
Abstract
Posttranscriptional control of mRNA subcellular localization, stability, and translation is a central aspect of gene regulation and expression. Much of this control is mediated via recognition of a given mRNA transcript’s 3′ untranslated region (UTR) by microRNAs and RNA-binding proteins. Here we describe how a novel, scalable piggyBac-based vector, pBUTR, can be utilized for analysis of 3′ UTR-mediated posttranscriptional gene regulation (PTGR) both in vitro and in vivo. This vector is specifically designed to express a selection marker, a control reporter, and an experimental reporter from three independent transcription units. Expression of spliced reporter transcripts from medium-copy non-viral promoter elements circumvents several potential confounding factors associated with saturation and stability, while stable integration of these reporter and selection elements in the context of a DNA transposon facilitates experimental reproducibility.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Posttranscriptional gene regulation
- PTGR
- 3′-UTR
- pBUTR
- piggyBac
- Reporter
- miRNA sensors
- RNA-binding proteins
- mRNA stability
1 Introduction
Coordinated regulation of gene expression is fundamentally important for all aspects of cellular function. Historically, the most widely utilized practice in assessing coordinated regulation of gene expression has been via analysis of mRNA steady-state expression using either microarray [1] or next-generation sequencing approaches [2, 3]. Both approaches provide powerful information about genome wide changes in transcript abundance. However, these approaches fail to provide any information in regard to whether mRNA that has been transcribed is indeed being actively utilized by the translation machinery to produce protein. Emerging evidence strongly suggests that regulation of gene expression at the translational level contributes as much, if not more, to gene expression than transcription [4–6]. In fact, a reasonable amount of evidence suggests that coordinated changes in posttranscriptional regulatory networks occur during cellular differentiation and/or response to stimulus, and that these networks may profoundly alter cellular phenotype and behavior [7–10].
A significant amount of the control of mRNA subcellular localization , translation, and stability is mediated via cis-regulatory elements in the 3′ untranslated region (UTR) of target transcripts. These elements may be recognized by specific microRNAs and RNA-binding proteins [8, 11, 12]. Dysregulation of posttranscriptional control by microRNAs and RNA-binding proteins underlies distinct steps of pathogenesis in a wide spectrum of human diseases [12]. In some systems, 3′ UTR identity is itself sufficient to confer appropriate temporospatial gene expression in vivo [11]. Both alternative splicing [13] and alternative cleavage and polyadenylation [14] can alter 3′ UTR identity, and thus the visibility of related gene products to the posttranscriptional regulatory machinery. However, as compared to other facets of gene regulation, the contributions of these phenomena to gene regulation remain largely unexplored. Given that mutations within the 3′ UTRs of certain genes can significantly impact human health [15–17], it is of great interest to determine if and how genomic variations within the 3′ UTR, uncovered via genome-wide association studies and next-generation sequencing surveys, impact the pathology of the disease or phenotype with which they are associated. It is for this reason that we were motivated to develop a scalable and robust reporter system explicitly designed to model 3′ UTR-mediated regulation.
To these ends, we engineered a novel, scalable piggyBac transposon-based reporter system that we have named pBUTR (p iggy B ac-based 3′ UnTranslated Region reporter) [18]. We chose a DNA transposon-based system in consideration of our specific purpose. The integration of the pBUTR vector into the DNA of the target cell is essentially a stable transfection, which is generally superior to transient transfection in regards to experimental reproducibility and reduction of “noise.” That the vector is DNA based allows for the inclusion of multiple independent transcription units. Thus, a control reporter may be expressed completely independently of the experimental reporter. Also, the use of a DNA-based vector allows the inclusion of splice junctions in each of the transcription units encoded within the vector. It is well established that transcripts that have not undergone splicing do not express as well as those that have, due in some part to the fact that the lack of an exon junction complex (EJC) marks unspliced transcripts as substrates for the nonsense-mediated decay (NMD) pathway [19]. Retro- and lentiviral vector systems do not have these features, and thus often times include a stability sequence such as the woodchuck hepatitis virus post-translationally regulated element (WPRE) element [20], which would be expected to confound native post-transcriptional regulation. In addition to these limitations, the long terminal repeat (LTR) elements of retro- and lentiviral vectors may be recognized as foreign and silenced by the cell over time, a problem that is particularly observed in the context of transgenesis [21–23].
The pBUTR vector system is comprised of three independent transcription units—a G418 selection cassette, a control turboGFP reporter gene driven by PGK promoter, and a Gateway® [24] recombineering cassette under the control of the ubiquitin C (UBC) promoter (Fig. 1). These promoters were chosen because they drive expression at low-to-medium levels, and therefore are less likely to overwhelm any endogenous regulatory mechanisms. The pBUTR destination vector is generated via four-part Gateway® recombineering using an attL1/L2-flanked coding sequence of interest, an attR2/attL4 flanked 3′ UTR element, and an attR4/attL5-flanked minimal polyadenylation sequence [25] followed by a unique 24-nucleotide barcode. Upon recombination of these three elements into the parent vector, a bi-fluorescent reporter is produced that can be employed in both in vitro and in vivo model systems.

