
Abstract
Faithful genome maintenance is essential to an organism’s growth and survival. To preserve genome fidelity, the DNA Damage Response (DDR) pathway has evolved to coordinate the surveillance and repair of genomic DNA, damaged by normal metabolic or environmental insults [1]. DDR surveillance mechanisms scan for discontinuities and structural changes in the DNA double helix. Upon detection of any damage to the DNA molecule, these surveillance sensors activate signal transduction cascades to amplify the damage signal, and coordinate the arrest of proliferation for proper DNA repair [1–4]. Alternatively, apoptosis may be initiated if repair is not possible. The abrupt termini of linear eukaryotic chromosomes pose specific challenges to DDR surveillance, as these natural ends are indistinguishable from damaged double-stranded DNA. In most eukaryotic organisms with linear chromosomes, phylogenetically conserved nucleoprotein structures, known as telomeres, differentiate chromosome ends from nonspecific DNA breaks [5–7]. Telomeres mask the ends of chromosomes from DDR surveillance sensors and protect the chromosome ends from inappropriate repair by DDR mechanisms [8].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Overview
Faithful genome maintenance is essential to an organism’s growth and survival. To preserve genome fidelity, the DNA Damage Response (DDR) pathway has evolved to coordinate the surveillance and repair of genomic DNA, damaged by normal metabolic or environmental insults [1]. DDR surveillance mechanisms scan for discontinuities and structural changes in the DNA double helix. Upon detection of any damage to the DNA molecule, these surveillance sensors activate signal transduction cascades to amplify the damage signal, and coordinate the arrest of proliferation for proper DNA repair [1–4]. Alternatively, apoptosis may be initiated if repair is not possible. The abrupt termini of linear eukaryotic chromosomes pose specific challenges to DDR surveillance, as these natural ends are indistinguishable from damaged double-stranded DNA. In most eukaryotic organisms with linear chromosomes, phylogenetically conserved nucleoprotein structures, known as telomeres, differentiate chromosome ends from nonspecific DNA breaks [5–7]. Telomeres mask the ends of chromosomes from DDR surveillance sensors and protect the chromosome ends from inappropriate repair by DDR mechanisms [8].
Over the past two decades, we have learned a great deal about the structure of telomeres, their homeostatic maintenance, and the cellular consequences of their dysfunction. We know that while telomeres suppress the erroneous activation of the DDR pathways by chromosome ends, the structural and functional integrity of these structures are dependent on the activities of the same DDR pathways. In this chapter, we describe the protein and nucleic acid components of telomeres, both stable and transient. We then describe the physiological mechanisms of telomere maintenance by the enzyme telomerase, its biogenesis and regulation, and how this reverse transcriptase might be utilized in anticancer chemotherapy.
2 Telomeres
2.1 Telomere Structure
At the ends of most eukaryotic chromosomes are highly conserved, tandem DNA repeats. These highly repetitive sequences are associated with their specific binding proteins, and together, these chromosome-end structures are known as telomeres. Telomeres cap chromosome ends and protect them from nonspecific nuclease digestion, as well as preventing them from being recognized as double-stranded DNA breaks. In the absence of telomeres, erroneous DNA repair can lead to chromosomal end-to-end fusions and genetic recombination [5]. The length of telomeric DNA repeats vary between species, ranging from ∼300 to 600 bp in yeast [9], to ∼150 kb in mice [10]. Human telomeres measure ∼5–15 kb in length [11, 12]. In all vertebrate chromosomes, telomeres are made up of a G-rich hexanucleotide sequence (TTAGGG)n [13]. Telomere repeats run 5′-3′, terminating in a single-stranded 3′ overhang of the G-rich strand [14, 15]. The length of this overhang is also species-specific, measuring ∼50–100 nucleotides in length in mouse and human telomeres [16].
Mammalian telomeres were previously thought to be linear. However, electron microscopy analysis of psoralene cross-linked telomeric DNA from human and mouse were visualized to end as large duplex loops [17]. At the molecular level, double-stranded telomeric DNA folds back onto itself to form a lariat structure termed the telomeric loop (Fig. 1a). This allows for the G-rich 3′ overhang to invade the duplex section of telomeric repeats, thereby forcing the formation of a single-stranded DNA displacement loop [18]. The resulting higher order chromatin structure is distinct from damaged DNA and thus serves to differentiate the normal chromosomal termini, preventing them from being recognized as double strand breaks. This differentiation mechanism is crucial in preventing the initiation of DNA damage checkpoint responses [5, 6, 16].

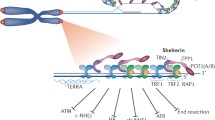
Human telomeres. (a) Telomere repeats at chromosome ends fold back to form a lariat structure (t-loop). The 3′ telomeric DNA overhang invades the double-stranded DNA region of telomeric repeats to form a displacement-loop (d-loop). (b) Shelterin protein complex aids in t-loop formation and stabilization: TRF1 and TRF2 interact with double-stranded telomeric repeats, recruiting the other four shelterin proteins, POT1, TIN2, TPP1, and Rap1, to the telomere end. TIN2 links TRF1 to TRF2, contributing to the stabilization of these proteins on the telomere. POT1, which has strong binding specificity for single-stranded telomeric repeats, together with its heterodimeric partner TPP1 associates with TRF1 and TRF2 through a bridge formed by TIN2. Rap1 is recruited by TRF2, forming a TRF2-Rap complex. (c) The human telomere 3′ overhangs exist in two structural forms. Shelterin components POT1-TPP1 bind single-stranded telomeric DNA with high sequence specificity. Recently, the human homologs of yeast CST complex have been identified to associate with single-stranded telomeric DNA structure, with low sequence specificity and in the absence of shelterin
A six-member protein complex, termed shelterin, associates with telomeric DNA in a sequence specific manner (Fig. 1b, Table 1). This complex facilitates formation of the telomeric loop to protect chromosome ends from DNA damage surveillance mechanisms, as well as to functionally maintain telomere length. The shelterin complex is composed of six distinct proteins: telomere repeat binding factors 1 and 2 (TRF1 and TRF2), protection of telomeres 1 (POT1), TRF1-and TRF2-interacting nuclear protein 2 (TIN2), repressor/activator protein 1 (Rap1), and TPP1 (formally known as PTOP, PIP1, or TINT1) [7, 19]. TRF1 and 2 are sequence specific telomeric DNA binding proteins that recruit the other four proteins to the telomeres [19]. Both TRF1 and TRF2 contain a C-terminal SANT/Myb-type DNA binding domain that binds to the 5′-TTAGGG-3′ sequence in duplex DNA, making the entire shelterin complex highly specific for telomeric repeats [20, 21].
TRF1 is a homodimeric protein that aids in telomeric loop formation and stabilization [22]. Its binding to arrays of telomeric repeats induces shallow bends and results in the formation of DNA loops, demonstrating the protein’s architectural role on telomeres [20]. This protein has also been shown to affect telomere length. Over-expression of TRF1 results in telomere shortening while expression of a dominant negative TRF1 mutant, lacking the Myb type domain, causes telomere lengthening. This suggests a negative correlation between TRF1 function and telomere length [23, 24]. On the other hand, accumulations of TRF1 and TRF2 at telomere ends were shown to positively correlate with telomere length [23, 24]. This led to a protein counting theory of telomere length regulation, which proposed that a feedback mechanism mediated by protein interactions with TRF1 is responsible for steady-state telomere length maintenance [23].
Like TRF1, TRF2 also binds to double-stranded telomeric DNA as a homodimer [25] and plays a role in telomeric loop assembly. In contrast to TRF1, TRF2 is believed to bind near the loop-tail junction where it stabilizes the G-rich single-stranded telomeric overhang at the displacement loop by facilitating strand invasion and preventing the single-stranded sequence from being recognized as a DNA break [17, 26]. In corroboration to this model, electron microscopy of a telomere DNA track containing ∼2 kb of telomeric repeats at the end of a linearized DNA plasmid and terminating in a 3′ single-stranded overhang, revealed the specific binding location of TRF2 at the telomeric loop junction [27]. Like TRF1, TRF2 also serves as a negative regulator of telomere length. TRF2 over-expression results in shortened telomeres and induces senescence in telomerase negative cells [28].
POT1 is the most highly conserved component of shelterin, and has a strong specificity for single-stranded 5′-(T)TAGGGTTAG-3′ sites [29, 30]. Following DNA replication, single-stranded telomeric overhangs initially associate with replication protein A (RPA). Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) binding displaces RPA binding, while the increase in TERRA expression levels (see below) following S phase removes hnRNPA1 from telomeric DNA, allowing for sequence-specific binding by POT1 [31]. POT1 accumulation at chromosome ends is believed to regulate telomerase activity by relaying telomere length information from the double-stranded region of the telomeric loop to the single-stranded region through its interaction with TRF1 [32]. Studies have also demonstrated that POT1 plays a positive role in telomere length maintenance, as ectopic expression of POT1 results in an increase in telomeric DNA [33, 34].
TPP1, the heterodimeric partner of POT1 [35, 36], enhances POT1 affinity for single-stranded telomeric DNA [36]. Most of the POT1-TPP1 complexes are associated with TRF1 and TRF2 through a bridge formed by TIN2, which functions to stabilize the interactions between these proteins [37]. In addition to the protection of telomere ends, the TPP1-POT1 complex also serves as a regulator of telomere length maintenance. Through its oligonucleotide- and oligosaccharide-binding fold, TPP1 has been suggested to regulate telomerase activity and the enzyme’s access to single-stranded telomeric DNA, both negatively and positively in a context dependent manner [36].
TIN2 co-localizes with TRF1 on metaphase chromosomes [38]. TIN2 forms bridges that join POT1 to TRF1 and TRF2 and also TRF1 to TRF2, contributing to the stabilization of these proteins at telomeres [39, 40]. The binding of TRF1 to TIN2 leads to the compaction of telomeric DNA and telomeric loop stabilization. Both events limit the accessibility of telomerase to telomere ends and thereby functions as a negative regulator of telomere length [38]. Genetic lesions of TIN2 underlie a subpopulation of autosomal dominant form of dyskeratosis congenita, representing the only shelterin protein associated with this genetic disease of telomere dysfunction [41].
Rap1 is recruited to the telomere by protein interactions with TRF2, forming a TRF2-Rap1 complex [42]. Rap1 affects telomere length homeostasis through its interactions with telomere length regulator proteins Rif1 and Rif2. Like the other shelterin proteins, Rap1 is a negative regulator of telomere length. Over-expression of Rap1 leads to telomere shortening, while expression of dominant negative mutants results in the gain of telomere length [43]. In addition, RAP1 binds to non-telomeric sequences and is implicated in diverse cellular activities including gene silencing and the transcriptional regulation of gene targets involved in adhesion, metabolism, and cancer [44].
In Saccharomyces cerevisiae, the cdc13-stn1-ten1 (CST) complex binds single-stranded telomeric DNA in place of POT1-TPP1 [45, 46]. Recently, the human version of this protein complex has been identified. The human ctc1-stn1-ten1 (CST) complex contains two human homologs of the ScCST complex (Stn1 and Ten1), and a third component, the conserved telomere maintenance component 1 (ctc1) [47]. Similar to the ScCST complex, human CST binds single-stranded G-rich telomeric DNA, in the absence of POT1-TPP1. Unlike ScCST, human CST does not exhibit sequence specificity for telomeric repeats, and likely associates with other single-stranded DNA in a manner analogous to the binding of single-stranded DNA by replication protein A (Fig. 1c, Table 1). A significant increase in the G-strand overhang was observed in Stn1 depleted human cells, indicating a role of the CST complex in single-strand telomeric DNA regulation [47]. Whether this newly identified CST complex functionally interacts with the shelterin complex is currently under investigation.
In addition to the binding of telomere-specific shelterin and the single-stranded DNA-specific CST complexes, heterochromatin formation via the epigenetic regulation of telomeric chromatin is also observed. DNA methylation of subtelomeric regions [48], together with histone methylation of the telomeric chromatin [49], are postulated to negatively regulate gene transcription, suppress homologous recombination and prevent telomerase access for telomere elongation [50]. Telomere-repeat containing RNAs (TERRA) are long UUAGGG-repeat containing noncoding RNA transcripts that have been recently identified [51, 52]. TERRAs are transcribed starting from the subtelomeric region, using the C-rich strand of the telomere as a template. TERRAs are found to be stably associated with telomeric chromatin and cellular machinery responsible for the nonsense mediated decay of dysfunctional RNA transcripts. Current models of these noncoding RNA functions prescribe a role in the induction of heterochromatin formation, serving as a negative regulator of telomerase access to telomeres [53]. Cell-cycle phase-specific changes in TERRA expression levels are also postulated to mediate the switch from RPA to POT1 binding at single-stranded telomeric DNA termini [31].
2.2 Telomere Function: End Replication Problem and the Hayflick Limit
Besides structurally protecting the ends of chromosomes, telomeres also serve as a solution to the end-replication problem. Because DNA polymerases fail to completely copy chromosomes to the very end, the placement of telomeres at the extreme ends of chromosomes allows them to buffer gene coding sequence from being eroded [11, 54]. Instead, telomeric DNA is lost after every round of DNA replication. Telomeric DNA loss is cumulative and with continual proliferation; telomeres will eventually reach a critical short length. At this point, genome surveillance mechanisms will trigger replicative senescence, an irreversible cellular growth arrest state where cells can no longer divide into daughter progeny, but remain metabolically active (Fig. 2) [5, 55–57]. This short telomere checkpoint serves as a “mitotic clock” which counts down the number of cell divisions in each cell lineage. Leonard Hayflick first described this relationship by observing the replicative potential of human primary fibroblasts in culture in 1965 [58]. Termed the Hayflick Limit, the number of times a cell lineage could divide before short telomere-induced proliferative arrest was determined by the structural integrity of telomeres and the activities of biological pathways responsible for maintaining the length of these specialized DNA tracts [59, 60]. Incidentally, this process can be viewed as a tumor suppressive mechanism: by limiting the number of cell divisions that can occur in a particular cell lineage, one can reduce the accumulation of deleterious mutations that precede cellular transformation [61].

