
Abstract
It is estimated that chronic inflammation contributes to nearly 25% of human cancers. Inflammation of the gastric mucosa is dependent on various modulatory components such as microbes, environment, and host predisposition. Helicobacter pylori (H. pylori) can initiate and sustain gastric inflammation by its virulence factors as well as by altered cellular pathways that are involved in the restoration of the tissue homeostasis after infection. Indeed, in an attempt to repair injured mucosa, immune system may contribute to gastric cancer development through its physiological pro-inflammatory and anti-inflammatory activities which, particularly in a setting of chronic antigenic stimulation, can turn in pro-tumorigenic effects. In this chapter some of the mechanisms connected to H. pylori-related inflammation will be depicted, also focusing on microenvironmental cellular and soluble driving factors recently highlighted in gastric cancer promotion.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Microenvironment
- Inflammation
- Helicobacter pylori
- CagPAI
- VacA
- HP-NAP
- GGT
- ROS
- Myeloid-derived suppressor cells
- T helper
- Cytokine
- Field cancerization
Introduction
Great improvements in molecular and cellular technologies and decades of in-depth studies were needed so that inflammation was added to the hallmarks of cancer, and pioneering observations of Virchow and Coley became widely accepted perspectives to be pursued for translation in cancer cures [1,2,3]. Inflammation is a coordinated response following infection or tissue damage by exogenous or endogenous agents, which involves innate and adaptive immune system cells and soluble factors. Macrophages, neutrophils, eosinophils, mast cells, dendritic cells (DCs), and natural killer (NK) cells represent the antigen-independent first line of immunological defense against homeostatic perturbation of tissue microenvironment. These cell subsets initiate inflammatory response by sensing pathogen-associated molecular patterns, which are present during microbial infections, and danger-associated molecular patterns, which are components of the host cells released during cell damage or death. At early stages of inflammation, tissue antigens are processed and transported to lymphoid organs by specialized antigen-presenting cells, which allow the activation and expansion of B and T lymphocyte-specific immune responses. In this scenario, intracellular regulatory pathways are activated, which ultimately lead to the secretion of reactive oxygen and reactive nitrogen species (ROS and RNS), of diffusible growth factors, of inflammatory cytokines, and of matrix-remodeling enzymes. These elements induce mobilization and infiltration of additional leucocytes in the affected field and magnify the inflammatory reaction until the resolution of the injury or infection.
The tumorigenic fate of the immune response largely depends on the physiological state of the epithelial, stromal, and vascular microenvironment and on the immune cell profile that are part of it, hence from the signals conveyed toward autophagy/death, differentiation, proliferation, and angiogenetic circuits and from the cross talk between them. The duration of the inflammation is another key feature affecting the outcome of the immune responses. This is strictly linked to the presence of host immunogenetic predisposition and/or ongoing chemical, physical, or biological irritation. In the case of gastric mucosa, infection with persistent microorganisms bearing oncogenic potential such as Epstein-Barr virus and Helicobacter pylori (H. pylori) can initiate local inflammation and, after elusion of immune clearance mechanisms, may cause chronic inflammation. Specific or non-specific viral and bacterial virulence factors in conjunction with immunity defects can cause aberrant interactions between microbes and gastric epithelial cells. This condition can drive premalignancy, implementing the inflammatory response with the accumulation of new genetic and epigenetic modifications in epithelial cells, actually favoring the establishment of a gastric cancerized field.
Helicobacter and Inflammation: The Two Facet Janus
H. pylori is a Gram-negative, spiral-shaped, microaerophilic bacterium colonizing the human stomach. From a biological and evolutionary point of view, H. pylori has coevolved with humans for at least 50,000 years to be transmitted from person to person and become a commensal of the stomach [4, 5]. An homeostatic equilibrium between bacterial effectors and host responses allows microbial persistence, but also confers the risk of gastric neoplasia. In 1994 H. pylori was classified as a class I human carcinogen by the International Agency for Research on Cancer working group for its association with an increased risk for gastric cancer, in particular non-cardia gastric cancer, and mucosa-associated lymphoid tissue (MALT) lymphoma [6]. Since that time, H. pylori infection is considered the primary cause of gastric neoplasms [7], although the etiology is multifactorial. One of the first mechanisms by which H. pylori may express its pathogenetic potential is inflammation-related and refers to the production of autoreactive immunoglobulins; these may cause complement-dependent cell lysis and small immune complexes formation that may promote local damage [8]. Autoantibodies originate through molecular mimicry of host epitopes by lipopolysaccharide (LPS) structures of H. pylori [9]. These observations prompt to evaluate the inflammation-related carcinogenic potential of the structural components of a broad range of microbial populations colonizing the gastric environment.
Both the undifferentiated and the differentiated gastric cancer types (named diffuse-type carcinoma and intestinal-type adenocarcinoma, respectively) are associated with H. pylori. However, only the pathogenesis of the intestinal cancer seems to significantly involve the chronic inflammation, which directs the abnormal differentiation of the normal gastric mucosa toward the precancerous gastric lesions. This can be done according to the cascade model hypothesized by Correa, which involves the evolution of the forms of non-atrophic gastritis, toward multifocal atrophic gastritis without intestinal metaplasia, intestinal metaplasia, dysplasia, and finally cancer [10]. All these lesions occur in a setting of inflammation and in a complex milieu of diffusible factors. Despite the variable, but significant, prevalence of H. pylori infection in various countries [11], it is estimated that 1–3% of infected people will develop non-cardia gastric cancer and lymphoma [12, 13]. Indeed, besides the environmental factors, such as smoking and diet, and the commensal microbes, the clinical outcome of the infection is conditioned by virulence factors of H. pylori, by its high phenotypic and genomic heterogeneity within the gastric niche [14] as well as by genetic susceptibility and immune profile of the host [15].
Each host is not colonized by a single type of H. pylori, but by a multitude of genetically closely related microorganisms similar to quasispecies, which interfere with signaling pathways influencing host cell growth and death [16, 17]. From an ecological and teleological point of view, the diversity is originated by the bacterium in an attempt to persist in the microenvironment, notwithstanding the oxidative stress directly caused by H. pylori virulence factors and indirectly by inflammatory response. Pro-inflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)1-β and IL-8, sustain inflammation in gastric mucosa, but anti-inflammatory cytokines, such as IL-10, tend to turn it off. They are released by several components of the immune system as well as by cells immersed in the stromal microenvironment, such as fibroblasts, epithelial, and endothelial cells. They can accomplish pleiotropic effects on a wide range of cell types, including immune and epithelial cells. Since variations in genotypes heightening cytokine levels have been associated with an increased risk of gastric cancer [18,19,20,21,22], cytokines are believed to enhance overall rather than attenuate the pathogenicity of the bacterium. However, it’s elemental to highlight that the cellular composition of the microenvironment might deeply influence the cancer risk; the own different CD4+ T cell subsets can secrete different cytokine and chemokine types, which in turn can stimulate different signal transduction pathways and activation of transcription factors, leading to pro-inflammatory reactive or anti-inflammatory suppressive responses.
Helicobacter pylori-Specific Determinants Affecting Inflammation and Tumorigenesis
A plenty of virulence factors have been described in H. pylori infection. Some of them are highly studied and specifically involved in inflammatory response after infection. Moreover, they cooperate to the inflammation-related tumorigenic process. Among the most mentioned virulence determinants for their relevance in colonization, persistence, and oxidative stress induction, there are the H. pylori neutrophil-activating protein (HP-NAP), the γ-glutamyl-transpeptidase (GGT), the cytotoxin-associated gene pathogenicity island (CagPAI), and the vacuolating cytotoxin A (VacA).
While GGT and HP-NAP are constitutively expressed and show little genetic variability among H. pylori isolates, perhaps indicating a structural function or a lack of immune selection for diversification [23], on the other hand, vacA and CagPAI show plasticity, being apt to genetic modifications which modulate their virulence [24,25,26]. The characteristics and modalities of action of these different virulence factors are summarized in the following paragraphs.
H. pylori Neutrophil-Activating Protein
H. pylori neutrophil-activating protein (HP-NAP) has probably evolved as a pro-inflammatory molecule to sustain the production of reactive oxygen intermediates by human neutrophils, functional to the release of nutrients, which can speed H. pylori growth [27]. It has been described that HP-NAP can trigger inflammation in conjunction with other bacterial and host-derived factors [28], but also as an only molecule. Indeed, several studies sustain a model in which HP-NAP represents a critical element in initiating the inflammatory process. HP-NAP is probably released after cell lysis in the infected mucosa of the stomach, and, after its transfer through gastric epithelial lining, it activates subepithelial resident mast cells and macrophages [29]. Consequently, these innate immune components release biochemical mediators and, in particular, the pleiotropic cytokine TNF-α. Overall, soluble factors attract and stimulate the adhesion and extravasation of polymorphonucleates (PMN) and lympho-monocytes through the endothelium lining the vessels, as suggested by the TNF-α-induced upregulation of adhesion molecules V-CAM and I-CAM on the surface of endothelial cells and by in vitro and in vivo experiments on animal models [30, 31]. PMN and monocytes produce and secrete ROS through the HP-NAP-induced increase of cytoplasmic Ca2+ and phosphorylation of proteins, leading to assembly of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase on plasma membrane. Moreover, PMN and monocytes are activated to secrete cytokines and chemokines which amplify the inflammatory state. Among these, IL-12 and IL-23 contribute to differentiate the monocytes into a mature dendritic phenotype and the T- lymphocytic response toward a cytotoxic T- helper type 1 (Th1) phenotype producing interferon-γ (IFN-γ), TNF-β, IL-12, IL-18, IL-17, and TNF-α [32]. Preclinical studies demonstrate that HP-NAP inhibits the differentiation of Th0 into Th2 profile [23].