Schematic representation of the pBUTR vector. The pBUTR destination vector is functionalized by four-part Gateway® recombineering using an att L1/L2-flanked tRFP (can be substituted with any coding sequence of interest), an att R2/att L4 flanked 3′ UTR element, and an att R4/att L5-flanked minimal polyadenylation sequence followed by a unique 24-nucleotide barcode. The inclusion of unique barcode elements with the minimal polyadenylation signal was made to allow analyses within pooled cell populations via flow cytometry and cell sorting. attXN, Gateway® recombination site; tRFP, turboRFP; UTR, untranslated region; mPA, minimum polyadenylation signal; BC, 24 nt barcode; PAS, polyadenylation signal; SV40 (left), SV40 early promoter region. Neo, neomycin resistance gene; Bgh, bovine growth hormone polyadenylation signal; UBC, ubiquitin C promoter element; CmR, chloramphenicol-resistance gene; PGK, murine phosphoglycerate kinase 1 promoter; tGFP, turboGFP; SV40, SV40 late polyadenylation signal. Features not to scale
The pBUTR vector was functionalized with Gateway® technology to allow high-dimensionality screening and validation applications. Given that Gateway® recombineering is scalable—meaning multiple individual 3′ UTR elements can be cloned into the vector in bulk—an inclusive, aggregate set of 3′ UTRs of interest can be rapidly generated and tested for contextual regulatory activity in pooled or arrayed format. Here we discuss how the pBUTR reporter can be used to study 3′ UTR-mediated gene regulation in vitro (in both arrayed and pooled format) and in vivo.
The E-cadherin transcriptional repressors ZEB1 and ZEB2 play established roles in epithelial to mesenchymal transition, both during tumor metastasis and during embryogenesis [26]. The mRNA transcripts of both of these gene products are characterized by multiple, validated miR-200 family recognition elements in their respective 3′ UTRs [26]. Cells with an epithelial phenotype express high relative levels of the miR-200b microRNA, which enforces posttranscriptional repression of the ZEB1 and ZEB2 mRNA transcripts. However, as cells undergo EMT, for example in response to transforming grown factor-beta (TGF-β), relative levels of miR-200b are reduced, allowing increased expression of ZEB1 and ZEB2 proteins and transcriptional repression of the CDH1 (E-cadherin) gene. Previously described [26] wild-type and mutant (where each miR-200b-binding site has been ablated via site-directed mutagenesis) ZEB2 3′ UTR elements were recombineered into the pBUTR destination vector so as to confer regulation upon tRFP expression in the assembled reporter. We initially discuss how to study microRNA (miR-200b in this case)-mediated repression in a cell-based model of epithelial to mesenchymal transition (EMT), and then how to assess this regulation in vivo during embryogenesis. Entirely similar strategies can be employed to use the pBUTR reporter to study in vitro and in vivo 3′ UTR-mediated gene regulation in the context of siRNA/microRNA sensor activity, and posttranscriptional gene regulation (PTGR) by RNA-binding proteins, in both arrayed and pooled screening approaches.
2 Materials
2.1 BP Recombination Reaction
-
1.
attB-flanked PCR products:
-
(a)
Turbo-RFP (tRFP—Evrogen) amplified with:
attB1-tRFP-forward oligonucleotide primer –
5′—GGGGACAAGTTTGTACAAAAAAGCAGGCTCGCCACCATGAGCGAGCTG—3′, and
attB2-tRFP-reverse oligonucleotide primer –
5′—GGGGACCACTTTGTACAAGAAAGCTGGGTAGATCCTACACATTGATCCTAGCAGAAGC—3′.
-
(b)
Amplify 3′ UTRs or siRNA/miRNA sensor elements with: attB2r-forward primer—5′—GGGGACCCAGCTTTCTTGTACAAAGTGGTNNNN…NNNN—3′ and attB4-reverse primer—5′—GGGGACAACTTTGTATAGAAAAGTTGGGTGNNNN…NNNN—3′, where “N” is the specific priming sequence (see Notes 1 and 2 ). Note that the length of the specific priming sequence should be 18–21 nucleotides as for any other standard PCR amplification.
-
(c)
Amplify the minimum polyadenylation/barcode element with attBr4-forward primer—5′-GGGGACAACTTTTCTATACAAAGTTGAACTAGTAATAAAGG—3′ and attB5-reverse primer—5′—GGGGACAACTTTGTATACAAAAGTTGCG- 3′ from a synthetic attBr4_mPA_barcode_attL5 oligo: (ACAACTTTTCTATACAAAGTTGAACTAGTAATAAAGGATCCTTTATTTTCATTGGATCCGTGTGTTGGTTTTTTGTGTNNNNNNNNNNNNNNNNNNNNNNNNCGCAACTTTTGTATACAAAGTTGT), where the N sequence represents the 24-nucleotide barcode (see Note 3 ).
-
(a)
-
2.
A pDONR223 attP1-attP2 plasmid to generate the tRFP entry clone, a pDONR223 attP2r-attP4 plasmid to generate the 3′ UTR or siRNA/miRNA sensor entry clone, and a pDONR223 attP4r-attP5 plasmid to generate the minimum polyadenylation/barcode entry clone.
-
3.
BP Clonase II enzyme mix (see Note 4 ).
-
4.
2 μg/μl Proteinase K in 50 mM Tris, pH 8.0, 3 mM CaCl2, 50 % Glycerol.
-
5.
