
Abstract
The discovery of small regulatory noncoding RNAs revolutionized our thinking on gene regulation. The class of microRNAs (miRs), a group of small noncoding RNAs (20–22 nt in length) that bind imperfectly to the 3′-untranslated region of target mRNA, has been insistently implicated in several pathological conditions including cancer. Indeed, major hallmarks of cancer, such as cell differentiation, cell proliferation, cell cycle, cell survival, and cell invasion, has been described as being regulated by miRs. Recent studies have also implicated miRs in cancer drug resistance. Regardless of the several studies done until now, drug resistance still is a burden for cancer therapy and patients’ outcome, often resulting in more aggressive tumors that tend to metastasize to distant organs. Hence, with this review, we aim to summarize the miRs that influence molecular pathways that are involved in cancer drug resistance, such as drug metabolism, drug influx/efflux, DNA damage response (DDR), epithelial-to-mesenchymal transition (EMT), and cancer stem cells.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
MicroRNAs (miRs) were discovered by Victor Ambros and colleagues [1] in 1993, who observed that the C. elegans lin-4 gene coded for a pair of small RNAs with antisense complementary to multiple sites on the 3′-UTR of lin-14 gene. This small RNA substantially reduced the amount of LIN-14 protein without noticeably changing the level of lin-14 mRNA. This landmark study showed that small RNAs possessed regulatory functions and soon the presence of other regulatory RNAs (e.g., let-7) was observed in other species namely humans [2]. This group of regulatory RNAs was called microRNAs (miRs) [3], an evolutionary conserved class of small RNAs that was found to control many developmental and cellular processes in eukaryotic organisms. The latest version (June 2014) of the miRBase database (miRbase 21) listed 24 521 miRs loci from 206 species, processed to produce 30,424 mature miR products. Of these, 1881 sequences belonged to the human genome [4].
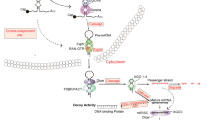
MiRs posttranscriptionally modulate gene expression by binding to their target mRNAs. miRs can be intergenic or intragenic and are produced from endogenous hairpin transcripts named pri-miR. Then, the nuclear Drosha/DGCR8 heterodimer cleaves pri-miR hairpin stem, producing the pre-miR (60–100 nucleotides) which is exported to the cytoplasm by Exportin5 and RAN-GTP. The pre-miR is then processed by the RNAse III endonuclease Dicer and its TRBP (HIV transactivating response RNA-binding protein) partner, releasing a duplex with 22–25 nucleotides. This duplex associates with the Argonaute protein forming a RNA-induced silencing complex (RISC). The mature miR stays in the complex and the passenger strand is degraded. The RISC complex is the functional complex that will interact with mRNA and trigger the regulatory effect [5]. Due to their small size, miRs are capable of binding to several regions in the 3′-UTR region of several mRNAs and in turn mRNAs can be targeted by several miRs. Consequently there is a biological redundancy in gene regulation executed by miRs. Thus, their action is extremely broad and their involvement in gene expression and cellular phenotype is well established. Although miR binding sites have also been found in 5′-UTR and in the coding sequences of mRNAs [6], they preferentially interact with seed-matching sequences in the 3′-UTR of mRNA. Several studies have shown that miRs could regulate cell differentiation [7–9], cell proliferation [10, 11], cell cycle [12, 13], cell survival [14, 15], and cell invasion [16–18]. Therefore, any misexpression of miRs can lead to altered cell phenotypes and consequently cancer initiation and progression [19]. Many miRs are located at fragile sites on chromosomes known for having common alterations (i.e., amplification, deletion, and rearrangements) in cancer [20]. MiRs that inhibit translation of proto-oncogenes are considered tumor suppressor miRs, and are usually downregulated in cancer. Other miRs are upregulated in cancer and may act as oncogenic miRs by downregulating tumor suppressor genes [21]. Recent studies have highlighted the intratumoral heterogeneity in expression of miRs [22]. This might explain the different miR expression profiles described by several groups for the same types of cancer and underlines the importance in analyzing numerous sample locations of the primary tumor in order to obtain an accurate profile of miR expression.
As stated in previous chapters, drug resistance is frequently classified into two broad types: intrinsic and acquired. Intrinsic drug resistance is not essentially a genetic attribute of the cancer cells, but can be defined as preexisting to the therapeutical challenge endowing the cancer cell with competence to survive treatment, thus rendering therapy potentially ineffective from the beginning. More often than not, intrinsic resistance could be conceived as the result of the pharmacogenetic/pharmacogenomic configuration of the host of the tumor. On the other hand, acquired drug resistance is developed during therapy and usually due to adaptive processes, such as compensatory signaling pathways, drug inactivation, increased expression of drug target, alterations in drug targets, increased expression of drug efflux pumps, cell death inhibition, epigenetic phenomena, tumor microenvironment, and DNA damage response and repair augmentation [23–26]. Drug resistance usually results in a more aggressive tumor and cancer cells often tend to metastasize to distant organs.
Within the molecular complexity of the cancer cells and their readily capacity to change the circuitry of molecular regulation, the discovery of miRs and their roles in gene expression quickly led to studies that assessed the influence of miRs in drug resistance. As a consequence, many groups have focused on the role of these small regulatory RNAs in the development of cancer drug resistance. Several studies have shown that drug resistance can also be influenced by miRs, since they can regulate drug resistance-related genes, alter drug targets, change drug concentrations, influence therapeutic-induced cell death, regulate angiogenesis, and be involved in the development of tumor stem cells.
2 MicroRNAs in Cancer Drug Resistance
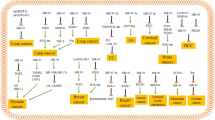
As stated above, miRs have been linked to several hallmarks of cancer in tumor cells. Differential expression of miRs in tumor cells before treatment has been associated with response to chemotherapy , while changes in miR expression have been observed in cancer cells following treatment. Table 1 summarizes the studies that showed a regulation of drug resistance by miRs. The table is divided into the main categories of drug resistance pathways and the respective regulator miR. Thus, we elaborate on miRs influencing on drug metabolism, drug transporters , DNA repair, epithelial to mesenchymal transition (EMT), and cancer stem cells . Recent studies have attempted to identify single nucleotide polymorphisms either in miR loci or target loci and correlate their presence with altered therapeutic response [27, 28].
2.1 Drug Metabolism
Drug metabolism is a complex pathway of xenobiotic detoxification that involves multiple proteins, and can be divided in three main phases: modification, conjugation, and excretion. Xenobiotics are foreign compounds (such as drugs) that are not normally produced or expected to be present in an organism. Concerted actions of drug-metabolizing enzymes (DME) and drug transporters lead primarily to an increase in the polarity of xenobiotics, called Phase I reactions, followed by conjugation reactions (Phase II reactions) that increase their polarity but block the reactivity of polar groups introduced in Phase I reactions. Thereafter the transmembrane transport of the resulting metabolites is performed by membrane transporter proteins, essentially ABC transporters (Phase III reactions).
Although extensive studies have been performed on transcriptional regulation of the DMEs, there is a lack of understanding of their posttranscriptional regulation [29]. Recent studies have shown that miRs also control the expression of some DME [30–32]. However few studies have shown a direct involvement of miRs and DME with drug resistance. One of the key players of the Phase I (modification) are cytochrome P450 (CYP) enzymes that catalyze oxidation reactions of the xenobiotics and occasionally reduction reactions [33]. More than 90 % of the reactions involved in the metabolism of all chemicals, whether general chemicals, natural, physiological compounds, and drugs, are catalyzed by P450s [34]. Three-fourths of the human CYP reactions can be accounted for by a set of five CYPs: 1A2, 2C9, 2C19, 2D6, and 3A4, with the largest fraction of the CYP reactions being catalyzed by CYP 3A enzymes. The importance of CYP 3A4 in metabolic reactions of drugs varies from 13 % for general chemicals to 27 % for drugs [34]. Therefore the regulation of DMEs is crucial to drug efficacy and may be related to drug failure or drug resistance.
