
Abstract
A number of Gram-positive bacteria are important players in industry as producers of a diverse array of economically interesting metabolites and proteins. As discussed in this overview, several Gram-positive bacteria are valuable hosts for the production of heterologous proteins. In contrast to Gram-negative bacteria, proteins secreted by Gram-positive bacteria are released into the culture medium where conditions for correct folding are more appropriate, thus facilitating the isolation and purification of active proteins. Although seven different protein secretion pathways have been identified in Gram-positive bacteria, the majority of heterologous proteins are produced via the general secretion or Sec pathway. Not all proteins are equally well secreted, because heterologous protein production often faces bottlenecks including hampered secretion, susceptibility to proteases, secretion stress, and metabolic burden. These bottlenecks are associated with reduced yields leading to non-marketable products. In this chapter, besides a general overview of the different protein secretion pathways, possible hurdles that may hinder efficient protein secretion are described and attempts to improve yield are discussed including modification of components of the Sec pathway. Attention is also paid to omics-based approaches that may offer a more rational approach to optimize production of heterologous proteins.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Lactic Acid Bacterium
- Protein Secretion
- Heterologous Protein
- Recombinant Protein Production
- Signal Peptidase
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Biotechnology involves the use of living organisms and their products for the benefit of mankind in different areas. The biotechnological manufacturing of products of biomedical interest such as antibiotics, vaccines, antibodies, and other biopharmaceuticals are termed red biotechnology. On the other hand, the production of industrial enzymes for the sustainable processing and production of chemicals, materials, and fuels is classified as white biotechnology, while green biotechnology serves agriculture and involves, for example, the development of pest-resistant plants and the control of food and feed. Recently, blue biotechnology gained interest, whereby marine organisms and their products are used for the making of valuable compounds including drugs and bioenergy. Molecules of interest can be synthetized by the original producing organism, but in many cases they are heterologously produced. This means that the necessary genes derived from other organisms are cloned into the host cell of choice with the intention to express the molecules encoded by the cloned DNA so that the host produces the molecules that it normally does not make. The global biotechnology market size is estimated to have a value of more than US$398 billion in 2015 and a growth expectation at a compound annual growth rate of 12.3 %. To be profitable in this vast market, not only the value of the molecules produced counts, but the cost-effectiveness and environmental-friendliness of the production process are at least as important. The main challenge therefore is to have the most effective production process for lowering the cost of the final product to obtain a commercially viable process. Optimization of microbes in production processes has been done over the past decades using, for example, random mutagenesis and selection, identification of metabolic reactions whose activities should be modified to achieve the desired cellular objective, genome-scale modeling of metabolism, and by fermentation optimization. More recently, these approaches are enhanced by synthetic biology tools.
To produce heterologous molecules, a variety of different expression systems has been described, each with its own advantages and disadvantages. The most popular host is Escherichia coli, as proven by the fact that nearly half of the approved recombinant biopharmaceuticals are synthesized using this host. Reasons why E. coli is so popular in recombinant protein production and as a workhorse in the laboratories of academia and the biopharma industry are obvious: its genetics are far better understood than those of other microorganisms; there are many genetic tools available, and proteins, if expressed, can be obtained in high production yields. As other bacteria, E. coli grows quickly on cheap media to high cell density. A drawback, however, is that E. coli are Gram-negative bacteria inherently having an outer membrane bilayer (OM) composed of lipopolysaccharides (LPS). The OM acts as an effective permeability barrier hindering secreted proteins from being released into the extracellular medium. LPS contains the endotoxin lipid A, that if released in the blood can cause septic shock, a systemic inflammatory response syndrome. When overexpressed in E. coli, many proteins become misfolded and accumulate in the cytoplasm as inclusion bodies. To become active, these inclusion bodies need to be solubilized, and proteins refolded into bioactive molecules, an often cumbersome process, with poor recovery and accounting for the major cost in the production process of recombinant proteins. Therefore, new solubilization techniques have been proposed, as well as genetic approaches to make E. coli a better host, for example, engineering E. coli strains that possess an oxidative cytoplasmic environment that favors disulfide bond formation, overexpression of different chaperones or combinations thereof, the fusion of appropriate tags to the N- or C-terminus of the overexpressed protein, secretion in the medium using the alpha-hemolysin secretion system. These improvements can be helpful, but are not used at an industrial scale, and depending on the proteins to be obtained other host cells derived from Gram-positive bacteria might be a better option.
An important asset is that Gram-positive bacteria are monoderm with a cell envelop that surrounds the cytoplasmic membrane with a thick peptidoglycan layer and associated teichoic acids. This structure protects the cell from mechanical or osmolytic lysis and is an anchor place for proteins, glycopolymers, and cations. Notwithstanding its complex structure, the cell envelop of Gram-positive bacteria is permeable to proteins as it does not contain an outer membrane. Consequently, secreted proteins will be released into the culture medium, where they can obtain their native conformation simplifying downstream processing. This is an advantage for the industrial production of heterologous proteins.
In this review, a survey will be presented about the protein secretion pathways in Gram-positive bacteria together with possible applications for specific species. Complementary to the beneficial properties of heterologous expression in Gram-positive bacteria, strategies for enhancing heterologous protein production are developed to acquire commercially acceptable production and yield. This review will further give a brief compilation of approaches tackling the bottlenecks at the level of expression up to metabolic fluxes.
2 Protein Secretion Pathways in Gram-Positive Bacteria
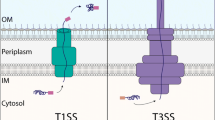
Protein secretion is a vital process for all organisms, since about 35 % of all proteins made in a cell are either membrane-embedded or secreted (Orfanoudaki and Economou 2014). To do so bacteria have different secretion pathways at their disposal. Whereas at least 7 diverse secretion systems (type I–VII) have been identified for Gram-negative bacteria, 6 protein translocation systems were reported for Gram-positive bacteria [for an overview, see Forster and Marquis (2012)] as explained below for Gram-positive bacteria. Only two of them are used for biotechnology purposes (Fig. 1).

Overview of protein secretion pathways of Gram-positive bacteria and their possible use in biotechnology
2.1 General Secretion (Sec) System
The most important protein secretion pathway is the general secretion (Sec) system, which directs proteins to the cytoplasmic membrane for their insertion into or translocation across the membrane. Proteins destined for secretion are in general synthesized as preproteins with an N-terminally extended sequence, named the signal peptide. The primary sequences of signal peptides are not homologous, although they do have 3 common structural features: a net positively charged N-terminus, a hydrophobic core region (H-region), and a polar C-terminal end containing the signal peptidase recognition site. In bacteria, the signal peptide is between 20 and 30 residues long, but can contain even more than 50 amino acids residues depending on the species and the protein to be secreted. The role of the signal peptide is to guide the protein to the secretion channel following binding to soluble targeting factors. The involvement of chaperones to target newly synthesized proteins to the translocation pathways, either co- or posttranslationally, is best studied for E. coli, as summarized hereafter. The chaperones trigger factor (Tf), DnaK/DnaJ/GrpE (DnaKJE) and GroEL do not only play a major role in the folding of newly synthesized cytosolic proteins, but are also important for posttranslational protein targeting (Grady et al. 2012; Castanie-Cornet et al. 2014). Besides Tf, DnaKJE and GroEL, of prime importance for secretion in proteobacteria, is SecB, which keeps proteins in an unfolded secretion-competent state and delivers them to the Sec translocon SecYEG via its interaction with SecA, the dimeric ATPase subunit of bacterial protein translocase (Karamanou et al. 1999). The peripherally associated motor protein SecA drives then the protein translocation step by repeated cycles of ATP-binding and hydrolysis resulting in SecA membrane insertion/deinsertion and stepwise exportation of the preprotein through the channel (Chatzi et al. 2013). Additional energy promoting translocation, when the preprotein is detached from SecA, comes from the proton motive force [PMF; (Schiebel et al. 1991)]. During or following translocation, the signal peptide is cleaved off by the membrane-embedded signal peptidase I or II (the latter specializing on secreted lipoprotein signal peptides) at the signal peptidase recognition site located in its C-terminal end. This recognition site is often a canonical A-X-A motif, but other residues are permitted as well. It was recently shown by computational analyses of ~1500 genomes that numerous major evolutionary clades have replaced the canonical signal peptide sequence with novel motifs (Payne et al. 2012).
Besides the Sec pathway , there is also the signal recognition pathway (SRP) for cotranslational secretion that in bacteria mainly deals with membrane protein insertion and to a lesser extent with protein secretion. SRP binds to particularly hydrophobic N-terminal signal sequences or hydrophobic transmembrane segments as they emerge from the ribosome. The SRP/RNC (ribosome nascent chain) complex interacts with the membrane-bound SRP Receptor (SR) and the delivery of the RNC to the translocation channel SecYEG in the membrane finally leads to the dissociation of the SRP/SR complex, whereupon the preprotein is driven across the translocation channel with the help of continuing translation and/or SecA.
It is generally assumed that the secretion of proteins in Gram-positive bacteria follows similar steps as they occur in E. coli since genes involved in the Sec-dependent protein secretion pathway are identified in the genome of Gram-positive bacteria. Nonetheless SecB is absent from the genomes of Gram-positive bacteria, although some SecB-like genes are present. For example, SecB-like protein (Rv1957) is present in the Mycobacterium tuberculosis genome where it specifically controls a stress-responsive toxin–antitoxin system. Experiments suggest that Rv1957 could play a role in protein export of M. tuberculosis (Sala et al. 2014), but its role is completely different from that of SecB in Gram-negative bacteria. In Bacillus subtilis, the SecB chaperone function has been attributed to CsaA (Shapova and Paetzel 2007). How Sec-dependent secretory proteins are kept in a Sec-secretion-competent way, and which chaperones are involved is not clear yet.
2.2 Twin-Arginine Transport Pathway (Tat)
A special characteristic of the Tat pathway is that proteins are transported across the cytoplasmic membrane in a folded state, and the energy for translocation comes from the proton motive force (PMF). Many Tat substrates receive cofactors and fold prior to translocation. Based on genome sequence analysis of prokaryotes, the Tat pathway is present in nearly 80 % of all bacteria, also in Archaea (Simone et al. 2013). Results indicate that the Tat pathway is utilized to highly varying extents. It operates in parallel with the Sec pathway. Signal peptides that target proteins to the Tat pathway resemble Sec signal peptides, but with a conserved S/T-R-R-x-FLK consensus motif at the end of the N-region, where the twin-arginines are invariant and normally essential for efficient export by the Tat pathway (Stanley et al. 2000). However, the Tat-specific signal sequence with two arginine residues may not be an absolute prerequisite for the Tat pathway (Watanabe et al. 2009). The Tat translocon comprises two kinds of small membrane proteins: TatC, a highly hydrophobic protein with 6 predicted transmembrane helices, and with its N- and C-termini at the cytoplasmic face of the membrane. The Tat translocon contains also one or two members of the TatA protein family, named TatA and TatB, sequence-related proteins with a common structure, each predicted to comprise a membrane-spanning α-helix at the N-terminus, immediately followed by an amphipathic helix located at the cytoplasmic side of the membrane and a C-terminal region of variable length. TatB and TatC form an oligomeric, multivalent receptor complex that binds Tat substrates, while multiple protomers of TatA assemble at substrate-bound TatBC receptors to facilitate substrate transport (Cleon et al. 2015). Minimal Tat systems contain only one type of TatA and one type of TatC. When the signal peptide of a Tat-dependent protein is recognized, it will be bound by a multi-subunit TatBC complex located in the membrane and Tat secretion is initiated. This binding event triggers the PMF-dependent recruitment and oligomerization of TatA protomers from a pool in the membrane to form the active TatABC-containing translocation site (Berks 2015). Possible cross talk between the Tat- and Sec-dependent protein secretion pathways has been reported (Goosens et al. 2014). This assumed interaction, however, needs to be further investigated.
In actinomycetes including Streptomyces lividans and other streptomycetes, Mycobacterium (McDonough et al. 2005) and Corynebacterium (Oertel et al. 2015), TatA, TatB, and TatC are the components for this pathway, similarly to Gram-negative bacteria. Of the Tat components in Streptomyces, TatC is essential whereas TatA and TatB are individually dispensable and are, next to the membrane-embedded localization, also found as active soluble complexes in the cytoplasm (De Keersmaeker et al. 2007). In contrast to the majority of Tat-containing organisms in which the Sec pathway is the major route for protein transport, the Tat pathway seems to be an important protein secretion route in Streptomyces. It is estimated that nearly 20 % of proteins of the extracellular proteome is secreted via the Tat pathway (Widdick et al. 2006). Using enhanced green fluorescent protein (eGFP) and mCherry fusions of the proteins of the Tat machinery, Willemse et al. (2012) tried to determine their subcellular localization in Streptomyces coelicolor throughout the complex life cycle of this organism. They showed that TatA, TatB, and TatC dynamically co-localize in the vegetative hyphae, with a strong preference for apical sites in growing hyphae. For Corynebacterium, secretion is mainly studied in Corynebacterium glutamicum (Kikuchi et al. 2006). A functional C. glutamicum Tat system requires TatA and TatC, while the TatB protein seems to be dispensable, but it is important for maximal efficiency, and it was also found to be essential for the secretion of a heterologous Tat-dependent model protein into the C. glutamicum culture supernatant (Oertel et al. 2015). It was further shown that TatB (in combination with TatA and TatC) is strictly required for unimpaired aerobic growth (Oertel et al. 2015).
Both M. tuberculosis and Mycobacterium smegmatis have a functional Tat pathway. As for other actinomycetes, the Tat translocon consists of TatA, TatB, and TatC (McDonough et al. 2005). In M. tuberculosis, the Tat pathway is essential for growth (Saint-Joanis et al. 2006) as concluded from the inability to obtain viable deletion mutants. This contrasts with M. smegmatis for which viable Tat mutants could be obtained, although these mutants showed growth defects. In addition, increased sensitivity to β-lactam antibiotics was also noticed, this as a consequence of reduced export of β-lactamase BlaS, a protein with a predicted Tat signal peptide (Saint-Joanis et al. 2006).
