
Abstract
Purpose of review: Heat shock factor 1 (HSF1) is the master transcriptional regulator of the heat shock response (HSR) in mammalian cells and is a critical element in maintaining protein homeostasis. HSF1 functions at the center of many physiological processes like embryogenesis, metabolism, immune response, aging, cancer, and neurodegeneration. However, the mechanisms that allow HSF1 to control these different biological and pathophysiological processes are not fully understood. This review focuses on Huntington’s disease (HD), a neurodegenerative disease characterized by severe protein aggregation of the huntingtin (HTT) protein. The aggregation of HTT, in turn, leads to a halt in the function of HSF1. Understanding the pathways that regulate HSF1 in different contexts like HD may hold the key to understanding the pathomechanisms underlying other proteinopathies. We provide the most current information on HSF1 structure, function, and regulation, emphasizing HD, and discussing its potential as a biological target for therapy.
Data sources: We performed PubMed search to find established and recent reports in HSF1, heat shock proteins (Hsp), HD, Hsp inhibitors, HSF1 activators, and HSF1 in aging, inflammation, cancer, brain development, mitochondria, synaptic plasticity, polyglutamine (polyQ) diseases, and HD.
Study selections: Research and review articles that described the mechanisms of action of HSF1 were selected based on terms used in PubMed search.
Results: HSF1 plays a crucial role in the progression of HD and other protein-misfolding related neurodegenerative diseases. Different animal models of HD, as well as postmortem brains of patients with HD, reveal a connection between the levels of HSF1 and HSF1 dysfunction to mutant HTT (mHTT)-induced toxicity and protein aggregation, dysregulation of the ubiquitin-proteasome system (UPS), oxidative stress, mitochondrial dysfunction, and disruption of the structural and functional integrity of synaptic connections, which eventually leads to neuronal loss. These features are shared with other neurodegenerative diseases (NDs). Currently, several inhibitors against negative regulators of HSF1, as well as HSF1 activators, are developed and hold promise to prevent neurodegeneration in HD and other NDs.
Conclusion: Understanding the role of HSF1 during protein aggregation and neurodegeneration in HD may help to develop therapeutic strategies that could be effective across different NDs.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Summary
Living organisms experience acute or chronic exposure to different endogenous and environmental insults during life, including elevated temperature, oxidative stress, and proteotoxic conditions. Cells have developed different self-defense mechanisms against such stressors to ensure cell survival. One of the most potent and successful mechanisms is the so-called heat shock response (HSR), characterized by the rapid induction of a group of molecular chaperones and heat shock proteins (Hsp) that fight the harmful effects of different stressors on proteins structure and function (Akerfelt et al. 2010; Anckar and Sistonen 2011). There is a regulation in the transcriptional activation of Hsp genes by a family of specialized heat shock transcription factors (HSFs) in eukaryotes. HSFs participate in many processes, including protein homeostasis, aging, innate immunity, and metabolism, and have a fundamental role in physiology and diseases like cancer and neurodegeneration. This chapter will focus on HSF1, the main HSF responsible for coordinating and executing the HSR in mammals. We will discuss fundamental features of HSF1, including structure, regulatory mechanisms, and physiological functions that go beyond the HSR, with particular emphasis on the neurodegenerative disease Huntington’s disease (HD).
2 Introduction
HSF1 is a multifaceted factor, traditionally known for coordinating the cellular response to internal and external stimuli that disrupt cellular protein homeostasis (Akerfelt et al. 2010; Gomez-Pastor et al. 2018). This response is usually mediated by the transcriptional regulation of several Hsp that have the task to prevent protein misfolding, refold misfolded proteins, and target damaged proteins for degradation (Ellis 2007). However, in the last few years, we have learned that HSF1 is much more complex, and it participates in numerous biological processes in both physiology and disease (Gomez-Pastor et al. 2018).
