
Abstract
Cancer predisposition syndromes affecting the gastrointestinal (GI) tract represent a small proportion of GI cancers and may arise in the background of a polyposis syndrome. The molecular mechanisms underlying these syndromes have been instrumental in our understanding of the molecular basis of development and progression of the more frequent counterpart sporadic neoplasms, sharing many common molecular features. Syndromic hereditary cancers can involve any segment of the GI tract but predominantly involve the colon, and the most common cancers are colorectal adenocarcinomas (CRC). The most frequent inheritable GI cancer syndromes are those associated with germline mutations in the DNA mismatch repair (MMR) genes, in which case cancers do not arise in a polyposis background, and those attributed to underlying germline mutations in the APC or MYH genes in patients who manifest an adenomatous polyposis phenotype in the intestine. In addition to the well-characterized cancer syndromes, there are families with clustering of colon cancer, including patients with colon cancers before age 50, for whom the susceptibility gene loci have not been identified.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Germline Mutation
- Familial Adenomatous Polyposis
- Adenomatous Polyposis Coli
- Lynch Syndrome
- Familial Adenomatous Polyposis Patient
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Cancer predisposition syndromes affecting the gastrointestinal (GI) tract represent a small proportion of GI cancers and may arise in the background of a polyposis syndrome (Table 7.1). The molecular mechanisms underlying these syndromes have been instrumental in our understanding of the molecular basis of development and progression of the more frequent counterpart sporadic neoplasms, sharing many common molecular features. Syndromic hereditary cancers can involve any segment of the GI tract but predominantly involve the colon, and the most common cancers are colorectal adenocarcinomas (CRC). The most frequent inheritable GI cancer syndromes are those associated with germline mutations in the DNA mismatch repair (MMR) genes, in which case cancers do not arise in a polyposis background, and those attributed to underlying germline mutations in the APC or MYH genes in patients who manifest an adenomatous polyposis phenotype in the intestine. In addition to the well-characterized cancer syndromes, there are families with clustering of colon cancer, including patients with colon cancers before age 50, for whom the susceptibility gene loci have not been identified.1 – 3
Hereditary Non-Polyposis Colorectal Cancer–Lynch Syndrome
Clinical Features
Hereditary Non-Polyposis Colorectal Cancer, also known as Lynch syndrome (HNPCC/LS) is diagnosed on the basis of a germline mutation in one of the DNA MMR genes that results in deficient DNA mismatch repair (dMMR).4 – 6 The deficiency of DNA mismatch repair results in increased mutation accumulation in the genome and higher risk of neoplastic transformation. HNPCC/LS represents an estimated 3–6% of all colorectal cancer cases.4 , 7 HNPCC/LS is inherited as an autosomal dominant cancer predisposition syndrome, and is characterized by early onset of colorectal cancers, and increased frequency of cancers in the small bowel, stomach, biliary tract, pancreas, endometrium, ovary, urothelium (urinary bladder, ureter, and renal pelvis), brain, sebaceous gland adenomas, and keratoacanthomas.5 – 9 The extra-gastrointestinal neoplasms associated with HNPCC/LS will be described in further detail later in this chapter. Sebaceous gland adenomas and keratoacanthomas are part of the Muir-Torre syndrome, whereas tumors of the brain, usually glioblastomas, occur in the Turcot syndrome.5 , 9
The average age of presentation of colorectal cancer in HNPCC patients is 45 years of age, but the disease can be discovered in older patients.10 , 11 Tumors are located in the proximal colon in two-thirds of cases, but can occur in any segment of the gastrointestinal tract.10 , 11 Tumors are often multiple or associated with other synchronous or metachronous neoplasms of the HNPCC-cancer spectrum.5 , 7 , 9 , 10 HNPCC/LS patients are at higher lifetime risk for colorectal cancer than the general population, developing CRC in up to 80% and endometrial carcinoma in up to 60% of mutation carriers.5
Pathologic Features
Unique histopathologic features may be seen in colorectal adenocarcinomas with underlying dMMR that suggest the possibility of HNPCC/LS (reviewed in Gologan et al11 , 12), (Fig. 7.1). These features are not specific for HNPCC/LS cancers, and are also seen frequently in sporadic CRC with dMMR, as well as in some tumors that are proficient in DNA MMR.11 , 13 Three major histopathologic groups of dMMR colorectal cancers can be recognized (reviewed in Gologan et al12 (Fig. 7.1): (1) poorly differentiated adenocarcinomas, including medullary-type carcinomas, in which the neoplastic epithelium is infiltrated with high numbers of lymphocytes (tumor-infiltrating lymphocytes/TILs), and carcinomas with signet ring cell features; (2) mucinous adenocarcinomas, and (3) well to moderately differentiated adenocarcinomas. The presence of prominent intratumoral lymphocytes, with at least four lymphocytes infiltrating the tumor epithelium in a single 400× microscopic field,11 is the most predictive histologic finding of dMMR in CRC (Fig. 7.1). TILs may be particularly numerous not only in poorly differentiated cancers but also occur in the other morphologic types of HNPCC/LS-associated cancers. The intratumoral lymphocytes are CD3-positive T cells and most are CD8-positive cytotoxic T lymphocytes. Peritumoral lymphocytic inflammation and lymphoid aggregates forming a Crohn-like reaction are also frequent in carcinomas with dMMR.11 , 12 , 14 – 18

(a) Poorly differentiated adenocarcinoma with medullary features, showing prominent TILs; (b) Immunohistochemistry for MLH1 shows loss of expression of the protein in tumor cell nuclei, whereas MLH1 remains positive in the nucleus of infiltrating lymphocytes and surrounding stromal cells. (c) Moderately differentiated CRC with prominent TILs. (d) Immunohistochemistry for MSH2 shows loss of expression in tumor cell nuclei, whereas MSH2 remains positive in the nucleus of infiltrating lymphocytes and surrounding stromal cells. (e) Mucinous adenocarcinoma (H&E). (f) Immunohistochemistry for MLH1 shows loss of expression in tumor cell nuclei. Original magnifications ×200
Patients with HNPCC/LS typically develop a small number of colonic adenomas of traditional type (tubular or tubulovillous adenomas) at earlier age (mean 42–43, range 24–62 years) as compared to noncarriers of DNA mismatch repair gene mutations.11 , 14 – 16 , 19 – 21 Progression from adenoma to invasive adenocarcinoma occurs rapidly, in many patients within less than 3 years, in contrast to an average of 15 years in patients without HNPCC/LS.19 , 22 HNPCC/LS adenomas arise most frequently in the proximal colon and often contain high-grade dysplasia.19 , 23
Molecular Mechanisms of Cancer Development and Progression in HNPCC/LS
As introduced in Chap. 1 and further discussed in Chap. 8, deficiencies of DNA mismatch repair have been implicated in the development and progression of CRC in two contexts: (1) in HNPCC/LS patients with inhered DNA MMR gene mutations and (2) in patients with sporadic colorectal cancers, where deficient MMR is nearly always attributed to epigenetic silencing of the DNA MMR MLH1 gene.24
The two most frequent genes underlying MMR deficiency in HNPCC/LS are MLH1 and MSH2. Although the reported proportions vary in different studies, HNPPC/LS patients inherit germline mutations affecting the coding regions of MLH1 (approximately 40%), MSH2 (approximately 40%), MSH6 (approximately 10%), and PMS2 (approximately 5%).12 , 25 – 35 In addition to inherited germline mutations, germline epimutation of the promoter regions of MLH1 or MSH2 have been reported in rare cases of HNPCC/LS35 – 37 (Table 7.1).
Deficient DNA MMR occurs when both alleles of one of the MMR genes are inactivated. During normal DNA replication, errors such as base mismatches and insertion or deletion loops, especially in repetitive regions, are corrected by the DNA mismatch repair proteins. MMR deficiency allows for replication-associated errors to be propagated in newly synthesized DNA strands (Fig. 7.2). In HNPCC/LS, pathogenic mutations of MMR genes are inherited and are present in constitutional DNA such as is found in peripheral blood; however, dMMR only develops in somatic tissues, especially in colonic epithelium and a few other cellular targets, when a second hit affects the MMR gene and leads to its loss of function. It has been shown that the second hits in HNPCC/LS can be caused by large or small chromosomal deletions or by mitotic recombination-mediated gene conversion, occurring in up to 46% of tumors.3 Somatic methylation of the promoter of the wild-type allele occurs in some cases, affecting MLH1 more often than MSH2. 3 Somatic point mutations are thought to be the least common second-hit mechanism.3 The double allelic dMMR leads to a mutational phenotype that affects primarily repetitive nucleotides, known as microsatellite regions, with resulting microsatellite instability (MSI) seen in more than 90% of CRC cases in HNPCC/LS patients10 (Fig. 7.3), as well as mutations in target genes that may contribute to neoplastic development.

DNA mismatch repair proteins and mechanisms of repair. MutS heterodimers (MSH2/MSH6 or MSH2/MSH3) recognize a DNA strand loop or base mismatch and recruit MutL (MLH1/PMS2) heterodimers to the site. Other molecules required for strand discrimination, helicase, endonuclease activity, resynthesis, and ligation are recruited in the complex with correction and maintenance of the native DNA sequence

Microsatellite instability detected by amplification of markers of the NCI panel. PCR amplification of the BAT26 locus reveals the appearance of novel alleles (arrows), of smaller size, in the tumor DNA as compared to non-neoplastic colonic DNA. The dinucleotide repeat marker D5S346 shows microsatellite instability characterized by a novel allele (arrow), of larger size, in the tumor DNA
The DNA MMR genes encode the MutS proteins MSH2, MSH3, and MSH6, and the MutL proteins MLH1, PMS1, PMS2, and MLH338 , 39 (Fig. 7.2). In the process of DNA MMR, the MMR proteins form heterodimers (MutS and MutL)38 , 40 – 43 (Fig. 7.2). The MSH2 protein can interact with MSH6, forming the MutS-alpha complex, or with MSH3, forming the MutS-beta complex. MutS-alpha and MutS-beta heterodimers recognize and bind to post-DNA replication mismatched sequences. The MutS-alpha heterodimers are involved in the repair of single base mispairs and small insertion or deletion mispairs, whereas MutS-beta heterodimers primarily are involved in the correction of insertion or deletion mispairs.44 – 46 MutL heterodimers bind MutS-alpha or MutS-beta.38 Of the two possible MutL heterodimers, only MutL-alpha (MLH1 and PMS2) heterodimers are involved in DNA MMR, whereas the MutL-beta heterodimers (MLH1 and PMS1) do not appear to be significantly involved in DNA mismatch repair functions.47 , 48 The MMR proteins are stabilized by heterodimerization, and loss of the heterodimer protein(s) may result in their degradation. Since MSH2 and MLH1 have two possible proteins to heterodimerize with (Fig. 7.2), their expression is only lost when their respective genes are primarily inactivated by a mutation or epigenetic silencing. However, MSH6 only heterodimerizes with MSH2, and PMS2 only heterodimerizes with MLH1; thus, loss of MSH6 expression can be secondary to primary loss of MSH2 and loss of PMS2 expression can be secondary to primary loss of MLH1 (Figs. 7.2, 7.4, and 7.5).