Telomere dynamics and cancer development. With each cell division, approximately 50–100 bp of telomeric DNA is lost from chromosome ends. With continual proliferation, telomeres will eventually reach a critical short length and are triggering replicative senescence. Inactivation of genome surveillance mechanisms mediated by the tumor suppressor genes p53 and Rb allow continual cell divisions, further depleting telomeric DNA leading to rampant genomic instability and the induction of apoptosis. A rare cell (∼1 in 10 million) can be forced to reactivate telomerase, allowing the cell to replace lost telomeric repeats, prevent further genomic instability and confer the unlimited proliferative capacity required for the formation of a malignant tumor cell
In rare cases, some somatic cells are able to bypass this short telomere checkpoint by inactivating the genome surveillance mechanisms mediated by the tumor suppressor genes p53 and retinoblastoma protein (Rb) (Fig. 2). Further cell divisions in p53/Rb-inactivated cells continue to deplete telomeric DNA, leading to the disruption of the telomere structure [62]. Uncapped telomeres are recognized by cellular repair mechanisms as damaged DNA, resulting in cells attempting to repair these damages. Erroneous repair leads to chromosome end fusions and rampant genomic instability. When this happens, a second checkpoint termed “crisis” [57] is activated and cells are triggered to undergo apoptosis. Under this extreme selective pressure, most cells will die. In extremely rare cases (∼1 in 10 million cells), genomic instability can lead to the reactivation of a specialized cellular reverse transcriptase, termed telomerase, which is capable of adding telomeric repeats to chromosome ends [63]. Telomerase expression allows cells to replace lost telomeric repeats and prevent further chromosome instability. In these cases of forced reactivation of telomerase enzyme expression, constitutive telomerase activity confers the unlimited proliferative capacity required for the formation of a malignant tumor cell (Fig. 2).
2.3 Dysfunctional Telomere Capping Is Recognized as DNA Damage
Dysfunctional telomeres are created when telomeric sequences are shortened beyond a critical length allowing for the formation of a higher order chromatin structure [5], or by the genetic deletion or protein dysfunction of key shelterin components [28, 56]. Uncapped telomeres expose the ends of chromosomes, thereby inducing the DDR and resulting in the cascade of genomic instability through erroneous chromosome end repair. Genetic deletion of different shelterin components in mice resulted in overlapping yet distinct phenotypes, underscoring the complexity and the distinct roles of shelterin components in chromosome end capping [64–67]. In parallel, cellular biological experiments using human cell models have demonstrated that TRF2 and POT1 have independent roles in the normal suppression of distinct DDR pathways, and that their dysfunctions cause severe molecular cytogenetic phenotypes [19, 56, 68].
Expression of dominant negative TRF2 that cannot bind DNA leads to the induction of a potent DDR mediated by the Mre11-Rad50-NBS (MRN) complex and the ataxia telangiectasia mutated (ATM) protein [26, 69]. As a protein kinase, ATM activates the DNA repair machinery through a cascade of phosphorylation activity, including targets such as the histone variant H2AX and the p53 binding protein (p53BP) [70]. Using immunofluorescent labeling, the MRN complex, ATM, gH2AX, and p53BP can all be seen to form DNA damage foci at uncapped telomeres resulting from the loss of TRF2 binding [56]. Known as telomere dysfunction induced foci (TIF), these structures contain DDR factors similar to those found in double-stranded DNA damage foci. Thus, part of the normal function of TRF2 binding to the telomere is the suppression of the ATM DDR pathway [19, 69].
Reducing the expression of POT1, or its binding partner TPP1, activates the ataxia telangiectasia and Rad3-related (ATR) kinases [69, 71]. Together with its obligate subunit, ATR interacting protein (ATRIP), ATR phosphorylates DDR effectors responsible for diverse DNA damages, such as those induced by UV exposure, exposure to nucleophilic crosslinking agents or resulting from collapsed replication forks [72]. In vitro biochemical experiments have shown that ATR is activated by replication protein A (RPA)-coated single-stranded DNA. This is reminiscent of the single-stranded telomeric G-rich overhang left vacant by the removal of POT1 binding. Thus, part of the normal function of POT1 binding to the telomeric terminai is the suppression of the ATR DDR pathway [19, 64, 69].
2.4 Key DDR Players Are Required for Normal Telomere Maintenance
The relationship between telomeric chromatin and the DDR machinery extends beyond simple antagonism. DDR components are known to play positive roles in the normal homeostatic maintenance of telomeres, in the absence or presence of telomerase activity. Evidence that key DDR components have important roles in normal telomere homeostasis comes from studies of inherited human diseases and from animal models of these diseases. Genetic disorders such as Ataxia telangiectasia, Nijmegen break syndrome, Bloom syndrome and Werner syndrome all exhibit molecular phenotypes of accelerated telomere shortening [70, 73]. Initially believed to be the function of an increased rate of telomere attrition due to the higher cellular turnover, animal models and cellular biology studies later revealed the normal functions of these proteins in telomere homeostasis. DDR mediators are activated each time telomeric DNA undergoes replication [78, 79]. Normal replication through telomere ends requires the resolution of the telomere loop, followed by leading and lagging strand synthesis through the ends. This requires the action of RecQ helicases, such as Bloom and Werner, the elective de novo synthesis of G-rich telomeric DNA when telomerase is active, nuclease trimming by Apollo, XPF or the MRN complex to create the correct telomeric DNA terminus and overhangs, and the reformation of the telomeric loop structures through the actions of Rad51D, RPA and other homologous recombination pathway effectors. In addition, the steps necessary to “open up” a telomere for DNA replication machinery access predicts that the transient recognition of these “open” telomere structures by DDR sensors. Indeed, both ATM-MRN and ATR-ATRIP complexes are found at functional telomeres during the DNA replication phase of the cell cycle [78, 79]. Despite the data supporting these models, detailed mechanisms of how telomeric binding proteins coordinate with transient DDR signals to direct telomere formation, instead of promoting erroneous DNA repair at these sites, still need to be elucidated.
3 Telomerase
3.1 Telomerase Structure and Biogenesis
The human telomere terminal transferase enzyme, more commonly referred to as telomerase, is a ribonucleoprotein (RNP) responsible for the de novo synthesis of telomere repeats. This unique reverse transcriptase extends chromosome ends by utilizing an integral RNA subunit as a template to synthesize the TTAGGG telomeric DNA repeats. The core components of this enzyme complex consist of the telomerase reverse transcriptase catalytic subunit (TERT) and the telomerase RNA (TER), which contains the template sequence for telomere synthesis. In the human enzyme, RNA-binding proteins such as the H/ACA proteins dyskerin, Nop10, and Nhp2 are also found to associate with the core enzyme complex (Fig. 3, Table 2). Other proteins transiently associate with the core enzyme complex and play important roles in the regulation of the catalytic activity, enzyme stability, cellular localization and intracellular trafficking of the enzyme (Table 3) [80–82].

Human Telomerase. Schematic depiction of the human telomerase enzyme. Telomerase is a specialized reverse transcriptase carrying its own RNA template (TER). Telomerase RNA serves multiple functions. The template domain allows sequence specific alignment of the linear chromosome ends into the catalytic site and provides the 6nt template sequence for RT. Other domains of the RNA serve structural and catalytic functions, RT activity is provided by the protein subunit, telomerase reverse transcriptase (TERT). Together, TER and TERT comprise the minimal functional unit that can be reconstituted in vitro for telomerase activity. The in vivo accumulation and stability of TER requires the association of RNA with two sets of H/ACA proteins. Other protein factors involved in the regulation of enzyme functions through cellular localization (TCAB1), assembly (Pontin and Reptin) and other mechanisms associate with the holoenzyme complex in a transient manner
TER and TERT were identified as the catalytic core of this complex by virtue of their ability to form a complex and elongate telomeres in vitro, in the absence of other protein factors [83]. However, in vivo, telomerase employs an intricate biogenesis pathway involving specific factors for enzyme assembly, trafficking and the subcellular localization of the holoenzyme complex. TER transcription is ubiquitous in all human cells. The stability of TER is dependent on biogenesis protein factors Shq1 and NAF-1 mediated complex formation with the H/ACA proteins (dyskerin, Nhp2, and Nop10). TER association with the H/ACA complex results in the formation of a stable but inactive telomerase RNP. Assembly of this inactive telomerase RNP with TERT is required for catalytic activity [84]. Telomerase enzyme assembly is cell cycle and subcellular localization dependent [85]. Numerous biogenesis factors, including staufen, L22, SmB/SmD3, PinX1, 14-3-3, nucleolin, pontin, reptin, Hsp90, p23, and telomerase Cajal body protein 1 (TCAB1) have all been demonstrated to play important roles in TER/TERT subcellular localization and enzyme assembly. Finally, following the formation of a functional telomerase enzyme, additional trafficking factors, such as TCAB, hEST1A, and hnRNPs, are required for proper transport of the active enzyme to chromosome ends.
3.2 Telomerase RNA
TER is a noncoding RNA that serves as a template for TERT-dependent addition of telomeric repeats. Ubiquitously expressed, human TER is synthesized by RNA polymerase II (pol II) and processed into a mature 451-nucleotide (nt) product with a 5′ trimethyl guanosine cap that lacks a polyadenosine tail at its 3′ end [86, 87]. TER contains a 341-nt pol II-type promoter region upstream of the transcription start site [88]. Nuclear factor-Y (NF-Y), Sp1 and Sp3 are essential regulators of TER promoter function.
Primary and secondary structural elements of TER contain many motifs that are essential for telomerase activity as well as cellular accumulation of mature TER. The 11-nt telomeric repeat template sequence is contained within the 5′ portion of TER in the pseudoknot domain (nt 1–209). The H/ACA motif (nt 275–451) is essential for TER association with the chaperone H/ACA protein complex. Association with the H/ACA proteins is crucial for cellular accumulation and 3′ end processing of TER. The 3′ terminal hairpin domain (CR7; nt 408–422) contains a Cajal body specific localization signal (CAB box) necessary for the accumulation of TER to the Cajal bodies (CBs), as well as a biogenesis box (BIO box), which is necessary for in vivo accumulation of TER (Fig. 4a) [89, 90].

Human TER and TERT organization. (a) Secondary structure of TER. The 451-nt RNA includes the 11-nt template region in addition to conserved regions: pseudoknot domain (nt 1–209), CR4/CR5 (nt 214–330), CR7 3′ terminal hairpin domain, which contains the CAB box and BIO box, and H/ACA domain (275–441). (b) Functional organization of TERT protein. The reverse transcriptase (RT) domain is flanked by an N-terminal domain which is subdivided into an RNA binding domain (TRBD/RID2) and a TERT essential N-terminal (TEN/RID1) domain. The seven universally conserved RT motifs are illustrated as purple boxes
3.3 The Hinge/ACA Proteins (H/ACA)
The H/ACA proteins dyskerin, Nop10, and Nhp2 form the core trimer that acts as a chaperone to promote the in vivo accumulation of TER. The binding of these proteins with TER immediately following transcription is essential for its cellular accumulation, processing and stability [93]. In contrast to other protein factors described in the later sections, H/ACA proteins associate with TER throughout the enzyme’s life span and are considered stable components of the telomerase holoenzyme, as illustrated by affinity purification experiments [92, 93].
Two sets of H/ACA proteins bind to the distal and proximal stem loops of the TER H/ACA motif (nt 275–441) [80]. Mutations in the H/ACA motif in TER, as well as in the members of the H/ACA core trimer complex (dyskerin, Nhp2, and Nop10), are associated with genetic diseases with the common etiology of telomerase deficiencies and overlapping clinical presentations of premature tissue aging phenotypes [94–98].
3.4 Other TER-Associating Factors
RNA binding proteins, staufen and L22, have been shown to independently associate with TER in vivo and are involved in TER processing, localization and telomerase assembly [99]. The Sm-fold proteins, SmB and SmD3, have also been shown to associate with TER and are involved in its subcellular localization to Cajal bodies. SmB and SmD3 both interact with the CAB box sequence on TER, located in the CR7 domain, through an extended C-terminal tail modified with symmetric dimethyl-arginine. Deletion of this modified C-terminal sequence disrupts their association with TER [94]. However, it is not known whether this association is mediated through direct interactions between Sm proteins and TER or through the interactions with a tether protein. Mutations in TER’s CAB box result in a significant decrease in SmB and SmD3 association and a loss of CB localization [100, 101]. Notably, the novel RNA binding protein TCAB1 was also shown to bind TER at the CAB box [103]. It is currently unknown whether SmB/SmD3 and TCAB1 proteins coexist on the same telomerase molecule, or if associations with these specific protein factors occur at different stages of the telomerase enzyme’s maturity.
3.5 Telomerase Reverse Transcriptase
Catalytic activation of the telomerase complex requires the transcriptional activation of TERT. The TERT gene, located on chromosome 5p15.33, is composed of 16 exons and encompasses more than 37 kb [103, 104]. The GC-rich promoter region is located 1,100 bp upstream from the ATG start codon [104, 105]. This region lacks both TATA and CAAT boxes [103] and was found to be hypermethylated in somatic cells which correlates with its transcriptional inactive state. The TERT promoter contains numerous c-myc, as well as other oncogenic transcription factors, such as c-Jun and c-fos binding sites, which have been demonstrated to mediate TERT transcriptional activation in transformed cells [105]. Transcription activation of the TERT locus produces a full length TERT-mRNA, as well as a variety of alternative spliced forms. TERT alternative splicing is believed to regulate the levels of functional telomerase in a development stage specific manner [107]. Following protein translation of the full length 125 kDa polypeptide [108, 109], TERT associates with chaperones Hsp90 and p23, and is transported to the nucleus via its nuclear localization signal where it is assembled with the TER-H/ACA complex to form the fully functional telomerase enzyme [110].
TERT contains a central reverse transcriptase (RT) domain that is flanked by a N-terminal region and a C-terminal domain. The TERT N-terminal region is further subdivided into two domains: an RNA binding domain (TRBD) and a TERT essential N-terminal (TEN) domain. A large non-conserved linker region separates the two N-terminal domains (Fig. 4b) [112].
The RT domain contains the seven universally conserved RT motifs (1, 2, A, B′, C, D, and E) [113]. An invariant trio of aspartic acids (found in motifs A and C) is directly involved in catalysis, as mutations of these residues results in abolished catalytic activity in vitro and in vivo [84, 114–117]. Mutations of other amino acid residues in any of the conserved RT motifs were also found to reduce or eliminate telomerase reverse transcriptase activity (Fig. 4b) [84, 115, 117].
The high affinity RNA binding domain (TRBD), also known as the RNA interacting domain 2 (RID 2), contains telomerase specific motifs CP, QFP, and T, also referred to as domains II, III, and IV, respectively [118–120]. These motifs mediate TER recognition and have a relatively high binding affinity to structured RNA stem loops, interacting with the CR4/CR5 domain of TER [121]. This domain plays a role in promoting stable enzyme assembly, as mutations in these motifs result in severe defects in TER–TERT association (Fig. 4b) [122].
The TERT essential N-terminal (TEN) domain or RNA interacting domain 1 (RID 1), contains the non-conserved extreme N-terminus motif [123] and moderately conserved GQ motif (also referred to as domain I) [112, 122]. The GQ motif is further divided into domains IA and IB, separated by a DAT (dissociates activities of telomerase) domain [124]. The TEN domain interacts with the TER pseudoknot-template domain [121], but is not considered a major TER binding surface as mutations in this region only result in modest reductions of TER–TERT association [122]. This region also displays high single-stranded telomeric DNA binding affinity, suggesting an important role in substrate recognition and primer binding (Fig. 4b) [121, 124–126].
