mailander P (2013) Intravenous colistin in a patient with serious burns and borderline syndrome: the benefits of therapeutic drug monitoring. Int J Antimicrob Agents 42(4):357–360. https://doi.org/10.1016/j.ijantimicag.2013.06.009
Akers KS, Rowan MP, Niece KL, Stewart IJ, Mende K, Cota JM, Murray CK, Chung KK (2015) Colistin pharmacokinetics in burn patients during continuous venovenous hemofiltration. Antimicrob Agents Chemother 59(1):46–52. https://doi.org/10.1128/aac.03783-14
Athanassa ZE, Markantonis SL, Fousteri MZ, Myrianthefs PM, Boutzouka EG, Tsakris A, Baltopoulos GJ (2012) Pharmacokinetics of inhaled colistimethate sodium (CMS) in mechanically ventilated critically ill patients. Intensive Care Med 38(11):1779–1786. https://doi.org/10.1007/s00134-012-2628-7
Nakwan N, Lertpichaluk P, Chokephaibulkit K, Villani P, Regazzi M, Imberti R (2015) Pulmonary and systemic pharmacokinetics of colistin following a single dose of nebulized colistimethate in mechanically ventilated neonates. Pediatr Infect Dis J 34(9):961–963. https://doi.org/10.1097/inf.0000000000000775
Boisson M, Gregoire N, Cormier M, Gobin P, Marchand S, Couet W, Mimoz O (2017) Pharmacokinetics of nebulized colistin methanesulfonate in critically ill patients. J Antimicrob Chemother 72(9):2607–2612. https://doi.org/10.1093/jac/dkx167
Landersdorfer CB, Nguyen TH, Lieu LT, Nguyen G, Bischof RJ, Meeusen EN, Li J, Nation RL, McIntosh MP (2017) Substantial targeting advantage achieved by pulmonary administration of colistin methanesulfonate in a large-animal model. Antimicrob Agents Chemother 61(1). https://doi.org/10.1128/aac.01934-16
Gu WJ, Wang F, Tang L, Bakker J, Liu JC (2014) Colistin for the treatment of ventilator-associated pneumonia caused by multidrug-resistant Gram-negative bacteria: a systematic review and meta-analysis. Int J Antimicrob Agents 44:477–485. https://doi.org/10.1016/j.ijantimicag.2014.07.004
Valachis A, Samonis G, Kofteridis DP (2015) The role of aerosolized colistin in the treatment of ventilator-associated pneumonia: a systematic review and metaanalysis. Crit Care Med 43(3):527–533. https://doi.org/10.1097/ccm.0000000000000771
Liu D, Zhang J, Liu HX, Zhu YG, Qu JM (2015) Intravenous combined with aerosolised polymyxin versus intravenous polymyxin alone in the treatment of pneumonia caused by multidrug-resistant pathogens: a systematic review and meta-analysis. Int J Antimicrob Agents 46(6):603–609. https://doi.org/10.1016/j.ijantimicag.2015.09.011
Abdellatif S, Trifi A, Daly F, Mahjoub K, Nasri R, Ben Lakhal S (2016) Efficacy and toxicity of aerosolised colistin in ventilator-associated pneumonia: a prospective, randomised trial. Ann Intensive Care 6(1):26. https://doi.org/10.1186/s13613-016-0127-7
Jang JY, Kwon HY, Choi EH, Lee WY, Shim H, Bae KS (2017) Efficacy and toxicity of high-dose nebulized colistin for critically ill surgical patients with ventilator-associated pneumonia caused by multidrug-resistant Acinetobacter baumannii. J Crit Care 40:251–256. https://doi.org/10.1016/j.jcrc.2017.04.004
Jung SY, Lee SH, Lee SY, Yang S, Noh H, Chung EK, Lee JI (2017) Antimicrobials for the treatment of drug-resistant Acinetobacter baumannii pneumonia in critically ill patients: a systemic review and Bayesian network meta-analysis. Crit Care 21(1):319. https://doi.org/10.1186/s13054-017-1916-6
Vardakas KZ, Voulgaris GL, Samonis G, Falagas ME (2018) Inhaled colistin monotherapy for respiratory tract infections in adults without cystic fibrosis: a systematic review and meta-analysis. Int J Antimicrob Agents 51(1):1–9. https://doi.org/10.1016/j.ijantimicag.2017.05.016