In unstressed cells, HSF1 exists as an inactive monomer in the cytoplasm due to the interaction by several regulatory proteins, including Hsp70, Hsp40, Hsp90, and the chaperonin complex TRiC (Gomez-Pastor et al. 2018). When cells encounter stress, HSF1 is released from its repressors and is activated. This activation process requires trimerization, several post-translational modifications (PTMs), and translocation to the nucleus where it binds to target genes. The transcriptional response elicited by HSF1 will then depend on the type of stress and the different protein-protein interactions in which HSF1 participates (Gomez-Pastor et al. 2018; Prince et al. 2020; Burchfiel et al. 2020). HSF1 recognizes a specific sequence in its target genes, composed of inverted repeats of a nGAAn sequence, called heat shock element (HSE) (Akerfelt et al. 2010; Vihervaara et al. 2013). Different studies conducting HSF1 chromatin immunoprecipitation followed by sequencing (ChIP-seq) revealed that numerous target genes contain HSE in their promoter and intergenic regions, but a subset of them do not (Korfanty et al. 2014; Riva et al. 2012; Vihervaara et al. 2017). However, it is important to note that the presence of an HSE within any given gene does not assure HSF1 binding. Another layer of complexity is that the expression of target genes of HSF1, with or without specific HSEs, can also be influenced by epigenetic modification of chromatin (Guertin and Lis 2010; Vihervaara et al. 2017). These studies imply that the regulatory events that control which set of genes are induced by HSF1 at any given time are much more complicated than we have previously anticipated. HSF1 is known for its role as a transcriptional activator. However, numerous studies confirmed its role in repressing transcription, i.e., interleukin-6 (IL-6) involved in inflammation and the microtubule-associated protein Tau involved in synaptic dysfunction in different tauopathies (Inouye et al. 2007; Kim et al. 2017).
HSF1 has been studied widely in the context of the HSR and proteotoxic stress, but its role is not limited to just regulating Hsp expression (Gomez-Pastor et al. 2018). Recent evidence now points to HSF1 influencing the expression of multiple genes that are essential for cell cycle regulation, glucose metabolism, inflammatory response, and development and maintenance of neuronal, reproductive, and sensory organs (Akerfelt et al. 2007; Nakai 2009; Page et al. 2006; Singh and Hasday 2013). Therefore, defects in the activity and levels of HSF1 result in devastating consequences. More studies now confirmed the role of HSF1 in age-related and neurodegenerative disorders like Alzheimer’s (AD), Parkinson’s (PD), and HD and its potential as a therapeutic target (Goetzl et al. 2015; Gomez-Pastor et al. 2017; Jiang et al. 2013; Khalsa 2015; Kim et al. 2016; Kozuki et al. 2011; Lee et al. 2014; Neef et al. 2010; Pierce et al. 2013; Soncin et al. 2003). Different molecules, i.e., Hsp90 inhibitors and proteotoxic stress inductors, have shown efficacy in activating HSF1 and ameliorating some neurodegeneration features in mouse models. Unfortunately, our incomplete understanding of the roles of HSF1 in the brain and the lack of direct activators of HSF1 that can penetrate the blood-brain barrier have negatively affected the translational potential of HSF1. Future studies need to address this need.
In this chapter, we have specifically focused on the pathological role of HSF1 in HD, a devastating neurodegenerative disease caused by a CAG repeat expansion in the HTT gene (Bates et al. 2015; MacDonald et al. 1993; Novak and Tabrizi 2010). A selective vulnerability characterizes HD with degeneration and death of medium spiny neurons (MSN) in the striatum (Gonitel et al. 2008; Goula et al. 2012; Kennedy et al. 2003; Lee et al. 2011; Mitchell and Griffiths 2003; Pickrell et al. 2011; Shelbourne et al. 2007) and deficits in behavioral, cognitive, and motor features (Group 1996; Kieburtz et al. 2001; Novak and Tabrizi 2010). Cumulative evidence shows that HSF1 plays a crucial role in ameliorating disease progression in HD. Recent research has shown the inappropriate degradation of HSF1 in HD, exacerbates HTT aggregation and neuronal death (Gomez-Pastor et al. 2017). These studies also revealed a potential implication of HSF1 in regulating genes with synaptic functions and new avenues for controlling the levels of HSF1 in the brain.
