
Abstract
Phospholipase C (PLC) expression and activity have repeatedly been reported to be elevated in cardiomyocytes under pathological conditions, including ischemia/reperfusion, hypertrophy, and chamber dilatation. In recent studies the subtypes of PLC involved have been identified, paving the way for studies of the mechanisms by which PLC may be activated under pathological conditions and how this may contribute to disease progression. PLC subtypes are localized by subtype- and tissue-specific binding to scaffolding proteins providing the possibility of developing cardiac-specific therapies based on inhibition of the localization of particular PLC subtypes in cardiomyocytes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
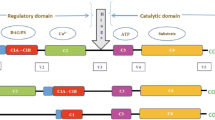
Phosphatidylinositol-specific phospholipases C (PLCs) are enzymes that cleave the plasma membrane phospholipid, phosphatidylinositol(4,5)bisphosphate (PIP2), to generate inositol(1,4,5)trisphosphate (Ins(1,4,5)P3), a Ca2+ releasing intracellular messenger, and sn-1,2-diacylglyerol (DAG), an activator of conventional subtypes of protein kinase C. The substrate lipid and the two products all have critical roles in regulating cellular responses and therefore PLCs are of central importance in the functioning of all cell types. Furthermore, perturbations in PLC activity may contribute substantially to disease phenotypes in a range of different tissues. As expected from a family of enzymes with such a central role in signaling, PLCs can be regulated in many different ways. PLCs are classified into six major classes (β, γ, δ, ε, ν, ζ), each of which includes multiple subtypes and splice variants (Fig. 17.1) [1]. PLCβ family members (PLCβ1-4) respond to G protein subunits activated downstream of seven transmembrane spanning receptors (also called G protein-coupled receptors, GPCR) [2]. PLCβ1 and PLCβ3 are expressed in cardiomyocytes, but PLCβ2 is not. PLCβ1 exists as two splice variants that differ only in their extreme C-terminal sequences, PLCβ1a (MW 150kD) and PLCβ1b (MW 140kDa, Fig. 17.2). Whilst both splice variants are expressed in neonatal rat cardiomyocytes [3], only PLCβ1b is expressed in adult human, rat, and mouse heart [4]. PLCγ members (PLCγ1 and PLCγ2) translocate to the plasma membrane subsequent to the activation of receptor tyrosine kinases, following stimulation with the appropriate growth factor [5]. Hearts express primarily PLCγ1 [6]. PLCδ subtypes are more sensitive to activation by Ca2+ than other subtypes, and hearts express PLCδ1, but the physiological importance of this has not been firmly established [7, 8]. PLCε regulation is complex involving a variety of activators including monomeric G proteins of the Ras family, as well as heterotrimeric G proteins of the G12/13 family and Gβγ [9]. Thus receptor activation can lead to PLCε activation by a variety of signaling mechanisms, often well downstream of receptor activation. There is only a single PLCε gene product, but this is expressed as two N-terminal splice variants [10]. Other PLC subtypes are not expressed in heart and will not be considered further.

Diagram showing the relationship between the different classes of the PLC family of proteins, emphasizing the structural motifs present

The splice variants of PLCβ1. Diagram showing the structures of PLCβ1a and PLCβ1b outlining the sequence differences in the C-terminal regions of the proteins. Proline-rich domains and PDZ-interacting domains are indicated. NLS is nuclear localization sequence
2 The Regulation of PLC Activity in Heart
Early studies showed that activation of α1-adrenergic receptors [11], M2 muscarininc cholinergic receptors [12] or endothelin receptors [13] resulted in generation of Ins(1,4,5)P3 and its metabolites. Subsequently, activation via purinergic receptors was reported [3]. All of these factors bind receptors coupled to Gq and would therefore be expected to activate PLCβ family members [14]. There have also been reports of activation via growth factor receptors that would be expected to activate PLCγ subtypes [15]. More recently the novel PLCε subtype has been identified in cardiomyocytes [16] and shown to be activated via thrombin (protease activated receptor 1, PAR1) and sphingosine1-phosphate (S1P) receptors [17]. In addition to activation by hormones and neurotransmitters, PLC in intact hearts and in cardiomyocytes in culture responds to acute stretch [18–21].
3 Localization of PLC Subtypes in Heart
To be active PLCs must be located close to their substrate PIP2, localized primarily or exclusively at the sarcolemma. It is now well recognized that PLC subtypes are specifically localized to particular membrane regions by binding scaffolding proteins. These scaffolds are selective for particular PLC subtypes and, in some cases, are also tissue specific.