Loss of expression of MLH1 (b) or MSH2 (e) is associated with loss of their MMR heterodimer proteins PMS2 (c) and MSH6 (f), respectively, exemplified in these two cases of moderately differentiated colonic adenocarcinoma. Note the prominent TILs in both cases, indicated by the arrows (c and d). Hematoxylin and Eosin stain (a) and (b).

Sebaceous adenoma in a patient with Muir-Torre syndrome (a) shows loss of expression of MSH2 (b) and MSH6 (c), suggestive of germline mutation in MSH2 and secondary loss of MSH6 due to inability to form stabilizing heterodimers. (a) H&E stain, (b and c) immunohistochemistry
The molecular mechanisms that underlie the neoplastic development and progression in HNPCC/LS characterize the so-called microsatellite instability (MSI pathway).32 , 49, 51 As in sporadic MSI-pathway carcinomas, dMMR may lead to accumulation of mutations in cancer-related genes, such as the TGF-beta receptor II, BAX, and MSH6 genes among others, in early pre-neoplastic cell populations as well as during the steps of neoplastic progression.52 In contrast to sporadic CRC, patients with germline mutations in DNA mismatch repair genes do not carry BRAF activating mutations in their tumors.53 BRAF V600E activating mutations have been reported in about 5% of all CRCs, 4–12% of CRC without microsatellite instability (MSS tumors), 40–70% of sporadic CRC with microsatellite instability (MSI-H), but are not seen in HNPCC/LS CRCs which nearly always show MSI-H.54 – 56 HNPCC/LS tumors, similar to sporadic CRCs with dMMR, show frequent aberrant nuclear beta-catenin, but aberrant p53 expression, 5q loss of heterozygosity, and KRAS mutations are uncommon.11
Criteria for Identification of HNPCC/ LS Patients and Molecular Testing Approaches
Historically, the first criteria to identify patients with HNPCC/LS, known as the Amsterdam criteria, were established in the early 1990s, with subsequent revisions.10 , 57 Later, more inclusive criteria, known as the Bethesda guidelines were established by a consensus group.5 , 35 , 58 – 60 According to Amsterdam II Criteria, patients are diagnosed with HNPCC when10:
-
1.
The family includes three or more relatives with an HNPCC-associated cancer, verified by pathological examination and:
-
(a)
One affected patient is a first-degree relative of the other two
-
(b)
two or more successive generations are affected
-
(c)
cancer in one or more affected relatives is diagnosed before the age of 50 years
-
(d)
familial adenomatous polyposis is excluded.
-
(a)
-
2.
Alternatively, patients meet one of the following modified Amsterdam criteria:
-
(a)
Very small families can be considered to have HNPCC with only two colorectal cancers in first-degree relatives if at least 2 generations have cancer and at least one case of CRC was diagnosed by the age of 55 years
-
(b)
In families with two first-degree relatives affected by colorectal cancer, the presence of a third relative with an unusual early onset neoplasm or endometrial cancer is sufficient
-
(c)
If an individual is diagnosed before the age of 40 years and does not have a family history that fulfills Amsterdam II or modified Amsterdam criteria, they are still considered as having HNPCC/LS.
-
(a)
The most recently revised Bethesda criteria recommend testing patients to rule out HNPCC/LS if there is one of the following criteria5 , 35 , 59 , 60:
-
1.
Patient is diagnosed with colorectal cancer before the age of 50 years.
-
2.
Patient has synchronous or metachronous colorectal cancer and another HNPCC-related tumor [stomach, urinary bladder, ureter and renal pelvis, biliary tract, brain (glioblastoma), sebaceous gland adenomas, keratoacanthomas, and small bowel], regardless of age.
-
3.
Colorectal cancers with histopathologic features suggestive of dMMR–microsatellite instability: abundant tumor infiltrating lymphocytes, Crohn-like lymphocytic reaction, mucinous or signet ring cell differentiation, or medullary growth pattern of poorly differentiated adenocarcinoma, diagnosed before the age of 60 years.
-
4.
Colorectal cancer patient with one or more first-degree relatives with CRC or other HNPCC-related tumors. One of the cancers must have been diagnosed before the age of 50 years.
-
5.
Colorectal cancer patient with two or more relatives with colorectal cancer or other HNPCC-related tumors, regardless of age.
The revised Bethesda guidelines resulted in an increased identification of patients with dMMR as compared to previous guidelines, identifying not only more HNPCC/LS cancers but also more sporadic-type cancers with dMMR.18 Given recent knowledge of the molecular changes underlying sporadic as compared to HNPCC-associated dMMR cancers, algorithms have been proposed to determine whether a patient has sporadic or HNPCC/LS cancers.61 Alternatively, as discussed later, universal testing for dMMR of all CRC has been proposed.3 , 62 – 64
Molecular Testing for HNPCC/LS
The diagnostic workup for dMMR either due to HNPCC/LS or due to sporadic CRC is initiated by testing cancer tissue for DNA for microsatellite instability (MSI) (Fig. 7.3), the functional end point of dMMR, and/or by performing immunohistochemistry (IHC) of tumor tissues for MSH2, MLH1, MSH6, and PMS2 DNA mismatch repair proteins, to evaluate for their preserved (in DNA MMR-proficient tumors) or loss of expression (in deficient DNA MMR) tumor cell nuclei. Usually, CRC tissue is used for testing but other tumors of the HNPCC/LS spectrum, such as endometrial carcinoma, can be used.
MSI DNA Test
The DNA-based test to assess for microsatellite instability (MSI) in tumor cells is based on the evaluation of instability in small 100–200 base pair DNA segments that consist of repetitive nucleotides, called microsatellite regions or short tandem repeats (STRs). The nucleotide sequences within the repetitive elements of marker loci used in the MSI test are mononucleotide repeats of adenine (A)n or cytosine–adenine (CA)n dinucleotide repeats. During DNA replication these repetitive sequences may undergo variations in length due to DNA strand slippage, leading to increased or reduced length of STRs. In normal cells, proficient DNA MMR proteins are able to correct these replication-associated DNA sequence errors; however, in dMMR cells with loss of function of both alleles, these mutations persist in the genome of daughter cells and in future cell generations in the tumor. The resulting changes in length of nucleotide repeats are known as microsatellite instability. Therefore, MSI can be detected in cells with dMMR, as occurs in patients with HNPCC/LS and sporadic dMMR cancers, as these mutations are not repaired and persist in the DNA of tumor cells. Tissue sections from formalin-fixed tumor tissue used for routine pathologic diagnosis and embedded in paraffin (FFPE) are adequate for the MSI test. Tumor enrichment by micro or macro-dissection is recommended. One of the most used sets of microsatellite markers was recommended by a NCI consensus group and consists of 5 loci: 2 poly(A) mononucleotide repeat markers (BAT25 and BAT26) and 3 poly(CA) dinucleotide repeat markers (D2S123, D5S346, and D17S250).35 , 58 The NCI panel of five microsatellite markers requires comparison of the tumor DNA profile with that of non-neoplastic/normal tissue, which can be obtained from non-neoplastic colonic mucosa and adjacent wall layers, away from the tumor. The results of the MSI test using the NCI panel are reported as MSI-high (MSI-H), MSI-low (MSI-L), or microsatellite stable (MSS). MSI-H tumors have MSI in at least two of the five markers, MSI-L tumors have MSI in only one marker, and MSS tumors do not have instability at any of the five markers. Figure 7.3 illustrates amplification of the microsatellite loci in a colorectal cancer with MSI-H. Alternative panels consisting of mononucleotide repeat markers that are highly monomorphic in the germline DNA of a wide spectrum of populations have been used, with the advantage that they perform well without the use of normal control DNA.65 , 66 One such panel includes five microsatellite loci (BAT-25, BAT-26, NR-21, NR-24, and NR-27) providing a pentaplex assay.66 – 68 The pentaplex assay data correlate well with MMR protein expression for MLH1, MSH2, and PMS2, but less robustly with MSH6 expression, similar to the NCI panel.
Testing for MSI-H with the DNA MSI test is highly sensitive for HNPCC/LS cancers; however, the test may yield about 5% false negative cases among overall CRC cases from patients with MMR gene mutations.4 Additionally, the MSI DNA test may detect MSI-H status in only 86% of the CRC cases from patients with germline mutations in MSH6.4
Immunohistochemistry of cancer tissues for DNA MMR proteins is a surrogate marker for the MSI status and when informative has the advantage of identifying the underlying deficient DNA mismatch repair gene based on the finding of which MMR protein is primarily lost in tumor cells. Overall, the sensitivity of immunohistochemistry to detect dMMR is about 95%.4 Limitations of immunohistochemistry result in part from occasional problems with tissue immunoreactivity and interpretation pitfalls.69 In MLH1-deficient tumors of sporadic type, where the MLH1 gene is silenced by promoter methylation, IHC shows complete loss of MLH1 expression in tumor cells, and this is accompanied by parallel loss of PMS2, since the latter proteins are unstable in the absence of MLH1.70 However, IHC may be difficult to interpret in cancers of HNPCC/LS patients with MLH1 missense mutations that result in nonfunctional protein and MSI-H phenotypes, where the mutant protein may be expressed and retain its immunoreactivity, at least partially, with variable levels of expression of PMS2 in parallel.70 – 72 In addition, MSI-H CRCs with preserved or variable expression of MLH1 protein, with associated loss or variably reduced PMS2 in tumor tissue, may represent a germline mutation in PMS2. IHC for MSH2 in HNPCC/LS patients with germline mutations in MSH2 usually cause a complete loss of gene expression in tumor tissues, accompanied by loss of MSH6 protein expression, but IHC heterogeneity has been reported in a rare case.70 Since both the MSI DNA test and IHC for DNA mismatch repair proteins will miss a small number of tumors in patients with underlying HNPCC/LS, it has been proposed to perform both tests upfront in the evaluation of CRC for potential HNPCC/LS.73
Genetic Testing for Constitutional Germline Mutations in DNA MMR Genes
If the tumor tissue reveals MSI-H and/or there is loss of expression of one of the DNA repair proteins by immunohistochemistry, constitutional germline testing should be performed for the gene encoding the deficient protein, after appropriate genetic counseling of the patient. If tissue testing is not feasible, or if there is sufficient clinical evidence of HNPCC/LS, germline analysis of the MSH2 and/or MLH1 genes can be done as first line testing.35 , 61 , 74 Mutation analysis can be performed by using a number of approaches, including single-strand conformational polymorphism analysis, denaturing gradient gel electrophoresis analysis, DNA sequencing, monoallelic expression analysis, Southern analysis, and quantitative polymerase chain reaction.35 Overall, the likelihood of finding a germline mutation in the MLH1 or MSH2 genes of patients with colorectal cancers that are not MSI-H is low.35 The germline mutations that occur in MSH2 and MLH1 are widely distributed throughout the two genes. More than 200 pathogenic mutations have been reported in MLH1 and in MSH2. 75
Two additional tests, MLH1 methylation and BRAF V600E activating mutation assays, may help discriminate between a sporadic MSI-tumor and HNPCC/LS tumor with loss of expression of MLH1 by immunohistochemistry but an undetected MLH1 mutation.61 Using quantitative methylation analysis, HNPCC/LS patients showed no or low level of MLH1 promoter methylation, in contrast to high levels of methylation (greater than a cutoff value of 18% methylation) in sporadic MSI-cancers.24 In addition, none of the patients with an unambiguous germline mutation in DNA mismatch repair genes demonstrated BRAF activating mutation.53 If no loss of expression of MSH2, MLH1, MSH6, or PMS2 is seen in MSI-H tumors or if the tumor is MSI-L or MSS but there is clinical suspicion of HNPCC/LS testing for germline constitutional mutations in the DNA MMR genes should be performed.35 , 76 Identification of a germline mutation in index cancer patients is important because it confirms the diagnosis of HNPCC/LS and the identified mutation may then be used to screen at risk relatives who may be mutation carriers and benefit from increased colonoscopic surveillance.