Here, we will explore the fundamental features of HSF1, including the structure and regulatory mechanisms, and its implication in physiological and pathological conditions, mainly focusing on the role in the regulation of Hsp expression, proteasome-mediated degradation of abnormal proteins, oxidative stress, mitochondrial dysfunction, excitotoxicity, and synaptic function in HD.
3 Introduction to Heat Shock Factor 1
3.1 HSF1: Structure, Function, and Regulation
The human HSF family consists of six members: HSF1, HSF2, HSF4, HSF5, HSFX, and HSFY (Gomez-Pastor et al. 2018). Each of them possesses specialized tissue distribution, functions, and regulation. Among them, HSF1 is the most studied HSF due to its relevance during the stress response and cell survival (Akerfelt et al. 2010). The human HSF1 gene is located on chromosome 8q24 and is translated into 529 amino acids with a predicted molecular weight of 57 kDa. However, due to several PTMs (discussed below), the actual molecular size reaches approximately 75 kDa. HSF1 is composed of 4 functional domains distributed from N-terminus to C-terminus as follows: DNA binding domain (DBD), oligomerization domains (OD) composed of 4 leucine zipper domains (LZ1–3 and LZ4), regulatory domain (RD), and a transactivation domain (TAD) (Fig. 1).

Diagram of structural domains, regulatory enzymes, and PTMs of human HSF1. HSF1 can be divided into different structural domains: DBD (DNA binding domain), HR-A/B (heptad repeat-A/B), RD (regulatory domain), and TAD (transactivation domain). PTMs located at the top part of HSF1 are modifications with positive regulatory properties (red arrow), whereas PTMs located at the (bottom) represent modifications with repressive properties (black arrow). The different enzymes responsible for positive PTMs are SIRT1 (sirtuin 1), PIM2 (proviral integrations of Moloney virus 2, Pim-2 proto-oncogene, Ser/Thr kinase), CK2 (casein kinase holoenzyme), p300 (histone acetyltransferase p300), MEK (mitogen-activated protein kinase kinase), AKT (protein kinase B), and PLK1 (polo-like kinase 1). The enzymes responsive for the repressive PTMs are p300, GCN5 (general control non-repressed protein 5 histone acetyltransferase), MK2 (MAPK activated protein kinase 2), AMPK (5′-AMP-activated protein kinase), PLK1, SAE1/2 (SUMO-activating enzyme), UBC9 (RING-type E3 SUMO transferase), GSK3β (glycogen synthase kinase 3β), CK2, CK2α’ (catalytic subunit CK2 holoenzyme), and ERK (extracellular signal-regulated kinase). Ac acetylation, P phosphorylation, and S sumoylation
The DBD, which is highly conserved within the human HSF family, binds to the major groove of the DNA by recognizing repeating units of a pentameric sequence motif (nGAAn, where n is any base) named HSE. Recent comprehensive ChIP-seq demonstrated that the architecture of HSEs is very diverse in the human genome, with deviations from the consensus sequence in the spacing, orientation, and extent of HSE repeats (Mahat et al. 2016; Pincus et al. 2018; Vihervaara et al. 2017; Vihervaara et al. 2013). These deviations can influence HSF1 DNA binding efficacy and the kinetics and magnitude of target gene expression. Several studies over the last decade have shown different types of HSE that can be classified as canonical and non-canonical HSE. Vihervaara et al. demonstrated that HSF1 prefers binding to triple inverted nGAAn pentamers (canonical HSE) in both mitotic and cycling human erythroleukemia K562 cells (Vihervaara et al. 2013). Studies in purified HSF1 using fluorescence polarization and thermal denaturation profiling showed that HSF1 prefers binding to extended HSE sequences. The HSE placement is on a head-to-head or tail-to-tail orientation (Jaeger et al. 2014). Studies from S. cerevisiae showed two types of non-canonical HSE, gap- and step-type HSE. In gap-type HSE, two nGAAn repeats are followed by a gap of 5 bp block and another repeat (nGAAnnTTCn(5 bp)nGAAn) (Morano et al. 2012), whereas, in step-type HSE, three of each nGAAn repeats are interrupted by 5-bp block (nGAAn(5 bp)nGAAn(5 bp)nGAAn) (Morano et al. 2012; Yamamoto et al. 2005). Recent studies using C. albicans found that GAAnnTTC and TTCn7TTC are another non-canonical binding site where HSF1 regulates virulence genes’ expression (Leach et al. 2016). Different mechanisms can explain preferred binding to different HSEs, including sequence orientation and oligomerization with different HSFs (Jaeger et al. 2014, 2016; Korfanty et al. 2014). HSF1 and HSF2 can form heterotrimers in vitro and in vivo and cooperate under different stressful and physiological conditions to bind different HSEs (Jaeger et al. 2016; Korfanty et al. 2014; Sandqvist et al. 2009). However, the full implications of different HSF1-HSF2 oligomers’ combination on tissue-specific genome regulation are still unknown.
