
Abstract
Neuroglial inflammation is a pathological hallmark of neuroimmunological disorders, such as multiple sclerosis, as well as neurodegenerative diseases, such as amyotrophic lateral sclerosis, Parkinson’s disease, and Alzheimer’s disease. Activated microglia and reactive astroglia accompany the loss of neurons and myelin in these conditions. Both microglia and astroglia can exert neuroprotective and neurotoxic functions, which are stage-dependent. Both cell types can switch from an anti-inflammatory/neuroprotective to a proinflammatory/neurotoxic phenotype according to the surrounding environmental stimuli. Deciphering glial dual actions may provide insights for the management of neuroglial inflammation and the future development of new drugs targeting glia in neuroimmunological and neurodegenerative diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Multiple Sclerosis
- Amyotrophic Lateral Sclerosis
- Amyotrophic Lateral Sclerosis Patient
- Experimental Allergic Encephalomyelitis
- Multiple Sclerosis Lesion
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
Demyelinating diseases, such as multiple sclerosis (MS) and neuromyelitis optica (NMO), are representative neuroimmunological diseases that affect the central nervous system (CNS). Demyelinating disorders are thought to be triggered by immune-mediated mechanisms although this has not been conclusively proven to date. MS and NMO subsequently develop neurodegeneration in addition to inflammatory demyelination. The accumulating disability and the resultant chronic disease progression in these conditions are likely determined by secondary neurodegeneration. In the chronic progressive phase of MS, T cell infiltration subsides while CNS-compartmentalized glial inflammation becomes dominant, which induces continuous tissue degeneration. Neurodegenerative disorders, such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (ALS), are triggered by neuronal abnormalities but are subsequently accompanied by neuroglial inflammation. Increasing evidence suggests that neuroglial inflammation determines disease progression, which is a clinical reflection of neurodegeneration. Therefore, neuroimmunological and neurodegenerative diseases share glial inflammation as an indispensable component of the disease processes, which underscores the importance of elucidating the mechanisms of glial inflammation. This review chapter describing neuroinflammation in neurological disease will focus on MS as a representative neuroimmunological disease afflicting the CNS, and ALS as a neurodegenerative disease accompanied with glial inflammation.
2.2 Neuroglial Inflammation in MS
2.2.1 Clinical Aspects Related to Neuroglial Inflammation and Neurodegeneration
Most MS patients initially develop a relapsing remitting disease course with a mean age of onset around 30 years of age (termed relapsing remitting MS, RRMS). After 10–20 years, approximately 50 % of RRMS patients enter a secondary progressive phase with or without superimposed relapses (termed secondary progressive MS, SPMS). Approximately 10–20 % of MS patients exhibit a relentlessly progressive disease course from the onset (termed primary progressive MS, PPMS). Clinical relapse is often accompanied or even preceded by appearance of contrast-enhanced magnetic resonance imaging (MRI) lesions in the CNS. Recent 7 T MRI studies clearly showed the presence of vessels in the center of MS lesions (Mistry et al. 2013), confirming that MS lesions develop around blood vessels. Pathologically, perivascular and diffuse lymphocytic infiltration is a common finding in active MS lesions. Thus, clinical relapse is likely to be caused by peripheral blood-borne inflammation around the blood vessels.
However, clinical relapses have only a weak effect on clinical progression (Confavreux et al. 2000). Irrespective of the initial disease course, a clinically progressive phase occurs in both SPMS and PPMS patients around 40 years of age and then proceeds at similar rate (Kremenchutzky et al. 2006). Large-scale epidemiological surveys revealed that MS patient disability, determined by Kurtzke’s Expanded Disability Status Scale (EDSS) scores (Kurtzke 1983), progresses at approximately the same rate until the EDSS scores reach four, even though the progression rates varied until the development of an EDSS score of four (Confavreux and Vukusic 2006). These findings suggest that common pathogenic mechanisms may underlie clinical disability progression. At the progressive stage of MS, none of the recent disease-modifying therapies (DMT) acting on the peripheral immune system are effective, even though they have high efficacy for reducing annualized relapse rates. Thus, disease progression may have distinct mechanisms from relapse caused by peripheral immune-mediated inflammation.
Many MRI studies have reported that the T2 lesion burden in the white matter modestly correlates with disability (Fisniku et al. 2008) and rather the degree of gray matter and spinal cord atrophy correlates best with accumulating disability (Fisniku et al. 2008; Fisher et al. 2008; Bonati et al. 2011). These observations suggest that disease progression could be attributable to neuroglial inflammation compartmentalized in the CNS behind the blood–brain barrier (BBB) (Reynolds et al. 2011), which causes neurodegeneration regardless of the initial inflammatory relapses.
2.2.2 White Matter Pathology and Oligodendroglia in MS Lesions
MS predominantly affects CNS white matter that is rich in myelin. Actively demyelinating lesions are destructive lesions that are densely and diffusely infiltrated with macrophages/activated microglia that phagocytose myelin debris, as identified by Luxol fast blue staining and immunohistochemistry for myelin proteins (Lassmann et al. 1998; Lucchinetti 2007). Such lesions are associated with perivascular lymphocyte cuffing. Chronic active lesions display a rim of macrophages and activated microglia while chronic inactive lesions show no increase in macrophage/activated microglia numbers throughout the lesions. A mild global inflammation containing microglial activation and a diffuse low-level of T cells is seen even in normal-appearing white matter. Such diffuse inflammatory changes are more prominent in SPMS and PPMS than RRMS. In chronic MS, leakage from the BBB is absent, which corresponds to a paucity of gadolinium-enhanced lesions in PPMS and SPMS. Thus, compartmentalized inflammation behind the BBB is suggested based on these pathological findings.
It is widely accepted that MS pathology shows heterogeneity. Lucchinetti et al. (2000) classified four demyelinating patterns of MS lesions, and proposed that an individual only develops one pattern, suggesting a single mechanism is operative in individual patients. All lesions have inflammatory infiltrates composed of T cells and macrophages/activated microglia, while each pattern has its own specific features as follows.
-
Pattern I: Active demyelination associated with the infiltration of T cells and macrophage/activated microglia in the absence of antibody and complement deposition. These lesions are centered around veins and venules.
-
Pattern II: Active demyelination associated with immunoglobulin and complement deposition. Prominent deposition of immunoglobulins (mainly IgG) and complement C9neo antigen are found in association with degenerating myelin at the active plaque edge. This pattern is also centered around blood vessels.
-
Pattern III: Distal oligodendrogliopathy characterized by selective myelin-associated glycoprotein (MAG) loss. A profound loss of oligodendroglia at the active plaque border, DNA fragmentation, and oligodendroglial apoptosis are observed with T cell and macrophage/activated microglial infiltration but without the deposition of immunoglobulins and complement. Such lesions are not centered around blood vessels and the margin is ill-defined.
-
Pattern IV: Oligodendroglial death with DNA fragmentation but without features of apoptosis in a small rim of periplaque white matter. A near complete loss of oligodendroglia in active and inactive lesions is observed without remyelination. The border is sharply demarcated.
However, we and others have observed heterogeneous demyelinating patterns even within one autopsied individual, indicating such patterns may represent stage-dependent heterogeneity but not disease heterogeneity (Matsuoka et al. 2011). Immunoglobulin and complement deposits are found in lesions from about 50 % of autopsied MS patients (pattern II) (Stadelmann et al. 2011), suggesting that antibody and complement-mediated myelin phagocytosis might become the dominant mechanism in established MS lesions (Breij et al. 2008). Currently, the idea that an individual only develops one demyelinating pattern or can develop more than one pattern is still controversial.
Oligodendroglial cells are especially vulnerable to oxidative stress and glutamate toxicity associated with inflammation. Oligodendroglia express the AMPA/kainite receptor in the cell body and NMDA receptors in the processes. Oligodendroglia also express excitatory amino acid transporter (EAAT)-1 and -2 and are regarded as the principal cells for glutamate clearance in the white matter (Benarroch 2009). The accumulation of glutamate has been demonstrated in MS lesions by biopsy (Werner et al. 2001) and magnetic resonance spectroscopy (Srinivasan et al. 2005) while EAAT-1 and EAAT-2 are reduced in oligodendroglia (Pitt et al. 2003). In these circumstances, oligodendroglia may be vulnerable to toxicity from glutamate secreted by activated microglia. Oligodendroglia contain a large pool of iron but have a low capacity for anti-oxidative mechanisms, which render the cell especially sensitive to oxidative stress (Benarroch 2009).
2.2.3 T Cells: A Key Player of the Effector Arm That Triggers CNS Inflammation
Multiple sclerosis (MS) is thought to be an autoimmune disease that targets myelin antigens. This has been suggested from studies demonstrating an increased frequency of T cells showing inter- and intramolecular epitopes spreading against myelin proteins, increased levels of interferon (IFN)-γ, interleukin (IL)-17, and downstream proinflammatory cytokines in the cerebrospinal fluid (CSF), exacerbation of disease following the administration of IFNγ, and the increased frequency of T helper 1 (Th1) cells secreting IFNγ and Th17 cells secreting IL-17, which support the involvement of Th1 and Th17 cells in MS, at least for the inflammatory aspects of the disease (Ishizu et al. 2005; Tanaka et al. 2008; Matsushita et al. 2013). CD4+ T cells are present mainly in the perivascular areas while parenchyma infiltrates largely consist of CD8+ T cells (Babbe et al. 2000). Interestingly, CD8+ T cells outnumber CD4+ T cells in MS lesions (Booss et al. 1983); however, the roles of CD8+ T cells remain unclear. In an animal model of MS, experimental allergic encephalomyelitis (EAE), the early events in the formation of inflammatory lesions, involve a predominantly CD4+ T cell-mediated process. B cells and plasma cells also exist in the perivascular areas, but represent a minor component of inflammatory infiltrates in the CNS parenchyma (Friese and Fugger 2009; Frischer et al. 2009).