For staphylococci, only some species contain a functional Tat system, including Staphylococcus carnosus (Meissner et al. 2007), Staphylococcus haemolyticus (Yamada et al. 2007), and Staphylococcus aureus (Biswas et al. 2009). The Tat system is composed of TatA and TatC and was proven to translocate iron-dependent peroxide FepB in S. aureus (Biswas et al. 2009). B. subtilis secretion system has been studied extensively (Goosens et al. 2014). Genes for two TatC (TatCd and TatCy) and three TatA components (TatAd, TatAy, and TatAc) have been identified in the B. subtilis genome. The core Tat complex consists of TatA and TatC, namely TatAy–TatCy, of which the latter is constitutively expressed and exports more substrates, including the Dyp-type peroxidase EfeB (YwbN), the Rieske iron-sulfur protein QcrA, and the alkaline phosphatase YkuE, while TatAdCd is only expressed under phosphate limitation (Pop et al. 2002). A third TatA-like protein TatAc can be combined as TatAcCd and TatAcCy. It is supposed to be an intermediate evolutionary step in TatA–TatB specialization (Goosens et al. 2015).
2.3 Type IV Secretion Systems (T4SSs)
T4SSs transport a diverse array of substrates from DNA to nucleoprotein complexes and effector proteins. They are multi-subunit, membrane-spanning translocation systems found in Gram-positive as well as Gram-negative bacteria and in some archaea (Chandran Darbari and Waksman 2015). They have evolved from a self-transmissible, single-stranded DNA conjugation system with VirB4-like AAA + ATPase to systems with an enormous diversity in their overall structure and the types of substrates secreted. T4SSs can be divided into 3 groups (Bhatty et al. 2013): (1) a conjugation system to deliver ssDNA and one or more proteins across the membrane to the bacterial or eukaryotic target cell whereby direct cell contact is required; (2) the effector translocation system by a contact-dependent mechanism to deliver proteins to the cytosol of eukaryotic target cells, and (3) a release/uptake system to export/import molecules from/to the extracellular milieu. As a consequence, T4SSs are involved in a variety of functions including type 4 pilus formation, toxin and other protein secretion, gene transfer, and biofilm formation. Secreted substrates are involved in pathogenesis and adaptation to the cellular host environment. T4SSs translocate also proteins that form pilin-like structures (Chen and Dubnau 2004). A typical characteristic of these proteins is the presence of a specialized leader peptide that is cleaved off by a cognate membrane-bound type 4 prepilin peptidase during the process of secretion. Only T4SS conjugation systems are known in Gram-positive bacteria and Archaea to date.
2.4 Type VII Secretion System (T7SS)
Recent studies have uncovered a T7SS or early secretory antigen 6-KDa (ESX) secretion system. Originally, it was detected via an in silico analysis of the M. tuberculosis virulence effectors ESAT-6 (early-secreted antigenic target, 6 kDa) and the associated 10-kDa culture filtrate protein (CFP-10, EsxB) encoded by the esxA and esxB genes, respectively. They were known to be secreted despite the lack of a recognizable secretion signal (Tekaia et al. 1999). Esx proteins are characterized by their small size (~100 residues) and a WXG motif in the middle of the protein that forms a hairpin bend (Pallen 2002). Therefore, an alternative name was proposed for T7SS, the WXG100 secretion system (Wss) (Sutcliffe 2011), because distant homologues of ESAT-6/CFP-10 identified in Gram-positive bacteria all share a central WXG motif. T7SSs are widespread in actinomycetes and Gram-positive bacteria and affect a range of bacterial processes including sporulation, conjugation, and cell wall stability (Sysoeva et al. 2014). The T7SS is a complex system with many components and substrates, at least in mycobacteria. M. tuberculosis has five T7S systems, designated ESX-1 through ESX-5 (Stoop et al. 2012), which show similarity in gene content and gene order. Of these T7S systems, 3 are important for survival in the host, namely ESX-3, responsible for the uptake of iron and zinc, and ESX-5, responsible for the secretion of immunomodulatory effector proteins, and ESX-1 is most crucial for virulence. First detected in M. tuberculosis (Stanley et al. 2003), it was shown afterward also to be present in the non-pathogenic species M. smegmatis (Converse and Cox 2005) and the fish pathogen Mycobacterium marinum (Abdallah et al. 2009). On the other hand, ESX-1 is absent in Mycobacterium bovis BCG, the attenuated vaccine strain. PE/PPE is also secreted by the T7SS. The PE/PPE protein family, which has a conserved signature motif proline–glutamate and proline–proline–glutamate residues near the start of their encoded proteins, affects mycobacterial interactions with the innate immune system, specifically inhibiting macrophage function (Ahmed et al. 2015). ESX-1 and ESX-5 have been implicated in major roles in the secretion of PE/PPE proteins (Abdallah et al. 2009). ESX secretion seems to be crucial for establishing and maintaining the infection for M. tuberculosis. A PSI-BLAST search on sequences retrieved from the NCBI or the ViruloGenome databases further evidenced the presence of ESAT-6 homologues in a number of low-GC Gram-positive bacteria, and also in several actinobacteria other than Mycobacterium (Pallen 2002), including all sequenced Streptomyces genomes such as S. coelicolor, S. lividans, and S. scabies. The biological importance of this pathway for streptomycetes is, so far, less well-known and begins only just to be revealed. For S. scabies no role in virulence for any of the T7SS components in any of the plant infection models tested could be detected, but it was demonstrated that components encoded by the T7SS gene cluster are required for the normal growth and development of S. scabies (Fyans et al. 2013). By mutagenesis analysis, it was shown that also proteins encoded by the esxBA operon and belonging to the WXG-100 superfamily play a role in morphogenesis in S. coelicolor (San Roman et al. 2010). In the sequenced genomes of other Gram-positive bacteria including B. subtilis, Bacillus anthracis, Clostridium acetobutylicum, Listeria monocytogenes, and S. aureus, ESAT-6 homologues were also discovered (Pallen 2002) and confirmed experimentally, for example, for S. aureus (Burts et al. 2005), B. subtilis (Sysoeva et al. 2014), B. anthracis (Fan et al. 2015).
2.5 Flagella Export Apparatus (FEA)
This specific protein export apparatus serves to secrete proteins that form the flagella hook, filament, and cap (Erhardt et al. 2010). Flagellar T3SS are present both in Gram-positive and Gram-negative bacteria, and it has been proposed that the type III secretion required for pathogenesis evolved from flagellar-specific T3SS (Hueck 1998). To transport proteins that form the flagella hook, filament, and cap to the distal growing end, the FEA utilizes ATP and PMF as the energy source (Paul et al. 2008). The flagellar export apparatus is thought to be the ancestor of all T3SS functions in the export of several components of the flagellum across the cytoplasmic membrane into the channel of the flagellum for assembly. Not much is known, however, about the FEA for Gram-positive bacteria. One report mentions that in B. subtilis FlgM is secreted by the flagellar export apparatus, consistent with the model of morphogenetic coupling proposed in Salmonella enterica (Calvo and Kearns 2015).
2.6 Holins
Originally holins were used to describe a group of phage-encoded pore forming membrane proteins that control access of phage-encoded endolysins to the peptidoglycan layer. During the phage lytic cycle, holins insert into the bacterial cell membrane to translocate phage-encoded cell wall hydrolases (Wang et al. 2000). Holins may also be important for a variety of other functions in Gram-positive phage-free bacteria (Saier and Reddy 2015) such as (i) spore morphogenesis and germination in B. subtilis (Real et al. 2005); (ii) biofilm formation and DNA release for S. aureus (Fischer et al. 2014) (iii) programmed cell death and acetate metabolism in S. aureus (Ahn et al. 2012); and (iv) biofilm formation and oxidative stress adaptation in Streptococcus mutans (Westbye et al. 2013). A number of practical applications have been described for the holin/lysin systems, for example, aiming to control bacterial or viral infections (Yan et al. 2013; Shi et al. 2012) or to deliver drugs, nucleic acids, and proteins to animal cells (Kuo et al. 2009).
2.7 Non-classically Secreted Proteins
Extracellular proteomic studies revealed that a number of proteins are found in the extracellular medium without any secretion signal (Tjalsma et al. 2004). As their secretion route is not known, they are indicated as “non-classically secreted proteins” (Wang et al. 2016). Although there is a debate if these proteins in the extracellular medium are not a consequence of cell lysis, evidence was given by Yang et al. (2011), who experimentally showed that the B. subtilis carboxylesterase Est55, and several other cytoplasmic proteins are secreted through a process in which the protein domain structure plays a contributing role. Furthermore, using enolase to which the heterologous protein GFP was fused it was shown that the intact long N-terminus including the hydrophobic helix domain is required to serve as a non-cleavable signal for the secretion of enolase (Yang et al. 2014). Moreover, signals of “non-classically secreted proteins” could be more generally used for the secretion of heterologous proteins (Chen et al. 2016).
Despite the presence in Gram-positive bacteria of a variety of different export systems, the industrial production of (heterologous) proteins has relied primarily on the Sec-dependent pathway and to a far lesser extent the Tat pathway. Below an overview is given for using these systems in a number of different Gram-positive host cells.
3 Gram-Positive Bacteria as Hosts for Heterologous Protein Production
Gram-positive bacteria are considered interesting hosts for the production of heterologous proteins. An important advantage they have is that secreted proteins are released into the culture medium in which the conditions are favorable for the correct folding of heterologous proteins. This contrasts to the reducing environment of the cytoplasm, in which secretory proteins that undergo oxidative folding, cannot fold. In addition, secreted proteins have the advantage that the mature protein has no methionine extension, but the authentic N-terminal amino acid sequence because of the cleavage by the signal peptidase. Therefore, several Gram-positive bacteria have been evaluated as hosts for the secretory production of heterologous proteins. Reasons why specific species have been tested are, for example, their industrial importance (or that of their relatives) and known fermentation technology, their proven secretion capacity, the absence of pathogenicity and toxicity and available tools for genetic manipulation. In Table 1, a number of possible advantages/disadvantages are compared for Gram-positive bacteria versus Gram-negative bacteria (E. coli).
3.1 Streptomyces
Streptomycetes belong to the phylum Actinobacteria, filamentous or rod-shaped bacteria, of which the filamentous forms tend to produce branching filaments. These Gram-positive soil bacteria are widespread in nature, they have a high guanine and cytosine content in their DNA (70–73 % GC) and a remarkably large genome size of up to 11.9 Mbps (S. bingchenggensis BCW-1; Accession ID: CP002047) (Wang et al. 2010) and with gene clusters from just a few to more than 30 pathways for the biosynthesis of a diverse range of secondary metabolites (Nett et al. 2009). Various Actinomycetales species are the richest source of natural products, they account for about 45 % of all microbial bioactive secondary metabolites with about 80 % of the 7600 compounds being produced by streptomycetes (Berdy 2005). Many of these secondary metabolites are of industrial and pharmaceutical value, including clinically important antibiotics for human and veterinary medicine or applied in agriculture, anticancer, and immunosuppressive agents, other pharmacologically active compounds, antiparasitic agents, and herbicides. Streptomycetes also play an important role in nature. Thanks to the large variety of enzymes they produce, such as cellulases and chitinases, they help to break down decaying vegetation as such playing an important role in the C- and N-cycle and replenishing the soil with nutrients. Typical for streptomycetes is their complex life cycle: Under suitable growth conditions, exospores germinate and subsequently develop into hyphae, which frequently become branched forming the vegetative mycelia that subsequently differentiate to aerial mycelia. Finally, aerial mycelia become divided into long chains of prespore compartments, which eventually mature to thick-walled exospores. In this phase of the life cycle, a large array of secondary metabolites is produced (van Wezel and McDowall 2011).
Strains of the class Actinobacteria include some of the most common soil, freshwater, and marine life. Other Actinobacteria inhabit plants and animals, including some pathogens such as M. tuberculosis, several strains of Corynebacterium, Nocardia, Rhodococcus spp. and a few Streptomyces species (Goodfellow 2012).
Over the last 20 years, some of these high-GC Gram-positive bacteria have been studied extensively as an alternative expression system [reviewed in Anné et al. (2014)]. Streptomyces is extremely well suited for the expression of DNA from other actinomycetes and genomes of high-GC content. Furthermore, due to its high innate secretion capacity, Streptomyces can be a better system than E. coli for the production of many extracellular proteins. The host of choice for secretory protein production of heterologous proteins using Streptomyces is S. lividans. The main reasons are its limited restriction–modification system as such avoiding the requirement to use non-methylated DNA for transformation or conjugation, and its low endogenous protease activity, when compared to many other streptomycetes (Butler et al. 1996). Based on whole-genome sequence analysis of S. lividans TK24 (Ruckert et al. 2015) and RNAseq analysis, S. lividans transcribes only a limited number of genes encoding proteases under standard growth conditions in minimal media. Only one-third of the genes encoding secreted proteases are transcribed at medium to high level (Tobias Busche and Jörn Kalinowski, personal communication). In case of cytoplasmic or membrane-bound proteases, 75 % of the encoding genes are transcribed at medium to high level.
A wide variety of host–vector systems have been developed, many of which are based on plasmid pIJ101, such as pIJ702 and pIJ486 (Kieser et al. 2000), but in addition, a large array of new vectors has been developed including replicative plasmid vectors, integrative plasmid and phage vectors, and special vectors for integrating DNA into the Streptomyces chromosome [for an overview see Rebets et al. (2016)]. As protoplast transformation with Streptomyces is time-consuming, conjugative plasmids are most often used for cloning purposes.
A number of heterologous prokaryotic and eukaryotic proteins have been successfully produced to economically interesting yields. For example, the L-Lysine α-oxidase (LysOX) gene from Trichoderma viride, a homodimeric 112-kDa flavoenzyme LysOX, was cloned and heterologously expressed in S. lividans TK24 with an enzyme activity up to 9.8 U/mL. Cel6A-(His)6 was secreted in S. lividans supernatant after 84 h of cultivation amounted to 5.56 U/mL. The maximum expression level of Cel6A-(His)6 in S. lividans supernatant reached up to 173 mg/L after 84 h of cultivation (Li et al. 2013). Using the promoter and signal sequence of subtilisin inhibitor of S. venezuelae CBS762.70 (Van Mellaert et al. 1998) yields of up to 300 mg/mL biologically active mouse TNFα could be obtained and monomeric red fluorescent protein yielded up to 500 mg/mL. In some cases, proteins which could hardly or not be produced in B. subtilis or E. coli such as e.g., xyloglucanase from Jonesia sp. (Sianidis et al. 2006) and CelA from Rhodothermus marinus (Halldórsdóttir et al. 1998) were successfully produced as secreted proteins with S. lividans. Mycobacterium Ag85A produced in S. lividans used in combination with rCFP-10, rESAT-6, rAPA, rPstS-1 obtained via E. coli heterologous production in an ELISA multi-antigen was shown to be an efficient, complementary tool for the diagnosis of active pulmonary tuberculosis (Ayala et al. 2015). In other cases, however, only low yields could be obtained, a phenomenon also experienced with other expression systems. For the examples mentioned above, the Sec-dependent secretion pathway was used. For a more complete overview of heterologous proteins secreted using recombinant S. lividans, see Anné et al. (2012). After the detection of the Tat-dependent pathway in bacteria, one was convinced that the latter pathway could be a solution for the production of heterologous proteins not or hardly produced via the Sec pathway. However, so far this hope has not been materialized. This does not mean, of course, that the Tat pathway cannot be used for the production of heterologous proteins, but it still has to be investigated at a larger scale. Surprisingly, Sec-dependent translocation in tat deletions mutants and especially in ΔtatB mutants showed an increase (Schaerlaekens et al. 2004). No real explanation for this phenomenon could be given up to now.