The OD is composed of two amphiphilic helices, heptad repeat HR-A/B or leucine zipper LZ1–3, which consist of a repeating pattern of seven hydrophobic and charged amino acid residues and it is critical in the activation of HSF1. HSF1 exists in monomeric and oligomeric (mostly trimer) states, and transitions between these two states are critical for HSF1 activation. Interactions between HR-A/B and other heptad repeats HR-C (or LZ4), located between the RD and TAD, regulate oligomerization of HSF1. Under non-stressful conditions, HR-A/B and HR-C are permanently bound by intramolecular coiled-coil interactions forcing the protein to be in a monomeric state (Chen et al. 1993; Nakai et al. 1997; Rabindran et al. 1993). Different conditions can disrupt the interaction between HR-A/B and HR-C, allowing HSF1 trimerization. Recent research has shown that HSF1 possesses an intrinsic capacity to sense temperature and that HSF1 monomers exposed to different temperatures can promote HR-A/B and HR-C dissociation (Hentze et al. 2016). However, other mechanisms described below have shown the ability to regulate oligomerization, such as PTMs and interactions with different regulatory proteins (reviewed in (Gomez-Pastor et al. 2018)). Trimerization is thought to be a prerequisite for the transcriptional activity of HSF1 and induction of Hsp (Farkas et al. 1998; Lu et al. 2008, 2009; Orosz et al. 1996). However, recent studies using a mutant form of HSF1 lacking the OD (HSF1Δ156–226) showed that, although HSF1 is unable to trimerize, this form was capable of protecting cells exposed to proteotoxic conditions (Qu et al. 2018; Verma et al. 2014), suggesting the existence of alternative mechanisms to control HSF1 activation not described yet.
The RD is a highly unstructured domain targeted by many different PTMs (Green et al. 1995; Guo et al. 2001), which allow the RD to provide activating or repressing functions to HSF1 (Newton et al. 1996). Finally, the TAD located in the C-terminus is responsible for the transcriptional activation of target genes (Green et al. 1995; Newton et al. 1996). Recent HSF1 and HSF2 crystal structure studies have revolutionized the way we think about HSF binding to DNA and have opened the door for new investigations regarding HSF DNA-binding regulation (Jaeger et al. 2016; Neudegger et al. 2016). Previous models suggested that the position of HSF1-DBD was so that the rest of the protein precluded access to the DBD once bound to the DNA, therefore limiting the interaction between the DBD and other potential regulatory proteins. Jaeger et al. and Neudegger et al. independently proposed a new HSF-DNA binding model in which the DBD wraps around the DNA, forcing the rest of the protein to be oriented in the opposite direction to the DBD and therefore leaving the DBD exposed for interaction with other potential regulators (Jaeger et al. 2016; Neudegger et al. 2016) (Fig. 2).