1× TE buffer: 10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0.
-
6.
37 °C water bath.
-
7.
Vortex.
-
8.
TOP10 competent E. coli cells.
-
9.
LB agar plates containing spectinomycin (50 μg/ml).
-
10.
Primers for colony PCR screening and sequencing: M13 forward (-20)—5′-GTAAAACGACGGCCAG-3′, M13 reverse—5′-CAGGAAACAGCTATGAC-3′.
-
11.
OneTaq DNA Polymerase or any other DNA polymerase with proof reading activity.
-
12.
Agarose.
-
13.
10 μg/ml ethidium bromide in double-distilled water. Use at a final concentration of 0.5 μg/ml.
-
14.
1× Tris-acetate EDTA buffer: 40 mM Tris-acetate, 1 mM EDTA.
2.2 LR Recombination Reaction
-
1.
Sequence verified attL1-attL2, attR2-attL4 and attR4-L5 donor plasmids.
-
2.
pBUTR destination vector containing 5′ attR1 and 3′ attR5 sites.
-
3.
LR Clonase II Plus enzyme mix (see Note 4 ).
-
4.
2 μg/μl Proteinase K in 50 mM Tris, pH 8.0, 3 mM CaCl2, 50 % glycerol.
-
5.
1× TE buffer: 10 mM Tris–HCl, pH 8.0, 1 mM EDTA, pH 8.0.
-
6.
37 °C water bath.
-
7.
Vortex.
-
8.
One Shot Mach1 T1 chemically competent E. coli cells.
-
9.
LB agar plates containing ampicillin (100 μg/ml) and kanamycin (100 μg/ml).
-
10.
Primers for PCR screening: UBC forward—5′-ATTGTCCGCTAAATTCTGGC-3′, PGK reverse—5′- TAAAGCGCATGCTCCAGAC -3′.
-
11.
OneTaq DNA Polymerase or any other DNA polymerase with proof reading activity.
-
12.
Agarose.
-
13.
10 μg/ml ethidium bromide in double-distilled water. Use at a final concentration of 0.5 μg/ml.
-
14.
1× Tris-acetate EDTA buffer: 40 mM Tris-acetate, 1 mM EDTA.
2.3 Cell Culture, Transfection, and Stable Clone Generation for In Vitro Experiments
-
1.
MCF10A cell line: Any appropriate adherent or suspension cell line can be similarly used.
-
2.
Growth media for MCF10A cells: DMEM/F12 medium, 5 % horse serum, 0.01 mg/ml bovine insulin, 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 20 ng/ml human EGF, and 100 U/ml penicillin and 0.1 mg/ml streptomycin.
-
3.
Mature pBUTR vector.
-
4.
Plasmid containing transposase (pCMV-HA-m7pB) [27].
-
5.
Lipofectamine-LTX or any other transfection reagent specific to the cell line being used.
-
6.
G418.
2.4 Flow Cytometry
-
1.
FACSCalibur system (BD Biosciences) or any other appropriate flow cytometry equipment.
-
2.
For cell sorting, BD FACS Aria II cell sorter (BD Biosciences) or any other appropriate cell sorter equipment.
2.5 Genomic DNA Isolation, Library Preparation, and Limited Next-Generation Sequencing
-
1.
Lysis buffer: 100 mM NaCl, 20-mM Tris, pH 7.6, 10-mM EDTA, pH 8.0, 0.5 % sodium dodecyl sulphate and 0.5 mg/ml proteinase K.
-
2.
60 % volume-saturated NaCl.
-
3.
Ethanol.
-
4.
Personal genome machine (PGM) manually barcoded forward primer: P-NNNNAGTTGAACTAGTAATAAAGGATCC and PGM barcoded reverse primer: P-NNNNTGACATGTTGTATGACGGTGTG (see Note 5 ).
-
5.
Ion Plus Fragment Library Kit (Life Technologies).
-
6.
Ion PGM Template OT2 200 Kit (Life Technologies).
-
7.
Ion PGM 200 Sequencing Kit (Life Technologies).
-
8.
Ion 314 chip (Life Technologies).
-
9.
PGM sequencing platform (Life Technologies).
2.6 Generation, Injection of Embryonic Stem Cells, Embryo Harvest, and Imaging for In Vivo Experiments
-
1.
V6.5 embryonic stem cells (ESCs) derived from F1 hybrid strain (C57BL/6 × 129/Sv) [28].
-
2.
ESC medium: DMEM, 15 % fetal bovine serum, 1000 U/ml LIF, 1 % β-mercaptoethanol, 1 % non-essential amino acids, 1 % l-glutamine, 0.5 % penicillin/streptomycin.
-
3.
1× phosphate-buffered saline (PBS): 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4.
-
4.
100 mm culture dishes with feeder cells.
-
5.
2 N (3.5 days postcoitus) C57BL/6 blastocysts.
-
6.
Pseudopregnant ICR recipient female mice—2.5 days postcoitus.
-
7.
4 % Paraformaldehyde.
-
8.
15 % and 30 % sucrose in 1× PBS.
-
9.
OCT compound.
-
10.
SuperFrost Plus slides.
-
11.
Vectashield.
-
12.
Confocal laser scanning microscope.
3 Methods
3.1 Construction of Donor Vectors
-
1.