3.2 Corynebacterium
Other Gram-positive bacteria with high-GC content are corynebacteria. Some species such as C. diphtheria are important pathogens, while the majority are not pathogenic and some are industrially very important as major producers of amino acids including glutamic acid, lysine, threonine and valine (Mitsuhashi 2014), nucleotides, and vitamins. In particular, C. glutamicum is of major industrial importance. Corynebacteria are also able to produce large amounts of extracellular proteins despite their diderm-mycolate cell wall. C. glutamicum has several attractive features, making it a potentially interesting host for the production of heterologous proteins at an industrial scale: it secretes few endogenous proteins, and no proteases in the culture filtrate are detected, although a proteome analysis revealed the presence of more than 40 proteins in the culture supernatant (Hermann et al. 2001). As a consequence, C. glutamicum has been shown to be a valuable host for the production of heterologous proteins including functionally active human epidermal growth factor (Date et al. 2006), thus demonstrating its potential for industrial-scale production of human proteins. Expressed proteins can be secreted through the Sec or the Tat pathway. For example, isomaltodextranase (IMD) of Arthrobacter globiformis and Streptomyces mobaraensis pro-transglutaminase (MTG) was produced via the C. glutamicum Tat pathway and yields could reach approximately 100 mg/L in flask cultures. This achievement implies a great potential for the industrial-scale production of proteins that are not efficiently secreted via other systems (Kikuchi et al. 2006).
3.3 Bacillus
Bacillus species are aerobic, endospore-forming, rod-shaped cells that are ubiquitously present in nature. Various Bacillus species including B. subtilis, Bacillus licheniformis, and Bacillus amyloliquefaciens can produce various enzymes including proteases, amylases, and lipases in amounts up to 25 g/L. These proteins are used in different industrial and household applications such as in the cleaning, paper, textile, food, and feed industry and also for bioremediation. Because of the efficient protein secretion of these bacilli, their secretion process is intensively investigated, in particular for B. subtilis which for this purpose is considered the model organism among Gram-positive bacteria. More insight in the fundamentals of the secretion process is meaningful to develop strains with superior secretion capacity. Whereas improvement of protein secretion was in general quite successful for homologous proteins, production of heterologous proteins was more cumbersome. The most important reasons therefore are a combination of the properties of the secretion pathway, the bacterial cell envelope, and the presence of a number of membrane-bound, cell-wall-bound, and secreted proteases (Westers et al. 2008). For example, the quality control proteases, WprA, HtrA, and HtrB, and feeding proteases, NprB, AprE, Epr, Bpr, NprE, Mpr, and VprA, quickly degrade slow-folding, or wrongly folded proteins (Pohl and Harwood 2010). Nevertheless, some heterologous prokaryotic and eukaryotic proteins could be well expressed, as quantities ranging from less than 10 µg up to more than 200 mg/L could be obtained [for an overview, see Schumann (2007), Kang et al. (2014)] Other reports mention the overproduction of α-amylase from B. licheniformis in a recombinant B. subtilis strain (Chen et al. 2015a); and the accumulation of biologically active hIL-3 in the growth medium in amounts of up to 100 mg/L (Westers et al. 2006).
3.4 Lactobacilli
Lactic acid bacteria (LAB) are a phylogenetically diverse group of Gram-positive, aerotolerant, non-spore-forming rods or cocci with a low-GC genome. They ferment carbohydrates with lactic acid as the major end-product. They are commonly used to ferment food and as probiotics. Lactobacilli are part of the normal flora of humans and animals. The reason why strains of LAB, mainly lactococci and lactobacilli, are chosen as cell factories are plentiful: (1) many species are generally recognized as safe (GRAS) organisms because they are traditionally used in food products; Lactobacillus infections occur very rarely; if so, they are opportunistic infections, especially in immunocompromised individuals (Schlegel et al. 1998); (2) genetic tools for manipulation of LAB are well-developed; (3) strains of Lactococcus lactis secrete relatively few proteins and express very few membrane-bound or secreted proteases. In such strains, HtrA is the only protease that has been characterized on the extracellular surface (Poquet et al. 2000). (4) A variety of constitutive and inducible vector systems have been developed including the well-known 2-component NIsin-Controlled gene Expression system (NICE). This system derives from the auto-induced expression of nisin, an antibacterial polycyclic peptide produced by some strains of L. lactis (Kuipers et al. 1998). When nisin binds to the receptor NisK, a membrane-associated protein kinase, NisR becomes phosphorylated. The activated NisR then induces the nisin promoter (Mierau and Kleerebezem 2005). Small amounts of nisin are sufficient to activate the promoter. The NICE system is widely used for the expression of heterologous proteins in L. lactis (Mierau and Kleerebezem 2005).
Several proteins could be efficiently secreted using L. lactis and Lactobacillus plantarum as hosts as illustrated hereafter with a few examples. S. aureus nuclease NucA was secreted in amounts of more than 200 mg/L culture medium (Tremillon et al. 2010; Karlskas et al. 2014); the C-terminal region of staphylococcal HtrA transmembrane proteins could efficiently be produced and secreted in L. lactis as correctly folded proteins (Samazan et al. 2015); L. lactis was shown to be a suitable host to express a variety of structurally different glycoside hydrolases of LAB in their native, multi-meric form (Schwab et al. 2010); B. subtilis oxalate decarboxylase (Anbazhagan et al. 2013); Thermobifida fusca cellulases and xylanases to convert biomass to biofuels using Lactobacillus plantarum as a host (Morais et al. 2013); recombinant L. lactis was able to secrete biologically active human interferon-γ inducible protein-10 (Villatoro-Hernandez et al. 2008). Chitosanase (CsnA) and a β-mannanase (ManB) from B. licheniformis and B. subtilis, respectively, were efficiently produced in L. plantarum (Sak-Ubol et al. 2016). More examples can be found in an overview given by Le Loir et al. (2005). For the expression and secretion of the heterologous proteins, mentioned in these examples, different plasmids (inducible), promoters, and signal peptides have been used.
An additionally interesting aspect of recombinant lactococci is that they can be used as live vectors for the delivery of antigenic or therapeutic proteins to mucosal surfaces in the framework of the treatment of allergic, infectious, and gastrointestinal diseases. This use has the potential to elicit antigen-specific secretory immunoglobin A responses at mucosal surfaces (Pontes et al. 2011; Bermudez-Humaran et al. 2011). L. lactis engineered to secrete bioactive molecules such as Interleukin-10 (IL-10), an anti-inflammatory cytokine, was shown to be beneficial in the treatment of inflammatory bowel disease (IBD). L. lactis producing IL-10 markedly reduced the pathology of colitis in several mouse models. Another strain expressing a Fab against TNF-α was also effective in the treatment of IBD (Vandenbroucke et al. 2010). A truncated version of the A2 antigen from Leishmania donovani expressed in L. lactis as cell wall anchored protein effectively gave induced high levels of antigen-specific serum antibodies (Yam et al. 2011). Subcutaneous immunization with live L. lactis expressing the LACK antigen anchored to the cell wall and L. lactis secreting IL-12 significantly delayed footpad swelling in Leishmania major infected BALB/c mice (Hugentobler et al. 2012). For a more extensive overview of protection studies with LAB vaccines, see among others in Wells and Mercenier (2008).
In addition, lactobacilli can be used for the delivery of DNA at the mucosal membrane. To improve the delivery, so-called invasive L. lactis strains were developed. These recombinant strains expressed S. aureus fibronectin-binding protein A or internalin A of Listeria monocytogenes (de Azevedo et al. 2015) or a mutated form thereof (Pontes et al. 2014) to increase the invasiveness of the strain and subsequent DNA delivery. Several examples showed the feasibility of this approach to elicit an immune response using DNA vaccination with L. lactis as a vector.
3.5 Clostridium and Bifidobacterium
These genera have in common that they are both anaerobic and Gram-positive. Clostridia are rod-shaped, endospore-forming bacteria with a low-GC content. Clostridium is mainly known for its pathogens like Clostridium tetani, Clostridium botulinum, and Clostridium perfringens, which secrete potent toxins leading to the life-threatening diseases tetanus, botulinum, and gangrene, respectively. Clostridium difficile is mainly a nosocomial pathogen and the causative agent of antibiotic-associated pseudomembranous enterocolitis. From the biotechnology point of view, C. acetobutylicum is an important producer of butanol and acetone. Pasteur was the first to report the fermentation process of butanol already in 1861 (Jones and Woods 1986). Stimulated by the First World War, the acetone and butanol fermentation gradually became a most important industrial fermentation processes until the 1950s. Then, the interest for the fermentative production of butanol and acetone wasted away because of cheap crude oil prices as raw material for their chemical synthesis. However, the acetone and butanol fermentation recently regained importance in the framework of renewable resources for biobutanol production. For this reason, several individual cellulosomal components and mini-cellulosomes from C. thermocellum and Clostridium cellulolyticum have been cloned and expressed in C. acetobutylicum and their gene products such as Cel5A, Cel8C, and Cel9M were successfully secreted into the medium. On the other hand, other cellulosomal component proteins such as Cel48F, Cel9G, and Cel9E could not be recombinantly obtained (Mingardon et al. 2011). The development of allele-coupled exchange (ACE) (Heap et al. 2012) for Clostridium allowed the generation of stable and iterative integrations within a relatively short period of time. As such three genes of C. thermocellum-derived cellulosome components inserted into the genome of C. acetobutylicum could be efficiently expressed, with subsequent secretion and complex formation (Kovacs et al. 2013).
Clostridium spp. came also in the focus of research for a totally different application, notably in the framework of anticancer therapy. As anaerobic bacteria survive and multiply only under anaerobic conditions, after intravenous administration they selectively colonize, if present, in the hypoxic/necrotic areas of solid tumor tissue, a consequence of inconsistent and insufficient blood flow within regions of the tumor. When administered to a tumor-bearing body, the hypoxic/necrotic zones in solid tumors are ideal niches for the growth of anaerobic bacteria, as other tissues in a body are well oxygenized. This selectivity is repeatedly demonstrated with experimental animals (Umer et al. 2012; Roberts et al. 2014). Strains tested during these experiments belong to the following species: C. acetobutylicum, C. sporogenes, an attenuated C. novyi-NT or C. beijrinckii and more recently also C. ghonii (Wei 2013). When multiplying in these tumor tissues, they destroy (part of) the tumor by the hydrolytic enzymes they produce. Besides, by combinatorial treatment antitumor activity can be increased by using recombinant strains in which genes for prodrug converting enzymes are cloned. Examples of such genes are nitroreductase that converts the CB1954 prodrug to an active antitumor drug (Theys et al. 2006; Heap et al. 2014) or cytosine deaminase (CDase) which converts 5-fluorocytosine to the cytotoxic drug 5-fluorouracil. Using appropriate signal peptides, CDase can be secreted in sufficient amounts to be of biological relevance as is also the case for cloned TNF-α or IL-10, cytokines with an antitumoral but also with an immune stimulating activity to combat the tumors [for an overview see Umer et al. (2012)].
Bifidobacteria are non-spore-forming, non-motile, often branched rod-shaped bacteria with a % GC value of circa 60. They are ubiquitously found in the intestines, and because they have a probiotic function they are often used in yogurt. Engineered Bifidobacterium adolescentis expressing endostatin (specifically inhibiting the proliferation of vessel endothelial cells stimulated by basic fibroblast growth factor, and hence also inhibiting tumor growth), when intravenously administered to tumor-bearing mice were found only in the tumor. They inhibited angiogenesis and tumor growth (Li et al. 2003). It must be mentioned that in this case endostatin was not secreted into the medium, but was expressed intracellularly. The above-mentioned results show the potential of using (recombinant) anaerobic bacteria as tumor-specific vectors to transport anticancer genes/proteins to tumor tissues.
4 Bottlenecks in Protein Secretion and Possible Remediation
To be economically interesting, recombinant strains should produce sufficient amounts of the protein of interest. However, in many cases concentrations are low or too low. Various reasons could be at the root of the problem (see Table 2). As a consequence, several approaches can be followed in an attempt to increase the yield: from strain engineering at several levels up to fermentation optimization and this using either rather empirical approaches up to more sophisticated ones, applying state-of-the-art technologies (Fig. 2). In the following paragraphs, several examples will be used to illustrate these possibilities.
4.1 Modulation of Components of the Protein Secretion Pathway
It is evident that the promoter is of utmost importance for high expression levels. Looking into the literature, a wide variety of promoters are available for different bacteria, either constitutive or inducible. Sources of promoters vary from native to synthetic promoter libraries. Promoter strength can be compared using reporter proteins such as β-glucuronidase (GusA) (Siegl et al. 2013), mCherry (Heiss et al. 2016) and others. To have a recent overview of different promoters used for Streptomycetes, we refer to Rebets et al. (2016).
The availability of strong promoters does not guarantee that the protein will be produced at sufficient levels, because bottlenecks are mainly at the secretion level and more downstream. Therefore, several approaches have been attempted related to the protein secretion pathway itself (Fig. 1).

Schematic overview of possible strategies for increased protein production
4.1.1 Signal Peptide Adaptation
It is not clear what determines the sequence of an “efficient” signal peptide; therefore, several approaches are being investigated including single amino acid replacements in the N-terminal region of the signal peptide (Lammertyn and Anné 1998) or testing large Sec-type signal peptide libraries (Mathiesen et al. 2009; Degering et al. 2010). Considerable differences exist between different signal peptides but also for different mature proteins for the same set of signal peptides and this independently of the host tested. Amino acid extension by which the amino acids in the neighborhood of the signal peptidase cleavage site are conserved could also be helpful (Sevillano et al. 2016).
