
Abstract
Phosphoinositide kinases (PIKs) are a group of lipid kinases that are important upstream activators of various signaling pathways that drive oncogenesis. Hyperactivation of the PI3K/AKT/mTOR pathways—either via mutations or genomic amplification—confers key oncogenic activity, essential for the development and progression of several solid tumors. Alterations in the PIK3CA gene are associated with poor prognosis of solid malignancies. Contradictory reports exist in the literature regarding the prognostic value of PIK3CA in aggressive cancers, but most available data highlights an important role of PIK3CA mutation in mediating tumorigenesis via increased signaling of the PI3K/AKT/mTOR survival pathway. Several inhibitors of PI3K/AKT/mTOR pathways have been investigated as potential therapeutic options in solid malignancies. This article reviews the role of PIK3CA mutations and inhibitors of the PI3K/AKT/mTOR pathway in cancer and examines association with the clinico-pathological parameters and prognosis.
Similar content being viewed by others


Introduction
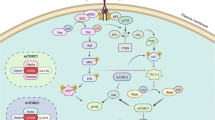
Phosphoinositide kinases (PIKs) are a group of lipid kinases that act as signal transducers in various signaling pathways. After a cell receives growth factors and extracellular signals through receptor tyrosine kinases and G-protein coupled receptors, PI3K is activated by GTP-bound RAS to propagate signaling by phosphorylating membrane-bound PIP2 to PIP3. This leads to the activation of AKT (protein kinase B) which promotes cellular growth by phosphorylating key cell cycle and metabolic proteins, including the mammalian target of rapamycin (mTOR) and glycogen synthase kinase 3 [1].
PI3K is a family of proteins grouped into various classes based on structure and function. Class IA PI3Ks have been extensively researched in cancer due to their high level of mutation and influence on cell growth and survival [2]. They contain one of several catalytic isoforms (p110α, p110β, p110δ, p110γ) and associated regulatory proteins (p85α, p85β, p55α, p55γ, p50α) [2]. The catalytic isoforms contain a catalytic domain, helical domain, RAS-binding domain, and a p85-binding domain. The p85 regulatory proteins contain a Src-Homology 2 (SH2) domain to allow for the binding of catalytic domain subunits [2]. PIK3CA is the gene transcribed into the p110 catalytic proteins of functional PI3K. Of the p110 catalytic proteins, p110α is frequently mutated, whereas the other three can be overexpressed but are rarely mutated [2]. In fact, when compared with 15 PI3K genes and PI3K-like genes, PIK3CA was the only one with somatic mutations [3].
PIK3CA mutations are almost exclusively gain of function missense mutations, and 80% of PIK3CA mutations are found at two locations: the H1047R helical domain at Glu-542, Glu-545, and Gln-546, and the kinase domain at His-1047 [4, 5]. Specifically, helical domain mutations allow for the accumulation of positive charge within the membrane binding domains, while kinase domain mutations lead to enzymatic hyperactivity [4]. Because these mutations can lead to functional changes, mutated PIK3CA can drastically alter neoplastic cell signaling, allowing cells to embody the inflammatory, angiogenic, and survival hallmarks seen in the multistep process of oncogenesis [6,7,8].
Identifying the location and prevalence of PIK3CA mutations by cancer
PIK3CA is located on chromosome 3q, and has five domains, including the adaptor-binding domain (ABD) of amino acid (AA) residues 1–108, Ras-binding domain (RBD) from AAs 190 to 291, C2 domain from AAs 330 to 480, helical domain from AAs 525 to 696, and kinase domain from AAs 697 to 1068 [9]. ABD is important for binding the regulatory partner p85a, C2 is implicated in binding cell membranes, the helical domain has unknown function, and the kinase domain is catalytic [9]. For the purposes of this paper, we have called AAs 109–189 interdomain (ID) 1, AAs 292–329 ID2, and AAs 481–524 ID3. The PIK3CA mutation rates seen at each domain of various cancers are represented in Fig. 1.

As previously stated, the most common PIK3CA mutations are present in genomic hotspots at the Glu-542, Glu-545, and Gln-546 locations on the helical domain and the His-1047 location of the kinase domain. The most common of the mutations found here are H1047R, E542K, and E545K [4, 5]. Not only is PIK3CA the third most mutated gene found in all cancers, but the E545K, H1047R, and E542K represent three of the top eight overall mutations [10].
Over the past several years, there has been an explosion in the availability of cancer genomics data, allowing for a greater understanding of the uniqueness of individual tumors, as well as the effects of gene alteration on the cancer growth and prognosis. While clinical and laboratory research into the significance of PIK3CA mutations in chemotherapeutic resistance has been somewhat limited up to this point, large-scale clinical genomics databases allow for a broader perspective comparing these data. Within the past decade, these genomic databases have grown, providing a more transparent molecular picture of different cancers for the future development of precision therapies. In order to determine the relative rate of PIK3CA mutations in solid malignancies, we have analyzed data from four of these cancer genomics projects: Catalogue of Somatic Mutations in Cancer (COSMIC), My Cancer Genome (MCG), the Cancer Genome Atlas (TCGA), and cBioPortal [11,12,13]. The patient data from these projects are not mutually exclusive, but provide a means of independent validation. The data from Fig. 1 show that the location of mutations varies somewhat between each of these solid cancers.