Generate the attB-flanked PCR products using OneTaq DNA polymerase or any other DNA polymerase with proofreading activity.
-
2.
For each BP recombination reaction between a given attB PCR product and donor vector, add the following components to 1.5 ml microcentrifuge tubes at room temperature and mix gently with a pipette:
-
3.
Quickly vortex the BP Clonase II enzyme mix, twice (2 s each time), and add 2 μl of BP Clonase II enzyme mix to each sample above. Mix well by vortexing briefly twice (2 s each time)
-
4.
Incubate reactions at 25 °C for 1 h (see Note 9 ).
-
5.
Add 1 μl of the Proteinase K solution to each reaction. Incubate for 10 min at 37 °C.
-
6.
Transform 2 μl of the transformation reaction to TOP10 competent E. coli cells (the remaining can be stored at −20 °C) and plate one-fifth of the transformants on LB agar spectinomycin plates.
-
7.
Incubate overnight at 37 °C.
-
8.
The following day, screen colonies using M13 forward and reverse primers. For colony PCR (see Note 10 ), determine the number of colonies intended to be screened. Set up a 96-well plate with 100 μl/well of LB media containing spectinomycin (100 μg/ml) and a similar number of PCR reactions with M13 forward (−20) and reverse primers and Taq polymerase. Using a pipette tip pick one colony, dip it in the PCR reaction cocktail containing OneTaq DNA polymerase, and then into the correspondingly labeled LB-containing well. Incubate the inoculated LB-containing plate at 37 °C.
-
9.
Set up a thermal cycler with the following conditions:
-
Initial denaturation at 94 °C for 30 s
-
25 cycles (see Note 11 ) at 94 °C for 30 s, 47 °C for 30 s, 68 °C for “n” seconds, where n = 60 s/kb
-
Final extension at 68 °C for 5 min
-
-
10.
Resolve PCR products on a 1 % agarose gel. Colonies without any insert are characterized by a background band of ~350 base pairs (bp). If an insert of “n” bp is expected then a band at “350+n” bp will show up (Fig. 2).
Fig. 2 
Representative agarose gel (1 %) electrophoresis image of colony PCR products to identify positive clones in pooled BP recombination reaction. Colony PCR was performed with M13 primers (see text) to screen for positive clones from BP recombination reaction done in two separate pools. Clones without any insert result in a ~350 bp product. Subtracting ~350 bp from the other inserts gives the approximate length of the amplified 3′ UTR inserts and an indication of their identity. Performing colony PCR helps to pick the right size inserts for sequence confirmation in comparison to sequencing in bulk to get the right inserts. A similar strategy can be adapted for screening post LR recombination reaction, but using UBC forward and PGK reverse primers instead (see text). L, ladder; C, BP clone
-
11.
Once a candidate insert has been identified, the corresponding inoculum can be used to seed miniprep culture, which can subsequently be sequence confirmed using the aforementioned M13 primers.

3.2 Construction of Expression Reporters
Complete expression reporters are generated via four part recombineering using the destination vector and the three donor plasmids—the tRFP entry clone, the donor plasmid containing the 3′ UTR/siRNA or miRNA sensor of interest, and the pool of donor plasmids containing the minimal polyadenylation signal and barcode.
-
1.
Use the following formula to convert femtomoles (fmol) to nanograms (ng) of DNA:
ng = [(X fmol) × (size of DNA in bp) × 660]/106
-
2.
For each LR recombination reaction between an appropriate attB PCR product and donor vector (see Note 12 ), add the following components to 1.5 ml microcentrifuge tubes at room temperature and mix gently with a pipette:
-
Entry clone (10 fmol each)—1–7 μl
-
Destination pBUTR vector (20 fmol)—1 μl
-
1× TE buffer, pH 8.0—to 8 μl
-
-
3.
Quickly vortex the LR Clonase II Plus enzyme mix, twice (2 s each time), and add 2 μl of LR Clonase II enzyme mix to each sample above. Mix well by vortexing briefly twice (2 s each time).
-
4.
Incubate reactions at 25 °C for 16 h (see Note 13 ).
-
5.
Add 1 μl of the Proteinase K solution to each reaction. Incubate for 10 min at 37 °C.
-
6.
Transform 2 μl of the transformation reaction to One Shot Mach1 T1R Competent E. coli cells (the remaining can be stored at −20 °C) and plate the entire transformants on LB agar ampicillin plus kanamycin plates.
-
7.
Incubate overnight at 37 °C.
-
8.
The following day, screen colonies using UBC forward and PGK reverse primers. For colony PCR, determine the number of colonies intended to be screened. Set up a 96-well plate with 100 μl/well of LB media containing ampicillin plus kanamycin (100 μg/ml each) and a similar number of PCR reactions with UBC forward and PGK reverse primers and OneTaq DNA polymerase. Using a pipette tip pick one colony, dip it in the PCR reaction cocktail, and then into the correspondingly labeled LB-containing well. Once done, run PCR (use the same conditions as Subheading 3.1, step 10, except for annealing temperature of 49 °C). Incubate the inoculated LB-containing plate at 37 °C.
-
9.
Resolve PCR products on an 1 % agarose gel. The right colonies can be identified based on the expected insert sizes of the 3′ UTR or siRNA/miRNA sensor elements.
-
10.