The smaller, less-conserved C-terminal domain (TEC or CDAT) plays several roles in telomerase function: it contributes to telomerase catalytic activity [121, 127], regulates the cellular localization of the enzyme, and plays a role in polymerase processivity [128, 129]. However, this domain is not essential for RNA binding, as mutations in this region were not found to impair TER–TERT association (Fig. 4b) [129].
3.6 TERT Chaperones and Localization Factors
Molecular chaperone proteins p23 and Hsp90 were identified as key factors in the assembly and functionality of the telomerase holoenzyme. Both were found to associate with TERT and aid in its nuclear import and localization. They were also demonstrated to be required for the assembly of active telomerase enzyme both in vitro and in vivo, as inhibition of either chaperone protein disrupts telomerase assembly leading to a reduction in enzyme activity ([110]; see geldanamycin mechanism below).
The nuclear retention of TERT is dependent on its association with the 14-3-3 proteins, a protein family involved in intracellular trafficking/targeting, cell cycle regulation, cytoskeleton structure, and transcription [129]. TERT and 14-3-3 interact via their respective C-termini. This interaction is required for the nuclear accumulation of TERT, as 14-3-3 proteins promote the nuclear retention of TERT by masking the nuclear export signal (NES)-like motif in the C-terminal region of TERT. Binding of 14-3-3 inhibits the binding of CRM1/exportin 1 to TERT NES, resulting in the nuclear accumulation of the reverse transcriptase.
Nucleolin is a phosphorprotein that binds to TERT through its RNA binding domain 4 and the carboxyl terminal RGG domain. RNA binding domain 1 may also be involved in the nucleolar localization of telomerase holoenzyme through its interactions with TER. Biochemical experiments had shown that the binding of TERT with the nucleolin-4R fragment, which lacks a nucleolar localization signal, resulted in the mislocalization of TERT in the cytoplasm, thereby implicating this protein in the subnuclear localization of TERT [130].
PINX1, a PIN2/TRF1 interacting protein, is involved in TERT nucleolar localization and has also been characterized as an inhibitor of telomerase activity and a negative regulator of telomere length. Inhibition of endogenous PINX1 resulted in an increase in telomerase activity, whereas over-expression of PINX1 decreases telomerase activity and shortens telomeres [131]. PINX1 was found to bind directly with TERT at its RNA binding domain and indirectly associate with TER through TERT [132].
3.7 TERT Post-translational Modifications
Telomerase activity is regulated via post-translational modifications of TERT. Several studies have demonstrated that the phosphorylation of TERT is required for the catalytic activity of the enzyme [133–136]. Protein kinase B (Akt) and protein kinase Cα have both been shown to interact with and phosphorylate TERT in vitro and in vivo [133–135], resulting in the increase in telomerase activity. Conversely, protein phosphatase 2A inhibits telomerase activity via the dephosphorylation of TERT directly [133, 137] or indirectly, through the dephosphorylation and inhibition of Akt. c-Abl protein tyrosine kinase associates with TERT and mediates TERT phosphorylation in vitro and in vivo. In contrast to the activation models above, c-Abl phosphorylation of TERT resulted in the inhibition of telomerase activity, making this kinase a negative regulator of TERT [138].
The E3 ubiquitin ligase MKRN1 was shown to have a negative role on telomere length homeostasis. MKRN1 is responsible for the ubiquitination of TERT, targeting TERT for protease degradation. Over-expression of MKRN1 results in the decrease of telomerase activity and subsequently in the shortening of telomere length [139].
3.8 TER–TERT Biogenesis/Assembly Factors
Pontin and reptin, members of the AAA+ family of DNA helicases [140], play pivotal roles in telomerase assembly. These helicases are found to bind to dyskerin and play a role in the formation of the TER-dyskerin complex. Subsequently, these helicases bind to endogenous TERT and mediate the assembly with TER-dyskerin complex to form the catalytically active telomerase enzyme [141]. The formation of the TERT-pontin-reptin complex is regulated by cell cycle stages, with the highest level of complex formation occurring during S-phase, providing evidence for another level of cell cycle dependent regulation of TERT.
Nucleoplasmic Cajal bodies (CBs) have been suggested as one of the sites for telomerase assembly. The novel RNA binding protein TCAB1 was shown to be required for telomerase localization to these sites. Knockdown of TCAB using retroviral shRNA and RNA interference resulted in a significant reduction in the percentage of cells with TER staining in CBs by microscopic analysis [102], indicating its role in CBs localization of telomerase. TCAB1 was found to associate with TER by binding specifically to the CAB-box sequence (CR7 motif). Inhibition of TCAB1 by shRNA also reduced the amount of TER at telomeres during S phase of the cell cycle, resulting in telomere shortening. This data suggested that TCAB1 plays a role in controlling the access of telomerase complex to telomeres, representing an additional level of enzyme activity regulation [102] and see below).
3.9 Targeting Telomerase Holoenzyme to Telomere
Newly assembled, catalytically active telomerase enzyme must travel to and associate with the limited number of telomere ends for its proper function. As illustrated with the earlier discussion on TCAB, the Cajal Bodies were suggested as sites where the delivery of the active enzyme to the telomeres occurs [142–144]. TER is found localized at CBs in cancer cells throughout the cell cycle [101, 142]. Mutations in the CAB box motif decrease the accumulation of TER in CBs as well as the frequency of TER association with telomeres, resulting in shorter telomere length [101, 144]. Recent analysis of genetic lesions responsible for the rare autosomal recessive isoforms of dyskeratosis congenita (AR-DC) identified TCAB1 compound heterozygous mutations in a small subpopulation of AR-DC. While TER accumulations were within the normal range, telomerase RNA was found to accumulate at nucleoli instead of Cajal bodies. Mislocalization of the telomerase holoenzyme prevented telomere access leading to a loss of telomere length maintenance. This data identified TCAB1 as a critical telomerase regulation factor, which recruits the holoenzyme complex to Cajal bodies for proper telomere access and synthesis [145].
The presence of TERT was also found to be necessary for the localization and accumulation of TER in CBs as well as trafficking of telomerase to telomeres during S phase of the cell cycle [142, 146, 147]. However, outside of S phase, TERT resides in subnuclear foci, termed TERT foci [143], indicating that these two components are not transported to CBs as an assembled complex. Inhibition of TERT resulted in a decrease of TER colocalized with CBs and telomeres without affecting the levels of TER in cells. Additionally, expression of TERT in telomerase negative cells resulted in the accumulation of TER at both sites [146]. These observations again suggest that CB localization of telomerase is connected to enzyme biogenesis and catalytic activity in transformed cells.
hEst1A has also been suggested to play an important role in telomere maintenance in a manner similar to its yeast homologue Est1p. Yeast Est1p interacts with TLC (yeast TER) and the yeast telomere binding protein Cdc13, thereby recruiting telomerase to the proximity of the telomeres [148, 149]. Using in silico methods, three human homologs of yeast telomerase telomere-recruitment factor Est1p were identified. Of these three proteins, Est1A shows the highest sequence homology with ScEst1p [150]. Over-expression of Est1A reduced the steady state telomere length, but co-expression of TERT and Est1A increases telomere length substantially, suggesting that Est1A’s role in telomere length regulation is completely telomerase dependent. The human Est1p homologs have recently been implicated in TERRA and telomeric chromatin regulation [52, 151].
hnRNPs are also implicated in the localization of telomerase to telomeric ends for the de novo synthesis of telomeric repeats. In vitro studies demonstrated that hnRNPs A1/UP1, A2, A3, C1/C2, and D bind to TER and single-stranded telomeric DNA [153–156], suggesting possible roles in the bridging and recruitment of telomerase holoenzyme to the telomeres. In parallel, hnRNP A1/UP1 was found at telomere ends in vivo and was suggested to stimulate telomerase activity through the disruption of G-quadruplex structures formed during telomere synthesis by telomerase [156].
The human homolog of yeast DNA helicase Pif1 negatively influences the regulation of telomere length, by modulating telomerase activity [157]. hPif1 reduces telomerase processivity at the telomere by binding to and unwinding the DNA substrate and RNA template hybrid, resulting in the removal of telomerase from chromosome ends. hPif1 expression is regulated by cell cycle progression, peaking at late S/G2 [158]. Over-expression of hPIF1 induces telomere shortening in human HT1080 cells through telomerase activity modulation [157].
4 Telomerase Catalytic Cycle
TERT directs the addition of deoxynucleotide triphosphates (dNTPs) to the ends of the G-rich strand of the chromosome by copying the last six nucleotide of the 11-nt telomere repeat template sequence of TER [159, 160]. This activity results in the de novo synthesis of a single, 6 nt repeat. Because the TER RNA template region is quite short, to generate multiple repeats within a single catalytic event, telomerase holoenzyme undergoes multiple rounds of transient dissociation from the DNA substrate, to reposition the enzyme-substrate complex. Telomerase relies on its unique ability to transiently move away from the active site after the addition of a single 6-nt repeat, translocate towards the 3′ end of the newly synthesized chromosome and mediate the realignment of the new chromosome end with the TER RNA template, in order to continue subsequent rounds of multiple telomeric repeat addition (Fig. 5). Following the addition of each telomeric repeat, the enzyme may either disassociate from the chromosome end, stay bound without continuing elongation, or translocate and continue additional cycles of repeat addition [124, 161]. Translocation of the enzyme requires the DNA substrate to remain bound to telomerase. This interaction is mediated through an “anchor site” within the N-terminal domain of TERT (Fig. 4b) [125, 162].

Telomere repeat synthesis. Due to its short RNA template sequence, telomerase relies on two movement behaviors to add multiple 6-nuleotide (nt) telomeric sequences to chromosome ends. Addition of each 6-nt repeat to the 3′ end of the template is followed by telomerase translocation. This mediates realignment of the chromosome end from the 5′ end to the 3′ end of the template to enable subsequent rounds of repeat addition. Telomerase’s ability to carry out these two movement behaviors is termed nucleotide addition processivity and repeat addition processivity, respectively
5 Alternative Lengthening of Telomeres
Telomere maintenance can also be achieved by a process named alternative lengthening of telomeres (ALT) [163, 164]. ALT was discovered in 1995 when telomere elongation was observed in immortal human cells without detectable telomerase activity [165]. In yeast, this process involves either a rolling circle recombination mechanism or a strand exchange recombination mechanism. ALT is believed to occur by similar processes in humans [166], as it requires the activity of many homologous recombination protein factors including Rad50, MRE11, and NBS1 [167, 168].
One of the defining characteristics of ALT cells is the presence of a special class of promyelocytic leukemia (PML) bodies, known as the ALT-PML bodies [169]. ALT-PML bodies are microscopically defined, multi-protein domains in the nucleus that associate with telomeres in a cell cycle-specific manner [170, 171]. Observation of the ALT cell line U2OS revealed that TRF1 and FANCD2, a member of the Fanconi anemia protein family, colocalized with ALT-PML bodies at the same stages of the cell cycle. Monoubiquitination of FANCD2 is essential for this association, as depletion of FANCA, a member of the ubiquitination complex, or FANCL, the E3 ubiquitin-protein ligase, resulted in the loss of FANCD2 signals in ALT-PML bodies [172]. Depletion of FANCA or FANCD2 also resulted in an increase in telomere-signal-free chromosome ends in ALT cells. Due to the heterogeneity of telomere length in ALT cells, there were no significant changes in the average telomere length corresponding to these events. However, examination of newly synthesized telomere ends revealed that in the absence of FANCA or FANCD2 there is a significant decrease in sister chromatid exchange, supporting a role for monoubiquitination of FANCD2 in the ALT-mechanism of telomere maintenance through homologous recombination. Mutations in the chromatin remodeling proteins, the alpha thalassemia/mental retardation syndrome X-linked protein (ATRX) and death-associated protein 6 (DAXX) were found to associate with the ALT phenotype in a panel of pancreatic neuroendocrine tumors. Loss of ATRX-DAXX function is postulated to compromise heterochromatin states at telomeres, leading to the development of ALT by providing a permissive environment for nonreciprocal homologous recombination [173].
Although detected in human cells, ALT is not considered to be the normal physiological process for the maintenance of telomeres in humans. It has only been observed in a small number of human tumors (carcinoma and osteocarcinoma) and some transformed cell types in culture (mainly fibroblasts) [165, 174]. Long-term telomerase inhibition could potentially select tumor cells for ALT activation, as recently described in a mouse model of inducible TERT expression [175]. The under-representation of ALT-positive tumors was puzzling, until a 2002 study proved that the ALT mechanism cannot fully substitute telomerase in tumorigenesis: expression of the oncogenic H-Ras allele in the immortal human fibroblast ALT cell line GM00847 did not result in malignant transformation when injected into nude mice. In contrast, the co-expression of TERT in these cells imparted a tumorigenic phenotype [176]. This tumorigenic phenotype was again observed with the introduction of a mutant TERT, TERT-HA, which retains its enzymatic activity in vitro but is incapable of maintaining telomere length in vivo. Additionally, recombinant telomerase expression in ALT models accelerates cell growth and promotes anchorage-independent growth. Telomerase-positive ALT cells pass through cell-division phases of the cell cycle more quickly, implying that the observed cell-growth advantage is cumulative over cycles of proliferation [177]. The ALT recombination mechanism was not able to completely replace telomerase in the process of cellular transformation, implicating an additional, tumor growth-promoting role of TERT, independent of it role in telomere length maintenance.
6 Telomerase and Cancer
Most normal human somatic cells do not express detectable levels of telomerase activity as TERT expression is rapidly down-regulated following embryonic development [178]. In some human cell types, such as germline cells and stem cells, where there is a high demand for proliferation, TERT transcription is periodically activated to allow for transient expression of the enzyme. In contrast, more than 85% of human tumors surveyed harbor robust telomerase activity [179]. In almost all cases, the transcriptional up-regulation of TERT is responsible for the increase in ectopic telomerase activity in tumor cells [180]. Proof-of-concept experiments showed that the inhibition of telomerase in human cancer cells resulted in telomere-induced crisis and apoptosis in cell culture models [181, 182].
Telomerase expression is not considered to be oncogenic, as it alone does not lead to the development of cancer [183]. Additionally, it has been shown that TERT expression alone is not sufficient for the immortalization of human mammary epithelial cells, keratinocytes [184], prostate epithelial cells [185], or airway epithelial cells [186]. Cooperation between TERT and other oncogenic factors are essential for the transformed phenotype [187].