After a germline mutation is identified or the patient is diagnosed with HNPCC/LS, relatives should be referred for genetic counseling and testing should be offered. If no mismatch repair gene mutation is found in a proband with an MSI-H tumor and/or a clinical history of HNPCC/LS, the genetic test result is noninformative. The patients and the relatives at-risk should be counseled as if HNPCC/LS was confirmed and high-risk surveillance should be performed.5 , 35 , 61
Testing dMMR in Adenomas
In HNPCC/LS a significant association was found between MSI-H and high-grade dysplasia in adenomas, with loss of either MLH1 or MSH2 expression.77 Based on these findings it was recommended that immunohistochemical staining/MSI testing of large adenomas with high-grade dysplasia in young patients (younger than 50 years) may be performed to help identify patients with suspected HNPCC/LS.19 , 77
Universal Testing of CRC for dMMR
During the past two decades, patients who presented with one of the tumors of the HNPCC/LS syndrome, in particular colon cancer, were offered testing for HNPCC/LS following guidelines based on family history (as described by the Amsterdam criteria) or based on a combination of family history, age of presentation, and histopathologic features of the tumor (as described by the Bethesda guidelines). However, more recently, studies have suggested a universal screening approach entailing the evaluation of all diagnosed colorectal cancers for markers of HNPCC/LS regardless of the family history.3 , 62 , 64 One of these studies evaluated more than 1,000 patients with colorectal cancer whose tumors were tested for MSI.78 Patients with MSI-positive tumors were tested for expression of MMR genes and germline mutations in MSH2, MLH1, MSH6, and PMS2 by genomic sequencing and deletion studies. A mutation causing HNPCC/LS was detected in 23 patients (2.2%), of whom as many as 22% would have been missed if Amsterdam or Bethesda criteria had been used alone. In families that meet strict clinical criteria for HNPCC/LS, germline mutations in MSH2 or MLH1 have been found in 45–70% of the families, and overall, germline mutations in these two genes account for 95% of HNPCC/LS cases with an identified mutation.31 , 74 These data show that despite extensive testing there is still a significant number of families without an identified germline mutation accounting for HNPCC/LS. It has been proposed that if an individual has a family history that is suggestive of HNPCC/LS, but does not fulfill the Amsterdam criteria, they are considered to represent an HNPCC variant, or familial colorectal cancer type X.79 , 80 Further, it was observed that nearly 40–60% of all cases who meet Amsterdam criteria for HNPCC/LS do not have characteristic MMR deficiency or germline mutation in a DNA MMR gene. The age at diagnosis of these MSS familial CRC patients is 6 years older on average and most tumors occur on the left side of the colon.2 The underlying genetic defect for these tumors is not yet known. Conversely there are also individuals who have tumors with MSI and germline mutation in a DNA MMR gene, but whose family history does not meet the Amsterdam criteria.3
In 2010, a consensus recommendation was published, stating that all CRCs should be screened using IHC for the four DNA MMR gene products, or alternatively by MSI, in order to evaluate for HNPCC/LS.3 Another point raised by this consensus group was that MSI and IHC testing should not be considered genetic tests and should be ordered by appropriate medical personnel as needed for medical care.3 Further, other investigators in the USA have advocated that all newly diagnosed colorectal and endometrial cancer patients should be screened for HNPCC/LS.62 , 81 – 83 Review of this topic by the Evaluation of Genomic Applications in Prevention and Practice (EGAPP) working group56 led to the recommendation that all newly diagnosed colorectal cancer patients should be screened for HNPCC/LS to reduce the morbidity and mortality from colorectal cancer in their at-risk unaffected relatives. A cost analysis of several possible screening methods indicated that the most cost-effective approach for screening all newly diagnosed colorectal cancer patients for HNPCC/LS would be to test tumor tissue with IHC followed by genetic testing in patients in whom any MMR protein was absent, after ruling out epigenetic (MLH1 CpG methylation) causes of protein absence to exclude sporadic dMMR cases.84 However, as reviewed earlier in this chapter, it should be cautioned that IHC alone will miss a small proportion of MSI-H cancers.73 A report from the Association for Molecular Pathology proposed a testing strategy combining IHC for the four DNA MMR proteins and MSI DNA test upfront, followed by BRAF and KRAS mutation testing, serving both the purposes of screening for HNPCC/LS as well as for evaluation for targeted anti-EGFR therapies.73
Extra-colonic Neoplasia in Lynch Syndrome
Early-onset colorectal cancer (CRC) is the hallmark of HNPCC/LS, but it has been estimated that extracolonic manifestations may be as common as CRC, particularly in women. MMR germline mutations collectively confer an 80% risk of CRC before age 70 in men and 30–50% in women.85 , 86 In comparison, the best characterized of the extracolonic manifestations is early-onset endometrial cancer, which occurs in an estimated 40–60% of at-risk patients.4 , 85 , 87
Weaker but definite associations10 have been made between LS and cancers of the ovary, stomach, hepatobiliary tract, and small bowel, transitional cell carcinoma of the ureter and renal pelvis, glioblastoma (Turcot variant of Lynch syndrome), and sebaceous adenoma/carcinoma (Muir-Torre variant of Lynch syndrome).88
Breast cancer has often been detected in patients with LS.89 – 95 The lifetime (to age 70) risk of breast cancer in LS patient series has been 2.2–5.4%.87 , 96 This risk is not convincingly higher than the 7.6% rate seen in the general population,97 in contrast to the dramatic enrichment seen for most LS-associated tumors.96 It remains unsettled whether breast cancer is part of the LS spectrum.
While there are no conclusive studies linking LS with other cancers, case reports have suggested potential associations of LS with cancer of the pancreas, adrenal cortical carcinoma,98 sarcomas (malignant fibrous histiocytoma,99 rhabdomyosarcoma,100 liposarcoma101 , 102), prostate cancer,103 and neurofibromatosis-like features including café-au-lait macules and plexiform neurofibromas.104
The prevalence of extracolonic tumors in HNPCC/LS is variable and reflects the incomplete penetrance of the underlying mutation. CRC is relatively more common in MLH1 germline mutants, while other cancers are more common in MSH2 mutation.88 MSH6 mutation also appears to favor endometrial cancer.88 PMS2 mutants tend to present with atypical tumors in childhood.88
Endometrial Cancer
Endometrial cancer (EC) is the most common extracolonic manifestation of HNPCC/LS and has been extensively studied and reviewed.105 – 110 The cumulative incidence of EC was 60% to age 70 in a study of 183 Finnish women with confirmed MMR mutations,87 compared with 54% for colorectal cancer in the same population. Other series of women with MMR mutations showed a 42% lifetime risk of EC and 30–54% lifetime risk of CRC.85 , 111
EC becomes all the more salient as a feature of Lynch syndrome when one notes that CRC is less common in women than in men with HNPCC/LS, for whom the lifetime cumulative incidence of CRC is 74–83%.85 , 111
Given that its cumulative incidence approaches or exceeds that of CRC, it is clear that EC may be the sentinel cancer in some Lynch syndrome patients and kindreds. In a series of 111 women meeting Amsterdam criteria and having both CRC and EC, the CRC presented first in 49 (median age of 40), EC presented first in 46 (median age of 45), and the tumors presented simultaneously in 12.112 When EC presented first in this series, the mean lead time before subsequent development of CRC was 11 years, allowing for follow-up and prevention if the Lynch phenotype is successfully identified.112 The converse situation, of course, also provides an opportunity for cancer prevention, since Lynch-associated CRC patients have a markedly elevated risk of subsequently developing endometrial cancer (10-year cumulative risk of 23.4% in LS patients, versus 1.6% in patients with sporadic CRC).113 The reported average lead time from CRC to the second malignancy (usually EC or ovarian carcinoma) was 8 years.112
LS-associated EC occurs at a younger age than sporadic cancer, and Lynch syndrome is more common in women presenting with EC at a young age. Functionally significant MMR gene mutations are present in 1.4–2.6% of EC overall,114 , 115 but 9% of EC occurring before age 50.116 Young age at presentation of EC has therefore been proposed as a trigger for further testing, usually either IHC testing for loss of MMR proteins or molecular testing for the MSI phenotype. Importantly, LS patients with EC are less likely than average women to fit the typical clinical profile of EC (obese, nulliparous), given that their tumors arise due to a genetic lesion. Thus, low BMI, family history suggestive of LS, and LS-associated histology (see below) have all been suggested as additional features to prompt further testing.117 Using age below 50 as the main trigger for further testing would, however, result in low sensitivity for detecting Lynch syndrome. In one study of 562 unselected EC patients, the 13 women with eventual discovery of LS had a mean age of 54.1 years at diagnosis.82 In another group of 7 women with Lynch syndrome related to inactivating MSH6 mutations, the mean age at diagnosis was 54.8 years, compared to 64.6 years in the overall population.118
There are conflicting data on the prognosis of LS-associated EC, with some studies showing no difference in 5-year survival between LS patients and matched controls.119 – 121 Other studies have variously shown either worse122 or better survival123 in MSI-H tumors. Taking these data together, it appears to be difficult to demonstrate a significant prognostic difference in LS-associated EC. Its prognostic importance lies, rather, in the likelihood of observing other associated malignancies in the patient and family members.
Pathologic Features
HNPCC/LS-associated endometrial carcinoma has a characteristic morphology.124 – 127 Typical features include numerous peritumoral and tumor-infiltrating lymphocytes (TILs). These features related to the inflammatory response are reminiscent of the Crohn-like reaction and dense TILs that are common in MSI-H CRC cases. Both features show a statistically significant association with MSI-H status, but neither of them is sensitive or specific enough to serve as a diagnostic marker.124 Although there appears to be no specific TIL density that sensitively and specifically identifies MSI-H cases,125 40 TILs/10 HPF (using a 400× high-power field) was suggested in one study as an analytically useful cutoff by which to define this feature.
