
Abstract
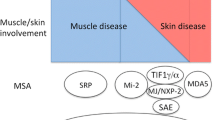
“Myositis-specific autoantibodies (MSA)” are distinctive in that myositis is their primary associated autoimmune disease despite other clinical features. Antisynthetase antibodies, anti-Mi-2, and anti-SRP have long been considered established MSAs. Autoantibodies such as anti-Ro/SSA or anti-U1RNP are termed “myositis-associated autoantibodies (MAA)” and occur in patients with myositis, but their primary association is with other autoimmune rheumatic diseases. Autoantibodies to aminoacyl-transfer RNA synthetase enzymes (ARS) are present in the cytoplasm and closely associated with “antisynthetase syndrome.” Anti-Mi-2 is strongly associated with dermatomyositis, which is glucocorticoid-responsive with a relatively good prognosis. Anti-SRP is associated with immune-mediated necrotizing myopathy. Anti-Ku and PM-SCL antibodies are MAA and associated with overlap syndromes. This chapter discusses traditional MSAs and MAAs in more detail.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Myositis-specific autoantibodies
- Myositis-associated autoantibodies
- Anti-Jo-1
- Antisynthetase antibodies
- Antisynthetase syndrome
- Anti-Mi-2 antibodies
- Anti-SRP antibodies
- Anti-PM-Scl
- Anti-Ku
-
Autoantibodies to aminoacyl-tRNA synthetases (ARS) with the presence of one or more associated clinical features including myositis, interstitial lung disease (ILD), inflammatory arthritis, or mechanic’s hands has been referred to as the antisynthetase syndrome. ILD can be more common than myositis and often clinically significant.
-
Eight ARS autoantibodies have been described. Anti Jo-1 is the most common. The titer of anti-Jo-1 may vary with disease activity. Antibodies to different ARS may differ in the relative frequency of the features of the antisynthetase syndrome.
-
Anti-Mi-2 is associated with dermatomyositis. Myositis in patients with anti-Mi-2 tends to be mild and glucocorticoid-responsive with a relatively good prognosis.
-
Anti-SRP is most commonly associated with immune-mediated necrotizing myopathy, without inflammation. Myositis is often rapid in onset, with very high CK levels, early muscle damage, severe weakness, treatment refractoriness, and multiple flares. Anti-SRP patients are more likely to require multiple immunosuppressive agents.
-
Anti-PM-Scl is most commonly associated with an overlap syndrome of polymyositis or dermatomyositis with scleroderma, often including polyarthritis. Anti-Ku is also associated with overlap syndromes involving myositis, as well as scleroderma and lupus.
-
Anti-Ro52 may occur in the absence of anti-Ro60 in myositis more often than in most other conditions and frequently occurs in association with other myositis autoantibodies, including anti-ARS, anti-PM-Scl, or anti-SRP. It may be a marker of more severe disease and a worse prognosis.
Introduction
Since the 1980s, the existence of certain autoantibodies strongly associated with myositis has been known [1,2,3]. Although no single autoantibody is present in a majority of myositis patients (low sensitivity), these “myositis-specific autoantibodies (MSA)” were distinctive in that most antibody-positive patients have myositis as their primary autoimmune disease (high specificity) despite other clinical features often being present. This was in contrast to autoantibodies such as anti-Ro/SSA or anti-U1RNP that could occur in patients with myositis, with potentially significant implications and potential diagnostic utility, but their primary association was typically with other autoimmune rheumatic diseases. Hence, these autoantibodies were termed “myositis-associated autoantibodies (MAA)” [4]. Thus, antisynthetase antibodies (anti-aminoacyl-tRNA synthetase or anti-ARS), anti-Mi-2, and anti-SRP have long been considered established MSAs [5]. Increasingly, anti-ARSs are more closely associated with unique features termed the “antisynthetase syndrome,” with or without clinical features of myositis [6]. Anti-PM-Scl and anti-Ku are usually considered to be MAAs but are commonly associated with overlap syndromes involving myositis [2, 7, 8].
Later, additional autoantibodies of importance were identified, particularly in association with dermatomyositis (DM) and necrotizing myopathy (NM). This increased the proportion of patients with myositis demonstrating an identifiable specific autoantibody (i.e., MSA) and increased the combined sensitivity of MSA testing in DM, NM, and polymyositis (PM). Together with newer myositis autoantibodies, the proportion of patients with PM, DM, or NM who have an identifiable autoantibody is now considerably higher, often >80% if comprehensive testing is done [9]. Although the traditional autoantibodies have been known for a long time, their value and importance in diagnosis and management has recently increased with better availability of testing and as collaborative studies involving larger patient groups have better defined the clinical and treatment considerations of autoantibody-defined groups. It is important to note, however, that differences in testing methods could affect the sensitivity, specificity, and clinical associations of these autoantibodies [9].
Myositis-Specific Autoantibodies (MSA): Autoantibodies that are highly specific for myositis and are associated with myositis as the primary autoimmune disease.
Myositis-Associated Autoantibodies (MAA): Autoantibodies associated with other autoimmune rheumatic diseases that could occur in patients with myositis or have myositis as a clinical feature.