In the case of the PLCβ family, such scaffolding interactions generally involve a C-terminal PDZ-interacting domain, present in all PLCβ1 subtypes except PLCβ1b. These PDZ-interacting domains associate with particular PDZ (postsynaptic density protein, Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein domain) proteins. PLCβ3, for instance, binds to cell polarity proteins, Par3 and Par6, in renal tubular epithelial cells, SH3 domain and ankyrin repeat protein 2 (Shank2) at glutamatergic synapses in neuronal tissues [22], and the sodium hydrogen exchange regulatory protein 2 (NHERF2) in Cos7 cells [23]. All of these interactions require PDZ domain interactions via the C-terminal sequence, NTQL. PLCβ3 is not localized to the sarcolemma, at least in neonatal rat cardiomyocytes [24], suggesting that suitable scaffolding proteins are not expressed, or are not associated with the sarcolemma. The C-terminal PDZ-interacting domain of PLCβ1a (DTPL) binds selectively to the first PDZ domain (nearest the N-terminal) of the scaffolding protein, sodium hydrogen exchange regulatory factor 1 (NHERF1), but not NHERF2, in HEK293 cells [25]. The first PDZ domain of NHERF1 also binds PLCβ2 via the sequence ESRL [26, 27]. NHERF1 is not expressed in heart providing an explanation for the cytoplasmic localization of PLCβ1a when expressed in cardiomyocytes [24]. As noted above, PLCβ1b does not have a C-terminal PDZ-interacting domain and so must target to membranes by a different mechanism from that used by other PLCβ subtypes. The presence of two proline-rich domains at the C-terminal end points to targeting by an SH3 domain- [28] or a WW domain-containing protein [29]. In cardiomyocytes the scaffolding protein for PLCβ1b was identified as Shank3. Shank3 is a high MW protein with multiple protein interaction motifs. Importantly, Shank3 has a type 1 SH3 domain suitable for binding the PPNP (1165–1168 in the human PLCβ1b sequence) proline-rich sequence in the extreme C-terminal region of PLCβ1b [30]. In addition to its SH3 domain, Shank3 has an N-terminal ankyrin-rich repeat sequence that binds α-fodrin, a PDZ domain, a long proline-rich sequence that binds the Homer family of proteins and cortactin, and finally a C-terminal sterile alpha motif (SAM) that facilitates dimerization. Association with fodrin likely localizes Shank3 close to the sarcolemma. Thus, association with Shank3 makes PLCβ1b part of a multi-protein system that may be critical for downstream signaling and cellular responses (Fig. 17.3). Importantly, Shank3 is expressed in only a limited number of tissues, primarily heart and glutamatergic neurons [30], and thus the binding of PLCβ1b (also with limited tissue distribution) to Shank3 provides a possible heart-specific drug target.

(a) PLCβ1b binding to a Shank3 complex localized below the sarcolemmal membrane. Shank3 forms homodimers via its C-terminal SAM domains and is bound to α-fodrin via ank repeats in the N-terminal sequence. Dimeric Homer proteins cross-link Shank3 to TrpC channels and to intracellular Ca2+ channels. (b) Diagram showing domain structure of Shank3
PLCδ1 is expressed in heart [4], although no function has unequivocally been ascribed. PLCδ subtypes have a high affinity PH domain that shows high selectivity for PIP2 and this is sufficient to localize these to the sarcolemma [31].
PLCγ family members are activated following phosphorylation by receptor tyrosine kinases and this facilitates binding to SH2 domains present in growth factor receptors localizing these PLCs close to the plasma membrane and their substrate PIP2 [5]. As with PLCβ subtypes, localization and activation of PLCγ members may also involve binding to other signaling proteins. PLCγ subtypes have been reported to bind to sodium-hydrogen exchanger 3 (NHE3), a plasma membrane-localized ion exchanger, and regulate its activity [32]. Interestingly, PLCγ1 has been shown to interact directly with canonical transient receptor 3 (TrpC3) to control its cell surface expression [33]. TrpC3 is implicated as contributing to pathological cardiomyocyte hypertrophy [34]; however, PLCγ1 has not been implicated in this response.
As outlined earlier, PLCε is structurally more complex that other PLCs and, as a consequence of this, its regulation also is multifactorial. Like other PLC subtypes, PLCε binds to a scaffolding protein via sequences in its C-terminal region, in this case its (Ras association 1) RA1 domain. The RA1 domain of PLCε binds to the first spectrin repeat domain of muscle A-kinase-anchoring protein β (mAKAPβ) localizing this PLC subtype principally to the nuclear envelope in cardiomyocytes [35]. mAKAPβ, like Shank and NHERF proteins, is a multidomain scaffold and thus PLCε probably functions as part of a large protein complex.
4 Pathological Responses in the Heart
The primary function of the heart is to supply blood to all tissues of the body at sufficient level to optimize their function. The pump function of the heart can be compromised by a loss of contractile function of the muscle that reduces cardiac output resulting in failure to adequately supply blood to the body, a condition known as heart failure. Ineffective pumping can also be caused by a loss in organization of the contraction of the individual muscle cells, a condition known as arrhythmia. Heart failure and arrhythmia often occur together, each worsens the other and both can result from chronic hypertrophic growth of the myocardium. Because of this, there is an interest in developing therapies targeted at reducing pathological hypertrophic cardiomyocyte growth, improving contractile function (inotropic agents), or reducing arrhythmia (anti-arrhythmic agents). Currently used pharmaceuticals commonly target cell surface receptors or ion channels, their ligands, or the downstream signaling pathways, including drugs that reduce the generation or the receptor binding of angiotensin II, blockers of β-adrenergic receptors, Ca2+ channel blockers, and agents that reduce the metabolism of cAMP [36]. There is clearly a need for the development of better tolerated therapies, particularly if they can be made relatively cardiac-specific.