Bladder
Bladder cancers are the ninth most common worldwide malignancy, and are described based on their originating epithelium and whether or not they have invaded the local muscle. Most bladder cancers originate from bladder urothelium, while less frequent subtypes include squamous cell and adenocarcinomas, among others [14]. At diagnosis, treatment can differ based on whether the cancer remains confined to the bladder as a non-muscle invasive (NMIBC) papillary tumor, as seen in ~75% of cases, or whether it has invaded the local muscle (MIBC) [14]. Patients with NMIBC are more likely to respond to treatment, whereas MIBC patients are more likely to develop metastasis [14]. While cisplatin-based chemotherapies are widely recognized as first line therapies, there are no other widely recognized options [15].
According to the genomics databases, PIK3CA is mutated in 18.7–22.5% of all bladder cancers (Table 1). Activating PIK3CA mutations occur in both NMIBC and MIBCs. In NMIBCs, they are often associated with FGFR3 mutations [14]. According to previous publications by TCGA and Zeng et al., PIK3CA mutations in bladder cancer are mainly located at the E545K and E542K location, with few mutations located at H1047R (2.2%) [16]. Similar data were seen in the cBioPortal search (Fig. 1).
As shown in Table 2, bladder cancers with hotspot mutations may be susceptible to PI3K \inhibitors. Malignant cells containing E545K mutations (in vitro) and H1047R (PDX model) have been shown to be more sensitive to pictilisib than WT cells. In vitro data additionally demonstrate that malignant cells with E542K mutations may be less sensitive than E545K mutations to cisplatin and dual inhibitors. In addition, target modulation and computer modeling shows that E545K binds drugs more tightly than E542K. Considering the prevalence of these specific mutations, these preclinical results offer potential targets in a large subset of cancer patients, but further clinical data is needed.
Breast
Breast cancer (BrC) is a heterogeneous disease, the majority appearing morphologically as ductal and lobular cancers, while having hormone-receptor positive (HR+) (75%), HER2 positive (15–20%), and triple negative (TNBC) molecular signatures [17]. HR positive BrCs are the most common subtype, and carry the best prognosis. According to the clinical genomics databases, PIK3CA is mutated in 19.1–36.7% of BrCs, and TCGA demonstrated that it is only one of three genes mutated in >10% of human breast tumors [18].
The rate of mutated PIK3CA differs between HR+ BrCs, HER2 BrCs, and TNBCs. In HR positive cancers, we have begun to target PIK3CA mutations for clinical benefit [19]. While benefit in early BrC when in combination with letrozole was limited, alpelisib has shown recent success in treating patients with HR+, Her2− advanced BrCs with H1047R or E545K mutations (ongoing Solar 1 Trial [20]). In these patients, alpelisib was shown to have a potent, low-dose effect, and increased progression-free survival (PFS) by 5.3 months as compared with placebo. Further work is being done testing the effects of alpelisib in other BrC subtypes in numerous clinical trials testing different drug combinations and indications for use [21]. Studies have shown that the addition of everolimus, an mTOR inhibitor, to hormone modulators can increase PFS, but the effects of PIK3CA mutations are still unknown [22]. PI3K is upstream of mTOR and activates AKT which in turn activates mTOR. Thus, inhibition of active mTOR may indirectly benefit patients with activating PIK3CA mutations. Several studies have shown that H1047R mutant PIK3CA in HER2 positive BrC cells provides a therapeutic window (Table 2). A recent retrospective study by Yi et al. suggest that H1047R may be a good marker for increased sensitivity to everolimus, leading to increased PFS in HR+ BrC [23]. However, these results have not been confirmed through prospective clinical trials. TNBC tends to have higher levels of amplified, but not mutated, PI3K pathway proteins [18].
In general, H1047R appears to be the most common mutation and is associated with less response to chemotherapy. Preclinical work has demonstrated that H1047R mutations are more resistant to trastuzumab or the combination of trastuzumab with lapatinib, pertuzumab, or paclitaxel. Helical domain mutations (including E545K) are more likely to have greater PFS and respond to everolimus and letrozole in HR+ BrC. Both mutations, however, show sensitivity to AKT inhibitors (Table 2). Due to the prevalence of H1047R mutations and its unfavorable prognosis, it is imperative to develop targeted therapies to cancers with this molecular signature.
Cervical
Over 300,000 people die every year from cervical cancer, most often due to human papillomavirus (HPV) infection [24]. Bivalent, quadrivalent, and nine-valent vaccines are now available for protection, and Papanicolaou smears help to detect asymptomatic cervical cancer and its precursor lesions. As seen in our search, PIK3CA is mutated in 9.6–26.5% of cervical cancers (Table 1).