Tsuchiya et al. [30] showed a direct association of miR-27b and CYP1B1 in breast cancer . The authors not only validated CYP1B1 as a mirR-27b target in cell lines but also showed that in tissue samples there is an inverse correlation between miR-27b expression and CYP1B1 protein expression. Indeed, the authors showed that miR-27b decreased in expression along the group staining of CYP1B1 by immunohistochemistry , being more expressed in the weak staining group and less expressed in the strong staining group. CYP1B1 is highly expressed in estrogen target tissues, and catalyzes the metabolic activation of various pro-carcinogens and the 4-hydroxylation of 17β-estradiol, and is also abundant in cancerous tissues. However, the authors did not show an association with drug resistance. Nevertheless, since deactivation of 4-hydroxy-tamoxifen, a biotransformation product of tamoxifen that has 100-fold increased affinity to estrogen receptors then tamoxifen itself, occurs via CYP1B1 [35], the increased expression of CYP1B1 in breast cancer cells could augment the resistance to tamoxifen, a widely used drug in breast cancer treatment.
CYP2E1 is the fourth most abundant isoform (approximately 7 % of total P450 protein) after CYP3A4 (30 % of total P450), CYP2C (20 % of total P450), and CYP1A2 (approximately 13 % of total P450). CYP2E1 catalyzes the metabolism of numerous low-molecular-weight xenobiotics, including organic solvents (e.g., ethanol, acetone, carbon tetrachloride, chloroform, vinyl chloride, glycerol, hexane, and toluene), and several procarcinogens, such as N-nitrosodimethylamine and N-nitrosomethylethylamine. Interestingly, the ectopic expression of CYP2E1 induced ROS generation, affected autophagy, and inhibited migration in breast cancer cells, thus potentially being involved in breast cancer metastasis [36]. Mohri et al. identified a possible miR-responsive element (MRE378) in the 3′-UTR of human CYP2E1 mRNA, and luciferase assays using HEK293 cells confirmed that miR-378 functionally recognized this region [37]. The overexpression of miR-378 significantly decreased the CYP2E1 protein level and enzyme activity in cells expressing CYP2E1 including 3′-UTR, but not in the cells expressing CYP2E1 excluding 3′-UTR, indicating that the 3′-UTR plays a role in the miR-378-dependent repression. However, the presence of miR-378 did not facilitate the degradation of the CYP2E1 mRNA. Therefore, according to the authors, the downregulation of CYP2E1 by miR-378 would mainly be due to the translational repression, not mRNA degradation. Additionally the relationship between the expression levels of miR-378, CYP2E1 mRNA and protein as well as enzyme activity was assessed using a panel of 25 human livers. CYP2E1 protein levels were significantly correlated with the enzymatic activities but were inversely correlated with CYP2E1 mRNA levels, while miR-378 levels showed a significant inverse correlation with the CYP2E1 protein levels [37]. In another study, Pan et al. [38] showed that miR-27b interacts with the 3′-UTR of CYP3A4, thus regulating its expression. Moreover an overexpression of miR-27b in PANC1 Human pancreas cancer cells led to a lower sensitivity to cyclophosphamide, indicating that miR-27b can alter CYP3A4-catalyzed drug activation, and consequently impact on drug response and resistance.
Regarding Phase II reactions even fewer studies have linked miR-mediated regulation and drug resistance. One example is the sulfotransferase isoform 1A1 (SULT1A1), a member of the sulfotransferase (SULT) family of phase II detoxification enzymes that catalyze the transfer of the sulfonyl group from 3′-phosphoadenosine 5′-phosphosulfate (PAPS) to nucleophilic groups of a variety of xenobiotic and endogenous compounds, thus increasing their solubility and excretion [39]. SULT1A1 is the most highly expressed SULT in the liver. Several therapeutic agents, including 4-hydroxytamoxifen, are substrates for SULT1A1, and variability in the activity levels of the enzyme can markedly influence the efficacy of these drugs and consequently drug resistance [40]. Interestingly, a common single nucleotide polymorphism (SNP) in the coding region of SULT1A1, several proximal promoter SNPs, and copy number variation (CNV) are associated with altered enzymatic activity, but these variants do not fully account for the observed variation of SULT1A1 activity in human populations. Thus, Yu et al. [41] looked for SNPs in the 3′-UTR region of this gene. In silico analyses predicted that the 973C→T SNP would influence the binding of miR-631 to the SULT1A1 3′-UTR. Accordingly, in vitro luciferase reporter assays and overexpression of miR inhibitors in ZR75-1, MCF7, and MCF10A breast cell lines confirmed that SULT1A1 is a direct target of miR-631 [41].
Finally, Moriya et al. [42] found that miR-133a was a potential regulator of GSTP1. Transfection of miR-133a repressed GSTP1 expression at both mRNA and protein levels in several different cell lines. The functional significance of miR-133a was investigated using head and neck Squamous Cell Carcinoma (SCC), esophageal SCC, and bladder cell lines, and the authors showed that restoration of miR-133a expression inhibited cancer cell proliferation, invasion, and migration, suggesting that miR-133a may function as a tumor suppressor. GSTP1 is a member of the GST enzyme superfamily, and catalyzes the conjugation of electrophiles to glutathione in phase II detoxification reactions, including platinum drugs such as cisplatin and carboplatin [43]. GSTP1 has several critical roles in both normal and neoplastic cells, including phase II xenobiotic metabolism, stress responses, signaling, and apoptosis . Overexpression of GSTP1 has been observed in many types of cancer and in human tumor cell lines either inherently or made resistant to chemotherapy drugs, including cisplatin and various alkylating agents [44]. For example, GSTP1 knockdown selectively influenced cisplatin and carboplatin chemosensitivity; cell cycle progression was unaffected, but cell invasion and migration was significantly reduced [45]. The reduced expression of miR-133a may thus lead to an increased expression of GSTP1, contributing to drug resistance.
In spite of these results, miR-dependent regulation of expression in DMEs does not seem to be the most important mode of regulation as few miR-binding regions are found in the 3′-UTR of DME genes. Furthermore, the miR binding sites described for most of the DMEs are poorly conserved, leading one to speculate that other forms of regulation are more important.
2.2 Drug Transport
Drug transport through cell membranes is a critical step in allowing access of pharmacologic agents to intracellular targets. The involvement of drug transport is probably amongst the most studied mechanisms in cancer drug resistance [46]. Multidrug resistance (MDR) is frequently linked to overexpression of one or more of drug transport proteins present in the cytoplasmic membrane. The ABC transporters have an important cellular role in the efflux and influx of several substrates necessary to the cell and also in the efflux of toxic endogenous molecules and xenobiotics (See chapters by Mitra, Viverios, and Gromicho, in this book). Up to now, 49 different ABC transporters were identified and classified in seven families from ABCA through ABCG [47, 48]. The relevance of miRs in regulating the expression of ABC transporters has been recently reviewed [31, 49].
One of the most well-known ABC transporters is ABCB1 , also known as MDR1 or P-gp transporter. In chemotherapeutic-resistant cancer cell lines, ABCB1 is often observed to be upregulated. The increased expression of ABCB1 leads to an increased resistance of several chemotherapeutics, such as taxanes (e.g., paclitaxel and docetaxel), epipodophyllotoxins derivates (e.g., etoposide and teniposide), anthracyclines (e.g., doxorubicin), antibiotics (e.g., actinomycin D), vinca alkaloids (e.g., vinblastine and vincristine), and tyrosine kinase inhibitors (e.g., imatinib and erlotinib) [47, 50]. To date, several authors have published data about misexpression of miRs and ABCB1 [51–54]. Kovalchuck and colleagues [51] showed that the ABCB1 gene is highly expressed in the MCF-7/DOX breast tumor cell lines resistant to doxorubicin when compared with wild type MCF-7. Conversely, miR-451 expression is undetected, showing a negative correlation between ABCB1 and miR-451 expression. These authors then showed that miR-451 targets the ABCB1 3′-UTR regulatory region which consequently leads to a depletion of the drug transporter and increased sensitivity to doxorubicin. Transfection of miR-451 reestablished the sensitivity of the MCF-7/DOX cells to doxorubicin. Similarly, Chen and colleagues [52] showed the same pattern but with miR-200c. The authors also showed a correlation of miR-200c with poor response to neoadjuvant chemotherapeutics using breast cancer tissues. Low expression of miR-200c leads to poor neoadjuvant therapeutic outcomes. However, they did not follow ABCB1 gene and protein expression in the patients. Although published studies suggest a decreased expression of miR-451 correlated with higher expression of ABCB1 in drug resistant cells [51, 55], in a human ovarian cancer cell line, and its multidrug resistant counterpart, as well as in a human cervix carcinoma cell line and its multidrug resistant variant, expressions of miR-27a and miR-451 were upregulated in multidrug resistant cells as compared with their parental lines, downregulating expression of the ABCB1 gene [56]. These results seem to point that the involvement of specific miRs in drug resistance should be cautiously taken at the moment, since the results could depend on various factors, including the cell lines under study. Bao et al. [53] used a different breast tumor cell line, MDA-MB-231, to show that miR-298 regulates ABCB1 gene expression and increases resistance to doxorubicin. Remarkably, the authors also showed that the miR processing is altered in the resistant cell lines, due to the fact that DICER is weakly expressed and higher levels of miR-298 precursor was detected instead of mature form. Other authors also demonstrated a regulation of ABCB1 by miR-145 [57] in intestinal epithelial cells, and mir-381 and miR-495 in leukemia K562 cells resistant to adriamycin (K562/ADM cells) [58]. In this last study, functional analysis indicated that restoring expression of miR-381 or miR-495 in K562/ADM cells was correlated with reduced expression of the ABCB1 gene and its protein product and increased drug uptake by the cells [58].