5 How Might PLC Activation Contribute to Pathology?
PLC enzymes hydrolyze the sarcolemmal phospholipid, PIP2, to generate Ins(1,4,5)P3 that can release Ca2+ from intracellular stores [37] and sn-1,2-diacylglycerol (DAG), an activator of conventional PKC subtypes [38], PKD [39] and some TrpC channels [40]. Each of these factors, individually and in concert, can have critical effects on cellular responses.
5.1 Ins(1,4,5)P3
Ins(1,4,5)P3 binds and activates IP3-R localized on intracellular Ca2+ stores [41]. The expression level of IP3-R in cardiomyocytes is low compared with that in most other tissues and compared with the highly expressed ryanodine receptors [42] that are primarily responsible for the intracellular Ca2+ cycling that regulates the heart beat. Furthermore, IP3-R in ventricular myocytes are localized around the nuclear membrane [43], seemingly distal from the site of generation of Ins(1,4,5)P3 following activation of cell surface receptors. These nuclear membrane-localized IP3-R(2) may supply the localized Ca2+ signals required to activate calmodulin-activated protein kinases (CaMKII) involved in transcriptional regulation [44]. Ins(1,4,5)P3 has been suggested to be involved in arrhythmogenesis [45–47] and in hypertrophy [48], although direct evidence for either of these is lacking.
5.2 DAG
The other product generated by PLC, DAG, has a complex spectrum of activities, all of which could contribute to pathology. DAG was initially discovered as an activator of PKC [38], particularly the “conventional” PKC subtypes (PKC α, β, γ, δ ε, η, θ) [49]. DAG also activates some TrpC channels [50] and protein kinase D directly [39], in addition to actions dependent on PKC. In contrast to the controversy surrounding the contribution of Ins(1,4,5)P3 and IP3-R to cardiac physiology/pathophysiology, DAG and the PKC family are well accepted as a contributor to cardiac regulation. The contribution of PKC to regulation in the heart is complex, varying with the PKC subtype, the stage of development, and the mechanism of activation. PKCα activation serves to suppress contractility [51], but can have profound pathological consequences when the regulatory domain that limits catalytic activity is removed by calpain cleavage under conditions of ischemia and reperfusion [52]. PKCβ subtypes have been shown to be involved in diabetic cardiomyopathy [53]. PKCδ has been considered an important contributor to cardiac pathology and cardiac remodeling, apparently related to activation of mitochondrial apoptotic responses [54]. PKCε primarily has a protective role in heart and is a component of preconditioning mechanism that reduces subsequent ischemic damage, discussed in more detail subsequently [55]. A recent review provides detailed information about PKC contribution to cardiac signaling under physiological and pathological conditions [56].
5.3 PIP2
The process of PLC activation depletes PIP2 as it generates Ins(1,4,5)P3 and DAG. Reductions in PIP2 are often localized and transient with the PIP2 being replaced immediately, presumably by phosphorylation of PIP [57, 58]. However, PLC-induced localized changes in PIP2 regulate ion channels and exchangers that are critical in maintaining heart rhythm [59], for a review see [40]. PIP2 is also critical for maintaining the cytoskeleton via its association with actin-binding proteins [60] and PIP2 is essential for localizing proteins to the plasma membrane [61].
6 PLC Involvement in Ischemia and Post-ischemia Reperfusion
Cardiac ischemia occurs when there is an interruption in the blood supply to the heart, depriving it of oxygen and nutrients, a condition associated with arrhythmia and cardiomyocyte death. The reintroduction of flow, reperfusion, also is associated with arrhythmia, cell death and contractile dysfunction. A number of studies have reported increased activity of PLC in animal models of acute cardiac ischemia [62–64]. Substantially increased PLC activation has been reported in early post-ischemic reperfusion following a brief period of ischemia [65–67], and inhibition of PLC under these conditions successfully prevents reperfusion arrhythmias [45, 46, 68] in addition to improving functional recovery [69]. However, the subtypes of PLC activated by ischemia/reperfusion are unknown as are the mechanisms leading to the heightened PLC response.
Increased expression of PLCβ, as well as of activating G proteins, has been reported in border zone and remote myocardium following myocardial infarction in humans, suggesting the likelihood of enhanced PLC activation [70] and pointing to a possible involvement in the heart’s responses to chronic ischemia. Other studies reported that protection from chronic ischemic damage by ethanol is mediated by elevation of PLC activity, but the subtype of PLC was not identified [71].
Defining contributions of PLC, its substrate and products, to ischemic or reperfusion responses is confounded by the likelihood that one or other of these might contribute to preconditioning, a phenomenon that can provide protection from arrhythmia and infarction following an ischemic insult [72]. Preconditioning involves subjecting hearts to brief periods of ischemia and reperfusion prior to the main ischemia/reperfusion procedure. This pretreatment procedure is sufficient to limit PLC activation in early post-ischemic reperfusion [73]. Preconditioning protection can be mimicked by activation of some of the PKC subtypes that are activated downstream of PLC, and to further complicate the situation, different PKC subtypes can have opposing effects on preconditioning [74]. Overexpression of either subtype of α1-adrenergic receptors (α1A- or α1B-) results in heightened PLC responses to endogenous or exogenous norepinephrine. However, whilst PLC activity in these overexpressing transgenic strains was heightened in normoxia, the exaggerated response during early reperfusion was eliminated, along with the reperfusion arrhythmias [75, 76]. Presumably, this apparent contradiction is related to activation of preconditioning pathways possibly initiated by PKC activation. Taken together, these studies imply that factors downstream of PLC, most likely PKC-initiated responses, effectively precondition the myocardium, and that preconditioning reduces PLC activation.