MSA and MAA can be seen in >80% of PM, NM, DM, and their associated phenotypes.
Antisynthetase Antibodies (Anti-ARS)
The aminoacyl-transfer RNA synthetase enzymes catalyze the formation of aminoacyl-transfer RNA, in which an amino acid is bound to a transfer RNA for that amino acid [1]. The transfer RNA can then contribute this amino acid to a growing polypeptide chain forming at the ribosome, at the proper position as defined by the messenger RNA. It is crucial to the accuracy of this process that each amino acid is attached to a transfer RNA specific for that amino acid. Thus, it is not surprising that there is a separate synthetase enzyme for each amino acid that is immunologically distinct from others. Autoantibodies to these enzymes do not cross-react with each other. Most patients have autoantibodies to only one of these enzymes (with the exception of those who have autoantibodies to isoleucyl-tRNA synthetase (OJ), which is part of a multienzyme complex of synthetases; although isoleucyl-tRNA synthetase is usually the primary target, they may also react with other enzymes in the complex [10]). That is, the anti-ARS is generally mutually exclusive. These enzymes localize to the cytoplasm where protein synthesis usually takes place [2, 11, 12].
Autoantibodies have been described reacting with at least eight of these enzymes: anti-Jo-1 (anti-histidyl); anti-PL-7 (anti-threonyl); anti-PL-12 (anti-alanyl); anti-OJ (anti-isoleucyl); anti-EJ (anti-glycyl); anti-KS (anti-asparaginyl); anti-ZO (anti-phenylalanyl); and anti-Ha (anti-tyrosyl). Patients with autoantibodies to any of the aminoacyl-tRNA synthetases have a generally similar set of clinical features including myositis, interstitial lung disease (ILD), inflammatory arthritis, Raynaud phenomenon, a hyperkeratotic rash on the fingers referred to as mechanic’s hands, and fever [5, 13]. The occurrence of one or more of these clinical features in the presence of anti-ARS is defined as the antisynthetase syndrome. Although myositis is often a major component, it may not be present. ILD can be more common than myositis and often clinically significant [14]. However, the frequency of these features may differ with different anti-ARS [13, 15, 16]. For example, there is generally a higher frequency of myositis with anti-Jo-1 (anti-histidyl-tRNA synthetase) than with anti-PL-12 (anti-alanyl-tRNA synthetase) or anti-PL-7 (anti-threonyl), where ILD is more common than myositis [15,16,17]. The frequency of different anti-ARS differs widely, with anti-Jo-1 the most frequent anti-ARS and the most common myositis autoantibody in most populations (20–30% of adult myositis patients). Each other anti-ARS (non-Jo-1 anti-ARS) is much less frequent (<5%), particularly among those with prominent myositis, but collectively non-Jo-1 anti-ARS constitutes up to 20–40% of any antisynthetase cohort. Autoantibodies to certain others such as tyrosyl-tRNA synthetase are very rare (<1%) and autoantibodies to some ARS have not been described. There may also be differences in the frequency of different anti-ARS in different ethnic or geographic populations [18] (Table 20.1).
Antisynthetase syndrome
One or more clinical features given below with one of the anti-ARS
-
Myositis
-
Interstitial lung disease
-
Polyarthritis
-
Mechanic’s hands
-
Raynaud phenomenon
-
Fever
Laboratory Testing: Although anti-Jo-1 testing is widely available using enzyme immunoassay methodology and immunodiffusion, specific testing for other ARSs requires more specialized methodology, which is not only less available and reliable but requires more time to accurately complete. Protein and RNA immunoprecipitation (IP) have traditionally been used for this purpose (Figs. 20.1 and 20.2). Most ARSs show both a distinctive protein and a distinctive set of transfer RNAs by IP, which was an important factor leading to the identification of the enzymes as the antigenic targets [1, 19, 20]. Direct methods are becoming available allowing more rapid clinical identification of these antibodies [9, 21,22,23,24,25,26,27]. However, most of these panels do not include all the ARSs that can occur and must be validated against the gold standard of IP. The presence of cytoplasmic staining in a consistent clinical setting would be a clue to pursue further testing (Fig. 20.3).