Once an insert has been identified, the corresponding inoculum can be used to seed bacterial growth cultures, which would subsequently be sequence confirmed using the UBC forward and PGK reverse primers described above (see Note 14 ).
3.3 Transfection of Cells and Generation of Stable Clones
-
1.
Transfect cells with plasmids containing transposase (pCMV-HA-m7pB) and transposon (respective pBUTR vector) at a ratio of 1:2 using appropriate transfection reagent (see Note 15 ). The pBUTR can be used for a wide spectrum of cell types (see Note 16 ). For MCF10A cells, seed 4 × 104 cells into each of the desired number of wells in a 24-well plate. Twenty-four hours after cell seeding, transfect the cells in each well with 333.3 ng of pBUTR-wild-type-ZEB2 or pBUTR-mutant-ZEB2 along with 166.7 ng of pCMV-HA-m7pB.
-
2.
Forty-eight hours after transfection, split cells 1:10 and select with G418 (1000 μg/ml for MCF10A) for approximately 2 weeks (see Note 17 ).
3.4 Flow Cytometric Analysis of Reporter Expression in Arrayed Format
-
1.
Following G418 selection, split each stably transduced cell line in replica plates, and then treat one or more replicates with the experimental stimulus while leaving another replicate plate untreated as a control. For MCF10A cells, treat with TGF-β or vehicle for 72 h (see Note 18 , and Fig. 3a).
Fig. 3 
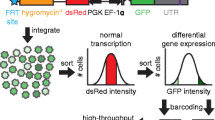
Schematic of work-flow for pBUTR vector-mediated high-dimensionality screening and validation applications in arrayed (a) or pooled format (b). One caveat associated with DNA transposon-based screening approaches relative to a retro- or lentiviral approach is that stable transfection of cells in bulk with a pool of vectors is not straightforward. For this reason, initial transfection and selection should be performed in an arrayed format. The inclusion of unique barcode elements with the minimal polyadenylation signal will allow analysis of enrichment or depletion within pooled cell populations via flow cytometry and cell sorting and limited next-generation sequencing
-
2.
Perform multicolor flow cytometry to assess the expression of the turboGFP (tGFP) (excitation/emission max = 482/502 nm) and tRFP (excitation/emission max = 553/574 nm) under the different experimental conditions.
-
3.
Determine the ratio of tRFP and tGFP expression as assessed via median fluorescence intensity (MFI) and calculate fold changes as follows:
Fold change = log2 (tRFP/tGFP)C1/(tRFP/tGFP)C2, where C1 and C2 are two different experimental conditions.
-
4.
A positive fold change will indicate 3′ UTR-mediated posttranscriptional induction or up-regulation of gene expression . Conversely, a negative fold change will indicate posttranscriptional repression or downregulation of gene expression (see Notes 19 and 20 ).

Treatment of MCF10A cells with TGF-β will result in decreased miR-200b and E-cadherin protein expression. In cells stably transfected with the wild-type ZEB2 reporters, these decreased levels will coincide with marked increases in tRFP fluorescence. The levels of tRFP fluorescence, as assessed via median fluorescent intensity (MFI) in the TGF-β treated MCF10A cells will be similar to those observed in untreated MCF10A cells transfected with mutant ZEB2 reporters (see Note 21 ).
3.5 Flow Cytometric Analysis of Reporter Expression in Pooled Format
-
1.
The pBUTR vectors containing the 3′-UTR elements of interest are individually transfected in arrayed format and selected with G418 exactly as described in Subheading 3.3 (see Note 22 ).
-
2.
Following selection for approximately 2–4 weeks, the individual transfectants expressing the different pBUTRs are pooled (see Note 23 , and Fig. 3b).
-
3.
The pooled stable transfectant lines are split to replica plates and treated according to experimental design.
-
4.
Cells from the different experimental conditions are sorted using multicolor flow cytometry based on tRFP expression (see Notes 24 and 25 , and Fig. 4).
Fig. 4 
Recommended subdivision of population prior to screening for more sensitivity in pooled approaches. Shown is a schematized example of the range of basal tRFP expression of a pool of pBUTRs in a given physiological context. The original pool may be sorted into two or more subpools (vertical dotted lines) on the basis of baseline tRFP expression (e.g., tRFPlo, tRFPmed, tRFPhi) prior to the experiment. Each subpool may then be individually treated and sorted. Please also refer to Note 25 . tRFP, turbo-red fluorescent protein; lo, low; me, medium; hi, high
-
5.
Isolate genomic DNA from the different pools of sorted tRFP+ positive cells using overnight proteinase K digestion at 55 °C before salting out with 60 % volume saturated NaCl and precipitating with ethanol.
-
6.
Use distinctly barcoded PGM PCR primer pairs to amplify 3′ UTR-correlated barcode elements from each of the sorted populations.
-
7.
Use these barcoded elements to template an Ion Torrent adapter-ligated library using Life Technology’s Ion Plus Fragment Library Kit protocol (#4471252, Revision 3.0). Perform sample emulsion PCR using the Ion PGM Template OT2 200 Kit (#4480974, Revision 5.0) following Life Technology's instructions. Prepare the samples for sequencing using the Ion PGM 200 Sequencing Kit (#4474004, Revision C). Load the completed samples on an Ion 314 chip and sequence on the PGM platform.
