
Abstract
Blast-induced neurotrauma (BINT) has increased in incidence over the past decades and can result in cognitive issues that have debilitating consequences. The exact primary and secondary mechanisms of injury have not been elucidated and appearance of cellular injury can vary based on many factors, such as blast overpressure magnitude and duration. Many methodologies to study blast neurotrauma have been employed, ranging from open-field explosives to experimental shock tubes for producing free-field blast waves. While there are benefits to the various methods, certain specifications need to be accounted for in order to properly examine BINT. Primary cell injury mechanisms, occurring as a direct result of the blast wave, have been identified in several studies and include cerebral vascular damage, blood–brain barrier disruption, axonal injury, and cytoskeletal damage. Secondary cell injury mechanisms, triggered subsequent to the initial insult, result in the activation of several molecular cascades and can include, but are not limited to, neuroinflammation and oxidative stress. The collective result of these secondary injuries can lead to functional deficits. Behavioral measures examining motor function, anxiety traits, and cognition/memory problems have been utilized to determine the level of injury severity. While cellular injury mechanisms have been identified following blast exposure, the various experimental models present both concurrent and conflicting results. Furthermore, the temporal response and progression of pathology after blast exposure have yet to be detailed and remain unclear due to limited resemblance of methodologies. This chapter summarizes the current state of blast neuropathology and emphasizes the need for a standardized preclinical model of blast neurotrauma.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
- Blast
- Neurotrauma
- Traumatic brain injury
- In vivo
- Methodology
- Preclinical models
- Neuropathology
- Shock wave
- Shock tube
1 Introduction
Blast-induced neurotrauma (BINT) has become the center of military health concern because of the increasing incidence of blast-induced traumatic brain injury (bTBI) in combatants over the past two decades [1]. Cognitive impairments that impede function and performance can be sustained from bTBI. Upon detonation, improvised explosive devices (IEDs) create a blast wave that involves a rapid overpressure change as high-speed energy moves through ambient air, impacting surrounding war fighters. In blast-induced injuries, the resulting insult can be categorized into four different modes: primary, secondary, tertiary, and quaternary. Primary injury is caused directly by the blast overpressure (BOP) wave and any barotrauma to the organs can be attributed to this exposure. Secondary injury is a result of blunt impact from any missiles, such as shrapnel, that are projected towards the body due to the blast. These insults can cause external, penetrating injuries including, but not limited to, bruises and lesions. Tertiary injury is due to the impact of the body with a rigid surface as a result of the blast wind. When soldiers are “thrown” by the blast wind, there is a potential for broken bones or even a concussion, which can compound bTBI. Quaternary injury is a result of any chemical or burn exposure that occurs from the explosion itself. These exposures, which could include infectious agents, can have subsequent systemic implications, such as lung injuries, which then exacerbate the brain injury. For the scope of this chapter, the discussion will focus on primary blast injury to the brain.
The mechanical transmission of blast energy to the brain has not been fully elucidated. However, while several mechanisms of primary blast injury to the brain have been hypothesized, only the “skull dynamics” theory is supported by computational modeling and experimental evidence. This theory is based on the fact that the skull is not perfectly rigid and it must flex/deform, especially when subjected to impulsive loading. Thus, the shock wave invokes different skull dynamic response modes which cause injury [2–9]. While there is an undeniable theory as well as independent experimental validations of the “skull dynamics” mechanism, how the skull dynamic response modes lead to injury at the cellular level still needs to be elucidated. The imparted stress waveforms from the blast energy can lead to localized stress and strains on the cells. Morphological deficits including cytoskeletal and axonal damage can lead to necrosis if injury thresholds are reached. Arguably, more emphasis has been placed on determining the time course of secondary cellular injury mechanisms. These include regulation of neuroinflammation through cytokine signaling, glial activation, and oxidative stress through accumulation of reactive oxygen species (ROS). Delayed apoptosis can result from these cascades and has a potential link to chronic behavioral and functional deficits. Many cellular cues have been explored, and yet the exact mechanism of injury at the cellular level has not been determined.
A significant limitation of evolvement within the blast neurotrauma community is the lack of standardized preclinical models. Many sources of data variability come from variances induced by different experimental setups. This has become such a significant concern that the NIH/NINDS has recently developed a list of common data elements (CDEs) for preclinical TBI research to promote the use of standard reporting and facilitate comparisons across studies [10]. Thus, the goal of this chapter is to present and compare blast neurotrauma methodology and subsequent data from the current literature to stress the importance of implementing standardizing preclinical methods.
1.1 Current Methodologies for Small Animal Preclinical Testing
Several experimental approaches are reported within the current blast neurotrauma literature. Various methods may contribute to inconsistent findings surrounding BINT. For those interested in studying the effects of BOP on the brain, isolation of the primary blast wave is crucial (see Note 1 ). However, some studies have examined the effect of composite injuries involving multiple injury modes [11]. Others have studied the combined effects of primary and quaternary blast injuries in what is referred to as a burn-blast model [12]. Knowing the exact blast parameters used to cause injury is crucial for comparison between research models.
Prior to experimentation, understanding blast physics, or working with experts in this field, is critical to designing appropriate environments to simulate a particular blast condition. The previous chapter (Chapter 7) by VandeVord et al. presents a detailed review of blast physics fundamentals. Briefly, BOP, defined as a deviation from atmospheric pressure, can be classified as static or dynamic. Static overpressure is considered the crushing force of the blast wave and can be measured perpendicular to the flow direction such that it does not impede the flow. Dynamic pressure is called the “blast wind” and for certain conditions is the major cause for blast displacement of objects as opposed to the static pressure, which causes crushing. In BINT studies, duration and magnitude of static overpressure are usually reported and most studies aim to represent a simplistic blast wave, the Friedlander waveform (Fig. 1) [13]. A comprehensive review of testing methods found in literature reporting primary blast brain injuries is summarized in Fig. 2. All pressure measurements reported within the review are static overpressures given by the cited article (see Note 2 ).

Classical free-field blast wave, referred to as the “Friedlander waveform,” is a simplified representation of the variation through time of static pressure for a classical free-field blast wave as would be measured at a fixed location passed by the blast. It does not account for reflection against the ground or other naturally occurring anomalies. In general, the classical free-field blast wave is characterized by a single high-pressure pulse, or shock front, followed by a rapid exponential decrease of the overpressure (positive phase) and terminating with a period of negative overpressure (negative phase) before returning to ambient conditions

Schematic of various methodologies which are used for BINT identified from literature. Defining characteristics, such as animal positioning and how the shock wave is generated, serve as distinctive features
1.2 Detonation of Explosives
Open-field detonations use an explosive to induce BINT. An explosion is a phenomenon that results in a sudden release of energy and creates a blast wave which propagates outward from the explosion. Trinitrotoluene (TNT) is commonly used in varying amounts depending on the magnitude of the detonation required for animal testing. One advantage of conducting open-field testing is that there is no flow hindrance of the blast wave, which allows for testing of either larger animal models or multiple small animal models (Fig. 3A, B) [14–16]. On the other hand, administering anesthesia and controlling for bacterial exposure to survival animals are more difficult in the harsh outside environment (see Note 3 ).