γ-Glutamyl-Transpeptidase
γ-Glutamyl-transpeptidase (GGT) is a virulence factor virtually associated with all wild-type H. pylori strains, although strain-to-strain variations in GGT expression among clinical isolates from patients with different disease statuses have been observed. GGT is related to ROS production from the epithelium and to oxidation of DNA and membrane lipids by using the body’s master antioxidant glutathione (GSH), which is catabolized by GGT itself. Besides pro-apoptotic and necrotic effects evoked by ROS compounds and potentially sustained by other virulence factors such as VacA, GGT shows anti-apoptotic activities by activation of p38 mitogen-activated protein kinases (MAPKs), protein kinase B (AKT), and nuclear factor k-light-chain-enhancer of activated B cell (NF-kB) signaling pathways; the subsequent production of inducible nitric oxide synthase (iNOS), DNA damage, IL-8, and prostaglandin synthase cyclooxygenase-2 (COX-2) enhances the inflammatory reaction and induces epithelial cell proliferation [33]. Additionally, GGT suppresses T cell proliferation by inducing cell cycle arrest through the disruption of the Ras signaling pathway [34]. In vitro and in vivo studies suggest that GGT contributes to DC tolerization and directs the T cell response toward a regulatory immunosuppressive phenotype [35]. A suppressive milieu inhibits lymphocyte activation and favors H. pylori escape and persistent infection.
Cytotoxin-Associated Gene A Pathogenicity Island and Vacuolating Cytotoxin A
Cytotoxin-associated gene A pathogenicity island (CagPAI) is a 40,000 base pairs sequence containing coding regions for virulence determinants and several proteins participating to the assembly of a specialized syringe machinery called type IV secretion system. Through this structure, H. pylori is able to inject into cells inflammation- and tumorigenesis-related bacterial components, such as the cytotoxin-associated gene A (CagA), peptidoglycans, and methyltransferases. Proteins encoded by CagPAI genes induce inflammation by using the host signaling pathways essential for maintenance of the normal gastric mucosa homeostasis [36]. In the case of CagA, after translocation into epithelial cells, it acts through direct interaction with intracellular receptors in a phosphorylation-dependent or phosphorylation-independent manner. In the first case, CagA becomes phosphorylated by members of the Src and Abl family kinases at specific amino acidic motifs in the C-terminus of the protein (Glu-Pro-Ile-Tyr-Ala, EPIYA). This phosphorylation allows CagA binding to SH2 domain-containing proteins, such as SHP2 tyrosine phosphatase, causing its activation and subsequent induction of the extracellular signal-regulated kinases (ERK)-MAPK pathway, which leads to mitogenic response and cellular migration [37]. In the second case, CagA is translocated, but not phosphorylated, and it determines altered activation of β-catenin, disruption of apical junctional complexes, and loss of cellular polarity. Moreover, non-phosphorylated CagA targets a series of adhesion, enzymatic, and transducer molecules, which leads to mitogenic and pro-inflammatory responses [38,39,40,41]. CagA also interacts with tumor suppressor proteins, such as Runt-related transcription factor 3 (RUNX3) and protein 53 (p53) leading to their proteasomal degradation [37]. It has been reported that translocated CagA into the host cell is degraded by oxidative stress-dependent autophagy and, hence, short-lived, except when it enters CD44v9+ gastric cancer stem-like cells, that show oxidative stress resistance due to their high GSH content [42]. The expression of the CD44 homing receptor can be induced upon chronic inflammation [43], is involved in the upregulation of GSH synthesis, contributes to the progression of precancerous gastric lesions in patients with H. pylori infection, and correlates positively with recurrence of gastric cancer [44,45,46]. These observations suggest that the accumulation of alterations due to ROS and the cell survival through protection against ROS may play a considerable role for the generation of cancer cells in the infected gastric mucosa.
CagPAI-codified type IV secretion system can also deliver peptidoglycans into host cells, where they are recognized by the nucleotide-binding oligomerization domain-containing protein 1 (NOD1). The subsequent activation of NF-kB, p38, and extracellular ERK signaling induces the production of pro-inflammatory cytokines macrophage inflammatory protein (MIP)-2, β-defensins, and IL-8. Additionally, the interaction between NOD1 and post-translational-modified peptidoglycans modulates the production of type I interferons which are involved in the activation of DCs and of T cell cytotoxic effector functions [47,48,49].
CagA and other H. pylori molecules can be injected not only into gastric epithelial cells, but also into B lymphoid cells and DCs. As a consequence, host’s immune responses can be suppressed through the reduction in the secretion of pro-inflammatory cytokines, such as IL-12p40, and the increase in the expression of suppressive cytokines, such as IL-10 [50]. This highlights the existence of pro-inflammatory and anti-inflammatory effects produced by the same virulence component in dependence on the cellular metabolic status and composition of the microenvironment.
VacA is a pore-forming protein which is secreted by H. pylori through a type V auto-transport secretion system. It exerts multiple effects on epithelial and immune cells in synergy with other virulence determinants. VacA can be internalized into the host cells by endocytosis; afterward it accumulates in different cellular compartments inducing apoptosis. In parallel, it contributes to the successful colonization of the gastric niche disrupting epithelial cell tight connections and allowing the access of bacterial molecules and H. pylori to the lamina propria. This function is shared with CagA that is able to bind and inhibit PAR1b, a protein essential for the establishment and maintenance of cell polarity. Once in the innermost layers of the gastric mucosa, VacA encounters granulocytes and T cells recruited to the sites of infection by the triggered inflammation program. Herein, VacA is capable of inducing an influx of Ca2+, probably NF-kB activation, and consequent inflammation through generation of oxidative stress and IL-8 secretion [51, 52]. On the other hand, it modulates the inflammatory response restricting T lymphocytes proliferation and effector functions [53]. In vitro and in vivo experiments demonstrate that VacA, in cooperation with GGT, contributes critically and non-redundantly to H. pylori tolerizing effects on murine DCs allowing persistence of the bacterium [35, 54].
Helicobacter pylori Affects Early Phases of Inflammation
Several evidences point to an involvement of H. pylori in the first phases of the carcinogenesis while long lasting molecular changes in epithelial cells, which result from the initial infection with virulent H. pylori strains, contribute to tissue damage progression [55, 56]. Indeed, the reversibility of oxidative and nitrosative stress processes, one of the crucial initial steps of the inflammatory reaction contributing to carcinogenesis in gastric mucosa, has been documented after H. pylori eradication [57]. Moreover, prospective studies show that H. pylori eradication by antibiotics reduces the incidence of precancerous lesions, and it is effective in reversing atrophic gastritis, but not intestinal metaplasia [58, 59]. Finally, H. pylori eradication does not decrease the risk of gastric cancer in patients with more advanced metaplastic or dysplastic mucosal lesions [60].
Inflammation and H. pylori-Mediated Oxidative and Nitrosative Stresses
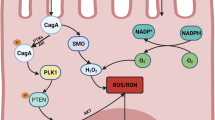
RNS are produced mainly by neutrophils and macrophages, but also by gastric epithelial cells through the action of the nitric oxide synthase (NOS) and, especially, of iNOS. Nitric oxide (NO) is sufficiently long-lived to diffuse through the extracellular matrix and enter the nucleus of epithelial cells infected by H. pylori and those surrounding them within the gastric pit. ROS, such as superoxide (O2−), is active in this biochemical pathway. The source of effective ROS is the epithelial cell itself, since ROS generated by neutrophils and macrophages are not sufficiently long-lived to diffuse through extracellular matrix and penetrate epithelial cell membranes. Here, NO and O2− react to form peroxynitrite (ONOO−), which causes DNA damage through guanine nitration and, finally, mutations, impairment of DNA repair enzymes and genomic instability [61,62,63]. Changes in lipid and protein expression consequent to oxidative stress have been observed [64, 65]. Last but not least, induced NO production interferes with transcriptional modulation by promoting DNA hyper-methylation both in noncoding and coding sequences for clincher proteins of the carcinogenetic intracellular pathways, such as p53, the cyclin-dependent kinase inhibitors (CDKN2A/CDKN2B), the epithelial cadherin-1 (CDH1) or mutL homolog 1 (MLH1), and many others. It’s worth noting that passenger genes, namely, genes that are not directly causally involved in gastric carcinogenesis, are even significantly subjected to silencing by aberrant methylation in the cancerized field [55, 66, 67]. These observations point to methylation rather than silencing of genes by mutation as the main mechanism for inactivation of driver and passenger tumor suppressor genes, indicating that gastric cancer is an epigenetic disease [56].
ROS accumulation in differentiated and stem gastric cells can be directly and indirectly induced by H. pylori. Due to its poor immunogenicity, LPS helps the bacteria to develop a chronic infection and, following activation of epithelial Toll-like receptor (TLR) 4 signaling, contributes to epithelial cell ROS production [68, 69]. Moreover, especially highly virulent CagA+ H. pylori strains can cause pro-oxidant activities through induction of NADPH oxidase or spermidine oxidase activity in host gastric cells [70,71,72]. ROS generation is indirectly induced by H. pylori infection through interaction of TNF-α-receptor on mucosal cell surface with TNF-α released by inflammatory cells in response to the infection. Epigenetic modifications can be directly induced by H. pylori possessing a functional type IV secretion system [36]. Indeed, through this structure, specific methyltransferases encoded by H. pylori may be injected into the host cell [73]. However, studies in gerbil-based models of carcinogenesis evidenced a major role of H. pylori-induced inflammation rather than a unique direct role of H. pylori-specific virulence factors in DNA methylation modulation. Indeed, increases of iNOS, IL1-β, TNF-α, and CXCL2 transcription, which are consequent to and synergistic with H. pylori infection immunopathologic effects, were shown to parallel the DNA methylation levels in gastric mucosa [74, 75]. Further experiments in animal models suggest that infiltrating mucosa monocytes are central components for H. pylori-dependent methylation induction and that the specificity of aberrant gene methylation in target cells is conditioned from their genomic architecture and from epigenetic elements already present in the cells where methylation is activated [56].