The majority of LS-associated EC is of endometrioid histology, but these endometrioid cases show some propensity to demonstrate a biphasic morphology in which a typical, rather well-differentiated endometrioid adenocarcinoma (FIGO grade 1 or 2) is admixed with an undifferentiated component.124 , 125 The resulting “dedifferentiated” carcinoma must, by definition, contain at least 10% of each component. 21% of MSI-H cases showed this phenotype versus 6% of non-MSI-H cases (p = 0.06).124
LS-associated EC also shows a propensity toward nonendometrioid histology. One series of 23 patients with germline MMR mutations consisted of 57% endometrioid tumors and 43% nonendometrioid ones, while sporadic controls were 96% endometrioid.128 Two other reports have shown endometrioid histology in 86–87% of LS-associated cases, as compared to >96% in both sporadic controls129 and MLH1 hypermethylated cases.118 , 129 The nonendometrioid tumors were predominantly serous, clear cell, and carcinosarcoma in these reports.
It has been reported that the prevalence of LS is higher in patients with carcinoma of the lower uterine segment (29%) as compared to the general population with endometrial cancer.130
Molecular Features
EC can be associated with mutation in any of the MMR genes (MSH2, MLH1, MSH6, PMS2). However, the lifetime risk of EC appears to be highest in MSH6 mutation, followed by MSH2 and MLH1. 131 This is the inverse of the behavior seen for CRC in women, for which the lifetime risk is highest for MLH1 mutants and lowest for MSH6. Indeed, while MSH6 mutation has been considered something of a hypomorph from the viewpoint of CRC, it is potentially the most significant of the MMR proteins from the viewpoint of EC. Data on EC risk in PMS2 mutants are relatively scarce, but in one series of 61 confirmed mutants, the cumulative risk of EC was 15% to age 70 years, showing the penetrance of PMS2 mutation to probably be lower than the other three MMR genes.132
Ovarian Cancer
An estimated 5–10% of epithelial ovarian cancers (EOC) are hereditary, consisting mainly of those associated with BRCA1 and BRCA2 mutations.110 , 133 LS patients make up most of the remaining 10–15% of hereditary epithelial ovarian cancer cases.110 , 134 Although these figures suggest that approximately 1% of ovarian cancer is associated with LS, the MSI-H phenotype is present in approximately 12% of all EOC cases,135 indicating that this defect probably arises de novo and plays a pathogenetic role in many sporadic tumors.
LS-associated ovarian cancers present at an earlier age than sporadic tumors (mean 49.5 versus 60.9 years).134 , 136 These cases are also identified at earlier stages than sporadic tumors. In one study, the stage at diagnosis was FIGO I for LS-associated tumors, but FIGO III for sporadic ones.136 Both age and stage differences, when they occur, probably reflect more active screening in the Lynch patients. However, a stage difference was not seen in a series of 37 hereditary ovarian cancers that included predominantly BRCA mutants, although these would presumably also have been heavily screened (in an era, however, predating the appreciation of the role of the fallopian tube in EOC carcinogenesis).134
Pathologic Features
EOCs in general exhibit several histologic patterns. In sporadic cases, the most common histology is serous (accounting for as many as 78% of all EOCs), followed by endometrioid, mucinous, clear cell, and malignant Brenner tumor in decreasing order of frequency.137 , 138 Carcinomas of mixed or indeterminate (i.e., poorly differentiated) histology also occur. LS-associated and MMR-deficient EOCs appear to be enriched in nonserous histologies, which in one meta-analysis constituted 57% of LS-associated ovarian cancers (128 patients in 6 studies).139 In this meta-analysis, 25% of LS-associated EOCs were endometrioid, 17% were clear cell, and 16% were mucinous. Similar proportions were found in MSI-H tumors occurring outside the context of Lynch syndrome, suggesting that the relative scarcity of serous tumors somehow reflects the biology of the MMR-deficient state.86
The prognosis of EOC in Lynch patients appears to be similar to that of non-Lynch controls. In one report, there was a small and statistically insignificant decrement in overall survival for Lynch patients,136 conceivably attributable to these patients’ risk of other malignancies. This situation contrasts with that for CRC, where survival is better in Lynch-associated tumors versus sporadic ones.4
Gastric Cancer
Gastric cancer was present, along with CRC and EC, in the original “family G of Warthin” in whom LS was initially described. A study of 60 families from a Brazilian registry found a history of gastric cancer in 12/1,040 = 1.1% of men and 12/1,055 = 1.1% of women meeting Amsterdam I or II criteria.89 The prevalence of gastric cancer in a Dutch LS registry population was similar (21/948 = 2.2% of men, 11/1,066 = 1.0% of women); Kaplan-Meier analysis in this population gave an estimated incidence of 6.2% in men and 2.0% in women to age 70.140 In the Dutch series, gastric cancer occurred only in MSH2 and MLH1 mutant kindreds. MSH6 mutation was well represented in the sample, but was not associated with any gastric cancers. PMS2 mutants had no gastric cancers, but represented only two of the 236 families.
The histology of Lynch-associated gastric cancers is predominantly intestinal type in the Lauren classification (at least two-thirds of patients), the remainder being of diffuse type.140 , 141 This finding is somewhat unexpected, given that the diffuse type is statistically more likely to have a primary genetic etiology (e.g., in hereditary diffuse gastric carcinoma).142 , 143 Intestinal-type tumors are numerically more common than diffuse type in the genetically normal background population (51% intestinal type, 37% diffuse type).144 H. pylori was noted in only 20% of LS-associated gastric cancers,141 a fraction that is lower relative to the general gastric cancer population and reflects the presence of an inherited cancer predisposition. The histologic pattern of LS-associated gastric cancer is not distinctive enough to be used to triage patients for further testing in the absence of contributory personal or family history.
Pancreatobiliary Carcinoma
The cumulative lifetime risk of pancreatobiliary carcinoma in confirmed and presumed MMR gene mutation carriers has been estimated at 2–4% to age 70,87 , 96 compared with 0.2% in the general population. These tumors appear to be almost entirely cholangiocarcinomas arising at various sites along the biliary tract, including common bile duct (7/18 in a series of 315 Finnish subjects), ampulla of Vater (4/18), intrahepatic biliary tree (4/18), and pancreas (3/18).145 A mucinous cholangiocarcinoma has been reported in a patient with Muir-Torre syndrome related to MSH2 mutation.146 LS does not seem to have an association with gallbladder cancer.
Small Intestinal Carcinoma
Carcinomas of the small bowel are approximately 100 times more common in LS than in the general population, with a cumulative lifetime risk of 4%.147 This is roughly similar to the relative risk in Crohn disease or familial adenomatous polyposis.148 The cancer risk is higher in MSH2 and MLH1 mutants, and lower in MSH6 and PMS2. 149 Small bowel tumors in LS are adenocarcinomas, have up to 3:1 male:female bias,147 have no particular site of predilection within the small bowel, and may occur synchronously at several sites.148 The median age at diagnosis is 40–50, at least a decade younger than the general population with small bowel cancer.147 , 148 As in the colon, the prognosis for patients with small bowel carcinoma in the setting of Lynch syndrome may be slightly better than for the general population.148
Transitional Cell Carcinoma
Lynch syndrome carries a 22-fold increased risk of transitional cell carcinoma (TCC) of the upper urinary tract (renal pelvis and ureter),150 occurring in approximately 4% of the Lynch syndrome population.87 , 111 , 151 There is no increase in TCC of the bladder in LS.150 , 152 The tumors present around age 56, a decade earlier than in the background population.150 They have an MSI phenotype,153 confirming that they are part of the Lynch syndrome spectrum. Special histologic features of these tumors have not been described.
CNS Tumors in Turcot Syndrome
The eponym “Turcot syndrome” has been used to describe the association of colorectal and CNS tumors. It is now apparent that this association occurs in two distinct sets of patients with fundamentally different genetic lesions.154 The larger group has APC mutations, with the characteristic florid pancolonic polyposis, and with medulloblastoma as the CNS manifestation. A smaller group of patients has MMR mutations, typical Lynch-type CRC, and glioblastoma as the CNS manifestation.155 Intriguingly, much as CRC associated with Lynch syndrome has a favorable prognosis, the survival of Turcot patients with glioblastoma appears to be better than that of patients with sporadic glioblastoma, although the number of patients reported is small.155
Sebaceous Adenoma/Carcinoma in Muir-Torre Syndrome
The Torre syndrome,156 simultaneously reported by Muir,157 was initially believed to represent the co-occurrence of cutaneous lesions with visceral malignancies, without mention of any familial association. It was subsequently noted that several of these patients belonged to families with what was then known as the cancer family syndrome, now HNPCC/LS.158 The cutaneous lesions are frequently sebaceous adenomas, sebaceous carcinomas, or keratoacanthomas, i.e., low-grade squamous cell carcinomas arising from the pilosebaceous unit.
Further study has confirmed Muir-Torre syndrome (MTS) to be a variant of Lynch syndrome159 defined by the presence of sebaceous skin neoplasms. The skin lesions of MTS have MMR mutations, usually in MSH2, 160 and a MSI-H phenotype.161 , 162 The presence of sebaceous neoplasms is variably penetrant, having been reported in 28% of families and 9.2% of individuals meeting criteria for LS. There were 42% of kindreds with MSH2 mutations, and 44% of kindreds with MLH1 mutations, that had at least one individual with MTS, while MTS was not found in kindreds with MSH6 or PMS2 mutation.
HNPCC/LS: Clinical Management
Colorectal Cancer
Carriers of DNA mismatch repair gene mutations seen in HNPCC/LS are recommended to have colonoscopic surveillance staring at early age.61 Colonoscopy is recommended every 1–2 years starting at age 20–25 years (age 30 years for those with MSH6 mutations). For individuals who will undergo surgical resection of colon cancer, subtotal colectomy may be favored.
Endometrial and Ovarian Cancer
Patients Without Known Lynch Syndrome
Although identification of a CRC meeting Amsterdam or Bethesda criteria has been suggested as a cue to screen for EC, it appears that EC is more common than CRC in these patients, indicating that EC with appropriate features may equally well be taken as a sentinel event to identify Lynch syndrome patients and families.127 Examining the converse situation, in a prospective study of 100 women with endometrial cancer diagnosed before age 50, nine carried germline MMR mutations (seven MSH2 mutants, one MLH1 mutant, and one MSH6 mutant).116
MMR testing by immunohistochemistry, MSI testing, or gene sequencing is commonly recommended when faced with a new diagnosis of colon cancer in an appropriate clinicopathologic setting. Similar reasoning dictates that EC patients of sufficiently low age be tested for MMR mutations (with age <50 years often suggested as a cutoff). As has been mentioned, the prevalence of such mutations is in the neighborhood of 9%.116 As age rises, the relative number of Lynch-associated mutations would be expected to fall, as sporadic tumors begin to predominate, but in absolute terms, Lynch cases continue to accumulate. Furthermore, with rising age, sporadic hypermethylation begins to predominate as a cause of MLH1 silencing, causing an apparent dMMR/MSI phenotype in a patient with no germline mutation and no increased tumor risk at other sites. Thus the relative diagnostic utility of immunohistochemistry as compared to other methods can be expected to change with age.