Studies from EAE demonstrated that myelin-specific CD4+ T cells could be transferred to naïve mice to induce a CNS demyelinating disease. Thus, it was hypothesized that in MS, naïve T cells are sensitized by myelin antigens in the peripheral lymph nodes, such as deep cervical lymph nodes, and differentiate to myelin antigen-specific Th1 or Th17 cells. These peripherally activated Th1 or Th17 cells express adhesion molecules that allow them to pass through the BBB and enter the CNS (Fig. 2.1). In EAE, adoptively transferred myelin antigen-specific T cells require several days to accumulate in the CNS. It was recently shown that such encephalitogenic T cells reside in bronchus-associated lymphoid tissue (Odoardi et al. 2012) and become eligible to enter the CNS.

Th17/Th1 cells are primed in the periphery and restimulated in the central nervous system (CNS). Naïve T cells (Th0) differentiate to Th1 or Th17 cells upon antigenic stimulation in the peripheral lymph nodes and enter the CNS via the blood–brain barrier (BBB). Perivascular macrophages present antigens to Th1 or Th17 cells in the perivascular space, and restimulated Th1 and Th17 cells traffic to the CNS parenchyma, secrete IFNγ and IL-17, and recruit macrophages and neutrophils, respectively. Microglia can produce either toxic or protective factors, sensing either “kill me” or “help me” signals from neurons. The destruction of astrocyte endfeet or decreased production of IL-25 from endothelial cells can cause the BBB to become “leaky”
T cells egress from postcapillary venules (high endothelial venules) and enter into the Virchow-Robins space (perivascular space) in the CNS. Here, activated T cells can firmly adhere to the surface of vascular endothelial cells via interactions between α4β1 integrin expressed on activated T cells and vascular cell adhesion molecule 1 (VCAM-1) on endothelial cells lining the BBB. Anti-α4β1 integrin antibody, natalizumab, effectively blocks firm adhesion of T cells, thereby markedly suppressing MS relapses (Coisne et al. 2009). Thus, peripherally activated T cells can invade across endothelial cells and the endothelial basement membrane on the abluminal side and remain in the perivascular space delineated by the endothelial basement membrane and the parenchymal basement membrane, which is an extension of the subarachnoid space (Ransohoff and Engelhardt 2012). T cells require restimulation by perivascular macrophages to further traffic into the CNS parenchyma across the glia limitans perivascularis composed of parenchymal basement membrane and astrocyte endfeet (Ransohoff and Engelhardt 2012). Perivascular macrophages continuously repopulated from the peripheral blood can engulf CNS antigens in the perivascular space where myelin antigens are conveyed by the CSF flow pathway to the subarachnoid space, and present these antigens to T cells (Ransohoff and Engelhardt 2012). Subsequently, T cells restimulated by perivascular macrophages secrete matrix metalloproteinase (MMP)-2 and -9, which disrupt the basement membrane leading to destabilization of astrocyte endfeet anchored to the parenchymal basement membrane, and promote their entry into the CNS parenchyma (Bechmann et al. 2007; Tran et al. 1998). Once in the CNS parenchyma, T cells secrete proinflammatory cytokines and chemokines that further recruit macrophages, neutrophils, and activating resident microglia, which serve as effectors for tissue destruction, at least during the relapse phase.
The ability of natalizumab to markedly suppress relapses implies the critical importance of T cell involvement in CNS inflammation at relapse. However, according to MS pathology, there is considerable debate as to whether T cell infiltration is a primary event or secondary to oligodendroglial apoptosis and subsequent microglial activation. Barnett and Prineas (2004) observed oligodendroglial apoptosis without lymphocyte infiltration in autopsied cases with very early MS, and proposed that oligodendroglial apoptosis preceded the formation of all MS lesions and BBB “leakiness,” and that microglial activation and T cell infiltration were secondary events. The source of the substantial debates regarding these issues is partly derived from the fact that factors causing initial oligodendroglial apoptosis remain unknown.
2.2.4 B Cells: Another Important Cell in the Effector Phase
Few plasma cells are observed in the CNS during the early stages of MS, but become increasingly prominent as disease progresses. In addition, there is an increased prevalence of oligoclonal IgG bands (OBs) in the CSF as the disease duration increases, which persist stably (Meinl et al. 2006). B cells exist in the perivascular areas and leptomeninges during all disease stages, but rarely in the CNS parenchyma (Magliozzi et al. 2007). Autoantibody and complement-mediated myelin phagocytosis are assumed the dominant mechanism in established MS lesions, as mentioned in Sect. 2.2 (Breij et al. 2008). In the leptomeninges, ectopic lymphoid follicle-like structures have been observed in approximately 40 % of postmortem SPMS cases (Magliozzi et al. 2007, 2010). These follicle-like structures consist mainly of CD20+ B cell aggregates interspersed with CD21+ CD35+ follicular dendritic cells (FDCs), CD4+ T cells, and CD8+ T cells. They are predominantly present in the deep cerebral sulci (Magliozzi et al. 2007, 2010). The majority of such meningeal lymphoid follicle-like structures are closely associated with large subpial demyelination (Magliozzi et al. 2007, 2010). MS cases with meningeal lymphoid-like structures showed a younger age at disease onset, a shorter time to wheelchair-bound disability, and a shorter time to progression than those without meningeal lymphoid-like structures (Magliozzi et al. 2007, 2010).
The importance of B cells in MS is clearly indicated by the fact that rituximab, that targets CD20 molecules expressed on B cells but not plasma cells, is highly efficacious in suppressing MS relapses (Hauser et al. 2008). In rituximab trials, B cell numbers decreased in parallel with the reduction of relapses, whereas total antibody levels did not decrease significantly. It is thus proposed that B–T cell interactions including antigen presentation or proinflammatory cytokine secretion by B cells is the critical step inhibited by rituximab, but not the inhibition of autoantibodies themselves. Interestingly, rituximab is also effective in NMO, where there is selective optic nerve and spinal cord demyelination in the presence of specific antibodies against astrocyte water channel protein, aquaporin-4 (AQP4) (Lennon et al. 2004, 2005), but without reducing anti-AQP4 antibody levels (Pellkofer et al. 2011). Therefore, B–T cell interactions and B cell cytokines are also thought to be critical in NMO. Highly specific autoantibodies in MS pathology remain to be identified. Autoantibodies against myelin oligodendrocyte glycoprotein (MOG) have been detected in children with atypical demyelinating disease (Brilot et al. 2009), but not in adult cases. The significance of anti-glycolipid antibodies and a recently described autoantibody against KIR4.1, an ATP-sensitive inward rectifying potassium channel expressed in astroglial endfeet and oligodendroglia (Srivastava et al. 2012), needs further confirmation in large-scale independent cohorts.
2.2.5 Gray Matter Lesions
Recently, gray matter lesions have gained much attention because they closely correlate with disability and disease progression. The introduction of double inversion recovery (DIR) MRI demonstrated that cortical lesions and cortical atrophy are present from the early stage of RRMS and become more prominent in SPMS (Fisniku et al. 2008; Fisher et al. 2008; Kutzelnigg et al. 2005; Vercellino et al. 2005). The absence of MRI evidence for noticeable inflammation suggests that neurodegeneration may take place in cortical lesions. Cortical lesion loads and atrophy are significantly associated with clinical progression (Geurts et al. 2005) whereas white matter atrophy does not correlate with increasing disability (Fisher et al. 2008). Thus, cortical lesions may play a major role in the development of both physical and cognitive disability (Calabrese et al. 2010).
Pathologically, demyelination is present in the spinal cord and cerebral and cerebellar cortex but also in the deep gray matter, including the thalamus, basal ganglia, and hypothalamus, to varying degrees (Bö et al. 2003; Peterson et al. 2001). Cortical lesions are classified into three types: type I lesions are leukocortical lesions affecting both subcortical white matter and the lower layer of gray matter; type II lesions are entirely intracortical; and type III lesions involve the subpial gray matter regions (subpial demyelination) (Peterson et al. 2001). Frontal and temporal cortices, cingulate gyrus, and hippocampus are most frequently involved (Reynolds et al. 2011), and may explain the correlation between cognitive impairment and cortical pathology. In hippocampal demyelinated lesions, a reduction of synaptic density has also been reported (Dutta et al. 2011). However, cortical demyelination does not correlate with white matter pathologic changes (Bö et al. 2003), suggesting independent mechanisms may be operative.