In a recent Japanese study, there was limited efficacy of PIK3CA mutation status as a biomarker for tumor response to PI3K inhibitor treatment. However, preclinical work shows that PI3K inhibitors can decrease oncogenic activity in E545K mutated cells. Further research should look to see whether it is possible to treat this mutation in particular, due to its prevalence in cervical cancers.
Colorectal
Over one million people are diagnosed with colorectal cancer (CRC) each year [25]. Most CRCs are sporadic cancers that display genetic, morphologic, and anatomic heterogeneity, while often following a common sequential pattern of genetic mutations [25,26,27]. There are several well-described molecular mechanisms associated with a diverse array of CRCs, including chromosome instability (CIN), microsatellite instability (MSI), and CpG island methylator phenotype (CIMP) pathways. Morphological development commonly occurs among two specific pathological staging processes: most develop from adenomas to adenocarcinomas, while 5–10% develop from serrated polyps to CRC [25]. In addition, CRCs can have varying signatures based on anatomic location (i.e., left or right sided colon, rectum).
Based on the genomics databases surveyed, PIK3CA is mutated in 12.7–18.5% of CRC tumors (Table 1). Of the different genetic backgrounds, PIK3CA mutations are seen at highest levels in genomically stable cancers. Of the morphologically different CRCs, PIK3CA mutations play a role in the later development following adenoma or serrated precancerous disease [25]. The incidence of PIK3CA mutations varies on the anatomical site–cecal cancers have the highest PIK3CA mutation rate (30%), while non-rectal colon cancers have 18% mutation rate and rectal cancers have only 9% (Table 1). In addition, while PIK3CA mutations are implicated in CRC oncogenesis, PIK3CA amplification was not found to be a common mechanism of oncogenesis [3].
Preclinical data demonstrate that H1047R mutations cause resistance to 5-FU therapy and FOLFOX, which can be restored with perifosine/LY294002 or PI3K/AKT inhibitor, respectively [28]. In addition, treating H1047R mutant CRC stem cells with dual PI3K/mTOR inhibitor can slow tumor growth. Further clinical data regarding the effect of E545K mutation on treatment in CRCs is needed.
Endometrial
Endometrial cancer (EC) is the most common gynecologic malignancy in the United States [29]. Most ECs can be cured with surgery alone, while radiation, chemotherapy, and hormonal therapies may be used to treat more advanced cancers. Histologically, ECs can be broken down into Type 1, hormone-driven endometrioid cancers, or Type 2, serous, clear cell, or high grade ECs, which present with a worse prognosis [30]. There are few options for patients with high risk, early stage cancers, recurrent cancers, or advanced Type 2 cancers [29].
Of the genomics databases surveyed, ECs contain the highest rate of PIK3CA mutations of any cancer, ranging from 21.8 to 45.0% (Table 1). Of the most common EC histological subtypes, clear-cell carcinoma, adenocarcinoma, endometrioid, and serous each contains different PIK3CA expression rates. TCGA lists PIK3CA as a commonly mutated gene in both Type I and Type II cancers [31].
What is different about ECs as compared with the other cancers we have surveyed is the frequency of EC mutations within the ABD. ECs show significantly more PIK3CA percentage mutation in the ABD, and significantly less in the helical and kinase domains as compared with the other cancers studied. This correlates with previously published findings [32].
While there have been advancements in treating patients with HER2 positive uterine serous cancers [33], subsequent in vitro studies have suggested that the PIK3CA H1047R and E545K activating mutations are associated with resistance to HER2 specific therapies in gynecological cancers [34]. However, some preclinical data suggest that E542K can be a biomarker for cancers that can be treated with PI3K pathway inhibitors. Further clinical analysis needs to assess the clinical significance of these mutations in treatment and the extent that these two mutations affect resistance to trastuzumab.
Head and neck
Head and neck squamous cell carcinomas (HNSCCs) affect over 600,000 people worldwide each year, and approximately half will die of the disease. These cancers are most associated with smoking and human papillomavirus (HPV) infection, and each carries distinct molecular signatures [35]. Of these, HPV negative, smoking associated cancers comprise the majority of the cases [36]. In addition, HNSCCs can be categorized into gene expression subtypes of basal, mesenchymal, atypical, and classical within oral and laryngeal anatomic groupings. In both oral and laryngeal cancers, the classical subtype had the worst overall survival [36].
Previous research has demonstrated that nearly one-third of all HSNCC tumors contain PI3K pathway mutations, with PIK3CA being the most commonly mutated (and amplified) of these [37]. MCG identifies PIK3CA as the third most common mutation in HNSCCs (TP53, CDKN2A). The most common HNSCC PIK3CA mutations are seen in the same helical and kinase domain hotspots as other cancers [37, 38]. TCGA demonstrated, using data from COSMIC searches, that PIK3CA is the only known oncogene present in HNSCCs with statistically significant mutation levels [38]. In addition, TCGA noted that approximately one-quarter of PIK3CA-mutated HNSCCs displayed concurrent amplification [38]. From reviewing the genomics databases, HNSCCs have PIK3CA mutation rates between 7.2 and 20.7% (Table 1).