ABCG2 is another ABC transporter that, in normal tissues, functions as a defense mechanism against toxins and xenobiotics, with expression in the gut, bile canaliculi, placenta, blood–testis and blood–brain barriers. ABCG2 recognizes and transports a variety of chemotherapeutic drugs out of cancer cells, thereby resulting in reduced drug concentration, and subsequent drug resistance. Consequently ABCG2 plays a critical role in the development of MDR in breast cancer [59]. Increased ABCG2 expression has been found in breast cancer cells that exhibit resistance to mitoxantrone (MX), topotecan, and 7-ethyl-10-hydroxycamptothecin (SN-38) [60]. Upregulation of ABCG2 also confers resistance to tamoxifen in breast cancer cells [61]. In addition, ABCG2 expression correlates with chemotherapeutic response to anthracycline in patients with breast cancer [62]. Jiao et al. [63], performed microarray analysis to determine the differential expression patterns of miRs that target ABCG2 between the MX resistant breast cancer cell line MCF-7/MX and its parental MX sensitive cell line MCF-7. MiR-181a was found to be the most significantly downregulated miRNA in MCF-7/MX cells. Overexpression of miR-181a downregulated ABCG2 expression, and sensitized MX-resistant MCF-7/MX cells to MX. Moreover, in a nude mouse xenograft model, intratumoral injection of miR-181a mimics inhibited ABCG2 expression, and enhanced the antitumor activity of MX. Other authors have shown that ABCG2 is regulated by other miRs, including miR-328 [64] and 487a [65], and can influence MX resistance. miR-519c and miR-328 were also described as ABCG2 regulators and Li et al. [66] showed intracellular accumulation of MX in cells lacking ABCG2 expression. Interestingly, the authors also showed differences in expression of this miRs in stem-like ABCG2+ cells and their ABCG2− counterparts. Thus, further investigation of miR regulation in stem cells may provide new insights into chemoresistance .
Another well-known ABC transporter is ABCC1, also known as MRP1. The main subtracts of ABCC1 are vincristine and etoposide and ABCC1 also confers resistance to anthracyclines (doxorubicin, daunorubicin, epirubicin), mitoxantrone, flutamide, and methotrexate. Curiously, many drugs are only transported in the presence of glutathione [67]. Regarding ABCC1, three reports were published showing a regulation by miRs [68–70]. Pogribny and colleagues [68] revealed that miR-345 and miR-7 increases sensitivity to cisplatin through a negative correlation with ABCC1. For that, the authors used a MCF-7 cell line resistant to cisplatin which expresses high levels of ABCC1 and lower levels of miR-345 and miR-7. Liang et al. [69] showed that miR-326 represses ABCC1 expression and sensitizes VP-16 resistant MCF-7 cells to VP-16 and doxorubicin. Pan et al. [70] reported that miR-1291 targets the 3’UTR of ABCC1 and consequently regulates its expression. This has impact in drug disposition and consequently in drug resistance. Interestingly, miR-1291 was described by these authors as being originated from a small nucleolar RNA, SNORA34.
ABCC2, also known as MRP2, and ABCC1 share a 49 % amino acid identity. As ABCC1, this efflux pump needs the presence of glutathione and can transport methotrexate, cisplatin, irinotecan, paclitaxel, and vincristine. ABCC2 is expressed in some solid tumors from the kidney, colon, breast, lung, ovary, and as well as in cells from patients with acute myelogenous leukemia [71]. Regarding ABCC2, to our knowledge, only one article has been published associating miR misexpression and ABCC2. Xu et al. [72] showed that miR-297 targets the 3′ UTR region of ABCC2 transcripts and consequently downregulates its expression. They also showed an inverse correlation between both molecules in colorectal carcinoma cell lines. Moreover, cell lines resistant to oxaliplatin and vincristine were sensitized when miR-297-mimics were transfected into these cells, in vitro and in vivo.
Intestinal epithelial cells are responsible for the absorption of most cancer drugs, and they express a variety of influx transporters specific for drugs, amino acids, peptides, organic anions, organic cations, and other nutrients. Peptide transporter 1 (PEPT1/SLC15A1), organic cation/carnitine transporter 2 (SLC22A5), organic anion transporting polypeptide 2B1 (SLCO2B1), and monocarboxylate transporter 1 (MCT1/SLC16A1) are expressed at the brush-border membrane, whereas organic cation transporter 1 (SLC22A1) is mainly expressed at the basolateral membrane in the small intestine [31]. Recent studies have indicated that the regional differences in the expression of these transporters are dependent on the differentiation of intestinal epithelial cells [73]. Hence, misexpression of miRs could have a marked impact on absorption of cancer drugs. There are a limited number of reports on the SLC transporters regulated by miRNAs (Table 1). Dalmasso et al. [74] showed for the first time that SLC15A1 is regulated by a miR, namely miR-92b, causing diminished influx activity. Moreover, it suppresses bacterial peptide-induced proinflammatory responses in intestinal epithelial cells by inhibiting SLC15A. Pullen et al. [75] showed that miR-29a, miR-29b, and miR-124 can target SLC16A1, resulting in decreased expression at the protein level. The authors also refer that this regulation mechanism is not the main regulator but complements other transcriptional mechanisms and mutations that alter SLC16A1 expression.
2.3 DNA Repair
DNA damage by endogenous or exogenous agents elicits a powerful cellular response called the DNA Damage Response (DDR), which call up concerted molecular pathways to detect, repair, induce cell cycle arrest to allow repair, or in cases of high numbers of DNA lesions or irreparable damage, apoptosis , or cellular senescence (permanent cell cycle arrest) [76–79]. In the past few years evidence has accumulated that drug resistance is also linked to alterations in these pathways [26, 80–85]. The DDR pathways include DNA tolerance mechanisms by error-prone polymerases, the direct reversal of lesions, essentially de-alkylation of alkylated bases by O6-methyl-guanine-DNA methyltransferase (MGMT), alkylation repair homolog 2 (ALKBH2) and alkylation repair homolog 3 (ALKBH3); nucleotide excision repair (NER); base excision repair (BER); mismatch repair (MMR); and the double strand break repair by homologous recombination (HR) and nonhomologous end joining (NHEJ) [86, 87]. Besides these signaling cascades, the DDR also elicits the induction of several noncoding RNAs, including miRs. A large number of miRs are transcriptionally induced upon DNA damage and the level of induction is variable depending on cell type and the nature and the intensity of DNA damage and time after DNA damage [88–93]. Conversely many miRs target DDR genes, thus controlling feed-back and feed-forward loops to fine-tune the response (for a review see refs. 88, 94, 95). Wouters et al. found that 74 (52 %) mammalian DNA repair and DNA damage checkpoint genes contain conserved microRNA target sites predicted in their 3′-UTR by the algorithms Targetscan, Miranda, or both [95].
One of the first indications that implicated miR-mediated regulation of the DDR was knockdown of the miR biogenesis pathway (Dicer and Ago2), which resulted in increased sensitivity to UV and altered cell cycle after UV damage [90]. Following this study many reports have shown that different DNA damaging agents induce different patterns of miR expression [95]. Thus it is conceivable that alterations in miRs are involved in tumor response to anticancer agents.