7 PLC in Acute and Chronic Dilatation of the Myocardium
The myocardium responds to acute stretch by increasing cardiac output in order to manage the increase in blood volume. Thus, acute stretch results in increased rate and force of contraction. Acute stretch of the right atrium causes substantial release of atrial natriuretic peptide, possibly to facilitate a lowering of blood volume [77]. As noted earlier, in addition to activation by ligand receptor binding, PLC in heart can be activated acutely by stretch [18–21]. In perfused rat heart preparations, right atrial stretch caused PLC activation that correlated with release of atrial natriuretic peptide [78]. Stretch activation of PLC requires Gq and may involve angiotensin II receptors (AT1) acting in a ligand-independent manner [21]. The involvement of Gq and AT1 receptors implicates PLCβ subtypes as major contributors to the response to acute stretch.
Chronically increased wall tension results in chamber dilatation and wall thinning that eventually limit contractile performance and these are the hallmarks of dilated cardiomyopathies. Dilatation of the atria is observed in patients with valve diseases and is also seen in association with ventricular failure. Interestingly, substantially heightened PLC activity was observed in the dilated atria of patients suffering from valvular heart disease, as well as in atria from a mouse model of dilated cardiomyopathy that has severe atrial enlargement together with conduction block and a sensitivity to atrial fibrillation [4, 79]. Furthermore, in both humans and mice, PLC activity correlated with atrial volume, suggesting that PLC activation was either a cause or a consequence of dilatation. Dilated atrial tissue from both humans and mice showed increased expression of only one PLC subtype, PLCβ1b, providing suggestive evidence that PLCβ1b is selectively involved in the response to chronic dilatation. There were no changes in expression of PLCβ3, PLCδ1, or PLCγ1 associated with atrial dilatation [4]. PLCε was not measured in these studies and a role for this subtype, therefore, cannot be discounted. PLCβ1a, although expressed in neonatal rat cardiomyocytes, was not expressed at measurable levels in adult human myocardium. The two splice variants of PLCβ1, PLCβ1a and PLCβ1b, differ only in their extreme C-terminal sequences as shown in Fig. 17.2. Whilst the catalytic domains and the Gαq-binding regions are identical, the differences in the C-terminal sequences would be expected to result in different localization, and consequently different activities.
Overexpression of a constitutively active Gαq is sufficient to cause severe chamber enlargement together with heightened PLC activity [80], but there are conflicting opinions about the role of PLC in promoting atrial dilatation in these Gαq-overexpressing models. Overexpression of a Gαq mutant with reduced ability to activate PLCβ, unlike the wild-type, did not result in chamber dilatation [81], providing powerful evidence for a requirement for PLC activity for the pathological responses initiated by Gq. Other studies showed that atrial remodeling in Gαq-overexpressing mice was reversed by co-expression of diacylglycerol (DAG) kinase ζ, an enzyme that depletes DAG, one of the immediate products of PLC activation [82], supporting a critical role for PLC and its immediate product, DAG, in atrial dilatation. However, in contrast to these findings, studies comparing two different Gαq-expressing transgenic lines reported that the degree of dilatation did not correlate with the extent of PLC activation [83]. These apparent discrepancies might be accounted for if there was a maximal level of PLC activation, above which further increases produced no greater effect on chamber dilatation.
At the cellular level, chamber dilatation and wall thinning are thought to involve loss of functional myocytes by apoptotic and non-apoptotic mechanisms. The ability of activated mutants of Gαq to induce apoptosis in cardiomyocytes is well documented [84], and more recently overexpression of wild-type PLCβ1b has also been shown to cause cardiomyocyte apoptosis [85]. Thus, heightened PLCβ1b activity could contribute to a dilated phenotype by promoting apoptotic death of cardiomyocytes. In summary, there is evidence for an involvement of PLC, and in particular PLCβ1b, in responses to acute and chronic dilatation of the myocardium, but the mechanisms involved remain to be established.
8 PLC Involvement in Cardiac Hypertrophy
Early studies using isolated cardiomyocytes or genetically modified mice pointed to a role for Gq family members in pathological growth and remodeling of the heart. Overexpression of Gαq, either the wild-type [86] or a constitutively active mutant [80], was sufficient to cause cardiomyocyte hypertrophy, and when expressed in vivo, Gαq promoted hypertrophy and heart failure [84]. More importantly, Gq inhibitors expressed in the heart were found to substantially reduce hypertrophic growth in response to the clinically relevant challenges of pressure or volume overload [87–89]. The apparent central role of Gq in these pathological responses suggests mediation by PLCβ subtypes, as these are the best understood effectors of Gq [90]. However, members of the Rho family of monomeric G proteins are activated downstream of Gq [91] and these may also contribute to hypertrophic responses [92].