Paradoxically, early neoplastic lesions typically have undetectable or low telomerase activity, when compared to advanced malignant lesions that over-express the enzyme [188, 189]. This suggests that initiation of tumor development may require the absence of telomerase activity. Indeed, data from tumor cytogenetic studies have demonstrated that telomere length from precancerous lesions are much shorter than in normal tissues [12, 190, 191]. Several studies have reported critically short telomeres as a common early feature of many human cancers, such as colon [189], lung [192], breast [193], pancreatic [194], and prostate [195]. The telomere dysfunction model of carcinogenesis suggested that rampant chromosome instability following the uncapping of dysfunctional, short telomeres contributes to the eventual development of aneuploidy, a genetic signature of cellular transformation and carcinogenesis [57, 196]. Telomere dysfunction is thus recognized as a late event in the process of cancer initiation. After which, telomerase activity has to be induced to prevent further chromosome instability that hinder cancer growth, and provide a mechanism for the indefinite proliferation and immortality phenotype in malignant tumors [197] (Fig. 2).
7 NON-telomere Maintenance Roles of Telomerase
Besides its role in telomere maintenance, there is growing evidence pointing to telomerase’s additional role in the cancer biology. Higher mRNA levels of several DNA repair and chromatin modifying genes, as well as better double-stranded break repair kinetics, were observed in human foreskin fibroblast cells expressing TERT as compared to cells lacking ectopic expression of the enzyme [198]. Importantly, these effects occurred rapidly before any significant telomere lengthening was observed. A transcriptome study done by Smith and colleagues [199] demonstrated that the ectopic expression of telomerase in human mammary epithelial cells reduced the need for exogenous mitogens for cellular proliferation, correlating with the telomerase dependent induction of gene expression that promotes cell growth and survival. This latter study provided evidence for a role of telomerase in cellular proliferation by affecting the expression profiles of growth and survival-related genes. In corroboration of this model, TERT is shown to act as a transcriptional co-activator of the beta-catenin transcriptional complex in mice [200], a function that is independent of its reverse transcriptase activity and its association with the telomerase RNA [200, 201]. These data have been recently corroborated by Masutomi and Hahn’s model implicating human TERT in the promotion of TWIST expression and the resultant epithelial–mesenchymal transition. In conformity with the mouse models, TERT was found to complex with the BRG1, a SWI/SNF-related chromatin-remodeling factor, in transformed human cells. Distinct from mouse models, human BRG1-TERT complexes with additional nucleolar proteins nucleostemin (NS) and/or GNL3L [202].
Aside from transcriptional regulation, TERT activity is also implicated in optimal mitochondrial function, independently of TER [203, 204]. Although direct molecular proof of TERT’s functionality in the mitochondrion has not yet been established, TERT has intriguingly been shown to exhibit a RNA-dependent RNA polymerase activity when partnered with a mitochondrial RNA, RMRP [203]. DePinho’s group showed that a switch to the ALT mechanism of telomere maintenance in mouse T-cell lymphoma, through the inhibition of TERT expression, was accompanied by a specific induction of mitochondrial enzymes that reduce oxidative damage. This supports the hypothesis that TERT harbors mitochondrial functions, independent of TER [175].
In addition to transcription co-activator functions, constitutive TERT expression is also involved in enhanced DNA repair. Normal, diploid human fibroblasts over-expressing TERT were found to be more resistant to apoptosis and necrosis induced by DNA damages, but equally susceptible to the cytotoxic effects of oxidative agents as normal fibroblasts without TERT expression [205]. This suggested that telomerase is involved in enhancing cellular survival following genotoxic stress. Direct evidence implicating telomerase’s role as a regulator of the DNA damage response pathway was provided by a cell biology study [206]. By suppressing endogenous TERT expression in diploid human fibroblasts using either an TERT-coding sequence specific shRNA or an TERT 3′ untranslated region-specific shRNA (TERT 3′ UTR shRNA), it was shown that TERT participates in DNA damage responses and chromatin maintenance in a manner that is separate from its role in telomere length maintenance. Following ionizing radiation (IR), irinotecan, or etoposide treatment, phosphorylation of H2AX and the ataxia telangiectasia mutated (ATM) protein was greatly impaired in telomerase knock-down cells as compared to control cells expressing normal levels of TERT. As a direct consequence, the phosphorylation of BRCA1 tumor suppressor proteins was not observed and protein levels of p53 were not up-regulated. These results indicate impaired DNA damage responses in cells lacking TERT. Telomerase knock-down cells also exhibited increased sensitivity to IR as shown by the decreased relative survival in clonogenic growth assays. When wildtype recombinant TERT was introduced into cells expressing the TERT 3′ UTR shRNA, which does not target these recombinant copies of TERT, the cells ability to respond to DNA damage was restored. The molecular mechanism of how TERT perform this role to promote DNA damage survival remains unclear, but is suggested to be associated with TERT’s chromatin remodeling activities. In agreement with these data, our laboratory showed that transient telomerase inhibition synergistically increased the cytotoxicity of double-stranded DNA-damaging agents, in a cell-cycle phase-specific manner. This short-term telomerase inhibition was not predicted to significantly reduce telomere length, and the synergistic cellular toxicity may be ascribed to the inhibition of a non-telomere-related telomerase function in tumor cell growth [207].
8 Targeting Telomeres and Telomerase in Anticancer Chemotherapy
Uncapped telomeres induce a dramatic DDR response culminating in cell cycle arrest and programmed cell death [19, 56, 69]. While targeted disruptions of the telomere structure could have been a viable strategy in anticancer therapy, the therapeutic index would be extremely low, considering that the same DDR activation will be induced in cancer and normal cells alike. Conversely, the apparent lack of TERT expression in normal somatic cells, and the growing evidence for TERT’s additional roles in cancer biology, makes telomerase an ideal target for anticancer therapies. Telomerase is constitutively over-expressed in over 85% of all human cancers [179]. Early proof-of-principle experiments demonstrated that the expression of a dominant negative form of TERT completely inhibited telomerase activity and substantially reduced telomere length in several cancer models [180]. The resulting telomere dysfunctions led to the formation of dicentric chromosomes and other types of chromosome fusions, resulting in the loss of cellular viability and apoptosis. This inhibition of TERT was demonstrated to limit tumorigenicity of mouse xenograft models of cancer [181].
Following these proof-of-principle experiments, numerous strategies targeting the telomerase holoenzyme components are described. In the following sections, we discuss some of the more notable strategies of telomerase inhibition in targeted therapy against cancers.
9 Telomerase Catalytic Activity Inhibitors
9.1 BIBR1532
BIBR1532 is a small molecule, non-nucleoside inhibitor that interferes with telomeric DNA repeat addition by telomerase through the targeting of the catalytic component TERT [208]. Treatment of cancer cells with low doses BIBR1532 reduces their growth capacity and sensitizes them to other chemotherapeutic drugs, in a telomere length-dependent manner [209]. At high doses of BIBR1532, cells exhibited off target cytotoxic effects independent of telomerase’s catalytic function [210, 211]. Leukemia cells, but not normal hematopoietic stem cells, treated with 30–80 μM BIBR1532 displayed an immediate reduction in proliferative capacity. In particular, telomere dysfunctions are manifested as increases in telomere signal free ends, formation of chromosome end-to-end fusions, and an increase in phosphorylation of p53 and a loss of TRF2 signals at the telomere. However, BIBR1532 induced cytotoxic effects may not be confined to the formation of dysfunctional telomeres and this off-target effect hampers its further development as an anticancer therapeutic agent.
9.2 3′-Azido-2′, 3′-Dideoxythymidine (AZT)
AZT is a reverse transcriptase inhibitor used in the highly active antiretroviral therapy (HAART) against HIV infection and in the treatment of virus-associated cancers. As a thymidine analog, AZT has been shown to inhibit telomerase in vitro and in vivo. Upon its activation through phosphorylation by thymidine kinase, this nucleoside analog is incorporated into telomeric DNA as a chain terminator, blocking further reverse transcription and telomere elongation [212, 213]. Prolonged treatment of adult T-cell leukemic cells with AZT results in telomere attrition, accompanied by increased expression of p14ARF and activation of the p53-dependent apoptotic pathway [214]. This leads to an increase in the p53 target p21WAF expression and the accumulation of p27KIP, to induce cell cycle arrest or program cell death of the tumor cells. In combination with chemotherapy agents such as 5-fluorouracil, AZT has been shown to increase treatment toxicity in colorectal cancer cell model, most likely in a synergistic manner.
9.3 Oligonucleotide-Based Specific Inhibitors of Telomerase
Oligonucleotide-based inhibitors of telomerase designed to target the TER template may provide a highly specific, telomerase-based antitumor therapy [86, 215, 216]. GRN163L is a 13-base, lipid modified N3′-P5′ thiophosphoramidate oligomer, complementary to the template region of TER. GRN163L binds with high affinity to telomerase [217, 218] and has been demonstrated to effectively inhibit the enzyme, resulting in telomere length shortening and subsequent growth arrest. The 5′-lipid palmitoyl domain facilitates cellular and tissue penetration, as well as makes this agent more acid resistant than other anti-telomerase oligonucleotides, thereby increasing the cellular uptake and bioavailability of the drug [220]. GRN163L shows antitumor effects in several cancers, including breast, liver, lung, and multiple myeloma, both in vitro and in vivo [219–223]. This drug is currently undergoing clinical trials in patients with chronic lymphocytic leukemia, multiple myeloma, solid tumor malignancies, locally recurrent or metastatic breast cancer and advanced or metastatic non-small cell lung cancer [224].
10 Telomerase Expression, Biogenesis and Assembly Inhibitors
10.1 Costunolide
Costunolide is a sesquiterpene lactone isolated from Magnolia sieboldii. Reported to harbor anti-inflammatory, antifungal, and antiviral properties [225–228], it was also shown to suppress cell proliferation and induce apoptosis in several tumor cell lines, including breast cancer and leukemia cells [229, 230]. Costunolide exerts its anticancer properties through transcription regulation of TERT. A decrease in c-Myc or Sp1 binding to their cognate DNA binding sites on the TERT promoter was observed after costunolide treatment, in a dose-dependent manner [229]. Corresponding to the decrease in TERT mRNA levels, there is a reduction in telomerase activity resulting in an inhibition of cell growth and an increase in apoptosis.
10.2 Geldanamycin
Geldanamycin is a benzoquinone ansamycin antibiotic and inhibits the binding of cofactor ATP and partner p23 to the molecular chaperone Hsp90 [231, 232]. The Hsp90-p23 complex is a molecular chaperone that binds to and stabilizes cytoplasmic TERT at intermediate stages for folding, assembly and movement across nuclear membranes. Geldanamycin blocks the assembly of active telomerase both in vitro and in vivo [110] by disrupting the Hsp90-p23-telomerase interaction. Geldanamycin actions result in the ubiquitination and proteosome degradation of TERT and the reduction of telomerase activity [139]. However, since Hsp90 and p23 form chaperone complexes that have integral roles in numerous biological processes, geldanamycin mediated inhibition of Hsp90 function lacks specificity for the telomerase pathway. Given that many of the Hsp90-p23 binding partners are key players in cancer progression, such as v-Src, Bcr-Abl, Raf-1, and ErbB2 [233–236], geldanamycin promiscuous activities might be beneficial in anticancer chemotherapy. The utility of geldanamycin disruption of Hsp90-p23 formation should be revisited in specific cancer types, based on the molecular etiology of the disease.
11 Telomerase Immunotherapy
Telomerase is tested as a novel target for cancer immunotherapy. In telomerase-positive cancers, TERT peptides are presented as epitopes on the tumor cell surface by the major histocompatibility complex (MHC) class I pathway. TERT antigen presentation was demonstrated to produce cytotoxic T lymphocyte responses [237–239]. Two first-generation vaccines have been developed: GRNVAC1 and GV1001. Telomerase cancer vaccine, GRNVAC1, uses an ex vivo process where mature dendritic cells are isolated from the patient’s blood and transfected with TERT mRNA. These cells are then returned to the body where they stimulate the production of CD4+ and CD8+ T-cells specific for TERT [240]. GV1001 is a peptide vaccine derived from the active functional domain of telomerase. GV1001 binds multiple human leukocyte antigen (HLA) class II molecules and harbors putative HLA class I epitopes, and also illicit CD4+ and CD8+ T-cell responses specific for TERT [240]. Both vaccines were test successful in phase I/II clinical trials for efficacy in producing telomerase specific CD4+ and CD8+ T-lymphocytes [240, 241]. GV1001 is currently in two phase III clinical trials for the treatments of pancreatic cancer while GRNVAC1 is being investigated in a phase II clinical trial in patients with acute myeloid leukemia [242].
12 Telomerase-Telomere Recruitment Inhibitors
12.1 Tankyrase1 Inhibitors
Poly(ADP-ribose) polymerase (PARPs) is a large family of enzymes that use NAD+ as a substrate to generate ADP-ribose polymers onto glutamic acid residues on protein acceptors [243–245]. Tankyrase 1 and 2 are PARP family members specifically known for their telomeric poly (ADP-ribosyl) polymerase activities. Tankyrase 1 and 2 ribosylates TRF1, preventing TRF1 from binding to telomeric DNA, and leading to TRF1’s proteolytic degradation [246]. Over-expression of tankyrase 1 reduces TRF1 binding to the telomere, enables telomerase access at the telomere ends and the corresponding telomere elongation. Conversely, inhibition of tankyrase 1 induces telomere shortening and cell death through a telomere length independent mechanism: in the absence of tankyrase 1, cells undergoing mitosis are unable to resolve sister telomeres cohesion and were arrested at the mitotic phase [248, 249].
Small molecule inhibitors of tankyrase 1’s PARP activities have been shown to complement telomerase inhibition to enhance the rate of telomere attrition [249]. However, given PARPs are known to mediate the ribosylation of multiple protein acceptors, the likelihood of off-target effects by these small molecules PARP inhibitors is high.
12.2 G-Quadruplex Stabilizers
G-quadruplexes are stable 4-stranded DNA structures made up of G-rich sequences where the guanine residues form square arrangements. The 3′ telomeric DNA overhang is guanine rich and can form these higher order molecular structures, in addition to the normal telomeric DNA structures. Small molecule, non-nucleoside compounds such as telomestatin, BRACO-19, TMPyP4, and carbcynanine dyes, are predicted to bind within the grooves [250] or intercalate [251] G-quadruplex DNA, to stabilize these structures. Compounds that intercalate into the DNA to stabilize the G-quadruplex tend to have large, flat aromatic surfaces and are cationically charged to allow for π-stacking interactions. Examples of such molecules are porphyrins and cisplatin [252]. These older platinum containing complexes are shown to potently inhibit telomerase, leading to telomere shortening, arrested cell growth and subsequent cell death. Newer platinum (II) containing structures are also reported to inhibit telomerase in vitro, with distinct covalent linkage that could lock the G-quadruplex structure irreversibly [253].