A consensus statement of the Society of Gynecologic Oncologists instructs practitioners to identify women who have a 20–25% chance of having an inherited predisposition to endometrial, colorectal, and related cancers, for whom genetic risk assessment is “recommended,” and those with a 5–10% chance of the same, for whom genetic risk assessment “may be helpful”.163 The SGO statement does not specify the form that the risk assessment should take, but underlines the need for it to be integrated with genetic counseling and education. Since LS-associated malignancies are uncommon before age 21, it is suggested that the benefits of testing do not outweigh its potential adverse effects in this age group.
Strategies for risk assessment in the patient with EC and suspected Lynch syndrome resemble those available in CRC, with immunohistochemistry, MSI testing, and gene sequencing representing the state of the art in approximately increasing order of cost.117 Application of the Amsterdam criteria to triage patients for testing increases specificity at the cost of decreased sensitivity. As in CRC, immunohistochemical staining of the endometrial tumor for MSH2, MLH1, MSH6, and PMS2 is a simple gatekeeper strategy that can serve as a useful initial screen. When sequencing is used, nonsense mutations are relatively simple to interpret, but missense mutations are heterogeneous and may or may not be of clinical significance.82
Several heuristics116 , 163 or specific algorithms4 , 105 , 117 , 124 have been proposed to combine clinicopathologic features with stepwise diagnostic testing in order to achieve sensitivity and specificity while minimizing cost.117 While the relative merits of each specific testing strategy will depend on the cost–benefit tradeoffs one chooses to make, the lowest cost-effectiveness ratio appears to be achieved with four-protein immunohistochemistry, followed by confirmatory sequencing of any putatively affected gene.117 When cost represents a major consideration, an initial panel consisting only of MSH6 and PMS2 IHC can provide clues regarding dMMR status at a reduced cost.164
Patients with Suspected Lynch Syndrome
Once a presumptive diagnosis of Lynch syndrome has been established, further management can involve a combination of surveillance and, potentially, prophylactic surgery. For women, in whom EC and EOC are the main malignancies to exclude, proposed surveillance models usually involve imaging, usually by ultrasound, and periodic endometrial sampling.105 , 165 – 169
Other HNPCC/LS Associated Carcinomas
Patients Without Known Lynch Syndrome
Endometrial and ovarian cancers are markedly more common than the other extracolonic Lynch-associated tumors and have been the focus of most of the research on screening. However, when other tumors associated with the syndrome present at a young age or with suspicious family history, tumor testing for features of Lynch syndrome is broadly recommended. As for endometrial cancer, IHC testing for loss of MMR proteins or molecular testing for MSI are both appropriate initial modalities, without clear consensus to favor one over the other.152 DNA sequencing should be reserved for later steps of the investigation due to its expense and complexity.170 For clinicians, it is important to emphasize the details of family history and to inform the pathologist of these details so that testing can be initiated or justified.
Patients with Suspected Lynch Syndrome
The heterogeneity in extracolonic tumors between Lynch kindreds and between patients within a given kindred,111 combined with the relatively low frequency of any given extracolonic tumor, makes it difficult to recommend any specific screening protocol in patients with suspected or confirmed Lynch syndrome. For some tumors, such as glioblastoma, no specific screening method seems practical. In the case of the small bowel, screening by capsule endoscopy or double-balloon enteroscopy has been suggested; rough calculations with rather favorable assumptions suggest a reasonable cost per QALY if screening is performed once above age 40.147 Urine cytology is a straightforward screening method for transitional cell carcinoma. None of these methods have been explicitly adopted or endorsed, and screening of the Lynch patient therefore seems to currently require an individualized approach.
Polyposis Syndromes of the Gastrointestinal Tract
Polyposis syndromes are characterized by the development of variable numbers of polypoid lesions throughout the colon, small intestine, and stomach and include developmental polypoid abnormalities or hamartomatous polyps, inflammatory-type polyps, and adenomas. The risk of adenocarcinoma is highest in the syndromes that develop adenomas, but increased risk of malignancy also characterizes the other polyposis syndromes. A summary of the gastrointestinal polyposis syndromes is depicted in Table 7.1.
Familial Adenomatous Polyposis
Clinical Features, Pathology, and Natural History
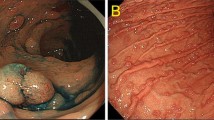
Familial adenomatous polyposis (FAP) is the most frequent gastrointestinal polyposis syndrome. Patients develop numerous adenomas preferentially involving the colorectum (Fig. 7.6), but also affecting the small intestine, in particular the duodenum and periampullary region.171 – 173 The stomach typically develops numerous fundic gland polyps, although adenomas may also occur.173 – 178 If the disease progresses through its natural history, 100% of affected individuals eventually develop colorectal cancer at early age179 (Fig. 7.6). Overall, FAP patients represent less than 1% of all colorectal carcinoma cases in the United States, affecting 1 in 8,000–10,000 individuals.180 FAP patients are also at higher risk of developing duodenal and ampullary carcinomas and have a slightly increased risk of gastric adenocarcinoma.181

Colonic adenomas and progression to adenocarcinoma in a patient with FAP. (a) Numerous polyps are identified at colonoscopy (image provided by courtesy of Dr. David Metz, University of Pennsylvania); (b and c) Histologic features of representative tubular adenomas. (d) Adenocarcinoma (arrow) arising in the background of numerous colonic polyps. (e and f) Histologically, the tumor is a moderately differentiated adenocarcinoma extending into the subserosal tissue
The majority of FAP patients inherit germline mutations in the adenomatous polyposis coli (APC) gene on 5q21–22 chromosome locus,182 – 184 but up to a third of FAP cases present as de novo germline mutations.180 , 185
In addition to the characteristic gastrointestinal polyposis, FAP patients manifest other lesions, including congenital hypertrophy of retinal pigmented epithelium (CHRPE) in 70–80% of FAP patients, desmoid tumors in 15% of cases, thyroid carcinoma (papillary and follicular types, including the characteristic cribriform-morular variant) in 1–2% of FAP patients,174 and hepatoblastoma in young children.174
In addition to the classic FAP syndrome, inherited mutations in the APC gene are associated with several variants of FAP that include Gardner Syndrome, Turcot Syndrome, and Attenuated FAP (AFAP), summarized in Table 7.1.173 , 180 , 181 , 186 , 187
Gardner syndrome: These patients develop a number of extra-colonic manifestations including osteomas, desmoid tumors, dental abnormalities, ophthalmologic abnormalities including congenital hypertrophy of retinal pigment epithelium (CHRPE) and cutaneous cysts.
Turcot syndrome: These patients develop colorectal polyposis and brain tumors, most commonly medulloblastoma (see also above).
Attenuated FAP: These patients develop a smaller number of colonic polyps as compared to classic FAP, but with sufficient colonic polyposis (frequently over 15), suggesting an underlying polyposis syndrome.181
Genetic and Molecular Features
FAP provides the basis to understand the molecular pathways underlying the most common mechanisms of stepwise progression in colorectal cancer.188 APC codes for a 312 kDa protein that is expressed in many tissues and is thought to participate in several cellular functions including Wnt-mediated signaling and transcriptional regulation, cell adhesion, cell migration, and chromosomal segregation. The APC gene product is a key mediator in the Wnt signaling pathway for cellular growth and proliferation. In the absence of a Wnt-mediated growth signal, APC occurs in a complex with beta-catenin, leading to beta-catenin phosphorylation and targeting it for destruction by the proteasome. When the Wnt pathway is activated by ligand binding, the APC–beta-catenin protein complex is disrupted, beta-catenin phosphorylation cannot occur, and this results in stability and nuclear localization of beta-catenin. Pathogenic mutations in the APC gene result in a disrupted APC–beta-catenin protein complex, leading to constitutive activation of the Wnt pathway.188 – 190
The APC protein contains multiple functional domains, including an oligomerization domain, an “armadillo” domain region which is thought to be involved in binding of APC to proteins related to cell morphology and motility, a beta-catenin-binding domain, an axin-binding domain, and a microtubule-binding domain.174 , 191
The majority of germline mutations associated with FAP are either frameshift or nonsense mutations that result in a truncated protein product, leading to disruption of interaction with beta-catenin, stabilization of beta-catenin with nuclear accumulation of the protein, and a phenotype of constitutive activation of the Wnt pathway.191 , 192 The most common germline mutations occur at codons 1061 and 1309 and account for 17% and 11% of all germline APC mutations, respectively. The region between codons 1286 and 1513 is known as the “mutation cluster region” (MCR), and many of the identified APC mutations occur in this segment of the gene.193 Some associations between the location of the germline APC mutation and the clinical phenotype have been found.194 Mutations within the MCR are associated with extensive polyposis of typical FAP patients, whereas mutations in the 5′ end of the gene (exons 4 and 5), and mutations in the alternatively spliced form of exon 9 or the 3′ distal end of the gene, are seen in attenuated polyposis. An intermediate phenotype is observed in patients with mutations between codon 157 and 1249 and between 1465 and 1595. The association of FAP with desmoid tumor formation has been correlated to mutations downstream of codon 1400.181 , 194 , 195 Mutations beyond codons 934, 1395, and within codons 564–1465 may be associated with upper gastrointestinal tumors. CHRPE is associated with mutations that occur between codons 311 and 1444.
Molecular Diagnosis
Molecular testing for germline mutations in the APC gene is recommended in specific settings as summarized by the American Gastroenterological Association.171 The primary indications include high clinical suspicion for FAP (>100 colorectal adenomas), first-degree relatives of FAP patients, >20 cumulative colorectal adenomas (suspected AFAP), and first-degree relatives of patients with AFAP.171 , 196
Different testing approaches are used in various laboratories, dictated by factors such as the volume of testing, platforms, and experience available in the laboratory. Sequencing of the entire coding region is the gold standard for diagnosis. Other methods include protein truncation tests and mutation scanning.
As numerous pathogenic mutations have been described in APC, including large exonic or whole gene deletions, a comprehensive approach to molecular diagnosis is required.197 If the patient has a phenotype suggestive of FAP but no mutations are found in the APC gene, underlying mutations in the MUTYH gene should be considered (see also below).198 When a mutation is identified, targeted genetic testing can be offered to family members.