A few examples indicate indeed that misexpression of miR is associated with drug responsiveness [96, 97]: members of the let-7 family of miRs are rapidly downregulated upon ionizing radiation in A549 lung cancer cells. Interestingly, the let-7 family of miRs regulates expression of oncogenes, such as RAS, and is specifically downregulated in many cancer subtypes. Low levels of let-7 predict a poor outcome in lung cancer. Overexpression of the let-7 family leads to radiosensitization in vitro of lung cancer cells and in vivo in a Caenorhabditis elegans model of radiation-induced cell death, whereas decreasing their levels causes radioresistance. In C. elegans, this was shown to occur partly through control of the proto-oncogene homologue let-60/RAS and genes in the DNA damage response pathway [96].
In another example, miR-138 was shown to target the ERCC1 gene, involved in NER, and to increase the sensitivity of A549/DDP cells to cisplatin in vitro and augmented apoptosis , suggesting that miR-138 could play an important role in the development of cisplatin resistance [98].
Valeri et al. [99] showed that MMR proteins MSH2 and MSH6 are inhibited by miR-21 overexpression causing a reduction in 5-fluorouracil (5-FU) induced G2/M damage arrest and apoptosis , in vitro. Moreover, xenograft studies demonstrate that miR-21 overexpression reduced the therapeutic efficacy of 5-FU.
REV1, an error-prone Y-family DNA polymerase required for translesion synthesis across interstrand crosslinks, was validated as a target of miR-96. Overexpression of miR-96 promoted cellular hypersensitivity to cisplatin in vitro and in vivo and enhanced sensitivity to the PARP inhibitor AZD2281. This miR also targets RAD51, a recombinase that promotes HR repair of double strand breaks (DSBs) and interstrand DNA crosslink (ICLs) [100]. RAD51 is also targeted by miR-155 in human breast cancer cells and affects the cellular response to ionizing radiation (IR). Due to this interaction, the efficiency of HR repair is reduced and sensitivity to IR augmented in vitro and in vivo. Indeed, overexpression of miR-155 was related with low levels of RAD51 and with better overall survival of patients with triple-negative breast cancers (TNBC) [101]. This emphasizes the possibility of how personalized therapy in TNBC patients could be used, knowing the miR-155 levels.
BRCA1 is an important component of the DDR pathway. BRCA1 encodes a nuclear phosphoprotein and primarily functions to maintain genomic stability via critical roles in DNA repair, cell cycle checkpoint control, transcriptional regulation, apoptosis , and mRNA splicing [102]. Mutations in BRCA1 are associated with an increased risk of developing breast and ovarian cancer. BRCA1 is also a target of miRNA-182 [103], indeed, the authors showed that high expression of this miR in multiple breast tumor cell lines influences BRCA1 levels and sensitivity to PARP1 inhibition. MiRNA-146a and miRNA-146-5p also bind to the same site in the 3′-UTR of BRCA1 and downregulate its expression. In breast tumors, levels of these miRs are inversely correlated with that of the BRCA1 protein and these miRs are overexpressed in triple negative breast cancers , a common type of breast cancer in women with BRCA1 mutations [104].
In another study, although the authors did not show specific targets, miR-296-5p and miR-193a-3p overexpression induced resistance to cisplatin, whereas miR-183 overexpression induced sensitivity. This study was done in breast cancer cells and also showed that miR-296-5p overexpression led to doxorubicin and paclitaxel resistance. These authors also examined whether overexpression of miR-16, miR-21, and miR-382 in Human Small Airway Epithelial progenitor (HSAEpCs) cells could modulate chemotherapy sensitivity. Thus, they found that miR-382 and miR-21 had no effect in resistance, while miR-16 promoted sensitivity to cisplatin and doxorubicin [91].
2.4 Epithelial to Mesenchymal Transition
Metastasis is the ultimate cause of death in most cancer patients. The growth of cancer cells at distant organs of a different tissue requires complex processes of detaching from the original tissue; invasion through the basement membrane; movement in the bloodstream or lymphatic system; and anchorage in other organs. The initial process is called epithelial-to-mesenchymal transition (EMT) and is characterized by a phenotypic change of the tumor cells from cell–cell adhesion and polarity to motility, invasiveness, and some of the features of stem cells. This process not only enable the spread of the tumor cells but also their anchorage in distant organs, since tumor cells that undergo EMT can reverse this characteristic acquiring the epithelial phenotype again, in a process called mesenchymal-to-epithelial transition (MET). In EMT, cells lose the expression of E-cadherin and gain the expression of vimentin, N-cadherin, and fibronectin, markers of mesenchymal phenotype. Presumably, EMT is sustained by transient molecular changes and not by permanent genetic alterations. Indeed, the reversible nature of EMT must be associated with reversible epigenetic mechanisms, which allows stable but reversible modifications that do not directly affect the DNA primary sequence [105–107].
MiRs, as posttranscriptional regulators, are good candidates as EMT regulators and, as with epigenetic mechanisms, do not affect the DNA primary sequence and can press tumor cells to acquire an EMT phenotype in the tumor microenvironment. The most studied case is the miR-200 family that targets at least two transcriptional repressors of E-cadherin, ZEB1, and ZEB2.
It is known that the sensitivity to some cancer drugs like etoposide, taxol, and epidermal growth factor receptor inhibitors is increased with restoration of E-cadherin expression. Chen et al. [108] showed that miR-200c increases drug sensitivity of breast cancer cells to doxorubicin through the E-cadherin-mediated upregulation of PTEN. Similarly, Manavalan et al. [109] showed that an increased expression of miR-200b and miR-200c enhances the sensitivity to growth inhibition by 4-hydroxytamoxifen (4-OHT) and fulvestrant in breast cancer cells. Although it is known that miR-200 family regulates EMT through ZEB1 and E-cadherin, the real mechanism through which the miR-200 family regulates drug resistance is not known, and thus further studies are necessary to understand these phenomena. In order to answer this question, Bai et al. [110] published interesting data about miR-200c and feedback circuits of miR-200c/ZEB1 and miR-200c/ZNF217/TGF-β/ZEB1. The authors showed that these circuits contribute to trastuzumab resistance and metastasis of breast cancers. Interestingly, this feedback circuits might be related with reverse EMT in metastasis formation, since ZEB1 can inhibit miR-200c expression. The authors also showed that low levels of miR-200c activate the TGF-β signaling pathway and consequently trastuzumab resistance in breast cancer cells. Indeed, restoring miR-200c was sufficient to resensitize cells to trastuzumab and reverse the mesenchymal phenotype by inhibiting TGF-β signaling and ZEB1 expression. Similarly, Izumchenko et al. [111] reported that a high MIG6 expression and a suppression of miR-200c expression is a consequence of TGF-β-induced EMT and a signature for resistance to erlotinib.
Kitamura and colleagues [112] also showed, in lung adenocarcinoma, the importance of TGF-β signaling in drug resistance and EMT, namely, they showed that miR-134/miR-487b/miR-655 cluster promotes the EMT through TGF-β signaling and induces resistance to gefitinib by directly targeting MAGI2, whose suppression is encompassed by loss of PTEN stability [112].
Another example is the overexpression of miR-147, which alone induced reversal of EMT and consequently reversal of the native drug resistance of the colon cancer cell line HCT116 to gefitinib. Although the specific mechanism of action of miR-147 is still unknown, the authors found that miR-147 significantly upregulates CDH1 and represses ZEB1, known EMT markers, and inhibited TGF-β1 expression and also repressed Akt phosphorylation, leading to gefitinib sensitivity [113]. Jiang et al. [114] reported that miR-489 is underexpressed in a MCF7 breast cancer cell line resistant to doxorubicin, a cell line that shows mesenchymal phenotype. On the contrary, SMAD3, involved in TGF-β-induced EMT, is overexpressed in the same cell line. Ectopic expression of mir-489 not only reversed mesenchymal features, as well as sensitized the breast cell line to doxorubicin, through inhibition of SMAD3. No matter what miR and the respective target might be deregulated, all these studies show a point in common that is TGF-β signaling. This enhances the importance of TGF-β signaling in EMT and the regulation of EMT influenced drug resistance by miRs. miR-223 was also associated with drug resistance and EMT in pancreatic cancer . miR-223 is upregulated in gemcitabine resistant pancreatic cancer cells, thus acting as an oncogene, most probably, through inhibition of Fbw7 which consequently overexpresses Notch-1. The authors also showed that by inhibiting miR-223, pancreatic cancer cells were sensitized to gemcitabine [115].