Cortical lesions are accompanied with mild, if any, inflammatory infiltrates, but with increased numbers of activated microglia (Magliozzi et al. 2010; Bö et al. 2003; Peterson et al. 2001). Other differences between cortical and white matter lesions include the lack of significant leakage of plasma proteins, suggesting the BBB is preserved, and the absence of complement activation (Reynolds et al. 2011). In extensive subpial demyelination, increased numbers and activation status of microglia, increased axonal injury, and neuronal loss are greatest close to the pial surface (Magliozzi et al. 2007, 2010), implying that secretion of proinflammatory cytokines into the CSF from lymphocytes in the follicles may be responsible for the activation of microglia, cortical demyelination, and neuronal damage. In these cases, the loss of layer III and V pyramidal neurons exceeded 40 % and 50 %, respectively, and was accompanied by loss of interneurons in other cortical layers (Magliozzi et al. 2010). Cortical neuronal loss was also reported to occur diffusely even in normal-appearing gray matter (Magliozzi et al. 2010), suggesting that demyelination and neuronal loss may not be directly linked (Reynolds et al. 2011). Neuronal apoptosis and mitochondrial damage were thought to be responsible for the neuronal loss (Reynolds et al. 2011; Campbell et al. 2011; Dutta et al. 2006). However, other research groups did not confirm such a relation between meningeal lymphoid follicles and cortical demyelination (Kooi et al. 2009), and more studies are required to establish the roles of meningeal lymphoid follicles in MS.
2.2.6 Mechanisms of Axonal Injury
Acute axonal damage is accompanied by active focal inflammatory demyelination and is most prominent during the early stages of MS (Ferguson et al. 1997; Trapp et al. 1998) but decreases with disease progression (Frischer et al. 2009; Kornek and Lassmann 1999; Kuhlmann et al. 2002), suggesting that inflammation plays a significant role in axonal loss. Cumulative axonal loss and resultant brain and spinal cord atrophy are significantly correlated with permanent disability (Frischer et al. 2009; Kuhlmann et al. 2002; Bjartmar et al. 2000). Acute damage can be detected by the presence of accumulated amyloid precursor protein (APP)-positive spheroids that reflect impaired axonal transport (Ferguson et al. 1997). APP-positive spheroids are more extensive during the first year of disease onset, and the number of acutely injured axons decrease with increasing disease duration (Kuhlmann et al. 2002). The extent of axonal loss correlates well with numbers of CD8+ T cells and macrophages/activated microglia that are present in close proximity (Kuhlmann et al. 2002) and numerous CD8+ T cells that infiltrate into CNS parenchyma transect axons possibly via major histocompatibility complex (MHC) class I-mediated self-antigen recognition (Trapp et al. 1998). Furthermore, reactive oxygen and nitrogen species and proinflammatory cytokines secreted by these cells may suppress axonal functions and cause mitochondrial damage (Dutta et al. 2006).
In limited numbers of MS cases, autoantibodies against nodal and paranodal antigens, such as neurofascin, contactin-2, and TAG-1, have been reported (Mathey et al. 2007; Derfuss et al. 2009). For example, anti-neurofascin antibody was found in one-third of MS patients, with higher prevalence in chronic progressive MS than in RRMS (Mathey et al. 2007). Neurofascin 186 expressed on the axolemma at the node of Ranvier concentrates voltage-gated sodium channels at the node while neurofascin 155 expressed on oligodendroglial membranes connects them to axons via binding to contactin-1 and Caspr1 (Ratcliffe et al. 2001; Sherman et al. 2005). Autoantibodies to nodal and paranodal antigens can induce axonal dysfunction in vivo and may be involved in axonal damage in MS (Desmazières et al. 2012).
2.2.7 Microglia and Monocyte/Macrophage in Demyelinating Diseases
The mononuclear phagocyte system in the CNS, including peripheral blood-borne monocytes/macrophages and resident microglia, plays major roles in the effector arm of demyelinating diseases by restimulating T cells within the CNS and by damaging and repairing CNS tissue.
2.2.7.1 Roles of Microglia and Monocytes/Macrophage in MS
The origin of microglia has long been a matter of debate, but recent studies indicated microglia are derived from extraembryonic yolk sac myeloid cells. Colony stimulating factor 1 receptor (CSFIR) is a cell-surface receptor for the cytokines CSF-1 and IL-34. CSF1R is usually expressed on monocytes and macrophages in the peripheral blood as well as on the surface of microglia in the CNS (Ransohoff and Cardona 2010). During fetal development, yolk sac myeloid cells colonize in the CNS due to IL-34 signaling through CSF1R. Once colonized, such cells lose surface markers characteristic of mononuclear phagocytes, and are assumed to become microglia in adults (Ransohoff and Cardona 2010). CSF1 on CSF1R signaling is associated with survival, proliferation, regulation, and differentiation of microglia (Wang et al. 2012). In adult CNS, microglia consist of more than 10 % of all cells. In the resting state, microglia have a small body with extensively branched processes and are termed “ramified microglia.” Microglia expressing CD11b, ionized calcium-binding adapter molecule 1 (Iba1), and CD68 constantly monitor the CNS environment (Ransohoff and Cardona 2010). Upon activation, the soma is enlarged while processes are retracted and these cells are termed “amoeboid microglia.” Myeloid cell markers are enhanced on “amoeboid microglia.”
Microglia are the only cells that express CX3CR1 in the CNS. CX3CR1-deficient mice develop severe EAE and increased neuron loss in a transgenic model of ALS (Cardona et al. 2006). Its ligand CX3CL1 is produced by neurons and down-regulates microglial neurotoxicity. A lack of CX3CL1 input from neurons rapidly activates microglia (Ransohoff and Cardona 2010). Furthermore, plasma fibrinogen extravasated from disrupted BBB also can activate microglia (Ransohoff and Cardona 2010). Activated microglia produce numerous cytokines/chemokines, growth factors, reactive oxygen and nitrogen species via oxidative burst, and inducible nitric oxide synthase (iNOS). Activated microglia can express MHC class II molecules and costimulatory molecules. However, they never traffic to the draining lymph node, unlike dendritic cells in other tissues.
In the CNS, peripheral blood-borne perivascular and meningeal macrophages play a major role in antigen presentation to restimulate T cells. Without restimulation by relevant antigens, T cells do not survive in the CNS. The recruitment of monocytes/macrophages is mediated by CCL2–CCR2 signaling. Hypertrophic astrocytes in active MS lesions express CCL2 while its receptor, CCR2, is expressed on monocytes/macrophages (Mahad and Ransohoff 2003). CSF CCL2 levels are decreased in MS (Mahad et al. 2002), which is presumably because of its consumption by infiltrating cells (Mahad et al. 2002). CCR2-deficient mice develop mild EAE with neutrophil infiltration (Yamasaki et al. 2012). Thus, macrophages play major roles in antigen presentation and tissue destruction while microglia induce tissue damage. However, microglia may also have neuroprotective properties through phagocytizing tissue debris and producing neurotrophic substances.
2.2.7.2 Hereditary Microgliopathy Showing Widespread Myelin and Neuroaxonal Loss
Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS), a rare autosomal dominant disease characterized by cerebral white matter degeneration with axonal spheroids presenting cognitive decline, depression, and motor impairment, is caused by mutations in the CSF1R gene (Rademakers et al. 2012). As mentioned in Sect. 2.2.7.1, IL-34 signaling through CSF1R is related to microglial migration into the CNS during the embryonic period, while CSF1 signaling is associated with survival, proliferation, and differentiation of microglia (Wang et al. 2012). We recently observed that microgliopathy caused by CSF1R mutation in HDLS causes myelin and axonal loss in the CNS, where CD4+ and CD8+ T cell infiltration occurs possibly through the actions of cytokines/chemokines produced by reactive microglia (Saitoh et al. 2013). In addition, a primary microglial disease known as Nasu–Hakola disease (NHD) is characterized by white matter degeneration and bone cysts. The recessive loss of function mutations in the gene encoding triggering receptor expressed on myeloid cells 2 (TREM2) and transmembrane adaptor signaling protein DAP12 that transduces TREM2 signals in NHD causes a lack of microglia and osteoclasts (Paloneva et al. 2000, 2002; Kondo et al. 2002). The TREM2–DAP12 protein complex is crucial for proliferation and survival of mononuclear phagocytes and is related to CSF1R signaling (Otero et al. 2009). That genetic mutation of indispensable molecules in microglial development and function causes diffuse myelin and axon degeneration underscores the critical roles of microglia in the maintenance of myelin and neurons. Thus, in HDLS and NHD, it has been suggested that microglial maintenance of myelin turnover is disrupted. Therefore, the disruption of normal microglial maintenance functions may exacerbate neurodegenerative process in neuroglial inflammation.
2.2.8 Roles of Astrocytes in Demyelinating Disease
Astrocytes normally have neuroprotective functions while in inflammatory circumstances they become neurotoxic. Such biphasic behavior of astrocytes makes neuroglial inflammation more complex.
2.2.8.1 Neuroprotective Aspects
Astroglia extend numerous processes, forming highly organized domains with little overlap between adjacent cells. Astroglia appose each other and interconnect via Cx43 gap junction channels to form functional networks. Highly ramified protoplasmic astrocytes in the gray matter ensheath synapses, forming tripartite synapses, while fibrous astrocytes in the white matter cover the nodes of Ranvier (Miller and Raff 1984). Astrocyte endfeet also have close contact with parenchymal basement membrane around vessels and contribute to maintenance of the BBB through the induction of tight junctions between endothelial cells (Janzer and Raff 1987). Astroglia also produce components of extracellular matrix, such as collagens, laminins, fibronectins, hyaluronan, chondroitin sulfate, and heparin sulfate (Zimmermann and Dours-Zimmermann 2008; van der Laan et al. 1997; van Horssen et al. 2007), which constitute the basal lamina around vessels. Astroglia constitutively express the membrane bound death ligand, CD95L, and can induce CD95L-mediated apoptosis of infiltrating T cells (Bechmann et al. 1999, 2002). Astroglia also secrete tissue inhibitor of metalloproteinase (TIM), thereby limiting disruption of basement membrane and extracellular matrix by MMPs secreted by infiltrating T cells (Miljković et al. 2011). Ablation of proliferating astroglia exacerbates EAE and is associated with the massive infiltration of macrophages and T cells (Voskuhl et al. 2009), suggesting critical roles of astroglia in preventing the expansion of inflammation. We demonstrated that CSF levels of angiotensin and angiotensin-converting enzymes produced and secreted from astrocyte endfeet were significantly decreased in NMO (Matsushita et al. 2010) and MS (Kawajiri et al. 2008, 2009) patients, suggesting that injury of astrocyte endfeet and dampening of astrocytic barrier functions may occur in both MS and NMO.