Inflammation and H. pylori-Mediated Alteration of DNA Repair Mechanisms
H. pylori may affect activation-induced cytidine deaminase (AID), which is an inducible enzyme, physiologically responsible for editing the human genome, e.g., for generating genomic diversity within the variable regions of immunoglobulin genes in activated B lymphocytes through somatic hypermutation and class switch recombination. AID is not expressed in normal gastric mucosa, but it is overexpressed in a proportion of H. pylori-infected gastric epithelium and in gastric cancer tissues, especially in the presence of mononuclear cell infiltration and intestinal metaplasia [76]. Most importantly, AID expression decreases after H. pylori eradication, suggesting a cause-effect link with the bacterium [77]. CagPAI+, but not cagPAI− H. pylori isolates, are able to stimulate aberrant AID expression in epithelial cell lines, causing chromosomal aberrations and somatic point mutations in tumor suppressor genes such as the aforementioned p53 and CDKN2A/CDKN2B [76, 78]. Moreover, also pro-inflammatory cytokines, such as TNF-α, indirectly increase the expression of AID through NF-kB intracellular pathway activation [55].
In vitro experiments on infected gastric epithelial cells demonstrate H. pylori-induced downregulation of proteins that are sequentially involved in the mechanism of base excision repair (BER) and which mediate the removal of incorrect single base residues [79]. Also the proteins of the DNA base mismatch repair are downregulated by H. pylori infection in gastric epithelial cells as well as in a H. pylori-infected mouse model and in H. pylori-positive patients with chronic gastritis [80, 81].
In gastric cell lines and primary gastric epithelial cells, it has been demonstrated that H. pylori infection prompts downregulation of several components engaged in double-strand DNA break (DSB) repair pathway, which generate carcinogenetic lesions if they are not appropriately restored [61]. Indeed, DNA damage affecting chromosomal ends resulting in telomere shortening and chromosomal instability has been reported [61]. Even if the precise mechanisms by which these events occur are not completely understood and elucidated, ex vivo and in vitro studies demonstrate that H. pylori infection is associated with alteration in DNA repair by direct host-pathogen contact, and prolonged infection may result in unrepaired breaks [82]. A genome-wide screening in gastric epithelial cell lines suggests an involvement of a type IV secretion system-dependent injection of XPF/XPG endonucleases together with NF-kB activation in DSB induction [83].
Toward Cellular Autophagy or Death
The molecular damage in gastric epithelial and immune cells, consequent to the activation of oxidative stress pathways by H. pylori-induced inflammation, stimulates caspase-mediated autophagy or apoptosis, with a raise in cell turnover during the initial steps of the infection [84, 85]. Autophagy is an intrinsic cytoprotective mechanism by self-eating and recycling of cellular components. Hence, autophagy can suppress tumor initiation by preserving normal cells and inhibiting inflammation. However, it can promote proliferation of damaged cells with precancerous characteristics by favoring inflammatory cell growth and providing sufficient oxygen and nutrients [86]. Hence, an increase in cell survival and proliferation may be induced in infected and in neighboring cells, adding on the possibility of malignant characteristics acquirement thanks to accumulation of mutagenic DNA lesions, altered methylation, and block of the DNA repair machinery [87]. VacA is important in autophagy induction through the formation of autophagosome, a double membrane structure encapsulating intracellular and pathogen-derived damaged organelles and proteins, among them VacA itself and CagA, whose activities are modulated [84, 88]. As the chronic infection establishes and progresses, DNA damage may determine aberrations in autophagy-associated proteins, such as the oncoprotein p62/SQSTM1, which is overexpressed in gastric lesions and has been found to promote tumorigenesis through the NF-kB signaling transduction pathway [89, 90].
In vitro studies demonstrate that autophagy and apoptosis are molecular mechanisms which could cross talk between them and may control the cell fate in autonomous or cooperative ways [91, 92]. The selected pathway seems to be dependent on the cellular surface receptor status, such as the presence of TRAIL or CD95, on Bcl-2 as a central regulator of autophagy and apoptosis, and on the intracellular signaling milieu [93,94,95]. A key intracellular component driving death and autophagy is the inflammasome, a cytosolic multiprotein oligomer containing caspases, whose exact composition depends on the activator which initiates inflammasome assembly. Inflammasome has dual opposite roles in the oncogenesis: one in the anti-tumor inflammatory response by eliminating precancerous precursors through apoptosis and, on the other hand, a pro-tumorigenic effect by stimulating production of trophic factors for precancerous cells and stroma [96]. Anti-tumorigenic and pro-tumorigenic properties are largely determined by the types of cells, tissues, and organs involved [97]. For instance, some cells with DNA damage elicited by CagA+ H. pylori strains are less likely to undergo apoptosis, and thus they are at high risk of malignant transformation [72]. This highlights that the interplay between the host and different H. pylori strains with differentially expressed virulence determinants is complex and may strongly influence the progression of the disease.
The Progression of H. pylori-Induced Precancerous Lesions: A Continuous Tolerizing Relationship
Beyond the biochemical, genetic, and molecular mechanisms triggered in the early phases of H. pylori infection and of inflammation, cellular and soluble factors deeply influence the relationships between H. pylori and the gastric microenvironment, sustaining bacterial persistence and survival of those cells altered from inflammation and that are shaped toward a precancerous lesion. Pro-inflammatory factors derived from damaged cells, such as IL-1β, and from activated T lymphocytes, such as IFN-γ, IL-4, IL-10, and IL-12, trigger immunosuppressive pathways from myeloid cells [98]. In addition, CD4+ T cells recruited in the inflamed microenvironment secrete pleiotropic chemokines and cytokines, which play a fundamental role in the final clinical outcome of the infection and of the cancerized field, through the activation of many pathways, such as those leading to epithelial-to-mesenchymal transition or development of gastric cancer stem cells [99,100,101,102,103].
Mechanisms of H. pylori Attenuation and Evasion from Immune Surveillance
Despite the activation of a strong immune response, H. pylori is able to sustain the infection for several years or throughout lifetime. H. pylori survives oxidative stress by the production of enzyme oxidase and superoxide catalase, thus determining its persistence in the gastric mucosa and the further enhancement of the oxidative burst. Strains with high virulence levels and carriers of bacterial determinants with toxic activity seem to account for a high risk of gastric cancer development [26, 104]. However, the H. pylori gastric niche harbors bacterial strains with differential virulence acquired by genetic recombination as a strategy for survival and persistence in the site of infection. Indeed, DNA damage induced by inflammation in epithelial and stromal cells can involve not only the host genetic background, but also the H. pylori genome. Homologous recombination can act as a repair pathway of DNA breaks, prompting antigenic variation in H. pylori [105]; for instance, rearrangements in the genes encoding post-translational modifying enzymes, such as alpha-fucosyltransferases or peptidoglycan deacetylases, can determine changes in their activity with modulation of bacterial cell wall antigenic specificity [106, 107].
Molecular biology studies suggest functional relationships between different genomic traits of H. pylori. In particular, the composition of the CagPAI greatly affects bacterial motility, survival capacity in different gastric microenvironments, production of pro-inflammatory cytokines, and antimicrobial susceptibility [108,109,110,111,112]. It has been highlighted that a single infective H. pylori strain may include variable proportions of subtypes with different CagPAI genotypes, a phenomenon consistent with host-induced adaptive changes of the bacterial population infecting the stomach [113, 114]. Indeed, heterogeneous genomic and proteomic profiles of H. pylori strains and subtypes have been described showing a tendency to an association with different precancerous or pathologic conditions [26, 115,116,117]. Deletions of CagPAI genes are more frequently detected among individuals with metaplasia and atrophic gastritis than non-atrophic gastritis or duodenal ulcers [118, 119]. These mechanisms entail virulence attenuation favoring colonization and persistence, but also modify the interaction capacity of the bacterium favoring the escape from the immunosurveillance.
Myeloid and Lymphoid Cellular and Soluble Factors Affecting the Clinical Outcome of H. pylori Infection
The secretion of inflammatory cytokines from healthy and damaged cells can be promoted by interaction of bacterial LPS, flagellins, toxins, and cellular products with membrane receptors, such as TLRs, or cytosolic components, such as inflammasomes. A paradigmatic example of membrane receptor is TLR4. It is expressed in immune as well as epithelial and stromal cells, where it can activate MyD88-dependent pathways, with the transcription of genes encoding for pro-inflammatory cytokines and chemokines (TNF, IL-1β, IL-18), immunosuppressive cytokines (IL-10 and transforming growth factor (TGF)-β), and angiogenic mediators (vascular endothelial growth factor (VEGF), epidermal growth factor (EGF)). In addition, TLR4 has been detected in tumoral cells, where it is capable of activating mitogen-activated protein kinases (MAPK) and NF-kB, suggesting its direct role in apoptosis inhibition and proliferation stimulation [120]. Inflammasomes are predominantly expressed in macrophages and can promote cytokine and chemokine production as well, especially IL-18 and IL-1β. The CagPAI-encoded type IV secretion system, LPS, VacA, and bacterial urease B subunit seem to play a role in inflammasome activation. Recent studies highlight that the H. pylori-induced inflammasome activation and consequent IL-18 and IL-1β secretion need the coordinated cooperation between TLR-2, Nod-like receptor family pyrin domain-containing 3 (NLRP3) and caspase-1 [121].
IL-18 is a multifactorial chemokine, which intervenes directly activating CD8+ cytotoxic T lymphocytes and CD4+ naïve T cells that acquire a Th1-IFN-γ-secreting phenotype under the synergic action of IL-12 [99, 122]. Besides this anti-inflammatory action, mucosa integrity protection, and anti-cancer effect, IL-18 manifests pro-cancer properties [123]. This effect seems to be related to an impaired NK cell function through a PD-1-dependent mechanism, as it has been evidenced by in vitro and murine models [124]. However, IL-18 role in gastric cancer is not clearly understood in the clinical settings [125]. Overexpression of IL-1β is involved in the pathogenesis of gastric cancer through an immune-tolerizing effect of the mucosal gastric microenvironment guided by the mobilization of myeloid-derived suppressor cells (MDSCs) to the stomach [126]. MDSCs are one of the representative immune suppressive cells having the capacity to increase T cell apoptosis and suppress T cell responses, directing the result of the infection toward evasion from immune system and pathology [127]. MDSC levels are significantly increased in cancer patients and correlate with cancer clinical stages and poor prognosis [128,129,130], to such an extent that they have been mentioned as possible prognostic biomarkers of gastric cancer together with macrophages, neutrophils, and DCs [131, 132]. Finally, IL-1β production by MDSCs may induce secretion of IL-17 by CD4+ T cells [102].