Of the PLCβ family, only PLCβ1b causes hypertrophy when overexpressed in cardiomyocytes, and this selectivity depends on its sarcolemmal localization facilitated by selective association of the splice variant-specific C-terminal sequence with the scaffolding protein Shank3 [24, 85]. Furthermore, inhibition of PLCβ1b binding to Shank3 prevented hypertrophy in response to Gq activation [85], suggesting that the sarcolemmal targeting of PLCβ1b might provide a novel target to limit hypertrophy and chamber dilatation. Both PLCβ1b and Shank3 have a limited tissue distribution opening up the possibility of cardiac-specific therapy. In addition to cardiomyocytes, Shank3 is expressed primarily in postsynaptic density fractions from central glutamatergic neurons [30], where PLCβ1b is not expressed. In neurons, Shank3 acts as a scaffold facilitating interactions between receptors and early signaling proteins [93]. In heart, Shank3 appears to function similarly, binding fodrin [94] and Homer1c [95] in addition to its association with the C-terminal sequence of PLCβ1b. Homer1c forms homodimers that can cross-link Shank3 to form large molecular scaffolds [96]. Homers promote crosstalk between intracellular Ca2+ channels, IP3-R and ryanodine receptors, and cell surface canonical transient receptor potential channels (TrpC) and thus are regulators of local Ca2+ responses [97]. Expression of PLCβ1b in cardiomyocytes results in increased expression of Homer1c as well as its translocation to the Shank3/PLCβ1b complex [95]. The mechanisms involved in these responses are unknown, but they appear to be critical for the hypertrophic response.
The possibility that PLCε was involved in cardiac pathology was first suggested when elevated expression was reported in failed human left ventricle [16]. This idea was supported by studies showing that PLCε−/− mice exhibited exacerbated hypertrophic responses leading to the idea that PLCε, in contrast to PLCβ1b, was protective to the myocardium by inhibiting hypertrophic signaling. However, subsequent studies in isolated cardiomyocytes have questioned this conclusion. These studies found that treatment with si-RNA to knockdown PLCε inhibited hypertrophy in response to endothelin or α1-adrenergic agonists [35], implying an involvement in Gq-initiated hypertrophy that other studies have shown involves PLCβ1b [85]. Importantly, PLC activity was absolutely required for this contribution of PLCε to hypertrophy, an important finding given the multiple functions of this complex PLC subtype. In cardiomyocytes, PLCε is localized onto the nuclear membrane by association with muscle A-kinase-activating protein (mAKAPβ, AKAP5) [35]. Such localization is suggestive of a role downstream of early signaling responses, such as initiated by PLCβ1b. In agreement with this, knockdown of PLCε inhibited hypertrophy in response to multiple stimuli, including both Gq hypertrophy that models pathological hypertrophy and hypertrophy caused by IGF treatment, considered a model of physiological hypertrophy that is independent of Gq [35]. This contrasts to PLCβ1b, where inhibition selectively prevented Gq-mediated hypertrophy [85]. There is clearly substantial evidence for an involvement of PLC in hypertrophy of the myocardium, with current data supporting roles for PLCβ1b and PLCε, most likely at different stages in the signaling response.
9 Conclusions
Under physiological conditions the functioning of the heart is regulated primarily by pathways that are independent of PLC activation. However, PLC expression and activity have been shown to increase under a range of pathological conditions, including ischemia/reperfusion, hypertrophy, and dilatation and it is likely that PLC contribute to the progression of these diseases.
References
Rhee SG (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70:281–312
Exton JH (1994) Phosphoinositide phospholipases and G proteins in hormone action. Annu Rev Physiol 56:349–369
Arthur JF, Matkovich SJ, Mitchell CJ et al (2001) Evidence for selective coupling of α1-adrenergic receptors to phospholipase Cβ1 in rat neonatal cardiomyocytes. J Biol Chem 276:37341–37346
Woodcock EA, Grubb DR, Filtz TM et al (2009) Selective activation of the “b” splice variant of phospholipase Cβ1 in chronically dilated human and mouse atria. J Mol Cell Cardiol 47:676–683
Gresset A, Hicks SN, Harden TK, Sondek J (2010) Mechanism of phosphorylation-induced activation of phospholipase Cγ isozymes. J Biol Chem 285:35836–35847
Shen E, Fan J, Chen R et al (2007) Phospholipase Cγ1 signalling regulates lipopolysaccharide-induced cyclooxygenase-2 expression in cardiomyocytes. J Mol Cell Cardiol 43:308–318
Allen V, Swigart P, Cheung R et al (1997) Regulation of inositol lipid-specific phospholipase Cδ by changes in Ca2+ ion concentrations. Biochem J 327:545–552