When germline mutations are not detected in patients clinically suspicious for FAP and related syndromes, other mechanisms of gene deficiency may be interrogated. These alternatives include alterations in epigenetic regulation of the APC gene, contribution of genes encoding other proteins involved in the beta-catenin pathway such as axin, alterations of allelic mRNA ratios, and somatic APC mosaicism.199 Germline hypermethylation of the APC gene has been shown not to be a significant cause of FAP in APC mutation-negative cases.200 In cases in which no other discrete mutation exists, unbalanced APC allelic mRNA expression, resulting in reduced “dosage” of APC and functional haploinsufficiency has been reported.201
Individuals who are carriers of an APC gene germline mutation are recommended to have regular surveillance starting early in life with annual sigmoidoscopy or colonoscopy, beginning at 10–12 years of age. Prophylactic colectomy is recommended, usually by the time the patient is in his or her early 20s. Regular esophagogastroduodenoscopy (EGD) is also recommended for identification of gastric, duodenal, and periampullary lesions. Additional recommendations are directed at extra-colonic manifestations and include annual palpation of the thyroid, serum alpha-fetoprotein (AFP) levels, and abdominal palpation every 6 months in young children of FAP families to detect hepatoblastoma.
MUTYH-Associated Polyposis (MAP)
MUTYH-associated polyposis is transmitted as an autosomal recessive colorectal cancer syndrome, occurring in patients with colorectal polyposis and underlying germline mutations in the DNA repair gene MYH 202 – 204 (Table 7.1). Patients with MAP usually present with fewer polyps, but they may show no polyps or even numerous polyps as seen in FAP.204 Colorectal cancer arising in the background of MAP represents an estimated 0.5–1% of all colorectal cancers, with a lifetime risk of CRC reaching 80%, and as in FAP the risk of duodenal cancer is also increased.204
The MUTYH gene encodes a protein with glycosylase functions, involved in the base excision repair (BER) system critical to the repair of DNA damage caused by oxidative stress. The oxidized base 7,8-dihydroxy-8-oxoguanine (8-oxo-G) is often mistakenly paired with adenine (A), resulting in the appearance of guanine:cytosine > thymine:adenine (G:C > T:A) transversions at the next round of DNA replication, as the detection of stable 8-oxo-G:A base-pairs is missed by the replicative DNA polymerases.205 , 206 The DNA damage-specific glycosylases OGG1, MUTYH, and MTH1 function by recognizing and facilitating the removal of 8-oxo-G adducts.206 Consequently, deficient MUTYH function is associated with an increased frequency of G:C > T:A transversions,207 which occur in regions of cancer-related genes such as APC, KRAS, and BRCA1/2. Mutations in two hotspots, Y165C and G382D, account for 70% of all MUTYH mutations in Caucasian patients. Germline mutational testing is indicated in individuals with greater than 10 adenomatous polyps (particularly with family history of colon cancer consistent with recessive inheritance) and significant polyposis similar to AFAP/FAP who test negative for mutations in APC. 204 Screening is recommended to begin at age 18–20 years with colonoscopies every 1–2 years. Colectomy can be considered in cases with larger numbers of polyps mimicking FAP. Guidelines for the screening of duodenal cancers in FAP/AFAP should be applied to MAP patients as well.204
References
Boland CR. Evolution of the nomenclature for the hereditary colorectal cancer syndromes. Fam Cancer. 2005;4:211–8.
Llor X, Pons E, Xicola RM, et al. Differential features of colorectal cancers fulfilling Amsterdam criteria without involvement of the mutator pathway. Clin Cancer Res. 2005;11:7304–10.
Boland CR, Shike M. Report from the Jerusalem workshop on Lynch syndrome-hereditary nonpolyposis colorectal cancer. Gastroenterology. 2010;138(2197):e1–7.
Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18.
Umar A. Lynch syndrome (HNPCC) and microsatellite instability. Dis Markers. 2004;20:179–80.
Peltomaki P. Deficient DNA, mismatch repair: a common etiologic factor for colon cancer. Hum Mol Genet. 2001;10:735–40.
Lynch HT, Lanspa SJ, Boman BM, et al. Hereditary nonpolyposis colorectal cancer–Lynch syndromes I and II. Gastroenterol Clin North Am. 1988;17:679–712.
Terdiman JP. HNPCC: an uncommon but important diagnosis. Gastroenterology. 2001;121:1005–8.
Lin KM, Shashidharan M, Thorson AG, et al. Cumulative incidence of colorectal and extracolonic cancers in MLH1 and MSH2 mutation carriers of hereditary nonpolyposis colorectal cancer. J Gastrointest Surg. 1998;2:67–71.
Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–6.
Young J, Simms LA, Biden KG, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001;159:2107–16.
Gologan A, Sepulveda AR. Microsatellite instability and DNA mismatch repair deficiency testing in hereditary and sporadic gastrointestinal cancers. Clin Lab Med. 2005;25:179–96.
Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145:148–56.
Alexander J, Watanabe T, Wu TT, Rashid A, Li S, Hamilton SR. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158:527–35.
Dolcetti R, Viel A, Doglioni C, et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999;154:1805–13.
Greenson JK, Bonner JD, Ben-Yzhak O, et al. Phenotype of microsatellite unstable colorectal carcinomas: Well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am J Surg Pathol. 2003;27: 563–70.
Jass JR. Pathology of hereditary nonpolyposis colorectal cancer. Ann N Y Acad Sci. 2000;910:62–73. discussion 73–4.
Gologan A, Krasinskas A, Hunt J, Thull DL, Farkas L, Sepulveda AR. Performance of the revised Bethesda guidelines for identification of colorectal carcinomas with a high level of microsatellite instability. Arch Pathol Lab Med. 2005;129:1390–7.
De Jong AE, Morreau H, Van Puijenbroek M, et al. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology. 2004;126:42–8.
Lindgren G, Liljegren A, Jaramillo E, Rubio C, Lindblom A. Adenoma prevalence and cancer risk in familial non-polyposis colorectal cancer. Gut. 2002;50:228–34.
Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–34.
Vasen HF, den Hartog Jager FC, Menko FH, Nagengast FM. Screening for hereditary non-polyposis colorectal cancer: a study of 22 kindreds in The Netherlands. Am J Med. 1989;86:278–81.
Rijcken FE, Hollema H, Kleibeuker JH. Proximal adenomas in hereditary non-polyposis colorectal cancer are prone to rapid malignant transformation. Gut. 2002;50:382–6.
Bettstetter M, Dechant S, Ruemmele P, et al. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res. 2007;13:3221–8.
Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225.
Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261.
Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80.
Berends MJ, Wu Y, Sijmons RH, et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am J Hum Genet. 2002;70:26–37.
Kariola R, Raevaara TE, Lonnqvist KE, Nystrom-Lahti M. Functional analysis of MSH6 mutations linked to kindreds with putative hereditary non-polyposis colorectal cancer syndrome. Hum Mol Genet. 2002;11:1303–10.
Buttin BM, Powell MA, Mutch DG, et al. Penetrance and expressivity of MSH6 germline mutations in seven kindreds not ascertained by family history. Am J Hum Genet. 2004;74:1262–9.
Liu B, Parsons R, Papadopoulos N, et al. Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med. 1996;2:169–174.
Peltomaki P. DNA mismatch repair and cancer. Mutat Res. 2001;488:77–85.
Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21:1174–9.
Liu B, Nicolaides C, Markowitz S, et al. Mismatch repair defects in sporadic colorectal cancers with microsatellite instability. Nat Genet. 1995;9:48–55.
Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–8.
Lynch HT, Lynch JF. Lynch syndrome: history and current status. Dis Markers. 2004;20:181–98.
Goel A, Nguyen TP, Leung HC, et al. De novo constitutional MLH1 epimutations confer early-onset colorectal cancer in two new sporadic Lynch syndrome cases, with derivation of the epimutation on the paternal allele in one. Int J Cancer. 2010;128:869–78.
Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9:89–96.
Lipkin SM, Wang V, Jacoby R, et al. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet. 2000;24:27–35.
Kolodner R. Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev. 1996;10:1433–42.
Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J Biol Chem. 1998;273:19895–901.
Umar A, Risinger JI, Glaab WE, Tindall KR, Barrett JC, Kunkel TA. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics. 1998;148:1637–46.
Palombo F, Gallinari P, Iaccarino I, et al. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–4.
Marsischky GT, Filosi N, Kane MF, Kolodner R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996;10:407–20.
Sia EA, Kokoska RJ, Dominska M, Greenwell P, Petes TD. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol Cell Biol. 1997;17:2851–8.
Lu A-L. Biochemistry of mammalian DNA mismatch repair. Humana Press, 1998
Leung WK, Kim JJ, Wu L, Sepulveda JL, Sepulveda AR. Identification of a second MutL DNA mismatch repair complex (hPMS1 and hMLH1) in human epithelial cells. J Biol Chem. 2000;275:15728–15732.
Raschle M, Marra G, Nystrom-Lahti M, Schar P, Jiricny J. Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J Biol Chem. 1999;274:32368–75.
Lynch HT, Boman B, Fitzgibbons RJ Jr, Lanspa SJ, Smyrk TC. Hereditary nonpolyposis colon cancer: (Lynch syndrome I and II). A challenge for the clinician. Nebr Med J. 1989;74:2–7.
Lynch HT, Drouhard T, Lanspa S, et al. Mutation of an mutL homologue in a Navajo family with hereditary nonpolyposis colorectal cancer. J Natl Cancer Inst. 1994;86:1417–9.
Lynch HT, Lynch JF. 25 years of HNPCC. Anticancer Res. 1994;14:1617–24.
Lynch HT, Lynch JF. Hereditary cancer: family history, diagnosis, molecular genetics, ecogenetics, and management strategies. Biochimie. 2002;84:3–17.
Bessa X, Balleste B, Andreu M, et al. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol. 2008;6:206–14.
De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62.
Deng G, Bell I, Crawley S, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:191–5.
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11:42–65.
Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum. 1991;34:424–5.
Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–57.
Syngal S, Fox EA, Eng C, Kolodner RD, Garber JE. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37:641–5.
Umar A, Risinger JI, Hawk ET, Barrett JC. Testing guidelines for hereditary non-polyposis colorectal cancer. Nat Rev Cancer. 2004;4:153–8.
Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: a systematic review. JAMA. 2006;296:1507–17.
Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005;352:1851–60.
Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35–41.
Hampel H, de la Chapelle A. The search for unaffected individuals with Lynch syndrome: do the ends justify the means? Cancer Prev Res (Phila). 2011;4:1–5.
Suraweera N, Duval A, Reperant M, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123: 1804–11.
Goel A, Xicola RM, Nguyen TP, et al. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2010;138:1854–62.
Bacher JW, Flanagan LA, Smalley RL, et al. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Dis Markers. 2004;20:237–50.
Murphy KM, Zhang S, Geiger T, et al. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn. 2006;8:305–11.
Baudhuin LM, Burgart LJ, Leontovich O, Thibodeau SN. Use of microsatellite instability and immunohistochemistry testing for the identification of individuals at risk for Lynch syndrome. Fam Cancer. 2005;4:255–65.
Watson N, Grieu F, Morris M, et al. Heterogeneous staining for mismatch repair proteins during population-based prescreening for hereditary nonpolyposis colorectal cancer. J Mol Diagn. 2007;9:472–8.