Th17
Th17 and Th1 are the predominant subsets during the inflammatory phases of H. pylori infection, with Th17 response involved at earlier stages of infection than Th1 response [133]. In particular, CagA+ strains stimulate DCs to IL-1β and IL-23 production. In the presence of antigen presentation, DCs activate CD4+ naïve T cells to differentiation toward a Th17 phenotype. At intracellular level, the process is controlled by signal transducers, such as Signal Transducer and Activator of Transcription-3 (STAT-3), and by the transcription factors retinoic acid receptor-Related Orphan Receptors (ROR) γt and α. TGF-β, BAFF, and IL-6 secreted by DCs may be additional important factors for Th17 differentiation. They act through STAT-3 and NF-kB pathways [134]. In particular, TGF-β induces the expression of both RORγt and of forkhead box P3 (FoxP3) in naïve T cells; the latter molecule is a transcription factor capable of suppressing the activation of RORγt by a physical interaction and of deviating the differentiation of naïve T cells toward an immunosuppressive T regulatory (Treg) signature [135]. IL-6 links the differentiative pathways of Th17 and Treg, by activating STAT-3 pathway and down-modulating FoxP3 expression, finally unbalancing the ratio between these two subsets in favor of Th17 . IL-6 expression is high in H. pylori-infected subjects as well as in physiological aging, where this cytokine is involved in the maintenance of a low level of systemic and local chronic inflammation, that can unbalance immune system functions toward tolerance and senescence, with a high risk of morbidity [136, 137].
Th17 cell subsets are able to release several chemokines and cytokines, namely, IL-17A, IL-17F, IL-21, IL-22, IL-23, IL-26, TNF-α, CCL20, and GM-CSF, although not all are Th17 specific. Epithelial cells and fibroblasts are stimulated by Th17 cytokines/chemokines toward pro-inflammatory soluble factors secretion, further recalling infiltration of macrophages, activated monocytes, T cells, and DCs in the microenvironment. Functional for tissue remodeling, but of relevance for re-localization of cells with malignant or premalignant characteristics, Th17 cells stimulate epithelial cells to produce matrix metalloproteinases (MMP) that disrupt microenvironment architecture. Although IL-17 responses are downregulated by immunosuppressive enzymes, such as indoleamine 2,3-dioxygenase (IDO), or by reduced expression of co-stimulatory receptors on the Th17 surface, the activity of this T cell subset continues also after disappearance of the bacterium thanks to the action of IL-1β, which levels remain elevated in the gastric mucosa [138].
Th1
Th1 cells are involved primarily in defense against intracellular pathogens and in the isotypic switch of immunoglobulins to isotypes with complement-activation properties. H. pylori colonization of gastric mucosa seems to be directly proportional to Th1 immune response, since an insufficiency in this lineage is associated with enhanced bacterial density [139]. Outer membrane proteins of H. pylori induce NK and DC activation and maturation with predominant production of IL-12, IL-18, and IFN-γ. The synergic action of DCs and NK cells and their soluble mediators induces the expression of the transcription factor T-box expressed in T cells (T-bet) in T cell receptor (TCR)-engaged naïve CD4+ T cells, leading to their differentiation in Th1 secreting at least IL-2 and IFN-γ. Hence, through an autocrine mechanism, IFN-γ enforces the Th1 polarization operated by NK, while IL-2 stimulates the progression of target cells from G0 to G1 phase, initiating the process of clonal expansion of activated T, B, and NK cells. Moreover, the Th1 cytokines cause further recruitment of macrophages into the infection site [140], emphasizing Th1 hyperactivation and reinforcing gastric inflammation finalized to the decrease of bacterial density.
During the early phases of infection, T lymphocytes from the H. pylori-infected gastric mucosa are not able to secrete Th2 cytokines. Indeed, IL-4 from basophils and mast cells stimulates the expression of the Th2 cell-specific master transcription factor GATA-binding protein-3 (GATA-3) in TCR-engaged naïve T cells. GATA-3 have reciprocal antagonistic activity with T-bet, and both transcription factors are involved in attenuating the harmful effects of Th1 response to maintain an healthy homeostasis [141]. Moreover, H. pylori, through HP-NAP and other virulence determinants, plays a central role in inhibiting the pathway of Th2 differentiation, promoting IL-12 and IL-23 secretion by neutrophils and monocytes, which support the polarization of Th1 and Th17 against H. pylori, respectively. However, negative feedbacks down-modulating Th1 responses can be exerted by some H. pylori virulence factors and by components of the inflammatory milieu. For instance, bacterial molecules such as GGT or Lewis-antigens on LPS can activate tolerogenic DC subsets unable to foster a Th1 differentiation and response [142, 143]. In addition, IDO, high levels of COX-2 and prostaglandin-2 (PGE-2) modify the Th1/Th2 balance in favor of the Th2 response [144,145,146].
Th2
The Th2 cytokine profile includes IL-4, IL-5, and IL-13, which are involved in a paracrine and autocrine self-activation and self-maintenance circuit. These cell subsets are important for the production of H. pylori-specific IgG, IgM, and IgA, which intervene in systemic and local antibody-mediated protection against the bacterium. Especially IgA are relevant in inhibiting the bacterial colonization of the mucosa [147]. Th1 immune responses are more efficient than Th2 responses against bacteria [148], but, when mechanisms down-modulating Th1 expression occur, Th2 and Th17 seem to prevail, and an imbalance toward Th2 responses is shown. Patients with precancerous gastric lesions and gastric cancer express a predominant Th2 signature [149, 150]. One of the mechanisms which links Th2 profile to worse prognosis is represented by the ability of GATA-3 to down-modulate onco-suppressor genes [151].
T Regulatory (Treg)
Treg subsets, together with MDSCs, play a clincher role in H. pylori immune escape, since they can suppress DCs and effector T cells by cell to cell contact and production of TGF-β, IL-10, and IL-35, which limit the inflammatory responses. They are delegated to maintain self-tolerance and physiological conditions avoiding autoimmunity. Two kinds of Tregs have been described with different ontogeny but some common features: the natural Tregs (nTregs) and the induced Tregs (iTregs). While nTregs are generated within the thymus from lymphoid precursors, the naïve CD4+ T cells residing in peripheral lymphoid organs and stimulated by the antigen can differentiate into iTregs in the presence of TGF-β and IL-2. Commonly both kinds of Tregs are defined by the intracytoplasmic expression of the transcription factor FoxP3.
Triggering of TLR-2 signaling pathway through H. pylori components LPS or HP-NAP is an important mechanism for Treg activation accompanied by Th1 inhibition. The Treg-induced onset of immunologically tolerant gastric microenvironment modulates the survival and persistence of H. pylori and directs the disease to a worse outcome [152, 153]. The increase in Tregs levels within gastric mucosa seems to be associated with increased expression of programmed death-ligand 1 (PD-L1) on epithelial cells in the site of infection. The binding of PD-L1 to inhibitory receptors present on the surface of CD4+ T lymphocytes, such as PD-1 or B7.1, transmits inhibitory signals which reduce the effector capacity of these subsets [154]. Hence, globally, an immunological anergy is established in the field of infection and cancerization, favoring the immune evasion of the bacterium and transformed cells. It has been observed that H. pylori-induced DCs stimulate proliferation of Treg possessing a reduced suppressive function due to the H. pylori-dependent IL-1β secretion by DCs itself, suggesting an attempt to maintain or restore an inflammatory milieu with effector proprieties [155]. The transition to anergic or reactive immunity also depends on the balance between the signaling pathways conveyed toward Tregs or Th17 subset differentiation. In particular, the absence or the presence of IL-6, together with the activation of IL-6/STAT-3 axis in naïve CD4+ T cells, prompts or suppresses the expression of FoxP3, determining the fate toward the differentiation of suppressive or reactive T cells, respectively [156, 157].
Besides the essential immunological components described in this paragraph and their basic relationships participating to an evolving immune profile within the gastric precancerous lesion, other cellular subsets, such as Th9 and Th22, are strictly interrelated and committed in the progression/regression of the infection and of the field cancerization. They are elegantly reviewed elsewhere [102, 103]. Furthermore, host genetic factors related to immunological and regulatory elements composing the mucosal milieu and entangled in bacterial interactions may play a pivotal role in addressing the outcome of H. pylori infection.
Host Factors Affecting Inflammation and Its Clinical Outcome
Functional polymorphisms that influence the level or the quality of the expression of genes encoding for intracellular and extracellular receptors, enzymes, cytokines, and chemokines modulating the inflammatory response have been associated with increased risk of gastric cancer [158]. The clinical significance of these associations is dependent from ethnicity, which is an important confounding factor in epidemiological studies [159, 160]. Interestingly, a correlation between the presence of certain single nucleotide substitutions (SNPs) and a high proportion of highly virulent H. pylori strains has been found, suggesting the existence of a selective pressure exerted by the host on the microorganism subtypes. This possible synergistic interaction could lead to the progression or regression of precancerous lesions [20, 161]. Individual genetic predisposition to exacerbate or dampen the effects of H. pylori infection may concern several steps of the interplay between the bacterium and the host (Table 1.1).
Accumulation of DNA damage following oxidative stresses can be worsen by SNPs present in the host genes coding for DNA repair enzymes, which may unbalance the relationship between apoptosis and cellular proliferation. Poly-ADP-ribose polymerase 1 (PARP-1) is a component of the BER system whose polymorphisms have been mentioned to be associated with gastric cancer in some studies [161, 173]. However, some investigations report no relationship between worse prognosis and this mutated enzyme [205], while only combined effect of genetic and H. pylori profile covariates shows significant associations with gastric cancer in other studies [206].
One of the intracellular receptors which plays a pivotal role in the transformation of the infected cells is the tyrosine phosphatase Src homology region 2 domain-containing phosphatase-2 (SHP-2), which is first intercepted by the phosphorylated CagA and which was found to induce cell morphological and physiological modifications. SHP-2 is coded by the PTPN11 gene, whose polymorphisms have been associated to increased risk of atrophic gastritis in Chinese population with, but not without, H. pylori infection. This effect is probably due to a different strength of signal transduction through the CagA-SHP-2 complex [165]. Although wide association studies focusing on hundreds of SNPs possibly involved in CagA interaction have identified new susceptibility loci for gastric cancer, the insufficient statistical power of these studies does not allow to assess the exact relationship between the selected SNPs and gastric cancer risk, providing only clues on the mechanisms entailing CagA function [207, 208].