Woodcock EA, Mitchell CJ, Biden TJ (2003) Phospholipase Cδ1 does not mediate Ca2+ responses in neonatal rat cardiomyocytes. FEBS Lett 546:325–328
Kelley GG, Reks SE, Ondrako JM, Smrcka AV (2001) Phospholipase Cε: a novel Ras effector. EMBO J 20:743–754
Sorli SC, Bunney TD, Sugden PH et al (2005) Signaling properties and expression in normal and tumor tissues of two phospholipase Cε splice variants. Oncogene 24:90–100
Woodcock EA, White LBS, Smith AI, McLeod JK (1987) Stimulation of phosphatidylinositol metabolism in the isolated, perfused rat heart. Circ Res 61:625–631
Brown SL, Brown JH (1983) Muscarinic stimulation of phosphatidylinositol metabolism in atria. Mol Pharmacol 24:351–356
Kuraja IJ, Tanner JK, Woodcock EA (1990) Endothelin stimulates phosphatidylinositol turnover in rat right and left atria. Eur J Pharmacol 189:299–306
Wu D, Lee C, Rhee S, Simon M (1992) Activation of phospholipaseC by the α subunits of the Gq and G11 proteins in transfected cos-7 cells. J Biol Chem 25:1811–1817
Ibarra C, Estrada M, Carrasco L et al (2004) Insulin-like growth factor-1 induces an inositol 1,4,5-trisphosphate-dependent increase in nuclear and cytosolic calcium in cultured rat cardiac myocytes. J Biol Chem 279:7554–7565
Wang H, Oestreich EA, Maekawa N et al (2005) Phospholipase Cε modulates β-adrenergic receptor dependent cardiac contraction and inhibits cardiac hypertrophy. Circ Res 97:1305–1313
Kelley GG, Reks SE, Smrcka AV (2004) Hormonal regulation of phospholipase Cε through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem J 378:129–139
von Harsdorf R, Lang R, Woodcock EA (1989) Dilatation of the right atrium stimulates phosphatidylinositol turnover. Clin Exp Pharmacol Physiol 16:341–344
von Harsdorf R, Lang R, Fullerton M, Woodcock EA (1989) Myocardial stretch stimulates phosphatidylinositol turnover. Circ Res 65:494–501
Sadoshima J, Izumo S (1993) Mechanical stretch rapidly activates multiple signal transduction pathways in cardiac myocytes—potential involvement of an autocrine/paracrine mechanism. EMBO J 12:1681–1692
Storch U, Schnitzler MMY, Gudermann T (2012) G protein-mediated stretch reception. Am J Physiol 302:H1241–H1249
Hwang JI, Kim HS, Lee JR et al (2005) The interaction of phospholipase Cβ3 with Shank2 regulates mGluR-mediated calcium signal. J Biol Chem 280:12467–12473
Hwang JI, Heo K, Shin KJ et al (2000) Regulation of phospholipase Cβ3 activity by Na+/H+ exchanger regulatory factor 2. J Biol Chem 275:16632–16637
Grubb DR, Vasilevski O, Huynh H, Woodcock EA (2008) The extreme C-terminal region of phospholipase Cβ1 determines subcellular localization and function; the “b” splice variant mediates α1-adrenergic receptor responses in cardiomyocytes. FASEB J 22:2768–2774
Tang Y, Tang J, Chen Z et al (2000) Association of mammalian Trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J Biol Chem 275:37559–37564
Mahon MJ, Segre GV (2004) Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase Cβ, and actin increases intracellular calcium in opossum kidney cells. J Biol Chem 279:23550–23558
Suh PG, Hwang JI, Ryu SH et al (2001) The roles of PDZ-containing proteins in PLCβ-mediated signaling. Biochem Biophys Res Commun 288:1–7
Kaneko T, Li L, Li SS (2008) The SH3 domain—a family of versatile peptide- and protein-recognition module. Front Biosci 13:4938–4952
Schlundt A, Sticht J, Piotukh K et al (2009) Proline-rich sequence recognition. Mol Cell Proteomics 8:2474–2486
Lim S, Naisbitt S, Yoon J et al (1999) Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J Biol Chem 274:29510–29518
Vasilevski O, Grubb DR, Filtz TM et al (2008) Ins(1,4,5)P3 regulates phospholipase Cβ1 expression in cardiomyocytes. J Mol Cell Cardiol 45:679–684
Zachos NC, van Rossum DB, Li XH et al (2009) Phospholipase Cγ binds directly to the Na+/H+ exchanger 3 and is required for calcium regulation of exchange activity. J Biol Chem 284:19437–19444
van Rossum DB, Patterson RL, Sharma S et al (2005) Phospholipase Cγ1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature 434:99–104
Onohara N, Nishida M, Inoue R et al (2006) TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J 25:5305–5316
Zhang L, Malik S, Kelley GG et al (2011) Phospholipase Cε scaffolds to muscle-specific A kinase anchoring protein (mAKAPβ) and integrates multiple hypertrophic stimuli in cardiac myocytes. J Biol Chem 286:23012–23021
Rauch H, Motsch J, Bottiger BW (2006) Newer approaches to the pharmacological management of heart failure. Curr Opin Anaesthesiol 19:75–81
Streb H, Bayerdorffer E, Haase W et al (1984) Effect of inositol-1,4,5-trisphosphate on isolated subcellular fractions of rat pancreas. J Membr Biol 81:241–253
Nishizuka Y (1984) Protein kinases in signal transduction. Trends Biochem Sci 9:163–166
Rybin VO, Guo J, Harleton E et al (2012) Regulatory domain determinants that control PKD1 activity. J Biol Chem 287:22609–22615