Several different G-quadruplex inhibitors have been shown to disrupt the binding of telomere-associated proteins, inhibit telomerase activity and induce apoptosis in vitro [254–258]. However, as G-quadruplex binding agents, these compounds are predicted to bind elsewhere in the genome and disrupt their local structure, leading to altered functions. For example, telomestatin can bind to non-telomeric G-rich DNA found in the promoter region of the c-myc oncogene [259, 260]. Expression of myc is reduced by telomestatin binding, which stabilizing the G-quartet structure in its promoter and prevent transcription factor access. In addition to these off-target effects, another major problem with G-quadruplex stabilizer is their inability to penetrate the cell membrane. Optimal delivery protocol for these types of drugs has yet to be developed.
13 Genetic Therapy Against Telomerase
13.1 TER with Mutant Template
The expression of mutant-template human telomerase RNA (MT-TER), in telomerase positive cells, has been tested as an anticancer gene therapy. MT-TER assemble with endogenous TERT and the recombinant enzyme then erroneously adds DNA repeats with mutant sequence to chromosome ends. A few copies of mutant DNA repeats are enough to disrupt the binding of telomeric proteins. The resulting compromised telomere structure leads to a loss in cellular viability by inducing apoptosis [260–262]. Even though mutant TER is dominant over endogenously expressed wild-type TER, it can only be expressed at low levels, thereby limiting its cytotoxic efficiency in cancer cells. To overcome this deficiency, co-expression of siRNA against endogenous TER, as well as lentiviral expression of mutant TER, has proven to increase the therapeutic efficacy of MT-TER [262].
13.2 TERT-Promoter Driven Suicidal Gene Therapy
Based on the selective activation of the TERT promoter in cancer cells, several groups reported the use of recombinant DNA vectors, with TERT promoter driving the expression of cytotoxic transgenes, including the herpes simplex virus thymidine kinase, Bcl2-associating X protein, caspase 8 and bacterial nitro-reductase, delivering suicidal enzymatic activities in a cancer cell specific manner [263–271]. While these proof-of-principle experiments provided the framework for a cancer specific targeting strategy, more work is still needed for the development, delivery, and clinical validity of these cancer gene therapies.
13.3 Hammerhead Ribozyme
Hammerhead ribozymes targeting either the RNA component or reverse transcriptase component of telomerase are shown to be effective strategies in several cancer models. Colon and gastric carcinoma cells treated with retrovirus delivered ribozyme targeted TERT displayed a significant decrease in telomerase activity and rapid induction of apoptosis [272]. In endometrial and hepatocellular carcinoma cells, ribozyme targeting of TER resulted in a dose dependent decrease in telomerase activity [273, 274]. Up to 90% inhibition of telomerase activity could be achieved at relatively low concentrations of the ribozyme. As with other genetic means of telomerase activity inhibition, the current lack of efficient delivery protocols hamper their use in clinical settings.
13.4 Zinc Finger Proteins
Zinc Finger Proteins are synthetic peptides designed to target specific chromosomal loci and alter their functionality or sequence identity. Transcription activation of TERT in tumor cells relies on the activation at multiple transcription factor binding sites on TERT’s promoter, including that for SP1, c-MYC, ER, E2F-1, WT-1 and MZF-2 [105, 275, 276]. Conceivably, ZFP designed to target these chromosomal loci will interfere with TERT transcription activities. Recently, a ZFP that recognizes a 12 bp sequence within the core TERT promoter fused to a KRAB repressor domain has been described [277]. In vitro expression of this ZFP resulted in >80% reduction of TERT expression. Cancer cell lines engineered to express this ZFP are shown to have significantly lower endogenous TERT mRNA levels, a decrease in telomerase activity and inhibition of cell proliferation within 8–12 days. Longer-term repression of endogenous TERT transcription in human cancer cell lines expressing this ZFP in a stable fashion mirrored these results and displayed shortened telomeres. Despite the positive laboratory data, several issues such as ZFP’s treatment efficacy, target efficiency and specificity, as well as the availability of appropriate delivery protocols will need to be addressed before the adoption of these novel therapeutic options into clinical applications.
14 Therapeutic Considerations
14.1 Combination Chemotherapies
Despite the demonstrations of several successful strategies targeting telomeres and telomerase in cancer cells, their usefulness in the clinics has been marred by several deficiencies. The timeline of inducing cytotoxicity by telomerase inhibition relies completely on the kinetics of telomere shortening to a critically short length. As telomere length decreases at a rate of 50–100 bp per cell division, this process can be quite long, and tumor specific. This time lag can range from weeks to months of continual telomerase inhibition therapy. However, prolonged inhibition of the telomerase enzyme could affect normal human cells that are also dependent on transient telomerase activity for their functionality [61]. In these cases, telomere erosion in off-target cells from telomerase inhibition therapy could precipitate adverse treatment effects in these normal cell types. Premature telomere shortening translate to the accelerated rate of tissue aging. If these cells were allowed to divide beyond the short telomere check point, due to the inactivation of tumor suppressive mechanism, new rounds of chromosome instability cycles could trigger the development of secondary tumors. This paradox, in addition to the lack of proper delivery methods for genetic-based inhibition of TERT function, argues that telomerase inhibition on its own is not efficacious as an anticancer therapy.
On the other hand, telomerase inhibition has been demonstrated to increase the sensitivity to chemotherapeutic agents by overwhelming the DNA repair mechanism, with the creation of unprotected chromosome ends. For example, telomere dysfunction in late generation TERC−/− mice, lacking the mouse telomerase RNA gene, resulted in decreased cellular survival after exposure to IR [278]. At the cellular level, the rate of apoptosis in gastrointestinal crypt cells and primary thymocytes was higher in telomerase deficient mice as compared to control. These TER−/− cells also displayed delayed DNA break repair kinetics, as well as persistent chromosomal breaks, complex chromosomal aberrations and massive fragmentation.
Reduction of telomerase activity also resulted in increased cell sensitivity to topoisomerase inhibitors. The MCF-7 breast cancer cell line and HBL-100 immortal breast cell line expressing an anti-TERT ribozyme, which cleaves human telomerase mRNA, resulted in inhibition of telomerase activity, decreased telomere length and induced apoptosis. Additionally, an increased sensitivity to the topoisomerase II inhibitor doxorubicin was also observed in these cell lines. In parallel, when exogenous TERT was introduced into telomerase-negative human fibroblasts, there was a decrease in the sensitivity of these cell lines to doxorubicin, as well as two other topoisomerase inhibitors: mitoxantrone and etoposide [279].
Telomerase inhibition via the ectopic expression of dominant negative-TERT (DN-TERT) in human cancer cells resulted in telomere shortening, growth arrest and apoptosis [181, 182]. Expression of recombinant DN-TERT in BCR-ABL positive leukemia cells completely inhibited endogenous telomerase activity and resulted in an increase in apoptosis following treatment with the tyrosine kinase inhibitor imatinib [280].
Telomerase inhibition was also demonstrated to increase telomerase positive pharynx Fadu tumor cell’s sensitivity to paclitaxel [281]. Telomerase inhibition was achieved using either antisense TER, which blocks the template for telomere synthesis, or 3′-azido-3′deoxythymidine (AZT), a nucleoside analog reverse transcriptase inhibitor. The combination of AZT and paclitaxel resulted in decreased tumor size, increased apoptosis, and prolonged survival in FaDu xenograft tumor mice models. This effect was not observed in telomerase negative human osteocarcinoma Saos-2 cells, indicating that the increase in sensitivity to paclitaxel was due to telomerase inhibition [282].
Knockdown of telomerase activity in human cells can also be achieved via retroviral transfer of siRNA targeting TERT. These telomerase knockdown cells displayed increased sensitivity to IR and chemotherapeutic agents etoposide, bleomycin, and doxorubicin [283]. In addition, the combination therapy using the TERT siRNA increased the apoptotic effect of cisplatin, a platinum-based chemotherapeutic agent, on the hepatocellular cell line SMMC7721 in vitro and also greatly reduced SMMC7721 and HepG2 tumor growth in the mouse xenograft model as compared to cisplatin monotherapy [284].
In 2005, Ward and Autexier reported the effects of telomerase inhibition on drug resistant leukemia and breast cancer cells by the non-nucleosidic small molecule inhibitor BIBR1532, a proprietary formulation from Boehringer Ingelheim [285]. This drug impairs telomere elongation by affecting telomerase translocation or promoting the disassociation of the enzyme from the telomere end [208]. They observed an increase in chemotherapy sensitivity when drug resistant leukemia and breast cancer cells were concurrently treated with BIBR1532. Continuous BIBR1532 treatment was found to decrease the proliferative capacity of these cells. as the number of population doublings with BIBR1532 increased these cells are progressively sensitized cells to the chemotherapeutic agents. This observation suggested that the effects of BIBR1532 treatment were telomere length dependent [211].
Combination chemotherapy studies demonstrated synergistic effects of GRN163L in combination with ionizing radiation [286]. Enhanced radiation sensitivity by GRN163L application was observed following long-term (42 days) drug treatment, with no significant differences in short-term (2 and 9 days) and intermediate inhibition (20 days) [286]. Accordingly, this synergistic effect was attributed to the generation of critically short telomeres following long-term telomerase inhibition. With breast cancer models, previous studies have also demonstrated that GRN163L in combination with the microtubule stabilizing agent paclitaxel [287], and tratsuzumab, a monoclonal antibody against the HER-2 receptor [288], has synergistic treatment effects, in a telomere-length dependent manner.
Combination studies have provided genetic and biological evidence linking telomere dysfunction and increased sensitivity to chemotherapeutic agents, making telomerase inhibition an effective therapeutic option for many different types of cancers. Many of these studies concluded that the observed increase in sensitivity of cancer cells to cytotoxic agents was telomere length dependent [209, 222, 286]. However, telomere shortening caused by the continuous inhibition of telomerase may affect normal human cell types that also require telomerase activity for growth and proliferation [61]. Recent data from our laboratory demonstrated that a transient inhibition of telomerase activity, at the time of the induction of DNA damage, also elicit a synergistic cytotoxicity response in breast and colon cancer cells. This potentiation of cytotoxicity is dependent on the timing and mode of action of the genotoxic agents, as only S/G2 specific DNA damage inducers are observed with increased cytotoxicity in combination with telomerase inhibition [207]. Even though the exact mechanism by which telomerase inhibition increases cellular toxicity in this manner, independent of telomere length, is not known, our work also demonstrated that inhibiting the ATM kinase, in conjunction with telomerase inhibition, synergistically increases the cytotoxicity of these S/G2 specific double-stranded DNA-damaging agents, suggesting a role for telomerase in DNA repair [207].
14.2 Transient Telomerase Activation as a Genome Maintenance Mechanism
Higher telomere attrition rates are often seen with chronic inflammation [289]. Accelerated telomere shortening in these conditions is associated with increased cellular turnover, leading to premature loss of tissue renewal capacity, and an increased risk of genomic instability. Proof-of-principle experiments showed that telomerase activation extends the replication lifespan of tissues with high turnover [290, 291]. Spurred by these early reports, telomerase activation strategies using small-molecule transcription activators are being heavily pursued. One of these agents, TAT2 (cycloastragenol) is extracted from the root of Astragalus membranaceus, a flowering plant used extensively in traditional Chinese herbal medicine [292]. TAT2 has been shown to activate telomerase in cell culture models, through the induction of TERT transcription. Short-term TAT2 treatment (12–18 days) improves the proliferative capacity of CD8+ T-lymphocytes from HIV-infected individuals by moderately increasing telomerase activity. By delaying the onset of immunosenescence of these T-cell models, TAT2 treatment increases the cytokine/chemokine production and antiviral (HIV) activity of T-cells in vitro. This effect is blocked by the addition of a specific telomerase inhibitor, GRN163L, confirming that the improvement of immune function by TAT2 is mediated by telomerase activation [291].
There is a strong negative correlation between mean telomere length and chronological age in humans [293, 294]. Conceivably, strategies to stimulate telomerase-dependent telomere maintenance in later life not only will contribute to boost tissue renewal capacity but will also help preserve the stability and integrity of the genome. This protection against genomic alterations which are frequently associated with cancers could be invaluable to older individuals, as age is one of the biggest risk factors for cancer [197]. Several studies have been initiated to test this. In one such study, TA-65, a compound related to TAT2, also isolated from Astragalus membranaceus, is being given as one of the active components of a dietary supplement. Other active ingredients include standard vitamins and trace minerals. An interim (1-year) report of this study revealed moderate improvement in participants’ immune system profiles with a continuous regimen of TA-65 in low doses [295]. The long-term utility of these strategies in health promotion including an improved tissue renewal capacity and cancer prevention, as well as the off-target/untoward effects of such therapies require further investigation.
14.3 Concluding Remarks
The integrity of telomere function has a paramount role in promoting chromosome stability. Loss of telomere function is implicated in the replicative aging of human tissues, and also has a major effect on cellular transformation related to carcinogenesis [1, 197]. The relationship between telomere structural maintenance and DDR pathways is an illustration of functional adaptation. Telomeres exist to protect the ends of chromosomes from being recognized by DDR sensors and undergoing erroneous repair by DDR mechanisms [7, 19, 69]. Yet, normal homeostatic maintenance of this nucleoprotein structure relies on many of the same DDR factors that need to be kept in check [78, 79]. How do these simple 6nt DNA repeats and their protein-binding partners accomplish these conflicting tasks? What are the regulatory/signal transduction events that allow the same DDR machinery to adapt its functions for the specific requirement of maintaining telomeres?
Interindividual variations in telomere maintenance capacity are an understudied area of telomere biology [289]. TERT A1062T non-synonymous single nucleotide polymorphism (SNP) has recently been discovered to associate, with a high prevalence, to acute myeloid leukemia (AML). Patients diagnosed with AML had three times higher prevalence of the 1062T-TERT isoforms compared to controls. 1062T-TERT was found to exhibit decreased telomerase activity compared to wild type (1062A) [296]. Conceivably, a decreased telomere maintenance capacity could accelerate the rate of telomeric DNA loss leading to the premature exhaustion of replicative cell pools and the precipitation of genomic instability. With increased availability of data from large-scale epidemiology studies, it is expected that genetic variations in telomerase and other telomere pathway components could also be named as risk factors in other types of malignancies, as well as idiosyncratic and orphaned tissue failure syndromes [289].
To maintain an immortal phenotype, cancer cells need to replenish lost telomere repeats. Accordingly, high telomerase activity is observed in more than 85% of all human cancers [179]. Even though telomerase inhibition alone has limited clinical efficacy as an anticancer treatment, chemotherapy regimens targeting telomerase, when combined with other cytotoxic stress, are reported to be effective against multiple types of malignancies. Several Phase I and II clinical studies with non-small cell lung cancer and breast cancer patients are currently underway [297, 298]. The results of these trials may provide new clinical anticancer strategies.