-
8.
Process the data from the PGM runs initially using the bam2fastq [29] to generate the fastq files and custom Perl scripts to bin based on barcodes and trim adapter sequences. Determine the percent representation of the different barcodes in the indicated populations.
-
9.
Enriched and depleted barcodes will reveal identity of gene products that are being regulated at the post-transcriptional level by their 3′ UTR elements under different experimental conditions.

3.6 Using pBUTR for In Vivo Monitoring of 3′ UTR-Mediated PTGR
-
1.
On the day of electroporation, trypsinize, count and aliquot 5 × 106 V6.5 ESCs.
-
2.
Spin the tube containing the aliquoted cells at 190 × g for 3 min. Aspirate as much media off as possible, wash cell pellet with 10 ml 1× PBS, and spin again for 3 min at 190 × g. Aspirate once again.
-
3.
Add 1 μg each of pBUTR vector and pCMV-HA-m7pB transposase to the cell pellet.
-
4.
Add 700 μl 1× PBS to the pellet. Suspend cells and DNA by pipetting multiple times and transfer to 0.4 cm gap electroporation cuvette. Replace cap on cuvette (see Note 26 ).
-
5.
Place cuvette in GenePulser shockpod and electroporate the cells at 240v and 500uF (see Note 27 ).
-
6.
Post-electroporation, let the cuvette sit inside a laminar air flow hood for 10 min at room temperature.
-
7.
Add 1 ml of ESC media to the cuvette and mix. Transfer cells to a 15 ml conical tube in sterile condition (see Note 28 ).
-
8.
Rinse cuvette with 1 ml of ESC media and add to the cells in the conical media. Resuspend cells ensuring there are no clumps.
-
9.
Plate 10 % of electroporated cells onto 100 mm culture dishes with a feeder layer by gently dripping the cells over the feeder layer.
-
10.
Tip dishes in “X” pattern (do not swirl cells to the periphery of the plate).
-
11.
At least 20 h post-electroporation, start selection with G418 (300 μg/ml) for 8 days, verify resulting ESC colonies for tGFP and tRFP expression using a microscope, pick the colonies, expand, and make freezer stocks.
-
12.
Inject ES cell clones into 2 N (3.5 days postcoitus) C57BL/6 blastocysts and subsequently transfer to the uterine horns of 2.5 days postcoitus pseudopregnant ICR recipient female mice.
-
13.
Sacrifice pregnant females by carbon dioxide asphyxiation on appropriate postcoitus day based on what developmental phase is being studied (see Note 29 ).
-
14.
Dissect out embryos and fix in 4 % paraformaldehyde (PFA) for 1 h, before incubation in 15 % and 30 % sucrose (each for 16 h), and finally embed in OCT compound.
-
15.
Cut 5 μm sections including desired physiological structure and mount on SuperFrost Plus slides using Vectashield.
-
16.
Obtain images documenting domains of tRFP and tGFP expression using a confocal laser scanning microscope.
-
17.
TurboGFP expression would be constitutively observed in all areas where the pBUTR has been internalized. On the other hand, the tRFP expression will be reliant on a particular 3′ UTR’s ability to confer correct temperospatial expression of the gene product during murine development.
4 Notes
-
1.
For generating the siRNA or miRNA sensors, the attB2r and attB4 flanked siRNA/sensors sequence may be commercially synthesized. For example, for a “2×” CXCR4 siRNA sensor [30] the attB2r-CXCR4-attB4 sequence is—5′ –GGGGACCCAGCTTTCTTGTACAAAGTGGTAAGTTTTCACTCCAGCTAACACCGGATCGGCATAAGTTTTCACTCCAGCTAACACCGGCACCCAACTTTTCTATACAAAGTTGTCCCC—3′ (the underlined portion is the CXCR4 sensor sequence). This commercially synthesized oligonucleotide may then be amplified with oligonucleotides corresponding only to the attB2r and attB4 sites, rather than chimeric oligonucleotides as described.
-
2.
For generating an attB-flanked entire 3′ UTR, design the forward primer with the 5′ end corresponding to the base immediately after the stop codon of the coding sequence and the 5′ end of the reverse primer corresponding to the nucleotide immediately preceding the poly (A) signal. The use of a synthetic polyadenylation signal for all clones to be analyzed removes any influence of the native polyadenylation signal (e.g., efficiency) on gene expression, which may confound analysis of 3′ UTR-mediated effects.
-
3.
The composition of our own barcodes, generated via mixed nucleotide synthesis, was informed by the average nucleotide composition of the 24 base pairs following the G/U-rich region of native polyadenylation sequences in the human genome. The inclusion of unique barcode elements with the minimal polyadenylation signal was made to allow analyses within pooled cell populations via flow cytometry, cell sorting, and limited next-generation sequencing analysis.
-
4.
The BP Clonase II and LR Clonase II Plus enzyme mixes should be kept at −20 °C until immediately before use; however, the Proteinase K solution can be thawed and kept on ice until use.
-
5.
The number of different ‘NNNN’ combinations to be used will depend on the experimental conditions. For example, if only a control and experimental conditions are being compared then two variants of NNNN like ACTG and AGTC will be used. On the other hand, if a time course experiment is being done for 0, 24, and 48 h, then three variants of NNNN like ACTG, TGAC, and AGTC may be used. These primer pairs should be phosphorylated at the 5′ end.