Astroglia also produce a variety of growth factors that promote oligodendrocytes to form myelin (Moore et al. 2011) by influencing oligodendroglial progenitor cells (OPCs) (Gallo and Armstrong 2008). IL-6 and transforming growth factor (TGF)-β produced by activated astrocytes may promote neuroprotection (Allaman et al. 2011). A recent study showed that ablation of astroglia in glial fibrillary acidic protein (GFAP)-thymidine kinase transgenic mice with ganciclovir caused a failure of damaged myelin removal through decreased microglial activation during cuprizone-induced demyelination (Skripuletz et al. 2013). Thus, astroglia can deliver signals to microglia to clear myelin debris, thereby contributing to the regenerative process.
2.2.8.2 Neurotoxic Aspects
Activated astroglia morphologically demonstrate hypertrophy and increase expression of GFAP. Activated astroglia produce cytokines/chemoattractants as well as adhesion molecules for lymphocyte trafficking. For example, astroglia produce various proinflammatory cytokines, such as IL-1, IL-6, IL-12, IL-15, IL-23, IL-27, IL-33, CCL2 (MCP-1), CCL5 (RANTES), CXCL8 (IL-8), CXCL10 (IP-10), and CXCL12 (SDF-1). IL-12, IL-23, and IL-27 are essential for inducing Th1 and Th17 cells (Xu and Drew 2007; Kroenke et al. 2008; Markovic et al. 2009) while IL-15 is crucial for the activation of encephalitogenic CD8+ T cells (Saikali et al. 2010). CCL2 is a critical chemokine that attracts peripheral blood macrophages into the CNS (Yamasaki et al. 2012). Moreover, astroglia can express VCAM-1 and fibronectin CS-1, which are up-regulated in MS lesions (van Horssen et al. 2005; Engelhardt 2010). α4β1 integrin expressed on T cells interacts with its receptors, VCAM-1 and CS-1, thereby enabling T cells to traffic from the perivascular areas deep into the CNS parenchyma (Gimenez et al. 2004). Astroglia can express MHC class II and costimulatory molecules such as B7-1, B7-2, and CD40 (Chastain et al. 1812) dependent on the presence of IFNγ, TNF, and IL-1β (Dong and Benveniste 2001). Thus, astroglia may present autoantigens to T cells in the context of MHC class II molecules. Astroglia also produce iNOS via effects of endoplasmic reticulum stress chaperones (Saha and Pahan 2006), leading to the production of superoxide anion and peroxynitrite, which can damage oligodendrocytes harboring low antioxidant levels (Antony et al. 2004).
2.2.8.3 Hereditary Astrogliopathy
Alexander disease is caused by mutations in the GFAP gene (Brenner et al. 2001; Li et al. 2005), and thus is regarded as a primary astrocytic disease. Alexander disease shows leukodystrophy with macrocephaly. Its characteristic feature is the widespread presence of Rosenthal fibers containing mutant GFAP, heat shock protein 27, and αB-crystallin that are exclusively found in astrocyte foot processes and cell bodies (Quinlan et al. 2007). Rosenthal fibers are plentiful, especially in the astrocytic foot processes around blood vessels, and in subpial, subependymal, and periventricular zones (Liem and Messing 2009). Mutant GFAP may act as a toxic gain-of-function (Liem and Messing 2009). Alexander disease develops frontal dominant white matter degeneration while postnatal myelination progresses from the central sulcus to the occipital, frontal, and temporal poles. This suggests that Alexander disease is not dysmyelinogenic (Sawaishi 2009). Megalencephalic leukodystrophy with subcortical cysts (MLC) is another example of leukodystrophy caused by mutations in the MCL1 gene that encodes a protein exclusively expressed in astrocytes (Leegwater et al. 2001). MLC1 is normally expressed in astrocyte endfeet around the blood vessels (Boor et al. 2005). These examples indicate astrocytes are important for the maintenance of myelin in the CNS and astrocytic endfoot dysfunction may induce widespread myelin loss.
2.2.9 Oligodendrocyte Precursor Cells and Remyelination in MS
Remission results from the resolution of inflammation, redistribution of ion channels along demyelinated axons, and remyelination. Many studies have demonstrated that demyelination in the MS brain and spinal cord can be followed by remyelination to a variable extent (Patrikios et al. 2006; Patani et al. 2007; Prineas et al. 1993). Remyelination is more prominent in the early stages of disease while chronic lesions have less or no remyelination. PPMS brains had a lower number of inflammatory active lesions and more complete remyelination than SPMS brains (Reynolds et al. 2011). Remyelination requires new oligodendroglia. OPCs expressing neuron glia 2, an integral membrane chondroitin sulfate proteoglycan, and platelet-derived growth factor receptor α, were shown to exist even in chronic MS lesions (Chang et al. 2000, 2002). Nonetheless, in chronic lesions demyelination tends to persist. These observations suggest that remyelination failure in MS is not attributable to the absence of OPCs, but rather the blockade of OPC differentiation to myelinating oligodendroglia. This blockade is explained by the existence of extracellular inhibitors or intrinsic intracellular blocking mechanisms. Examples of extracellular inhibitors include Jagged 1 expressed by astrocytes, which activates Notch 1 receptors on oligodendroglia to promote expression of Hes5 (John et al. 2002), a transcriptional inhibitor that blocks differentiation. LINGO-1 expressed on astrocytes and macrophages (Satoh et al. 2007; Mi et al. 2005), PSA-NCAM abnormally expressed on demyelinated axons (Charles et al. 2002), and myelin debris (Kotter et al. 2006) can inhibit the differentiation of OPCs to myelinating oligodendroglia. A recent report showed that aggregates of fibronectin produced by astrocytes and leaked from damaged BBB inhibited oligodendroglial differentiation and remyelination (Stoffels et al. 2013). Increased expression of fibronectin in active MS lesions (Stoffels et al. 2013) may also inhibit remyelination. Furthermore, OPCs expressing Notch1 were stimulated by contactin expressed by demyelinated axons in chronic MS lesions while over-expression of TAT-interacting protein 30 kDa, a direct inhibitor of importin B, in OPCs blocked the translocation of Notch1-intracellular domains produced by cleavage of Notch1 receptor engagement (Nakahara et al. 2009). Thus, signals required for myelinogenesis are not operative in chronic MS lesions.
2.2.10 Connexins
Connexins (Cxs) form homotypic or heterotypic gap junctions between astrocytes, or between astrocytes and oligodendrocytes. Gap junctions appose two cells and form channels for direct intercellular communication through which intracellular second messengers such as calcium ions and other small molecules are exchanged. Astrocytic Cx43 and Cx30, oligodendrocytic Cx32 and Cx47, and astrocytic Cx43 and oligodendrocytic Cx32 double-knockout mice showed diffuse demyelination (Lutz et al. 2009; Magnotti et al. 2011; Menichella et al. 2003). Notably, Ezan et al. (2012) demonstrated that mice lacking Cx43/Cx30 in GFAP-positive astrocytes displayed astrocyte endfeet edema and a partial loss of AQP4. Thus, astrocytic and oligodendrocytic Cxs may play critical roles in maintaining CNS myelin.
Recently, we showed the extensive loss of Cxs43, 32, and 47 in demyelinated and myelin-preserved layers of acute lesions from patients with Baló’s concentric sclerosis, a rare extremely severe variant of MS (Masaki et al. 2012). In the leading edge areas, where the expression of MAG was partly diminished with other myelin proteins well preserved, compatible with distal oligodendrogliopathy, astrocytic Cx43 was totally lost. Similar changes were also observed in MS and NMO cases culminating in death within 2 years after the disease onset (Masaki et al. 2013). It was reported that a significant reduction of Cx32 and Cx47 occurs in active lesions of MOG-induced EAE (Markoullis et al. 2012). In MOG- and myelin basic protein (MBP)-induced EAE (Brand-Schieber et al. 2005), astrocytic Cx43 was also diminished in active lesions, suggesting that myelin antigen-specific T cells may down-modulate Cx expression in oligodendrocytes and astrocytes. In the healthy state, Cx43 on astrocytes apposes Cx47 on oligodendrocytes, forming astrocyte–oligodendrocyte (A/O) gap junctions. A/O gap junctions are important for intercellular communication through this channel. Disruption of A/O gap junctions may cause the loss of glia syncytium, thereby inducing oligodendroglial damage and myelin loss (Fig. 2.2).