References
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
You Z, Bailis JM (2010) DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol 20:402–409
Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S (2000) The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 19:463–471
Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 10:886–895
de Lange T (2002) Protection of mammalian telomeres. Oncogene 21:532–540
de Lange T (2004) T-loops and the origin of telomeres. Nat Rev Mol Cell Biol 5:323–329
de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19:2100–2110
Blackburn EH (2001) Switching and signaling at the telomere. Cell 106:661–673
Chan CS, Tye BK (1983) Organization of DNA sequences and replication origins at yeast telomeres. Cell 33:563–573
Kipling D, Cooke HJ (1990) Hypervariable ultra-long telomeres in mice. Nature 347:400–402
Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345:458–460
de Lange T, Shiue L, Myers RM, Cox DR, Naylor SL, Killery AM et al (1990) Structure and variability of human chromosome ends. Mol Cell Biol 10:518–527
Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD et al (1988) A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA 85:6622–6626
Makarov VL, Hirose Y, Langmore JP (1997) Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 88:657–666
McElligott R, Wellinger RJ (1997) The terminal DNA structure of mammalian chromosomes. EMBO J 16:3705–3714
Greider CW (1999) Telomeres do D-loop-T-loop. Cell 97:419–422
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H et al (1999) Mammalian telomeres end in a large duplex loop. Cell 97:503–514
Klobutcher LA, Swanton MT, Donini P, Prescott DM (1981) All gene-sized DNA molecules in four species of hypotrichs have the same terminal sequence and an unusual 3′ terminus. Proc Natl Acad Sci USA 78:3015–3019
Palm W, de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42:301–334
Bianchi A, Stansel RM, Fairall L, Griffith JD, Rhodes D, de Lange T (1999) TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J 18:5735–5744
Court R, Chapman L, Fairall L, Rhodes D (2005) How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep 6:39–45
Shay JW (1999) At the end of the millennium, a view of the end. Nat Genet 23:382–383
van Steensel B, de Lange T (1997) Control of telomere length by the human telomeric protein TRF1. Nature 385:740–743
Smogorzewska A, van Steensel B, Bianchi A, Oelmann S, Schaefer MR, Schnapp G et al (2000) Control of human telomere length by TRF1 and TRF2. Mol Cell Biol 20:1659–1668
Broccoli D, Smogorzewska A, Chong L, de Lange T (1997) Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet 17:231–235
Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T (1999) p53- and ATM- dependent apoptosis induced by telomeres lacking TRF2. Science 283:1321–1325
Stansel RM, de Lange T, Griffith JD (2001) T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J 20:5532–5540
Karlseder J, Smogorzewska A, de Lange T (2002) Senescence induced by altered telomere state, not telomere loss. Science 295:2446–2449
Lei M, Podell ER, Cech TR (2004) Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol 11:1223–1229
Loayza D, Parsons H, Donigian J, Hoke K, de Lange T (2004) DNA binding features of human POT1: a nonamer 5′-TAGGGTTAG-3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J Biol Chem 279:13241–13248
Flynn RL, Centore RC, O’Sullivan RJ, Rai R, Tse A, Songyang Z et al (2011) TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471:532–536
Loayza D, De Lange T (2003) POT1 as a terminal transducer of TRF1 telomere length control. Nature 423:1013–1018
Colgin LM, Baran K, Baumann P, Cech TR, Reddel RR (2003) Human POT1 facilitates telomere elongation by telomerase. Curr Biol 13:942–946
Armbruster BN, Linardic CM, Veldman T, Bansal NP, Downie DL, Counter CM (2004) Rescue of an hTERT mutant defective in telomere elongation by fusion with hPot1. Mol Cell Biol 24:3552–3561
Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR et al (2007) The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 445:506–510
Xin H, Liu D, Wan M, Safari A, Kim H, Sun W et al (2007) TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 445:559–562
O’Connor MS, Safari A, Xin H, Liu D, Songyang Z (2006) A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc Natl Acad Sci USA 103: 11874–11879
Kim SH, Kaminker P, Campisi J (1999) TIN2, a new regulator of telomere length in human cells. Nat Genet 23:405–412
Liu D, O’Connor MS, Qin J, Songyang Z (2004) Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem 279:51338–51342
Ye JZ, de Lange T (2004) TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 36:618–623
Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP (2008) TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82:501–509
Li B, Oestreich S, de Lange T (2000) Identification of human Rap1: implications for telomere evolution. Cell 101:471–483
Li B, de Lange T (2003) Rap1 affects the length and heterogeneity of human telomeres. Mol Biol Cell 14:5060–5068
Martinez P, Blasco MA (2011) Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer 11:161–176
Surovtseva YV, Churikov D, Boltz KA, Song X, Lamb JC, Warrington R et al (2009) Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell 36:207–218
Wellinger RJ, The CST (2009) complex and telomere maintenance: the exception becomes the rule. Mol Cell 36:168–169
Miyake Y, Nakamura M, Nabetani A, Shimamura S, Tamura M, Yonehara S et al (2009) RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Mol Cell 36:193–206
Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M et al (2006) DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8:416–424
Garcia-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA (2004) Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet 36:94–99
Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8: 299–309
Redon S, Reichenbach P, Lingner J (2010) The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res 38:5797–5806
Luke B, Lingner J (2009) TERRA: telomeric repeat-containing RNA. EMBO J 28:2503–2510
Feuerhahn S, Iglesias N, Panza A, Porro A, Lingner J (2010) TERRA biogenesis, turnover and implications for function. FEBS Lett 584:3812–3818
Olovnikov AM (1973) A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol 41:181–190
Smogorzewska A, de Lange T (2002) Different telomere damage signaling pathways in human and mouse cells. EMBO J 21:4338–4348
Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13:1549–1556
Wright WE, Shay JW (1992) The two-stage mechanism controlling cellular senescence and immortalization. Exp Gerontol 27:383–389
Hayflick L (1965) The limited in vitro lifetime of human diploid strains. Exptl Cell Res 37:614–636
Effros RB, Pawelec G (1997) Replicative senescence of T cells: does the Hayflick limit lead to immune exhaustion? Immunol Today 18:450–454
Shay JW, Wright WE (2000) Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol 1:72–76
Fleisig HB, Wong JM (2007) Telomerase as a clinical target: current strategies and potential applications. Exp Gerontol 42:102–112
Rajaraman S, Choi J, Cheung P, Beaudry V, Moore H, Artandi SE (2007) Telomere uncapping in progenitor cells with critical telomere shortening is coupled to S-phase progression in vivo. Proc Natl Acad Sci USA 104:17747–17752
Wright WE, Shay JW (1992) Telomere positional effects and the regulation of cellular senescence. Trends Genet 8:193–197
Chan SS, Chang S (2010) Defending the end zone: studying the players involved in protecting chromosome ends. FEBS Lett 584:3773–3778
Tejera AM, Alcontres M, Thanasoula M, Marion RM, Martinez P, Liao C et al (2010) TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev Cell 18:775–789
Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T (2010) Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 327:1657–1661
Hockemeyer D, Daniels JP, Takai H, de Lange T (2006) Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell 126:63–77
Hockemeyer D, Sfeir AJ, Shay JW, Wright WE, de Lange T (2005) POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J 24:2667–2678
Denchi EL, de Lange T (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448:1068–1071
Lavin MF (2008) Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 9:759–769
He H, Wang Y, Guo X, Ramchandani S, Ma J, Shen MF et al (2009) Pot1b deletion and telomerase haploinsufficiency in mice initiate an ATR-dependent DNA damage response and elicit phenotypes resembling dyskeratosis congenita. Mol Cell Biol 29:229–240
Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9:616–627
Pandita TK (2001) The role of ATM in telomere structure and function. Radiat Res 156: 642–647
Pandita TK (2002) ATM function and telomere stability. Oncogene 21:611–618
Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI et al (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64:9152–9159
Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D et al (2004) Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet 36:877–882
Crabbe L, Jauch A, Naeger CM, Holtgreve-Grez H, Karlseder J (2007) Telomere dysfunction as a cause of genomic instability in Werner syndrome. Proc Natl Acad Sci USA 104:2205–2210
Verdun RE, Crabbe L, Haggblom C, Karlseder J (2005) Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell 20:551–561
Verdun RE, Karlseder J (2006) The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127:709–720
Mitchell JR, Cheng J, Collins K (1999) A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol Cell Biol 19:567–576
Dragon F, Pogacic V, Filipowicz W (2000) In vitro assembly of human H/ACA small nucleolar RNPs reveals unique features of U17 and telomerase RNAs. Mol Cell Biol 20:3037–3048
Wang C, Meier UT (2004) Architecture and assembly of mammalian H/ACA small nucleolar and telomerase ribonucleoproteins. EMBO J 23:1857–1867
Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE et al (1997) Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet 17:498–502
Harrington L (2003) Biochemical aspects of telomerase function. Cancer Lett 194:139–154
Wong JM, Kusdra L, Collins K (2002) Subnuclear shuttling of human telomerase induced by transformation and DNA damage. Nat Cell Biol 4:731–736
Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP et al (1995) The RNA component of human telomerase. Science 269:1236–1241
Zaug AJ, Linger J, Cech TR (1996) Method for determining RNA 3′ ends and application to human telomerase RNA. Nucleic Acids Res 24:532–533
Zhao JQ, Hoare SF, McFarlane R, Muir S, Parkinson EK, Black DM et al (1998) Cloning and characterization of human and mouse telomerase RNA gene promoter sequences. Oncogene 16:1345–1350
Fu D, Collins K (2003) Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol Cell 11:1361–1372
Chen JL, Blasco MA, Greider CW (2000) Secondary structure of vertebrate telomerase RNA. Cell 100:503–514
Dez C, Henras A, Faucon B, Lafontaine DLJ, Caizergues-Ferrer M, Henry Y (2001) Stable expression in yeast of the mature form of human telomerase RNA depends of its association with the box H/ACA small nucleolar RNP proteins Cbf5p, Nhp2p and Nop10p. Nucleic Acids Res 29:598–603
Cohen SB, Graham ME, Lovrecz GO, Bache N, Robinson PJ, Reddel RR (2007) Protein composition of catalytically active human telomerase from immortal cells. Science 315: 1850–1853
Fu D, Collins K (2007) Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol Cell 28:773–785
Collins K, Mitchell JR (2002) Telomerase in the human organism. Oncogene 21:564–579
Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ et al (1998) X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet 19:32–38
Mitchell JR, Wood E, Collins K (1999) A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402:551–555
Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ et al (2001) The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413:432–435
Wong JM, Collins K (2003) Telomere maintenance and disease. Lancet 362:983–988
Le S, Sternglanz R, Greider CW (2000) Identification of two RNA-binding proteins associated with human telomerase RNA. Mol Biol Cell 11:999–1010
Fu D, Collins K (2006) Human telomerase and Cajal body ribonucleoproteins share a unique specificity of Sm protein association. Genes Dev 20:531–536
Jady BE, Bertrand E, Kiss T (2004) Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J Cell Biol 164:647–652
Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD et al (2009) A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323:644–648
Cong YS, Wen J, Bacchetti S (1999) The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet 8:137–142
Wick M, Zubov D, Hagen G (1999) Genomic organization and promoter characterization of the gene encoding the human telomerase reverse transcriptase (hTERT). Gene 232:97–106
Horikawa I, Cable PL, Afshari C, Barrett JC (1999) Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res 59:826–830
Wu KJ, Grandori C, Amacker M, Simon-Vermot N, Polack A, Lingner J et al (1999) Direct activation of TERT transcription by c-MYC. Nat Genet 21:220–224
Yi X, White DM, Aisner DL, Baur JA, Wright WE, Shay JW (2000) An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2:433–440
Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD et al (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90:785–795
Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A et al (1999) Human endothelial cell life extension by telomerase expression. J Biol Chem 274:26141–26148
Holt SE, Aisner DL, Baur J, Tesmer VM, Dy M, Ouellette M et al (1999) Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev 13:817–826
Kelleher C, Teixeira MT, Forstemann K, Lingner J (2002) Telomerase: biochemical considerations for enzyme and substrate. Trends Biochem Sci 27:572–579
Xiong Y, Eickbush TH (1990) Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J 9:3353–3362
Harrington L, Zhou W, McPhail T, Oulton R, Yeung DS, Mar V et al (1997) Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev 11:3109–3115
Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276:561–567
Beattie TL, Zhou W, Robinson MO, Harrington L (1998) Reconstitution of human telomerase activity in vitro. Curr Biol 8:177–180
Nakayama J, Tahara H, Tahara E, Saito M, Ito K, Nakamura H et al (1998) Telomerase activation by hTRT in human normal fibroblasts and hepatocellular carcinomas. Nat Genet 18:65–68
Xia J, Peng Y, Mian IS, Lue NF (2000) Identification of functionally important domains in the N-terminal region of telomerase reverse transcriptase. Mol Cell Biol 20:5196–5207
Bryan TM, Goodrich KJ, Cech TR (2000) A mutant of Tetrahymena telomerase reverse transcriptase with increased processivity. J Biol Chem 275:24199–24207
Bosoy D, Peng Y, Mian IS, Lue NF (2003) Conserved N-terminal motifs of telomerase reverse transcriptase required for ribonucleoprotein assembly in vivo. J Biol Chem 278: 3882–3890
Lai CK, Mitchell JR, Collins K (2001) RNA binding domain of telomerase reverse transcriptase. Mol Cell Biol 21:990–1000
Moriarty TJ, Huard S, Dupuis S, Autexier C (2002) Functional multimerization of human telomerase requires an RNA interaction domain in the N terminus of the catalytic subunit. Mol Cell Biol 22:1253–1265
Lue NF (2004) Adding to the ends: what makes telomerase processive and how important is it? Bioessays 26:955–962