RNA immunoprecipitation: Results of polyacrylamide gel electrophoresis (PAGE) for the detection of RNAs of immunoprecipitates using antisynthetase antibodies, anti-SRP, and others. Anti-SRP serum immunoprecipitates a strong band in the 7S region representing the 7SL RNA of the SRP, not seen with other myositis autoantibodies. The distinctive transfer RNAs associated with different aminoacyl-tRNA synthetase antigens are seen, which are distinguishable from each other. The anti-PL-12 serum shown also contains anti-Ro/SSA and anti-La/SSB and immunoprecipitates the characteristic associated RNAs. Anti-PM-Scl and anti-Mi-2 do not immunoprecipitate nucleic acids. Serum with anti-U1RNP and anti-U2RNP precipitates these small nuclear RNAs, while anti-Sm precipitates those along with U4, U5, and U6 RNAs. Anti-Ku has affinity for DNA and precipitates a heterogeneous DNA smear. Anti-Ro immunoprecipitates the hY1 (highest) through hY5 (lowest) RNAs. NL normal serum, TNA total nucleic acid. The position of the 5.8S and 5.0S RNAs, as well as the transfer RNAs (tRNAs), is shown. Mas serum precipitates a very weak unidentified RNA. (This figure was published as Figure 1, page 863, in Targoff IN. Immune manifestations of inflammatory muscle disease. Rheu Dis Clin N Am. 20(4):857–80, Copyright Elsevier 1994)

Protein immunoprecipitation: The figure shows immunoprecipitation with S35-labeled HeLa cells using sera from patients with various different myositis-specific and myositis-associated autoantibodies. Anti-Mi-2 precipitates the 240 kDa main antigenic protein, which is part of a complex of proteins (the NuRD complex) (lane 6). Several other components of that complex can also be seen (at molecular weights of 150, 75, 65, and 63 kDa). In contrast, the anti-SRP serum immunoprecipitates a strong band at 54 kD and a weaker band at 72 kDa (lanes 2 and 18). The signal recognition particle (SRP) is a complex containing a unique RNA (“7SL”) and specific proteins. The combination of RNA-immunoprecipitation (see Fig. 20.1) and S35-immunoprecipitation results consistent with anti-SRP results provides very specific identification of the presence of these autoantibodies. The presence of anti-Mi-2 can be sensitively detected and specifically identified by this method and can be clearly distinguished from other myositis autoantibodies (lane 6). The antisynthetases precipitate characteristic proteins and, along with RNA immunoprecipitation (Fig. 20.1), can be distinguished from other autoantibodies. In lane 3, anti-PL-7 (anti-threonyl-tRNA synthetase) shows a strong protein of approximately 80 kDa, and in lane 5, anti-PL-12 (anti-alanyl-tRNA synthetase) shows a strong protein of approximately 110 kDa. A protein of approximately 50kDa is precipitated by anti-Jo-1 in lane 12. Results for anti-PM-Scl are shown in lane 9. This antibody immunoprecipitates the multiple proteins of the exosome, including the major antigens of approximately 100 and 75 kDa. In lane 10, anti-Ku shows the typical strong proteins of 72 and 86 kDa. (Images Courtesy: Alpini Claudia, Angela Ceribelli and Lorenzo Cavagna, University of Pavia, Italy)

Indirect immunofluorescence with anti-Jo-1, anti-SRP, anti-Mi-2, and anti-PM-Scl antibodies: An antinuclear antibody (ANA) test was done using indirect immunofluorescence testing on Hep-2 cell substrate with a serum that contained anti-Jo-1 (top right), anti-PM-Scl (top left), anti-SRP (bottom left), and anti-Mi-2 (bottom right) autoantibodies, respectively. Anti-PM-Scl shows a nucleolar pattern with additional nucleoplasmic staining. With anti-Jo-1, cytoplasmic fluorescence is seen. Anti-SRP autoantibodies give a characteristic cytoplasmic pattern, consistent with the known location of the SRP in the cytoplasm of the cell. Anti-Mi-2 shows a pure nuclear pattern that spares the nucleolus. (Images Courtesy: Alpini Claudia, Angela Ceribelli and Lorenzo Cavagna, University of Pavia, Italy)
Serum Levels of Anti-ARS: Quantitative measurements of the antibody have suggested that the titer of anti-Jo-1 may vary with disease activity and decrease with improvement in disease status over time [23, 24]. Although detectable antibody typically persists in most patients after treatment, the occasional disappearance of the antibody has been associated with remission [28]. These results suggest that MSA, especially anti-Jo-1, could serve as potential biomarkers [23, 29].
Serum levels of anti-Jo1 may be associated with disease activity in myositis.
The myositis in patients with anti-ARSs is generally similar to that seen with other polymyositis or dermatomyositis patients, although recurrences may be more likely. Histologically, a distinctive pattern of involvement was noted in patients with anti-Jo-1 , with perimysial inflammation similar to that seen with dermatomyositis, but without the concomitant capillary loss [30]. There was also evidence of fasciitis with perimysial connective tissue fragmentation in muscle from patients with anti-Jo-1 [30, 31]. In another study of myositis histological findings among 50 patients with anti-ARSs, myofiber necrosis in the perifascicular region was observed in about 50% of patients [32]. Necrosis, not restricted to the perifascicular area, seen most commonly with anti-OJ antibodies, was associated with more severe muscle involvement. The myositis may also be more responsive to rituximab treatment than for those without the antibodies [29].
Antisynthetase patients have distinct muscle histopathology: perimysial inflammation and perifascicular myofiber pathology, without the vascular changes of dermatomyositis.
There may be a delay in making a diagnosis in patients with non-Jo-1 ARSs compared to those with anti-Jo-1 , which may relate in part to wider availability of testing for anti-Jo-1 [13, 33]. This may be a factor in decreased survival. For nearly half of non-Jo-1 anti-ARS patients, the initial diagnosis is an overlap syndrome or undifferentiated connective tissue disease, while those with anti-Jo-1 are more likely to have an initial diagnosis of myositis (83%) [16, 33].