Loss of astrocytic Cx43 and disruption of astroglia–oligodendroglia gap junction channels in the leading edge areas of Baló’s disease lesions. (a) In the normal state, astrocyte–oligodendrocyte gap junction channels are formed by Cx43 and Cx43. (b) In the outer portion of the leading edge areas (dagger), astrocytic Cx43 is preferentially lost and therefore oligodendrocytic Cx47 forms a oligodendroglia
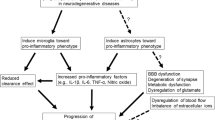
2.3 Neuroglial Inflammation in ALS
ALS is a progressive, fatal neurodegenerative disease where the loss of motor neurons in the spinal cord, brainstem, and motor cortex causes progressive motor paralysis. ALS usually develops sporadically; however, around 10 % of cases have a family history. A variety of susceptibility genes for familial ALS have been identified including mutations in Cu 2+ Zn 2+ superoxide dismutase (SOD1) gene (20 % of cases). Mutations in TADRBP, FUS, ANG, VCP, OPTN, and C9ORF72 genes are less frequent (Ince et al. 2011; Philips and Robberecht 2011). Although glutamate excitotoxicity, oxidative stress, neuroinflammation, and neurotrophic factor failure have been proposed to explain the pathogenesis of ALS, the mechanisms underlying ALS remain to be elucidated.
The appearance of reactive microglia and astroglia is a characteristic feature of ALS and other neurodegenerative diseases, such as Parkinson’s disease and Alzheimer’s disease. In this section, the review will focus on microglia and astroglia that are thought to have key roles in neuroinflammation in neurodegenerative diseases. However, recent studies have revealed that oligodendroglia have close connections with neurons and support the metabolism of neuronal cells (Philips et al. 2013).
2.3.1 Non-cell Autonomous Cell Death
Neuroinflammation characterized by activated microglia and infiltrating T cells is one of the prominent pathological features of ALS (Philips and Robberecht 2011; Kawamata et al. 1992). Motor neuron-specific expression of mutant SOD1 (mSOD1) does not result in ALS-like disease (Pramatarova et al. 2001), and wild-type non-neuronal cells extend the survival of mSOD1-positive motor neurons in a mSOD1 transgenic ALS mouse model (Clement et al. 2003). The selective lowering of mSOD1 levels in microglia or astroglia slows disease progression, while reducing mSOD1 levels in neurons delays disease onset (Boillée et al. 2006; Yamanaka et al. 2008). These results suggest that mSOD1 in motor neurons determines disease onset while its expression in microglia and astroglia determines disease progression. Thus, motor neurons do not die by intrinsic mechanisms and motor neuron death is now regarded as non-cell autonomous cell death.
2.3.2 Alterations of Cytokines/Chemokines and Growth Factors Influencing Neuroinflammation and Neurodegeneration in ALS
We and others have demonstrated that proinflammatory cytokines/chemokines, such as CCL2 (MCP1), CCL4 (MIP1β), CXCL8 (IL-8), CXCL10 (IP10), IL-1β, IL-7, IL-9, IL-12 (p70), IL-17, IFNγ, and TNFα, were elevated in CSF from ALS patients (Tanaka et al. 2006; Mitchell et al. 2009; Tateishi et al. 2010). These proinflammatory cytokines were also elevated in spinal cord tissues (Weydt et al. 2004; Meissner et al. 2010). Interestingly, CCL2 and CXCL8 levels in CSF showed a significant positive correlation with disease severity while those of CCL4 and CXCL10 had a negative correlation with disease severity (Tateishi et al. 2010). Thus, the relationship of proinflammatory cytokines/chemokines with disease severity is not uniform, suggesting that CCL2 and CXCL8 may be neurotoxic while CCL4 and CXCL10 may be neuroprotective.
We also found increased granulocyte colony stimulating factor (G-CSF) levels in the CSF of ALS cases (Tanaka et al. 2006). G-CSF was expressed in reactive astroglia from ALS cases but not controls, while G-CSF receptor expression was significantly decreased in motor neurons in ALS cases (Tanaka et al. 2006). Thus, the neuroprotective effects of G-CSF secreted by astroglia do not occur because of the down-modulation of G-CSF receptors in motor neurons (Tanaka et al. 2006).
2.3.3 Microglia in ALS
Activated microglia produce a variety of proinflammatory cytokines/chemokines and oxidative molecules, such as NO and O2. In contrast, they can also secrete neurotrophic factors and anti-inflammatory cytokines. Microglia producing proinflammatory cytokines are termed M1 microglia and exert deleterious effects and those that secrete anti-inflammatory cytokines are termed M2 microglia and exhibit neuroprotective activity. However, the clear-cut separation of M1 and M2 microglia may be difficult and the surrounding environment may modify the dual features of microglia. Another issue of note is that the classification of activated microglia residing in the CNS and infiltrating macrophages from the peripheral blood is often difficult when staining for cell-surface markers, and therefore immunohistochemical results should be interpreted cautiously.
Microglial activation is observed in the anterior horns, along the corticospinal tract, and in the motor cortex of postmortem ALS samples (Kawamata et al. 1992). However, some studies have reported a more widespread infiltration of activated microglia (Hayashi et al. 2001). Intriguingly, [11C]-PK11195 PET imaging observed the in vivo activation of microglia in motor cortices, dorsolateral prefrontal cortices, and thalami from ALS patients (Turner et al. 2004). In a G93A mSOD1 transgenic ALS mouse model, microglial activation occurred well before motor neuron loss and was present even at preclinical stages (Yamasaki et al. 2010; Kawamura et al. 2012; Nagara et al. 2013). In the sciatic nerves, macrophage infiltration occurs early, probably reflecting that the mSOD1 ALS model has characteristics of distal axonopathy (Fischer et al. 2004). Microglial activation is frequently associated with CD4+ and CD8+ T cell infiltration in ALS (Engelhardt et al. 1993), suggesting interactions between these two cell types. Activated microglia in the spinal cord from ALS patients up-regulate dendritic cell markers, such as CD11c and CD86 (Henkel et al. 2004). Similar up-regulation of antigen-presenting markers is also seen in the late stage of the mSOD1 transgenic ALS mouse model (Henkel et al. 2005). Therefore, microglia may present antigens to T cells (Henkel et al. 2004), similar to dendritic cells, during the late stages of ALS, while T cells may potentiate neuroinflammation (Chiu et al. 2008) or act neuroprotectively (Yamasaki et al. 2010; Beers et al. 2008). Although activated microglia may be toxic in the progressive stages, in mSOD1 ALS mice, motor neuron loss was not altered by ablation of proliferating microglia (Gowing et al. 2006). However, the same procedure worsened animal models of Alzheimer’s disease (Simard et al. 2006), suggesting the roles of activated microglia are not similar among various neurodegenerative disorders.
In mSOD1-Tg mice, microglial proliferation and T cell infiltration occurred relatively early in the spinal cord (Alexianu et al. 2001), but not in the brainstem (Yamasaki et al. 2010). Furthermore, relatively preserved motor neurons were observed even in the disease progression stage (Chiu et al. 1995). Thus, we studied the actions of microglia following acute and chronic motor neuronal insults in mSOD1 ALS mice (Yamasaki et al. 2010; Kawamura et al. 2012) by unilateral hypoglossal nerve axotomy at young (8 weeks) and adult (17 weeks) ages. On day 21 following hypoglossal axotomy, the numbers of surviving neurons were markedly reduced in mSOD1 mice than non-transgenic littermates at 17 weeks of age but the same difference was not seen at 8 weeks, suggesting increased vulnerability of hypoglossal motor neurons to neuronal injury in adult mice (Yamasaki et al. 2010). On day 3 after axotomy, the number of microglia expressing glial cell-derived neurotrophic factor (GDNF) and insulin-like growth factor (IGF-1) that surrounded axotomized hypoglossal neurons was significantly lower in mSOD1-Tg mice than in non-transgenic mice at 17 weeks but not at 8 weeks, despite the good preservation of hypoglossal neurons. Infiltration of CD3+ T cells, mostly expressing CD4, occurred on day 7 (Kawamura et al. 2012). The migratory ability of cultured microglia from mSOD1 mice to MCP-1 was decreased compared with those from non-transgenic littermates (Yamasaki et al. 2010). G-CSF significantly restored the impaired migratory ability of mSOD1 microglia in vitro, while in vivo it increased the number of microglia surrounding axotomized neurons and improved neuronal survival following axotomy in mSOD1 ALS mice. Moreover, chronic administration of G-CSF significantly increased the numbers of GDNF-positive microglia surrounding spinal motor neurons in the anterior horns and increased the life span of mSOD1 ALS mice (Yamasaki et al. 2010). These findings suggest that microglia act protectively through the production of growth factors, and that decreased microglial protective ability of mSOD1 mice can be restored by appropriate growth factors, such as G-CSF. It is interesting to note that CNS microglia and peripheral blood monocytes/macrophages show decreased chemotactic activity in mSOD1 mice (Yamasaki et al. 2010). Decreased mobilization of microglia into the damaged CNS and macrophages into the peripheral nerves may explain the acceleration of motor neuron death, because these myeloid cells produce neuroprotective factors.
2.3.4 Astroglia in ALS
Astroglia usually have a neuroprotective function but can become neuroinflammatory upon interactions with their surrounding microenvironment. Reactive astroglia, characterized by increased expression of GFAP, exist in the anterior and posterior horns in the spinal cord from ALS patients and are present in the gray and subcortical white matter, not being limited to the motor cortex (Nagy et al. 1994; Kushner et al. 1991; Schiffer et al. 1996). Reactive astroglia can overexpress neurotoxic factors such as iNOS, which in turn produces reactive oxygen and nitrogen species (Sasaki et al. 2001).