Examples of different blast experimental setups. (A) Open-field testing (Rubovitch et al. [15]) (B) Bunker setup (Kaur et al.). (c1) Shock tube with animal outside (Long et al. [75]). (c2) Shock gun with animal on-axis (Svetlov et al.). (d) Shock gun with animal off-axis (Svetlov et al.). (e) Shock tube with an open end (Nambier et al.). (f) Advanced blast simulator (VandeVord et al. [54]). (g) Combined explosive in tube (Saljo et al.)
While detonations are arguably the most successful in recreating battlefield explosions, a limitation is that repetition of the same testing conditions is nearly impossible given variability between explosive devices and reflections of the blast wave. This makes reproducing results and testing set parameters of blast overpressure and duration a formidable task. Though this may mimic IED warfare in theatre, consistent exposure is crucial for obtaining statistical significance within research data. Static overpressure profiles do not resemble the ideal Friedlander waveform and positive durations have been measured up to 18 ms [14]. Obtaining the clearance to perform open-field blast studies in an approved facility is difficult. Yet, some researchers have used explosives in conjunction with blast tubes to replicate battlefield explosions in an experimental environment, eliminating the need for open space (Fig. 3G) [17]. Another drawback is that secondary injuries, such as those sustained from debris, often occur with explosives setups, thus precautions (e.g., animal shielding) are required in order to limit studies to primary injuries (see Note 4 ). While it is difficult to isolate primary blast wave, animal protection such as containment within a cage has been utilized for open-field blasts [15, 18].
1.3 Shock Guns
Since live detonation testing is not optimal for research laboratories, several devices have been constructed to recreate shock waves . The shock gun method consists of a narrow tube (usually vertical) that contains a driver and driven tube separated by a diaphragm. The bursting of the diaphragm creates a shock wave that is transmitted to a specimen positioned outside of the tube. In this setup, positioning of the animal is crucial in terms of achieving exposure to static overpressure without exposure to the dynamic winds (see Note 4 ). The effects of positioning the animal perpendicular to the shock wave have been studied, and it was found that this orientation causes head accelerations atypical of blast TBI (Fig. 3c2) [19]. Placing the animal directly under the tube causes very high dynamic overpressure exposure that is not representative of open-field blast exposure. The fast expansion of the wave leaving the narrow tube causes rapid dissipation of energy and can therefore make it difficult to produce an accurate free-field blast wave. The combination of the emerging shock wave and venting gas causes the formation of a vortex and high-flow-velocity region called end-jet. As such, researchers using this device have modified their methodology so that the animal is offset from the end-jet and not exposed to reflections (Fig. 3d) [11, 20]. One advantage of using this method is that it is possible to use on a laboratory bench top due to the smaller size of the device. However, appropriate measurements need to be collected to verify that the conditions resemble an appropriate blast environment.
1.4 Conventional Shock Tubes (ST) and Advanced Blast Simulators (ABS)
Historically, conventional shock tubes (ST) have been used to mimic blast conditions within the laboratory setting. This method allows for manipulation of the shock wave within a controlled environment with high repeatability. Most recently, modifications to the ST have led to the design of the advanced blast simulator (ABS), which was designed to intrinsically replicate all the key features of blast wave flow conditions, including the negative phase and secondary shock as described previously by VandeVord et al. (Chapter 7). The ST is composed of two separate chambers: the driver, where the pressure is created by means of an air compressor system or other gas, and the driven, where the shock wave propagates through the test section [21]. Because the wave is produced by compressed gas bursting a membrane instead of an actual chemical explosion, the term shock wave is used instead of blast wave. It is important to understand how the shock wave develops within the tube and how end-of-tube rarefaction leads to an imbalance of high dynamic pressures and yet reduced static pressure conditions, amounting to extremely adverse effects. Thus, experiments staged with a specimen near the end of the tube, where the static pressure decreases and dynamic pressure increases, should be avoided (Fig. 3C1; see Note 4 ). In order to create a more accurate blast environment for animal testing, the ABS was designed. There are three chambers in the ABS device: a driver, driven, and end-wave eliminator (EWE) (Fig. 3f). The EWE consists of a dump tank that can contain the expanding gases from entering the lab space and at the same time creates some overpressure reflection that counteracts the rarefaction wave. Baffling is incorporated into the EWE to break up the venting shock front and prevents the waves from traveling back up the device, in contrary to the ST which has an open end causing a reflection of the wave and exposing the animal to multiple extraneous shocks that do not exist in real blast conditions (Fig. 3e). A disadvantage of using either the ST or ABS designs is that shock flow constraints require less than 20 % restriction of specimen in the device in order to recreate the most accurate blast flow conditions. Large animal studies would require a much larger chamber (approximately 16 square foot cross section) or the addition of an expansion section for optimal flow specifications, which ultimately leads to laboratory space concerns.
2 Materials
2.1 Animals
Male adult Sprague Dawley rats (250–300 g) from Harlan Labs (San Diego, CA, USA) are used for these experiments. Animals are acclimated 12-h light/dark cycle with food and water provided ad lib in an Association for Assessment and Accreditation of Laboratory Animal Care-approved facility.
2.2 Anesthesia
The animals are anesthetized with a continuous flow of 3 % isoflurane and 60 % oxygen for 5 min.
2.3 Materials for Testing
-
1.
Compressed gas (helium preferred).
-
2.
ABS (driver section, test section, end-wave eliminator).
-
3.
Membrane (Acetate, Mylar, Vinyl, Metal; Grafix Plastics, Cleveland, OH, USA).
-
4.
Pressure sensors (PCB Piezotronics Inc., Depew, NY, USA).
-
5.
Signal Conditioner (PCB Piezotronics Inc., Depew, NY, USA).
-
6.
Dash 8HF data acquisition system (Astro-Med, Inc, West Warwick, RI, USA).
-
7.
Animal sling holder (Custom made from mesh).
-
8.
High-speed video camera (Phantom Miro eX2, Vision Research, Wayne, NJ, USA).
-
9.