4.1.2 Signal Peptidase Overexpression
As explained above, several proteins constitute the protein secretion pathway with proteins different for the Sec- and Tat-dependent pathway. What they have in common is the signal peptidase type I, an enzyme needed to release the signal peptide from the mature protein upon translocation. Most Gram-negative bacteria have only 1 chromosomally encoded signal peptidase I, but some have more. For example, Pseudomonas aeruginosa has two (LepB and PA1303), each with a different role in virulence and physiology (Waite et al. 2012). On the other hand, with the exception of i.a. Streptococcus pneumoniae, M. tuberculosis, and S. aureus, many Gram-positive bacteria have more than 1 SPase I with a maximum for B. subtilis which contains 5 chromosomally encoded SPase I genes (sip); namely sipT, sipS, sipU, sipV, and sipW. In addition, various B. subtilis strains contain in addition 2 plasmid encoded (sipP) SPase I genes (van Roosmalen et al. 2004). SipS and SipT are key to preprotein processing, while SipU, SipV, and SipW appear to play minor roles in protein secretion (Tjalsma et al. 1998). Overexpression of signal peptidases resulted in an increased level of secretion (Pummi et al. 2002). Similarly, in Bacillus megaterium MS941, co-overexpression of its unique signal peptidase SipM increased the heterologously expressed Leuconostoc mesenteroides dextransucrase (Malten et al. 2005).
S. lividans has 4 chromosomally encoded SPases I (SipW, SipX, SipY, and SipZ) (Parro et al. 1999). None of the individual SPases I was found to be essential for cell viability, indicating they have an overlapping substrate specificity. Nevertheless, SipY was shown to be the major SPase as the secretome of a SipY-deficient strain is severely affected (Palacin et al. 2002) on growth as well as on morphology (Gullón et al. 2012). Moreover, in particular cases the SipY mutant was shown to have some interesting advantages compared to the wild-type S. lividans for the overproduction of extracellular agarose probably as a consequence of the diminished extracellular proteolytic activity (Gabarró et al. 2016). Alternatively, co-overexpression of all 4 sip genes led to the highest increase in total preprotein processing capacity of the cell, and also to a higher amount of extracellular human CC16. It can thus be concluded that for S. lividans both overexpression and inactivation of individual Sip proteins can be advantageous for yield improvement of secretory proteins (Geukens 2002).
4.1.3 Overexpression of Chaperones and Foldases
When heterologous proteins are expressed, they ought to obtain their correct conformation, both for activity and stability, as incorrectly folded proteins are more prone to proteases and aggregation. Correct conformation is obtained with the help of chaperones. Folding facilitators are, for example, the DnaK chaperone (DnaK, DnaJ, and the nucleotide-exchange factor GrpE) and GroEL/ES (mainly studied in E. coli) which assist the folding of newly synthesized proteins and prevent protein aggregation. Following secretion, peptidyl-prolyl cis/trans isomerases (PPIases) and disulfide bond formation proteins (Dsb) are needed for formation and rearrangement of disulfide bonds. Six Dsb (A-G) have been identified in E. coli. In Gram-positive bacteria, this folding process is hardly investigated except for Bacillus. The main components responsible for secretory protein folding and quality control in B. subtilis are summarized in Sarvas et al. (2004). The lipoprotein PrsA, a putative peptidyl-prolyl cis/trans isomerase, plays a major role in protein secretion by helping the posttranslocational extracellular folding of several secreted proteins. The presence of the extracytoplasmic enzymes thiol-disulfide oxidoreductases (TDOR) in B. subtilis were identified based on data searches. They were named BdbA (YolI), BdbB (YolK), BdbC (YvgU), and BdbD (YvgV) (Kouwen and van Dijl 2009). It was shown that BdbB and BdbC are involved in the folding of tested proteins including PhoA and A13i-Bla (Bolhuis et al. 1999). Overexpression of chaperones is considered an attractive approach to increase yield of heterologous proteins (Mogk et al. 2002). Overexpression of prsA in Bacillus, for example, increased the secretion of α-amylases, recombinant protective antigen, and a protease (Williams et al. 2003; Vitikainen et al. 2005; Chen et al. 2015b). Overexpression of the B. subtilis TDOR genes, however, did not improve the folding of the secreted heterologous proteins as investigated with PhoA. On the other hand, overexpression of the DsbA from S. aureus or the S. carnosus DsbA allowed the secretion of active PhoA at elevated levels (Kouwen and van Dijl 2009). Folding modulators in other Gram-positive bacteria and their impact on heterologous protein production have not yet been investigated. Based on homology searches in the genome of the S. lividans TK24, chaperones and peptidyl-prolyl cis/trans isomerases have been identified (Tobias Busche and Jörn Kalinowski, personal communication), but their effect on (heterologous) protein secretion has still to be investigated.
4.1.4 Sec Components
The Sec translocase consists of the integral membrane complex SecYEG, the ATPase SecA, and two additional membrane proteins that promote the release of the mature peptide across the cytoplasmic membrane (SecD and SecF). In E. coli, SecD and SecF are two separate membrane proteins, whereas in B. subtilis they are present as one polypeptide, named SecDF (Bolhuis et al. 1998). It is required to maintain a high capacity for secretion. It is not essential, but its deletion results in low-temperature sensitivity, aberrant cell division, and impaired protein secretion. The secDF deletion mutant exhibits a reduced level of secreted proteins (Vorös et al. 2014). Few attempts have been made to modulate specific Sec proteins to improve protein secretion. One example is the co-expression in B. subtilis of the E. coli SecB and a hybrid SecA of B. subtilis in which the 32 C-terminal amino acids end was replaced by the corresponding SecA fragment of E. coli (Diao et al. 2012). This artificial protein targeting pathway led to a significant increase in the secretion of 2 model proteins tested, mutant maltose binding protein (MalE11) and alkaline phosphatase (PhoA), which B. subtilis could hardly export using the native secretion pathway. Kakeshita et al. (2010) deleted 61 amino acids of the C-terminus of SecA, a region, known to bind SecB in E. coli. In Gram-positive bacteria; however, SecB is absent, and the C-terminal region is not essential for protein secretion nor for growth. Moreover, the 61 amino acid deletion dramatically increased the extracellular production of the heterologous proteins alkaliphilic Bacillus sp. thermostable alkaline cellulase (Egl-237) and human interferon a (hIFN-a2b) in B. subtilis. Differential expression of SecA demonstrates that various precursors may exhibit major differences in their dependency on the amount of functional SecA in the cell (Leloup et al. 1999). In some cases, therefore, SecA overexpression or mutation might be beneficial for improved protein secretion as shown for cutinase in B. subtilis (Brockmeier 2006).
4.1.5 Tat Translocon Overexpression
The Tat pathway represents an alternative pathway for the production of secreted recombinant proteins, in particular for proteins prefolded in the cytoplasm. Notwithstanding this particular property, this pathway is so far not much explored for the industrial production of (heterologous) proteins. Several reasons account for this: Protein yield of Tat-exported proteins is in general substantially lower than of Sec-secreted proteins, and much of the synthesized proteins is retained in the cytoplasm (DeLisa et al. 2004). This might be a consequence of the fact that the export machinery becomes easily saturated not only by overexpressed target proteins, but even for native Tat-exported proteins (Barrett et al. 2003). The saturation of the export machinery can partially be relieved by co-expression of proteins of the Tat translocon. The stoichiometry of the TatABC components seems, however, critical for export function. For example, in E. coli overexpression of tatB resulted in complete loss of Tat transport, overexpression of tatA has a less severe but nonetheless significant effect on translocation (Sargent et al. 1999), while high expression of tatC can relieve saturation of the Tat pathway (DeLisa et al. 2004). Therefore, most attempts to relieve the saturation problem of the Tat translocon have been done by the coordinated overexpression of TatABC. This can certainly have a positive effect on the secretion of Tat-dependent proteins as illustrated for different organisms both Gram-positive and Gram-negative bacteria. When TatABC were overproduced in S. lividans, a fivefold increased xylanase C secretion was noticed. Surprisingly, the overproduction of TatABC in S. lividans caused a strong reduction in the secretion of the monitored Sec-dependent substrates (De Keersmaeker et al. 2006), suggesting a possible cross talk between the Tat- and Sec-dependent protein secretion pathway. Also for C. glutamicum overexpression of Tat components dramatically increased the secretion of Chryseobacterium proteolyticum pro-protein glutaminase (pro-PG) and Streptomyces mobaraensis pro-transglutaminase (pro-TG). The amounts of secreted pro-PG were more than threefold higher when TatC or TatAC was overexpressed, and there was a further threefold increase when TatABC were overexpressed (Kikuchi et al. 2009). More recently, Albiniak et al. (2013) investigated the ability of an E. coli tat null mutant containing B. subtilis TatAdCd system to export the Tat-dependent model protein GFP. These cells do indeed export GFP to the periplasm with high efficiency; moreover, the protein was subsequently released into the extracellular medium during batch fermentation. The latter property was a consequence of the fact that the E. coli tat null mutant strain has impaired outer membrane integrity (Ize et al. 2003). Such an example shows that within Gram-positive bacteria, similar approaches can be tested to optimize secretion yield of (heterologous) proteins and to broaden the spectrum of proteins that can be produced by Gram-positive bacteria.
Tat-dependent secretion could be further increased if the phage shock protein (PspA) was overexpressed as shown for S. lividans (Vrancken et al. 2007), which was also true for the Gram-negative E. coli (DeLisa et al. 2004). The beneficial effect of PspA overproduction could be a consequence of its effector role in the maintenance of the integrity of the cytoplasmic membrane and proton motive force (Darwin 2005), the latter providing the energy in Tat-dependent protein translocation.
4.2 Omics Approaches for Enhanced Protein Secretion
Thanks to the new and fast techniques of DNA sequencing for genome analysis and RNAseq for transcriptome analysis, the availability of new methods for proteome analyses and the massive amounts of data and intelligent bioinformatics tools, it has now become more easy to have an insight at the systems-level burden caused by the overproduction of proteins, by the presence of plasmids and the biosynthesis and secretion of heterologous proteins. Stepping closer to the observed phenotype, metabolites and metabolic fluxes matter most and can be investigated using metabolomics and fluxomics techniques. Omics approaches are new drivers for rational engineering of host strains for improved fitness and increased productivity. Despite the availability of these new resources and their potentialities, so far not much research has been done in this field.
4.2.1 Transcriptomics and Proteomics
One of the first studies in which transcriptomics and proteomics studies were combined to understand the physiological and metabolic changes that occurred in high cell density cultivation (in order to obtain higher yield) was done with E. coli (Yoon et al. 2003). A recent study with B. licheniformis investigated the early responses to physical stress and nutrient starvation using integrated transcriptomics and proteomics (Voigt et al. 2014). With this approach, they were able to identify general and specific marker proteins for different stress and starvation conditions including high protein secretion. Such markers might be interesting to follow the production process, and when needed to adapt it accordingly.
Because of its improved Tat-dependent protein secretion, the transcriptional profile of the S. lividans pspA mutant (see Sect. 4.1.5) was compared with the wild-type strain to see whether genes were differentially expressed in the pspA mutant. A number of genes were shown to be up- or downregulated in the mutant strain using a microarray screen containing all genes of S. lividans (Anné et al. 2014). Sixty-seven genes were twofold or more upregulated in the pspA mutant, while 117 genes were down-regulated. Among the genes encoding proteins for which a function is known or predicted, there are several which are linked to stress regulation (cold shock proteins, sigma factors), while others are involved in metabolic processes such as energy production and conversion and general metabolism. Among others, an increased expression of sco6996 in the pspA mutant was identified. The corresponding protein SCO6996 shows some homology to the RNA polymerase sigma factor RpoE and experiments in Salmonella Typhimurium previously showed that RpoE can (at least partially) compensate for the lack of PspA (Becker et al. 2005). Loss of either pspA or rpoE leads to a depolarization of the membrane potential, indicating that both can affect the PMF. Moreover, PspA overproduction could partially compensate for the loss of RpoE in a Salmonella Typhimurium ΔrpoE strain. Furthermore, Gordon et al. (2008) recently showed that overexpression of one particular sigma factor (SigU) in S. coelicolor could lead to a significant alteration in the secretome. The SigU-overproducing strain secreted a much greater quantity and diversity of proteins than the wild-type strain, revealing that modification of the sigma factor expression in S. lividans might also affect protein secretion. Overexpression of sco6996 led to an increased secretion of the tested proteins (XylC, eGFP) through the Tat pathway. This increase was far less pronounced than in the case of PspA overexpression, but still yielded a 20 % increase in final protein yield, which is still highly interesting. In another study, transcriptomics expression profiles of S. lividans TK24 strains producing the heterologous proteins human/mouse tumor necrosis factor alpha (hTNFα/mTNFα), monomeric red fluorescent protein, and xyloglucanase were compared to the corresponding control strain containing the empty vector only. Based on these analyses, a number of genes showed a significant twofold change in the recombinant strains overproducing the heterologous proteins. One gene, encoding a phosphoenolpyruvate carboxykinase (PEP carboxykinase) involved in the tricarboxylic acid (TCA) cycle and gluconeogenesis, was selected for further investigation. Overexpression of this gene in S. lividans TK24 hTNFα and xyloglucanase C production strains increased almost twofold the yield of recombinant hTNFα (Lule et al. 2012) and XylC in comparison with the initial production strains. Overall, these results show that a transcriptomics-based approach represents a useful tool for a rational optimization of heterologous protein secretion in S. lividans.
4.2.2 Metabolomics and Fluxomics
Metabolomics refers to the comprehensive analysis of small molecules produced by cellular metabolism. Metabolites inside as well as outside the cell (referred to as the endo- and exometabolome) can help to understand phenotypic behavior of recombinant strains, can be used for metabolic flux estimation, and can assist in strain development when combined with other omics data. Analysis is mostly done with mass spectrometry (MS) preceded by chromatographic separation, for which the technique of choice depends on the depth of analysis, the targeted metabolites, and the type of application [e.g., Garcia-Ochoa and Gomez (2009)]. Metabolomics, however, does not reach the same high resolution as RNAseq- or MS-based proteome analysis [e.g., Goodacre et al. (2004)]. From the vast pool of small molecules, some hundreds of metabolites can be detected in untargeted analysis but less than a hundred metabolites can usually be identified and quantified in a targeted metabolome analysis. When analyzing for intracellular metabolites, rapid quenching is required (e.g., in cold methanol, in liquid nitrogen) since their metabolite levels can quickly change (e.g., order of seconds in the central carbon metabolism) upon sampling. Quenched cells are then separated from the culture broth (e.g., centrifugation), and metabolites are extracted from the cell pellet (e.g., freeze-thawing cycles, ethanol boiling). Final derivatization follows when using GC-MS analysis. All stages need to be carefully evaluated and optimized to avoid leakage during quenching, to ensure complete extraction of metabolites, and to minimize loss of metabolites. Protocols can be found in literature but require validation prior to their application. Some examples of exo- and endometabolome analysis for Gram-positive hosts for heterologous protein expression are given in the next paragraph.