Antisynthetase syndrome with non-Jo-1 anti-ARS has a worse prognosis than that with anti-Jo-1 partly due to delay in diagnosis and partly to a higher frequency of ILD.
Although dermatomyositis skin involvement (such as Gottron changes or a heliotrope rash) may occur in patients with anti-ARSs, some studies have found that clinical polymyositis is more common with anti-Jo-1 [34, 35]. Dermatomyositis may be more common with non-Jo-1 anti-ARSs than with anti-Jo-1 [35]. Patients with antisynthetase syndrome may have “mechanic’s hands,” a hyperkeratotic rash with splitting or cracking on the edges of the fingers [5, 36]. Antisynthetase syndrome may occur in children [37, 38], where a syndrome similar to that in adults may be seen [38], but anti-ARSs are less common in children, while DM-related autoantibodies such as anti-MJ/NXP2 and anti-p155/140 are more common [38, 39]. Patients with cancer-associated myositis usually do not have anti-ARSs [40], although occasional cases have been described.
Antisynthetase syndrome patients can have clinical dermatomyositis (including amyopathic forms) or polymyositis.
ILD is an important and common feature of antisynthetase syndrome in view of its clinical implications and impact on survival and prognosis [41,42,43,44,45]. The severity and progression can be variable [46]. It may be the presenting feature, while some patients have ILD without clinical myositis [6, 15, 47, 48]. Non-Jo-1 anti-ARS may be associated with a higher frequency and more severe lung disease, and certain anti-ARSs, including anti-PL-12, anti-PL-7, anti OJ, and anti-KS, appear to be more likely to present with ILD rather than myositis [16, 17, 49].
ILD is the most common and important clinical feature of the antisynthetase syndrome.
Non-Jo-1 anti-ARS has a higher frequency (80–100%) and severity of ILD than anti-Jo-1 (70–80%).
CT scan features at the time of diagnosis include nonspecific interstitial pneumonia with organizing pneumonia, either isolated or in combination [6, 47, 48, 50]. In one study of 14 antisynthetase patients, CT scans revealed ground-glass opacities with peribronchovascular interstitial thickening and traction bronchiectasis and consolidation. Honeycombing is less frequent. NSIP and predominantly organizing pneumonia with focal NSIP are common histopathological patterns [51, 52]. Consolidations decrease or disappear in most cases, but the disease may progress to fibrosis in more than one third of patients. The main cause of death among patients with anti-ARS is pulmonary fibrosis followed by pulmonary hypertension. The extent of lung inflammation on high-resolution CT scanning and forced vital capacity can be factors in predicting survival in antisynthetase syndrome patients [53].
Anti-Ro52 is more likely to be present in patients with anti-ARSs than in other myositis patients [54, 55] and tends to be associated with more severe disease [56, 57]. The presence of anti-Ro52 predicts rapidly progressive ILD. Other prognostic indicators for poor outcomes of patients with the antisynthetase syndrome include malignancy and hyperferritinemia.
Anti-Ro52 is commonly seen with anti-ARS and is associated with worse prognosis.
The arthritis in association with anti-ARS can also be significant and similar to rheumatoid arthritis (symmetric polyarthritis of the small joints of the hand) [58] and may be the presenting feature. It can sometimes be deforming and may be more likely in patients with rheumatoid factor and/or anti-CCP antibody positivity, which can make the diagnosis of the antisynthetase syndrome versus rheumatoid arthritis very challenging [32].
Anti-Mi-2
Anti-Mi-2 was described using immunodiffusion [59] but later found to immunoprecipitate a multiprotein complex [60, 61] that was identified as the nucleosome remodeling deacetylase (NuRD) complex [62, 63] (Fig. 20.2), which is involved in transcriptional regulation by chromatin remodeling and histone deacetylation. There are two forms of the major antigen of the Mi-2 complex, a 240 kD and 200 kD protein, which are DNA-helicases [64]. They include Mi-2α [CHD (chromatin organization modifier, helicase, and DNA binding) 3 and Mi-2ß (CHD4)]. Most Mi-2-positive patient sera react with both proteins.
The sera usually show a fine-speckled nuclear pattern by ANA testing (Fig. 20.3) [2]. Interestingly, sera with anti-Mi-2 will sometimes react with TIF-1 proteins targeted by other patients with DM [2, 65]. Immunoprecipitation blotting may show precipitation of TIF1α, and there may be low-level cross-reaction with TIF1g by ELISA [65]. It is usually clear which antibody predominates in a patient serum.
Anti-Mi-2 was the first autoantibody to be strongly associated with myositis, particularly dermatomyositis, using immunoprecipitation or immunodiffusion [59]. These methods appear to identify binding to a particular conformational epitope, and immunoblotting assays may give results that are less specific for dermatomyositis [9]. Although found in both adult and juvenile myositis, it may be more common in adults. Malignancy has been reported, but it does not appear to have the increased frequency in cancer-associated myositis as seen with anti-TIF1g. Cutaneous involvement is typically that of classic DM with Gottron changes, heliotrope, and the “V sign” (involvement of the portion of the upper chest around the neck) and “shawl” sign (involvement of the upper back in the area covered by a shawl) [5].