Peltomaki P. Lynch syndrome genes. Fam Cancer. 2005;4:227–32.
Salahshor S, Koelble K, Rubio C, Lindblom A. Microsatellite Instability and hMLH1 and hMSH2 expression analysis in familial and sporadic colorectal cancer. Lab Invest. 2001;81:535–41.
Funkhouser WK Jr, Lubin IM, Monzon FA, et al. Relevance, pathogenesis, and testing algorithm for mismatch repair-defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn. 2012;14:91–103.
Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124:1574–94.
Mutations MaM. http://www.insight-group.org/ Accessed September 2004.
Akiyama Y, Sato H, Yamada T, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3.
Iino H, Simms L, Young J, et al. DNA microsatellite instability and mismatch repair protein loss in adenomas presenting in hereditary non-polyposis colorectal cancer. Gut. 2000;47:37–42.
Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129:415–21.
Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005;293:1979–85.
Guillem JG, Wood WC, Moley JF, et al. ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol. 2006;24:4642–60.
Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26:5783–8.
Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7.
Hampel H, Panescu J, Lockman J, et al. Comment on: screening for lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2007;67:9603.
Mvundura M, Grosse SD, Hampel H, Palomaki GE. The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med. 2010;12:93–104.
Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997;6:105–10.
Watson P, Butzow R, Lynch HT, et al. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2001;82:223–8.
Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–8.
Lynch HT, Boland CR, Gong G, et al. Phenotypic and genotypic heterogeneity in the Lynch syndrome: diagnostic, surveillance and management implications. Eur J Hum Genet. 2006;14:390–402.
da Silva FC, de Oliveira LP, Santos EM, et al. Frequency of extracolonic tumors in Brazilian families with Lynch syndrome: analysis of a hereditary colorectal cancer institutional registry. Fam Cancer. 2010;9:563–70.
Vasen HF, Morreau H, Nortier JW. Is breast cancer part of the tumor spectrum of hereditary nonpolyposis colorectal cancer? Am J Hum Genet. 2001;68:1533–5.
Westenend PJ, Schutte R, Hoogmans MM, Wagner A, Dinjens WN. Breast cancer in an MSH2 gene mutation carrier. Hum Pathol. 2005;36:1322–6.
Risinger JI, Barrett JC, Watson P, Lynch HT, Boyd J. Molecular genetic evidence of the occurrence of breast cancer as an integral tumor in patients with the hereditary nonpolyposis colorectal carcinoma syndrome. Cancer. 1996;77:1836–43.
Nelson CL, Sellers TA, Rich SS, Potter JD, McGovern PG, Kushi LH. Familial clustering of colon, breast, uterine, and ovarian cancers as assessed by family history. Genet Epidemiol. 1993;10:235–44.
de Leeuw WJ, van Puijenbroek M, Tollenaar RA, et al. Exclusion of breast cancer as an integral tumor of hereditary nonpolyposis colorectal cancer. Cancer Res. 2003;62:1014–1019. Cancer Res 2003;63:1148–9.
Boyd J, Rhei E, Federici MG, et al. Male breast cancer in the hereditary nonpolyposis colorectal cancer syndrome. Breast Cancer Res Treat. 1999;53:87–91.
Watson P, Vasen HF, Mecklin JP, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–9.
Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26.
Broaddus RR, Lynch PM, Lu KH, Luthra R, Michelson SJ. Unusual tumors associated with the hereditary nonpolyposis colorectal cancer syndrome. Mod Pathol. 2004;17:981–9.
Sijmons R, Hofstra R, Hollema H, et al. Inclusion of malignant fibrous histiocytoma in the tumour spectrum associated with hereditary non-polyposis colorectal cancer. Genes Chromosomes Cancer. 2000;29:353–5.
den Bakker MA, Seynaeve C, Kliffen M, Dinjens WN. Microsatellite instability in a pleomorphic rhabdomyosarcoma in a patient with hereditary non-polyposis colorectal cancer. Histopathology. 2003;43:297–9.
Kawaguchi K, Oda Y, Takahira T, et al. Microsatellite instability and hMLH1 and hMSH2 expression analysis in soft tissue sarcomas. Oncol Rep. 2005;13:241–6.
Suwa K, Ohmori M, Miki H. Microsatellite alterations in various sarcomas in Japanese patients. J Orthop Sci. 1999;4:223–30.
Soravia C, van der Klift H, Brundler MA, et al. Prostate cancer is part of the hereditary non-polyposis colorectal cancer (HNPCC) tumor spectrum. Am J Med Genet. 2003;121A:159–62.
Gallinger S, Aronson M, Shayan K, et al. Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology. 2004;126:576–85.
Meyer LA, Broaddus RR, Lu KH. Endometrial cancer and Lynch syndrome: clinical and pathologic considerations. Cancer Control. 2009;16:14–22.
Garg K, Soslow RA. Lynch syndrome (hereditary non-polyposis colorectal cancer) and endometrial carcinoma. J Clin Pathol. 2009;62:679–84.
Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Curr Opin Obstet Gynecol. 2009;21:31–8.
Resnick KE, Hampel H, Fishel R, Cohn DE. Current and emerging trends in Lynch syndrome identification in women with endometrial cancer. Gynecol Oncol. 2009;114:128–34.
Taylor N, Mutch DG. Gynecologic manifestations of hereditary nonpolyposis colorectal cancer. From inherited to sporadic disease. Oncology (Williston Park). 2006;20:85–94.
Prat J, Ribe A, Gallardo A. Hereditary ovarian cancer. Hum Pathol. 2005;36:861–70.
Watson P, Lynch HT. Cancer risk in mismatch repair gene mutation carriers. Fam Cancer. 2001;1:57–60.
Lu KH, Dinh M, Kohlmann W, et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet Gynecol. 2005;105:569–74.
Obermair A, Youlden DR, Young JP, et al. Risk of endometrial cancer for women diagnosed with HNPCC-related colorectal carcinoma. Int J Cancer. 2010;127:2678–84.
Chadwick RB, Pyatt RE, Niemann TH, et al. Hereditary and somatic DNA mismatch repair gene mutations in sporadic endometrial carcinoma. J Med Genet. 2001;38:461–6.
Banno K, Susumu N, Yanokura M, et al. Association of HNPCC and endometrial cancers. Int J Clin Oncol. 2004;9:262–9.
Lu KH, Schorge JO, Rodabaugh KJ, et al. Prospective determination of prevalence of lynch syndrome in young women with endometrial cancer. J Clin Oncol. 2007;25:5158–64.
Resnick K, Straughn JM Jr, Backes F, Hampel H, Matthews KS, Cohn DE. Lynch syndrome screening strategies among newly diagnosed endometrial cancer patients. Obstet Gynecol. 2009;114:530–6.
Goodfellow PJ, Buttin BM, Herzog TJ, et al. Prevalence of defective DNA mismatch repair and MSH6 mutation in an unselected series of endometrial cancers. Proc Natl Acad Sci USA. 2003;100:5908–13.
Boks DE, Trujillo AP, Voogd AC, Morreau H, Kenter GG, Vasen HF. Survival analysis of endometrial carcinoma associated with hereditary nonpolyposis colorectal cancer. Int J Cancer. 2002;102:198–200.
MacDonald ND, Salvesen HB, Ryan A, Iversen OE, Akslen LA, Jacobs IJ. Frequency and prognostic impact of microsatellite instability in a large population-based study of endometrial carcinomas. Cancer Res. 2000;60:1750–2.
Basil JB, Goodfellow PJ, Rader JS, Mutch DG, Herzog TJ. Clinical significance of microsatellite instability in endometrial carcinoma. Cancer. 2000;89:1758–64.
Caduff RF, Johnston CM, Svoboda-Newman SM, Poy EL, Merajver SD, Frank TS. Clinical and pathological significance of microsatellite instability in sporadic endometrial carcinoma. Am J Pathol. 1996;148:1671–8.
Maxwell GL, Risinger JI, Alvarez AA, Barrett JC, Berchuck A. Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obstet Gynecol. 2001;97:417–22.
Garg K, Leitao MM Jr, Kauff ND, et al. Selection of endometrial carcinomas for DNA mismatch repair protein immunohistochemistry using patient age and tumor morphology enhances detection of mismatch repair abnormalities. Am J Surg Pathol. 2009;33:925–33.
Shia J, Black D, Hummer AJ, Boyd J, Soslow RA. Routinely assessed morphological features correlate with microsatellite instability status in endometrial cancer. Hum Pathol. 2008;39: 116–25.
van den Bos M, van den Hoven M, Jongejan E, et al. More differences between HNPCC-related and sporadic carcinomas from the endometrium as compared to the colon. Am J Surg Pathol. 2004;28:706–11.
Walsh MD, Cummings MC, Buchanan DD, et al. Molecular, pathologic, and clinical features of early-onset endometrial cancer: identifying presumptive Lynch syndrome patients. Clin Cancer Res. 2008;14:1692–700.
Carcangiu ML, Radice P, Casalini P, Bertario L, Merola M, Sala P. Lynch syndrome–related endometrial carcinomas show a high frequency of nonendometrioid types and of high FIGO grade endometrioid types. Int J Surg Pathol. 2010;18:21–6.
Broaddus RR, Lynch HT, Chen LM, et al. Pathologic features of endometrial carcinoma associated with HNPCC: a comparison with sporadic endometrial carcinoma. Cancer. 2006;106:87–94.
Westin SN, Lacour RA, Urbauer DL, et al. Carcinoma of the lower uterine segment: a newly described association with Lynch syndrome. J Clin Oncol. 2008;26:5965–71.
Ramsoekh D, Wagner A, van Leerdam ME, et al. Cancer risk in MLH1, MSH2 and MSH6 mutation carriers; different risk profiles may influence clinical management. Hered Cancer Clin Pract. 2009;7:17.
Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135:419–28.
Greggi S, Genuardi M, Benedetti-Panici P, et al. Analysis of 138 consecutive ovarian cancer patients: incidence and characteristics of familial cases. Gynecol Oncol. 1990;39:300–4.
Bewtra C, Watson P, Conway T, Read-Hippee C, Lynch HT. Hereditary ovarian cancer: a clinicopathological study. Int J Gynecol Pathol. 1992;11:180–7.
Pal T, Permuth-Wey J, Sellers TA. A review of the clinical relevance of mismatch-repair deficiency in ovarian cancer. Cancer. 2008;113:733–42.
Crijnen TE, Janssen-Heijnen ML, Gelderblom H, et al. Survival of patients with ovarian cancer due to a mismatch repair defect. Fam Cancer. 2005;4:301–5.
Seidman JD, Horkayne-Szakaly I, Haiba M, Boice CR, Kurman RJ, Ronnett BM. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int J Gynecol Pathol. 2004;23:41–4.
Quirk JT, Natarajan N. Ovarian cancer incidence in the United States, 1992–1999. Gynecol Oncol. 2005;97:519–23.
Pal T, Permuth-Wey J, Kumar A, Sellers TA. Systematic review and meta-analysis of ovarian cancers: estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin Cancer Res. 2008;14:6847–54.