Astroglia produce a variety of neurotrophic factors, such as GDNF, brain-derived neurotropic factor (BDNF), and IGF-1. Astroglia also produce VEGF that induces vascular proliferation and enhances neuronal survival (Jin et al. 2002; Tolosa et al. 2008). However, VEGF receptor 1 and 2 are decreased during disease progression, and, therefore, the over-expression of VEGF is unable to exert its protective effects in human ALS (Nagara et al. 2013). Astroglial expression of glutamate transporter EAAT2 and GLT1 is decreased in ALS patients and the mSOD1 ALS mice, respectively (Rothstein et al. 1992, 1995; Howland et al. 2002). Neurons can increase astroglial glutamate transporter expression through the induction of kappa B-motif binding phosphoprotein factor to the GLT1 promoter (Yang et al. 2009), thereby reinforcing the clearance of glutamate. Decreased neuronal regulation may in turn accelerate excitotoxic neuronal death by overstimulation of AMPA and NMDA receptors.
2.3.5 T Cells in ALS
T cells are rarely seen in the normal CNS, which is an immunologically privileged site, while infiltrating T cells were observed in postmortem tissues of ALS patients (Engelhardt et al. 1993). Depletion of T cells in mSOD1 mice crossbred with RAG2−/− or CD4−/− mice resulted in accelerated motor neuron degeneration (Beers et al. 2008) where CD11b+ microglia were markedly reduced. In ALS models, depletion of T cells induces CNS neurotoxic substances, such as TNFα, and reduces neuroprotective factors, such as IGF-1, GDNF, and BDNF (Chiu et al. 2008; Beers et al. 2008). The adoptive transfer of regulatory or effector T cells from wild-type mice to mSOD1 Tg mice delayed disease progression (Banerjee et al. 2008) and the passive transfer of CD4+ CD25+ FoxP3+ regulatory T cells into mSOD1 mice ameliorated ALS (Beers et al. 2011). IL-4 produced by regulatory T cells suppressed the toxic properties of microglia (Beers et al. 2011). T cells from ALS CNS tissues may be neuroprotective; however, their protective function appears to be insufficient.
2.3.6 Experimental Therapy in ALS
The presence of glial inflammation in ALS encouraged the use of anti-inflammatory and immunomodulatory drugs, and several case reports have described beneficial effects of anti-inflammatory therapies, even in familial ALS cases (Saiga et al. 2012). This suggested that some inflammatory components are involved in human ALS pathogenesis. However, clinical trials investigating these drugs demonstrated no efficacy. Trials of nonspecific immunosuppression with cyclophosphamide or cyclosporin, whole body irradiation, or bone-marrow stem cell transplantation have all been unsuccessful (Appel et al. 2008). Expression of cyclooxygenase 2 (COX2), which is critical in producing proinflammatory prostaglandins, is increased in ALS patients and mSOD1 mice (Almer et al. 2001). However, although the COX2 inhibitor celecoxib is effective at delaying the disease course in ALS model mice, it has no effect in human ALS (Drachman et al. 2002; Pompl et al. 2003). Minocycline down-modulated microglial activation and improved the survival rate of mSOD1 ALS mice (Zhu et al. 2002), but it was not effective in humans with ALS (Scott et al. 2008). To date, no single anti-inflammatory drug has been successful in human ALS even though some are effective in animal models. Not all inflammatory components are toxic and some might act protectively. This may explain in part why general anti-inflammatory procedures are not beneficial in human ALS. Either TNFα (Gowing et al. 2008) or IL-1β gene knockout (Nguyen et al. 2001) did not ameliorate disease in mSOD1 ALS mice. Thus, it is unlikely that a single cytokine is responsible for the exacerbation of motor neuron degeneration in ALS, but rather the combined effects of such proinflammatory cytokines may accelerate motor neuron death.
Although the administration of neurotrophic factors, such as GDNF, BDNF, IGF-1, and VEGF, increased the survival of mSOD1 Tg mice (Kaspar et al. 2003; Azzouz et al. 2004; Storkebaum et al. 2005), these factors could not sufficiently delay clinical progression in human ALS patients. This may be explained by the differences in bioavailability and dosage between mice and humans. In our study, mSOD1-harboring microglia increased GDNF expression upon stimulation with G-CSF while concurrently such GDNF-positive microglia also expressed iNOS, a potential neurotoxic factor (Yamasaki et al. 2010; Kawamura et al. 2012). These findings support the idea that M1 and M2 microglia are not clearly separable and that microglia may simultaneously produce both protective and toxic substances. Thus, the nonselective targeting of microglia may not be successful in future clinical trials for neurodegenerative diseases.
2.4 Conclusion and Future Perspectives
Neuroglial inflammation has dual actions; protective and toxic. Neuroglial inflammatory reactions are distinct between neuroimmunological and neurodegenerative diseases and even among neurodegenerative disorders. Detailed analyses of glial activation states may increase our understanding and influence how we can decipher and control glial dual actions, thereby enabling new drug development targeting glia in humans.
References
Alexianu ME, Kozovska M, Appel SH (2001) Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 57:1282–1289
Allaman I, Bélanger M, Magistretti PJ (2011) Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci 34:76–87
Almer G, Guegan C, Teismann P et al (2001) Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol 49:176–185
Antony JM, van Marle G, Opii W et al (2004) Human endogenous retrovirus glycoprotein-mediated induction of redox reactants causes oligodendrocyte death and demyelination. Nat Neurosci 7:1088–1095
Appel SH, Engelhardt JI, Henkel JS et al (2008) Hematopoietic stem cell transplantation in patients with sporadic amyotrophic lateral sclerosis. Neurology 71:1326–1334
Azzouz M, Ralph GS, Storkebaum E et al (2004) VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 429:413–417
Babbe H, Roers A, Waisman A et al (2000) Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 192:393–404
Banerjee R, Mosley RL, Reynolds AD et al (2008) Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One 3:e2740
Barnett MH, Prineas JW (2004) Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468
Bechmann I, Mor G, Nilsen J et al (1999) FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia 27:62–74
Bechmann I, Steiner B, Gimsa U et al (2002) Astrocyte-induced T cell elimination is CD95 ligand dependent. J Neuroimmunol 132:60–65
Bechmann I, Galea I, Perry VH (2007) What is the blood-brain barrier (not)? Trends Immunol 28:5–11
Beers DR, Henkel JS, Zhao W, Wang J, Appel SH (2008) CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A 105:15558–15563
Beers DR, Henkel JS, Zhao W et al (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 134:1293–1314
Benarroch EE (2009) Oligodendrocytes. Susceptibility to injury and involvement in neurologic disease. Neurology 72:1779–1785
Bjartmar C, Kidd G, Mörk S, Rudick R, Trapp BD (2000) Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol 48:893–901
Bö L, Vedeler CA, Nyland H, Trapp BD, Mörk SJ (2003) Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler 9:323–331
Boillée S, Yamanaka K, Lobsiger CS et al (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392
Bonati U, Fisniku LK, Altmann DR et al (2011) Cervical cord and brain grey matter atrophy independently associate with long-term MS disability. J Neurol Neurosurg Psychiatry 82:471–472
Boor PK, de Groot K, Waisfisz Q et al (2005) MLC1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol 64:412–419
Booss J, Esiri MM, Tourtellotte WW, Mason DY (1983) Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J Neurol Sci 62:219–232
Brand-Schieber E, Werner P, Iacobas DA et al (2005) Connexin43, the major gap junction protein of astrocytes, is down-regulated in inflamed white matter in an animal model of multiple sclerosis. J Neurosci Res 80:798–808
Breij EC, Brink BP, Veerhuis R et al (2008) Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann Neurol 63:16–25
Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet 27:117–120
Brilot F, Dale RC, Selter RC et al (2009) Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol 66:833–842
Calabrese M, Filippi M, Gallo P (2010) Cortical lesions in multiple sclerosis. Nat Rev Neurol 6:438–444
Campbell GR, Ziabreva I, Reeve AK et al (2011) Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 69:481–492
Cardona AE, Pioro EP, Sasse ME et al (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924
Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD (2000) NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20:6404–6412
Chang A, Tourtellotte WW, Rudick R, Trapp BD (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346:165–173
Charles P, Reynolds R, Seilhean D et al (2002) Re-expression of PSA-NCAM by demyelinated axons: an inhibitor of remyelination in multiple sclerosis? Brain 125:1972–1979
Chastain EM, Duncan DS, Rodgers JM, Miller SD (1812) The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta 2011:265–274
Chiu AY, Zhai P, Dal Canto MC, Peters TM, Kwon YW, Prattis SM, Gurney ME (1995) Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci 6:349–362
Chiu IM, Chen A, Zheng Y et al (2008) T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A 105:17913–17918
Clement AM, Nguyen MD, Roberts EA et al (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117
Coisne C, Mao W, Engelhardt B (2009) Cutting edge: natalizumab blocks adhesion but not initial contact of human T cells to the blood-brain barrier in vivo in an animal model of multiple sclerosis. J Immunol 182:5909–5913
Confavreux C, Vukusic S (2006) Age at disability milestones in multiple sclerosis. Brain 129:595–605
Confavreux C, Vukusic S, Moreau T, Adeleine P (2000) Relapses and progression of disability in multiple sclerosis. N Engl J Med 343:1430–1438
Derfuss T, Parikh K, Velhin S et al (2009) Contctin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci U S A 106:8302–8307