2.5 Cancer Stem Cells and Drug Resistance
Somatic stem cells are typically slowly cycling cells capable of self-renewing mitotic divisions in which one or both of the daughter cells are faithful reproductions of the parent stem cell. The experimental observation that certain minority subpopulations of primary human acute myeloid leukemias (AMLs) could propagate the disease in immunodeficient mouse hosts at higher frequencies than the bulk populations of leukemic cells, led to the basis of what was later called the stem cell hypothesis. These cells made up the so-called side population (SP) cells, described as a subset of cells highly expressing ABC transporters and exhibiting cancer stem cell (CSC)-like phenotypes. Initially they were isolated by fluorescence-activated cell sorting (FACS) techniques based on Hoechst 33342 efflux. The SP cells were first isolated from the hematopoietic system but were then identified in normal tissues and several solid tumors.
Although it is accepted that most tumors arise from a single mutated cell, i.e., their origin is monoclonal, the tumor itself is a sum of several types of cells, due to the heterogeneity derived from a continuous evolution of the primitive cancer cell. Not all of these cells will display characteristics of cancer cells, such as metastization or unlimited replication potential. Operationally, (CSC) make up subpopulations of neoplastic cells within a tumor that have an elevated ability to seed new tumors upon experimental implantation in appropriate animal hosts [116]. They share many of the features of normal stem cells, including the capacity for self-renewal and differentiation, although their ability to differentiate into more than a few cell types has not been unequivocally proven, besides leukemias [117]. Although CSCs have been well characterized in hematological malignancies, their existence in other tissues has been much debated (for a review see Ref. [118]). Over the past few years CSC have been identified using stem cell specific markers in several solid tumors including breast, brain, colon, prostate, and pancreatic cancer [119–122]. It is often difficult to strictly define CSCs by associating them with traits beyond their tumor-initiating capability [118, 123]. Moreover, the possible existence of CSCs within tumors is intimately linked to tumor heterogeneity and tumor dedifferentiation. Nevertheless, several miRs have been shown to regulate stemness, or what we consider as properties of tumor-initiating and maintaining cancer cells, of different cancer types.
Recent studies showed differential expression of certain miRs between CSC and their differentiated counterparts [6, 124, 125], suggesting that miRs could also be involved in the regulation of CSC. For example, miR-200c and miR-34 have been shown to regulate CSC properties by targeting Bmi1 and downregulating Bcl2 and Notch, respectively [125, 126]. Additionally, miR-134, miR-296, and miR-470 modulate embryonic stem cell differentiation by suppressing the expression of the stem cell transcription factors Nanog, Oct4, and Sox2 [6]. Therefore miRs may impact on cancer drug resistance and several miRs have been reported to regulate stem cell properties and drug resistance concomitantly [127].
Yu et al. showed that let-7a expression was significantly decreased and Nanog/Oct4 expression was increased in head and neck cancer (HNC) tissues as compared to adjacent normal cells [128]. HNC–ALDH1+ cells displayed a decreased level of let-7a than HNC–ALDH1− cells. The overexpression of let-7a in vitro and in vivo showed that the self-renewal , resistance to cisplatin, and tumor initiation properties were significantly suppressed in let7a-overexpressing HNC–ALDH1+ cells, suggesting that the resistance of HNC–ALDH1+ cells to chemotherapy is partially due to the preferential activation of let-7a miRNA gene expression.
In another study, expression of miR-145, a tumor-suppressive miR, was shown to be inversely correlated with the levels of Oct4 and Sox2 in glioblastoma-CD133+ (GBM-CD133+) cells and malignant glioma specimens [129]. CD133 is a putative CSC marker in glioblastomas. The authors subsequently showed that miR-145 negatively regulates GBM tumorigenesis by targeting Oct4 and Sox2 in GBM-CD133+ cells. miR-145 delivery to GBM-CD133+ cells using polyurethane-short branch polyethylenimine (PU-PEI) significantly inhibited their tumorigenic and CSC-like abilities and facilitated their differentiation into CD133−-non-CSCs. Moreover, PU-PEI-miR145-treated GBM-CD133+ cells suppressed the expression of stemness (Nanog, c-Myc, and Bmi-1), drug-resistance (ABCG2, ABCC5, ABCB1), and anti-apoptotic genes (Bcl-2, Bcl-xL) and increased the sensitivity of the cells to radiation and temozolomide. The in vivo delivery of PU-PEI-miR145 alone significantly suppressed tumorigenesis with stemness, and synergistically improved the survival rate when used with radiotherapy and temozolomide in orthotopic GBM-CD133+-transplanted immunocompromised mice [129].
Some miRs possess the ability to promote the generation of CSC by downregulating tumor suppressors. In hepatocellular carcinoma, miR-130b was shown to be associated with CSC growth that leads to worse overall survival and more frequent recurrence of cancer in patients. The increased miR-130b occurs in parallel with the reduction of tumor protein 53-induced nuclear protein 1, a known miR-130b target. Moreover, cells transfected with miR-130b presented a higher resistance to doxorubicin [130].
Similarly, other studies have revealed a regulation of stem cell properties through stem cell factors, including the p53–Nanog axis. For example, Xu et al. [131] showed that miR-214 regulates ovarian cancer cell stemness and chemoresistance towards cisplatin and doxorubicin treatment by targeting p53–Nanog, and expression of p53 abrogated miR-214-induced ovarian CSC properties.
Bitarte et al. [132] prepared colonospheres with CSCs properties from different colon carcinoma cells, and after performing miR profiling observed that miR-451 was downregulated in colonospheres versus parental cells. Expression of miR-451 caused a decrease in self-renewal , tumorigenicity, and chemoresistance to irinotecan, through a downregulation of the ABCB1 transporter.
Bourguignon et al. [133] observed that human head and neck squamous cell carcinoma (HNSCC) derived HSC-3 cells contain a subpopulation of (CSCs) characterized by high levels of CD44v3 and aldehyde dehydrogenase-1 (ALDH1) expression. These tumor cells also expressed stem cell markers (Oct4, Sox2, and Nanog) and displayed the hallmark CSC properties of self-renewal/clonal formation and the ability to generate heterogeneous cell populations. Hyaluronan (HA) activation of CD44v3 (an HA receptor) lead to nuclear accumulation of oncogenic transcription factors (Nanog, Oct4, Sox2), and CSCs in HNSCC display upregulated miR-302 expression which, in turn, upregulates several survival proteins responsible for clonal formation, self-renewal, and cisplatin resistance. MiR-302 is controlled by an upstream promoter containing Oct4-Sox2-Nanog binding sites, while stimulation of miR-302 expression by HA-CD44 is Oct4-Sox2-Nanog-dependent in HNSCC-specific CSCs. This process results in suppression of several epigenetic regulators (AOF1/AOF2 and DNMT1) and the upregulation of several survival proteins (cIAP-1, cIAP-2, and XIAP) leading to self-renewal, clonal formation, and cisplatin resistance [133].
Several of these studies have used cell lines in vitro that express stem cell markers; however, one must keep in mind that these cell lines have vastly altered karyotypes (e.g., several translocations, insertions, and deletions) that will obviously alter their biological behavior. Therefore, caution must be exercised in interpreting the results described.