Physiological monitoring system (Nonin PulseSense VET Pulse Oximeter, Henry Schein Inc. Melville, NY, USA).
3 Primary Mechanisms of Blast Neurotrauma
In all of the described methods for inducing blast neurotrauma, the mechanical insult from a blast wave ultimately causes direct damage to cells in terms of morphological defects. While there are several hypotheses regarding which component of blast loading is most closely correlated to injury, it is accepted that brain cells are exposed to high-speed mechanical impulses. bTBI is a diffuse injury due to the global exposure to the entire skull. This widespread injury differentiates bTBI from blunt impact TBI, which is a focal insult accompanied by head rotational acceleration [22]. Even though energy translation leading to injury differs between blunt and blast TBI, cortical, and hippocampal injuries have been observed with both mechanisms, as well as similar clinical symptoms [23–25]. Shearing and stretching of cells occur due to the imparted wave stress at the microscopic level. Since the methodology of inducing BINT may lead to differing injuries, the following sections aim to provide a comparison of the primary injury mechanisms of BINT resulting from the various experimental blast models. Identifying specific biomarkers of primary injury mechanisms can serve to differentiate between the experimental methods of BINT.
3.1 Cytoskeletal Damage
The cytoskeleton is the supporting network of fibers and filaments in the cytoplasm of a cell and has various functions, including deformation resistance through supporting structure, cell signaling, and intracellular transport. Damage to the cytoskeleton has been shown to elicit downstream abnormalities in acute brain injury [26]. Due to cellular shearing during exposure to blast, cytoskeletal breakage can occur, impairing cytoskeletal functions. A study of explosive-induced blast neurotrauma reported an increase of phosphorylated neurofilament proteins (p-NFH) in the cortex and hippocampus following blast exposure at 240 kPa at 18 h post-blast [17]. These changes were found during acute stages and were resolved by 21 days after blast [17]. Cytoskeletal degradation in the hippocampus as well as the cortex was observed in several low-level (11.5 kPa) shock gun studies at 12 h post-exposure with some recovery [27, 28]. The ST model produced a decrease of actin (microfilament protein) in the nucleus accumbens at 3 days post-blast [29], as well as an increased amount of cytoskeletal enzymes, which contribute to the degradation of structural cytoskeletal fibers, in the cortex and cerebellum [30]. These changes were observed at 1 and 7 days following BOP exposure. While there was natural resolution of cytoskeletal impairments in an explosive model at 240 kPa by 21 days, there was faster recovery (by 7 days) in an 11.5 kPa BOP shock gun model. These results show that cytoskeletal degradation is a common outcome occurring throughout the brain; however, the extent of degradation and recovery time differ based on methodology used to induce injury and magnitude of BOP.
3.2 Blood–Brain Barrier Dysfunction
The blood–brain barrier (BBB) is a selective membrane that is essential to creating a controlled environment suitable for brain function. The BBB functions to protect the brain from pathogens and other harmful molecules through selective transport and permeability. The loss of integrity of this barrier may lead to neurological dysfunction and disease. Following TBI, it is known that the BBB loses integrity which enhances subsequent damage through neuroinflammation and increased permeability [31]. Similarly, BBB disruption has been reported following BINT. BBB permeability/integrity is identified by evaluating the quantity of endothelial cells in models of BINT. While secondary mechanisms ensue following injury and can affect BBB integrity, BBB disruption, including acute microlesions, has been reported as the result of the mechanical insult. In a shock gun study which utilized a rifle barrel to generate the shock wave , BBB disruption was observed to be dependent on the magnitude of the blast overpressure from 145 to 323 kPa but independent of the time point of assessment [20]. BBB breakdown, characterized by immunoglobulin (IgG) extravasation, was also reported in open-ended ST studies to be present in the cortex at 3 and 24 h post-blast but not at longer time points [32, 33]. Skotak et al. [34] found BBB degradation from over 190 kPa BOP in an open-ended ST experiment at 24 h post-exposure. However, these changes were found throughout the brain and were not localized to the cortex. BBB disruption appears to occur immediately in high-severity blasts and is possibly the best correlate of cellular damage along with primary blast level [20, 32].
3.3 Neurovascular Disruption
Cerebral vasculature includes arteries and veins that supply the brain with oxygen and nutrients necessary for brain function and transport of blood back to the heart. Any disruption of these vessels could have catastrophic consequences on brain health. In concussive brain injury, traumatic cerebral vascular injury plays a large role in pathophysiology [35]. Abnormalities in vascular structure and impaired vascular integrity are seen in morphological stains and can be due to the shearing effects at density interfaces during injury. Rapid overpressure is reported to cause mechanical damage at the brain-blood interface, which can be identified by vascular damage [36]. In open-field explosive studies, narrowed and permeable vasculature were found in a mouse mode of BINT with pressures ranging from 48 to 77 kPa and recovery up to 4 days post-exposure [14]. Cerebral microvascular lesions and downregulation of type IV collagen (basal lamina component) are commonly reported in studies using a ST with the rodent near the end of the tube, demonstrating a diffuse response throughout the brain [37–40]. Gama Sosa et al. [38] reported shear-related injuries, such as microhemorrhage and degeneration in cerebral microvessels, in cortical vessels in a repeated-exposure (74.5 kPa) ST model at 24 h and 10 months post-exposure. Kamnaksh et al. [40] reported increased plasma levels of vascular endothelial growth factor (VEGF) in the repeated-exposure group at 2 h post-exposure and in the single-blast group at 22 days post-exposure. Damage of cerebral vasculature was observed with all blast methodologies but the time course of recovery is debatable, depicting the diffuse nature of BINT resulting in lasting damage. Multiple blast exposures can also contribute to early presentation of vascular damage.
3.4 Axonal Injury
The axon is an extension of the neuron that transmits impulses away from the soma to the axonal terminal to relay information. Axons can signal over long distances; thus, having axonal integrity is crucial for optimal brain activity. In TBI, axonal injury, characterized by axonal swelling and cytoskeletal damage, is commonly found due to the susceptibility of axons to deformation [41]. In BINT, axonal injury has been identified in some modalities. Axonal injury is often diagnosed using beta-amyloid precursor protein (β-APP), but gross morphology through histological analysis can also be a predictor. β-APP is an integral membrane protein that gives rise to beta-amyloid following posttranslational modifications. In explosive open-field modalities, morphological axonal abnormalities quantified using diffusion tensor imaging (DTI) at 7 and 30 days post-blast have been observed [15] as well as increased β-APP at 24 h post-blast for 49 and 77 kPa exposures [14]. An abnormal distribution of p-NFH, indicating damage of axons, was seen in a model with an explosive driver at 240 kPa [17]. Axonal injury (increased β-APP) at acute stages has also been observed for experimental tests outside the ST using explosives to generate the shock wave [36]. The reports suggest that nonuniform, uncontrolled shock waves from explosive devices will result in high injury severity that culminates in axonal disruption.