Importantly, TCGA notes that PIK3CA alterations represent the most common potential target for therapeutics in both HPV positive and negative patients. In addition, previous work has demonstrated that HNSCCs with H1047R mutations may be sensitive to BYL719 (alpelisib) and BEZ235. However, E545K has responsiveness of unknown significance. Alpelisib has shown preclinical success in treating HNSCCs and clinical success in lengthening PFS in patients with E545K or H1047R mutations in certain BrCs, and its efficacy is currently being measured in several clinical trials for HNSCCs [39]. While some of the preclinical data on targeting cells with kinase domain mutations is promising, helical domain mutations are far more common in HNSCCs (Fig. 1). Further research is necessary to determine how to target E545K mutant HNSCCs and how to make the H1047R mutated cancers targetable in the clinic.
Ovarian
Ovarian carcinomas are the second most common malignancies of the female genital tract, and are the fifth leading causes of cancer in women. Most ovarian carcinomas are epithelial in origin, including serous, endometrioid, clear-cell, and mucinous histological subtypes. Among these, there were noticeable differences in PIK3CA mutation rates [40]. Clear-cell and endometrioid ovarian carcinomas had high rates of PIK3CA mutations, whereas serous had far lower rates. Overall, PIK3CA was mutated in 8.1–15.9% of cases from the genomics databases (Table 1). Of note, ovarian carcinomas contain the second highest frequency of PIK3CA amplifications in any cancer, with amplifications seen in 18–29% of tumors [8].
There is a lack of clinical and preclinical data available regarding the effects of PIK3CA mutations on therapy resistance and sensitivity (Table 2). Future work should look to determine how to target ovarian cancers with these mutational signatures.
Inhibitors of the PI3K signaling pathway: current status, challenges and future directions
As discussed earlier, hyperactivation of PI3K signaling is a hallmark of cancer and activating mutations in this pathway are common in solid malignancies [3]. With this observation in mind, and given the multiple cellular processes controlled by the pathway (metabolism, motility, growth, and proliferation), it is no surprise to see the development of a plethora of PI3K pathway antagonists. More than 40 different inhibitors of different components of this pathway have reached various stages of clinical development, including PI3K and AKT inhibitors, as well as allosteric mTOR and catalytic mTOR inhibitors [41]. Yet, and despite initial enthusiasm, only five have been approved by FDA for clinical use in cancer patients: the mTOR inhibitors temsirolimus and everolimus, and the PI3K inhibitors idelalisib, copanlisib and alpelisib. In this section, we will focus on current inhibitors approved for clinical use, briefly discuss the trials leading to approval, as well as the challenges facing the clinical development of PI3K inhibitors. We will also discuss potential strategies to improve the impact of PI3K/AKT/mTOR inhibitors in solid tumors, and the potential role of PIK3CA mutation as a marker of sensitivity to PI3K inhibitors.
Temsirolimus
Temsirolimus is an allosteric inhibitor of mTORC1. It was approved in 2007 by the FDA for advanced-stage renal cell carcinoma (RCC) based on the Global ARCC phase III trial [42]. In this multicenter trial, 626 untreated patients with poor-prognosis metastatic RCC were randomized to receive temsirolimus monotherapy, or interferon-alpha (INF-α), or a combination of both. The trial compared overall survival (OS; the primary endpoint) between the three groups. Results were consistent with prolonged OS (10.9 months vs. 7.3 months; HR for death, 0.73; CI: 0.58–0.92; P = 0.008) and PFS in the temsirolimus monotherapy group (P < 0.001) compared with INF-α monotherapy, with no significant difference between the combination group and the INF-α group. In terms of toxicities, rash, hyperglycemia and hyperlipidemia were common in the temsirolimus group with few patients with serious adverse events (SAEs) in the temsirolimus monotherapy group vs. interferon group (P = 0.02).
Everolimus
Everolimus is also a derivative of rapamycin and acts as an allosteric inhibitor of mTORC1. Based on multiple clinical trials, it is currently approved for advanced-stage RCC, HR+/HER2− breast cancer in postmenopausal women, in pancreatic neuroendocrine tumors (NETs), adult renal angiolipoma (associated with tuberous sclerosis complex), as well as pediatric and adult ependymal giant cell astrocytoma [43,44,45,46]. As a monotherapy for previously treated advanced RCC (n = 272), everolimus was associated with prolonged PFS compared with placebo (n = 138; 4.0 months vs. 1.9 months; HR 0.3; CI 0.22–0.40; P < 0.0001) without translating into significant improvement in OS or in ORR, in the phase III RECORD-I trial [44]. Stomatitis, rash, and fatigue were the most commonly reported AEs, but were mostly mild or moderate in severity. Pneumonitis was also an AE more common in the everolimus group. Everolimus is also FDA indicated for patients with advanced-stage pancreatic NETs after data supporting prolonged PFS in the randomized phase III RADIANT-3 trial (mPFS 11.0 months vs. 4.6 months; HR 0.35; CI: 0.27–0.45; P < 0.001) without clear ORR benefit in the everolimus arm [46]. Similar data were demonstrated for well-differentiated, non-functional gastrointestinal, or lung NETs in the randomized RADIANT-4 trial with significant prolongation in PFS in the everolimus arm compared with placebo (11.0 months vs. 3.9 months; HR 0.48, CI: 0.35–0.67; P < 0.00001) [45]. Last, results from another randomized trial provided supported for FDA approval of everolimus in HR+/HER2− advanced breast cancer previously treated with aromatase inhibitors (AIs; letrozole or anastrozole): in BOLERO-2, everolimus plus AI was associated with significantly higher ORR (9.5% vs. 0.5% P < 0.001) and a longer mPFS (6.9 months vs. 2.8 months; HR 0.43; CI: 0.35–0.54; P < 0.001) compared with placebo plus AI [43].