References
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854
Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, Spring J, Srinivasan A, Fishman M, Finnerty J, Corbo J, Levine M, Leahy P, Davidson E, Ruvkun G (2000) Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature 408:86–89
Lee RC, Ambros V (2001) An extensive class of small RNAs in Caenorhabditis elegans. Science 294:862–864
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Di Leva G, Garofalo M, Croce CM (2014) MicroRNAs in cancer. Annu Rev Pathol 9:287–314
Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I (2008) MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455:1124–1128
Deng L, Shang L, Bai S, Chen J, He X, Trevino RM, Chen S, Li X, Meng X, Yu B, Wang X, Liu Y, McDermott SP, Ariazi AE, Ginestier C, Ibarra I, Ke J, Luther TK, Clouthier SG, Xu L, Shan G, Song E, Yao H, Hannon GJ, Weiss SJ, Wicha MS, Liu S (2014) MicroRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res 74(22):6648–6660
Kang IH, Jeong BC, Hur SW, Choi H, Choi SH, Ryu JH, Hwang YC, Koh JT (2014) MicroRNA-302a stimulates osteoblastic differentiation by repressing COUP-TFII expression. J Cell Physiol 230:911–921
Lazare SS, Wojtowicz EE, Bystrykh LV, de Haan G (2014) microRNAs in hematopoiesis. Exp Cell Res 329(2):234–238
Janaki Ramaiah M, Lavanya A, Honarpisheh M, Zarea M, Bhadra U, Bhadra MP (2014) miR-15/16 complex targets p70S6 kinase1 and controls cell proliferation in MDA-MB-231 breast cancer cells. Gene 552:255–264
Zhong K, Chen K, Han L, Li B (2014) microRNA-30b/c inhibits non-small cell lung cancer cell proliferation by targeting Rab18. BMC Cancer 14:703
Lerner M, Lundgren J, Akhoondi S, Jahn A, Ng HF, Akbari Moqadam F, Oude Vrielink JA, Agami R, Den Boer ML, Grander D, Sangfelt O (2011) MiRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle 10:2172–2183
Liang LH, He XH (2011) Macro-management of microRNAs in cell cycle progression of tumor cells and its implications in anti-cancer therapy. Acta Pharmacol Sin 32:1311–1320
Zhou L, Zhang WG, Wang DS, Tao KS, Song WJ, Dou KF (2014) MicroRNA-183 is involved in cell proliferation, survival and poor prognosis in pancreatic ductal adenocarcinoma by regulating Bmi-1. Oncol Rep 32:1734–1740
Floyd DH, Zhang Y, Dey BK, Kefas B, Breit H, Marks K, Dutta A, Herold-Mende C, Synowitz M, Glass R, Abounader R, Purow BW (2014) Novel anti-apoptotic microRNAs 582-5p and 363 promote human glioblastoma stem cell survival via direct inhibition of caspase 3, caspase 9, and Bim. PLoS One 9, e96239
Li R, Yuan W, Mei W, Yang K, Chen Z (2014) MicroRNA 520d-3p inhibits gastric cancer cell proliferation, migration, and invasion by downregulating EphA2 expression. Mol Cell Biochem 396:295–305
Li W, Zang W, Liu P, Wang Y, Du Y, Chen X, Deng M, Sun W, Wang L, Zhao G, Zhai B (2014) MicroRNA-124 inhibits cellular proliferation and invasion by targeting Ets-1 in breast cancer. Tumour Biol 35(11):10897–10904
Zhang R, Luo H, Wang S, Chen Z, Hua L, Wang HW, Chen W, Yuan Y, Zhou X, Li D, Shen S, Jiang T, You Y, Liu N, Wang H (2014) miR-622 suppresses proliferation, invasion and migration by directly targeting activating transcription factor 2 in glioma cells. J Neurooncol 121(1):63–72
Melo SA, Esteller M (2011) Dysregulation of microRNAs in cancer: playing with fire. FEBS Lett 585:2087–2099
Calin G, Sevignani C, Dumitru C, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M (2004) Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101:2999–3004
Shenouda SK, Alahari SK (2009) MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev 28:369–378
Raychaudhuri M, Schuster T, Buchner T, Malinowsky K, Bronger H, Schwarz-Boeger U, Hofler H, Avril S (2012) Intratumoral heterogeneity of microRNA expression in breast cancer. J Mol Diagn 14:376–384
Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, Sarkar S (2014) Drug resistance in cancer: an overview. Cancers 6:1769–1792
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG (2013) Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 13:714–726
Longley DB, Johnston PG (2005) Molecular mechanisms of drug resistance. J Pathol 205:275–292
Rodrigues AS, Dinis J, Gromicho M, Martins C, Laires A, Rueff J (2012) Genomics and cancer drug resistance. Curr Pharm Biotechnol 13:651–673
Rukov JL, Shomron N (2011) MicroRNA pharmacogenomics: post-transcriptional regulation of drug response. Trends Mol Med 17:412–423
Manikandan M, Munirajan AK (2014) Single nucleotide polymorphisms in microRNA binding sites of oncogenes: implications in cancer and pharmacogenomics. Omics 18:142–154
Urquhart BL, Tirona RG, Kim RB (2007) Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J Clin Pharmacol 47:566–578
Tsuchiya Y, Nakajima M, Takagi S, Taniya T, Yokoi T (2006) MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res 66:9090–9098
Ikemura K, Iwamoto T, Okuda M (2014) MicroRNAs as regulators of drug transporters, drug-metabolizing enzymes, and tight junctions: implication for intestinal barrier function. Pharmacol Ther 143:217–224
Koturbash I, Beland FA, Pogribny IP (2012) Role of microRNAs in the regulation of drug metabolizing and transporting genes and the response to environmental toxicants. Expert Opin Drug Metab Toxicol 8:597–606
Rodriguez-Antona C, Ingelman-Sundberg M (2006) Cytochrome P450 pharmacogenetics and cancer. Oncogene 25:1679–1691
Rendic SP, Guengerich FP (2015) Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of chemicals. Chem Res Toxicol 28(1):38–42
Crewe HK, Notley LM, Wunsch RM, Lennard MS, Gillam EM (2002) Metabolism of tamoxifen by recombinant human cytochrome P450 enzymes: formation of the 4-hydroxy, 4′-hydroxy and N-desmethyl metabolites and isomerization of trans-4-hydroxytamoxifen. Drug Metab Dispos 30:869–874
Leung T, Rajendran R, Singh S, Garva R, Krstic-Demonacos M, Demonacos C (2013) Cytochrome P450 2E1 (CYP2E1) regulates the response to oxidative stress and migration of breast cancer cells. Breast Cancer Res 15:R107
Mohri T, Nakajima M, Fukami T, Takamiya M, Aoki Y, Yokoi T (2010) Human CYP2E1 is regulated by miR-378. Biochem Pharmacol 79:1045–1052
Pan YZ, Gao W, Yu AM (2009) MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab Dispos 37:2112–2117
Duffel MW, Marshal AD, McPhie P, Sharma V, Jakoby WB (2001) Enzymatic aspects of the phenol (aryl) sulfotransferases. Drug Metab Rev 33:369–395
Mercer KE, Apostolov EO, da Costa GG, Yu X, Lang P, Roberts DW, Davis W, Basnakian AG, Kadlubar FF, Kadlubar SA (2010) Expression of sulfotransferase isoform 1A1 (SULT1A1) in breast cancer cells significantly increases 4-hydroxytamoxifen-induced apoptosis. Int J Mol Epidemiol Genet 1:92–103
Yu X, Dhakal IB, Beggs M, Edavana VK, Williams S, Zhang X, Mercer K, Ning B, Lang NP, Kadlubar FF, Kadlubar S (2010) Functional genetic variants in the 3′-untranslated region of sulfotransferase isoform 1A1 (SULT1A1) and their effect on enzymatic activity. Toxicol Sci 118:391–403
Moriya Y, Nohata N, Kinoshita T, Mutallip M, Okamoto T, Yoshida S, Suzuki M, Yoshino I, Seki N (2012) Tumor suppressive microRNA-133a regulates novel molecular networks in lung squamous cell carcinoma. J Hum Genet 57:38–45
McLellan LI, Wolf CR (1999) Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resist Updat 2:153–164
Shea TC, Kelley SL, Henner WD (1988) Identification of an anionic form of glutathione transferase present in many human tumors and human tumor cell lines. Cancer Res 48:527–533
Sawers L, Ferguson MJ, Ihrig BR, Young HC, Chakravarty P, Wolf CR, Smith G (2014) Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br J Cancer 111:1150–1158
Gottesman MM, Fojo T, Bates SE (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58
Kathawala RJ, Gupta P, Ashby CR Jr, Chen Z (2014) The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat 18:1–17