D’Huys et al. (2011) performed a comprehensive exometabolome profiling of wild-type, empty plasmid-containing and mTNFα-producing S. lividans. Metabolite profiles revealed that glutamate and aspartate are two important growth-determining amino acids. Cometabolization with glucose results in a high growth rate, although this fast biomass accumulation did not coincide with the highest mTNFα to biomass yield. Overflow of alanine and organic acids was typical for the fast growth phase and pointed out the imbalance in carbon and nitrogen metabolism. After depletion of aspartate and glutamate, growth slows down and mTNFα yield increases. Entering the stationary growth phase after glucose depletion, a diauxic shift toward consumption of overflow metabolites can be observed and mTNFα yield was maximal. Fed-batch processing is proposed as a strategy for tackling overflow metabolism. Based on the protocol for endometabolome analysis developed in Kassama et al. (2010), Muhamadali et al. (2015) performed a complementary endometabolome analysis which confirmed the intracellular metabolic shifts and observed organic acids and sugar overflow inside mTNFα-producing S. lividans.
A first example of using metabolome profiling for debottlenecking heterologous protein production is described in Korneli et al. (2012). Green fluorescent protein (GFP) production by B. megaterium was investigated. Large-scale bioreactor conditions are mimicked in small-scale bioreactors by intermitted feeding of substrate, hereby inducing periods of feast and famine and resulting in a reduced process performance and product yield. Detailed time course of intracellular metabolites uncovered limitations in particular amino acids which could be resolved by supplementation of these amino acids during fermentation.
To fully understand the nature of metabolic bottlenecks and associated metabolome profiles, however, one needs to investigate metabolic fluxes in metabolic reaction networks. Fluxomics refers here to any technique applicable for this metabolic flux analysis. A genome-wide analysis of metabolic fluxes uses constrained-based metabolic modeling approaches [e.g., Lewis et al. (2012) and cited references therein], in which flux balance analysis (FBA) forms a central methodology. This FBA method is based on measured exchange rates of substrates and products, a genome-scale stoichiometric network model, steady-state assumption for intracellular metabolites, reaction flux constraints, and the optimization of a cellular objective function such as biomass growth or redox potential. This technique is tractable because of its genome-wide scope and commonly used for testing metabolic capacity of strains and for development of in silico strain engineering programs [e.g., Kim and Reed (2010), Schellenberger et al. (2011) and Wiechert (2001)]. However, exact knowledge of parallel reactions, bidirectional reactions, cycles, and flux split ratios requires 13C-based metabolic flux analysis [e.g., Wiechert (2001); Zamboni et al. (2009)]. 13C-based fluxomics is particularly suited for accurate flux calculations in the central carbon metabolism. Fluxes are estimated from intracellular mass isotopomer distributions in free intracellular metabolites or proteinogenic amino acids observed after feeding a 13C-labeled carbon source. 13C-labeling distributions will be determined by the actual reaction rates. Published flux maps are usually snapshots taken during a specific growth phase adopting a pseudo steady-state condition, but transient profiles of metabolic fluxes can also be modeled using dynamic FBA [e.g., Hjersted and Henson (2009)] or in stationary 13C-based flux analysis [e.g., Wiechert and Nöh (2013)].
Genome-scale FBA was applied by D’Huys et al. (2012) to get understanding in the metabolome profiles and growth of S. lividans in a complex medium. In contrast to the maximum biomass formation capacity predicted from the complex medium, S. lividans shows suboptimal growth illustrating that rich media do not necessarily support maximum biomass growth. Overflow metabolism could not be predicted but needed to be imposed by constraints. Uptake of amino acids clearly contributed to biomass growth by augmenting the pool of available amino acids and by increasing the fluxes in the tricarboxylic (TCA) cycle. Genome-scale analysis of metabolic fluxes during human growth hormone production with B subtilis also showed metabolic shifts during batch fermentation on a minimal medium as well as shifts in the number of reactions that carried fluxes (Özdamar et al. 2010). A 13C-based fluxomics was performed by Umakoshi et al. (2011) on batch cultures of C. glutamicum secreting heterologous transglutaminase (TGase). An increased flux through the pentose phosphate pathway for NADPH generation and also an increased flux through the TCA cycle augmenting the NADH/NAD and ATP/ADP ratios could be observed. This inspired the authors to increase the NADH/NAD ratio by promoting lactate production. Elevation of the pH from 6.2 to 7.0 gave a small yet notably 1.4-fold increase in product yield.
Fluxomics can also form the foundation for rational strain engineering, i.e., for the identification of interesting gene knockouts redirecting fluxes and leading to higher yields of the desired product. Advantage of fluxomics-based strain design is that the interconnected nature of the cellular metabolic reactions is taken into account. In the broader context of LAB-based vaccine production (Oddone et al. 2009), for example, applied dynamic genome-scale FBA to identify targets for enhanced heterologous protein production in L. lactis. Green fluorescent protein (GFP) expression (as a model protein) could be increased with 15 % by implementing predicted gene targets.
A recent trend to increase production performance of microbial host cells is genome reduction where large segments of the genomic DNA are removed with the intension of removing metabolic ballast and increasing resources of product formation. Genome reduction efforts often focus on production of secondary metabolites [e.g., Gomez-Escribano and Bibb (2011)], but their application to heterologous protein production has also been reported by Toya et al. (2014) and Lieder et al. (2015). The genome-reduced Pseudomonas putida strain created exhibits a 40 % increased GFP production (Lieder et al. 2015). Toya et al. (2014) transformed a genome-reduced B. subtilis (Morimoto et al. 2008) to produce heterologous cellulase and investigated fluxes in the central carbon metabolism using 13C fluxomics. A 1.7-fold increase in specific cellulase production rate as compared to the parental strain with empty plasmid was attained and flux maps reflects higher pentose phosphate pathway flux and thus NADPH generation, which seems to be a general requirement for enhanced recombinant protein production.
5 Fermentation
Development of a recombinant protein production process starts under laboratory conditions in small volume shake flasks. Many screening experiments for strain selection, testing of vectors, promoters and signal peptidases, and medium optimization are required. Laboratory-scale bioreactor experiments are performed for defining optimal culture conditions such as pH, dissolved oxygen, stirrer speed, and process operation (mostly batch or fed-batch). Screening can be greatly speed up by using high-throughput microbioreactor platforms. A first proof of principle for filamentous bacteria is reported for S. coelicolor by Sohoni et al. (2012). Rohe et al. (2012) developed a milliliter bioreactor screening platform and validated this setup for heterologous protein secretion of Fusarium solani pisi cutinase by C. glutamicum. Multiple cultures are run in parallel in a microtiter plate cultivation system (Biolector®) in which each well is stirred and dissolved oxygen, pH and biomass are monitored online. A liquid-handling robot enables swift media preparation, online dosing (e.g., for optimization of inducer concentration and time) and sampling. Auxiliary devices can be added for sample handling and online assaying (Unthan et al. 2015). Scalability for C. glutamicum was proven excellent to 1-L bioreactors and even up to 20-L bioreactors. This advanced automated platform of Rohe et al. (2012) proved also be applicable for Streptomyces, a species with a more complex growth physiology including clump formation (J. Koepff and M. Oldiges, Forschungszentrum Jülich GmbH, personal communication).
Yield of secreted heterologous proteins and productivity are affected by the medium constitution, growth phase and fermentation time. Optimization of the medium composition can be done randomly but a more rational approach is guided by design of experiments techniques [e.g., Mandenius and Brundin (2008)]. A satisfactory recombinant protein production typically requires nutrient-rich media containing amino acids. Pozidis et al. (2001), for example, illustrate fermentation upscaling and medium selection for murine tumor necrosis factors alfa (mTNF-α) production with S. lividans TK24 and tested different amino acid rich media. Final biomass and heterologous protein concentration show no consistency and protein yields are better in a less efficient growth medium, even with the use of a constitutive promotor. D’Huys et al. (2011) further demonstrated that the yield of mTNF-α increases after glutamate and aspartate depletion from the nutrient-rich medium and when the biomass growth rate slows down. Secreted protein yield becomes highest in the stationary phase but the fermentation must be stopped when degradation by extracellular protease activity is observed. Although complex media are commonly used in Streptomyces cultivations, cells do not exploit their nutritional resources optimally toward biomass formation and by-product formation is usually observed (D’Huys et al. 2012). Media can also be defined and identification of the most essential amino acids can be a tedious job. Nowruzi et al. (2008), for example, screened for the impact of different amino acids and defined mixtures of them on the heterologous expression of recombinant human interleukin 3 (rHuIL-3) in S. lividans 66.
Batch and fed-batch cultivation are both industrially relevant modi operandi in heterologous protein production. Batch operation is simple and flexible, but controlled substrate addition in fed-batch fermentations enables metabolic control and high density growth by avoiding by-product formation related to overflow metabolism. Minimization of acetate overflow by restricting the specific biomass growth rate is a common practice in E. coli recombinant protein production [e.g., Eiteman and Altman (2006)]. After reaching the high cell density at the end of the substrate feeding phase, heterologous protein expression is started by inducer addition. Heterologous protein production by Gram-positive bacteria can also be favored by fed-batch operation, although the fed-batch control strategy depends on the expressed protein, the promotor used and other factors. No standardized approaches as those established for E. coli seem to exist. A good illustration of the diversity and complexity in fed-batch operation strategies is given in Oztürk et al. (2016). Fed-batch processes for homologous and heterologous expression by Bacillus are reviewed and associated fed-batch operation strategies are derived. No consensus fed-batch operation strategy could be found and feeding strategies largely depended on promoter choice.
From a practical and financial point of view, microorganisms growing as single cells are more favorable for easy fermentation. A filamentous growth morphology increases the power input requirements in aerated bioreactors, increases cooling requirements and results into clump formation which introduces diffusion limitations and biomass heterogeneity. Not all cells in pellets and clumps of Streptomyces are biologically active and metabolically equal (Manteca et al. 2008; Rioseras et al. 2014). Large and dense clump formation can be partially counteracted by addition of hydrophilic polymers like polyethylene glycol [e.g., Kieser et al. (2000)]. Clump formation, mycelial differentiation and programmed cell death have been linked to the production of antibiotics and are important factors for secondary metabolite production by Streptomyces [Rioseras et al. (2014) and cited references therein]. Studies linking heterologous protein secretion and cellular morphology are limited, but van Wezel and coworkers found that overexpression of ssgA in Streptomyces leads to more fragmented growth without substantial clump formation and increases heterologous protein yields in S. lividans 1326 (van Wezel et al. 2006; Sevillano et al. 2016). Morphology engineering could be a strategy for enhanced protein secretion.
Upscaling from laboratory-scale to industrial-scale bioreactors generally reduces the final product yields due to spatial gradients, oxygen transfer limitations, shear stress, medium differences, etc. Guidelines for upscaling of bioreactor processes are described in many handbooks on bioreactor process engineering or fermentation technology [e.g., Doran (2013) and McNeil (2008)]. Many empirical relations have been established to estimate important parameters like oxygen transfer coefficients in large-scale fermenters, but upscaling generally remains a trial and error process based on some generally accepted rules of thumbs [e.g., Garcia-Ochoa and Gomez (2009)]. Keeping a constant dissolved oxygen concentration for aerobic processes with non-filamentous organisms or keeping a constant impeller tip speed for filamentous organisms are good starting points for upscaling. Oxygen supply to the production biomass is of key importance, and spatial differences are common in large bioreactors. Oscillations in oxygen availability can lead to temporary metabolic shifts, by-product formation, and eventually multi-substrate growth. Kass et al. (2014) characterize these effects for C. glutamicum and strain robustness can be guaranteed when temporary gradients are limited to the scale of a few minutes. Metabolic robustness toward spatial gradients in large bioreactors is a desired property of a robust production strain, but effects on heterologous protein secretion are not yet characterized.
6 Conclusion
Bacteria are fast-growing organisms able to be replicate in cheap culture media. This property makes them potentially attractive cell factories in biotechnological processes such as for the production of heterologous proteins. For well-known reasons, E. coli remains the host of choice but cannot meet all expectations as not all proteins are equally well produced in this Gram-negative host. Therefore, several Gram-positive bacteria are being explored and used as alternative bacterial hosts for the production of heterologous proteins. An important motivation therefore is that Gram-positive bacteria secrete proteins in the extracellular medium allowing correct folding, a problem encountered with E. coli in which case proteins often are precipitated in inclusion bodies impeding the purification process of the proteins to correctly folded active compounds.
The use of Gram-positive bacteria for heterologous proteins in secreted form shows mixed successes. While some proteins are produced in industrially viable amounts as secreted proteins, others give only small or disappointingly low yields. The new techniques that in recent years became available, including next-generation sequencing (NGS), RNA-seq, proteomics, metabolomics, and fluxomics combined with more advanced bioinformatics, and the improved understanding of the protein secretion pathways help to understand the cellular background that underlies production yield. Using this understanding allows rational strain engineering, possibly in combination with synthetic biology tools, and will undoubtedly broaden the applicability of Gram-positive bacteria for efficient use in protein secretion biotechnology.