Copanlisib
Copanlisib is the only pan-PI3K inhibitor to have been approved for clinical use, with the ability to inhibit the catalytic activity of all four PI3K class I isoforms (α, β, ɣ, and δ). Despite its pan-isoforms coverage, it retains a preferential activity against the p110α and p110δ isoforms [47]. The first-in-human (FIH) trial of copanlisib in advanced solid tumors revealed 2 CRs and 7 PRs, thus highlighting potential effectiveness against solid tumors [48]. Interestingly, 1/2 solid tumor patients with CRs harbored a PIK3CA mutation, pointing toward possible sensitivity of PIK3CA-mutants to copanlisib, especially considering preferential activity against the p100α subunit. The relationship between a PIK3CA mutational status and response to PI3K inhibitors has been described in previous studies: In a sample of 140 patients with breast, cervical, endometrial, and ovarian cancer, around 18% (n = 25) had PIK3CA mutations. A total of 23 of those patients treated with an inhibitor of the PI3K/AKT/mTOR pathway demonstrated higher response than patients without mutations: 6 had SD, with 2/6 achieving prolonged SD (> 6 months), and 7 patients had a PR. Similarly, another study has shown that combination of endocrine therapy and PI3K inhibitor had a significantly better outcome in PIK3CA-mutated ER+/HER2− breast cancer patients, compared with wild-type (WT) PIK3CA, reaching median PFS (mPFS) of 9 months (vs. 4.7 months in WT). Last, in a sample of 217 patients (endometrial, ovarian, CRC, breast, cervical, and head and neck), 11.5% (25/217) harbored the PIK3CA mutation. Of those, 17/25 received a PI3K/AKT/mTOR pathway inhibitor, 6/17 (35%) achieved PR (vs. 6% in WT PIK3CA), while 6/17 achieved SD (6–20 weeks). With that in mind, results from this phase I study were revisited and biomarker analysis was carried in solid tumor patients with and without PIK3CA mutations: 10/25 solid tumor patients (40%) harbored the mutation and three of these patients (30%) had an objective response or extended (≥4 cycles) SD when treated with copanlisib.
Copanlisib is not currently approved outside refractory follicular lymphoma (FL) based on data generated in the phase II CHRONOS-1 trial: in this study 142 patients (relapsed/refractory B-cell non-Hodgkin lymphoma-NHL; lymphoplasmocytoid or Waldenstrom macroglobulinemia) were treated with copanlisib monotherapy. Therapy resulted in ORR of 59% (12% CR and 47% PR), with a mPFS of 11.2 months [47].
Idelalisib
Idelalisib is a selective inhibitor of the PI3K catalytic subunit δ [49]. It was the first PI3K inhibitor to be approved for clinical use and is currently indicated in combination with rituximab in patients with relapse chronic lymphocytic leukemia (CLL) and as a monotherapy for patients with relapsed follicular B-cell NHL or relapsed small lymphocytic leukemia (SLL). The FDA approved Idelalisib on the basis of results of a phase III trial comparing Idelalisib plus rituximab to placebo plus rituximab (NCT01539512; [50]), involving 220 patients with relapsed CLL. The study demonstrated significant increase in ORR (81% vs. 13%), improved PFS at 24 weeks (93% vs. 46%; P < 0.001), and importantly, improved 1-year OS (92% vs. 80%; P = 002). The approval of idelalisib for recurrent B-NHL and SLL was based on the results of a multicenter single-arm trial (NCT01282424; [51]), involving 125 patients; The ORR was 54% in FL patients and 58% in SLL. A common aspect to both studies was the frequency and high grade of AEs: these included grade 3 or 4 diarrhea and rash. Idelalisib was also shown to confer a risk of SAEs involving immune-mediated hepatitis and pneumonitis, as well as pneumonia, pyrexia, sepsis, febrile neutropenia [50, 51].