Structural insights into HSF-DNA interaction topology. (a, b) Crystal structure of the DNA binding domain (DBD) of HSF1 and HSF2 bound to a two-site HSE as a dimer. These independently solved structures revealed that a previously unknown carboxy-terminal helix (red) that is conserved in both HSF1 and HSF2 directs these HSFs to wrap around the HSE DNA, resulting in a topology where the DBD and the remainder of the HSF1 protein (not present in the crystal structure) occupy opposite sides of the DNA. (c) A new model of the HSF-DNA interaction. Structural studies support a model in contrast with the previous model for the topology of DNA-bound HSF oligomers. In the old model (left), the oligomerization domains (light blue) were positioned on top of the DBD, such that the rest of the protein buried the free surface of the DBD (shown in red, in contrast to the DNA-bound portion of the DBD shown in blue). In the new model (right), this free surface of the DBD is solvent-exposed, making it available for interactions with regulatory proteins and accepting PTMs. (Figure adapted from (Gomez-Pastor et al. 2018) with authors’ permission)
As stated above, the HSF1 activation/attenuation mechanism is highly influenced by its structural conformation and different protein-protein interactions (Fig. 3). Under non-stressful conditions, HSF1 exists in the cytoplasm as an inactive monomeric form due to intramolecular interactions between the HR-A/B and HR-C and direct protein-protein interactions with several inhibitory complexes. One of them is an auto-regulatory complex induced by HSF1-regulated proteins such as Hsp90, Hsp70, Hsp40, and T-complex protein-1 ring complex (TRiC)/chaperonin containing TCP-1 (CCT) (Akerfelt et al. 2010; Anckar and Sistonen 2011; Neef et al. 2014; Zheng et al. 2016). Previous research has shown that Hsp90 can inhibit HSF1 oligomerization and DNA binding (Zou et al. 1998), whereas Hsp70 and its co-chaperone Hsp40 regulate HSF1 transactivation by interacting with the TAD (Gomez et al. 2008; Shi et al. 1998). The cytosolic chaperonin TRiC/CCT also inhibits HSF1 activation by direct interaction with HSF1, although the exact mechanism by which TRiC/CCT inhibits HSF1 is not fully characterized (Akerfelt et al. 2010; Neef et al. 2014). Other repressive hetero-complexes include 14–3-3, histone deacetylase 6 (HDAC6), and the valosin-containing protein (VCP) that ultimately tune HSF1 activation (Pernet et al. 2014). Upon stress, there is a liberation of HSF1 from these repressive complexes allowing HSF1 trimerization and accumulation into the nucleus where HSF1 interacts with a different set of regulatory proteins that assist HSF1-DNA binding and transcriptional activation of its target genes (Amin et al. 1988; Pelham 1982; Sorger and Pelham 1988). HSF1 transcriptional activation results in increased levels of Hsp and other repressive proteins that hinder HSF1 activation by a negative feedback mechanism after the stress has subsided. Among these additional repressive proteins, filamin A interacting protein 1-like (FILIP-1 L) promotes HSF1 poly-ubiquitination and degradation by recruiting hHR23A, a ubiquitin receptor protein functioning as a transferrer of ubiquitinated proteins to the 19S proteasome (Hu and Mivechi 2011). In mammalian cells, FILIP-1 L forms complexes with HSF1 and Hsp72, and its ectopic overexpression reduces HSF1 protein levels leading to inhibition of HSF1-mediated transcription. However, the biological function of FILIP-1 L and the regulatory mechanisms responsible for HSF1-FILIP-1 L interaction are still unclear. Below we will discuss other regulatory events responsible for controlling HSF1 activity and stability in different contexts.

