
Abstract
The mechanisms by which tumor cells resist the action of multiple anticancer drugs, often with widely different chemical structures, have been pursued for more than 30 years. The identification of P-glycoprotein (P-gp), a drug efflux transporter protein with affinity for multiple therapeutic drugs, provided an important potential mechanism and further work, which identified other members of ATP-binding cassette (ABC) family that act as drug transporters. Several observations, including results of clinical trials with pharmacological inhibitors of P-gp, have suggested that mechanisms other than efflux transporters should be considered as contributors to resistance, and in this review mechanisms of anticancer drug resistance are considered more broadly. Cells in human tumors exist is a state of continuous turnover, allowing ongoing selection and “survival of the fittest.” Tumor cells die not only as a consequence of drug therapy but also by apoptosis induced by their microenvironment. Cell death can be mediated by host immune mechanisms and by nonimmune cells acting on so-called death receptors. The tumor cell proliferation rate is also important because it controls tumor regeneration. Resistance to therapy might therefore be considered to arise from a reduction of several distinct cell death mechanisms, as well as from an increased ability to regenerate. This review provides a perspective on these mechanisms, together with brief descriptions of some of the methods that can be used to investigate them in a clinical situation.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
The appearance of resistance to cancer therapy is hugely distressing for cancer patients; it may occur at the outset of drug treatment, as is frequently the case with tumors such as glioblastoma and pancreatic cancer , or may develop following initial response to successful first-line therapy. A common clinical experience is that the chance of response to a further drug or drug combination decreases with each relapse . In some cases, the mechanisms of resistance can be identified in molecular terms; for example, resistance to the cytotoxic drug temozolomide may be a consequence of expression of the DNA repair enzyme MGMT [1], and resistance to a targeted drug acting on a mutant BRAF protein may be a consequence of expression of alternative signaling proteins in the RAF pathway [2]. In most clinical cases, the basis of resistance is not clearly defined and tumor progression often appears to be accompanied by resistance to all available drugs. The term “multidrug resistance” (MDR) , which is also applied to multidrug-resistant microbial infections [3], has often been used to describe this situation. An enormous amount of work on cancer resistance is currently being undertaken, with over 1000 new publications each month, and this review can provide only a perspective on the field.
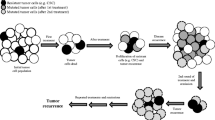
There are many possible reasons for resistance to cancer treatment and, as summarized in Fig. 1, they can be divided into two broad (and partially overlapping) categories. The first, which we have called “intrinsic resistance,” involves a decreased ability of a therapeutic agent to induce cellular damage that is potentially cytostatic or cytotoxic to cancer cells. The second category reflects a dynamic response, i.e., the life and death responses of cancer cells that govern the repopulation of the tumor following therapy. Two hypothetical examples of resistance are illustrated diagrammatically in Fig. 2; here hypothetical tumor populations have potential population-doubling times of either 14 days (Fig. 2a, b) or 7 days (Fig. 2c). For the sensitive tumor population (Fig. 2a), each cycle of treatment (administered weekly in this example) reduces the viable population by 90 %, and the surviving population cannot completely regenerate in the interval between successive therapies. Thus, after five cycles, the surviving population is reduced by 99.9 %. For an intrinsically resistant tumor population (Fig. 2b), each cycle of treatment reduces the population by 30 % rather than 90 %; now, the surviving population can regenerate during the interval between successive cycles of treatment and no lasting therapeutic effect is observed. For a dynamically resistant population (Fig. 2c), the cancer cell population remains intrinsically sensitive to therapy, as in Fig. 2a, but the tumor population can repopulate more effectively because the population-doubling time is 7 days rather than 14 days. Thus, the tumor cell population can again regenerate during the interval between successive treatment cycles and no lasting therapeutic effect is observed (Fig. 2c). These concepts have been discussed previously [4, 5].

Examples of resistance to therapy. (Left-hand side) Tumor cells may be non-responsive to either individual drugs or to groups of drugs because of a lack of expression of appropriate drug targets, or because of expression of cellular transport mechanisms that restrict access of the drug to the target. Tumor cells can also reside in “pharmacological sanctuaries” where diffusion limits access of the drug to the tumor. (Right-hand side) Tumor cells may be damaged by therapy but have a reduced rate of cell death. Alternatively they may be killed but surviving cells regenerate more effectively

Models of multiple drug resistance. (a) Tumor is sensitive to therapy and multiple applications result in the progressive reduction of the tumor population. (b) Tumor is partially resistant to therapy but the tumor population can regenerate in the time between successive therapeutic doses. (c) Tumor is sensitive to therapy but has an increased ability to regenerate between successive therapeutic doses, negating the therapeutic effect. See text for details
Most previous reviews on multidrug resistance have focused on resistance of tumor cells to cytotoxic drugs. With the development of targeted cancer therapies, it is important to examine a broader range of resistance mechanisms and to determine which are most relevant to human cancer. This review commences with a description and discussion of the resistance mechanisms and continues by examining some of the experimental protocols that can be used to study these mechanisms.
2 Resistance Involving Altered Drug Pharmacokinetics
Resistance can also occur because of increased drug clearance, or by decreased diffusion, in both cases leading to a reduced amount of drug entering the cell, although resistance may apply only to one or a small number of anticancer drugs. An early finding was that patients with acute myeloblastic leukemia who failed to respond to the drug cytosine arabinoside had a shorter plasma half-life, apparently because of increased expression of the drug-metabolizing enzyme cytidine deaminase [6]. This led to the consideration of pharmacological factors in the optimization of treatment with this drug [7]. The same principle has been applied to patients treated with the drug cisplatin; individual variations in the pharmacokinetics of this drug have been allowed for in treatment protocols by basing dose on the area under the plasma concentration-time curve (AUC) [8]. Tumor pharmacokinetics of anticancer drugs in solid tumors are highly dependent on both drug diffusion rates and drug diffusion distances; these can be modeled experimentally using a three-dimensional matrix based on solid tumor imaging [9]. Diffusion barriers within tumor tissue, caused for instance by a low vascular density, can give rise to a pharmacokinetic “sanctuary” and to pockets of resistant cancer cells.