Alpelisib
Alpelisib is the first oral PI3K inhibitor to selectively target the class I PI3K α-isoform (IC50 = 4.6 nM). The FIH Phase Ia study of alpelisib either as a single agent in patients with advanced solid tumors, or combined with fulvestrant in HR + breast cancer specifically enrolled patients with PIK3CA alterations (mutation or amplification), and demonstrated a more favorable safety profile with mostly on-target AEs such as hyperglycemia, nausea, and diarrhea. Importantly, it showed promising single-agent antitumor activity in PIK3CA-altered advanced tumors, with 15/131 patients demonstrating confirmed PRs with a mPFS of 166 days (vs. no PRs and mPFS of 7–61 days in PIK3CA WT patients later enrolled in dose-expansion phase) [52]. The recent phase III SOLAR-1 trial of alpelisib + fulvestrant vs. fulvestrant + placebo has also shown that PIK3CA-mutant cancers (n = 341) were more likely to response to alpelisib (mPFS 11 vs. 5.7 months in control) compared with PIK3CA WT (n = 231; mPFS 7.4 vs. 5.6 months in control) [53].
Although copanlisib, idelalisib and alpelisib are currently the only three FDA-approved PI3K inhibitors, several other ones have been studied in clinical trials, and further examined a potential role of PIK3CA mutations in increasing sensitivity to therapy. Here we describe three: buparlisib, alpelisib, and pictilisib.
Buparlisib
is another oral pan-class I PI3K inhibitor that targets the p110α, -β, -δ, and -ɣ subunits, with significantly higher activity against the α subunit (IC50 of 52 nM vs. 116–262 nM for other subunits). Phase I FIH study of buparlisib demonstrated tolerability up to the MTD of 100 mf/day with common treatment related AEs such as decreased appetite, rash, and hyperglycemia. In the phase III BELLE-2 trial, PIK3CA mutations detected in plasma-derived cell-free DNA (cf-DNA) correlated with longer PFS duration when treated with buparlisib, compared with WT PIK3CA (7.0 vs. 3.2 months) [54]. Similar results were obtained in the BELLE-3 trial using cf-DNA: a positive PIK3CA mutational status showed greater prolongation of mPFS in patients harboring the mutation (4.7 vs. 1.6 months with placebo), compared with WT PIK3CA (3.7 vs. 1.01 months with placebo) [55]. However, PIK3CA alteration did not demonstrate increased sensitivity to buparlisib when combined with chemotherapy: in the BELLE-4 study involving advanced-stage breast cancer patients, administration of buparlisib in combination with paclitaxel vs. paclitaxel monotherapy, did not demonstrated statistically significant difference in PFS between PIK3CA-mutant and WT tumors (9.1 vs. 9.2 months) [56].
Pictilisib
Pictilisib is a pan-PI3K inhibitor that exhibits equipotent inhibition of the p110α and -δ PI3K isoforms. In a phase I FIH study, pictilisib was safely administered with most frequent grade 3/4 AEs including rash, hyperglycemia, and pneumonitis. PR was demonstrated in 1/60 patients and SD in 2/60 patients (4 and 7 months) [57]. However, the phase 2 FERGI study that evaluated pictilisib in postmenopausal women with advanced AI-resistant, HR+/HER2− breast cancer (n = 229), did not show any benefit in terms of PFS, ORR, or OS even after stratification by PIK3CA status [58].
Finally, an important aspect to be kept in mind when discussing PI3K inhibitors is toxicity. In fact, drug-related toxicity linked to inhibition of the PI3K/mTOR/AKT pathway has been a significant limitation in achieving sustained target inhibition. The inability to achieve optimal drug-target blockage while avoiding undue toxicities in patients has been a major hurdle that well explains the limited success PI3K inhibitors have had despite a promising therapeutic rational [59]. These side effects are most pronounced with pan-PI3K inhibitors that present a broad toxicity profile compared with subunit-specific inhibitors, and include rash, fatigue, hyperglycemia/hyper-triglyceridemia, hepatotoxicity, pneumonitis, and diarrhea [59, 60].