Demirdal T, Sari US, Nemli SA (2016) Is inhaled colistin beneficial in ventilator associated pneumonia or nosocomial pneumonia caused by Acinetobacter baumannii? Ann Clin Microbiol Antimicrob 15(1):11. https://doi.org/10.1186/s12941-016-0123-7
Bargiacchi O, Rossati A, Car P, Brustia D, Brondolo R, Rosa F, Garavelli PL, De Rosa FG (2014) Intrathecal/intraventricular colistin in external ventricular device-related infections by multi-drug resistant Gram negative bacteria: case reports and review. Infection 42:801–809. https://doi.org/10.1007/s15010-014-0618-0
Imberti R, Cusato M, Accetta G, Marino V, Procaccio F, Del Gaudio A, Iotti GA, Regazzi M (2012) Pharmacokinetics of colistin in cerebrospinal fluid after intraventricular administration of colistin methanesulfonate. Antimicrob Agents Chemother 56(8):4416–4421. https://doi.org/10.1128/AAC.00231-12
Karaiskos I, Galani L, Baziaka F, Giamarellou H (2013) Intraventricular and intrathecal colistin as the last therapeutic resort for the treatment of multidrug-resistant and extensively drug-resistant Acinetobacter baumannii ventriculitis and meningitis: a literature review. Int J Antimicrob Agents 41(6):499–508. https://doi.org/10.1016/j.ijantimicag.2013.02.006
Karaiskos I, Galani L, Baziaka F, Katsouda E, Ioannidis I, Andreou A, Paskalis H, Giamarellou H (2013) Successful treatment of extensively drug-resistant Acinetobacter baumannii ventriculitis and meningitis with intraventricular colistin after application of a loading dose: a case series. Int J Antimicrob Agents 41(5):480–483. https://doi.org/10.1016/j.ijantimicag.2013.02.010
De Bonis P, Lofrese G, Scoppettuolo G, Spanu T, Cultrera R, Labonia M, Cavallo MA, Mangiola A, Anile C, Pompucci A (2015) Intraventricular versus intravenous colistin for the treatment of extensively drug resistant Acinetobacter baumannii meningitis. Eur J Neurol 23:68–75. https://doi.org/10.1111/ene.12789
Fotakopoulos G, Makris D, Chatzi M, Tsimitrea E, Zakynthinos E, Fountas K (2016) Outcomes in meningitis/ventriculitis treated with intravenous or intraventricular plus intravenous colistin. Acta Neurochir 158(3):603–610. https://doi.org/10.1007/s00701-016-2702-y
Inamasu J, Ishikawa K, Oheda M, Nakae S, Hirose Y, Yoshida S (2016) Intrathecal administration of colistin for meningitis due to New Delhi metallo-beta-lactamase 1(NDM-1)-producing Klebsiella pneumoniae. J Infect Chemother 22(3):184–186. https://doi.org/10.1016/j.jiac.2015.10.007
Landersdorfer CB, Wang J, Wirth V, Chen K, Kaye KS, Tsuji BT, Li J, Nation RL (2018) Pharmacokinetics/pharmacodynamics of systemically administered polymyxin B against Klebsiella pneumoniae in mouse thigh and lung infection models. J Antimicrob Chemother 73(2):462–468. https://doi.org/10.1093/jac/dkx409
Lakota EA, Landersdorfer CB, Nation RL, Li J, Kaye KS, Rao GG, Forrest A (2018) Personalizing polymyxin B dosing using an adaptive feedback control algorithm. Antimicrob Agents Chemother 62(7). https://doi.org/10.1128/aac.00483-18
Kwa AL, Lim TP, Low JG, Hou J, Kurup A, Prince RA, Tam VH (2008) Pharmacokinetics of polymyxin B1 in patients with multidrug-resistant Gram-negative bacterial infections. Diagn Microbiol Infect Dis 60(2):163–167. https://doi.org/10.1016/j.diagmicrobio.2007.08.008
Kwa AL, Abdelraouf K, Low JG, Tam VH (2011) Pharmacokinetics of polymyxin B in a patient with renal insufficiency: a case report. Clin Infect Dis 52(10):1280–1281. https://doi.org/10.1093/cid/cir137
Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Saitovitch D, Wang J, Forrest A, Nation RL, Zavascki AP, Li J (2013) Pharmacokinetics of polymyxin B in patients on continuous venovenous haemodialysis. J Antimicrob Chemother 68(3):674–677. https://doi.org/10.1093/jac/dks437
Thamlikitkul V, Dubrovskaya Y, Manchandani P, Ngamprasertchai T, Boonyasiri A, Babic JT, Tam VH (2017) Dosing and pharmacokinetics of polymyxin B in patients with renal insufficiency. Antimicrob Agents Chemother 61(1). https://doi.org/10.1128/aac.01337-16
Manchandani P, Thamlikitkul V, Dubrovskaya Y, Babic JT, Lye DC, Lee LS, Tam VH (2018) Population pharmacokinetics of polymyxin B. Clin Pharmacol Ther 104(3):534–538. https://doi.org/10.1002/cpt.981
Kubin CJ, Nelson BC, Miglis C, Scheetz MH, Rhodes NJ, Avedissian SN, Cremers S, Yin MT (2018) Population pharmacokinetics of intravenous polymyxin B from clinical samples. Antimicrob Agents Chemother 62(3):e01493–e01417. https://doi.org/10.1128/aac.01493-17. [pii]
Miglis C, Rhodes NJ, Avedissian SN, Kubin CJ, Yin MT, Nelson BC, Pai MP, Scheetz MH (2018) Population pharmacokinetics of polymyxin B in acutely ill adult patients. Antimicrob Agents Chemother 62(3):e01475–e01417. https://doi.org/10.1128/aac.01475-17. [pii]
Avedissian S, Miglis C, Kubin CJ, Rhodes NJ, Yin MT, Cremers S, Prickett M, Scheetz MH (2018) Polymyxin B pharmacokinetics in adult cystic fibrosis patients. Pharmacotherapy 38:730–738. https://doi.org/10.1002/phar.2129
Nelson BC, Eiras DP, Gomez-Simmonds A, Loo AS, Satlin MJ, Jenkins SG, Whittier S, Calfee DP, Furuya EY, Kubin CJ (2015) Clinical outcomes associated with polymyxin B dose in patients with bloodstream infections due to carbapenem-resistant Gram-negative rods. Antimicrob Agents Chemother 59(11):7000–7006. https://doi.org/10.1128/aac.00844-15
Roberts KD, Azad MA, Wang J, Horne AS, Thompson PE, Nation RL, Velkov T, Li J (2015) Antimicrobial activity and toxicity of the major lipopeptide components of polymyxin B and colistin: last-line antibiotics against multidrug-resistant Gram-negative bacteria. ACS Infect Dis 1(11):568–575. https://doi.org/10.1021/acsinfecdis.5b00085
Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, Suppes R, Feinstein D, Zanotti S, Taiberg L, Gurka D, Kumar A, Cheang M (2006) Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 34(6):1589–1596. https://doi.org/10.1097/01.ccm.0000217961.75225.e9
Luna CM, Aruj P, Niederman MS, Garzon J, Violi D, Prignoni A, Rios F, Baquero S, Gando S (2006) Appropriateness and delay to initiate therapy in ventilator-associated pneumonia. Eur Respir J 27(1):158–164. https://doi.org/10.1183/09031936.06.00049105
Spapen HD, Honore PM, Gregoire N, Gobin P, de Regt J, Martens GA, Pierard D, Couet W (2011) Convulsions and apnoea in a patient infected with New Delhi metallo-beta-lactamase-1 Escherichia coli treated with colistin. J Infect 63(6):468–470. https://doi.org/10.1016/j.jinf.2011.07.008
Kim EJ, Oh J, Lee K, Yu KS, Chung JY, Hwang JH, Nam EY, Kim HS, Kim M, Park JS, Song KH, Kim ES, Song J, Kim HB (2018) A short communication: pharmacokinetic characteristics and limited sampling strategies for therapeutic drug monitoring of colistin in patients with multidrug-resistant Gram-negative bacterial infections. Ther Drug Monit 41:102–106. https://doi.org/10.1097/ftd.0000000000000572