3 Intrinsic Multidrug Resistance Mechanisms
3.1 Resistance Mediated by P-gp
An important early step towards our understanding of drug resistance was made with the identification of a single protein, over-expression of which was accompanied by increased resistance to a variety of structurally unrelated anticancer drugs [10–12]. This protein was given the term P-glycoprotein (P-gp; MDR1; gene ABCB1) because chemical analysis showed it to contain multiple oligosaccharides attached to the protein. The development of an antibody to P-gp allowed the distribution of protein, both within single cells and in different organs of the body, to be studied. P-gp was initially found to be associated with the plasma membrane of resistant cultured cancer cells and further structural and biochemical studies led to the formulation of a molecular model where the protein actively transported a variety of drugs and other molecules, typically those containing hydrophobic and basically charged features, out of the cell. The P-gp transporter was embedded within the plasma membrane with the polysaccharide chains on the external cell surface and ATP-binding protein domains on the cytoplasmic side. P-gp had a tandemly duplicated structure with each half containing six potential lipophilic transmembrane domains and one nucleotide-binding site [13]. Molecular structural studies indicated that the protein could adopt two main conformations, a looser “open” form and a tightly twisted “closed” form, with the transition to the tightly twisted form driven by ATP hydrolysis. P-gp, like the protein albumin, has the ability to bind to a variety of small-molecular-weight molecules, which are generally categorized by the presence of lipophilic and/or basically charged motifs. A simple model for the action of P-gp in the membrane suggests that it functions a little like a floor mop; the open form, with its multiple transmembrane regions, can bind to a range of molecules, but a twist in conformation leads to a closed form that lacks these binding sites, allowing the protein to “squeeze out” attached molecules and discharge them from the membrane surface. A molecular model of P-gp, as previously proposed [14], is shown in Fig. 3.

Model for the action of P-gp, reproduced from the original article (Aller et al. [14]), with the permission of the publisher. P-gp protein is embedded in a membrane (either the plasma membrane or an organelle membrane) and has two conformations. In the “open” form it can interact and trap a variety of substrate molecules that enter from membrane sites. In the “closed” form it excludes these substrate molecules and the transition from the open to the closed form requires energy, which is derived by ATP hydrolysis. See text for further details
Subsequent research has indicated that P-gp is not always located on the plasma membrane; in some cell lines it is found in intracellular organelles including lysosomes, the Golgi apparatus, and the nuclear envelope [15, 16]. Here, P-gp acts on substrates to transport them from the cytoplasm to the lumen of an organelle, meaning that resistance is mediated by sequestration of drug into vesicles rather than direct outward transport. Sequestered drug can in turn be released from the cell by exocytosis.
A number of “classical” anticancer drugs, including anthracyclines such as doxorubicin, epirubicin, and daunorubicin; epipodophyllotoxins such as etoposide and teniposide; and taxanes such as paclitaxel and docetaxel, are substrates for P-gp-mediated transport [13]. The development of targeted anticancer therapies such as inhibitors of the epidermal growth factor receptor tyrosine kinase [17] raises the question of whether the efficacy of these drugs is also affected by expression of P-gp. Here the situation is complex because while such drugs can be substrates for P-gp [18] they may also antagonize P-gp function [19]. In one study, brain tissue AUC values for the drug erlotinib were found to increase by 3.8-fold in mice lacking expression of P-gp [20], consistent with a role of P-gp in intrinsic sensitivity.
3.2 Resistance Mediated by Other Transporters
The discovery of P-gp was followed by the identification of a second transporter, designated MRP1 (ABCC1), and its relative MRP2 (ABCC2), which were also associated with the MDR phenotype [21, 22]. As with P-gp, the subcellular distribution of these transporters has been found to extend to cellular organelles as well as the plasma membrane. MRP1 and MRP2 can be distinguished from P-gp in being able to couple drug transport to glutathione transport [22]. The action of these transporters is also coordinated with another type of resistance mechanism whereby a variety of cytotoxic agents, usually lipophilic, are metabolized by conjugation with hydrophilic molecules such as glucuronic acid [23]. Such conjugates not only have reduced activity as a cytotoxic species, but also have increased affinity for the transporter.
Subsequent studies on ABC transporters have identified the breast cancer resistance protein BCRP (ABCG2) as a further transporter associated with drug resistance. BCRP differs from P-gp, MRP1, and MRP2 in having two subunits rather than the tandemly repeated form of the other transporters. A superfamily of proteins, designated as ATP-binding cassette (“ABC”) transporters, has now been identified; it comprises the products of 48 genes and encompasses a broad variety of molecular structures [24]. Subfamily A includes P-gp, subfamily B includes MRP1 and MRP2, and subfamily G includes BCRP. Members of the ABC transporter family carry out a wide variety of functions, many essential, in normal tissue [22, 24] and only a few family members are well characterized in terms of mediating anticancer drug resistance. Molecules binding to MRP1 and MRP2 include a variety of natural products, such as vinblastine, vincristine, paclitaxel, docetaxel, doxorubicin, daunorubicin, epirubicin, and etoposide as well as the synthetic cytotoxic anticancer drugs mitoxantrone and methotrexate [13]. Targeted anticancer drugs can also be substrates for MRP1 and MRP2 [19] and cells transfected with the gene for MRP2 were found to exhibit 6.4-fold resistance to sorafenib but showed no change in susceptibility to the structurally related drug sunitinib [25]. In one study, brain tissue accumulation of erlotinib was found to be reduced in mice over-expressing BCRP [26].