Among polymorphisms concerning TLRs, two SNPs within TLR4 coding gene have been linked with susceptibility to chronic infection, atrophic gastritis, and gastric cancer in Caucasian population by more than one study [176,177,178]; moreover, an alteration in the ligand-binding receptor site with proven diminished LPS responsiveness has been underlined [209, 210].
In addition to TLR, NLRs are important in the recognition of H. pylori. Polymorphisms of NOD1 and NOD2 are the best characterized in manifold studies. Overall, they highlight that functional SNPs reducing NOD1-/NOD2-mediated immune response to H. pylori contribute to bacterial survival and persistence and that a subsequent over-activation of other inflammatory responses may result in inflammation-related carcinogenesis [211].
As already mentioned, IL-1β is an important pro-inflammatory cytokine and a powerful inhibitor of gastric acid secretion, hence an inducer of atrophy progression. Polymorphisms in the IL1B promoter region, together with those concerning the IL-1 receptor antagonist (IL1-RN), have been reported to modulate IL-1β levels and action and be associated with an increased risk of gastric cancer [19]. These associations have been partially confirmed for Caucasian subjects by meta-analyses [212, 213], even if slightly contrasting results have emerged due to different grouping of subjects with different allelic frequencies or different genetic models of analyses [214]. A meta-analysis including 36 studies to evaluate the effect of TNFA on genetic susceptibility to gastritis and gastric cancer has shown that the TNFA -308G>A polymorphism is a risk factor for developing gastric tumors in different ethnic groups, with significant results found in Caucasians, but no significant associations among East Asians or other ethnicities [159]. A meta-analysis on a total of 203 studies assessing associations between gastric cancers and 225 polymorphisms in 95 genes showed ambiguous effects for several gene polymorphisms between Asian and Caucasian populations. However, this study was able to confirm, through gene clusters, two panels of polymorphisms that were significantly associated with the risk of gastric cancer and able to specifically distinguish these two different ethnic groups [160].
The results of association studies between genetic determinants and H. pylori-related gastric carcinogenesis may suffer from bias linked not only to the selection of the analyzed subjects but also to the population sample size, to the interactions between several covariates that can have an impact on this system and cannot be all eligible or valuable, and to the intrinsic limitations of the statistical methods applied in these complex contexts. However, they may help in personalization of the surveillance if they are directed to specific patient populations.
Conclusions
Gastric tumorigenesis is a multifactorial process involving complex interactions between gastric microenvironment, inflammation, and colonizing microorganisms, with H. pylori being the most studied and well-known cancer determinant. In dependence on its genetic and phenotypic heterogeneity, H. pylori triggers a number of innate and adaptive immune responses entangled in tumor formation process. CagA+ strains present an increased risk of gastric cancer, and elevated levels of inflammatory cytokines have been observed in H. pylori-infected individuals. Through these mediators, several kinds of immune cells are stimulated to cooperate in the modulation of the oncogenic and anti-suppressive pathway activity. Methylation of tumor suppressor genes increases the risk of adenocarcinoma in the stomach. Autophagy and apoptosis processes may be hijacked toward cell growth and differentiation.
New technologies allow to discover additional elements, which can inflame the progression of the precancerous gastric lesions occurring in achlorhydria and atrophy settings. However, functional and mechanistic studies are needed to elucidate their specific activities within the evolution and dynamics of inflammation and their correlations with the pathogenesis of gastric cancer. Understanding of the mechanisms that regulate cancer-associated inflammation could open the way to new biomarkers able to distinguish patients with precancerous lesions that will remain indolent from those that will evolve, and to unexplored treatment opportunities influencing prevention and prognosis of therapeutic options.
References
Reese DM. Fundamentals-Rudolf Virchow and modern medicine. West J Med. 1998;169(2):105–8.
Orange M, Reuter U, Hobohm U. Coley’s lessons remembered: augmenting mistletoe therapy. Integr Cancer Ther. 2016;15(4):502–11.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113(3):321–33.
Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119(9):2475–87.
International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risks to humans. Schistosomes, liver flukes and helicobacter pylori, vol. 61. Lyon: International Agency for Research on Cancer; 1994. p. 177.
Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118(12):3030–44.
Chmiela M, Wadstrom T, Folkesson H, et al. Anti-Lewis X antibody and Lewis X-anti-Lewis X immune complexes in Helicobacter pylori infection. Immunol Lett. 1998;61(2–3):119–25.
Chmiela M, Gonciarz W. Molecular mimicry in Helicobacter pylori infections. World J Gastroenterol. 2017;23(22):3964–77.
Correa P, Piazuelo MB. The gastric precancerous cascade. J Dig Dis. 2012;13(1):2–9.
Burucoa C, Axon A. Epidemiology of Helicobacter pylori infection. Helicobacter. 2017;22 Suppl 1:1–5.
Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2(1):28–37.
Wroblewski LE, Peek RM Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–39.
Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–52.
Atherton JC. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol. 2006;1:63–96.
Saberi S, Douraghi M, Azadmanesh K, et al. A potential association between Helicobacter pylori CagA EPIYA and multimerization motifs with cytokeratin 18 cleavage rate during early apoptosis. Helicobacter. 2012;17(5):350–7.
Greenfield LK, Jones NL. Modulation of autophagy by Helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol. 2013;21(11):602–12.
El-Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124(5):1193–201.
El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404(6776):398–402. Erratum in: Nature 2001 Jul 5;412(6842):99.
Machado JC, Pharoah P, Sousa S, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology. 2001;121(4):823–9.
Lee WP, Tai DI, Lan KH, et al. The -251T allele of the interleukin-8 promoter is associated with increased risk of gastric carcinoma featuring diffuse-type histopathology in Chinese population. Clin Cancer Res. 2005;11(18):6431–41.
Taguchi A, Ohmiya N, Shirai K, et al. Interleukin-8 promoter polymorphism increases the risk of atrophic gastritis and gastric cancer in Japan. Cancer Epidemiol Biomark Prev. 2005;14(11 Pt 1):2487–93.
De Bernard M, D’Elios MM. The immune modulating activity of the Helicobacter pylori HP-NAP: friend or foe? Toxicon. 2010;56(7):1186–92.
Palframan SL, Kwok T, Gabriel K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front Cell Infect Microbiol. 2012;2:92.
Barrozo RM, Cooke CL, Hansen LM, et al. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathogens. 2013;9(2):e1003189.
Figura N, Marano L, Moretti E, et al. Helicobacter pylori infection and gastric carcinoma: not all the strains and patients are alike. World J Gastrointest Oncol. 2016;8(1):40–54.
Wang CA, Liu YC, Du SY, et al. Helicobacter pylori neutrophil-activating protein promotes myeloperoxidase release from human neutrophils. Biochem Biophys Res Commun. 2008;377(1):52–6.
Petersson C, Forsberg M, Aspholm M, et al. Helicobacter pylori SabA adhesin evokes a strong inflammatory response in human neutrophils which is down-regulated by the neutrophil-activating protein. Med Microbiol Immunol. 2006;195(4):195–206.
Montemurro P, Nishioka H, Dundon WG, et al. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a potent stimulant of mast cells. Eur J Immunol. 2002;32(3):671–6.
Brisslert M, Enarsson K, Lundin S, et al. Helicobacter pylori induce neutrophil transendothelial migration: role of the bacterial HP-NAP. FEMS Microbiol Lett. 2005;249(1):95–103.
Polenghi A, Bossi F, Fischetti F, et al. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J Immunol. 2007;178(3):1312–20.
Amedei A, Cappon A, Codolo G, et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J Clin Invest. 2006;116(4):1092–101.
Ricci V, Giannouli M, Romano M, et al. Helicobacter pylori gamma-glutamyl transpeptidase and its pathogenic role. World J Gastroenterol. 2014;20(3):630–8.
Schmees C, Prinz C, Treptau T, et al. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology. 2007;132(5):1820–33.
Oertli M, Noben M, Engler DB, et al. Helicobacter pylori γ-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A. 2013;110(8):3047–52.
Backert S, Tegtmeyer N, Fischer W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015;10(6):955–65.
Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014;15(3):306–16.
Churin Y, Al-Ghoul L, Kepp O, et al. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. J Cell Biol. 2003;161(2):249–55.
Mimuro H, Suzuki T, Tanaka J, et al. Grb2 is a key mediator of helicobacter pylori CagA protein activities. Mol Cell. 2002;10(4):745–55.
Murata-Kamiya N, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26(32):4617–26.
Saadat I, Higashi H, Obuse C, et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447(7142):330–3.
Tsugawa H, Suzuki H, Saya H, et al. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe. 2012;12(6):764–77.
Ishimoto T, Oshima H, Oshima M, et al. CD44+ slow-cycling tumor cell expansion is triggered by cooperative actions of Wnt and prostaglandin E2 in gastric tumorigenesis. Cancer Sci. 2010;101(3):673–8.
Garay J, Piazuelo MB, Majumdar S, et al. The homing receptor CD44 is involved in the progression of precancerous gastric lesions in patients infected with Helicobacter pylori and in development of mucous metaplasia in mice. Cancer Lett. 2016;371(1):90–8.
Hirata K, Suzuki H, Imaeda H, et al. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br J Cancer. 2013;109(2):379–86.
Wakamatsu Y, Sakamoto N, Oo HZ, et al. Expression of cancer stem cell markers ALDH1, CD44 and CD133 in primary tumor and lymph node metastasis of gastric cancer. Pathol Int. 2012;62(2):112–9.
Watanabe T, Asano N, Fichtner-Feigl S, et al. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest. 2010;120(5):1645–62.
Wang G, Lo LF, Forsberg LS, et al. Helicobacter pylori peptidoglycan modifications confer lysozyme resistance and contribute to survival in the host. MBio. 2012;3(6):e00409–12.