In an open-end ST model, Valiyaveettil, et al. [30] found diffuse axonal injury following repeated blasts on mice, while Koliatsos et al. [42] reported axonal injury with single exposures of 68–183 kPa static overpressure. Damage was localized to the cerebellum and brainstem. In these studies, multiple blast exposures could have an effect on progression of axonal injury throughout the brain. Conversely, using a model with animals placed within the ST [43], β-APP was not found to be elevated within the hippocampus; thus, no axonal injury was identified. While pressure magnitude likely affects the results, one possible explanation is that a reflection wave due to an open end produces an upstream shock that causes multiple or enhanced shock exposures that would cause uncharacteristic shearing of axons in specific regions of the brain. Garman et al. [33] reported that exposure in an ST produced deep axonal injury in the cerebellum and brainstem, with tendencies of diffuse axonal injury. However, the animal was placed in a body shielding device in which the head will undergo rapid accelerations (brainstem damage is an indicator of acceleration-based injury) while the body is shielded, giving an inaccurate representation of true primary blast exposure and the possibility of the tertiary mode of blast injury .
4 Secondary Mechanisms of Blast Neurotrauma
Following the initial mechanical insult, secondary injury pathologies ensue through the activation of various cellular cascades. The cellular response to blast injury encompasses but is not limited to chronic inflammation and oxidative stress. Similar responses have been observed in other neurological disorders and have been linked with cognitive impairments [44, 45]. While the roles of pro-inflammatory and pro-oxidative pathways have been investigated, their temporal appearance and subsequent progression in BINT remains unknown. Since various methods of inducing BINT have led to differing pathologies, the following section aims to provide a comparison of the secondary mechanism of BINT resulting from different experimental blast models.
4.1 Inflammation
Brain injury, including BINT, is followed by a chronic inflammatory response characterized by a sustained activation of glial cells including microglia and astrocytes [46, 47]. The response from glial cells is important to cell survival and neuroprotective efforts; however if not controlled, it can contribute to sustained brain injury. Microglia are the resident immune cells of the central nervous system (CNS) and their activation following injury involves the release of several inflammatory molecules. Several studies have observed microglia activation as a result of BOP exposure both in the acute and chronic stages of recovery [18, 32, 48–50]. Microglia activation has not been investigated as a result of the shock gun method but has been demonstrated in both open-field and ST experimental setups. In the open-field setup, Kaur et al. [49] found lasting microglia activation for 14 days in the pineal gland. Similarly, with animals placed inside an ST, increased activation of microglia was observed for up to 30 days in the hippocampus and brainstem [48]. An increase in the microglia population, observed using Iba-1 and ox-42, has been reported for open-field, ABS , and ST testing [14, 49–51]. These changes were reported in the hippocampus and corpus callosum and have been shown to correlate with injury severity [14, 51]. This indicates a diverse response of microglia with varied pressure magnitudes. Such diversity may imply that microglial response may be suitable for use as a biological pressure sensor in which activation of certain molecular pathways depends on the severity of the blast exposure.
Alterations in astrocyte activation have also been observed. Astrocytes are a key player in brain homeostasis and their activation occurs through a mechanism of hypertrophy coupled with an increase of intermediate filaments such as glial fibrillary acidic protein (GFAP) . Widespread changes to astrocyte intensity, through swelling and proliferation, have been observed in several brain regions following BINT with various experimental set-ups. Increases in GFAP following blast have been reported in the hippocampus [43, 48, 50–54], cortex [50, 53–55], amygdala [53, 54], and other brain regions [29, 48] and appear to correlate with elevated levels of GFAP in the blood [11, 40]. Both acute (less than 3 days) and chronic changes (lasting up to 21 days) in GFAP expression and protein levels have been reported following BINT in rodent models. Using a shock gun method, Svetlov et al. [52] found no changes to GFAP levels in the cortex but found increased GFAP levels in the hippocampus 7 days after exposure. More prolonged GFAP changes have been observed in the open-field, ABS, and ST experimental setups. In the open-field setup, GFAP immunoreactivity increased both in the cortex and in the hippocampus for up to 21 days following injury [50]. Similar results were found in the hippocampus using an ST and ABS with the animal within the device [43, 48, 51, 54] and an open ST with the animal located just outside [53].
Inflammation is regulated by pro-inflammatory mediators called cytokines. Cytokines including various interleukins, tumor necrosis factor α (TNF-α), and interferon γ (IFN-γ) have been implicated in the pathology of BINT. The involvement of these molecules has been demonstrated in animal models and multiple experimental setups but their role in the pathology of the injury is not yet fully understood.
Interleukins are a family of cytokines that play a critical role in mediating the inflammatory and immune response, several of which have been found to change following BINT [53, 56, 57]. Interleukin 6 (IL-6) has been the most studied interleukin likely due to its involvement in the inflammatory response following brain injury [58]. Using a rat model, IL-6 protein levels were found to be increased in the hippocampus and amygdala [53]. The injury was administered using an open-ended ST with the animal placed at the end of the tube. Valiyaveettil et al. [57] showed that mRNA expression of IL-6 was increased in the midbrain of mice placed within the ST. These results indicate upregulation of IL-6 in several brain regions in the acute stages following BINT which differ from results observed in serum. Sajja et al. [56] found that IL-6 protein levels were decreased in the blood serum from rats 72 h following injury using an open-end ST with animals placed inside. These results suggest potential local release of IL-6 from glia and/or relocalization of IL-6 from circulation to the site of injury. However, the inflammatory response appears to be delayed.
Other interleukins have been investigated following BOP exposure in animal models. Using an open-ended ST with the animal inside the tube, mRNA expression of various interleukins has been observed to vary within various brain regions. IL-2 was decreased in the hippocampus, IL-28 was decreased in the cerebellum but IL-7 increased in the frontal cortex [57]. Cytokine expression changes have not been limited to the brain. Interleukins have been shown to be altered in the blood serum in the acute stages following BOP exposure [56]. A significant increase in IL-5 was observed in the nucleus accumbens, anterior motor cortex, prefrontal cortex, and anterior striatum [59]. Authors speculate that IL-5 could be a key regulator of inflammation after blast injury since the response was so diffuse in the brain. Other reported cytokines were found elevated within the brain, including GM-CSF, IL-1α, IL-10, and IL-1β [56, 59].