Myositis in patients with anti-Mi-2 tends to be mild and glucocorticoid responsive with a relatively good prognosis despite the CK being high initially [5]. In contrast to anti-MDA5 or anti-TIF1g, anti-Mi-2 patients are less likely to have amyopathic dermatomyositis. Further, there are less associated connective tissue disease features.
Anti-Mi-2 is associated with a steroid-responsive milder phenotype of DM including classic DM with Gottron changes, heliotrope, the V sign, and shawl sign.
It was observed that the frequency of anti-Mi-2 (and of DM itself) varied greatly in different populations, from 60% in Guatemala to 3.2% in Montreal [66]. The frequency of involvement appeared to correlate with greater exposure to UV light. A high frequency was observed in Mexico and Central America in particular, which could relate to a combination of genetic factors and environmental exposures.
Anti-Signal Recognition Particle Antibodies (Anti-SRP)
The signal recognition particle is a complex of an RNA (7SL) and six proteins involved in translocation, the process through which newly forming proteins are targeted to the endoplasmic reticulum for secretion or membrane expression. Patient antibodies usually react with the 54 and/or the 72 kD proteins [67, 68]. The antibody is easily, specifically, and sensitively identified by immunoprecipitation, which can show the RNA and the protein complex (Figs. 20.1 and 20.2) [68]. Other methods have been used, which may be less specific with resulting differences in observed clinical associations. Anti-SRP autoantibodies give a characteristic cytoplasmic pattern on ANA testing (indirect immunofluorescence) (Fig. 20.3). Most patients with this antibody previously had a diagnosis of PM [5, 68], but the more common recent association is with immune-mediated necrotizing myopathy [2, 69, 70]. In typical cases, the distinctive feature is severe muscle weakness, often greater than with typical polymyositis [69]. It can be relatively acute, or rapid in onset, with very high CK levels compared to usual PM, leading to early muscle damage, treatment refractoriness, and multiple flares [5]. Often the myositis responds incompletely and is more likely to require multiple immunosuppressive agents. However, this distinctive presentation is not uniformly seen, as some patients have better responses [71]. Increased cardiac involvement noted in early reports [68] has not consistently been observed in subsequent reports.
Anti-SRP is associated with necrotizing myopathy presenting with acute onset of severe weakness, very high CPK, and refractory disease.
Histologically, the typical picture with this antibody is a necrotizing myopathy without inflammation. One study found vasculopathy with capillary loss and deposition of membrane attack complex as seen in DM, but without perifascicular atrophy [69].
Patients with this antibody may be less likely to show connective tissue disease overlap features such as interstitial lung disease, arthritis, or Raynaud phenomenon compared with antisynthetase-positive patients [68], but overlap features can certainly occur (and are more common than in patients with anti-HMGCR antibody-positive NM).
Anti-PM-Scl and Anti-Ku
Anti-PM-Scl is an MAA originally identified using immunodiffusion, as a clarification of the originally reported specificity of anti-PM-1 [7]. It was named for the clinical association of the antibody with an overlap syndrome with features of myositis and scleroderma. The antibody was found to show a series of at least 11 proteins by immunoprecipitation that are easily recognized and identified (Fig. 20.1), although there is no associated RNA [72,73,74]. It shows a combination of nucleolar and nuclear staining by IIF (Fig. 20.3). The proteins with apparent molecular weights of 100 and 75 kD are the major antigens. The PM-Scl complex was identified as the exosome, which is involved in RNA processing.
Although many patients with this antibody have the typical overlap syndrome of myositis and scleroderma, some patients show only myositis or only scleroderma [72]. The myositis is often associated with typical DM cutaneous involvement, and mechanic’s hands can occur. The scleroderma is most commonly limited in cutaneous involvement. However, there have been occasional reports of renal crisis [75]. There is often a significant associated inflammatory polyarthritis. The myositis tends to be responsive to treatment, often responding to lower doses of glucocorticoids than other forms of myositis, with scleroderma features remaining unchanged. This overlap syndrome has been referred to as “scleromyositis” [76]. In some populations, anti-PM-Scl may account for a substantial proportion of myositis-scleroderma overlap patients. However, there is a strong association with HLA DR3, which varies considerably among different ethnic populations, being infrequent in Japanese patients. The antibody tends to be mutually exclusive with MSAs or scleroderma antibodies, with occasional exceptions.
Anti-PM-Scl and anti-Ku are MAA associated with scleroderma-myositis overlap syndrome.
Anti-Ku autoantibodies were first described using immunodiffusion and can be seen by immunoprecipitation with two strong proteins of 72 and 86 kD, with associated heterogeneous nucleic acid [77]. There is an additional, high-molecular-weight DNA protein kinase component. The antigen is involved in DNA repair.
Anti-Ku is an MAA that has been associated with scleroderma-myositis overlap syndrome in Japanese patients [8] and is relatively frequent in patients with that condition. In the United States, it is more common in African-American than Caucasian patients [78] and is often associated with myositis or systemic lupus or overlap syndromes [77].