Woodcock EA, Kistler PM, Ju YK (2009) Phosphoinositide signalling and cardiac arrhythmias. Cardiovasc Res 82:286–295
Streb H, Irvine R, Berridge M, Schulz I (1983) Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306:67–68
Marks AR (2000) Cardiac intracellular calcium release channels: role in heart failure. Circ Res 87:8–11
Wu X, Bers DM (2006) Sarcoplasmic reticulum and nuclear envelope are one highly interconnected Ca2+ store throughout cardiac myocyte. Circ Res 99:283–291
Wu X, Zhang T, Bossuyt J et al (2006) Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest 116:675–682
Jacobsen AN, Du XJ, Dart AM, Woodcock EA (1997) Ins(1,4,5)P3 and arrhythmogenic responses during myocardial reperfusion: evidence for receptor specificity. Am J Physiol 42:H1119–H1125
Du X-J, Anderson K, Jacobsen A et al (1995) Suppression of ventricular arrhythmias during ischaemia-reperfusion by agents inhibiting Ins(1,4,5)P3 release. Circulation 91:2712–2716
Li X, Zima AV, Sheikh F et al (2005) Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate (IP3)-receptor type 2-deficient mice. Circ Res 96:1274–1281
Nakayama H, Bodi I, Maillet M et al (2010) The IP3 receptor regulates cardiac hypertrophy in response to select stimuli. Circ Res 107:659–666
Newton AC (2009) Lipid activation of protein kinases. J Lipid Res 50(suppl):S266–S271
Lemonnier L, Trebak M, Putney JW (2008) Complex regulation of the TRPC3, 6 and 7 channel subfamily by diacylglycerol and phosphatidylinositol-4,5-bisphosphate. Cell Calcium 43:506–514
Braz JC, Gregory K, Pathak A et al (2004) PKCα regulates cardiac contractility and propensity toward heart failure. Nat Med 10:248–254
Zhang Y, Matkovich SJ, Duan XJ et al (2011) Receptor-independent protein kinase Cα (PKCα) signaling by calpain-generated free catalytic domains induces HDAC5 nuclear export and regulates cardiac transcription. J Biol Chem 286:26943–26951
Inoguchi T, Battan R, Handler E et al (1992) Preferential elevation of protein kinase C isoform βII and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A 89:11059–11063
Murriel CL, Churchill E, Inagaki K et al (2004) Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage—a mechanism involving BAD and the mitochondria. J Biol Chem 279:47985–47991
Ping PP, Zhang J, Qiu YM et al (1997) Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res 81:404–414
Steinberg SF (2012) Cardiac actions of protein kinase C isoforms. Physiology 27:130–139
Nasuhoglu C, Feng SY, Mao YP et al (2002) Modulation of cardiac PIP2 by cardioactive hormones and other physiologically relevant interventions. Am J Physiol 283:C223–C234
Meyer T, WellnerKienitz MC, Biewald A et al (2001) Depletion of phosphatidylinositol 4,5-bisphosphate by activation of phospholipase C-coupled receptors causes slow inhibition but not desensitization of G protein-gated inward rectifier K+ current in atrial myocytes. J Biol Chem 276:5650–5658
Cho H, Kim YA, Yoon JY et al (2005) Low mobility of phosphatidylinositol 4,5-bisphosphate underlies receptor specificity of Gq-mediated ion channel regulation in atrial myocytes. Proc Natl Acad Sci U S A 102:15241–15246
Nebl T, Oh SW, Luna EJ (2000) Membrane cytoskeleton: PIP2 pulls the strings. Curr Biol 10:R351–R354
Falkenburger BH, Jensen JB, Dickson EJ et al (2010) Phosphoinositides: lipid regulators of membrane proteins. J Physiol 588:3179–3185
Schwertz D, Halverson J, Isaacson T et al (1987) Alterations on phospholipid metabolism in the globally ischemic rat heart: emphasis on phosphoilositide specific phospholipase C activity. J Mol Cell Cardiol 19:685–697
Corr PB, Yamada KA, DaTorre SD (1990) Modulation of α-adrenergic receptors and their intracellular coupling in the ischemic heart. Basic Res Cardiol 85(suppl 1):31–45
Woodcock E, Lambert K, Phan T, Jacobsen A (1997) Inositol phosphate metabolism during myocardial ischemia. J Mol Cell Cardiol 29:449–460
Anderson K, Dart A, Woodcock E (1995) Inositol phosphate release and metabolism during myocardial ischemia and reperfusion in rat heart. Circ Res 76:261–268
Lochner A, Tromp E, Mouton R (1996) Signal transduction in myocardial ischaemia and reperfusion. Mol Cell Biochem 161:129–136
Huisamen B, Mouton R, Opie LH, Lochner A (1994) Effects of ischaemia, reperfusion and α1-adrenergic receptor stimulation on the inositol trisphosphate receptor population in rat heart atria and ventricles. Mol Cell Biochem 140:23–30
Jacobsen AN, Du XJ, Lambert KA et al (1996) Arrhythmogenic action of thrombin during myocardial reperfusion via release of inositol 1,4,5-triphosphate. Circulation 93:23–26
Asemu G, Dhalla NS, Tappia PS (2004) Inhibition of PLC improves postischemic recovery in isolated rat heart. Am J Physiol 287:H2598–H2605