Wang G, Maier SE, Lo LF, et al. Peptidoglycan deacetylation in Helicobacter pylori contributes to bacterial survival by mitigating host immune responses. Infect Immun. 2010;78(11):4660–6.
Kalali B, Mejías-Luque R, Javaheri A, et al. H. pylori virulence factors: influence on immune system and pathology. Mediators Inflamm. 2014;2014:426309.
Kim JM, Kim JS, Lee JY, et al. Vacuolating cytotoxin in Helicobacter pylori water-soluble proteins upregulates chemokine expression in human eosinophils via Ca2+ influx, mitochondrial reactive oxygen intermediates, and NF-kappaB activation. Infect Immun. 2007;75(7):3373–81.
Takeshima E, Tomimori K, Takamatsu R, et al. Helicobacter pylori VacA activates NF-κB in T cells via the classical but not alternative pathway. Helicobacter. 2009;14(4):271–9.
Muller A, Oertli M, Arnold IC. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell Commun Signal. 2011;9(1):25.
Rizzuti D, Ang M, Sokollik C, et al. Helicobacter pylori inhibits dendritic cell maturation via interleukin-10-mediated activation of the signal transducer and activator of transcription 3 pathway. J Innate Immun. 2015;7(2):199–211.
Shimizu T, Chiba T, Marusawa H. Helicobacter pylori-mediated genetic instability and gastric carcinogenesis. Curr Top Microbiol Immunol. 2017;400:305–23.
Ushijima T, Hattori N. Molecular pathways: involvement of Helicobacter pylori-triggered inflammation in the formation of an epigenetic field defect, and its usefulness as cancer risk and exposure markers. Clin Cancer Res. 2012;18(4):923–9.
Pignatelli B, Bancel B, Plummer M, et al. Helicobacter pylori eradication attenuates oxidative stress in human gastric mucosa. Am J Gastroenterol. 2001;96(6):1758–66.
Mera R, Fontham ET, Bravo LE, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005;54(11):1536–40.
Massarrat S, Haj-Sheykholeslami A, Mohamadkhani A, et al. Precancerous conditions after H. pylori eradication: a randomized double blind study in first degree relatives of gastric cancer patients. Arch Iran Med. 2012;15(11):664–9.
Chen HN, Wang Z, Li X, Zhou ZG. Helicobacter pylori eradication cannot reduce the risk of gastric cancer in patients with intestinal metaplasia and dysplasia: evidence from a meta-analysis. Gastric Cancer. 2016;19(1):166–75.
Koeppel M, Garcia-Alcalde F, Glowinski F, et al. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 2015;11(11):1703–13.
Lee WP, Hou MC, Lan KH, et al. Helicobacter pylori-induced chronic inflammation causes telomere shortening of gastric mucosa by promoting PARP-1-mediated non-homologous end joining of DNA. Arch Biochem Biophys. 2016;606:90–8.
Kawanishi S, Ohnishi S, Ma N, et al. Crosstalk between DNA damage and inflammation in the multiple steps of carcinogenesis. Int J Mol Sci. 2017;18(8):E1808.
Baek HY, Lim JW, Kim H, et al. Oxidative-stress-related proteome changes in Helicobacter pylori-infected human gastric mucosa. Biochem J. 2004;379(Pt 2):291–9.
Huang FY, Chan AO, Rashid A, et al. Helicobacter pylori induces promoter methylation of E-cadherin via interleukin-1β activation of nitric oxide production in gastric cancer cells. Cancer. 2012;118(20):4969–80.
Hanada K, Uchida T, Tsukamoto Y, et al. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect Immun. 2014;82(10):4182–9.
Shimizu T, Marusawa H, Matsumoto Y, et al. Accumulation of somatic mutations in TP53 in gastric epithelium with Helicobacter pylori infection. Gastroenterology. 2014;147(2):407–17.e3.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
Yuan X, Zhou Y, Wang W, et al. Activation of TLR4 signaling promotes gastric cancer progression by inducing mitochondrial ROS production. Cell Death Dis. 2013;4:e794.
Cha B, Lim JW, Kim KH, et al. HSP90beta interacts with Rac1 to activate NADPH oxidase in Helicobacter pylori-infected gastric epithelial cells. Int J Biochem Cell Biol. 2010;42(9):1455–61.
Handa O, Naito Y, Yoshikawa T. CagA protein of Helicobacter pylori: a hijacker of gastric epithelial cell signaling. Biochem Pharmacol. 2007;73(11):1697–702.
Chaturvedi R, Asim M, Romero-Gallo J, et al. Spermine oxidase mediates the gastric cancer risk associated with Helicobacter pylori CagA. Gastroenterology. 2011;141(5):1696–708.e1–2.
Vitkute J, Stankevicius K, Tamulaitiene G, et al. Specificities of eleven different DNA methyltransferases of Helicobacter pylori strain 26695. J Bacteriol. 2001;183(2):443–50.
Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70(4):1430–40.
Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer. 2017;20(Suppl 1):8–15.
Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13(4):470–6.
Nagata N, Akiyama J, Marusawa H, et al. Enhanced expression of activation-induced cytidine deaminase in human gastric mucosa infected by Helicobacter pylori and its decrease following eradication. J Gastroenterol. 2014;49(3):427–35.
Matsumoto Y, Marusawa H, Kinoshita K, et al. Up-regulation of activation-induced cytidine deaminase causes genetic aberrations at the CDKN2b-CDKN2a in gastric cancer. Gastroenterology. 2010;139(6):1984–94.
Machado AM, Figueiredo C, Touati E, et al. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin Cancer Res. 2009;15(9):2995–3002.
Kim JJ, Tao H, Carloni E, et al. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123(2):542–53.
Park DI, Park SH, Kim SH, et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter. 2005;10(3):179–84.
Toller IM, Neelsen KJ, Steger M, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci U S A. 2011;108(36):14944–9.
Hartung ML, Gruber DC, Koch KN, et al. H. pylori-induced DNA Strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep. 2015;13(1):70–9.
Terebiznik MR, Raju D, Vázquez CL, et al. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5(3):370–9.
Wang YH, Wu JJ, Lei HY. The autophagic induction in Helicobacter pylori-infected macrophage. Exp Biol Med (Maywood). 2009;234(2):171–80.
Yang X, Yu DD, Yan F, et al. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015;5:14.
Polk DB, Peek RM Jr. Helicobacter pylori: gastric cancer and beyond. Nat Rev Cancer. 2010;10(6):403–14.
Raju D, Jones NL. Methods to monitor autophagy in H. pylori vacuolating cytotoxin A (VacA)-treated cells. Autophagy. 2010;6(1):138–43.
Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75.
Mohamed A, Ayman A, Deniece J, et al. P62/biquitin IHC expression correlated with clinicopathologic parameters and outcome in gastrointestinal carcinomas. Front Oncol. 2015;5:70.
Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 2011;21(7):387–92.
Eisenberg-Lerner A, Bialik S, Simon HU, et al. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16(7):966–75.
Xu MY, Lee DH, Joo EJ, et al. Akebia saponin PA induces autophagic and apoptotic cell death in AGS human gastric cancer cells. Food Chem Toxicol. 2013;59:703–8. https://doi.org/10.1016/j.fct.2013.06.059. Epub 2013 Jul 9.
Lim SC, Han SI. Ursodeoxycholic acid effectively kills drug-resistant gastric cancer cells through induction of autophagic death. Oncol Rep. 2015;34(3):1261–8.
Mukhopadhyay S, Panda PK, Sinha N, et al. Autophagy and apoptosis: where do they meet? Apoptosis. 2014;19(4):555–66.
Karki R, Man SM, Kanneganti TD. Inflammasomes and Cancer. Cancer Immunol Res. 2017;5(2):94–9.
Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–64.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
O’Keeffe J, Moran AP. Conventional, regulatory, and unconventional T cells in the immunologic response to Helicobacter pylori. Helicobacter. 2008;13(1):1–19.
Choi YJ, Kim N, Chang H, et al. Helicobacter pylori-induced epithelial-mesenchymal transition, a potential role of gastric cancer initiation and an emergence of stem cells. Carcinogenesis. 2015;36(5):553–63.
Mesali H, Ajami A, Hussein-Nattaj H, et al. Regulatory T cells and myeloid-derived suppressor cells in patients with peptic ulcer and gastric cancer. Iran J Immunol. 2016;13(3):167–77.
Bockerstett KA, DiPaolo RJ. Regulation of gastric carcinogenesis by inflammatory cytokines. Cell Mol Gastroenterol Hepatol. 2017;4(1):47–53.
Jafarzadeh A, Larussa T, Nemati M, et al. T cell subsets play an important role in the determination of the clinical outcome of Helicobacter pylori infection. Microb Pathog. 2018. pii: S0882–4010(16)30548–4.
González CA, Figueiredo C, Lic CB, et al. Helicobacter pylori cagA and vacA genotypes as predictors of progression of gastric preneoplastic lesions: a long-term follow-up in a high-risk area in Spain. Am J Gastroenterol. 2011;106(5):867–74.
Hanada K, Yamaoka Y. Genetic battle between Helicobacter pylori and humans. The mechanism underlying homologous recombination in bacteria, which can infect human cells. Microbes Infect. 2014;16(10):833–9.
Rubin EJ, Trent MS. Colonize, evade, flourish: how glyco-conjugates promote virulence of Helicobacter pylori. Gut Microbes. 2013;4(6):439–53.
Ferreira JA, Magalhães A, Gomes J, et al. Protein glycosylation in gastric and colorectal cancers: toward cancer detection and targeted therapeutics. Cancer Lett. 2017;387:32–45.
Karita M, Blaser MJ. Acid-tolerance response in Helicobacter pylori and differences between cagA+ and cagA- strains. J Infect Dis. 1998;178:213–9.
Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med. 2002;347:1175–86.
Figura N, Trabalzini L, Mini R, et al. Inactivation of Helicobacter pylori cagA gene affects motility. Helicobacter. 2004;9:185–93.
Basaglia G, Sperandio P, Tomasini ML, et al. Analysis of antimicrobial susceptibility and virulence factors in Helicobacter pylori clinical isolates. J Chemother. 2004;16(5):504–6.