Abbreviations
- FEA:
-
Flagella export apparatus
- GFP:
-
Green fluorescent protein
- GRAS:
-
Generally recognized as safe
- LAB:
-
Lactic acid bacteria
- mTNFα:
-
Mouse tumor necrosis factor α
- PMF:
-
Proton motive force
- PSPa:
-
Phage shock protein A
- Sec pathway:
-
General secretory pathway
- SPase I:
-
Signal peptidase type I
- T4SS:
-
Type 4 secretion system
- T7SS:
-
Type VII secretion system
- Tat:
-
Twin-arginine translocation
References
Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jimenez C, Parra M, Cadieux N, Brennan MJ, Appelmelk BJ, Bitter W (2009) PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol 73:329–340
Ahmed A, Das A, Mukhopadhyay S (2015) Immunoregulatory functions and expression patterns of PE/PPE family members: roles in pathogenicity and impact on anti-tuberculosis vaccine and drug design. IUBMB Life 67:414–427
Ahn SJ, Qu MD, Roberts E, Burne RA, Rice KC (2012) Identification of the Streptococcus mutans LytST two-component regulon reveals its contribution to oxidative stress tolerance. BMC Microbiol 12:187
Albiniak AM, Matos CFRO, Branston SD, Freedman RB, Keshavarz-Moore E, Robinson C (2013) High-level secretion of a recombinant protein to the culture medium with a Bacillus subtilis twin-arginine translocation system in Escherichia coli. FEBS J 280:3810–3821
Anbazhagan K, Sasikumar P, Gomathi S, Priya HP, Selvam GS (2013) In vitro degradation of oxalate by recombinant Lactobacillus plantarum expressing heterologous oxalate decarboxylase. J Appl Microbiol 115:880–887
Anné J, Maldonado B, Van Impe J, Van Mellaert L, Bernaerts K (2012) Recombinant protein production and streptomycetes. J Biotechnol 158:159–167
Anné J, Vrancken K, Van Mellaert L, Van Impe J, Bernaerts K (2014) Protein secretion biotechnology in Gram-positive bacteria with special emphasis on Streptomyces lividans. Biochim Biophys Acta 1843:1750–1761
Ayala JC, Pimienta E, Rodríguez C, Sarzo M, Jones J, Vallín C, Guerrero A, Milanés MT, Anné J, Mellaert LV, Huygen K (2015) Assessment of an ELISA for serodiagnosis of active pulmonary tuberculosis in a Cuban population. Asian Pac J Trop Dis 5:772–778
Barrett CM, Ray N, Thomas JD, Robinson C, Bolhuis A (2003) Quantitative export of a reporter protein, GFP, by the twin-arginine translocation pathway in Escherichia coli. Biochem Biophys Res Commun 304:279–284
Becker LA, Bang IS, Crouch ML, Fang FC (2005) Compensatory role of PspA, a member of the phage shock protein operon, in rpoE mutant Salmonella enterica serovar Typhimurium. Mol Microbiol 56:1004–1016
Berdy J (2005) Bioactive microbial metabolites. J Antibiot 58:1–26
Berks BC (2015) The twin-arginine protein translocation pathway. Annu Rev Biochem 84:843–864
Bermudez-Humaran LG, Kharrat P, Chatel JM, Langella P (2011) Lactococci and lactobacilli as mucosal delivery vectors for therapeutic proteins and DNA vaccines. Microb Cell Fact 10(Suppl 1):S4
Bhatty M, Laverde Gomez JA, Christie PJ (2013) The expanding bacterial type IV secretion lexicon. Res Microbiol 164:620–639
Biswas L, Biswas R, Nerz C, Ohlsen K, Schlag M, Schafer T, Lamkemeyer T, Ziebandt AK, Hantke K, Rosenstein R, Gotz F (2009) Role of the twin-arginine translocation pathway in Staphylococcus. J Bacteriol 191:5921–5929
Bolhuis A, Broekhuizen CP, Sorokin A, van Roosmalen ML, Venema G, Bron S, Quax WJ, van Dijl JM (1998) SecDF of Bacillus subtilis, a molecular Siamese twin required for the efficient secretion of proteins. J Biol Chem 273:21217–21224
Bolhuis A, Venema G, Quax WJ, Bron S, van Dijl JM (1999) Functional analysis of paralogous thiol-disulfide oxidoreductases in Bacillus subtilis. J Biol Chem 274:24531–24538
Brockmeier U (2006) New strategies to optimize the secretion capacity for heterologous proteins in Bacillus subtilis. Ruhr-Universität Bochum Bochum, Germany
Burts ML, Williams WA, DeBord K, Missiakas DM (2005) EsxA and EsxB are secreted by an ESAT-6-like system that is required for the pathogenesis of Staphylococcus aureus infections. Proc Natl Acad Sci USA 102:1169–1174
Butler MJ, Aphale JS, Binnie C, DiZonno MA, Krygsman P, Soltes G, Walczyk E, Malek LT (1996) Cloning and analysis of a gene from Streptomyces lividans 66 encoding a novel secreted protease exhibiting homology to subtilisin BPN’. Appl Microbiol Biotechnol 45:141–147
Calvo RA, Kearns DB (2015) FlgM is secreted by the flagellar export apparatus in Bacillus subtilis. J Bacteriol 197:81–91
Castanie-Cornet MP, Bruel N, Genevaux P (2014) Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim Biophys Acta 1843:1442–1456
Chandran Darbari V, Waksman G (2015) Structural biology of bacterial type IV secretion systems. Annu Rev Biochem 84:603–629
Chatzi KE, Sardis MF, Karamanou S, Economou A (2013) Breaking on through to the other side: protein export through the bacterial Sec system. Biochem J 449:25–37
Chen I, Dubnau D (2004) DNA uptake during bacterial transformation. Nat Rev Microbiol 2:241–249
Chen J, Fu G, Gai Y, Zheng P, Zhang D, Wen J (2015a) Combinatorial Sec pathway analysis for improved heterologous protein secretion in Bacillus subtilis: identification of bottlenecks by systematic gene overexpression. Microb Cell Fact 14:92
Chen J, Gai Y, Fu G, Zhou W, Zhang D, Wen J (2015b) Enhanced extracellular production of alpha-amylase in Bacillus subtilis by optimization of regulatory elements and over-expression of PrsA lipoprotein. Biotechnol Lett 37:899–906
Chen J, Zhao L, Fu G, Zhou W, Sun Y, Zheng P, Sun J, Zhang D (2016) A novel strategy for protein production using non-classical secretion pathway in Bacillus subtilis. Microb Cell Fact 15:69
Cleon F, Habersetzer J, Alcock F, Kneuper H, Stansfeld PJ, Basit H, Wallace MI, Berks BC, Palmer T (2015) The TatC component of the twin-arginine protein translocase functions as an obligate oligomer. Mol Microbiol 98:111–129
Converse SE, Cox JS (2005) A protein secretion pathway critical for Mycobacterium tuberculosis virulence is conserved and functional in Mycobacterium smegmatis. J Bacteriol 187:1238–1245
D’Huys PJ, Lule I, Van Hove S, Vercammen D, Wouters C, Bernaerts K, Anné J, Van Impe JF (2011) Amino acid uptake profiling of wild type and recombinant Streptomyces lividans TK24 batch fermentations. J Biotechnol 152:132–143
D’Huys PJ, Lule I, Vercammen D, Anné J, Van Impe JF, Bernaerts K (2012) Genome-scale metabolic flux analysis of Streptomyces lividans growing on a complex medium. J Biotechnol 161:1–13
Darwin AJ (2005) The phage-shock-protein response. Mol Microbiol 57:621–628
Date M, Itaya H, Matsui H, Kikuchi Y (2006) Secretion of human epidermal growth factor by Corynebacterium glutamicum. Lett Appl Microbiol 42:66–70
de Azevedo M, Meijerink M, Taverne N, Pereira VB, LeBlanc JG, Azevedo V, Miyoshi A, Langella P, Wells JM, Chatel JM (2015) Recombinant invasive Lactococcus lactis can transfer DNA vaccines either directly to dendritic cells or across an epithelial cell monolayer. Vaccine 33:4807–4812
De Keersmaeker S, Vrancken K, Van Mellaert L, Anné J, Geukens N (2007) The Tat pathway in Streptomyces lividans: interaction of Tat subunits and their role in translocation. Microbiology (Reading, England) 153:1087–1094
De Keersmaeker S, Vrancken K, Van Mellaert L, Lammertyn E, Anné J, Geukens N (2006) Evaluation of TatABC overproduction on Tat- and Sec-dependent protein secretion in Streptomyces lividans. Arch Microbiol 186:507–512
Degering C, Eggert T, Puls M, Bongaerts J, Evers S, Maurer KH, Jaeger KE (2010) Optimization of protease secretion in Bacillus subtilis and Bacillus licheniformis by screening of homologous and heterologous signal peptides. Appl Environ Microb 76:6370–6376
DeLisa MP, Lee P, Palmer T, Georgiou G (2004) Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J Bacteriol 186:366–373
Diao L, Dong Q, Xu Z, Yang S, Zhou J, Freudl R (2012) Functional implementation of the posttranslational SecB-SecA protein-targeting pathway in Bacillus subtilis. Appl Environ Microb 78:651–659
Doran PM (2013) Bioprocess engineering principles, 2nd edn. Academic Press, London
Eiteman MA, Altman E (2006) Overcoming acetate in Escherichia coli recombinant protein fermentations. Trends Biotech 24:530–536
Erhardt M, Namba K, Hughes KT (2010) Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harb Perspect Biol 2:a000299
Fan Y, Tan K, Chhor G, Butler EK, Jedrzejczak RP, Missiakas D, Joachimiak A (2015) EsxB, a secreted protein from Bacillus anthracis forms two distinct helical bundles. Protein Sci Publ Protein Soc 24:1389–1400
Fischer A, Kambara K, Meyer H, Stenz L, Bonetti EJ, Girard M, Lalk M, Francois P, Schrenzel J (2014) GdpS contributes to Staphylococcus aureus biofilm formation by regulation of eDNA release. Int J Med Microbiol 304:284–299
Forster BM, Marquis H (2012) Protein transport across the cell wall of monoderm Gram-positive bacteria. Mol Microbiol 84:405–413
Fyans JK, Bignell D, Loria R, Toth I, Palmer T (2013) The ESX/type VII secretion system modulates development, but not virulence, of the plant pathogen Streptomyces scabies. Mol Plant Pathol 14:119–130
Gabarró MlV, Gullón S, Vicente RL, Caminal G, Mellado RP, López-Santín J (2016) A Streptomyces lividans SipY deficient strain as a host for protein production: standardization of operational alternatives for model proteins. J Chem Technol Biotechnol doi:10.1002/jctb.4933
Garcia-Ochoa F, Gomez E (2009) Bioreactor scale-up and oxygen transfer rate in microbial processes: an overview. Biotechnol Adv 27:153–176
Geukens N (2002) Characterisation of Streptomyces lividans signal peptidases. KU Leuven, Leuven, Belgium
Gomez-Escribano JP, Bibb MJ (2011) Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb Biotechnol 4:207–215
Goodacre R, Vaidyanathan S, Dunn WB, Harrigan GG, Kell DB (2004) Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotech 22:245–252
Goodfellow M (2012) Phylum XXVI. Actinobacteria phyl. nov. In: Whitman WG, Goodfellow M. Kämpfer P, Busse H-J, Trujillo ME, Ludwig W, Suzuki K-i, Parte A (eds) Bergey’s manual® of systematic bacteriology, vol 5. The Actinobacteria, Part A and B, p 2028
Goosens VJ, De-San-Eustaquio-Campillo A, Carballido-Lopez R, van Dijl JM (2015) A Tat menage a trois—the role of Bacillus subtilis TatAc in twin-arginine protein translocation. Biochim Biophys Acta 1853:2745–2753
Goosens VJ, Monteferrante CG, van Dijl JM (2014) The Tat system of Gram-positive bacteria. Biochim Biophys Acta 1843:1698–1706
Gordon ND, Ottaviano GL, Connell SE, Tobkin GV, Son CH, Shterental S, Gehring AM (2008) Secreted-protein response to sigmaU activity in Streptomyces coelicolor. J Bacteriol 190:894–904
Grady LM, Michtavy J, Oliver DB (2012) Characterization of the Escherichia coli SecA signal peptide-binding site. J Bacteriol 194:307–316
Gullón S, Palomino C, Navajas R, Paradela A, Mellado RP (2012) Translocase and major signal peptidase malfunctions affect aerial mycelium formation in Streptomyces lividans. J Biotechnol 160:112–122
Halldórsdóttir S, Thórólfsdóttir ET, Spilliaert R, Johansson M, Thorbjarnardóttir SH, Palsdottir A, Hreggvidsson GO, Kristjánsson JK, Holst O, Eggertsson G (1998) Cloning, sequencing and overexpression of a Rhodothermus marinus gene encoding a thermostable cellulase of glycosyl hydrolase family 12. Appl Microbiol Biotechnol 49:277–284
Heap JT, Ehsaan M, Cooksley CM, Ng YK, Cartman ST, Winzer K, Minton NP (2012) Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res 40:e59
Heap JT, Theys J, Ehsaan M, Kubiak AM, Dubois L, Paesmans K, Mellaert LV, Knox R, Kuehne SA, Lambin P, Minton NP (2014) Spores of Clostridium engineered for clinical efficacy and safety cause regression and cure of tumors in vivo. Oncotarget 5:1761–1769
Heiss S, Hormann A, Tauer C, Sonnleitner M, Egger E, Grabherr R, Heinl S (2016) Evaluation of novel inducible promoter/repressor systems for recombinant protein expression in Lactobacillus plantarum. Microb Cell Fact 15:50
Hermann T, Pfefferle W, Baumann C, Busker E, Schaffer S, Bott M, Sahm H, Dusch N, Kalinowski J, Puhler A, Bendt AK, Kramer R, Burkovski A (2001) Proteome analysis of Corynebacterium glutamicum. Electrophoresis 22:1712–1723
Hjersted JL, Henson MA (2009) Steady-state and dynamic flux balance analysis of ethanol production by Saccharomyces cerevisiae. IET Syst Biol 3:167–179
Hueck CJ (1998) Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62:379–433
Hugentobler F, Yam KK, Gillard J, Mahbuba R, Olivier M, Cousineau B (2012) Immunization against Leishmania major infection using LACK- and IL-12-expressing Lactococcus lactis induces delay in footpad swelling. PLoS ONE 7:e30945
Ize B, Stanley NR, Buchanan G, Palmer T (2003) Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol 48:1183–1193
Jones DT, Woods DR (1986) Acetone-butanol fermentation revisited. Microbiol Rev 50:484–524
Kakeshita H, Kageyama Y, Ara K, Ozaki K, Nakamura K (2010) Enhanced extracellular production of heterologous proteins in Bacillus subtilis by deleting the C-terminal region of the SecA secretory machinery. Mol Biotechnol 46:250–257
Kang Z, Yang S, Du G, Chen J (2014) Molecular engineering of secretory machinery components for high-level secretion of proteins in Bacillus species. J Ind Microbiol Biotechnol 41:1599–1607