Desmazières A, Sol-Foulon N, Lubetzki C (2012) Changes at the nodal and perinodal axonal domains: a basis for multiple sclerosis pathology? Mult Scler 18:133–137
Dong Y, Benveniste EN (2001) Immune function of astrocytes. Glia 36:180–190
Drachman DB, Frank K, Dykes-Hoberg M et al (2002) Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol 52:771–778
Dutta R, McDonough J, Yin X et al (2006) Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 59:478–489
Dutta R, Chang A, Doud MK et al (2011) Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol 69:445–454
Engelhardt B (2010) T cell migration into the central nervous system during health and disease: different molecular keys allow access to different central nervous system compartments. Clin Exp Neuroimmunol 1:79–93
Engelhardt JI, Tajti J, Appel SH (1993) Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol 50:30–36
Ezan P, André P, Cisternino S et al (2012) Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab 32:1457–1467
Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120:393–399
Fischer LR, Culver DG, Tennant P et al (2004) Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240
Fisher E, Lee JC, Nakamura K, Rudick RA (2008) Gray matter atrophy in multiple sclerosis: a longitudinal study. Ann Neurol 64:255–265
Fisniku LK, Chard DT, Jackson JS et al (2008) Gray matter atrophy is related to long-term disability in multiple sclerosis. Ann Neurol 64:247–254
Friese MA, Fugger L (2009) Pathogenic CD8(+) T cells in multiple sclerosis. Ann Neurol 66:132–141
Frischer JM, Bramow S, Dal-Bianco A et al (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132:1175–1189
Gallo V, Armstrong RC (2008) Myelin repair strategies: a cellular view. Curr Opin Neurol 21:278–283
Geurts JJ, Pouwels PJ, Uitdehaag BM, Polman CH, Barkhof F, Castelijns JA (2005) Intracortical lesions in multiple sclerosis: improved detection with 3D double inversion-recovery MR imaging. Radiology 236:254–260
Gimenez MA, Sim JE, Russell JH (2004) TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol 151:116–125
Gowing G, Dequen F, Soucy G, Julien JP (2006) Absence of tumor necrosis factor-alpha does not affect motor neuron disease caused by superoxide dismutase I mutations. J Neurosci 26:11397–11402
Gowing G, Philips T, Van Wijmeersch B et al (2008) Ablation of proliferating microglia does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci 28:10234–10244
Hauser SL, Waubant E, Arnold DL et al (2008) B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358:676–688
Hayashi S, Sakurai A, Amari M, Okamoto K (2001) Pathological study of the diffuse myelin pallor in the anterolateral columns of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 188:3–7
Henkel JS, Engelhardt JI, Siklos L et al (2004) Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 55:221–235
Henkel JS, Beers DR, Siklos L, Apple SH (2005) The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol Cell Neurosci 31:427–437
Howland DS, Liu J, She Y et al (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 99:1604–1609
Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ (2011) Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 122:657–671
Ishizu T, Osoegawa M, Mei FJ et al (2005) Intrathecal activation of the IL-17/IL-8 axis in opticospinal multiple sclerosis. Brain 128:988–1002
Janzer RC, Raff MC (1987) Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 325:253–257
Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA (2002) Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A 99:11946–11950
John GR, Shankar SL, Shafit-Zagardo B et al (2002) Multiple sclerosis: re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med 8:1115–1121
Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH (2003) Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301:839–842
Kawajiri M, Mogi M, Osoegawa M et al (2008) Reduction of angiotensin II in the cerebrospinal fluid of patients with multiple sclerosis. Mult Scler 14:557–560
Kawajiri M, Mogi M, Higaki N et al (2009) Angiotensin-converting enzyme (ACE) and ACE2 levels in the cerebrospinal fluid of patients with multiple sclerosis. Mult Scler 15:262–265
Kawamata T, Akiyama H, Yamada T, McGeer PL (1992) Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol 140:691–707
Kawamura MF, Yamasaki R, Kawamura N et al (2012) Impaired recruitment of neuroprotective microglia and T cells during acute neuronal injury coincides with increased neuronal vulnerability in an amyotrophic lateral sclerosis model. Exp Neurol 234:437–445
Kondo T, Takahashi K, Kohara N et al (2002) Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology 59:1105–1107
Kooi EJ, Geurts JJ, van Horssen J, Bø L, van der Valk P (2009) Meningeal inflammation is not associated with cortical demyelination in chronic multiple sclerosis. J Neuropathol Exp Neurol 68:1021–1028
Kornek B, Lassmann H (1999) Axonal pathology in multiple sclerosis. A historical note. Brain Pathol 9:651–656
Kotter MR, Li WW, Zhao C, Franklin RJ (2006) Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 26:328–332
Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, Ebers GC (2006) The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain 129:584–594
Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM (2008) IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med 205:1535–1541
Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Brück W (2002) Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 125:2202–2212
Kurtzke JF (1983) Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33:1444–1452
Kushner PD, Stephenson DT, Wright S (1991) Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J Neuropathol Exp Neurol 50:63–277
Kutzelnigg A, Lucchinetti CF, Stadelmann C et al (2005) Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 128:2705–2712
Lassmann H, Raine CS, Antel J, Prineas JW (1998) Immunopathology of multiple sclerosis: report on an international meeting held at the institute of neurology of the University of Vienna. J Neuroimmunol 86:213–217
Leegwater PA, Yuan BQ, van der Steen J et al (2001) Mutations of MLC1 (KIAA0027) encoding a putative membrane protein cause megalencepahlic leukoencephalopathy with subcortical cysts. Am J Hum Genet 68:831–838
Lennon VA, Wingerchuk DM, Kryzer TJ et al (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112
Lennon VA, Kryzer TJ, Pittock SJ et al (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Li R, Johnson AB, Salomons G et al (2005) Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann Neurol 57:310–326
Liem RK, Messing A (2009) Dysfunctions of neuronal and glial intermediate filaments in disease. J Clin Invest 119:1814–1824
Lucchinetti C (2007) Multiple sclerosis pathology during early and late disease phases: pathogenic and clinical relevance. In: Zhang J (ed) Immune regulation and immunotherapy in autoimmune disease. Springer, New York, pp 214–264
Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717
Lutz SE, Zhao Y, Gulinello M, Lee SC, Raine CS, Brosnan CF (2009) Deletion of astrocyte connexins 43 and 30 leads to a dysmyelinating phenotype and hippocampal CA1 vacuolation. J Neurosci 29:7743–7752
Magliozzi R, Howell O, Vora A et al (2007) Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130:1089–1104
Magliozzi R, Howell OW, Reeves C et al (2010) A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 68:477–493
Magnotti LM, Goodenough DA, Paul DL (2011) Deletion of oligodendrocyte Cx32 and astrocyte Cx43 causes white matter vacuolation, astrocyte loss and early mortality. Glia 59:1064–1074
Mahad DJ, Ransohoff RM (2003) The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 15:23–32
Mahad DJ, Howell SJ, Woodroofe MN (2002) Expression of chemokines in the CSF and correlation with clinical disease activity in patients with multiple sclerosis. J Neurol Neurosurg Psychiatry 72:498–502
Markoullis K, Sargiannidou I, Gardner C, Hadjisavvas A, Reynolds R, Kleopa KA (2012) Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 60:1053–1066
Markovic M, Miljkovic D, Momcilovic M et al (2009) Strain difference in susceptibility to experimental autoimmune encephalomyelitis in rats correlates with T(H)1 and T(H)17-inducing cytokine profiles. Mol Immunol 47:141–146
Masaki K, Suzuki SO, Matsushita T et al (2012) Extensive loss of connexins in Baló’s disease: evidence for an auto-antibody-independent astrocytopathy via impaired astrocyte-oligodendrocyte/myelin interaction. Acta Neuropathol 123:887–900
Masaki K, Suzuki SO, Matsushita T et al (2013) Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS One (in press)
Mathey EK, Derfuss T, Storch MK et al (2007) Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med 204:2363–2372
Matsuoka T, Suzuki SO, Suenaga T, Iwaki T, Kira J (2011) Reappraisal of aquaporin-4 astrocytopathy in Asian neuromyelitis optica and multiple sclerosis patients. Brain Pathol 11:516–532
Matsushita T, Isobe N, Kawajiri M et al (2010) CSF angiotensin II and angiotensin-converting enzyme levels in anti-aquaporin-4 autoimmunity. J Neurol Sci 291:37–43
Matsushita T, Tateishi T, Isobe N, Yonekawa T, Yamasaki R, Murai H, Kira J (2013) Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS One 8(4):e61835
Meinl E, Krumbholz M, Hohlfeld R (2006) B lineage cells in the inflammatory central nervous system environment: migration, maintenance, local antibody production, and therapeutic modulation. Ann Neurol 59:880–892
Meissner F, Molawi K, Zychlinsky A (2010) Mutant superoxide dismutase 1-induced IL-1beta accelerates ALS pathogenesis. Proc Natl Acad Sci U S A 107:13046–13050
Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL (2003) Connexins are critical for normal myelination in the CNS. J Neurosci 23:5963–5973
Mi S, Miller RH, Lee X et al (2005) LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci 8:745–751
Miljković D, Timotijević G, Mostarica Stojković M (2011) Astrocytes in the tempest of multiple sclerosis. FEBS Lett 585:3781–3788
Miller RH, Raff MC (1984) Fibrous and protoplasmic astrocytes are biochemically and developmentally distinct. J Neurosci 4:585–592
Mistry N, Dixon J, Tallantyre E et al (2013) Central veins in brain lesions visualized with high-field magnetic resonance imaging: a pathologically specific diagnostic biomarker for inflammatory demyelination in the brain. JAMA Neurol 70:1–6