3.3 Pharmacokinetic Consequences of Expression of ABC Transporters
While many studies on ABC transporters have been carried out using cultured cells, it is important to consider transporter action in the context of tumor tissue. Tumors generally have a multicellular organization in which the majority of cells are not adjacent to the vascular endothelium. Drugs must diffuse from the bloodstream to tumor cells either through the extracellular compartment of tumor tissue or by uptake /efflux by cells comprising the tumor tissue. Drugs can cross the plasma membranes of normal and tumor cells within the tumor tissue by either transporter-mediated or passive diffusion. ABC transporters can act on intracellular drug molecules either by exporting them out of the cell again or by sequestering them into vesicles, the contents of which are subsequently released by exocytosis into the extracellular compartment. If exocytosis occurs in a polarized fashion in a direction that is distal to the vascular supply, it will effectively promote distribution of drugs to other cells within the tumor tissue. Thus, depending on their subcellular distribution, ABC transporters can either increase or decrease the distribution of a drug in tumor tissue.
A pharmacological study of SN 28049, a new DNA-binding topoisomerase II poison [27], illustrates how drug transporters might influence tissue pharmacokinetics. Tumor tissue AUC values were evaluated in two murine tumors and three human melanoma xenografts and found to vary by over two orders of magnitude, with the murine colon 38 (MCA38) tumor showing the highest value [28]. Cultured colon 38 tumor cells showed strongly positive staining for MRP1 expression in cytoplasmic bodies [29] and although other explanations are possible, MRP1-mediated sequestration of SN 28049 in cytoplasmic vesicles may contribute to the high AUC value and long tumor tissue half-life in colon 38 tumor tissue. The vesicles could constitute a depot form, slow release which enhances the overall activity of SN 28049 against the colon 38 tumor [29]. Among the murine tumors and human melanoma xenografts tested, antitumor activity was related to the observed tumor tissue pharmacokinetics, consistent with this hypothesis [28].
3.4 Use of Inhibitors of ABC Transporters in Combination Chemotherapy
Since the cellular action of ABC drug transporters involves the ATP-dependent transport of these drugs out of the cytoplasm either to the cell exterior or into subcellular vesicles, the concept of inhibiting the action of P-gp in order to increase the cytoplasmic (and nuclear) concentration of a substrate anticancer drug presented a promising therapeutic strategy. The concept was first suggested more than 30 years ago [30] and initial studies were carried out in rodents using drugs such as verapamil (a Ca2+ channel blocker), cyclosporine (an immunosuppressive agent), tamoxifen (a steroid receptor antagonist), and calmodulin antagonists in conjunction with cytotoxic agents [31]. Most preclinical studies were carried out using cultured cells but some in vivo studies were reported [32]. Clinical trials identified a number of problems including alteration of the pharmacokinetics of administered cytotoxic drugs and consequent increases in toxicity. Consequent changes in dose made the efficacy of the co-administered transporter inhibitor difficult to assess. Subsequent studies aimed at increasing affinity of the inhibitor for the transporter and increasing its dose potency led to so-called second-generation inhibitors such as dexverapamil (an analogue of verapamil), valspodar (an analogue of cyclosporine), and biricodar (a pipecolinate derivative). Further development sought to minimize interaction with cytochrome P450 and to optimize individual transporters, and led to “third-generation” inhibitors such as elacridar, tariquidar, zosuquidar, and laniquidar [5, 33]. Many clinical trials have been carried out with ABC transporter inhibitors but as yet no definitively increased therapeutic benefit has been demonstrated. A small Phase II clinical study in breast cancer patients showed that co-administration of tariquidar showed limited ability to increase response to doxorubicin, paclitaxel, or docetaxel [34]. A larger Phase II clinical study in breast cancer patients treated with docetaxel with or without zosuquidar concluded that there were no differences in progression-free survival, overall survival , or response rate between the two groups of patients [35]. Ongoing difficulties include selection of appropriate tumors and measurement of the effects of the ABC transporter inhibitor on the pharmacokinetics of the cytotoxic drug.
3.5 Multidrug Resistance Mechanisms not Mediated by ABC Transporters
Several additional classes of resistance to multiple anticancer drugs of differing structures have been defined. One involves the modification of the enzyme DNA topoisomerase II, a target protein for cytotoxic action. Cells were identified that lacked ABC transporter expression and yet were resistant to the drugs doxorubicin, etoposide, and amsacrine, which have widely differing structures. The phenomenon was termed “atypical” multidrug resistance and the cause was traced to reduced activity of DNA topoisomerase II and consequent induction of DNA damage , which was essential for the cytotoxic activity of these drugs [36].
A second class of resistance involves the increased expression of a DNA repair enzyme which attenuates the cytotoxic activity of multiple agents. The discovery of the antitumor activity of nitrogen mustard (mechlorethamine) in 1945 led to testing of a large number of clinical anticancer agents whose activity depends mainly on the O6-alkylation of the DNA constituent guanine [37]. These include melphalan, cyclophosphamide, ifosfamide, dacarbazine (DTIC), temozolomide, carmustine (BCNU), lomustine (CCNU), 1-(2-chloroethyl)-3-(4-methylcyclohexyl)-1-nitrosourea (MeCCNU), 1-(4-amino-2-methyl-pyrimidinyl)methyl-3(2-chloroethyl)-3-nitrosourea (ACNU); N-methyl-N-nitrosourea (MNU), N-ethyl-N-nitrosourea (ENU), procarbazine, and streptozotocin. A single enzyme, O 6-alkylguanine-DNA alkyltransferase (AGT), also known as methylguanine transferase (MGMT), acts to repair some of the DNA lesions induced by these drugs. An approach to overcoming this resistance is to co-administer an inhibitor of DNA O6-alkylation; trials of one such inhibitor, O 6-benzylguanine, are currently under way [37, 38].
A third class of resistance involves activation of an alternate signaling pathway for cell proliferation and survival. Most examples are found in the use of targeted therapies; one example is provided in the MAP kinase pathway. Some tumors express a mutant form of the BRAF enzyme, one of the components of the MAP kinase pathway, and since cells become dependent on signaling by this overactive enzyme, their proliferation and survival are compromised by inhibitors of the mutant enzyme [39]. Resistance to multiple agents, including vemurafenib and dabrafenib, can then be mediated by up-regulation of CRAF, which provides an alternative pathway for proliferation and survival [40].