Capelle LG, Van Grieken NC, Lingsma HF, et al. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands. Gastroenterology. 2010;138:487–92.
Aarnio M, Salovaara R, Aaltonen LA, Mecklin JP, Jarvinen HJ. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer. 1997;74:551–5.
Lehtola J. Family study of gastric carcinoma; with special reference to histological types. Scand J Gastroenterol Suppl. 1978;50: 3–54.
Lynch HT, Grady W, Suriano G, Huntsman D. Gastric cancer: new genetic developments. J Surg Oncol. 2005;90:114–33. discussion 133.
Lauren PA, Nevalainen TJ. Epidemiology of intestinal and diffuse types of gastric carcinoma. A time-trend study in Finland with comparison between studies from high- and low-risk areas. Cancer. 1993;71:2926–33.
Mecklin JP, Jarvinen HJ, Virolainen M. The association between cholangiocarcinoma and hereditary nonpolyposis colorectal carcinoma. Cancer. 1992;69:1112–4.
Vernez M, Hutter P, Monnerat C, Halkic N, Gugerli O, Bouzourene H. A case of Muir-Torre syndrome associated with mucinous hepatic cholangiocarcinoma and a novel germline mutation of the MSH2 gene. Fam Cancer. 2007;6:141–5.
Koornstra JJ, Kleibeuker JH, Vasen HF. Small-bowel cancer in Lynch syndrome: is it time for surveillance? Lancet Oncol. 2008;9:901–5.
Rodriguez-Bigas MA, Vasen HF, Lynch HT, et al. Characteristics of small bowel carcinoma in hereditary nonpolyposis colorectal carcinoma. International Collaborative Group on HNPCC. Cancer. 1998;83:240–4.
Schulmann K, Brasch FE, Kunstmann E, et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology. 2005;128:590–9.
Watson P, Lynch HT. The tumor spectrum in HNPCC. Anticancer Res. 1994;14:1635–9.
Maul JS, Warner NR, Kuwada SK, Burt RW, Cannon-Albright LA. Extracolonic cancers associated with hereditary nonpolyposis colorectal cancer in the Utah Population Database. Am J Gastroenterol. 2006;101:1591–6.
Roupret M, Yates DR, Comperat E, Cussenot O. Upper urinary tract urothelial cell carcinomas and other urological malignancies involved in the hereditary nonpolyposis colorectal cancer (lynch syndrome) tumor spectrum. Eur Urol. 2008;54:1226–36.
Catto JW, Azzouzi AR, Amira N, et al. Distinct patterns of microsatellite instability are seen in tumours of the urinary tract. Oncogene. 2003;22:8699–706.
Sarin S, Bernath A. Turcot syndrome (glioma polyposis): a case report. South Med J. 2008;101:1273–4.
Hamilton SR, Liu B, Parsons RE, et al. The molecular basis of Turcot’s syndrome. N Engl J Med. 1995;332:839–47.
Torre D. Multiple sebaceous tumors. Arch Dermatol. 1968;98: 549–51.
Muir EG, Bell AJ, Barlow KA. Multiple primary carcinomata of the colon, duodenum, and larynx associated with kerato-acanthomata of the face. Br J Surg. 1967;54:191–5.
Lynch HT, Lynch PM, Pester J, Fusaro RM. The cancer family syndrome. Rare cutaneous phenotypic linkage of Torre’s syndrome. Arch Intern Med. 1981;141:607–11.
South CD, Hampel H, Comeras I, Westman JA, Frankel WL, de la Chapelle A. The frequency of Muir-Torre syndrome among Lynch syndrome families. J Natl Cancer Inst. 2008;100:277–81.
Mangold E, Pagenstecher C, Leister M, et al. A genotype-phenotype correlation in HNPCC: strong predominance of msh2 mutations in 41 patients with Muir-Torre syndrome. J Med Genet. 2004;41:567–72.
Entius MM, Keller JJ, Drillenburg P, Kuypers KC, Giardiello FM, Offerhaus GJ. Microsatellite instability and expression of hMLH-1 and hMSH-2 in sebaceous gland carcinomas as markers for Muir-Torre syndrome. Clin Cancer Res. 2000;6:1784–9.
Mathiak M, Rutten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338–43.
Lancaster JM, Powell CB, Kauff ND, et al. Society of Gynecologic Oncologists Education Committee statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol. 2007;107:159–62.
Mojtahed A, Schrijver I, Ford JM, Longacre TA, Pai RK. A two-antibody mismatch repair protein immunohistochemistry screening approach for colorectal carcinomas, skin sebaceous tumors, and gynecologic tract carcinomas. Mod Pathol. 2011;24: 1004–14.
Dove-Edwin I, Boks D, Goff S, et al. The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer. 2002;94:1708–12.
Rijcken FE, Mourits MJ, Kleibeuker JH, Hollema H, van der Zee AG. Gynecologic screening in hereditary nonpolyposis colorectal cancer. Gynecol Oncol. 2003;91:74–80.
Renkonen-Sinisalo L, Butzow R, Leminen A, Lehtovirta P, Mecklin JP, Jarvinen HJ. Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer. 2007;120:821–4.
Lecuru F, Metzger U, Scarabin C, Le Frere Belda MA, Olschwang S, Laurent Puig P. Hysteroscopic findings in women at risk of HNPCC. Results of a prospective observational study. Fam Cancer. 2007;6:295–9.
Lecuru F, Le Frere Belda MA, Bats AS, et al. Performance of office hysteroscopy and endometrial biopsy for detecting endometrial disease in women at risk of human non-polyposis colon cancer: a prospective study. Int J Gynecol Cancer. 2008;18: 1326–31.
Lynch HT, Boland CR, Rodriguez-Bigas MA, Amos C, Lynch JF, Lynch PM. Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J Clin Oncol. 2007;25:3534–42.
Bonis PA, Ahnen DJ, Axell L. Screening strategies in patients and families with familial colon cancer syndromes. In: Basow DE (Ed) UpToDate. Waltham, MA. 2008.
Doxey BW, Kuwada SK, Burt RW. Inherited polyposis syndromes: molecular mechanisms, clinicopathology, and genetic testing. Clin Gastroenterol Hepatol. 2005;3:633–41.
Lynch HT, Lynch JF, Shaw TG. Hereditary gastrointestinal cancer syndromes. Gastrointest Cancer Res. 2011;4:S9-S17.
Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101:385–98.
Hofgartner WT, Thorp M, Ramus MW, et al. Gastric adenocarcinoma associated with fundic gland polyps in a patient with attenuated familial adenomatous polyposis. Am J Gastroenterol. 1999;94:2275–81.
Ahnen DJ, Axell L. Clinical features and diagnosis of familial adenomatous polyposis. UpToDate, Basow DS (Ed), Waltham, MA. 2008.
Abraham SC, Park SJ, Mugartegui L, Hamilton SR, Wu TT. Sporadic fundic gland polyps with epithelial dysplasia: evidence for preferential targeting for mutations in the adenomatous polyposis coli gene. Am J Pathol. 2002;161:1735–42.
Lam-Himlin D, Park JY, Cornish TC, Shi C, Montgomery E. Morphologic characterization of syndromic gastric polyps. Am J Surg Pathol. 2010;34:1656–62.
Lynch PM. Prevention of colorectal cancer in high-risk populations: the increasing role for endoscopy and chemoprevention in FAP and HNPCC. Digestion. 2007;76:68–76.
Lipton L, Tomlinson I. The genetics of FAP and FAP-like syndromes. Fam Cancer. 2006;5:221–6.
Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21:2525–38.
Bodmer WF, Bailey CJ, Bodmer J, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987;328:614–6.
Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–5.
Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600.
Schulmann K, Pox C, Tannapfel A, Schmiegel W. The patient with multiple intestinal polyps. Best Pract Res Clin Gastroenterol. 2007;21:409–26.
Ahnen DJ. The genetic basis of colorectal cancer risk. Adv Intern Med. 1996;41:531–52.
Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22.
Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51.
Aoki K, Aoki M, Sugai M, et al. Chromosomal instability by beta-catenin/TCF transcription in APC or beta-catenin mutant cells. Oncogene. 2007;26:3511–20.
Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007;56:417–25.
Senda T, Iizuka-Kogo A, Onouchi T, Shimomura A. Adenomatous polyposis coli (APC) plays multiple roles in the intestinal and colorectal epithelia. Med Mol Morphol. 2007;40:68–81.
Abdel-Rahman WM, Peltomaki P. Molecular basis and diagnostics of hereditary colorectal cancers. Ann Med. 2004;36:379–88.
Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7.
Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007;61:153–61.
Rozen P, Macrae F. Familial adenomatous polyposis: the practical applications of clinical and molecular screening. Fam Cancer. 2006;5:227–35.
Hegde MR, Roa BB (2006) Detecting mutations in the APC gene in familial adenomatous polyposis (FAP). Curr Protoc Hum Genet 2006; Chapter 10: Unit 10 8.
Michils G, Tejpar S, Thoelen R, et al. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): a Belgian study. Hum Mutat. 2005;25:125–34.
Nielsen M, Hes FJ, Nagengast FM, et al. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet. 2007;71:427–33.
Hes FJ, Nielsen M, Bik EC, et al. Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut. 2008;57:71–6.
Romero-Gimenez J, Dopeso H, Blanco I, et al. Germline hypermethylation of the APC promoter is not a frequent cause of familial adenomatous polyposis in APC/MUTYH mutation negative families. Int J Cancer. 2008;122:1422–5.
Renkonen ET, Nieminen P, Abdel-Rahman WM, et al. Adenomatous polyposis families that screen APC mutation-negative by conventional methods are genetically heterogeneous. J Clin Oncol. 2005;23:5651–9.
Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C– > T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–32.
Poulsen ML, Bisgaard ML. MUTYH Associated Polyposis (MAP). Curr Genomics. 2008;9:420–35.
Gala M, Chung DC. Hereditary colon cancer syndromes. Semin Oncol. 2011;38:490–9.
Cheadle JP, Sampson JR. MUTYH-associated polyposis–from defect in base excision repair to clinical genetic testing. DNA Repair (Amst). 2007;6:274–9.
Lu AL, Bai H, Shi G, Chang DY. MutY and MutY homologs (MYH) in genome maintenance. Front Biosci. 2006;11:3062–80.
Parker AR, Eshleman JR. Human MutY: gene structure, protein functions and interactions, and role in carcinogenesis. Cell Mol Life Sci. 2003;60:2064–83.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Hagemann, I.S., Sepulveda, A.R. (2013). Cancer Predisposition Syndromes of the Gastrointestinal Tract. In: Sepulveda, A., Lynch, J. (eds) Molecular Pathology of Neoplastic Gastrointestinal Diseases. Molecular Pathology Library, vol 7. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-6015-2_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6015-2_7
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-6014-5
Online ISBN: 978-1-4614-6015-2
eBook Packages: MedicineMedicine (R0)