De Paoli P, Tomasini ML, Basaglia G. The predictive value of Helicobacter pylori in-vitro metronidazole resistance. Clin Microbiol Infect. 2004;10(12):1105–6.
Tomasini ML, Zanussi S, Sozzi M, et al. Heterogeneity of cag genotypes in Helicobacter pylori isolates from human biopsy specimens. J Clin Microbiol. 2003;41(3):976–80.
Sozzi M, Crosatti M, Kim SK, et al. Heterogeneity of Helicobacter pylori cag genotypes in experimentally infected mice. FEMS Microbiol Lett. 2001;203(1):109–14.
Figura N, Valassina M, Moretti E, et al. Histological variety of gastric carcinoma and Helicobacter pylori cagA and vacA polymorphism. Eur J Gastroenterol Hepatol. 2015;27(9):1017–21.
Repetto O, Zanussi S, Casarotto M, et al. Differential proteomics of Helicobacter pylori associated with autoimmune atrophic gastritis. Mol Med. 2014;20:57–71.
Bernardini G, Figura N, Ponzetto A, et al. Application of proteomics to the study of Helicobacter pylori and implications for the clinic. Expert Rev Proteomics. 2017;14(6):477–90.
Sozzi M, Valentini M, Figura N, et al. Atrophic gastritis and intestinal metaplasia in Helicobacter pylori infection: the role of CagA status. Am J Gastroenterol. 1998;93(3):375–9.
Sozzi M, Tomasini ML, Vindigni C, et al. Heterogeneity of cag genotypes and clinical outcome of Helicobacter pylori infection. J Lab Clin Med. 2005;146(5):262–70.
Korneev KV, Atretkhany KN, Drutskaya MS, et al. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine. 2017;89:127–35.
Pachathundikandi SK, Müller A, Backert S. Inflammasome activation by Helicobacter pylori and its implications for persistence and immunity. Curr Top Microbiol Immunol. 2016;397:117–31.
Kohyama M, Saijyo K, Hayasida M, et al. Direct activation of human CD8+ cytotoxic T lymphocytes by interleukin-18. Jpn J Cancer Res. 1998;89(10):1041–6.
Palma G, Barbieri A, Bimonte S, et al. Interleukin 18: friend or foe in cancer. Biochim Biophys Acta. 2013;1836(2):296–303.
Terme M, Ullrich E, Aymeric L, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011;71(16):5393–9.
Yao J, Li ZH, Li YX, et al. Association between the −607 C > a polymorphism in interleukin-18 gene promoter with gastrointestinal cancer risk: a meta-analysis. Genet Mol Res. 2015;14(4):16880–7.
Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14(5):408–19.
Chen J, Ye Y, Liu P, et al. Suppression of T cells by myeloid-derived suppressor cells in cancer. Hum Immunol. 2017;78(2):113–9.
Diaz-Montero CM, Salem ML, Nishimura MI, et al. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59.
Mantovani A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur J Immunol. 2010;40(12):3317–20.
Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139.
Chang WJ, Du Y, Zhao X, et al. Inflammation-related factors predicting prognosis of gastric cancer. World J Gastroenterol. 2014;20(16):4586–96.
Shoji H, Tada K, Kitano S, et al. The peripheral immune status of granulocytic myeloid-derived suppressor cells correlates the survival in advanced gastric cancer patients receiving cisplatin-based chemotherapy. Oncotarget. 2017;8(56):95083–94.
Ricci V, Romano M, Boquet P. Molecular cross-talk between Helicobacter pylori and human gastric mucosa. World J Gastroenterol. 2011;17(11):1383–99.
Zhuang Y, Shi Y, Liu XF, et al. Helicobacter pylori infected macrophages induce Th17 cell differentiation. Immunobiology. 2011;216(1–2):200–7.
Ichiyama K, Yoshida H, Wakabayashi Y, et al. Foxp3 inhibits RORgammat-mediated IL-17A mRNA transcription through direct interaction with RORgammat. J Biol Chem. 2008;283(25):17003–8. https://doi.org/10.1074/jbc.M801286200. Epub 2008 Apr 23.
Caruso C, Lio D, Cavallone L, et al. Aging, longevity, inflammation, and cancer. Ann N Y Acad Sci. 2004;1028:1–13.
Zanussi S, Serraino D, Dolcetti R, et al. Cancer, aging and immune reconstitution. Anti Cancer Agents Med Chem. 2013;13(9):1310–24.
Serelli-Lee V, Ling KL, Ho C, et al. Persistent Helicobacter pylori specific Th17 responses in patients with past H. pylori infection are associated with elevated gastric mucosal IL-1beta. PLoS One. 2012;7(6):e39199.
Akhiani AA, Pappo J, Kabok Z, et al. Protection against Helicobacter pylori infection following immunization is IL-12-dependent and mediated by Th1 cells. J Immunol. 2002;169(12):6977–84.
Jager A, Kuchroo VK. Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation. Scand J Immunol. 2010;72(3):173–84.
Zhang Y, Zhang Y, Gu W, et al. TH1/TH2 cell differentiation and molecular signals. Adv Exp Med Biol. 2014;841:15–44.
Kabisch R, Semper RP, Wustner S, et al. Helicobacter pylori gamma-glutamyltranspeptidase induces tolerogenic human dendritic cells by activation of glutamate receptors. J Immunol. 2016;196(10):4246–52.
Bergman MP, Engering A, Smits HH, et al. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med. 2004;200(8):979–90.
Larussa T, Leone I, Suraci E, et al. Enhanced expression of indoleamine 2, 3-dioxygenase in Helicobacter pylori-infected human gastric mucosa modulates Th1/Th2 pathway and interleukin 17 production. Helicobacter. 2015;20(1):41–8.
Pellicanò A, Imeneo M, Leone I, et al. Enhanced activation of Cyclooxygenase-2 downregulates Th1 signaling pathway in Helicobacter pylori-infected human gastric mucosa. Helicobacter. 2007;12(3):193–9.
Toller IM, Hitzler I, Sayi A, et al. Prostaglandin E2 prevents Helicobacter-induced gastric preneoplasia and facilitates persistent infection in a mouse model. Gastroenterology. 2010;138(4):1455–67.
Forchielli ML, Walker WA. The role of gut-associated lymphoid tissues and mucosal defence. Br J Nutr. 2005;93(Suppl 1):S41–8.
Taylor JM, Ziman ME, Canfield DR, et al. Effects of a Th1-versus a Th2-biased immune response in protection against Helicobacter pylori challenge in mice. Microb Pathog. 2008;44(1):20–7.
Marotti B, Rocco A, De Colibus P, et al. Interleukin-13 mucosal production in Helicobacter pylori-related gastric diseases. Dig Liver Dis. 2008;40(4):240–7.
Yang P, Qiu G, Wang S, et al. The mutations of Th1 cell-specific T-box transcription factor may be associated with a predominant Th2 phenotype in gastric cancers. Int J Immunogenet. 2010;37(2):111–5.
Liu X, Cao K, Xu C, et al. GATA-3 augmentation down-regulates Connexin43 in Helicobacter Pylori associated gastric carcinogenesis. Cancer Biol Ther. 2015;16(6):987–96.
Sun X, Zhang M, El-Zataari M, et al. TLR2 mediates Helicobacter pylori-induced tolerogenic immune response in mice. PLoS One. 2013;8(9):e74595.
Nemati M, Larussa T, Khorramdelazad H, et al. Toll-like receptor 2: an important immunomodulatory molecule during Helicobacter pylori infection. Life Sci. 2017;178:17–29.
Das S, Suarez G, Beswick EJ, et al. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176(5):3000–9.
Mitchell P, Afzali B, Fazekasova H, et al. Helicobacter pylori induces in-vivo expansion of human regulatory T cells through stimulating interleukin-1β production by dendritic cells. Clin Exp Immunol. 2012;70(3):300–9.
Zhang C, Zhang X. ChenXH. Inhibition of the interleukin-6 signaling pathway: a strategy to induce immune tolerance. Clin. Rev. Allerg Immunol. 2014;47(2):163–73.
Zheng SG. Regulatory T cells vs Th17: differentiation of Th17 versus Treg, are they mutually exclusive? Afr J Clin Exp Immunol. 2013;2(1):94–106.
Hamajima N, Naito M, Kondo T, et al. Genetic factors involved in the development of Helicobacter pylori-related gastric cancer. Cancer Sci. 2006;97(11):1129–38.
Li M, Wang Y, Gu Y. Quantitative assessment of the influence of tumor necrosis factor alpha polymorphism with gastritis and gastric cancer risk. Tumour Biol. 2014;35(2):1495–502.
Loh M, Koh KX, Yeo BH, et al. Meta-analysis of genetic polymorphisms and gastric cancer risk: variability in associations according to race. Eur J Cancer. 2009;45(14):2562–8.
Silva-Fernandes IJ, da Silva TA, Agnez-Lima LF, et al. Helicobacter pylori genotype and polymorphisms in DNA repair enzymes: where do they correlate in gastric cancer? J Surg Oncol. 2012;106(4):448–55.
Goto Y, Ando T, Yamamoto K, et al. Association between serum pepsinogens and polymorphismof PTPN11 encoding SHP-2 among Helicobacter pylori seropositive Japanese. Int J Cancer. 2006;118(1):203–8.
Kawai S, Goto Y, Ito LS, et al. Significant association between PTPN11 polymorphism and gastric atrophy among Japanese Brazilians. Gastric Cancer. 2006;9(4):277–83.
Hishida A, Matsuo K, Goto Y, et al. Associations of a PTPN11 G/A polymorphism at intron 3 with Helicobactor pylori seropositivity, gastric atrophy and gastric cancer in Japanese. BMC Gastroenterol. 2009;9:51.
He C, Tu H, Sun L, et al. Helicobacter pylori-related host gene polymorphisms associated with susceptibility of gastric carcinogenesis: a two-stage case-control study in Chinese. Carcinogenesis. 2013;34(7):1450–7.
Wang GY, Lu CQ, Zhang RM, et al. The E-cadherin gene polymorphism 160C->A and cancer risk: a HuGE review and meta-analysis of 26 case-control studies. Am J Epidemiol. 2008;167(1):7–14.