Jelliffe RW, Schumitzky A, Van Guilder M, Liu M, Hu L, Maire P, Gomis P, Barbaut X, Tahani B (1993) Individualizing drug dosage regimens: roles of population pharmacokinetic and dynamic models, Bayesian fitting, and adaptive control. Ther Drug Monit 15(5):380–393
Luque S, Escano C, Sorli L, Li J, Campillo N, Horcajada JP, Salas E, Grau S (2017) Urinary concentrations of colistimethate and formed colistin after intravenous administration in patients with multidrug-resistant Gram-negative bacterial infections. Antimicrob Agents Chemother 61(8):e02595-16. https://doi.org/10.1128/aac.02595-16. [pii]
Oliveira MS, Prado GV, Costa SF, Grinbaum RS, Levin AS (2009) Polymyxin B and colistimethate are comparable as to efficacy and renal toxicity. Diagn Microbiol Infect Dis 65(4):431–434. https://doi.org/10.1016/j.diagmicrobio.2009.07.018
Tuon FF, Rigatto MH, Lopes CK, Kamei LK, Rocha JL, Zavascki AP (2014) Risk factors for acute kidney injury in patients treated with polymyxin B or colistin methanesulfonate sodium. Int J Antimicrob Agents 43(4):349–352. https://doi.org/10.1016/j.ijantimicag.2013.12.002
Rigatto MH, Oliveira MS, Perdigao-Neto LV, Levin AS, Carrilho CM, Tanita MT, Tuon FF, Cardoso DE, Lopes NT, Falci DR, Zavascki AP (2016) Multicenter prospective cohort study of renal failure in patients treated with colistin versus polymyxin B. Antimicrob Agents Chemother 60(4):2443–2449. https://doi.org/10.1128/aac.02634-15
Vardakas KZ, Falagas ME (2017) Colistin versus polymyxin B for the treatment of patients with multidrug-resistant Gram-negative infections: a systematic review and meta-analysis. Int J Antimicrob Agents 49(2):233–238. https://doi.org/10.1016/j.ijantimicag.2016.07.023
Zavascki AP, Nation RL (2017) Nephrotoxicity of polymyxins: is there any difference between colistimethate and polymyxin B? Antimicrob Agents Chemother 61(3):e02319–e02316. https://doi.org/10.1128/aac.02319-16. [pii]
Kenyon R (2015) Structural evidence for a new understanding of the nature of CMS. Paper presented at the 2nd international conference on polymyxins, 22–24 September, San Diego, USA
Tsuji BT, Pogue JM, Zavascki AP, Paul M, Daikos GL, Forrest A, Giacobbe DR, Viscoli C, Giamarellou H, Karaiskos I, Kaye D, Mouton JW, Tam VH, Thamlikitkul V, Wunderink RG, Li J, Nation RL, Kaye KS (2019) International consensus guidelines for the optimal use of the polymyxins: endorsed by the American College of Clinical Pharmacy (ACCP), European Society of Clinical Microbiology and Infectious Diseases (ESCMID), Infectious Diseases Society of America (IDSA), International Society for Anti-infective Pharmacology (ISAP), Society of Critical Care Medicine (SCCM), and Society of Infectious Diseases Pharmacists (SIDP). Pharmacotherapy 39(1):10–39
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
This article is dedicated to the memory of Alan Forrest, a friend of many and an inspiring researcher and teacher.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nation, R.L., Forrest, A. (2019). Clinical Pharmacokinetics, Pharmacodynamics and Toxicodynamics of Polymyxins: Implications for Therapeutic Use. In: Li, J., Nation, R., Kaye, K. (eds) Polymyxin Antibiotics: From Laboratory Bench to Bedside. Advances in Experimental Medicine and Biology, vol 1145. Springer, Cham. https://doi.org/10.1007/978-3-030-16373-0_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-16373-0_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16371-6
Online ISBN: 978-3-030-16373-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)