Dean M, Hamon Y, Chimini G (2001) The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res 42:1007–1017
Haenisch S, Werk AN, Cascorbi I (2014) MicroRNAs and their relevance to ABC transporters. Br J Clin Pharmacol 77:587–596
Gromicho M, Dinis J, Magalhães M, Fernandes A, Tavares P, Laires A, Rueff J, Rodrigues A (2011) Development of Imatinib and Dasatinib resistance: dynamics of the drug transporters expression ABCB1, ABCC1, ABCG2, MVP and SLC22A1. Leuk Lymphoma 52:1980–1990
Kovalchuk O, Filkowski J, Meservy J, Ilnytskyy Y, Tryndyak VP, Chekhun VF, Pogribny IP (2008) Involvement of microRNA-451 in resistance of the MCF-7 breast cancer cells to chemotherapeutic drug doxorubicin. Mol Cancer Ther 7:2152–2159
Chen J, Tian W, Cai H, He H, Deng Y (2012) Down-regulation of microRNA-200c is associated with drug resistance in human breast cancer. Med Oncol 29:2527–2534
Bao L, Hazari S, Mehra S, Kaushal D, Moroz K, Dash S (2012) Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am J Pathol 180:2490–2503
Gromicho M, Magalhaes M, Torres F, Dinis J, Fernandes AR, Rendeiro P, Tavares P, Laires A, Rueff J, Sebastiao Rodrigues A (2013) Instability of mRNA expression signatures of drug transporters in chronic myeloid leukemia patients resistant to imatinib. Oncol Rep 29:741–750
van Jaarsveld MT, Helleman J, Berns EM, Wiemer EA (2010) MicroRNAs in ovarian cancer biology and therapy resistance. Int J Biochem Cell Biol 42:1282–1290
Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu CG, Yang JM (2008) Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem Pharmacol 76:582–588
Ikemura K, Yamamoto M, Miyazaki S, Mizutani H, Iwamoto T, Okuda M (2013) MicroRNA-145 post-transcriptionally regulates the expression and function of P-glycoprotein in intestinal epithelial cells. Mol Pharmacol 83:399–405
Xu Y, Ohms SJ, Li Z, Wang Q, Gong G, Hu Y, Mao Z, Shannon MF, Fan JY (2013) Changes in the expression of miR-381 and miR-495 are inversely associated with the expression of the MDR1 gene and development of multi-drug resistance. PLoS One 8, e82062
Natarajan K, Xie Y, Baer MR, Ross DD (2012) Role of breast cancer resistance protein (BCRP/ABCG2) in cancer drug resistance. Biochem Pharmacol 83:1084–1103
Shiozawa K, Oka M, Soda H, Yoshikawa M, Ikegami Y, Tsurutani J, Nakatomi K, Nakamura Y, Doi S, Kitazaki T, Mizuta Y, Murase K, Yoshida H, Ross DD, Kohno S (2004) Reversal of breast cancer resistance protein (BCRP/ABCG2)-mediated drug resistance by novobiocin, a coumermycin antibiotic. Int J Cancer 108:146–151
Selever J, Gu G, Lewis MT, Beyer A, Herynk MH, Covington KR, Tsimelzon A, Dontu G, Provost P, Di Pietro A, Boumendjel A, Albain K, Miele L, Weiss H, Barone I, Ando S, Fuqua SA (2011) Dicer-mediated upregulation of BCRP confers tamoxifen resistance in human breast cancer cells. Clin Cancer Res 17:6510–6521
Burger H, Foekens JA, Look MP, Meijer-van Gelder ME, Klijn JG, Wiemer EA, Stoter G, Nooter K (2003) RNA expression of breast cancer resistance protein, lung resistance-related protein, multidrug resistance-associated proteins 1 and 2, and multidrug resistance gene 1 in breast cancer: correlation with chemotherapeutic response. Clin Cancer Res 9:827–836
Jiao X, Zhao L, Ma M, Bai X, He M, Yan Y, Wang Y, Chen Q, Zhao X, Zhou M, Cui Z, Zheng Z, Wang E, Wei M (2013) MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2). Breast Cancer Res Treat 139:717–730
Pan YZ, Morris ME, Yu AM (2009) MicroRNA-328 negatively regulates the expression of breast cancer resistance protein (BCRP/ABCG2) in human cancer cells. Mol Pharmacol 75:1374–1379
Ma MT, He M, Wang Y, Jiao XY, Zhao L, Bai XF, Yu ZJ, Wu HZ, Sun ML, Song ZG, Wei MJ (2013) MiR-487a resensitizes mitoxantrone (MX)-resistant breast cancer cells (MCF-7/MX) to MX by targeting breast cancer resistance protein (BCRP/ABCG2). Cancer Lett 339:107–115
Li X, Pan YZ, Seigel GM, Hu ZH, Huang M, Yu AM (2011) Breast cancer resistance protein BCRP/ABCG2 regulatory microRNAs (hsa-miR-328, -519c and -520h) and their differential expression in stem-like ABCG2+ cancer cells. Biochem Pharmacol 81:783–792
Cole SP (2014) Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 54:95–117
Pogribny IP, Filkowski JN, Tryndyak VP, Golubov A, Shpyleva SI, Kovalchuk O (2010) Alterations of microRNAs and their targets are associated with acquired resistance of MCF-7 breast cancer cells to cisplatin. Int J Cancer 127:1785–1794
Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang K, Wagar N, Yoon Y, Cho HT, Scala S, Shim H (2010) Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem Pharmacol 79:817–824
Pan YZ, Zhou A, Hu Z, Yu AM (2013) Small nucleolar RNA-derived microRNA hsa-miR-1291 modulates cellular drug disposition through direct targeting of ABC transporter ABCC1. Drug Metab Dispos 41:1744–1751
Chen ZS, Tiwari AK (2011) Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J 278:3226–3245
Xu K, Liang X, Shen K, Cui D, Zheng Y, Xu J, Fan Z, Qiu Y, Li Q, Ni L, Liu J (2012) miR-297 modulates multidrug resistance in human colorectal carcinoma by down-regulating MRP-2. Biochem J 446:291–300
McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR, Kaestner KH (2010) MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology 139:1654–1664, 1664–1651
Dalmasso G, Nguyen HT, Yan Y, Laroui H, Charania MA, Obertone TS, Sitaraman SV, Merlin D (2011) MicroRNA-92b regulates expression of the oligopeptide transporter PepT1 in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 300:G52–G59
Pullen TJ, da Silva Xavier G, Kelsey G, Rutter GA (2011) miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol Cell Biol 31:3182–3194
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
Harper JW, Elledge SJ (2007) The DNA damage response: ten years after. Mol Cell 28:739–745
Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FMG (2015) Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 15:166–180
d'Adda di Fagagna F (2008) Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 8:512–522
Kelley MR, Fishel ML (2008) DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem 8:417–425
Kelley MR (2011) DNA repair inhibitors: where do we go from here? DNA Repair (Amst) 10:1183–1185
Kelley MR (2012) Future directions with DNA repair inhibitors: a roadmap for disruptive approaches to cancer therapy (Chapter 14). In: Mark RK (ed) DNA repair in cancer therapy. Academic, San Diego, CA, pp 301–310. doi:10.1016/b978-0-12-384999-1.10014-9
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8:193–204
Dinis J, Silva V, Gromicho M, Martins C, Laires A, Tavares P, Rendeiro P, Torres F, Rueff J, Rodrigues A (2012) DNA damage response in imatinib resistant chronic myeloid leukemia K562 cells. Leuk Lymphoma 53:2004–2014
Rodrigues AS, Gomes BC, Martins C, Gromicho M, Oliveira NG, Guerreiro PS, Rueff J (2013) DNA repair and resistance to cancer therapy. In: Chen C (ed) DNA repair and resistance to cancer therapy, new research directions in DNA repair. Intech. doi:10.5772/53952
Hoeijmakers J (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374
Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361:1475–1485
Sharma V, Misteli T (2013) Non-coding RNAs in DNA damage and repair. FEBS Lett 587:1832–1839
Templin T, Paul S, Amundson SA, Young EF, Barker CA, Wolden SL, Smilenov LB (2011) Radiation-induced micro-RNA expression changes in peripheral blood cells of radiotherapy patients. Int J Radiat Oncol Biol Phys 80:549–557
Pothof J, Verkaik NS, Van IW, Ta VT, van der Horst GT, Jaspers NG, van Gent DC, Hoeijmakers JH, Persengiev SP (2009) MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J 28:2090–2099