4 Resistance Involving Altered Tumor Cytokinetics
Tumor cells in a solid cancer generally grow in a latticelike “cage” of blood vessels, which contains not only tumor cells but also host cells, typically fibroblasts, tumor-associated macrophages, other cells, and stromal/capsular components [41]. The volume-doubling times of the vascular cages reflect those of the tumor itself, which from imaging studies in human cancers cover a broad range with a median of about 4 months [42]. The vascular cage expands by a number of mechanisms including the generation of new vascular endothelial cells (angiogenesis), the co-option of existing blood vessels of normal tissue by tumor cells, the recruitment of circulating endothelial precursor cells into the vascular, and the phenotypic conversion (vasculogenic mimicry) of tumor cells to a vascular phenotype [43]. Because the potential doubling times of human tumor cells comprising the tumor, typically about 6 days [44], are much shorter than that of the tumor itself, tumor cells within the vascular cage exist in a constant stage of turnover; on average, for every 100 tumor cells dividing, approximately 90 cells are lost [45]. Resistance to multiple agents, or perhaps to all agents, can occur by modulation of the dynamic balance between tumor cell birth and death.
In vivo potential doubling times of human tumor cells vary over quite a wide range [44], raising the question of why the most rapidly growing cells are not selected for during tumor evolution. However, in a solid tumor microenvironment with extensive cell turnover, cell death mechanisms dominate the selection of tumor cells that are least susceptible to cell death mechanisms and that will therefore have a survival advantage. Cell loss from the tumors, even in the absence of therapy, involves a diverse variety of mechanisms as shown in Fig. 4, and there are a corresponding number of control mechanisms. In contrast, increases in the tumor cell population occur almost exclusively by cell division, apart from a small number of tumor cells migrating from other sites (Fig. 4).

Human tumors exhibit a high rate of cell turnover, which drives tumor evolution and also the development of resistance. One main mechanism (cell division) drives cell population increases but several mechanisms drive population decreases. Decreases in any of these mechanisms can therefore contribute to resistance. See text for further details
Apoptosis is likely to be the dominant mechanism for tumor cell loss and tumors can be characterized by their “apoptotic index” [46]. Tumor cells express so-called death receptors, such as Fas, DR4, and DR5 and interaction with the corresponding ligands (TRAIL/Apo2L and FasL), which are also expressed in both tumor cells, leads to apoptosis [47]; cell death may thus due to cell-cell proximity and thus occur as a consequence of crowding. Host immune mechanisms make an important potential contribution to tumor cell loss. Cells may be killed by host T-lymphocytes in a complex mechanism that combines the release of FasL to activate death receptors and the release of cytotoxic granules that are taken up by the target cells [48]. Tumor-associated macrophages and dendritic cells also play important roles in immune cell-mediated tumor cell loss [49]. Tumor cells may die of other programmed cell death mechanisms [50] and may also be lost by migration out of the tumor by coupling with macrophages and export along collagen fibers into the bloodstream [51].
4.1 Resistance Arising from an Increased Rate of Tumor Cell Proliferation
A high tumor cell proliferation rate is important for resistance because it allows more efficient regeneration in the intervals between successive cycles of therapy (Fig. 2). This argument applies to radiotherapy and even surgery in addition to cytotoxic or targeted therapy. Clinically, more rapid proliferation is indicated by a shorter Tpot value, as determined in vivo [44, 52], and there is clinical evidence that higher proliferation rates are associated with shorter survival [53], particularly in the radiotherapy of head and neck tumors [54]. Cytokinetic data can also be obtained using an in vitro approach where surgical cancer samples are cultured for a short time (1 week) and the estimated cell proliferation rates are compared to clinical outcome. In two studies, one of ovarian cancer and one of glioma, decreased patient survival was significantly related to shorter culture cell cycle time [55].
4.2 Resistance to Apoptosis
Resistance to apoptosis has been described as one of the hallmarks of cancer [56] and increased resistance to the induction of apoptosis is an obvious mechanism that could apply to both cytotoxic and targeted anticancer therapeutic agents. Early experiments with the Lewis lung transplantable murine carcinoma (3LL) sought to clarify the role of apoptosis in resistance. Two variants of this tumor are one which had been maintained in vivo and recently grown in culture, and one that had been adapted to culture conditions over a considerable period. When grown to high cell density in culture, the first variant maintained a high proportion of S-phase cells and showed a high rate of cell loss, while the second variant entered a state of reduced proliferation with a low proportion of S-phase cells. These properties were echoed by those shown in vivo when the lines were grown as subcutaneous tumors; the tumors containing the more slowly growing cells were resistant to the cytotoxic drugs tested [57, 58].
There are several clinical examples where a low proliferation rate is associated with resistance to cancer chemotherapy [59–61]. This seems to conflict with reports that a higher proliferation rate is associated with reduced survival [53, 54]. However, it is quite possible that both occur because they operate on different time scales; a lower proliferation rate can be associated with resistance to apoptosis while a higher proliferation rate is associated with increased tumor regeneration. These considerations may help to explain why there is no consistent reported relationship between tumor cell proliferation and sensitivity to therapy.
4.3 Resistance Because of Loss of Host Immunity Mechanisms
As shown in Fig. 4, host immunity contributes to mechanisms of cell loss within tumors. The extent of this contribution in individual tumors is still not clear but if it is a major contribution, then its loss will have major significance for the outcome of cancer therapy. Put another way, loss of tumor immunity can lead to tumor progression. The potential importance of antitumor tumor immunity in a murine system was illustrated by a study of tumor responses to the cytotoxic drug gemcitabine [62]. Here, the response of a series of murine tumors to this drug was found to be not related to the intrinsic sensitivity of tumor-derived cultured cell line to gemcitabine, but rather to biomarkers for the immunogenicity of the tumor. This study suggested not only that host immunity played a major role in tumor regression, but also that gemcitabine itself might trigger host immune responses. Subsequent work has indicated that a number of anticancer agents including gemcitabine, doxorubicin, cisplatin, and cyclophosphamide stimulate host immunity [63, 64], further supporting the hypothesis that immune mechanisms need to be considered in the context of chemotherapy and resistance [65].