TNF-α and IFN-γ are additional key cytokines found in the brain and are involved in the inflammatory response through transcriptional regulation [60, 61]. Dalle Lucca et al. [62] reported acute increases in TNF-α following injury using an open-ended ST with the animal placed inside the tube. Similarly, IFN-γ upregulation was demonstrated in the acute stages of recovery from BOP exposure. Cho et al. [63] found increased IFN-γ in the hippocampus resulting from the animal being placed within an ABS . Kamnaksh et al. [53] found a similar increase in the hippocampus as well as an increase in the amygdala following exposure from an open-end ST with the animal at the end. Upregulation of each of these cytokines may have a detrimental impact on cell survival. Many of the genes that are regulated by TNF-α are also regulated by IFN-γ and the combination of these cytokines can lead to a large increase in the transcription of these genes [64]. This transcriptional change may directly affect the chronic pathology of BINT.
4.2 Oxidative Stress
Oxidative stress occurs following the aberrant production of ROS, including hydrogen peroxide and superoxide anion. ROS are produced as by-products of normal cell metabolism, and play a role in cell signaling and homeostasis [65]. ROS levels are maintained by a dynamic equilibrium between their production during metabolism and degradation facilitated by antioxidants. Excess accumulation of ROS can cause neurotoxicity and neurodegeneration by prolonged upregulation of pro-inflammatory mediators [66] and has been shown to play a role in the secondary injury process of TBI and BINT. Increased levels of ROS have been found in both acute and chronic stages following blast injury [32, 63, 67–71]. The oxidative stress appears to be widespread following BINT as it has been observed in the hippocampus [63, 68], cortex [67, 69], and hypothalamus [70]. These changes have been found in rodent models using both the ST and shock gun experimental setups. The shock gun method has been shown to produce increased oxidative stress in the hypothalamus 6 h following injury [70], but longer time points have not been investigated. In the ABS setup (animal placed inside), increased oxidative stress was observed in the hippocampus from 4 h to 2 weeks post-injury [63]. Open-ended STs have shown similar increases in the hippocampus [32, 68] as well as increases in the cortex [67, 69]. The prolonged, widespread accumulation of ROS undoubtedly affects brain function, and has been shown to induce changes to the BBB that cause increased neuroinflammation [69]. This secondary effect was observed in an ST rat model [69] and an open-field explosion mouse model where BBB dysfunction was observed 1 month after injury [15]. This can potentially explain the chronic inflammatory response associated with the secondary injury mechanisms.
In order to ameliorate the pro-oxidative environment, superoxide dismutase 1 (SOD1) and superoxide dismutase 2 (SOD2) are natural antioxidants that function to restore the proper balance of superoxide radicals. These antioxidants have been shown to be altered following BINT. In rodent models, SOD1 and SOD2 expression and protein levels were increased following injury. Both open ST and ABS have shown elevated SOD1 in the hippocampus when the animal is placed within the device [51, 68, 72]. In the open-field setup, Rubovitch et al. [15] observed altered SOD2 levels in the areas surrounding vasculature. Cernak et al. [68] found transient SOD2 levels in the hippocampus following blast injury inside an open-end ST. SOD2 was significantly increased 5 days after injury but no change was found after 24 h. In contrast, Huber et al. [72] found increased SOD2 in the hippocampus 24 h after injury which subsided 30 days after injury. These results show that the antioxidant, SOD2, is upregulated in response to injury. This response may be triggered by the aberrant accumulation of ROS. Upregulation of antioxidant enzymes indicates potential neuroprotective efforts of glial cells to reduce the pro-oxidative environment. These efforts appear to decrease in the chronic stages as the antioxidant enzymes return to basal level expression. It has been well established that the accumulation of ROS is critical in BINT pathology. Therefore, natural methods to ameliorate the pro-oxidative environment become very critical to recovery.
5 Functional and Cognitive Outcomes Following Blast Neurotrauma
Cognitive and functional deficits have been observed clinically following blast-induced TBI [24]. Loss of motor function, memory deficits, and increased anxiety are some of the key features associated with BINT pathology. Experimental reproduction of blast injuries in animal models has shown similar outcomes. The changes have been observed predominantly in rodent models in controlled ST models, but other methodologies have been also used to characterize behavior after injury. This section begins to summarize some of the functional, behavioral, and cognitive changes observed following BINT in animal models.
5.1 Motor Function
As previously described, evidence of cellular disturbances in primary and secondary modes of blast injury has been observed within the cortex and can begin to explain the loss of motor function observed by BINT pathology. Loss of motor function in the rodent model has been well established and has been demonstrated in the shock gun, ST, and open-field experimental setups [28, 42, 48, 67, 73, 74]. In the open-field setup, animals showed decreased motor function up to 3 days following injury [74] but not at 7 or 30 days [15]. In a shock gun setup, Park et al. [27] found lasting motor function deficits until 9 days after exposure. Results from ST experiments show differing results. Some studies have shown that motor function deficits followed blast exposure and are apparent in the acute stages [42, 67, 73] but they have also been shown to last for 21 days following the injury [48]. However, other investigations have shown no deficits in motor function [54].
Motor function impairment appears to occur immediately following the blast injury and only be present temporarily [42, 48, 73, 75], which leads to a concern regarding anesthesia effects (see Note 3 ). While motor control may be diminished at the acute stage, the timeframe of impairment remains unclear. Using a shock tube paradigm, it has been shown that motor function returns to basal levels at 2 h following the injury according to the balance beam task [73]. While another study found that the motor function was not restored until 21 days later using the Rotarod task [48]. These differences may result from either the varied sensitivities of the performed tasks or the different magnitudes of BOP exposure (see Note 5 ).
5.2 Behavior
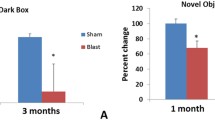
Anxiety-like behavior has been described as a symptom of BINT through animal models. Several behavioral tests have been performed to assess anxiety including an open-field test, light dark box, and the elevated plus maze. In all behavioral tests, anxiety following BINT has been observed [28, 40, 42, 48, 73]. While the shock gun, ST, and open-field experimental setups have been utilized to assess anxiety , only the shock gun and the ST setups produced anxiety-like behavior. The open-field experiment did not produce anxiety within 7 days following exposure [25]. Anxiety-like behavior has been found to persist in both the acute and chronic stages for up to 30 days following BOP exposure [48]. These changes likely result from primary injuries sustained to the amygdala and the activation of secondary cascades, which were noted and described previously.