-
6.
Anywhere between 15 and 150 ng of the attB PCR product can be used for the BP reaction.
-
7.
Set up a BP reaction with no attB PCR product as a negative control.
-
8.
A major advantage of the Gateway® system is the potential for scalability. Multiple donor vectors containing 3′ UTR elements to be assessed can be generated simultaneously if the attB2r and attB4 flanked PCR products corresponding to these 3′ UTRs are pooled for the BP reaction. For pooling of large groups of UTRs it is recommended to generate “subpools” such that individual inserts may be easily discerned following colony PCR screening and gel electrophoresis. For example if there are 3′ UTRs of length 150, 175, 200, 250, 500, 750, 1200, 1400 bp—then we would recommend two subpools, with one containing 3′ UTRs of length 150, 200, 500, and 1200 bp and the other containing the 175, 250, 750, and 1400 bp length 3′ UTRs. This facilitates identification of individual inserts during visualization after gel electrophoresis.
-
9.
Normally a 1-h incubation yields a sufficient number of donor vectors. However, the length of the recombination reaction can be extended up to a maximum of 18 h. For PCR products ≥5 kb, longer incubations will increase the yield of colonies and are recommended. Normally, an overnight incubation typically yields five to ten times more colonies than 1-h incubation.
-
10.
If just one donor vector is being constructed then the colonies can just be grown up for miniprep and subsequently sequenced. The colony PCR is especially beneficial when pooled BP reactions are being done since this precludes the need to sequence a large number of colonies to get the desired donor vectors.
-
11.
Normally 25 cycles of PCR is enough to view products on an agarose gel. The precise conditions for PCR will be informed by the choice of polymerase mix and the thermal cycler used.
-
12.
The expression constructs can be generated through pooled LR recombination reaction in a manner analogous to that described in see Note 8 . Again, we recommend a subpooling strategy based on the size of the 3′ UTR elements of interest (see Note 8 ) such that the positive clones can be easily identified through colony PCR.
-
13.
The reactions can be incubated up to 24 h at room temperature.
-
14.
The pBUTR reporters should be prepped with Endotoxin-free miniprep or maxiprep kits, depending on the number of projected downstream transfections.
-
15.
The total amount of DNA transfected depends on the cell numbers and the specific transfection reagent being used.
-
16.
Originally isolated from the genome of the cabbage looper moth Trichoplusia ni [31], the piggyBac transposon has distinct advantages. It has a large cargo size [31], and is highly active in many cell types [32, 33]. In addition, it has been shown to effect long-term expression in mammalian cells in vivo [34].
-
17.
Perform a kill curve with G418 for the particular cell line being used, then use the lowest concentration of G418 that effectively kills untransfected cells. Normally you get stable colonies of cells within 2 weeks and very distinct isolated colonies after 4 weeks of selection with G418. Successful selection can be confirmed by observing the cells under a fluorescence microscope and determining the approximate percentage of tGFP+ cells.
-
18.
Posttreatment with TGF-β for 72 h, MCF10A cells switch from polarized, tightly packed discoid epithelial cells to highly motile fibroblastic or mesenchymal phenotype, characteristics of distinct morphological changes associated with EMT [26], a reduction in E-cadherin protein expression concomitant with an induction of the mesenchymal cell marker N-cadherin [18]. These parameters can be used to verify that the answer obtained in the pBUTR experiment corroborates with the expected landmarks of a phenomenon.
-
19.
The observed changes in tRFP expression can be further validated by appropriately using miRNA mimics or antagomirs in the case of siRNA/miRNA sensors or miRNA-mediated regulation or siRNA/ectopic overexpression in case of RNA binding proteins.
-
20.
The relative reporter expression within this system does not differentiate between mechanisms impacting mRNA stability or translational repression, which will require additional downstream experimentation.
-
21.
Of note, even though endogenous promoters are used in the pBUTR vector, it is necessary to include appropriate control reporters with minimal or otherwise defined 3′ UTR elements to offset effect of promoter activity, if any.
-
22.
A drawback of DNA transposon-based approaches is that there is some risk in transfecting pooled reporters into a population of cells. In contrast to viral vector systems, where low multiplicities of infection can be used to ensure a single integrant per cell, DNA transposons necessitate the use of electroporation or cationic lipid-based delivery methods. Since both of the latter methods will deliver multiple vectors from a pool into a given cell, there is a very high risk of confounding results in any reporter-based screen. We thus strongly suggest that individual cell lines be generated in arrayed format and then pooled for screening approaches.
-
23.
Ideally, equal numbers of cells from each stably transduced line are mixed together. However, since a comparison of relative representation within control and experimental populations will be assessed, this is not essential in high-dimensionality screens.
-
24.
An advantage of flow cytometry based screening is that additional fluorophores may be simultaneously used as experimental controls, e.g., decreased surface expression of E-cadherin and increased surface expression of N-cadherin in the context of EMT.
-
25.