More recently, clinical studies have highlighted the role of immune checkpoints in cancer immunology. T-lymphocytes have clearly delineated mechanisms by which they can kill tumor cells, but their potential to kill normal cells in autoimmune reactions must also be carefully regulated. Some of the main mechanisms of regulation involve the so-called immune checkpoints, where cell-surface proteins such as CTLA-4 or PD-1 interact with CD80/86 or PD-L1/PD-L2, respectively, to suppress T-cell responses. The significance of these processes has been highlighted by recent studies with immune checkpoint inhibitors such as ipilimumab and nivolumab, which are engineered antibodies against CTLA-4 and PD-1, respectively, as therapeutic agents aimed at combatting resistance caused by reduced T-cell responses [66].
4.4 Tumor Tissue Heterogeneity and Resistance
Heterogeneity is a hallmark of human tumors [67] and is well illustrated by the analysis of renal cell carcinoma, where tumor subpopulations with distinct gene expression profiles, and consequently different predictions of clinical outcome, are obtained from different biopsies of the same tumor [68]. Tumor heterogeneity can also be discerned in established tumor cell lines, as shown for MCF-7, a typical human breast cancer line. Growth of this line in the absence of estrogen signaling causes an immediate cessation of culture growth, followed several months later by the outgrowth of hormone-resistant cell lines. Surprisingly, the G1-phase DNA content, median cell volume, and proliferation rates of the emerging variant lines were not the same as those of the parental cell line, strongly suggesting that they arose from expansion of pre-existing minor populations, rather than by metabolic adaptation of the parental population [69]. Changes in chromosome numbers, as well as chromosome translocations, fusions, and alterations caused by recombination events, are likely to lead to the continuous generation of genetically distinct variants during culture. These changes have been described specifically for the MCF-7 line [70].
As well as undergoing genetic variation, tumor cells can undergo reversible phenotypic switches that presumably arise from changes in the regulation of gene transcription. Two main categories of phenotypic switch have been described. The first reflects a change in the expression of stem cell characteristics and can be measured, for instance, by an increased ability of the cell population to proliferate indefinitely. The second switch, called the epithelial to mesenchymal transition (EMT), reflects a change towards more mesenchymal behavior and includes increased migratory and invasive potential. Within each category there may be differences in intrinsic cellular drug resistance [71, 72]. An important feature of these phenotypic switches is that they may be associated with multiple changes in resistance properties [71, 73].
5 General Protocols for Studying Resistance to Multiple Anticancer Drugs
The field of drug resistance is very broad and it would be impossible in a limited space to recommend protocols for aspects of resistance. The approach taken here is to review the general approaches that can be used to study resistance. Since the starting point should always be the cancer patient, discussion is directed towards protocols that might be applicable to clinical studies.
5.1 Assessment of Transport-Mediated Multidrug Resistance
The expression of ABC transporter proteins in biopsies of human tumor material can be investigated using standard histological techniques, but it should be kept in mind that staining intensity does not accurately reflect the activity of these proteins. Moreover, the subcellular location of these ABC transporters, as well as regulation by other signaling pathways, will have an effect on activity. The major challenge for the future is to develop robust in vivo methods to assess transport-related multiple drug resistance. In one approach [34], a technetium-labeled P-gp substrate, 99mTc-sestamibi (25–30 mCi per patient), was injected intravenously. Planar scintigraphic images of known tumor sites were taken after 10 min and 2 h to determine the rate of clearance. The same procedure was repeated after administration of the tariquidar, an inhibitor of ABC transporters, in order to determine its effect on 99mTc-sestamibi uptake. The tumor-to-background ratios were calculated for all tumor sites by measurement of sestamibi uptake within the visualized portion of the tumor, and comparing it with that of adjacent tissues that were without tumor involvement [34].
5.2 Assessment of Drug Resistance of Cultured Tumor Cells
Human tumor cell lines have provided the basis for a very large number of published studies on resistance mechanisms. It is important to realize that from the time they are isolated from surgical samples subjected to culture, tumor cells are subjected to severe selective pressures from their new environment, and can change their characteristics accordingly. One of the largest selective pressures is for rapid proliferation rate; the initial doubling times of surgical samples cultured from solid tumors have been measured over the first week of culture [55, 74] and cover a broad range (3 days to more than 2 months), which is similar to that of measurements of potential doubling times in vivo [44]. Development of cell lines from these surgical samples has been reported to be accompanied by a two- to threefold decrease in doubling times [45] consistent with selection of more rapidly growing variants. Another selective pressure is the presence in cultures of atmospheric oxygen concentrations; this leads to increased concentrations of reactive oxygen and consequent toxicity, but can be counteracted by the use of low-oxygen incubators in the derivation and maintenance of cell lines [75].
Not all surgical samples grow well in culture. Early studies showed that partially disaggregated samples of human metastatic melanoma grew with a high success rate in 96-well culture plates that had been coated with a thin layer of agarose to prevent proliferation of fibroblasts; the culture medium was supplemented with fetal bovine serum, insulin, transferrin, and selenite and cells were grown under 5 % oxygen [75]. Subsequent work showed that glioma cells and a range of carcinoma cells could be grown with moderate-to-good success rates using the culture medium, sometimes supplemented with growth factors. Tumor cell proliferation was assessed by uptake of 3H-labeled thymidine into DNA of proliferating cells. This technique has the advantage that these cultures have a variable number of host cells, which can distort the evaluation of drug effects on the total cell population [74].
A further approach, which has also used 96-well culture plate technology and has been reported to have higher success rates with carcinoma samples [76], is to utilize cultured fibroblasts as feeder cells [77]. In this case proliferation was assessed by counting cell density. Like the technique described in the previous paragraph, this method can be used to screen for activity of both conventional cytotoxic agents and targeted therapeutics.
5.3 Transplanted Tumors in Animals
As with culture systems, the majority of reported studies have growth-established cell lines, sometimes drug-resistant cell lines as xenografts in immunodeficient mice in order to gain an understanding of in vivo resistance. However, some early studies have utilized samples of surgically removed tumor material to establish xenografts [78] and more recent work has extended this to a number of genetically characterized tumor types. Samples representing 18 distinct cancer pathologies were implanted within 24 h of surgical resection and implanted into immune-compromised nude mice with an overall take rate of 27 %. Tumors were found to retain their differentiation patterns and supporting stromal elements were preserved. Genes downregulated specifically in the tumor xenografts were enriched for pathways involved in host immune response, consistent with the immune deficiency status of the host [79].