van Jaarsveld MT, Wouters MD, Boersma AW, Smid M, van Ijcken WF, Mathijssen RH, Hoeijmakers JH, Martens JW, van Laere S, Wiemer EA, Pothof J (2014) DNA damage responsive microRNAs misexpressed in human cancer modulate therapy sensitivity. Mol Oncol 8:458–468
d'Adda di Fagagna F (2014) A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol 24:171–178
Chowdhury D, Choi YE, Brault ME (2013) Charity begins at home: non-coding RNA functions in DNA repair. Nat Rev Mol Cell Biol 14:181–189
Bottai G, Pasculli B, Calin GA, Santarpia L (2014) Targeting the microRNA-regulating DNA damage/repair pathways in cancer. Expert Opin Biol Ther 14:1667–1683
Wouters MD, van Gent DC, Hoeijmakers JHJ, Pothof J (2011) MicroRNAs, the DNA damage response and cancer. Mutat Res 717:54–66
Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, Gillespie E, Slack FJ (2007) MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res 67:11111–11116
Blower PE, Chung JH, Verducci JS, Lin S, Park JK, Dai Z, Liu CG, Schmittgen TD, Reinhold WC, Croce CM, Weinstein JN, Sadee W (2008) MicroRNAs modulate the chemosensitivity of tumor cells. Mol Cancer Ther 7:1–9
Wang Q, Zhong M, Liu W, Li J, Huang J, Zheng L (2011) Alterations of microRNAs in cisplatin-resistant human non-small cell lung cancer cells (A549/DDP). Exp Lung Res 37:427–434
Valeri N, Gasparini P, Braconi C, Paone A, Lovat F, Fabbri M, Sumani KM, Alder H, Amadori D, Patel T, Nuovo GJ, Fishel R, Croce CM (2010) MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc Natl Acad Sci 107:21098–21103
Wang Y, Huang JW, Calses P, Kemp CJ, Taniguchi T (2012) MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res 72:4037–4046
Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, Carasi S, Palmieri D, Costinean S, Shapiro CL, Huebner K, Croce CM (2014) Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci U S A 111:4536–4541
Savage KI, Harkin DP (2015) BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J 282:630–646
Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, Gorospe M, Harris AL, Helleday T, Chowdhury D (2011) miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell 41:210–220
Garcia AI, Buisson M, Bertrand P, Rimokh R, Rouleau E, Lopez BS, Lidereau R, Mikaelian I, Mazoyer S (2011) Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol Med 3:279–290
Li L, Li W (2015) Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Ther 150:33–46
Zielinska HA, Bahl A, Holly JM, Perks CM (2015) Epithelial-to-mesenchymal transition in breast cancer: a role for insulin-like growth factor I and insulin-like growth factor-binding protein 3? Breast Cancer (Dove Med Press) 7:9–19
Lindsey S, Langhans SA (2014) Crosstalk of oncogenic signaling pathways during epithelial-mesenchymal transition. Front Oncol 4:358
Chen Y, Sun Y, Chen L, Xu X, Zhang X, Wang B, Min L, Liu W (2013) miRNA-200c increases the sensitivity of breast cancer cells to doxorubicin through the suppression of E-cadherin-mediated PTEN/Akt signaling. Mol Med Rep 7:1579–1584
Manavalan TT, Teng Y, Litchfield LM, Muluhngwi P, Al-Rayyan N, Klinge CM (2013) Reduced expression of miR-200 family members contributes to antiestrogen resistance in LY2 human breast cancer cells. PLoS One 8, e62334
Bai WD, Ye XM, Zhang MY, Zhu HY, Xi WJ, Huang X, Zhao J, Gu B, Zheng GX, Yang AG, Jia LT (2014) MiR-200c suppresses TGF-beta signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int J Cancer 135:1356–1368
Izumchenko E, Chang X, Michailidi C, Kagohara L, Ravi R, Paz K, Brait M, Hoque M, Ling S, Bedi A, Sidransky D (2014) The TGFbeta-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer Res 74:3995–4005
Kitamura K, Seike M, Okano T, Matsuda K, Miyanaga A, Mizutani H, Noro R, Minegishi Y, Kubota K, Gemma A (2014) MiR-134/487b/655 cluster regulates TGF-beta-induced epithelial-mesenchymal transition and drug resistance to gefitinib by targeting MAGI2 in lung adenocarcinoma cells. Mol Cancer Ther 13:444–453
Lee CG, McCarthy S, Gruidl M, Timme C, Yeatman TJ (2014) MicroRNA-147 induces a mesenchymal-to-epithelial transition (MET) and reverses EGFR inhibitor resistance. PLoS One 9, e84597
Jiang L, He D, Yang D, Chen Z, Pan Q, Mao A, Cai Y, Li X, Xing H, Shi M, Chen Y, Bruce IC, Wang T, Jin L, Qi X, Hua D, Jin J, Ma X (2014) MiR-489 regulates chemoresistance in breast cancer via epithelial mesenchymal transition pathway. FEBS Lett 588:2009–2015
Ma J, Fang B, Zeng F, Ma C, Pang H, Cheng L, Shi Y, Wang H, Yin B, Xia J, Wang Z (2015) Down-regulation of miR-223 reverses epithelial-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Oncotarget 6:1740–1749
Nguyen LV, Vanner R, Dirks P, Eaves CJ (2012) Cancer stem cells: an evolving concept. Nat Rev Cancer 12:133–143
Wiseman DH, Greystoke BF, Somervaille TC (2014) The variety of leukemic stem cells in myeloid malignancy. Oncogene 33:3091–3098
Pattabiraman DR, Weinberg RA (2014) Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov 13:497–512
Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM (2007) Identification of pancreatic cancer stem cells. Cancer Res 67:1030–1037
O'Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445:106–110
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 100:3983–3988
Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res 63:5821–5828
Antoniou A, Hebrant A, Dom G, Dumont JE, Maenhaut C (2013) Cancer stem cells, a fuzzy evolving concept: a cell population or a cell property? Cell Cycle 12:3743–3748
Liu C, Tang DG (2011) MicroRNA regulation of cancer stem cells. Cancer Res 71:5950–5954
Shimono Y, Zabala M, Cho R, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula S, Chiao E (2009) Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138:592–603
Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, Xiang D, Desano JT, Bommer GT, Fan D, Fearon ER, Lawrence TS, Xu L (2009) MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 4, e6816
Raza U, Zhang JD, Sahin O (2014) MicroRNAs: master regulators of drug resistance, stemness, and metastasis. J Mol Med (Berl) 92:321–336
Yu CC, Chen YW, Chiou GY, Tsai LL, Huang PI, Chang CY, Tseng LM, Chiou SH, Yen SH, Chou MY, Chu PY, Lo WL (2011) MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol 47:202–210
Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, Chang YL, Huang PI, Chen YW, Shih YH, Chen MT, Chiou SH (2012) Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials 33:1462–1476
Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, Ng I, Man K, Wong N, To KF, Zheng BJ, Lai PB, Lo CM, Chan KW, Guan XY (2010) miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 7:694–707
Xu CX, Xu M, Tan L, Yang H, Permuth-Wey J, Kruk PA, Wenham RM, Nicosia SV, Lancaster JM, Sellers TA, Cheng JQ (2012) MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog. J Biol Chem 287:34970–34978
Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, Lopez I, Javier Sola J, Alonso MM, Fortes P, Garcia-Foncillas J (2011) MicroRNA-451 is involved in the self-renewal, tumorigenicity, and chemoresistance of colorectal cancer stem cells. Stem Cells 29:1661–1671
Bourguignon LY, Wong G, Earle C, Chen L (2012) Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem 287:32800–32824
Acknowledgments
This work was supported by grant PEst-OE/SAU/UI0009/2014 from Fundação de Ciência e Tecnologia (FCT). B.C.G. was supported by SFRH/BD/64131/2009 from FCT.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Gomes, B.C., Rueff, J., Rodrigues, A.S. (2016). MicroRNAs and Cancer Drug Resistance. In: Rueff, J., Rodrigues, A. (eds) Cancer Drug Resistance. Methods in Molecular Biology, vol 1395. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3347-1_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3347-1_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3345-7
Online ISBN: 978-1-4939-3347-1
eBook Packages: Springer Protocols