5.3 Cognition
Primary and secondary mechanisms of blast neurotrauma have been reported to create biological changes in the hippocampus (which is crucial for various forms of memory). As a result, memory loss is an important outcome of BINT and brain injury in general. Memory deficits have been studied following BINT in the rodent model using a variety of cognitive tests. These tests have been conducted to investigate changes in recognition and recollection, associative memory, and spatial memory following BINT.
In order to investigate recognition memory, novel object recognition (NOR) tests have been performed using a rodent model of BINT [25, 63, 76]. This test capitalizes on the rodent’s natural instinct to explore new objects in their environment. Blast exposure has been shown to correlate with a decline in recognition memory observed at 7 days [25, 63] and 14 days [63] following the injury induced by an open-field environment and the ABS . Interestingly, memory changes were not observed at 72 h following injury which may indicate a delayed response and dependence on secondary injury mechanisms [76].
Avoidance tasks use certain cues associated with aversive stimuli to test associative memory, which entails the ability of the animal to associate cues with a stimulus. These tests have demonstrated deficits in associative memory immediately following blast exposure [73] and lasting at least 30 days [48]. These behavioral deficiencies were observed following blast exposure in an open-end ST setup with the animal inside the tube and were not observed in an open-field setup [25]. However, associative memory changes have been demonstrated to be dependent on injury severity and overpressure magnitude [48, 73] which may be the cause of these contradictions.
Behavioral tests like the Y-maze, MWM, and Barnes maze are useful in assessing changes to spatial memory following BINT. Spatial memory is challenged based on the animals’ ability to remember and perform in a previously explored environment. While deficits to spatial memory have been observed in rodents, the changes appear to be transient following BINT. Many studies have demonstrated deficits following BINT only at various time points in the acute stages following injury in the ST [42, 54, 73, 75, 77]. The results from these tests are inconsistent with open-field tests as lasting spatial memory deficits have been observed [15]. Still, other studies have found, both in the ST and in an open-field environment, little to no changes in spatial memory [25, 40]. The inconsistency between findings may result from the differences in the BOP exposure and subsequent injury severity which can cause differences in the primary and secondary mechanism of injury. Despite some contradictions, it appears that spatial memory is altered in the acute stages following BINT.
6 Notes
This chapter summarizes the various cellular and behavioral outcomes that are seen in different experimental setups of BINT. Common themes have been established, such as morphological defects, inflammation, and oxidative stress, but the specific time course at which these are presented differs with respect to the method of inducing bTBI. Differences in cognitive and functional outcomes are also seen between the various blast methodologies. There are many inconsistencies between researchers in the methodology of inducing bTBI. These inconsistencies manifest in altered cellular regulation due to the modes of injury and the injury severity. Some methods do not accurately recreate the free-field blast wave or use incorrect animal positioning, which can introduce secondary and tertiary forms of injury and are unrepresentative of BINT. In order to be able to more effectively to compare data between research groups, limitations need to be minimized. Such limitations include the following:
-
1.
Improper animal harnessing which exposes the animal to tertiary injury and head accelerations and thus may lead to different injury pathologies.
-
2.
Differences in pressure sensors and location of static pressure measurements which can lead to inconsistent relationships between peak static overpressure, dynamic pressure , and duration.
-
3.
The use of different anesthetics during testing, some of which are neuroprotectants and have various recovery rates leading to inconsistent results.
-
4.
Improper animal location which results in exposure to multiple waveforms or a dynamic blast wind injuring the live specimen.
-
5.
Biological and behavioral assessments that vary between laboratories and have different sensitivities to pathological outcomes.
References
Elder GA, Mitsis EM, Ahlers ST, Cristian A (2010) Blast-induced mild traumatic brain injury. Psychiatr Clin North Am 33:757–781
Clemedson C-J, Criborn CO (1955) Mechanical response of different parts of a living body to a high explosive shock wave impact. Am J Physiol 181:471
Romba J, Martin P (1961) The propagation of air shock waves on a biophysical model
Moss WC, King MJ, Blackman EG (2009) Skull flexure from blast waves: a mechanism for brain injury with implications for helmet design. Phys Rev Lett 103:108702
Alley MD, Schimizze BR, Son SF (2011) Experimental modeling of explosive blast-related traumatic brain injuries. Neuroimage 54(Suppl 1):S45–S54
Bauman RA, Ling G, Tong L, Januszkiewicz A, Agoston D, Delanerolle N, Kim Y, Ritzel D, Bell R, Ecklund J, Armonda R, Bandak F, Parks S (2009) An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma 26:841–860
Leonardi A (2011) An investigation of the biomechanical response from shock wave loading to the head. In: Biomedical engineering. Wayne State University, Detroit, MI
Leonardi AD, Bir CA, Ritzel DV, VandeVord PJ (2011) Intracranial pressure increases during exposure to a shock wave. J Neurotrauma 28:85–94
Bolander R, Mathie B, Bir C, Ritzel D, VandeVord P (2011) The contribution of skull flexure as a possible mechanism for neurotrauma in the rat when exposed to a shock wave. Ann Biomed Eng 39:2550–2559
Hicks R (2014) Common data elements (CDE) for preclinical TBI research, NIH/NINDS
Svetlov SI, Prima V, Glushakova O, Svetlov A, Kirk D, Gutierrez H, Serebruany V, Curley K, Wang KKW, Hayes RL (2012) Neuro-glial and systemic mechanisms of pathological responses in rat models of primary blast overpressure compared to ‘composite’ blast. Front Neurol 3:15