Anti-Ro/SSA, Anti-U1RNP
Although the primary clinical associations of anti-Ro/SSA are Sjogren syndrome and lupus, it can be found in some patients with myositis and thus would satisfy the definition of an MAA. Of interest is that anti-Ro52, which typically occurs in association with anti-Ro60 in most conditions, is more likely to occur in the absence of Ro60 in myositis than in other situations [55]. This could possibly have diagnostic implications. When occurring in myositis, it most often is seen in association with other autoantibodies, including anti-ARS, anti-PM-Scl, or anti-SRP [54]. In those patients, it may be a marker of more severe disease and a worse prognosis.
Anti-U1RNP is commonly seen in patients with lupus who have myositis as a component of their disease and thus would satisfy the definition of an MAA [79]. It can be seen by itself or with other myositis autoantibodies. Myositis may occur in patients with anti-U1RNP as part of the MCTD spectrum. The myositis in this situation may be milder or more responsive, but can be severe in some patients. Anti-U1RNP can occur in some patients with other MSAs.
Anti-U1RNP reacts with proteins of the U1 small nuclear ribonucleoprotein, which is involved in messenger RNA splicing. Patients may have autoantibodies that are specific for proteins that are unique to this particle, or patients with anti-Sm may have autoantibodies to proteins that are shared with other U small nuclear ribonucleoproteins involved in the splicing process. Of interest in myositis patients is the occasional occurrence of autoantibodies that react with proteins that are unique to U small nuclear RNPs other than the U1 particle. Anti-U2RNP [80, 81], anti-U5RNP [82], and anti-U4/6RNP [82, 83]have been observed. Usually, these patients have overlap connective tissue disease syndromes that may involve myositis.
References
Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin N Am. 2002;28(4):859–90, viii.
Satoh M, et al. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin Rev Allergy Immunol. 2017;52(1):1–19.
Gunawardena H. The clinical features of myositis-associated autoantibodies: a review. Clin Rev Allergy Immunol. 2017;52(1):45–57.
Targoff IN. Update on myositis-specific and myositis-associated autoantibodies. Curr Opin Rheumatol. 2000;12(6):475–81.
Love LA, et al. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine. 1991;70(6):360–74.
Watanabe K, et al. Detection of antisynthetase syndrome in patients with idiopathic interstitial pneumonias. Respir Med. 2011;105(8):1238–47.
Reichlin M, et al. Antibodies to a nuclear/nucleolar antigen in patients with polymyositis overlap syndromes. J Clin Immunol. 1984;4(1):40–4.
Mimori T, et al. Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap. J Clin Investig. 1981;68(3):611–20.
Koenig M, et al. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther. 2007;9(4):R78.
Targoff IN, Trieu EP, Miller FW. Reaction of anti-OJ autoantibodies with components of the multi-enzyme complex of aminoacyl-tRNA synthetases in addition to isoleucyl-tRNA synthetase. J Clin Investig. 1993;91(6):2556–64.
Satoh M, Ceribelli A, Chan EK. Common pathways of autoimmune inflammatory myopathies and genetic neuromuscular disorders. Clin Rev Allergy Immunol. 2012;42(1):16–25.
Rojas-Serrano J, et al. Prognostic factors in a cohort of antisynthetase syndrome (ASS): serologic profile is associated with mortality in patients with interstitial lung disease (ILD). Clin Rheumatol. 2015;34(9):1563–9.
Mahler M, Miller FW, Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev. 2014;13(4–5):367–71.
Chen F, et al. Clinical heterogeneity of interstitial lung disease in polymyositis and dermatomyositis patients with or without specific autoantibodies. Am J Med Sci. 2018;355(1):48–53.
Friedman AW, Targoff IN, Arnett FC. Interstitial lung disease with autoantibodies against aminoacyl-tRNA synthetases in the absence of clinically apparent myositis. Semin Arthritis Rheum. 1996;26(1):459–67.
Marie I, et al. Comparison of long-term outcome between anti-Jo1- and anti-PL7/PL12 positive patients with antisynthetase syndrome. Autoimmun Rev. 2012;11(10):739–45.
Targoff IN, Arnett FC. Clinical manifestations in patients with antibody to PL-12 antigen (alanyl-tRNA synthetase). Am J Med. 1990;88(3):241–51.
Hamaguchi Y, et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS ONE [Electronic Resource]. 2013;8(4):e60442.
Hirakata M, et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol. 1999;162(4):2315–20.
Targoff IN. Autoantibodies to aminoacyl-transfer RNA synthetases for isoleucine and glycine. Two additional synthetases are antigenic in myositis. J Immunol. 1990;144(5):1737–43.
Abe T, et al. Reliability and clinical utility of enzyme-linked immunosorbent assay for detection of anti-aminoacyl-tRNA synthetase antibody. Nihon Rinsho Meneki Gakkai Kaishi. 2016;39(2):140–4.