Mitchell RM, Freeman WM, Randazzo WT, Stephens HE, Beard JL, Simmons Z, Connor JR (2009) A CSF biomarker panel for identification of patients with amyotrophic lateral sclerosis. Neurology 72:14–19
Moore CS, Abdullah SL, Brown A, Arulpragasam A, Crocker SJ (2011) How factors secreted from astrocytes impact myelin repair. J Neurosci Res 89:13–21
Nagara Y, Tateishi T, Yamasaki R, et al (2013) Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1α in amyotrophic lateral sclerosis. Brain Pathol. doi: 10.1111/bpa.12040. [Epub ahead of print]
Nagy D, Kato T, Kushner PD (1994) Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J Neurosci Res 38:336–347
Nakahara J, Kanekura K, Nawa M, Aiso S, Suzuki N (2009) Abnormal expression of TIP30 and arrested nucleocytoplasmic transport within oligodendrocyte precursor cells in multiple sclerosis. J Clin Invest 119:169–181
Nguyen MD, Julien JP, Rivest S (2001) Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol 50:630–639
Odoardi F, Sie C, Streyl K et al (2012) T cells become licensed in the lung to enter the central nervous system. Nature 488:675–679
Otero K, Turnbull IR, Poliani PL et al (2009) Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol 10:734–743
Paloneva J, Kestilä M, Wu J et al (2000) Loss-of-function mutations in TYROBP (DAP12) results in a presenile dementia with bone cysts. Nat Genet 25:357–361
Paloneva J, Manninen T, Christman G et al (2002) Mutations in two genes encoding different subunits of a receptor signalling complex result in an identical disease phenotype. Am J Hum Genet 71:656–662
Patani R, Balaratnam M, Vora A, Reynolds R (2007) Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol 33:277–287
Patrikios P, Stadelmann C, Kutzelnigg A et al (2006) Remyelination is extensive in a subset of multiple sclerosis patients. Brain 129:3165–3172
Pellkofer HL, Krumbholz M, Berthele A et al (2011) Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 76:1310–1315
Peterson JW, Bö L, Mörk S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400
Philips T, Robberecht W (2011) Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol 10:253–263
Philips T, Bento-Abreu A, Nonneman A et al (2013) Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 136:471–482
Pitt D, Nagelmeier IE, Wilson HC, Raine CS (2003) Glutamate uptake by oligodendrocytes: implications for excitotoxicity in multiple sclerosis. Neurology 61:1113–1120
Pompl PN, Ho L, Bianchi M, McManus T, Qin W, Pasinetti GM (2003) A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 17:725–727
Pramatarova A, Laganière J, Roussel J, Brisebois K, Rouleau GA (2001) Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci 21:3369–3374
Prineas JW, Barnard RO, Revesz T, Kwon EE, Sharer L, Cho ES (1993) Multiple sclerosis. Pathology of recurrent lesions. Brain 116:681–693
Quinlan RA, Brenner M, Goldman JE, Messing A (2007) GFAP and its role in Alexander disease. Exp Cell Res 313:2077–2087
Rademakers R, Baker M, Nicholson AM et al (2012) Mutations in colony stimulating factor 1 (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 44:200–205
Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system parenchyma. Nature 468:253–262
Ransohoff RM, Engelhardt B (2012) The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 12:623–635
Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA (2001) Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154:427–434
Reynolds R, Roncaroli F, Nicholas R, Radotra B, Gveric D, Howell O (2011) The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol 122:155–170
Rothstein JD, Martin LJ, Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 326:1464–1468
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84
Saha RN, Pahan K (2006) Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal 8:929–947
Saiga T, Tateishi T, Torii T et al (2012) Inflammatory radiculoneuropathy in an ALS4 patient with a novel SETX mutation. J Neurol Neurosurg Psychiatry 83:763–764
Saikali P, Antel JP, Pittet CL, Newcombe J, Arbour N (2010) Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J Immunol 185:5693–5703
Saitoh B, Yamasaki R, Hayashi S et al (2013) A case of hereditary diffuse leukoencephalopathy with spheroids caused by a de novo mutation in CSFIR masquerading as primary progressive multiple sclerosis. Mult Scler (in press)
Sasaki S, Warita H, Abe K, Iwata M (2001) Inducible nitric oxide synthase (iNOS) and nitrotyrosine immunoreactivity in the spinal cords of transgenic mice with a G93A mutant SOD1 gene. J Neuropathol Exp Neurol 60:839–846
Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H (2007) TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol Appl Neurobiol 33:99–107
Sawaishi Y (2009) Review of Alexander disease: beyond the classical concept of leukodystrophy. Brain Dev 31:493–498
Schiffer D, Cordera S, Cavalla P, Migheli A (1996) Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139(suppl):27–33
Scott S, Kranz JE, Cole J et al (2008) Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler 9:4–15
Sherman DL, Tait S, Melrose S et al (2005) Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 48:737–742
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 16:489–502
Skripuletz T, Hackstette D, Bauer K et al (2013) Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 136:147–167
Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D (2005) Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 128:1016–1025
Srivastava R, Aslam M, Kalluri SR et al (2012) Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med 367:115–123
Stadelmann C, Wegner C, Brück W (2011) Inflammation, demyelination, and degeneration—recent insights from MS pathology. Biochim Biophys Acta 1812:275–282
Stoffels JM, de Jonge JC, Stancic M et al (2013) Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain 136:116–131
Storkebaum E, Lambrechts D, Dewerchin M et al (2005) Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci 8:85–92
Tanaka M, Kikuchi H, Ishizu T et al (2006) Intrathecal upregulation of granulocyte colony stimulating factor and its neuroprotective actions on motor neurons in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 65:816–825
Tanaka M, Matsushita T, Tateishi T et al (2008) Distinct CSF cytokine/chemokine profiles in atopic myelitis and other causes of myelitis. Neurology 71:974–981
Tateishi T, Yamasaki R, Tanaka M et al (2010) CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol 222:76–81
Tolosa L, Mir M, Asensio VJ, Olmos G, Llado J (2008) Vascular endothelial growth factor protects spinal cord motoneurons against glutamate-induced excitotoxicity via phosphatidylinositol 3-kinase. J Neurochem 105:1080–1090
Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T (1998) Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol 161:3767–3775
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285
Turner MR, Cagnin A, Turkheimer FE et al (2004) Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis 15:601–609
van der Laan LJ, De Groot CJ, Elices MJ, Dijkstra CD (1997) Extracellular matrix proteins expressed by human adult astrocytes in vivo and in vitro: an astrocyte surface protein containing the CS1 domain contributes to binding of lymphoblasts. J Neurosci Res 50:539–548
van Horssen J, Bö L, Vos CM, Virtanen I, de Vries HE (2005) Basement membrane proteins in multiple sclerosis-associated inflammatory cuffs: potential role in influx and transport of leukocytes. J Neuropathol Exp Neurol 64:722–729
van Horssen J, Dijkstra CD, de Vries HE (2007) The extracellular matrix in multiple sclerosis pathology. J Neurochem 103:1293–1301
Vercellino M, Plano F, Votta B, Mutani R, Giordana MT, Cavalla P (2005) Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol 64:1101–1107
Voskuhl RR, Peterson RS, Song B et al (2009) Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29:11511–11522
Wang Y, Szretter KJ, Vermi W et al (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13:753–760
Werner P, Pitt D, Raine CS (2001) Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 50:169–180
Weydt P, Yuen EC, Ransom BR, Moller T (2004) Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 48:179–182
Xu J, Drew PD (2007) Peroxisome proliferator-activated receptor-gamma agonists suppress the production of IL-12 family cytokines by activated glia. J Immunol 178:1904–1913
Yamanaka K, Chun SJ, Boillée S et al (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253
Yamasaki R, Tanaka M, Fukunaga M et al (2010) Restoration of microglial function by granulocyte-colony stimulating factor in ALS model mice. J Neuroimmunol 229:51–62
Yamasaki R, Liu LP, Lin J, Ransohoff RM (2012) Role of CCR2 in immunobiology and neurobiology. Clin Exp Neuroimmunol 3:16–29
Yang Y, Gozen O, Watkins A et al (2009) Presynaptic regulation of astroglial excitatory neurotransmitter transporter GLT1. Neuron 61:880–894
Zhu S, Stavrovskaya IG, Drozda M et al (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417:74–78
Zimmermann DR, Dours-Zimmermann MT (2008) Extracellular matrix of the central nervous system: from neglect to challenge. Histochem Cell Biol 130:635–653
Acknowledgements
This work was supported in part by a Health and Labour Sciences Research Grant on Intractable Diseases from the Ministry of Health, Labour and Welfare, Japan, and a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Conflict of interest Dr. Kira is an advisory board member for Merck Serono and a consultant for Biogen Idec Japan. He has received payment for lectures from Bayer Schering Pharma, Cosmic Cooperation, and Biogen Idec Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Kira, Ji. (2013). Neuroinflammation in Neurological Disorders. In: Suzumura, A., Ikenaka, K. (eds) Neuron-Glia Interaction in Neuroinflammation. Advances in Neurobiology, vol 7. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8313-7_2
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8313-7_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8312-0
Online ISBN: 978-1-4614-8313-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)