One of the problems of this approach, as it is with cell lines, is that there is competition for survival among the tumor cells and that the most rapidly growing cells are likely to dominate. Furthermore, since first-generation xenografts will generally have to be transplanted into further mice to provide a sufficient number of tumors for measurement of resistance to multiple drugs, further selection for a proliferation rate will be made. Because these experiments are carried out in immunosuppressed mice, possible contributions of immune cell-mediated killing cannot be assessed.
5.4 Contribution of Host Immune Mechanisms in Individual Human Tumors
There is great current interest in the clinical evaluation of immune checkpoint inhibitors and most current studies are using survival or other clinical parameters as the main index for patient comparison [80]. However, there is a need for robust assays of the contribution of T-lymphocytes or of other immune mechanisms to clinical outcome. Clinical studies are still at an early stage, but the formulation of suitable assays could lead to their use to assess immune cell activity in tumor tissue before and after therapy, providing an approach to estimate the contribution of immune effects to response and thus to resistance.
6 Conclusions
The last 30 years has seen progressive change in our appreciation of the diversity of mechanisms of drug resistance , particularly of multiple drug resistance, in human cancer. However we still do not know, for any individual patient, whether a lack of observed response to therapy is due to drug-specific resistance mechanisms, to selection of tumor cells that are resistant to the induction of apoptosis by both conventional and targeted therapies, or to a generalized breakdown of tumor immunity; perhaps all three mechanisms contribute. What we do know is that tumor heterogeneity and growth kinetics are of great importance in the transitions towards resistance. Heterogeneity will be generated in tumor populations by chromosomal instability, errors in chromosome partitioning, and other factors, and individual cells are likely to vary in cell division rate, degree of resistance to apoptosis, and susceptibility to immune responses. Since potential tumor population doubling times can be as short as 3.2 days [44], a minor population (5 %) can under conditions appropriate for selective survival become a major population (80 %) in as little as 13 days. It is easy to underestimate the potential of tumor cells that are resistant to chemotherapy or to specific immune responses to be selected on this basis. There are many reports showing that the presence of an oncogenic mutations leads to resistance to apoptosis, for instance that for c-kit mutation in leukemia [81]. Because of tumor cell turnover, such resistant cells are eventually likely to dominate because of natural selection. The way in which tumor heterogeneity can so rapidly lead to resistance perhaps paints a rather bleak picture, but it should be remembered that the emergence of a particular phenotype by such selection can also lead to an opportunity for selective chemotherapy. A major challenge for the future is to develop methods to identify such phenotypes in the course of clinical treatment, so that individualized treatment can be based on appropriate biomarkers.
References
Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A, Plass C, Hegi M, Platten M, Reifenberger G (2014) MGMT testing--the challenges for biomarker-based glioma treatment. Nat Rev Neurol 10:372–385
Spagnolo F, Ghiorzo P, Queirolo P (2014) Overcoming resistance to BRAF inhibition in BRAF-mutated metastatic melanoma. Oncotarget 5:10206–10221
Hughes D (2014) Selection and evolution of resistance to antimicrobial drugs. IUBMB Life 66:521–529
Durand RE (1993) Cell kinetics and repopulation during multifraction irradiation of spheroids: implications for clinical radiotherapy. Semin Radiat Oncol 3:105–114
Baguley BC (2010) Multidrug resistance in cancer. Methods Mol Biol 596:1–14
Baguley BC, Falkenhaug EM (1971) Plasma half-life of cytosine arabinoside (NSC-63878) in patients treated for acute myeloblastic leukemia. Cancer Chemother Rep 1(55):291–298
Momparler RL (2013) Optimization of cytarabine (ARA-C) therapy for acute myeloid leukemia. Exp Hematol Oncol 2:20
Newell DR, Eeles RA, Gumbrell LA, Boxall FE, Horwich A, Calvert AH (1989) Carboplatin and etoposide pharmacokinetics in patients with testicular teratoma. Cancer Chemother Pharmacol 23:367–372
Hicks KO, Pruijn FB, Secomb TW, Hay MP, Hsu R, Brown JM, Denny WA, Dewhirst MW, Wilson WR (2006) Use of three-dimensional tissue cultures to model extravascular transport and predict in vivo activity of hypoxia-targeted anticancer drugs. J Natl Cancer Inst 98:1118–1128
Juliano RL, Ling V (1976) A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 455:152–162
Ling V (1997) Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol 40(Suppl):S3–S8
Gottesman MM, Ling V (2006) The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 580:998–1009
Endicott JA, Ling V (1989) The biochemistry of P-glycoprotein-mediated multidrug resistance. Annu Rev Biochem 58:137–171
Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323:1718–1722
Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR (2013) P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem 288:31761–31771
Molinari A, Calcabrini A, Meschini S, Stringaro A, Crateri P, Toccacieli L, Marra M, Colone M, Cianfriglia M, Arancia G (2002) Subcellular detection and localization of the drug transporter P-glycoprotein in cultured tumor cells. Curr Protein Pept Sci 3:653–670
Roskoski R Jr (2014) The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 79:34–74
Agarwal S, Sane R, Gallardo JL, Ohlfest JR, Elmquist WF (2010) Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J Pharmacol Exp Ther 334:147–155