Nakashima R, Hosono Y, Mimori T. Clinical significance and new detection system of autoantibodies in myositis with interstitial lung disease. Lupus. 2016;25(8):925–33.
Aggarwal R, et al. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab.[Erratum appears in Rheumatology (Oxford). 2016 Sep;55(9):1710; PMID: 27383242]. Rheumatology. 2016;55(6):991–9.
Stone KB, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum. 2007;56(9):3125–31.
Ghirardello A, et al. Autoantibody testing in patients with myositis: clinical accuracy of a multiparametric line immunoassay. Clin Exp Rheumatol. 2017;35(1):176–7.
Nakashima R, et al. The multicenter study of a new assay for simultaneous detection of multiple anti-aminoacyl-tRNA synthetases in myositis and interstitial pneumonia. PLoS ONE [Electronic Resource]. 2014;9(1):e85062.
Bundell C, et al. Diagnostic performance of a commercial immunoblot assay for myositis antibody testing. Pathology. 2016;48(4):363–6.
Miller FW, et al. Origin and regulation of a disease-specific autoantibody response. Antigenic epitopes, spectrotype stability, and isotype restriction of anti-Jo-1 autoantibodies. J Clin Invest. 1990;85(2):468–75.
Aggarwal R, et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheum. 2014;66(3):740–9.
Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry. 2000;68(4):472–8.
Mescam-Mancini L, et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain. 2015;138(Pt 9):2485–92.
Noguchi E, et al. Skeletal muscle involvement in antisynthetase syndrome. JAMA Neurol. 2017;74(8):992–9.
Aggarwal R, et al. Patients with non-Jo-1 anti-tRNA-synthetase autoantibodies have worse survival than Jo-1 positive patients. Ann Rheum Dis. 2014;73(1):227–32.
Arnett FC, et al. Interrelationship of major histocompatibility complex class II alleles and autoantibodies in four ethnic groups with various forms of myositis. Arthritis Rheum. 1996;39(9):1507–18.
Targoff IN, et al. Antibodies to glycyl-transfer RNA synthetase in patients with myositis and interstitial lung disease. Arthritis Rheum. 1992;35(7):821–30.
Dugan EM, et al. Photoessay of the cutaneous manifestations of the idiopathic inflammatory myopathies. Dermatol Online J. 2009;15(2):1.
Rider LG, et al. A broadened spectrum of juvenile myositis. Myositis-specific autoantibodies in children. Arthritis Rheum. 1994;37(10):1534–8.
Rider LG, et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92(4):223–43.
Shah M, et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine. 2013;92(1):25–41.
Chinoy H, et al. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis. 2007;66(10):1345–9.
Shi J, et al. Clinical profiles and prognosis of patients with distinct Antisynthetase autoantibodies. J Rheumatol. 2017;44(7):1051–7.
Zamora AC, et al. Clinical features and outcomes of interstitial lung disease in anti-Jo-1 positive antisynthetase syndrome. Respir Med. 2016;118:39–45.
Marie I, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47:614.
Marie I, et al. Interstitial lung disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Care Res. 2013;65(5):800–8.
Stanciu R, et al. Antisynthetase syndrome with anti-Jo1 antibodies in 48 patients: pulmonary involvement predicts disease-modifying antirheumatic drug use. J Rheumatol. 2012;39(9):1835–9.
Yura H, et al. Clinical characteristics of patients with anti-aminoacyl-tRNA synthetase antibody positive idiopathic interstitial pneumonia. Respir Med. 2017;132:189–94.
Takato H, et al. Pulmonary manifestations of anti-ARS antibody positive interstitial pneumonia--with or without PM/DM. Respir Med. 2013;107(1):128–33.
Tanizawa K, et al. The long-term outcome of interstitial lung disease with anti-aminoacyl-tRNA synthetase antibodies. Respir Med. 2017;127:57–64.
Hirakata M, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum. 2007;56(4):1295–303.
Waseda Y, et al. Antisynthetase syndrome: pulmonary computed tomography findings of adult patients with antibodies to aminoacyl-tRNA synthetases. Eur J Radiol. 2016;85(8):1421–6.
Debray M-P, et al. Interstitial lung disease in anti-synthetase syndrome: initial and follow-up CT findings. Eur J Radiol. 2015;84(3):516–23.
Doyle TJ, et al. Rituximab in the treatment of interstitial lung disease associated with Antisynthetase syndrome: a multicenter retrospective case review. J Rheumatol. 2018;45:841.
Tanizawa K, et al. The prognostic value of HRCT in myositis-associated interstitial lung disease. Respir Med. 2013;107(5):745–52.
Frank MB, et al. The association of anti-Ro52 autoantibodies with myositis and scleroderma autoantibodies. J Autoimmun. 1999;12(2):137–42.
Rutjes SA, et al. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patients with idiopathic inflammatory myopathy. Clin Exp Immunol. 1997;109(1):32–40.
Marie I, et al. Short-term and long-term outcome of anti-Jo1-positive patients with anti-Ro52 antibody. Semin Arthritis Rheum. 2012;41(6):890–9.