Karamanou S, Vrontou E, Sianidis G, Baud C, Roos T, Kuhn A, Politou AS, Economou A (1999) A molecular switch in SecA protein couples ATP hydrolysis to protein translocation. Mol Microbiol 34:1133–1145
Karlskas IL, Maudal K, Axelsson L, Rud I, Eijsink VG, Mathiesen G (2014) Heterologous protein secretion in Lactobacilli with modified pSIP vectors. PLoS ONE 9:e91125
Kass F, Junne S, Neubauer P, Wiechert W, Oldiges M (2014) Process inhomogeneity leads to rapid side product turnover in cultivation of Corynebacterium glutamicum. Microb Cell Fact 13:6
Kassama Y, Xu Y, Dunn WB, Geukens N, Anné J, Goodacre R (2010) Assessment of adaptive focused acoustics versus manual vortex/freeze-thaw for intracellular metabolite extraction from Streptomyces lividans producing recombinant proteins using GC-MS and multi-block principal component analysis. Analyst 135:934–942
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical streptomyces genetics. The John Innes Foundation, Norwich
Kikuchi Y, Date M, Itaya H, Matsui K, Wu LF (2006) Functional analysis of the twin-arginine translocation pathway in Corynebacterium glutamicum ATCC 13869. Appl Environ Microb 72:7183–7192
Kikuchi Y, Itaya H, Date M, Matsui K, Wu LF (2009) TatABC overexpression improves Corynebacterium glutamicum Tat-dependent protein secretion. Appl Environ Microb 75:603–607
Kim J, Reed JL (2010) OptORF: optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst Biol 4:53
Korneli C, Bolten CJ, Godard T, Franco-Lara E, Wittmann C (2012) Debottlenecking recombinant protein production in Bacillus megaterium under large-scale conditions—targeted precursor feeding designed from metabolomics. Biotechnol Bioeng 109:1538–1550
Kouwen TR, van Dijl JM (2009) Applications of thiol-disulfide oxidoreductases for optimized in vivo production of functionally active proteins in Bacillus. Appl Microbiol Biotechnol 85:45–52
Kovacs K, Willson BJ, Schwarz K, Heap JT, Jackson A, Bolam DN, Winzer K, Minton NP (2013) Secretion and assembly of functional mini-cellulosomes from synthetic chromosomal operons in Clostridium acetobutylicum ATCC 824. Biotechnol Biofuels 6:117
Kuipers OP, de Ruyter PGGA, Kleerebezem M, de Vos WM (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64:15–21
Kuo CY, Sinha S, Jazayeri JA, Pouton CW (2009) A stably engineered, suicidal strain of listeria monocytogenes delivers protein and/or DNA to fully differentiated intestinal epithelial monolayers. Mol Pharm 6:1052–1061
Lammertyn E, Anné J (1998) Modifications of Streptomyces signal peptides and their effects on protein production and secretion. FEMS Microbiol Lett 160:1–10
Le Loir Y, Azevedo V, Oliveira SC, Freitas DA, Miyoshi A, Bermudez-Humaran LG, Nouaille S, Ribeiro LA, Leclercq S, Gabriel JE, Guimaraes VD, Oliveira MN, Charlier C, Gautier M, Langella P (2005) Protein secretion in Lactococcus lactis: an efficient way to increase the overall heterologous protein production. Microb Cell Fact 4:1
Leloup L, Driessen AJM, Freudl R, Chambert R, Petit-Glatron MF (1999) Differential dependence of levansucrase and alpha-amylase secretion on SecA (Div) during the exponential phase of growth of Bacillus subtilis. J Bacteriol 181:1820–1826
Lewis NE, Nagarajan H, Palsson BO (2012) Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat Rev Microbiol 10:291–305
Li J-X, Zhao L-M, Wu R-J, Zheng Z-J, Zhang R-J (2013) High-Level overproduction of Thermobifida Enzyme in Streptomyces lividans using a novel expression vector. Int J Mol Sci 14:18629–18639
Li X, Fu GF, Fan YR, Liu WH, Liu XJ, Wang JJ, Xu GX (2003) Bifidobacterium adolescentis as a delivery system of endostatin for cancer gene therapy: selective inhibitor of angiogenesis and hypoxic tumor growth. Cancer Gene Ther 10:105–111
Lieder S, Nikel PI, de Lorenzo V, Takors R (2015) Genome reduction boosts heterologous gene expression in Pseudomonas putida. Microb Cell Fact 14:23
Lule I, Maldonado B, D’Huys PJ, Van Mellaert L, Van Impe J, Bernaerts K, Anné J (2012) On the influence of overexpression of phosphoenolpyruvate carboxykinase in Streptomyces lividans on growth and production of human tumour necrosis factor-alpha. Appl Microbiol Biotechnol 96:367–372
Malten M, Nahrstedt H, Meinhardt F, Jahn D (2005) Coexpression of the type I signal peptidase gene sipM increases recombinant protein production and export in Bacillus megaterium MS941. Biotechnol Bioeng 91:616–621
Mandenius CF, Brundin A (2008) Bioprocess optimization using design-of-experiments methodology. Biotechnol Prog 24:1191–1203
Manteca A, Alvarez R, Salazar N, Yague P, Sanchez J (2008) Mycelium differentiation and antibiotic production in submerged cultures of Streptomyces coelicolor. Appl Environ Microbiol 74:3877–3886
Mathiesen G, Sveen A, Brurberg MB, Fredriksen L, Axelsson L, Eijsink VG (2009) Genome-wide analysis of signal peptide functionality in Lactobacillus plantarum WCFS1. BMC Genom 10:425
McDonough JA, Hacker KE, Flores AR, Pavelka MS Jr, Braunstein M (2005) The twin-arginine translocation pathway of Mycobacterium smegmatis is functional and required for the export of mycobacterial beta-lactamases. J Bacteriol 187:7667–7679
McNeil BH, Harvey LM (2008) Practical fermentation technology. Wiley, Chichester
Meissner D, Vollstedt A, van Dijl JM, Freudl R (2007) Comparative analysis of twin-arginine (Tat)-dependent protein secretion of a heterologous model protein (GFP) in three different Gram-positive bacteria. Appl Microbiol Biotechnol 76:633–642
Mierau I, Kleerebezem M (2005) 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol 68:705–717
Mingardon F, Chanal A, Tardif C, Fierobe HP (2011) The issue of secretion in heterologous expression of Clostridium cellulolyticum cellulase-encoding genes in Clostridium acetobutylicum ATCC 824. Appl Environ Microb 77:2831–2838
Mitsuhashi S (2014) Current topics in the biotechnological production of essential amino acids, functional amino acids, and dipeptides. Curr Opin Biotechnol 26:38–44
Mogk A, Mayer MP, Deuerling E (2002) Mechanisms of protein folding: molecular chaperones and their application in biotechnology. ChemBioChem 3:807–814
Morais S, Shterzer N, Grinberg IR, Mathiesen G, Eijsink VG, Axelsson L, Lamed R, Bayer EA, Mizrahi I (2013) Establishment of a simple Lactobacillus plantarum cell consortium for cellulase-xylanase synergistic interactions. Appl Environ Microb 79:5242–5249
Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N (2008) Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res 15:73–81
Muhamadali H, Xu Y, Ellis DI, Trivedi DK, Rattray NJ, Bernaerts K, Goodacre R (2015) Metabolomics investigation of recombinant mTNFalpha production in Streptomyces lividans. Microb Cell Fact 14:157
Nett M, Ikeda H, Moore BS (2009) Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep 26:1362–1384
Nowruzi K, Elkamel A, Scharer JM, Cossar D, Moo-Young M (2008) Development of a minimal defined medium for recombinant human interleukin-3 production by Streptomyces lividans 66. Biotechnol Bioeng 99:214–222
Oddone GM, Mills DA, Block DE (2009) A dynamic, genome-scale flux model of Lactococcus lactis to increase specific recombinant protein expression. Metab Eng 11:367–381
Oertel D, Schmitz S, Freudl R (2015) A TatABC-type Tat translocase is required for unimpaired aerobic growth of Corynebacterium glutamicum ATCC13032. PLoS ONE 10:e0123413
Orfanoudaki G, Economou A (2014) Proteome-wide subcellular topologies of E. coli polypeptides database (STEPdb). Mol Cell Proteomics 13:3674–3687
Özdamar TH, Şentürk B, Yılmaz ÖD, Kocabaş P, Çalık G, Çalık P (2010) Bioreaction network flux analysis for human protein producing Bacillus subtilis based on genome-scale model. Chem Eng Sci 65:574–580
Oztürk S, Calik P, Ozdamar TH (2016) Fed-batch biomolecule production by Bacillus subtilis: a state of the art review. Trends Biotech 34:329–345
Palacin A, Parro V, Geukens N, Anné J, Mellado RP (2002) SipY Is the Streptomyces lividans type I signal peptidase exerting a major effect on protein secretion. J Bacteriol 184:4875–4880
Pallen MJ (2002) The ESAT-6/WXG100 superfamily—and a new Gram-positive secretion system? Trends Microbiol 10:209–212
Parro V, Schacht S, Anné J, Mellado RP (1999) Four genes encoding different type I signal peptidases are organized in a cluster in Streptomyces lividans TK21. Microbiology (Reading, England) 145(Pt 9):2255–2263
Paul K, Erhardt M, Hirano T, Blair DF, Hughes KT (2008) Energy source of flagellar type III secretion. Nature 451:489–492
Payne SH, Bonissone S, Wu S, Brown RN, Ivankov DN, Frishman D, Pasa-Tolic L, Smith RD, Pevzner PA (2012) Unexpected diversity of signal peptides in prokaryotes. MBio 3:e00339-12
Pohl S, Harwood CR (2010) Heterologous protein secretion by bacillus species from the cradle to the grave. Adv Appl Microbiol 73:1–25
Pontes D, Azevedo M, Innocentin S, Blugeon S, Lefevre F, Azevedo V, Miyoshi A, Courtin P, Chapot-Chartier MP, Langella P, Chatel JM (2014) Immune response elicited by DNA vaccination using Lactococcus lactis is modified by the production of surface exposed pathogenic protein. PLoS ONE 9:e84509
Pontes DS, de Azevedo MS, Chatel JM, Langella P, Azevedo V, Miyoshi A (2011) Lactococcus lactis as a live vector: heterologous protein production and DNA delivery systems. Protein Expr Purif 79:165–175
Pop O, Martin U, Abel C, Muller JP (2002) The twin-arginine signal peptide of PhoD and the TatAd/Cd proteins of Bacillus subtilis form an autonomous Tat translocation system. J Biol Chem 277:3268–3273
Poquet I, Saint V, Seznec E, Simoes N, Bolotin A, Gruss A (2000) HtrA is the unique surface housekeeping protease in Lactococcus lactis and is required for natural protein processing. Mol Microbiol 35:1042–1051
Pozidis C, Lammertyn E, Politou AS, Anné J, Tsiftsoglou AS, Sianidis G, Economou A (2001) Protein secretion biotechnology using Streptomyces lividans: large-scale production of functional trimeric tumor necrosis factor alpha. Biotechnol Bioeng 72:611–619
Pummi T, Leskela S, Wahlstrom E, Gerth U, Tjalsma H, Hecker M, Sarvas M, Kontinen VP (2002) ClpXP protease regulates the signal peptide cleavage of secretory preproteins in Bacillus subtilis with a mechanism distinct from that of the Ecs ABC transporter. J Bacteriol 184:1010–1018
Real G, Pinto SM, Schyns G, Costa T, Henriques AO, Moran CP Jr (2005) A gene encoding a holin-like protein involved in spore morphogenesis and spore germination in Bacillus subtilis. J Bacteriol 187:6443–6453
Rebets Y, Kormanec J, Lutzhetskyy A, Bernaerts K, Anné J (2016) Cloning and expression of metagenomic DNA in Streptomyces lividans and its subsequent fermentation for optimized production. In: Streit W, Daniel R (eds) Metagenomics—methods and protocols, vol 1539. Springer, Berlin
Rioseras B, Lopez-Garcia MT, Yague P, Sanchez J, Manteca A (2014) Mycelium differentiation and development of Streptomyces coelicolor in lab-scale bioreactors: programmed cell death, differentiation, and lysis are closely linked to undecylprodigiosin and actinorhodin production. Bioresour Technol 151:191–198
Roberts NJ, Zhang L, Janku F, Collins A, Bai RY, Staedtke V, Rusk AW, Tung D, Miller M, Roix J, Khanna KV, Murthy R, Benjamin RS, Helgason T, Szvalb AD, Bird JE, Roy-Chowdhuri S, Zhang HH, Qiao Y, Karim B, McDaniel J, Elpiner A, Sahora A, Lachowicz J, Phillips B, Turner A, Klein MK, Post G, Diaz LA Jr, Riggins GJ, Papadopoulos N, Kinzler KW, Vogelstein B, Bettegowda C, Huso DL, Varterasian M, Saha S, Zhou S (2014) Intratumoral injection of Clostridium novyi-NT spores induces antitumor responses. Sci Transl Med 6:249ra111
Rohe P, Venkanna D, Kleine B, Freudl R, Oldiges M (2012) An automated workflow for enhancing microbial bioprocess optimization on a novel microbioreactor platform. Microb Cell Fact 11:144
Ruckert C, Albersmeier A, Busche T, Jaenicke S, Winkler A, Friethjonsson OH, Hreggviethsson GO, Lambert C, Badcock D, Bernaerts K, Anné J, Economou A, Kalinowski J (2015) Complete genome sequence of Streptomyces lividans TK24. J Biotechnol 199:21–22
Saier MH Jr, Reddy BL (2015) Holins in bacteria, eukaryotes, and archaea: multifunctional xenologues with potential biotechnological and biomedical applications. J Bacteriol 197:7–17
Saint-Joanis B, Demangel C, Jackson M, Brodin P, Marsollier L, Boshoff H, Cole ST (2006) Inactivation of Rv2525c, a substrate of the twin arginine translocation (Tat) system of Mycobacterium tuberculosis, increases beta-lactam susceptibility and virulence. J Bacteriol 188:6669–6679
Sak-Ubol S, Namvijitr P, Pechsrichuang P, Haltrich D, Nguyen TH, Mathiesen G, Eijsink VG, Yamabhai M (2016) Secretory production of a beta-mannanase and a chitosanase using a Lactobacillus plantarum expression system. Microb Cell Fact 15:81
Sala A, Bordes P, Genevaux P (2014) Multitasking SecB chaperones in bacteria. Front Microbiol 5:666
Samazan F, Rokbi B, Seguin D, Telles F, Gautier V, Richarme G, Chevret D, Varela PF, Velours C, Poquet I (2015) Production, secretion and purification of a correctly folded staphylococcal antigen in Lactococcus lactis. Microb Cell Fact 14:104
San Roman SA, Facey PD, Fernandez-Martinez L, Rodriguez C, Vallin C, Del Sol R, Dyson P (2010) A heterodimer of EsxA and EsxB is involved in sporulation and is secreted by a type VII secretion system in Streptomyces coelicolor. Microbiol-Sgm 156:1719–1729