Lainey E, Sebert M, Thepot S, Scoazec M, Bouteloup C, Leroy C, De Botton S, Galluzzi L, Fenaux P, Kroemer G (2012) Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle 11:4079–4092
de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JH, van Tellingen O (2012) Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drugs 30:443–449
Cole SP, Chanda ER, Dicke FP, Gerlach JH, Mirski SE (1991) Non-P-glycoprotein-mediated multidrug resistance in a small cell lung cancer cell line: evidence for decreased susceptibility to drug-induced DNA damage and reduced levels of topoisomerase II. Cancer Res 51:3345–3352
Cole SP (2014) Targeting multidrug resistance protein 1 (MRP1, ABCC1): past, present, and future. Annu Rev Pharmacol Toxicol 54:95–117
Tukey RH, Strassburg CP (2000) Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol 40:581–616
ter Beek J, Guskov A, Slotboom DJ (2014) Structural diversity of ABC transporters. J Gen Physiol 143:419–435
Shibayama Y, Nakano K, Maeda H, Taguchi M, Ikeda R, Sugawara M, Iseki K, Takeda Y, Yamada K (2011) Multidrug resistance protein 2 implicates anticancer drug-resistance to sorafenib. Biol Pharm Bull 34:433–435
Elmeliegy MA, Carcaboso AM, Tagen M, Bai F, Stewart CF (2011) Role of ATP-binding cassette and solute carrier transporters in erlotinib CNS penetration and intracellular accumulation. Clin Cancer Res 17:89–99
Deady LW, Rodemann T, Zhuang L, Baguley BC, Denny WA (2003) Synthesis and cytotoxic activity of carboxamide derivatives of benzo[b][1,6]naphthyridines. J Med Chem 46:1049–1054
Lukka PB, Chen YY, Finlay GJ, Joseph WR, Richardson E, Paxton JW, Baguley BC (2013) Tumour tissue selectivity in the uptake and retention of SN 28049, a new topoisomerase II-directed anticancer agent. Cancer Chemother Pharmacol 72:1013–1022
Chen YY, Lukka PB, Joseph WR, Finlay GJ, Paxton JW, McKeage MJ, Baguley BC (2014) Selective cellular uptake and retention of SN 28049, a new DNA-binding topoisomerase II-directed antitumor agent. Cancer Chemother Pharmacol 74:25–35
Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y (1981) Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 41:1967–1972
Beck WT (1991) Modulators of P-glycoprotein-associated multidrug resistance. Cancer Treat Res 57:151–170
Boesch D, Gaveriaux C, Jachez B, Pourtier-Manzanedo A, Bollinger P, Loor F (1991) In vivo circumvention of P-glycoprotein-mediated multidrug resistance of tumor cells with SDZ PSC 833. Cancer Res 51:4226–4233
Avendano C, Menendez JC (2002) Inhibitors of multidrug resistance to antitumor agents (MDR). Curr Med Chem 9:159–193
Pusztai L, Wagner P, Ibrahim N, Rivera E, Theriault R, Booser D, Symmans FW, Wong F, Blumenschein G, Fleming DR, Rouzier R, Boniface G, Hortobagyi GN (2005) Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 104:682–691
Ruff P, Vorobiof DA, Jordaan JP, Demetriou GS, Moodley SD, Nosworthy AL, Werner ID, Raats J, Burgess LJ (2009) A randomized, placebo-controlled, double-blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar (LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen. Cancer Chemother Pharmacol 64:763–768
Danks MK, Yalowich JC, Beck WT (1987) Atypical multiple drug resistance in a human leukemic cell line selected for resistance to teniposide (VM-26). Cancer Res 47:1297–1301
Pegg AE (2000) Repair of O(6)-alkylguanine by alkyltransferases. Mutat Res 462:83–100
Preuss I, Thust R, Kaina B (1996) Protective effect of O6-methylguanine-DNA methyltransferase (MGMT) on the cytotoxic and recombinogenic activity of different antineoplastic drugs. Int J Cancer 65:506–512
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867
Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J (2008) Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 68:4853–4861
Zhang J, Liu J (2013) Tumor stroma as targets for cancer therapy. Pharmacol Ther 137:200–215
Friberg S, Mattson S (1997) On the growth rates of human malignant tumors: implications for medical decision making. J Surg Oncol 65:284–297
Gasparini G, Longo R, Toi M, Ferrara N (2005) Angiogenic inhibitors: a new therapeutic strategy in oncology. Nat Clin Pract Oncol 2:562–577
Wilson GD, McNally NJ, Dische S, Saunders MI, Des Rochers C, Lewis AA, Bennett MH (1988) Measurement of cell kinetics in human tumours in vivo using bromodeoxyuridine incorporation and flow cytometry. Br J Cancer 58:423–431
Baguley BC (2011) The paradox of cancer cell apoptosis. Front Biosci (Landmark Ed) 16:1759–1767
Diaz D, Prieto A, Reyes E, Barcenilla H, Monserrat J, Alvarez-Mon M (2008) Flow cytometry enumeration of apoptotic cancer cells by apoptotic rate. Methods Mol Biol 414:23–33
Micheau O, Shirley S, Dufour F (2013) Death receptors as targets in cancer. Br J Pharmacol 169:1723–1744
Fan Z, Zhang Q (2005) Molecular mechanisms of lymphocyte-mediated cytotoxicity. Cell Mol Immunol 2:259–264
Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, Pamer EG, Li MO (2014) The cellular and molecular origin of tumor-associated macrophages. Science 344:921–925
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK (2012) Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 45:487–498
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266
Meyer JS, He W (1993) Cell proliferation measurements by bromodeoxyuridine or thymidine incorporation: clinical correlates. Semin Radiat Oncol 3:126–134
Laing JH, Wilson GD, Martindale CA (2003) Proliferation rates in human malignant melanoma: relationship to clinicopathological features and outcome. Melanoma Res 13:271–277
Begg AC, Haustermans K, Hart AA, Dische S, Saunders M, Zackrisson B, Gustaffson H, Coucke P, Paschoud N, Hoyer M, Overgaard J, Antognoni P, Richetti A, Bourhis J, Bartelink H, Horiot JC, Corvo R, Giaretti W, Awwad H, Shouman T, Jouffroy T, Maciorowski Z, Dobrowsky W, Struikmans H, Wilson GD et al (1999) The value of pretreatment cell kinetic parameters as predictors for radiotherapy outcome in head and neck cancer: a multicenter analysis. Radiother oncol 50:13–23