As a general rule, one would collect the 10 % of cells expressing the highest level of tRFP in each condition and compare these populations. However, this strategy may miss several posttranscriptional regulatory events. Each 3′ UTR has its own baseline level of expression, which from our experience may vary over an order of magnitude from other 3′ UTRs in the population. For example, consider that the basal tRFP fluorescence intensity of a pooled population ranges from 10 to 100 (arbitrary units). An individual reporter may have a fluorescence intensity of 10 in the control state and 80 in the experimental state—an impressive eightfold induction. However, since one is merely collecting the top 10 % of events in each population (fluorescence intensities of 90–100) this induction would be missed. To increase the sensitivity in a screening experiment, the original pool may be sorted into two or more subpools on the basis of baseline reporter expression (e.g., tRFPlo, tRFPmed, tRFPhi) prior to the experiment. Each subpool may then be individually treated and sorted.
-
26.
While loading the cuvette, be careful not to touch the sides, especially the metallic surface.
-
27.
Confirm that the Time Constant from the GenePulser was between 7.0 and 8.0 during electroporation.
-
28.
The mixing is done best with Pasteur pipettes.
-
29.
Although we describe a transient transgenesis approach, depending on the depth and breadth of the planned analysis, a better strategy may be to let the fetuses come to term and screen pups for tGFP expression upon birth [35]. TGFP+ pups may then be used as founders for a line of reporter mice that may be used to extensively characterize 3′ UTR-mediated gene regulation throughout embryogenesis and adulthood.
References
Schena M, Shalon D, Davis RW et al (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470
Cloonan N, Forrest AR, Kolle G et al (2008) Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5:613–619
Mortazavi A, Williams BA, McCue K et al (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
Chen G, Gharib TG, Huang CC et al (2002) Discordant protein and mRNA expression in lung adenocarcinomas. Mol Cell Proteomics 1:304–313
Vogel C, Abreu RS, Ko D et al (2010) Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol Syst Biol 6:400
Schwanhausser B, Busse D, Li N et al (2011) Global quantification of mammalian gene expression control. Nature 473:337–342
Jansen RP (2001) mRNA localization: message on the move. Nat Rev Mol Cell Biol 2:247–256
de Moor CH, Meijer H, Lissenden S (2005) Mechanisms of translational control by the 3′ UTR in development and differentiation. Semin Cell Dev Biol 16:49–58
Garneau NL, Wilusz J, Wilusz CJ (2007) The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8:113–126
Keene JD (2007) RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8:533–543
Merritt C, Rasoloson D, Ko D et al (2008) 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol 18:1476–1482
Matoulkova E, Michalova E, Vojtesek B et al (2012) The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol 9:563–576
Kornblihtt AR, Schor IE, Allo M et al (2013) Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14:153–165
Elkon R, Ugalde AP, Agami R (2013) Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet 14:496–506
Conne B, Stutz A, Vassalli JD (2000) The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat Med 6:637–641
Shibayama A, Cook EH Jr, Feng J et al (2004) MECP2 structural and 3′-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet B Neuropsychiatr Genet 128B:50–53
Chatterjee S, Pal JK (2009) Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol Cell 101:251–262
Chaudhury A, Kongchan N, Gengler JP et al (2014) A piggyBac-based reporter system for scalable in vitro and in vivo analysis of 3′ untranslated region-mediated gene regulation. Nucleic Acids Res 42, e86
Nakano K, Ando T, Yamagishi M et al (2013) Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: implications for retroviral replication. Microbes Infect 15:491–505
Zufferey R, Donello JE, Trono D et al (1999) Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol 73:2886–2892
Jahner D, Stuhlmann H, Stewart CL et al (1982) De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 298:623–628
Yoder JA, Walsh CP, Bestor TH (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13:335–340
Jones S, Peng PD, Yang S et al (2009) Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum Gene Ther 20:630–640
Hartley JL, Temple GF, Brasch MA (2000) DNA cloning using in vitro site-specific recombination. Genome Res 10:1788–1795
Xia H, Mao Q, Davidson BL (2001) The HIV Tat protein transduction domain improves the biodistribution of beta-glucuronidase expressed from recombinant viral vectors. Nat Biotechnol 19:640–644
Gregory PA, Bert AG, Paterson EL et al (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Yusa K, Zhou L, Li MA et al (2011) A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A 108:1531–1536
Eggan K, Akutsu H, Loring J et al (2001) Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A 98:6209–6214
http://www.hudsonalpha.org/gsl/information/software/bam2fastq.
Doench JG, Petersen CP, Sharp PA (2003) siRNAs can function as miRNAs. Genes Dev 17:438–442
Cary LC, Goebel M, Corsaro BG et al (1989) Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 172:156–169
Li MA, Turner DJ, Ning Z et al (2011) Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res 39, e148
Ding S, Wu X, Li G et al (2005) Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122:473–478
Wu SC, Meir YJ, Coates CJ et al (2006) piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci U S A 103:15008–15013
Nakanishi H, Higuchi Y, Kawakami S et al (2010) piggyBac transposon-mediated long-term gene expression in mice. Mol Ther 18:707–714
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Chaudhury, A., Neilson, J.R. (2016). Use of the pBUTR Reporter System for Scalable Analysis of 3′ UTR-Mediated Gene Regulation. In: Dassi, E. (eds) Post-Transcriptional Gene Regulation. Methods in Molecular Biology, vol 1358. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3067-8_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3067-8_7
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3066-1
Online ISBN: 978-1-4939-3067-8
eBook Packages: Springer Protocols