Gao L, Nieters A, Brenner H. Meta-analysis: tumour invasion-related genetic polymorphisms and gastric cancer susceptibility. Aliment Pharmacol Ther. 2008;28(5):565–73.
Wang Q, Gu D, Wang M, et al. The E-cadherin (CDH1) -160C>A polymorphism associated with gastric cancer among Asians but not Europeans. DNA Cell Biol. 2011;30(6):395–400.
Chen B, Zhou Y, Yang P, et al. CDH1 -160C>A gene polymorphism is an ethnicity-dependent risk factor for gastric cancer. Cytokine. 2011;55(2):266–73.
Cui Y, Xue H, Lin B, et al. A meta-analysis of CDH1 C-160A genetic polymorphism and gastric cancer risk. DNA Cell Biol. 2011;30(11):937–45.
Li YL, Tian Z, Zhang JB, et al. CDH1 promoter polymorphism and stomach cancer susceptibility. Mol Biol Rep. 2012;39(2):1283–6.
Wang L, Wang G, Lu C, et al. Contribution of the -160C/A polymorphism in the E-cadherin promoter to cancer risk: a meta-analysis of 47 case-control studies. PLoS One. 2012;7(7):e40219.
Miao X, Zhang X, Zhang L, et al. Adenosine diphosphate ribosyl transferase and x-ray repair cross-complementing 1 polymorphisms in gastric cardia cancer. Gastroenterology. 2006;131(2):420–7.
Fan H, Liu D, Qiu X, et al. A functional polymorphism in the DNA methyltransferase-3A promoter modifies the susceptibility in gastric cancer but not in esophageal carcinoma. BMC Med. 2010;8:12.
Ravishankar Ram M, Goh KL, Leow AH, et al. Polymorphisms at locus 4p14 of toll-like receptors TLR-1 and TLR-10 confer susceptibility to gastric carcinoma in Helicobacter pylori infection. PLoS One. 2015;10(11):e0141865.
Rigoli L, Di Bella C, Fedele F, et al. TLR4 and NOD2/CARD15 genetic polymorphisms and their possible role in gastric carcinogenesis. Anticancer Res. 2010;30(2):513–7.
Hold GL, Rabkin CS, Chow WH, et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132(3):905–12.
Santini D, Angeletti S, Ruzzo A, et al. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in gastric cancer of intestinal and diffuse histotypes. Clin Exp Immunol. 2008;154(3):360–4.
Jing JJ, Li M, Yuan Y. Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms in cancer: a meta-analysis. Gene. 2012;499(2):237–42.
Hishida A, Matsuo K, Goto Y, et al. Toll-like receptor 4 +3725 G/C polymorphism, Helicobacter pylori seropositivity, and the risk of gastric atrophy and gastric cancer in Japanese. Helicobacter. 2009;14(1):47–53.
De Oliveira JG, Silva AE. Polymorphisms of the TLR2 and TLR4 genes are associated with risk of gastric cancer in a Brazilian population. World J Gastroenterol. 2012;18(11):1235–42.
Tahara T, Arisawa T, Wang F, et al. Toll-like receptor 2–196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci. 2007;98(11):1790–4.
Tahara T, Arisawa T, Wang F, et al. Toll-like receptor 2 (TLR) -196 to 174del polymorphism in gastro-duodenal diseases in Japanese population. Dig Dis Sci. 2008;53(4):919–24.
Zeng HM, Pan KF, Zhang Y, et al. Genetic variants of toll-like receptor 2 and 5, helicobacter pylori infection, and risk of gastric cancer and its precursors in a chinese population. Cancer Epidemiol Biomark Prev. 2011;20(12):2594–602.
Hofner P, Gyulai Z, Kiss ZF, et al. Genetic polymorphisms of NOD1 and IL-8, but not polymorphisms of TLR4 genes, are associated with Helicobacter pylori-induced duodenal ulcer and gastritis. Helicobacter. 2007;12(2):124–31.
Kara B, Akkiz H, Doran F, et al. The significance of E266K polymorphism in the NOD1 gene on Helicobacter pylori infection: an effective force on pathogenesis? Clin Exp Med. 2010;10(2):107–12.
Wang P, Zhang L, Jiang JM, et al. Association of NOD1 and NOD2 genes polymorphisms with Helicobacter pylori related gastric cancer in a Chinese population. World J Gastroenterol. 2012;18(17):2112–20.
Kim EJ, Lee JR, Chung WC, et al. Association between genetic polymorphisms of NOD 1 and Helicobacter pylori-induced gastric mucosal inflammation in healthy Korean population. Helicobacter. 2013;18(2):143–50.
Li ZX, Wang YM, Tang FB, et al. NOD1 and NOD2 genetic variants in association with risk of gastric Cancer and its precursors in a Chinese population. PLoS One. 2015;10(5):e0124949.
Castaño-Rodríguez N, Kaakoush NO, Goh KL, et al. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS One. 2014;9(6):e98899.
Companioni O, Bonet C, Muñoz X, et al. Polymorphisms of Helicobacter pylori signaling pathway genes and gastric cancer risk in the European Prospective Investigation into Cancer-Eurgast cohort. Int J Cancer. 2014;134(1):92–101.
Wex T, Ebert MP, Kropf S, et al. Gene polymorphisms of the NOD-2/CARD-15 gene and the risk of gastric cancer in Germany. Anticancer Res. 2008;28(2A):757–62.
Hnatyszyn A, Szalata M, Stanczyk J, et al. Association of c.802C>T polymorphism of NOD2/CARD15 gene with the chronic gastritis and predisposition to cancer in H. pylori infected patients. Exp Mol Pathol. 2010;88(3):388–93.
Angeletti S, Galluzzo S, Santini D, et al. NOD2/CARD15 polymorphisms impair innate immunity and increase susceptibility to gastric cancer in an Italian population. Hum Immunol. 2009;70(9):729–32.
Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–7.
Salucci V, Rimoldi M, Penati C, et al. Monocyte-derived dendritic cells from Crohn patients show differential NOD2/CARD15-dependent immune responses to bacteria. Inflamm Bowel Dis. 2008;14(6):812–8.
Ying HY, Yu BW, Yang Z, et al. Interleukin-1B 31 C>T polymorphism combined with Helicobacter pylori-modified gastric cancer susceptibility: evidence from 37 studies. J Cell Mol Med. 2016;20(3):526–36.
Park MJ, Hyun MH, Yang JP, et al. Effects of the interleukin-1β-511 C/T gene polymorphism on the risk of gastric cancer in the context of the relationship between race and H. pylori infection: a meta-analysis of 20,000 subjects. Mol Biol Rep. 2015;42(1):119–34.
Persson C, Canedo P, Machado JC, et al. Polymorphisms in inflammatory response genes and their association with gastric cancer: a HuGE systematic review and meta-analyses. Am J Epidemiol. 2011;173(3):259–70.
Lu W, Pan K, Zhang L, et al. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor {alpha} and risk of gastric cancer in a Chinese population. Carcinogenesis. 2005;26(3):631–6.
Cheng D, Hao Y, Zhou W, et al. Positive association between Interleukin-8 -251A > T polymorphism and susceptibility to gastric carcinogenesis: a meta-analysis. Cancer Cell Int. 2013;13(1):100.
Wu MS, Wu CY, Chen CJ, et al. Interleukin-10 genotypes associate with the risk of gastric carcinoma in Taiwanese Chinese. Int J Cancer. 2003;104(5):617–23.
Yang JP, Hyun MH, Yoon JM, et al. Association between TNF-α-308 G/A gene polymorphism and gastric cancer risk: a systematic review and meta-analysis. Cytokine. 2014;70(2):104–14.
Yu JY, Li L, Ma H, et al. Tumor necrosis factor-α 238 G/A polymorphism and gastric cancer risk: a meta-analysis. Tumour Biol. 2013;34(6):3859–63.
Qin Q, Lu J, Zhu H, et al. PARP-1 Val762Ala polymorphism and risk of cancer: a meta-analysis based on 39 case-control studies. PLoS One. 2014;9(5):e98022.
Zhang WH, Wang XL, Zhou J, et al. Association of interleukin-1B (IL-1B) gene polymorphisms with risk of gastric cancer in Chinese population. Cytokine. 2005;30(6):378–81.
Yang JJ, Cho LY, Ma SH, et al. Oncogenic CagA promotes gastric cancer risk via activating ERK signaling pathways: a nested case-control study. PLoS One. 2011;6(6):e21155.
Yang JJ, Cho LY, Ko KP, et al. Genetic susceptibility on CagA-interacting molecules and gene-environment interaction with phytoestrogens: a putative risk factor for gastric cancer. PLoS One. 2012;7(2):e31020.
Arbour NC, Lorenz E, Schutte BC, et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25(2):187–91.
Rallabhandi P, Bell J, Boukhvalova MS, et al. Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J Immunol. 2006;177(1):322–32.
Mommersteeg MC, Yu J, Peppelenbosch MP, et al. Genetic host factors in Helicobacter pylori-induced carcinogenesis: emerging new paradigms. Biochim Biophys Acta. 2018;1869(1):42–52.
Camargo MC, Mera R, Correa P, et al. Interleukin-1B and interleukin-1 receptor antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomark Prev. 2006;15:1674–87.
Kamangar F, Cheng C, Abnet CC, et al. Interleukin-1B polymorphisms and gastric cancer risk—a meta-analysis. Cancer Epidemiol Biomark Prev. 2006;15:1920–8.
Camargo MC, Mera R, Correa P, et al. IL1B polymorphisms and gastric cancer risk. Cancer Epidemiol Biomark Prev. 2007;16(3):635; author reply 635–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zanussi, S., Casarotto, M., Pratesi, C., Paoli, P.D. (2019). Gastric Tumorigenesis: Role of Inflammation and Helicobacter pylori. In: Canzonieri, V., Giordano, A. (eds) Gastric Cancer In The Precision Medicine Era. Current Clinical Pathology. Humana, Cham. https://doi.org/10.1007/978-3-030-04861-7_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-04861-7_1
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-04860-0
Online ISBN: 978-3-030-04861-7
eBook Packages: MedicineMedicine (R0)