Chai JK, Liu W, Deng HP, Cai JH, Hu QG, Zou XF, Shen CA, Yin HN, Han YF, Zhang XB, Chi YF, Ma L, Sun TJ, Feng R, Lan YT (2013) A novel model of burn-blast combined injury and its phasic changes of blood coagulation in rats. Shock 40:297–302
Kinney GF (ed) (1962) Explosive shocks in air. Springer, New York, NY
Pun PB, Kan EM, Salim A, Li Z, Ng KC, Moochhala SM, Ling EA, Tan MH, Lu J (2011) Low level primary blast injury in rodent brain. Front Neurol 2:19
Rubovitch V, Ten-Bosch M, Zohar O, Harrison CR, Tempel-Brami C, Stein E, Hoffer BJ, Balaban CD, Schreiber S, Chiu WT, Pick CG (2011) A mouse model of blast-induced mild traumatic brain injury. Exp Neurol 232:280–289
Axelsson H, Hjelmqvist H, Medin A, Persson JK, Suneson A (2000) Physiological changes in pigs exposed to a blast wave from a detonating high-explosive charge. Mil Med 165:119–126
Saljo A, Bao F, Haglid KG, Hansson HA (2000) Blast exposure causes redistribution of phosphorylated neurofilament subunits in neurons of the adult rat brain. J Neurotrauma 17:719–726
Kaur C, Singh J, Lim MK, Ng BL, Yap EP, Ling EA (1995) The response of neurons and microglia to blast injury in the rat brain. Neuropathol Appl Neurobiol 21:369–377
Gullotti DM, Beamer M, Panzer MB, Chen YC, Patel TP, Yu A, Jaumard N, Winkelstein B, Bass CR, Morrison B, Meaney DF (2014) Significant head accelerations can influence immediate neurological impairments in a murine model of blast-induced traumatic brain injury. J Biomech Eng 136:091004
Yeoh S, Bell ED, Monson KL (2013) Distribution of blood-brain barrier disruption in primary blast injury. Ann Biomed Eng 41:2206–2214
Celander H, Clemedson C-J, Ericsson UA, Hultman HI (1955) The use of a compressed air operated shock tube for physiological blast research. Acta Physiol Scand 33:6–13
Stemper BD, Pintar FA (2014) Biomechanics of concussion. Prog Neurol Surg 28:14–27
Belanger HG, Proctor-Weber Z, Kretzmer T, Kim M, French LM, Vanderploeg RD (2011) Symptom complaints following reports of blast versus non-blast mild TBI: does mechanism of injury matter? Clin Neuropsychol 25:702–715
Mac Donald CL, Johnson AM, Wierzechowski L, Kassner E, Stewart T, Nelson EC, Werner NJ, Zonies D, Oh J, Fang R, Brody DL (2014) Prospectively assessed clinical outcomes in concussive blast vs nonblast traumatic brain injury among evacuated US military personnel. JAMA Neurol 71:994–1002
Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, Hoffer BJ, Pick CG, Greig NH (2013) Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol Dis 54:1–11
Fitzpatrick MO, Dewar D, Teasdale GM, Graham DI (1998) The neuronal cytoskeleton in acute brain injury. Br J Neurosurg 12:313–317
Park E, Gottlieb JJ, Cheung B, Shek PN, Baker AJ (2011) A model of low-level primary blast brain trauma results in cytoskeletal proteolysis and chronic functional impairment in the absence of lung barotrauma. J Neurotrauma 28:343–357
Park E, Eisen R, Kinio A, Baker AJ (2013) Electrophysiological white matter dysfunction and association with neurobehavioral deficits following low-level primary blast trauma. Neurobiol Dis 52:150–159
Sajja VS, Galloway M, Ghoddoussi F, Kepsel A, VandeVord P (2013) Effects of blast-induced neurotrauma on the nucleus accumbens. J Neurosci Res 91:593–601
Valiyaveettil M, Alamneh YA, Wang Y, Arun P, Oguntayo S, Wei Y, Long JB, Nambiar MP (2014) Cytoskeletal protein α–II spectrin degradation in the brain of repeated blast exposed mice. Brain Res 1549:32–41
Alves JL (2014) Blood-brain barrier and traumatic brain injury. J Neurosci Res 92:141–147
Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR, McCarron RM, Sullivan PG (2010) Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res 88:3530–3539
Garman RH, Jenkins LW, Switzer RC 3rd, Bauman RA, Tong LC, Swauger PV, Parks SA, Ritzel DV, Dixon CE, Clark RS, Bayir H, Kagan V, Jackson EK, Kochanek PM (2011) Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma 28:947–959
Skotak M, Wang F, Alai A, Holmberg A, Harris S, Switzer RC, Chandra N (2013) Rat injury model under controlled field-relevant primary blast conditions: acute response to a wide range of peak overpressures. J Neurotrauma 30:1147–1160
DeWitt DS, Prough DS (2003) Traumatic cerebral vascular injury: the effects of concussive brain injury on the cerebral vasculature. J Neurotrauma 20:795–825
Kuehn R, Simard PF, Driscoll I, Keledjian K, Ivanova S, Tosun C, Williams A, Bochicchio G, Gerzanich V, Simard JM (2011) Rodent model of direct cranial blast injury. J Neurotrauma 28:2155–2169
Gama Sosa MA, De Gasperi R, Paulino AJ, Pricop PE, Shaughness MC, Maudlin-Jeronimo E, Hall AA, Janssen WG, Yuk FJ, Dorr NP, Dickstein DL, McCarron RM, Chavko M, Hof PR, Ahlers ST, Elder GA (2013) Blast overpressure induces shear-related injuries in the brain of rats exposed to a mild traumatic brain injury. Acta Neuropathol Commun 1:51
Gama Sosa MA, De Gasperi R, Janssen PL, Yuk FJ, Anazodo PC, Pricop PE, Paulino AJ, Wicinski B, Shaughness MC, Maudlin-Jeronimo E, Hall AA, Dickstein DL, McCarron RM, Chavko M, Hof PR, Ahlers ST, Elder GA (2014) Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol Commun 2:67
Bir C, Vandevord P, Shen Y, Raza W, Haacke EM (2012) Effects of variable blast pressures on blood flow and oxygen saturation in rat brain as evidenced using MRI. Magn Reson Imaging 30:527–534
Kamnaksh A, Kwon SK, Kovesdi E, Ahmed F, Barry ES, Grunberg NE, Long J, Agoston D (2012) Neurobehavioral, cellular, and molecular consequences of single and multiple mild blast exposure. Electrophoresis 33:3680–3692
Smith DH, Meaney DF, Shull WH (2003) Diffuse axonal injury in head trauma. J Head Trauma Rehabil 18:307–316
Koliatsos VE, Cernak I, Xu L, Song Y, Savonenko A, Crain BJ, Eberhart CG, Frangakis CE, Melnikova T, Kim H, Lee D (2011) A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J Neuropathol Exp Neurol 70:399–416
Sajja VS, Galloway MP, Ghoddoussi F, Thiruthalinathan D, Kepsel A, Hay K, Bir CA, VandeVord PJ (2012) Blast-induced neurotrauma leads to neurochemical changes and neuronal degeneration in the rat hippocampus. NMR Biomed 25:1331–1339
Amor S, Puentes F, Baker D, Van Der Valk P (2010) Inflammation in neurodegenerative diseases. Immunology 129:154–169
Uttara B, Singh AV, Zamboni P, Mahajan RT (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7:65–74