Armbruster BN, Banik SS, Guo C, Smith AC, Counter CM (2001) N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol Cell Biol 21: 7775–7786
Lee SR, Wong JM, Collins K (2003) Human telomerase reverse transcriptase motifs required for elongation of a telomeric substrate. J Biol Chem 278:52531–52536
Moriarty TJ, Ward RJ, Taboski MA, Autexier C (2005) An anchor site-type defect in human telomerase that disrupts telomere length maintenance and cellular immortalization. Mol Biol Cell 16:3152–3161
Bachand F, Autexier C (2001) Functional regions of human telomerase reverse transcriptase and human telomerase RNA required for telomerase activity and RNA-protein interactions. Mol Cell Biol 21:1888–1897
Hossain S, Singh S, Lue NF (2002) Functional analysis of the C-terminal extension of telomerase reverse transcriptase. A putative “thumb” domain. J Biol Chem 277:36174–36180
Huard S, Moriarty TJ, Autexier C (2003) The C terminus of the human telomerase reverse transcriptase is a determinant of enzyme processivity. Nucleic Acids Res 31:4059–4070
Aitken A (2006) 14-3-3 proteins: a historic overview. Semin Cancer Biol 16:162–172
Khurts S, Masutomi K, Delgermaa L, Arai K, Oishi N, Mizuno H et al (2004) Nucleolin interacts with telomerase. J Biol Chem 279:51508–51515
Zhou XZ, Lu KP (2001) The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell 107:347–359
Banik SS, Counter CM (2004) Characterization of interactions between PinX1 and human telomerase subunits hTERT and hTR. J Biol Chem 279:51745–51748
Li H, Zhao L, Yang Z, Funder JW, Liu JP (1998) Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem 273:33436–33442
Kang SS, Kwon T, Kwon DY, Do SI (1999) Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 274:13085–13090
Breitschopf K, Zeiher AM, Dimmeler S (2001) Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett 493:21–25
Haendeler J, Hoffmann J, Rahman S, Zeiher AM, Dimmeler S (2003) Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett 536:180–186
Li H, Zhao LL, Funder JW, Liu JP (1997) Protein phosphatase 2A inhibits nuclear telomerase activity in human breast cancer cells. J Biol Chem 272:16729–16732
Kharbanda S, Kumar V, Dhar S, Pandey P, Chen C, Majumder P et al (2000) Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol 10: 568–575
Kim JH, Park SM, Kang MR, Oh SY, Lee TH, Muller MT et al (2005) Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes Dev 19:776–781
Huber O, Menard L, Haurie V, Nicou A, Taras D, Rosenbaum J (2008) Pontin and reptin, two related ATPases with multiple roles in cancer. Cancer Res 68:6873–6876
Venteicher AS, Meng Z, Mason PJ, Veenstra TD, Artandi SE (2008) Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132: 945–957
Zhu Y, Tomlinson RL, Lukowiak AA, Terns RM, Terns MP (2004) Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol Biol Cell 15:81–90
Tomlinson RL, Ziegler TD, Supakorndej T, Terns RM, Terns MP (2006) Cell cycle-regulated trafficking of human telomerase to telomeres. Mol Biol Cell 17:955–965
Cristofari G, Adolf E, Reichenbach P, Sikora K, Terns RM, Terns MP et al (2007) Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol Cell 27:882–889
Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T et al (2011) Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev 25:11–16
Tomlinson RL, Abreu EB, Ziegler T, Ly H, Counter CM, Terns RM et al (2008) Telomerase reverse transcriptase is required for the localization of telomerase RNA to cajal bodies and telomeres in human cancer cells. Mol Biol Cell 19:3793–3800
Jady BE, Richard P, Bertrand E, Kiss T (2006) Cell cycle-dependent recruitment of telomerase RNA and Cajal bodies to human telomeres. Mol Biol Cell 17:944–954
Pennock E, Buckley K, Lundblad V (2001) Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 104:387–396
Evans SK, Lundblad V (2002) The Est1 subunit of Saccharomyces cerevisiae telomerase makes multiple contributions to telomere length maintenance. Genetics 162:1101–1115
Snow BE, Erdmann N, Cruickshank J, Goldman H, Gill RM, Robinson MO et al (2003) Functional conservation of the telomerase protein Est1p in humans. Curr Biol 13:698–704
Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J (2007) Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318:798–801
Ding J, Hayashi MK, Zhang Y, Manche L, Krainer AR, Xu R (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev 13:1102–1115
Eversole A, Maizels N (2000) In vitro properties of the conserved mammalian protein hnRNP D suggest a role in telomere maintenance. Mol Cell Biol 20:5425–5432
Ford LP, Suh JM, Wright WE, Shay JW (2000) Heterogeneous nuclear ribonucleoproteins C1 and C2 associate with the RNA component of human telomerase. Mol Cell Biol 20:9084–9091
Fiset S, Chabot B (2001) hnRNP A1 may interact simultaneously with telomeric DNA and the human telomerase RNA in vitro. Nucleic Acids Res 29:2268–2275
Dallaire F, Dupuis S, Fiset S, Chabot B (2000) Heterogeneous nuclear ribonucleoprotein A1 and UP1 protect mammalian telomeric repeats and modulate telomere replication in vitro. J Biol Chem 275:14509–14516
Zhang DH, Zhou B, Huang Y, Xu LX, Zhou JQ (2006) The human Pif1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res 34: 1393–1404
Mateyak MK, Zakian VA (2006) Human PIF helicase is cell cycle regulated and associates with telomerase. Cell Cycle 5:2796–2804
Shippen-Lentz D, Blackburn EH (1990) Functional evidence for an RNA template in telomerase. Science 247:546–552
Blackburn EH (1992) Telomerases. Annu Rev Biochem 61:113–129
Fulton TB, Blackburn EH (1998) Identification of Kluyveromyces lactis telomerase: discontinuous synthesis along the 30-nucleotide-long templating domain. Mol Cell Biol 18:4961–4970
Wyatt HD, Lobb DA, Beattie TL (2007) Characterization of physical and functional anchor site interactions in human telomerase. Mol Cell Biol 27:3226–3240
Reddel RR, Bryan TM, Colgin LM, Perrem KT, Yeager TR (2001) Alternative lengthening of telomeres in human cells. Radiat Res 155:194–200
Henson JD, Neumann AA, Yeager TR, Reddel RR (2002) Alternative lengthening of telomeres in mammalian cells. Oncogene 21:598–610
Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR (1995) Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 14:4240–4248
Cerone MA, Londono-Vallejo JA, Bacchetti S (2001) Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum Mol Genet 10:1945–1952
Bechter OE, Zou Y, Shay JW, Wright WE (2003) Homologous recombination in human telomerase-positive and ALT cells occurs with the same frequency. EMBO Rep 4:1138–1143
Jiang WQ, Zhong ZH, Henson JD, Neumann AA, Chang AC, Reddel RR (2005) Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol Cell Biol 25:2708–2721
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR (1999) Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res 59:4175–4179
Grobelny JV, Godwin AK, Broccoli D (2000) ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci 113(Pt 24):4577–4585
Wu G, Lee WH, Chen PL (2000) NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G2 phases in immortalized telomerase-negative cells. Implication of NBS1 in alternative lengthening of telomeres. J Biol Chem 275:30618–30622
Fan Q, Zhang F, Barrett B, Ren K, Andreassen PR (2009) A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res 37:1740–1754
Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C et al (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333:425
Heaphy CM, Subhawong AP, Hong SM, Goggins MG, Montgomery EA, Gabrielson E et al (2011) Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Pathol 179:1608–1615
Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M et al (2012) Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148:651–663
Stewart SA, Hahn WC, O’Connor BF, Banner EN, Lundberg AS, Modha P et al (2002) Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc Natl Acad Sci USA 99:12606–12611
Fleisig HB, Wong JM (2012) Telomerase promotes efficient cell cycle kinetics and confers growth advantage to telomerase-negative transformed human cells. Oncogene 31: 954–965
Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW (1996) Telomerase activity in human germline and embryonic tissues and cells. Dev Genet 18:173–179
Shay JW, Bacchetti S (1997) A survey of telomerase activity in human cancer. Eur J Cancer 33:787–791
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL et al (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266: 2011–2015
Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A et al (1999) Inhibition of telomerase limits the growth of human cancer cells. Nat Med 5:1164–1170
Zhang X, Mar V, Zhou W, Harrington L, Robinson MO (1999) Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev 13:2388–2399
Harley CB (2002) Telomerase is not an oncogene. Oncogene 21:494–502
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ (1998) Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 396:84–88
Berger R, Febbo PG, Majumder PK, Zhao JJ, Mukherjee S, Signoretti S et al (2004) Androgen-induced differentiation and tumorigenicity of human prostate epithelial cells. Cancer Res 64:8867–8875
Lundberg AS, Randell SH, Stewart SA, Elenbaas B, Hartwell KA, Brooks MW et al (2002) Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 21:4577–4586
Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA (1999) Creation of human tumour cells with defined genetic elements. Nature 400:464–468
Chadeneau C, Hay K, Hirte HW, Gallinger S, Bacchetti S (1995) Telomerase activity associated with acquisition of malignancy in human colorectal cancer. Cancer Res 55: 2533–2536
Engelhardt M, Drullinsky P, Guillem J, Moore MA (1997) Telomerase and telomere length in the development and progression of premalignant lesions to colorectal cancer. Clin Cancer Res 3:1931–1941
Miura N, Horikawa I, Nishimoto A, Ohmura H, Ito H, Hirohashi S et al (1997) Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet Cytogenet 93:56–62
Rudolph KL, Millard M, Bosenberg MW, DePinho RA (2001) Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet 28:155–159
Lantuejoul S, Soria JC, Morat L, Lorimier P, Moro-Sibilot D, Sabatier L et al (2005) Telomere shortening and telomerase reverse transcriptase expression in preinvasive bronchial lesions. Clin Cancer Res 11:2074–2082
Meeker AK, Hicks JL, Gabrielson E, Strauss WM, De Marzo AM, Argani P (2004) Telomere shortening occurs in subsets of normal breast epithelium as well as in situ and invasive carcinoma. Am J Pathol 164:925–935
van Heek NT, Meeker AK, Kern SE, Yeo CJ, Lillemoe KD, Cameron JL et al (2002) Telomere shortening is nearly universal in pancreatic intraepithelial neoplasia. Am J Pathol 161: 1541–1547
Meeker AK, Hicks JL, Platz EA, March GE, Bennett CJ, Delannoy MJ et al (2002) Telomere shortening is an early somatic DNA alteration in human prostate tumorigenesis. Cancer Res 62:6405–6409
Gisselsson D, Jonson T, Petersen A, Strombeck B, Dal Cin P, Hoglund M et al (2001) Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci USA 98:12683–12688
DePinho RA (2000) The age of cancer. Nature 408:248–254
Sharma GG, Gupta A, Wang H, Scherthan H, Dhar S, Gandhi V et al (2003) hTERT associates with human telomeres and enhances genomic stability and DNA repair. Oncogene 22:131–146
Smith LL, Coller HA, Roberts JM (2003) Telomerase modulates expression of growth-controlling genes and enhances cell proliferation. Nat Cell Biol 5:474–479
Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M et al (2009) Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460:66–72
Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W et al (2008) TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet 4:e10
Okamoto N, Yasukawa M, Nguyen C, Kasim V, Maida Y, Possemato R et al (2011) Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc Natl Acad Sci USA 108:20388–20393
Maida Y, Yasukawa M, Furuuchi M, Lassmann T, Possemato R, Okamoto N et al (2009) An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 461:230–235
Rosenbluh J, Nijhawan D, Chen Z, Wong KK, Masutomi K, Hahn WC (2011) RMRP is a non-coding RNA essential for early murine development. PLoS One 6:e26270
Gorbunova V, Seluanov A, Pereira-Smith OM (2002) Expression of human telomerase (hTERT) does not prevent stress-induced senescence in normal human fibroblasts but protects the cells from stress-induced apoptosis and necrosis. J Biol Chem 277:38540–38549
Masutomi K, Possemato R, Wong JM, Currier JL, Tothova Z, Manola JB et al (2005) The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci USA 102:8222–8227
Tamakawa RA, Fleisig HB, Wong JM (2010) Telomerase inhibition potentiates the effects of genotoxic agents in breast and colorectal cancer cells in a cell cycle-specific manner. Cancer Res 70:8684–8694
Pascolo E, Wenz C, Lingner J, Hauel N, Priepke H, Kauffmann I et al (2002) Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J Biol Chem 277:15566–15572
Ward RJ, Autexier C (2005) Pharmacological telomerase inhibition can sensitize drug-resistant and drug-sensitive cells to chemotherapeutic treatment. Mol Pharmacol 68:779–786
El Daly H, Martens UM (2007) Telomerase inhibition and telomere targeting in hematopoietic cancer cell lines with small non-nucleosidic synthetic compounds (BIBR1532). Methods Mol Biol 405:47–60
El-Daly H, Kull M, Zimmermann S, Pantic M, Waller CF, Martens UM (2005) Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532. Blood 105:1742–1749
Furman PA, Fyfe JA, St Clair MH, Weinhold K, Rideout JL, Freeman GA et al (1986) Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci USA 83:8333–8337
Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC et al (1985) 3′-Azido-3′-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci USA 82:7096–7100
Datta A, Bellon M, Sinha-Datta U, Bazarbachi A, Lepelletier Y, Canioni D et al (2006) Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood 108:1021–1029
Geary RS, Yu RZ, Levin AA (2001) Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr Opin Investig Drugs 2:562–573
Folini M, Brambilla C, Villa R, Gandellini P, Vignati S, Paduano F et al (2005) Antisense oligonucleotide-mediated inhibition of hTERT, but not hTERC, induces rapid cell growth decline and apoptosis in the absence of telomere shortening in human prostate cancer cells. Eur J Cancer 41:624–634
Akiyama M, Hideshima T, Shammas MA, Hayashi T, Hamasaki M, Tai YT et al (2003) Effects of oligonucleotide N3′–>P5′ thio-phosphoramidate (GRN163) targeting telomerase RNA in human multiple myeloma cells. Cancer Res 63:6187–6194
Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S et al (2003) A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res 63:3931–3939
Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D et al (2005) Lipid modification of GRN163, an N3′–>P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 24:5262–5268
Dikmen ZG, Gellert GC, Jackson S, Gryaznov S, Tressler R, Dogan P et al (2005) In vivo inhibition of lung cancer by GRN163L: a novel human telomerase inhibitor. Cancer Res 65:7866–7873
Gellert GC, Dikmen ZG, Wright WE, Gryaznov S, Shay JW (2006) Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res Treat 96:73–81