Conclusion and future directions
PIK3CA activating mutations are, with no doubts, scientifically and clinically relevant in developing targeted therapies against solid malignancies. Results support a sound rationale for targeting the PI3K/AKT/mTOR pathway, and the impressive advances in drug development have been made under critical FDA review and with NCCN guideline support. The correlation of PIK3CA mutational status with either clinical-pathological features, or clinical outcome is also encouraging but focused clinical testing based on target relationship has been limited. As discussed in the first section, some contradictory reports exist regarding the prognostic value of PIK3CA mutations. Clinically, several PI3K inhibitors have been, and are currently under investigation, although some have been discontinued owing to insufficient efficacy. Clinically manageable toxicities, have also been observed that sometimes limits use. Future efforts should thus be aimed at identifying and addressing the obstacles to effective targeting of PIK3CA-abbarent cancers: first, the inconsistent prognostic value of PIK3CA along with the variation in efficacy of PI3K inhibitors, point toward the need to appropriately select patients that could benefit from PI3K inhibition. The PI3K/AKT/mTOR pathway is known to cross talk with other oncogenic pathways in what is known as “oncogenic cooperativity”, an example of which was demonstrated in early preclinical work showing the need for PTEN loss and p53 mutations on top of a PIK3CA mutation to induce tumorigenesis in mice [59, 61, 62]. Similarly, the PI3K/AKT/mTOR pathway cross-talks with the EGFR and RAS/RAF/MEK/ERK pathways. Clinically, this was demonstrated by better clinical outcomes when combining buparlisib and trametinib (MEK inhibitor) in patients with a PIK3CA and RAS/RAF mutations, as reflected by ORR of 29% [63]. Similarly, improved outcomes have been achieved when combining alpelisib with AI or anti-estrogen in patients with high HR expression (hormones activate EGFR and other RTKs) [53]. This observation generates two implications: (i) there is a need for a comprehensive molecular approach and analysis, whereby the decision of whether or not a PI3K inhibitor is appropriate, takes into account the molecular background as a whole, and perhaps excludes those that have other mutations that can mediate resistance to PI3K inhibitors; and (ii) the need for a combination approach based on co-occurring alterations, or simply to limit adaptive responses by tumor cells. Another challenge related to proper patient selection, is the accuracy of the molecular assay for PIK3CA mutation identification: in the BELLE-2, for example, 307 patients were identified as PIK3CA WT based on sequencing of tumor tissue at diagnosis; however, using circulating cf-DNA, 64/307 (21%) had PIK3CA mutations, suggesting either higher sensitivity of the cf-DNA assay, or an actual evolution of cancer between the original tissue diagnosis and the development of metastatic disease, or both [54].
Besides proper patient selection and good molecular analysis of the complete mutational background, drug-related toxicities are also a major challenge facing the development of PI3K inhibitors, as discussed earlier. Besides the combinational approach discussed above, future efforts could be directed at developing inhibitors that are isoform specific, such as PIK3CA mutant-selective inhibitors that specifically target the PI3K-α subunit.
Thus, the success of future PIK3CA-target inhibition lies in a better understanding of the complex signaling pathways involving the PI3K/AKT/mTOR pathway, as well as in development of target-specific inhibitors, coupled with more sensitive assay to tumor analysis. With this, developing a therapeutic strategy (monotherapy vs. appropriate combination) that ensures selective targeting with maximal therapeutic effect and minimal toxicities becomes achievable.
References
Kim D, Dan HC, Park S, Yang L, Liu Q, Kaneko S, et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci. 2005;10:975–87.
Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
Gkeka P, Evangelidis T, Pavlaki M, Lazani V, Christoforidis S, Agianian B, et al. Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput Biol. 2014;10:e1003895.
Hart JR, Zhang Y, Liao L, Ueno L, Du L, Jonkers M, et al. The butterfly effect in cancer: a single base mutation can remodel the cell. Proc Natl Acad Sci USA. 2015;112:1131–6.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–91.
Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318:1744–8.
Koyama T, Rhrissorrakrai K, Parida L. Analysis on GENIE reveals novel recurrent variants that affect molecular diagnosis of sizable number of cancer patients. BMC Cancer. 2019;19:114.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018;47(D1):D941–D7.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Insitiute WS. COSMIC: Catalogue of Somatic Mutations in Cancer.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, et al. Bladder cancer. Nat Rev Dis Primers. 2017;3:17022.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–22.
Zeng SX, Zhu Y, Ma AH, Zhang H, Lin TY, Shi W, et al. The phosphatidylinositol 3-kinase pathway as a potential therapeutic target in bladder cancer. Clin Cancer Res. 2017;23:6580–91.
O’Sullivan CC, Loprinzi CL, Haddad TC. Updates in the evaluation and management of breast cancer. Mayo Clin Proc. 2018;93:794–807.
Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Omarini C, Filieri ME, Bettelli S, Manfredini S, Kaleci S, Caprera C, et al. Mutational profile of metastatic breast cancer tissue in patients treated with exemestane plus everolimus. Biomed Res Int. 2018;2018:3756981.
Markham A. Alpelisib: first global approval. Drugs. 2019;79:1249–53.
Cantley LC, Auger KR, Carpenter C, Duckworth B, Graziani A, Kapeller R, et al. Oncogenes and signal transduction. Cell. 1991;64:281–302.
Beaver JA, Park BH. The BOLERO-2 trial: the addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor-positive advanced breast cancer. Future Oncol. 2012;8:651–7.
Yi Z, Ma F, Liu B, Guan X, Li L, Li C, et al. Everolimus in hormone receptor-positive metastatic breast cancer: PIK3CA mutation H1047R was a potential efficacy biomarker in a retrospective study. BMC Cancer. 2019;19:442.
Cohen PA, Jhingran A, Oaknin A, Denny L. Cervical cancer. Lancet. 2019;393:169–82.
Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, et al. Colorectal cancer. Nat Rev Dis Primers. 2015;1:15065.
Bogaert J, Prenen H. Molecular genetics of colorectal cancer. Ann Gastroenterol. 2014;27:9–14.
Vogelstein B, Kinzler KW. The genetic basis of human cancer; 1998.