Ju H, Zhao S, Tappia PS et al (1998) Expression of Gqα and PLCβ in scar and border tissue in heart failure due to myocardial infarction. Circulation 97:892–899
Miyamae M, Domae N, Zhou HZ et al (2003) Phospholipase C activation is required for cardioprotection by ethanol consumption. Exp Clin Cardiol 8:184–188
Downey JM (1992) Ischemic preconditioning—nature’s own cardioprotective intervention. Trends Cardiovasc Med 2:170–176
Anderson KE, Woodcock EA (1995) Preconditioning of perfused rat heart inhibits reperfusion-induced release of inositol(1,4,5)trisphosphate. J Mol Cell Cardiol 27:2421–2431
Duquesnes N, Lezoualc’h F, Crozatier B (2011) PKCδ and PKCε: foes of the same family or strangers? J Mol Cell Cardiol 51:665–673
Harrison SN, Autelitano DJ, Wang BH et al (1998) Reduced reperfusion-induced Ins(1,4,5)P3 generation and arrhythmias in hearts expressing constitutively active α1B-adrenergic receptors. Circ Res 83:1232–1240
Amirahmadi F, Turnbull L, Du XJ et al (2008) Heightened α1A-adrenergic receptor activity suppresses ischaemia/reperfusion-induced Ins(1,4,5)P3 generation in the mouse heart: a comparison with ischaemic preconditioning. Clin Sci (Lond) 114:157–164
Lang RE, Tholken H, Ganten D et al (1985) Atrial natriuretic factor—a circulating hormone stimulated by volume loading. Nature 314:264–266
von Harsdorf R, Lang R, Fullerton M et al (1988) Right atrial dilatation increases inositol-(1,4,5)trisphosphate accumulation: implications for the control of atrial natriuretic peptide secretion. FEBS Lett 233:201–215
Pretorius L, Du XJ, Woodcock EA et al (2009) Reduced phosphoinositide 3-kinase (p110α) activation increases the susceptibility to atrial fibrillation. Am J Pathol 175:998–1009
Mende U, Kagen A, Cohen A et al (1998) Transient cardiac expression of constitutively active Gαq leads to hypertrophy and dilated cardiomyopathy by calcineurin-dependent and independent pathways. Proc Natl Acad Sci U S A 95:13893–13898
Lu Z, Jiang YP, Ballou LM et al (2005) Gαq inhibits cardiac L-type Ca2+ channels through phosphatidylinositol 3-kinase. J Biol Chem 280:40347–40354
Hirose M, Takeishi Y, Niizeki T et al (2009) Diacylglycerol kinase ζ inhibits Gαq-induced atrial remodeling in transgenic mice. Heart Rhythm 6:78–84
Mende U, Semsarian C, Martins DC et al (2001) Dilated cardiomyopathy in two transgenic mouse lines expressing activated G protein αq: lack of correlation between phospholipase C activation and the phenotype. J Mol Cell Cardiol 33:1477–1491
Adams JW, Sakata Y, Davis MG et al (1998) Enhanced Gαq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A 95:10140–10145
Filtz TM, Grubb DR, McLeod-Dryden TJ et al (2009) Gq-initiated cardiomyocyte hypertrophy is mediated by phospholipase Cβ1b. FASEB J 23:3564–3570
Sakata Y, Hoit BD, Liggett SB et al (1998) Decompensation of pressure-overload hypertrophy in Gαq-overexpressing mice. Circulation 97:1488–1495
Akhter SA, Luttrell LM, Rockman HA et al (1998) Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280:574–577
Esposito G, Rapacciuolo A, Naga Prasad SV et al (2002) Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 105:85–92
Wettschureck N, Rutten H, Zywietz A et al (2001) Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Gαq/Gα11 in cardiomyocytes. Nat Med 7:1236–1240
Smrcka AV, Hepler JR, Brown KO, Sternweis PC (1991) Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 251:804–807
Shankaranarayanan A, Thal DM, Tesmer VM et al (2008) Assembly of high order Gαq-effector complexes with RGS proteins. J Biol Chem 283:34923–34934
Rojas RJ, Yohe ME, Gershburg S et al (2007) Gαq directly activates p63RhoGEF and Trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J Biol Chem 282:29201–29210
Kreienkamp HJ (2008) Scaffolding proteins at the postsynaptic density: shank as the architectural framework. Handb Exp Pharmacol 186:365–380
Grubb DR, Iliades P, Cooley N et al (2011) Phospholipase C β1b associates with a Shank3 complex at the cardiac sarcolemma. FASEB J 25:1040–1047
Grubb DR, Luo JT, Yu YL, Woodcock EA (2012) Scaffolding protein Homer 1c mediates hypertrophic responses downstream of Gq in cardiomyocytes. FASEB J 26:596–603
Tu JC, Xiao B, Naisbitt S et al (1999) Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23:583–592
Yuan JP, Lee KP, Hong JH, Muallem S (2012) The closing and opening of TRPC channels by Homer1 and STIM1. Acta Physiol 204:238–247
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Woodcock, E.A. (2014). Phospholipase C Signaling in Heart Disease. In: Tappia, P., Dhalla, N. (eds) Phospholipases in Health and Disease. Advances in Biochemistry in Health and Disease, vol 10. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0464-8_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0464-8_17
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0463-1
Online ISBN: 978-1-4939-0464-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)