Hochreiter AE, Xiao H, Goldblatt EM, Gryaznov SM, Miller KD, Badve S et al (2006) Telomerase template antagonist GRN163L disrupts telomere maintenance, tumor growth, and metastasis of breast cancer. Clin Cancer Res 12:3184–3192
Djojosubroto MW, Chin AC, Go N, Schaetzlein S, Manns MP, Gryaznov S et al (2005) Telomerase antagonists GRN163 and GRN163L inhibit tumor growth and increase chemosensitivity of human hepatoma. Hepatology 42:1127–1136
Brower V (2010) Telomerase-based therapies emerging slowly. J Natl Cancer Inst 102:520–521
Chen HC, Chou CK, Lee SD, Wang JC, Yeh SF (1995) Active compounds from Saussurea lappa Clarks that suppress hepatitis B virus surface antigen gene expression in human hepatoma cells. Antiviral Res 27:99–109
Kang JS, Yoon YD, Lee KH, Park SK, Kim HM (2004) Costunolide inhibits interleukin-1beta expression by down-regulation of AP-1 and MAPK activity in LPS-stimulated RAW 264.7 cells. Biochem Biophys Res Commun 313:171–177
Park HJ, Jung WT, Basnet P, Kadota S, Namba T (1996) Syringin 4-O-beta-glucoside, a new phenylpropanoid glycoside, and costunolide, a nitric oxide synthase inhibitor, from the stem bark of Magnolia sieboldii. J Nat Prod 59:1128–1130
Wedge DE, Galindo JC, Macias FA (2000) Fungicidal activity of natural and synthetic sesquiterpene lactone analogs. Phytochemistry 53:747–757
Choi SH, Im E, Kang HK, Lee JH, Kwak HS, Bae YT et al (2005) Inhibitory effects of costunolide on the telomerase activity in human breast carcinoma cells. Cancer Lett 227:153–162
Kanno S, Kitajima Y, Kakuta M, Osanai Y, Kurauchi K, Ujibe M et al (2008) Costunolide-induced apoptosis is caused by receptor-mediated pathway and inhibition of telomerase activity in NALM-6 cells. Biol Pharm Bull 31:1024–1028
Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E et al (1997) The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J Biol Chem 272:23843–23850
Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1997) Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90:65–75
Gorre ME, Ellwood-Yen K, Chiosis G, Rosen N, Sawyers CL (2002) BCR-ABL point mutants isolated from patients with imatinib mesylate-resistant chronic myeloid leukemia remain sensitive to inhibitors of the BCR-ABL chaperone heat shock protein 90. Blood 100:3041–3044
Hartson SD, Matts RL (1994) Association of Hsp90 with cellular Src-family kinases in a cell-free system correlates with altered kinase structure and function. Biochemistry 33:8912–8920
Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N (2003) Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res 63: 2139–2144
Stancato LF, Chow YH, Hutchison KA, Perdew GH, Jove R, Pratt WB (1993) Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J Biol Chem 268:21711–21716
Vonderheide RH, Hahn WC, Schultze JL, Nadler LM (1999) The telomerase catalytic subunit is a widely expressed tumor-associated antigen recognized by cytotoxic T lymphocytes. Immunity 10:673–679
Minev B, Hipp J, Firat H, Schmidt JD, Langlade-Demoyen P, Zanetti M (2000) Cytotoxic T cell immunity against telomerase reverse transcriptase in humans. Proc Natl Acad Sci USA 97:4796–4801
Nair SK, Heiser A, Boczkowski D, Majumdar A, Naoe M, Lebkowski JS et al (2000) Induction of cytotoxic T cell responses and tumor immunity against unrelated tumors using telomerase reverse transcriptase RNA transfected dendritic cells. Nat Med 6:1011–1017
Su Z, Dannull J, Yang BK, Dahm P, Coleman D, Yancey D et al (2005) Telomerase mRNA-transfected dendritic cells stimulate antigen-specific CD8+ and CD4+ T cell responses in patients with metastatic prostate cancer. J Immunol 174:3798–3807
Bernhardt SL, Gjertsen MK, Trachsel S, Moller M, Eriksen JA, Meo M et al (2006) Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer 95:1474–1482
Shay JW, Keith WN (2008) Targeting telomerase for cancer therapeutics. Br J Cancer 98:677–683
Cook BD, Dynek JN, Chang W, Shostak G, Smith S (2002) Role for the related poly(ADP-Ribose) polymerases tankyrase 1 and 2 at human telomeres. Mol Cell Biol 22:332–342
Smith S, de Lange T (2000) Tankyrase promotes telomere elongation in human cells. Curr Biol 10:1299–1302
Smith S, Giriat I, Schmitt A, de Lange T (1998) Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 282:1484–1487
Chang W, Dynek JN, Smith S (2003) TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev 17:1328–1333
Dynek JN, Smith S (2004) Resolution of sister telomere association is required for progression through mitosis. Science 304:97–100
Seimiya H, Muramatsu Y, Ohishi T, Tsuruo T (2005) Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 7:25–37
Mergny JL, Duval-Valentin G, Nguyen CH, Perrouault L, Faucon B, Rougee M et al (1992) Triple helix-specific ligands. Science 256:1681–1684
Escude C, Nguyen CH, Kukreti S, Janin Y, Sun JS, Bisagni E et al (1998) Rational design of a triple helix-specific intercalating ligand. Proc Natl Acad Sci USA 95:3591–3596
Shi DF, Wheelhouse RT, Sun D, Hurley LH (2001) Quadruplex-interactive agents as telomerase inhibitors: synthesis of porphyrins and structure-activity relationship for the inhibition of telomerase. J Med Chem 44:4509–4523
Bertrand H, Bombard S, Monchaud D, Teulade-Fichou MP (2008) New platinum(II) complexes targeting the loops of the human telomeric G-quadruplex. Nucleic Acids Symp Ser (Oxf) 52:163–164
Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO et al (1997) Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem 40:2113–2116
Perry PJ, Gowan SM, Reszka AP, Polucci P, Jenkins TC, Kelland LR et al (1998) 1,4- and 2,6-disubstituted amidoanthracene-9,10-dione derivatives as inhibitors of human telomerase. J Med Chem 41:3253–3260
Perry PJ, Gowan SM, Read MA, Kelland LR, Neidle S (1999) Design, synthesis and evaluation of human telomerase inhibitors based upon a tetracyclic structural motif. Anticancer Drug Des 14:373–382
Perry PJ, Read MA, Davies RT, Gowan SM, Reszka AP, Wood AA et al (1999) 2,7-Disubstituted amidofluorenone derivatives as inhibitors of human telomerase. J Med Chem 42:2679–2684
Gowan SM, Heald R, Stevens MF, Kelland LR (2001) Potent inhibition of telomerase by small-molecule pentacyclic acridines capable of interacting with G-quadruplexes. Mol Pharmacol 60:981–988
Simonsson T, Pecinka P, Kubista M (1998) DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res 26:1167–1172
Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH (2002) Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA 99:11593–11598
Guiducci C, Cerone MA, Bacchetti S (2001) Expression of mutant telomerase in immortal telomerase-negative human cells results in cell cycle deregulation, nuclear and chromosomal abnormalities and rapid loss of viability. Oncogene 20:714–725
Kim MM, Rivera MA, Botchkina IL, Shalaby R, Thor AD, Blackburn EH (2001) A low threshold level of expression of mutant-template telomerase RNA inhibits human tumor cell proliferation. Proc Natl Acad Sci USA 98:7982–7987
Li S, Rosenberg JE, Donjacour AA, Botchkina IL, Hom YK, Cunha GR et al (2004) Rapid inhibition of cancer cell growth induced by lentiviral delivery and expression of mutant-template telomerase RNA and anti-telomerase short-interfering RNA. Cancer Res 64:4833–4840
Abdul-Ghani R, Ohana P, Matouk I, Ayesh S, Ayesh B, Laster M et al (2000) Use of transcriptional regulatory sequences of telomerase (hTER and hTERT) for selective killing of cancer cells. Mol Ther 2:539–544
Gu J, Kagawa S, Takakura M, Kyo S, Inoue M, Roth JA et al (2000) Tumor-specific transgene expression from the human telomerase reverse transcriptase promoter enables targeting of the therapeutic effects of the Bax gene to cancers. Cancer Res 60:5359–5364
Koga S, Hirohata S, Kondo Y, Komata T, Takakura M, Inoue M et al (2000) A novel telomerase-specific gene therapy: gene transfer of caspase-8 utilizing the human telomerase catalytic subunit gene promoter. Hum Gene Ther 11:1397–1406
Boyd M, Mairs RJ, Mairs SC, Wilson L, Livingstone A, Cunningham SH et al (2001) Expression in UVW glioma cells of the noradrenaline transporter gene, driven by the telomerase RNA promoter, induces active uptake of [131I]MIBG and clonogenic cell kill. Oncogene 20:7804–7808
Komata T, Kondo Y, Kanzawa T, Hirohata S, Koga S, Sumiyoshi H et al (2001) Treatment of malignant glioma cells with the transfer of constitutively active caspase-6 using the human telomerase catalytic subunit (human telomerase reverse transcriptase) gene promoter. Cancer Res 61:5796–5802
Koga S, Hirohata S, Kondo Y, Komata T, Takakura M, Inoue M et al (2001) FADD gene therapy using the human telomerase catalytic subunit (hTERT) gene promoter to restrict induction of apoptosis to tumors in vitro and in vivo. Anticancer Res 21:1937–1943
Koga S, Kondo Y, Komata T, Kondo S (2001) Treatment of bladder cancer cells in vitro and in vivo with 2-5A antisense telomerase RNA. Gene Ther 8:654–658
Majumdar AS, Hughes DE, Lichtsteiner SP, Wang Z, Lebkowski JS, Vasserot AP (2001) The telomerase reverse transcriptase promoter drives efficacious tumor suicide gene therapy while preventing hepatotoxicity encountered with constitutive promoters. Gene Ther 8:568–578
Plumb JA, Bilsland A, Kakani R, Zhao J, Glasspool RM, Knox RJ et al (2001) Telomerase-specific suicide gene therapy vectors expressing bacterial nitroreductase sensitize human cancer cells to the pro-drug CB1954. Oncogene 20:7797–7803
Hao ZM, Luo JY, Cheng J, Li L, He D, Wang QY et al (2005) Intensive inhibition of hTERT expression by a ribozyme induces rapid apoptosis of cancer cells through a telomere length-independent pathway. Cancer Biol Ther 4:1098–1103
Kanazawa Y, Ohkawa K, Ueda K, Mita E, Takehara T, Sasaki Y et al (1996) Hammerhead ribozyme-mediated inhibition of telomerase activity in extracts of human hepatocellular carcinoma cells. Biochem Biophys Res Commun 225:570–576
Yokoyama Y, Takahashi Y, Shinohara A, Lian Z, Wan X, Niwa K et al (1998) Attenuation of telomerase activity by a hammerhead ribozyme targeting the template region of telomerase RNA in endometrial carcinoma cells. Cancer Res 58:5406–5410
Lin SY, Elledge SJ (2003) Multiple tumor suppressor pathways negatively regulate telomerase. Cell 113:881–889
Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M et al (1999) Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res 59:551–557
Sohn JH, Yeh BI, Choi JW, Yoon J, Namkung J, Park KK et al (2010) Repression of human telomerase reverse transcriptase using artificial zinc finger transcription factors. Mol Cancer Res 8:246–253
Wong KK, Chang S, Weiler SR, Ganesan S, Chaudhuri J, Zhu C et al (2000) Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet 26:85–88
Ludwig A, Saretzki G, Holm PS, Tiemann F, Lorenz M, Emrich T et al (2001) Ribozyme cleavage of telomerase mRNA sensitizes breast epithelial cells to inhibitors of topoisomerase. Cancer Res 61:3053–3061
Tauchi T, Nakajima A, Sashida G, Shimamoto T, Ohyashiki JH, Abe K et al (2002) Inhibition of human telomerase enhances the effect of the tyrosine kinase inhibitor, imatinib, in BCR-ABL-positive leukemia cells. Clin Cancer Res 8:3341–3347
Multani AS, Li C, Ozen M, Imam AS, Wallace S, Pathak S (1999) Cell-killing by paclitaxel in a metastatic murine melanoma cell line is mediated by extensive telomere erosion with no decrease in telomerase activity. Oncol Rep 6:39–44
Mo Y, Gan Y, Song S, Johnston J, Xiao X, Wientjes MG et al (2003) Simultaneous targeting of telomeres and telomerase as a cancer therapeutic approach. Cancer Res 63:579–585
Nakamura M, Masutomi K, Kyo S, Hashimoto M, Maida Y, Kanaya T et al (2005) Efficient inhibition of human telomerase reverse transcriptase expression by RNA interference sensitizes cancer cells to ionizing radiation and chemotherapy. Hum Gene Ther 16:859–868
Guo X, Wang W, Zhou F, Lu Z, Fang R, Jia F et al (2008) siRNA-mediated inhibition of hTERT enhances chemosensitivity of hepatocellular carcinoma. Cancer Biol Ther 7:1555–1560
Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H et al (2001) A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J 20:6958–6968
Gomez-Millan J, Goldblatt EM, Gryaznov SM, Mendonca MS, Herbert BS (2007) Specific telomere dysfunction induced by GRN163L increases radiation sensitivity in breast cancer cells. Int J Radiat Oncol Biol Phys 67:897–905
Goldblatt EM, Gentry ER, Fox MJ, Gryaznov SM, Shen C, Herbert BS (2009) The telomerase template antagonist GRN163L alters MDA-MB-231 breast cancer cell morphology, inhibits growth, and augments the effects of paclitaxel. Mol Cancer Ther 8:2027–2035
Goldblatt EM, Erickson PA, Gentry ER, Gryaznov SM, Herbert BS (2009) Lipid-conjugated telomerase template antagonists sensitize resistant HER2-positive breast cancer cells to trastuzumab. Breast Cancer Res Treat 118:21–32
Trudeau M, Wong J (2010) Genetic variations in telomere maintenance, with implications on tissue renewal capacity and chronic disease pathologies. Curr Pharmacogenomics Person Med 8:7–24
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB et al (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279:349–352
Fauce SR, Jamieson BD, Chin AC, Mitsuyasu RT, Parish ST, Ng HL et al (2008) Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J Immunol 181:7400–7406
Zhu J, Lee S, Ho MK, Hu Y, Pang H, Ip FC et al (2010) In vitro intestinal absorption and first-pass intestinal and hepatic metabolism of cycloastragenol, a potent small molecule telomerase activator. Drug Metab Pharmacokinet 25:477–486
Benetos A, Okuda K, Lajemi M, Kimura M, Thomas F, Skurnick J et al (2001) Telomere length as an indicator of biological aging: the gender effect and relation with pulse pressure and pulse wave velocity. Hypertension 37:381–385
Cawthon RM, Smith KR, O’Brien E, Sivatchenko A, Kerber RA (2003) Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361:393–395
Harley CB, Liu W, Blasco M, Vera E, Andrews WH, Briggs LA et al (2011) A natural product telomerase activator as part of a health maintenance program. Rejuvenation Res 14:45–56
Calado RT, Regal JA, Hills M, Yewdell WT, Dalmazzo LF, Zago MA et al (2009) Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc Natl Acad Sci USA 106:1187–1192
Gryaznov SM (2010) Oligonucleotide n3′–>p5′ phosphoramidates and thio-phoshoramidates as potential therapeutic agents. Chem Biodivers 7:477–493
Harley CB (2008) Telomerase and cancer therapeutics. Nat Rev Cancer 8:167–179
Acknowledgment
We thank Dragony Fu, Suzanne Lee, and Naresh Thumati for reading this chapter and for their helpful comments. Research in Judy Wong’s laboratory is supported by the Canadian Institutes of Health Research, the Canadian Cancer Society, and the Leukemia and Lymphoma Society of Canada. JMYW is supported by the Canada Research Chair and Michael Smith Foundation of Health Research career development programs.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Tamakawa, R.A., Fleisig, H.B., Wong, J.M.Y. (2013). Telomeres, Telomerase, and DNA Damage Response in Cancer Therapy. In: Panasci, L., Aloyz, R., Alaoui-Jamali, M. (eds) Advances in DNA Repair in Cancer Therapy. Cancer Drug Discovery and Development, vol 72. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4741-2_11
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4741-2_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4740-5
Online ISBN: 978-1-4614-4741-2
eBook Packages: MedicineMedicine (R0)