Wang Q, Shi YL, Zhou K, Wang LL, Yan ZX, Liu YL, et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018;9:739.
Charo LM, Plaxe SC. Recent advances in endometrial cancer: a review of key clinical trials from 2015 to 2019. F1000Res. 2019;8.
Saso S, Chatterjee J, Georgiou E, Ditri AM, Smith JR, Ghaem-Maghami S. Endometrial cancer. BMJ. 2011;343:d3954.
Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73.
Rudd ML, Price JC, Fogoros S, Godwin AK, Sgroi DC, Merino MJ, et al. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin Cancer Res. 2011;17:1331–40.
Fader ARD, Siegel E, Buza N, Hui P, Abdelghany O, Chambers S, et al. Randomized Phase II trial of carboplatin-paclitaxel vesus carboplatin-paclitaxel-trastuzumab in uterine serous carcinomas that overexpress human epidermal growth factor receptor 2/neu. J Clin Oncol. 2018;36:7.
Bonazzoli E, Cocco E, Lopez S, Bellone S, Zammataro L, Bianchi A, et al. PI3K oncogenic mutations mediate resistance to afatinib in HER2/neu overexpressing gynecological cancers. Gynecol Oncol. 2019;153:158–64.
Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60.
Zevallos JP, Mazul AL, Walter V, Hayes DN. Gene expression subtype predicts nodal metastasis and survival in human papillomavirus-negative head and neck cancer. Laryngoscope. 2019;129:154–61.
Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3:761–9.
Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–82.
Medicine USNLo. https://clinicaltrials.gov/ct2/results?cond=&term=alpelisib&cntry=&state=&city=&dist=; 2019.
Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061.
Polivka J Jr., Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164–75.
Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81.
Baselga J, Campone M, Piccart M, Burris HA III, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. 2012;366:520–9.
Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. The Lancet. 2008;372:449–56.
Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. The Lancet. 2016;387:968–77.
Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514–23.
Dreyling M, Santoro A, Mollica L, Leppä S, Follows GA, Lenz G, et al. Copanlisib in patients with relapsed or refractory indolent b-cell lymphoma (CHRONOS-1). Hematol. Oncol. 2017;35:119–20.
Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann Oncol. 2016;27:1928–40.
Randis TM, Puri KD, Zhou H, Diacovo TG. Role of PI3Kdelta and PI3Kgamma in inflammatory arthritis and tissue localization of neutrophils. Eur J Immunol. 2008;38:1215–24.
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007.
Gopal AK, Kahl BS, de Vos S, Wagner-Johnston ND, Schuster SJ, Jurczak WJ, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370:1008–18.
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-kinase alpha-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36:1291–9.
Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A phase ib study of alpelisib (BYL719), a PI3Kalpha-specific inhibitor, with letrozole in ER+/HER2- metastatic breast cancer. Clin Cancer Res. 2017;23:26–34.
Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:904–16.
Di Leo A, Johnston S, Lee KS, Ciruelos E, Lonning PE, Janni W, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19:87–100.
Martin M, Chan A, Dirix L, O’Shaughnessy J, Hegg R, Manikhas A, et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2- advanced breast cancer (BELLE-4). Ann Oncol. 2017;28:313–20.
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86.
Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:811–21.
Hanker AB, Kaklamani V, Arteaga CL. Challenges for the clinical development of pi3k inhibitors: strategies to improve their impact in solid tumors. Cancer Discov. 2019;9:482–91.
Chia S, Gandhi S, Joy AA, Edwards S, Gorr M, Hopkins S, et al. Novel agents and associated toxicities of inhibitors of the pi3k/Akt/mtor pathway for the treatment of breast cancer. Current Oncol. 2015;22:33–48.
Wu R, Baker SJ, Hu TC, Norman KM, Fearon ER, Cho KR. Type I to type II ovarian carcinoma progression: mutant Trp53 or Pik3ca confers a more aggressive tumor phenotype in a mouse model of ovarian cancer. Am J Pathol. 2013;182:1391–9.
Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–33.
Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res. 2015;21:730–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Willis, O., Choucair, K., Alloghbi, A. et al. PIK3CA gene aberrancy and role in targeted therapy of solid malignancies. Cancer Gene Ther 27, 634–644 (2020). https://doi.org/10.1038/s41417-020-0164-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-0164-0
- Springer Nature America, Inc.
This article is cited by
-
Molecular substratification of endometrial carcinomas with no special molecular profile (NSMP) by using a limited NGS custom panel may facilitate effective patient selection for the PIK3CA-targeted therapy
Virchows Archiv (2024)
-
From the identification of actionable molecular targets to the generation of faithful neuroblastoma patient-derived preclinical models
Journal of Translational Medicine (2024)
-
Inferring early genetic progression in cancers with unobtainable premalignant disease
Nature Cancer (2023)
-
Cribriform Morular Thyroid Carcinoma – Ultimobranchial Pouch-Related? Deep Molecular Insights of a Unique Case
Endocrine Pathology (2023)
-
Cancer gene therapy 2020: highlights from a challenging year
Cancer Gene Therapy (2022)