Bauhammer J, et al. Rituximab in the treatment of Jo1 antibody-associated Antisynthetase syndrome: anti-Ro52 positivity as a marker for severity and treatment response. J Rheumatol. 2016;43(8):1566–74.
Oddis CV, Medsger TA Jr, Cooperstein LA. A subluxing arthropathy associated with the anti-Jo-1 antibody in polymyositis/dermatomyositis. Arthritis Rheum. 1990;33(11):1640–5.
Targoff IN, Reichlin M. The association between Mi-2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28(7):796–803.
Nilasena DS, Trieu EP, Targoff IN. Analysis of the Mi-2 autoantigen of dermatomyositis. Arthritis Rheum. 1995;38(1):123–8.
Ge Q, et al. Molecular analysis of a major antigenic region of the 240-kD protein of Mi-2 autoantigen. J Clin Investig. 1995;96(4):1730–7.
Zhang Y, et al. The dermatomyositis-specific autoantigen Mi-2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell. 1998;95(2):279–89.
Seelig HP, et al. The major Dermatomyositis-Specific Mi-2 Autoantigen is a presumed helicase involved in transcriptional activation. Arthritis Rheum. 1995;38(10):1389–99.
Seelig HP, et al. Two forms of the major antigenic protein of the dermatomyositis-specific Mi-2 autoantigen. Arthritis Rheum. 1996;39(10):1769–71.
Fujimoto M, et al. Enzyme-linked immunosorbent assays for detection of anti-transcriptional intermediary factor-1 gamma and anti-Mi-2 autoantibodies in dermatomyositis. J Dermatol Sci. 2016;84(3):272–81.
Shamim EA, et al. Differences in idiopathic inflammatory myopathy phenotypes and genotypes between Mesoamerican Mestizos and North American Caucasians: ethnogeographic influences in the genetics and clinical expression of myositis. Arthritis Rheum. 2002;46(7):1885–93.
Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with the signal-recognition particle. Proc Natl Acad Sci U S A. 1986;83(24):9507–11.
Targoff IN, Johnson AE, Miller FW. Antibody to signal recognition particle in polymyositis. Arthritis Rheum. 1990;33(9):1361–70.
Miller T, et al. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry. 2002;73(4):420–8.
Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol. 2011;23(6):595–604.
Kao AH, et al. Anti-signal recognition particle autoantibody in patients with and patients without idiopathic inflammatory myopathy. Arthritis Rheum. 2004;50(1):209–15.
Oddis CV, et al. Serum autoantibody to the nucleolar antigen PM-Scl. Clinical and immunogenetic associations. Arthritis Rheum. 1992;35(10):1211–7.
Ge Q, et al. Cloning of a complementary DNA coding for the 100-kD antigenic protein of the PM-Scl autoantigen. J Clin Investig. 1992;90(2):559–70.
Ge Q, et al. Analysis of the specificity of anti-PM-Scl autoantibodies. Arthritis Rheum. 1994;37(10):1445–52.
Schnitz W, et al. Anti-PM/Scl autoantibodies in patients without clinical polymyositis or scleroderma. J Rheumatol. 1996;23(10):1729–33.
Jablonska S, Blaszyk M. Scleromyositis (scleroderma/polimyositis overlap) is an entity. J Eur Acad Dermatol Venereol. 2004;18(3):265–6.
Reeves WH. Antibodies to the p70/p80 (Ku) antigens in systemic lupus erythematosus. Rheum Dis Clin N Am. 1992;18(2):391–414.
Wang J, et al. Increased prevalence of autoantibodies to ku antigen in African American versus white patients with systemic lupus erythematosus. Arthritis Rheum. 2001;44(10):2367–70.
Dayal NA, Isenberg DA. SLE/myositis overlap: are the manifestations of SLE different in overlap disease? Lupus. 2002;11(5):293–8.
Mimori T, et al. Autoantibodies to the U2 small nuclear ribonucleoprotein in a patient with scleroderma-polymyositis overlap syndrome. J Biol Chem. 1984;259(1):560–5.
Craft J, et al. The U2 small nuclear ribonucleoprotein particle as an autoantigen. Analysis with sera from patients with overlap syndromes. J Clin Investig. 1988;81(6):1716–24.
Okano Y, et al. Anti-U5 small nuclear ribonucleoprotein (snRNP) antibodies: a rare anti-U snRNP specificity. Clin Immunol Immunopathol. 1996;81(1):41–7.
Okano Y, Medsger TA. Newly identified U4/U6 snRNP-binding proteins by serum autoantibodies from a patient with systemic sclerosis. J Immunol. 1991;146(2):535–42.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vaseer, S., Targoff, I.N. (2020). Traditional Myositis Autoantibodies: Synthetase, Mi-2, SRP, Ku, PM-Scl, Ro, U1RNP. In: Aggarwal, R., Oddis, C. (eds) Managing Myositis. Springer, Cham. https://doi.org/10.1007/978-3-030-15820-0_20
Download citation
DOI: https://doi.org/10.1007/978-3-030-15820-0_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-15819-4
Online ISBN: 978-3-030-15820-0
eBook Packages: MedicineMedicine (R0)