Furneaux CE, Marshall ES, Yeoh K, Monteith SJ, Mews PJ, Sansur CA, Oskouian RJ, Sharples KJ, Baguley BC (2008) Cell cycle times of short-term cultures of brain cancers as predictors of survival. Br J Cancer 99:1678–1683
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Baguley BC, Finlay GJ, Wilson WR (1986) Cytokinetic resistance of Lewis lung carcinoma to cyclophosphamide and the amsacrine derivative CI-921. Prog Clin Biol Res 223:47–61
Finlay GJ, Wilson WR, Baguley BC (1987) Cytokinetic factors in drug resistance of Lewis lung carcinoma: comparison of cells freshly isolated from tumours with cells from exponential and plateau-phase cultures. Br J Cancer 56:755–762
Itamochi H, Kigawa J, Sugiyama T, Kikuchi Y, Suzuki M, Terakawa N (2002) Low proliferation activity may be associated with chemoresistance in clear cell carcinoma of the ovary. Obstet Gynecol 100:281–287
Bonetti A, Zaninelli M, Rodella S, Molino A, Sperotto L, Piubello Q, Bonetti F, Nortilli R, Turazza M, Cetto GL (1996) Tumor proliferative activity and response to first-line chemotherapy in advanced breast carcinoma. Breast Cancer Res Treat 38:289–297
Anjomshoaa A, Lin YH, Black MA, McCall JL, Humar B, Song S, Fukuzawa R, Yoon HS, Holzmann B, Friederichs J, van Rij A, Thompson-Fawcett M, Reeve AE (2008) Reduced expression of a gene proliferation signature is associated with enhanced malignancy in colon cancer. Br J Cancer 99:966–973
Suzuki E, Sun J, Kapoor V, Jassar AS, Albelda SM (2007) Gemcitabine has significant immunomodulatory activity in murine tumor models independent of its cytotoxic effects. Cancer Biol Ther 6:880–885
de Biasi AR, Villena-Vargas J, Adusumilli PS (2014) Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res 20:5384–5391
Bracci L, Schiavoni G, Sistigu A, Belardelli F (2014) Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ 21:15–25
Zitvogel L, Apetoh L, Ghiringhelli F, Andre F, Tesniere A, Kroemer G (2008) The anticancer immune response: indispensable for therapeutic success? J Clin Invest 118:1991–2001
Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480:480–489
Gerdes MJ, Sood A, Sevinsky C, Pris AD, Zavodszky MI, Ginty F (2014) Emerging understanding of multiscale tumor heterogeneity. Front Oncol 4:366
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366:883–892
Leung E, Kannan N, Krissansen GW, Findlay MP, Baguley BC (2010) MCF-7 breast cancer cells selected for tamoxifen resistance acquire new phenotypes differing in DNA content, phospho-HER2 and PAX2 expression, and rapamycin sensitivity. Cancer Biol Ther 9:717–724
Hampton OA, Den Hollander P, Miller CA, Delgado DA, Li J, Coarfa C, Harris RA, Richards S, Scherer SE, Muzny DM, Gibbs RA, Lee AV, Milosavljevic A (2009) A sequence-level map of chromosomal breakpoints in the MCF-7 breast cancer cell line yields insights into the evolution of a cancer genome. Genome Res 19:167–177
Kemper K, de Goeje PL, Peeper DS, van Amerongen R (2014) Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res 74:5937–5941
Vidal SJ, Rodriguez-Bravo V, Galsky M, Cordon-Cardo C, Domingo-Domenech J (2014) Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene 33:4451–4463
Hong IS, Lee HY, Nam JS (2015) Cancer stem cells: the ‘Achilles heel’ of chemo-resistant tumors. Recent Pat Anticancer Drug Discov 10:2–22
Marshall ES, Baguley BC, Matthews JH, Jose CC, Furneaux CE, Shaw JH, Kirker JA, Morton RP, White JB, Rice ML, Isaacs RJ, Coutts R, Whittaker JR (2004) Estimation of radiation-induced interphase cell death in cultures of human tumor material and in cell lines. Oncol Res 14:297–304
Marshall ES, Finlay GJ, Matthews JH, Shaw JH, Nixon J, Baguley BC (1992) Microculture-based chemosensitivity testing: a feasibility study comparing freshly explanted human melanoma cells with human melanoma cell lines. J Natl Cancer Inst 84:340–345
Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, Lee D, Kalsy A, Gomez-Caraballo M, Elamine L, Howe E, Hur W, Lifshits E, Robinson HE, Katayama R, Faber AC, Awad MM, Ramaswamy S, Mino-Kenudson M, Iafrate AJ, Benes CH, Engelman JA (2014) Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 346:1480–1486
Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, Haddad BR, Rhim JS, Dritschilo A, Riegel A, McBride A, Schlegel R (2012) ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180:599–607
Houghton JA, Taylor DM (1978) Maintenance of biological and biochemical characteristics of human colorectal tumours during serial passage in immune-deprived mice. Br J Cancer 37:199–212
Monsma DJ, Monks NR, Cherba DM, Dylewski D, Eugster E, Jahn H, Srikanth S, Scott SB, Richardson PJ, Everts RE, Ishkin A, Nikolsky Y, Resau JH, Sigler R, Nickoloff BJ, Webb CP (2012) Genomic characterization of explant tumorgraft models derived from fresh patient tumor tissue. J Transl Med 10:125
Wargo JA, Cooper ZA, Flaherty KT (2014) Universes collide: combining immunotherapy with targeted therapy for cancer. Cancer discov 4:1377–1386
Selimoglu-Buet D, Gallais I, Denis N, Guillouf C, Moreau-Gachelin F (2012) Oncogenic kit triggers Shp2/Erk1/2 pathway to down-regulate the pro-apoptotic protein Bim and to promote apoptosis resistance in leukemic cells. PLoS One 7, e49052
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Baguley, B.C. (2016). Classical and Targeted Anticancer Drugs: An Appraisal of Mechanisms of Multidrug Resistance. In: Rueff, J., Rodrigues, A. (eds) Cancer Drug Resistance. Methods in Molecular Biology, vol 1395. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3347-1_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3347-1_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3345-7
Online ISBN: 978-1-4939-3347-1
eBook Packages: Springer Protocols