Sargent F, Stanley NR, Berks BC, Palmer T (1999) Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J Biol Chem 274:36073–36082
Sarvas M, Harwood CR, Bron S, van Dijl JM (2004) Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim Biophys Acta 1694:311–327
Schaerlaekens K, Van Mellaert L, Lammertyn E, Geukens N, Anné J (2004) The importance of the Tat-dependent protein secretion pathway in Streptomyces as revealed by phenotypic changes in tat deletion mutants and genome analysis. Microbiology (Reading, England) 150:21–31
Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BO (2011) Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc 6:1290–1307
Schiebel E, Driessen AJ, Hartl FU, Wickner W (1991) Delta mu H+ and ATP function at different steps of the catalytic cycle of preprotein translocase. Cell 64:927–939
Schlegel L, Lemerle S, Geslin P (1998) Lactobacillus species as opportunistic pathogens in immunocompromised patients. Eur J Clin Microbiol Infect Dis 17:887–888
Schumann W (2007) Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol 62:137–189
Schwab C, Sorensen KI, Ganzle MG (2010) Heterologous expression of glycoside hydrolase family 2 and 42 beta-galactosidases of lactic acid bacteria in Lactococcus lactis. Syst Appl Microbiol 33:300–307
Sevillano L, Vijgenboom E, van Wezel GP, Diaz M, Santamaria RI (2016) New approaches to achieve high level enzyme production in Streptomyces lividans. Microb Cell Fact 15:28
Shapova YA, Paetzel M (2007) Crystallographic analysis of Bacillus subtilis CsaA. Acta Crystallogr Sect D Biol Crystallogr 63:478–485
Shi Y, Li N, Yan Y, Wang H, Li Y, Lu C, Sun J (2012) Combined antibacterial activity of phage lytic proteins holin and lysin from Streptococcus suis bacteriophage SMP. Curr Microbiol 65:28–34
Sianidis G, Pozidis C, Becker F, Vrancken K, Sjoeholm C, Karamanou S, Takamiya-Wik M, van Mellaert L, Schaefer T, Anné J, Economou A (2006) Functional large-scale production of a novel Jonesia sp. xyloglucanase by heterologous secretion from Streptomyces lividans. J Biotechnol 121:498–507
Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A (2013) Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng 19:98–106
Simone D, Bay DC, Leach T, Turner RJ (2013) Diversity and evolution of bacterial twin arginine translocase protein, TatC, reveals a protein secretion system that is evolving to fit its environmental niche. PLoS ONE 8:e78742
Sohoni SV, Bapat PM, Lantz AE (2012) Robust, small-scale cultivation platform for Streptomyces coelicolor. Microb Cell Fact 11:9
Stanley NR, Palmer T, Berks BC (2000) The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J Biol Chem 275:11591–11596
Stanley SA, Raghavan S, Hwang WW, Cox JS (2003) Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA 100:13001–13006
Stoop EJ, Bitter W, van der Sar AM (2012) Tubercle bacilli rely on a type VII army for pathogenicity. Trends Microbiol 20:477–484
Sutcliffe IC (2011) New insights into the distribution of WXG100 protein secretion systems. Antonie Van Leeuwenhoek 99:127–131
Sysoeva TA, Zepeda-Rivera MA, Huppert LA, Burton BM (2014) Dimer recognition and secretion by the ESX secretion system in Bacillus subtilis. Proc Natl Acad Sci USA 111:7653–7658
Tekaia F, Gordon SV, Garnier T, Brosch R, Barrell BG, Cole ST (1999) Analysis of the proteome of Mycobacterium tuberculosis in silico. Tubercle 79:329–342
Theys J, Pennington O, Dubois L, Anlezark G, Vaughan T, Mengesha A, Landuyt W, Anné J, Burke PJ, Durre P, Wouters BG, Minton NP, Lambin P (2006) Repeated cycles of Clostridium-directed enzyme prodrug therapy result in sustained antitumour effects in vivo. Br J Cancer 95:1212–1219
Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM (2004) Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev 68:207–233
Tjalsma H, Bolhuis A, van Roosmalen ML, Wiegert T, Schumann W, Broekhuizen CP, Quax WJ, Venema G, Bron S, van Dijl JM (1998) Functional analysis of the secretory precursor processing machinery of Bacillus subtilis: identification of a eubacterial homolog of archaeal and eukaryotic signal peptidases. Genes Dev 12:2318–2331
Toya Y, Hirasawa T, Morimoto T, Masuda K, Kageyama Y, Ozaki K, Ogasawara N, Shimizu H (2014) 13 C-metabolic flux analysis in heterologous cellulase production by Bacillus subtilis genome-reduced strain. J Biotechnol 179:42–49
Tremillon N, Issaly N, Mozo J, Duvignau T, Ginisty H, Devic E, Poquet I (2010) Production and purification of staphylococcal nuclease in Lactococcus lactis using a new expression-secretion system and a pH-regulated mini-reactor. Microb Cell Fact 9:37
Umakoshi M, Hirasawa T, Furusawa C, Takenaka Y, Kikuchi Y, Shimizu H (2011) Improving protein secretion of a transglutaminase-secreting Corynebacterium glutamicum recombinant strain on the basis of 13C metabolic flux analysis. J Biosci Bioeng 112:595–601
Umer B, Good D, Anné J, Duan W, Wei MQ (2012) Clostridial spores for cancer therapy: targeting solid tumour microenvironment. J Toxicol 2012:862764
Unthan S, Radek A, Wiechert W, Oldiges M, Noack S (2015) Bioprocess automation on a Mini Pilot Plant enables fast quantitative microbial phenotyping. Microb Cell Fact 14:32
Van Mellaert L, Lammertyn E, Schacht S, Proost P, Van Damme J, Wroblowski B, Anné J, Scarcez T, Sablon E, Raeymaeckers J, Van Broekhoven A (1998) Molecular characterization of a novel subtilisin inhibitor protein produced by Streptomyces venezuelae CBS762.70. DNA Seq 9:19–30
van Roosmalen ML, Geukens N, Jongbloed JD, Tjalsma H, Dubois JY, Bron S, van Dijl JM, Anné J (2004) Type I signal peptidases of Gram-positive bacteria. Biochim Biophys Acta 1694:279–297
van Wezel GP, Krabben P, Traag BA, Keijser BJ, Kerste R, Vijgenboom E, Heijnen JJ, Kraal B (2006) Unlocking Streptomyces spp. for use as sustainable industrial production platforms by morphological engineering. Appl Environ Microbiol 72:5283–5288
van Wezel GP, McDowall KJ (2011) The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat Prod Rep 28:1311–1333
Vandenbroucke K, de Haard H, Beirnaert E, Dreier T, Lauwereys M, Huyck L, Van Huysse J, Demetter P, Steidler L, Remaut E, Cuvelier C, Rottiers P (2010) Orally administered L. lactis secreting an anti-TNF Nanobody demonstrate efficacy in chronic colitis. Mucosal Immunol 3:49–56
Villatoro-Hernandez J, Loera-Arias MJ, Gamez-Escobedo A, Franco-Molina M, Gomez-Gutierrez JG, Rodriguez-Rocha H, Gutierrez-Puente Y, Saucedo-Cardenas O, Valdes-Flores J, Montes-de-Oca-Luna R (2008) Secretion of biologically active interferon-gamma inducible protein-10 (IP-10) by Lactococcus lactis. Microb Cell Fact 7:22
Vitikainen M, Hyyrylainen HL, Kivimaki A, Kontinen VP, Sarvas M (2005) Secretion of heterologous proteins in Bacillus subtilis can be improved by engineering cell components affecting post-translocational protein folding and degradation. J Appl Microbiol 99:363–375
Voigt B, Schroeter R, Schweder T, Jurgen B, Albrecht D, van Dijl JM, Maurer KH, Hecker M (2014) A proteomic view of cell physiology of the industrial workhorse Bacillus licheniformis. J Biotechnol 191:139–149
Vonderviszt F, Sajo R, Dobo J, Zavodszky P (2012) The use of a flagellar export signal for the secretion of recombinant proteins in Salmonella. Methods Mol Biol 824:131–143
Vorös A, Simm R, Slamti L, McKay MJ, Hegna IK, Nielsen-LeRoux C, Hassan KA, Paulsen IT, Lereclus D, Okstad OA, Molloy MP, Kolsto AB (2014) SecDF as part of the Sec-translocase facilitates efficient secretion of Bacillus cereus toxins and cell wall-associated proteins. PLoS ONE 9:e103326
Vrancken K, De Keersmaeker S, Geukens N, Lammertyn E, Anné J, Van Mellaert L (2007) pspA overexpression in Streptomyces lividans improves both Sec- and Tat-dependent protein secretion. Appl Microbiol Biotechnol 73:1150–1157
Waite RD, Rose RS, Rangarajan M, Aduse-Opoku J, Hashim A, Curtis MA (2012) Pseudomonas aeruginosa possesses two putative type I signal peptidases, LepB and PA1303, each with distinct roles in physiology and virulence. J Bacteriol 194:4521–4536
Wang G, Xia Y, Song X, Ai L (2016) Common non-classically secreted bacterial proteins with experimental evidence. Curr Microbiol 72:102–111
Wang IN, Smith DL, Young R (2000) Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol 54:799–825
Wang XJ, Yan YJ, Zhang B, An J, Wang JJ, Tian J, Jiang L, Chen YH, Huang SX, Yin M, Zhang J, Gao AL, Liu CX, Zhu ZX, Xiang WS (2010) Genome sequence of the milbemycin-producing bacterium Streptomyces bingchenggensis. J Bacteriol 192:4526–4527
Watanabe K, Tsuchida Y, Okibe N, Teramoto H, Suzuki N, Inui M, Yukawa H (2009) Scanning the Corynebacterium glutamicum R genome for high-efficiency secretion signal sequences. Microbiology (Reading, England) 155:741–750
Wei M (2013) Oncolytic clostridium ghonii strains, and methods of production and use. Google Patents
Wells JM, Mercenier A (2008) Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat Rev Microbiol 6:349–362
Westbye AB, Leung MM, Florizone SM, Taylor TA, Johnson JA, Fogg PC, Beatty JT (2013) Phosphate concentration and the putative sensor kinase protein CckA modulate cell lysis and release of the Rhodobacter capsulatus gene transfer agent. J Bacteriol 195:5025–5040
Westers L, Dijkstra DS, Westers H, van Dijl JM, Quax WJ (2006) Secretion of functional human interleukin-3 from Bacillus subtilis. J Biotechnol 123:211–224
Westers L, Westers H, Zanen G, Antelmann H, Hecker M, Noone D, Devine KM, van Dijl JM, Quax WJ (2008) Genetic or chemical protease inhibition causes significant changes in the Bacillus subtilis exoproteome. Proteomics 8:2704–2713
Whitaker N, Berry TM, Rosenthal N, Gordon JE, Gonzalez-Rivera C, Sheehan KB, Truchan HK, VieBrock L, Newton IL, Carlyon JA, Christie PJ (2016) Chimeric coupling proteins mediate transfer of heterologous type IV effectors through the Escherichia coli pKM101-encoded conjugation machine. J Bacteriol 198:2701–2718
Widdick DA, Dilks K, Chandra G, Bottrill A, Naldrett M, Pohlschroder M, Palmer T (2006) The twin-arginine translocation pathway is a major route of protein export in Streptomyces coelicolor. Proc Natl Acad Sci USA 103:17927–17932
Wiechert W (2001) 13C metabolic flux analysis. Metab Eng 3:195–206
Wiechert W, Nöh K (2013) Isotopically non-stationary metabolic flux analysis: complex yet highly informative. Curr Opin Biotechnol 24:979–986
Willemse J, Ruban-Osmialowska B, Widdick D, Celler K, Hutchings MI, van Wezel GP, Palmer T (2012) Dynamic localization of Tat protein transport machinery components in Streptomyces coelicolor. J Bacteriol 194:6272–6281
Williams RC, Rees ML, Jacobs MF, Pragai Z, Thwaite JE, Baillie LW, Emmerson PT, Harwood CR (2003) Production of Bacillus anthracis protective antigen is dependent on the extracellular chaperone, PrsA. J Biol Chem 278:18056–18062
Yam KK, Hugentobler F, Pouliot P, Stern AM, Lalande JD, Matlashewski G, Olivier M, Cousineau B (2011) Generation and evaluation of A2-expressing Lactococcus lactis live vaccines against Leishmania donovani in BALB/c mice. J Med Microbiol 60:1248–1260
Yamada K, Sanzen I, Ohkura T, Okamoto A, Torii K, Hasegawa T, Ohta M (2007) Analysis of twin-arginine translocation pathway homologue in Staphylococcus aureus. Curr Microbiol 55:14–19
Yan J, Fan X, Xie J (2013) Emerging biomedicines based on bacteriophages. Crit Rev Eukaryot Gene Expr 23:299–308
Yang CK, Ewis HE, Zhang X, Lu CD, Hu HJ, Pan Y, Abdelal AT, Tai PC (2011) Nonclassical protein secretion by Bacillus subtilis in the stationary phase is not due to cell lysis. J Bacteriol 193:5607–5615
Yang CK, Zhang XZ, Lu CD, Tai PC (2014) An internal hydrophobic helical domain of Bacillus subtilis enolase is essential but not sufficient as a non-cleavable signal for its secretion. Biochem Biophys Res Commun 446:901–905
Yoon SH, Han MJ, Lee SY, Jeong KJ, Yoo JS (2003) Combined transcriptome and proteome analysis of Escherichia coli during high cell density culture. Biotechnol Bioeng 81:753–767
Zamboni N, Fendt S-M, Ruhl M, Sauer U (2009) 13C-based metabolic flux analysis. Nat Protoc 4:878–892
Acknowledgments
Part of the research leading to the results described for Streptomyces has received funding from the European Commission’s Seventh Framework Program (FP7/2007–2013) under the grant agreement STREPSYNTH (Project No. 613877). Tobias Busche and Jörn Kalinowski (CeBiTec, Bielefeld, Germany), Joachim Koepff and Marco Oldiges (Forschungszentrum Jülich GmbH, Germany) are acknowledged for communication of results before publication).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Anné, J., Economou, A., Bernaerts, K. (2016). Protein Secretion in Gram-Positive Bacteria: From Multiple Pathways to Biotechnology. In: Bagnoli, F., Rappuoli, R. (eds) Protein and Sugar Export and Assembly in Gram-positive Bacteria . Current Topics in Microbiology and Immunology, vol 404. Springer, Cham. https://doi.org/10.1007/82_2016_49
Download citation
DOI: https://doi.org/10.1007/82_2016_49
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56012-0
Online ISBN: 978-3-319-56014-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)