Kou Z, VandeVord PJ (2014) Traumatic white matter injury and glial activation: from basic science to clinics. Glia 62:1831–1855
Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26:1191–1201
Cernak I, Merkle AC, Koliatsos VE, Bilik JM, Luong QT, Mahota TM, Xu L, Slack N, Windle D, Ahmed FA (2011) The pathobiology of blast injuries and blast-induced neurotrauma as identified using a new experimental model of injury in mice. Neurobiol Dis 41:538–551
Kaur C, Singh J, Lim MK, Ng BL, Ling EA (1997) Macrophages/microglia as ‘sensors’ of injury in the pineal gland of rats following a non-penetrative blast. Neurosci Res 27:317–322
Saljo A, Bao F, Hamberger A, Haglid KG, Hansson HA (2001) Exposure to short-lasting impulse noise causes microglial and astroglial cell activation in the adult rat brain. Pathophysiology 8:105–111
Sajja VS, Ereifej ES, VandeVord PJ (2014) Hippocampal vulnerability and subacute response following varied blast magnitudes. Neurosci Lett 570:33–37
Svetlov SI, Prima V, Kirk DR, Gutierrez H, Curley KC, Hayes RL, Wang KK (2010) Morphologic and biochemical characterization of brain injury in a model of controlled blast overpressure exposure. J Trauma 69:795–804
Kamnaksh A, Kovesdi E, Kwon SK, Wingo D, Ahmed F, Grunberg NE, Long J, Agoston DV (2011) Factors affecting blast traumatic brain injury. J Neurotrauma 28:2145–2153
VandeVord PJ, Bolander R, Sajja VSS, Hay K, Bir CA (2012) Mild neurotrauma indicates a range-specific pressure response to low level shock wave exposure. Ann Biomed Eng 40:227–236
de Lanerolle NC, Bandak F, Kang D, Li AY, Du F, Swauger P, Parks S, Ling G, Kim JH (2011) Characteristics of an explosive blast-induced brain injury in an experimental model. J Neuropathol Exp Neurol 70:1046–1057
Sajja VS, Tenn C, McLaws LJ, Vandevord PJ (2012) A temporal evaluation of cytokines in rats after blast exposure. Biomed Sci Instrum 48:374–379
Valiyaveettil M, Alamneh YA, Miller SA, Hammamieh R, Arun P, Wang Y, Wei Y, Oguntayo S, Long JB, Nambiar MP (2013) Modulation of cholinergic pathways and inflammatory mediators in blast-induced traumatic brain injury. Chem Biol Interact 203:371–375
Lenzlinger P, Morganti-Kossmann M-C, Laurer H, McIntosh T (2001) The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol 24:169–181
Sajja VSSS, Tenn C, Mclaws LJ, Vandevord P (2014) IL-5; a diffuse biomarker associated with brain inflammation after blast exposure. Biomed Sci Instrum 50:375
Feuerstein GZ, Liu T, Barone FC (1994) Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc Brain Metab Rev 6:341–360
Mühl H, Pfeilschifter J (2003) Anti-inflammatory properties of pro-inflammatory interferon-γ. Int Immunopharmacol 3:1247–1255
Dalle Lucca JJ, Chavko M, Dubick MA, Adeeb S, Falabella MJ, Slack JL, McCarron R, Li Y (2012) Blast-induced moderate neurotrauma (BINT) elicits early complement activation and tumor necrosis factor alpha (TNFalpha) release in a rat brain. J Neurol Sci 318:146–154
Cho HJ, Sajja VSSS, VandeVord PJ, Lee YW (2013) Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience 253:9–20
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75:163–189
Devasagayam TP, Tilak JC, Boloor KK, Sane KS, Ghaskadbi SS, Lele RD (2004) Free radicals and antioxidants in human health: current status and future prospects. J Assoc Physicians India 52:794–804
Lull M, Block M (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365
Wang Y, Wei Y, Oguntayo S, Wilkins W, Arun P, Valiyaveettil M, Song J, Long JB, Nambiar MP (2011) Tightly coupled repetitive blast-induced traumatic brain injury: development and characterization in mice. J Neurotrauma 28:2171–2183
Cernak I, Wang Z, Jiang J, Bian X, Savic J (2001) Ultrastructural and functional characteristics of blast injury-induced neurotrauma. J Trauma 50:695–706
Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N, Haorah J (2013) Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med 60:282–291
Tümer N, Svetlov S, Whidden M, Kirichenko N, Prima V, Erdos B, Sherman A, Kobeissy F, Yezierski R, Scarpace PJ, Vierck C, Wang KKW (2013) Overpressure blast-wave induced brain injury elevates oxidative stress in the hypothalamus and catecholamine biosynthesis in the rat adrenal medulla. Neurosci Lett 544:62–67
Ahmed FA, Kamnaksh A, Kovesdi E, Long JB, Agoston DV (2013) Long-term consequences of single and multiple mild blast exposure on select physiological parameters and blood-based biomarkers. Electrophoresis 34:2229–2233
Huber BR, Meabon JS, Martin TJ, Mourad PD, Bennett R, Kraemer BC, Cernak I, Petrie EC, Emery MJ, Swenson ER, Mayer C, Mehic E, Peskind ER, Cook DG (2013) Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J Alzheimers Dis 37:309–323
Ahlers ST, Vasserman-Stokes E, Shaughness MC, Hall AA, Shear DA, Chavko M, McCarron RM, Stone JR (2012) Assessment of the effects of acute and repeated exposure to blast overpressure in rodents: toward a greater understanding of blast and the potential ramifications for injury in humans exposed to blast. Front Neurol 3:32
Moochhala SM, Md S, Lu J, Teng CH, Greengrass C (2004) Neuroprotective role of aminoguanidine in behavioral changes after blast injury. J Trauma 56:393–403
Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA (2009) Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma 26:827–840
Sajja VSSS, Perrine SA, Ghoddoussi F, Hall CS, Galloway MP, VandeVord PJ (2014) Blast neurotrauma impairs working memory and disrupts prefrontal myo-inositol levels in rats. Mol Cell Neurosci 59:119–126
Saljo A, Svensson B, Mayorga M, Hamberger A, Bolouri H (2009) Low-level blasts raise intracranial pressure and impair cognitive function in rats. J Neurotrauma 26:1345–1352
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Bailey, Z.S., Hubbard, W.B., VandeVord, P.J. (2016). Cellular Mechanisms and Behavioral Outcomes in Blast-Induced Neurotrauma: Comparing Experimental Setups. In: Kobeissy, F., Dixon, C., Hayes, R., Mondello, S. (eds) Injury Models of the Central Nervous System. Methods in Molecular Biology, vol 1462. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3816-2_8
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3816-2_8
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3814-8
Online ISBN: 978-1-4939-3816-2
eBook Packages: Springer Protocols