HSF1 activation/attenuation cycle. In response to proteotoxic stress conditions, HSF1 is subject to a multi-step activation and attenuation cycle. Inactive HSF1 monomer is kept in the cytoplasm in a complex with regulatory proteins such as Hsp 40, 70, and 90, as well as the cytosolic chaperonin TCP1 ring complex (TRiC). Upon stress sensing, HSF1 is modified by several activating PTMs that promote DNA binding and transcriptional activation of target genes in concert with cofactor recruitment. HSF1 is then modified by different inhibitory PTMs and by p23 causing DNA dissociation, HSF1 inactivation, and HSF1 degradation (see Fig. 1) for PTMs details). It is currently unknown where HSF1 degradation occurs and the extent to which HSF1 is newly synthesized or recycled into the cytoplasm. Ultimately, after attenuation, HSF1 is maintained in the cytoplasm by an inhibitory protein complex in a negative feedback mechanism. Color code: DNA-binding domain (dark blue), leucine zipper oligomerization domain LZ1–3 (light blue), regulatory domain (gray-white), LZ4 (yellow), and activation domain (orange). (Figure adapted from (Gomez-Pastor et al. 2018) with authors’ permission)
3.2 HSF1 PTMs: Pathophysiological Implications
The levels of HSF1 do not usually vary during stress-induced activation. In contrast, HSF1 undergoes numerous PTMs, including phosphorylation, sumoylation, and acetylation, that establish a complex code responsible for controlling every step of the HSF1 activation/attenuation cycle. Overall, HSF1-PTMs are classified into two main functions: positive and negative regulatory PTMs (Fig. 1). It is essential to mention that the enzymes responsible for HSF1 modifications and their effects are entirely dependent on the context. One example of the versatile-PTMs and the effects that they cause on HSF1 is led by polo-like kinase 1 (PLK1). During early mitosis, HSF1 is phosphorylated at Ser216 by PLK1, leading to HSF1 ubiquitin-dependent degradation by the SCFβTrCP E3 ligase (Lee et al. 2008). This event is critical to ensure mitosis progression. However, under heat shock conditions, PLK1 phosphorylates Ser419 and regulates HSF1 nuclear translocation. Similarly, protein kinase CK2 (casein kinase holoenzyme) regulates Thr142 phosphorylation and HSF1 DNA binding activation under heat shock conditions, but it controls phosphorylation of Ser303/307 and HSF1 degradation in the presence of pathogenic huntingtin (HTT) aggregates (Gomez-Pastor et al. 2017; Soncin et al. 2003).
Dozens of HSF1-PTM descriptions have been looked at under different experimental conditions, especially under heat shock. They have recently been reviewed in Gomez-Pastor et al. (2018), and their summaries can be seen in Fig. 1. This chapter will focus on some of those HSF1 PTMs that have relevance in pathophysiological stages. A fundamental set of PTMs that seem to control HSF1 activity and stability in different pathological conditions is Ser303 and Ser307 phosphorylation. In many cancer cells, there is a dramatic alteration in the levels of HSF1, contributing to cell proliferation and tumorigenesis (Jiang et al. 2015; Vydra et al. 2014). In melanoma cancer cells, the ubiquitin E3 ligase complex (Skp1-Cul1-F box) formed by the substrate-targeting subunit F-box and WD repeat domain containing 7 (FBXW7) interacts with and ubiquitylates HSF1. This interaction depends on Ser303/307 phosphorylation mediated by glycogen synthase kinase 3β (GSK3β) and extracellular signal-regulated kinase 1 (ERK1), respectively (Kourtis et al. 2015). Kourtis et al. suggested that reduced levels of FBXW7 or loss of function mutations present in many tumors may lead to increased levels of HSF1. However, a recent study in breast cancer cells showed that FBXW7 knockdown does not enhance HSF1 levels in those cells (Yang et al. 2019). Instead, elevated proviral integrations of Moloney virus 2 (PIM2) kinase phosphorylates HSF1 at Thr120, disrupting HSF1, and FBXW7 and promoting HSF1 accumulation (Yang et al. 2019). On the other hand, a very recent study by Gomez-Pastor et al. found that protein kinase CK2 α prime (CK2α’) also phosphorylates Ser303/307 in HD (Gomez-Pastor et al. 2017). CK2α’ is upregulated in HD, leading to increased FBXW7-dependent HSF1 degradation. Recently Jin et al. generated a knock-in mouse model where Ser303/307 were mutated to Ala and showed that HSF1 levels were stabilized and increased compared to wild-type mice (Jin et al. 2018). However, these mice presented age-dependent obesity, fatty liver disease, and insulin resistance, suggesting that phosphorylation of Ser303/307 may exert a positive effect in specific situations.
In general, phosphorylation of residues with inactivation functions is reduced in tumor cells, contributing to the hyperactivation of HSF1 previously reported in cancer. Mitogen-activated protein kinase kinase (MEK)-mediated Ser326 phosphorylation causes the stabilization of HSF1 by preventing it from poly-ubiquitination and subsequent proteasomal degradation (Tang et al. 2015). Repressive Thr367 phosphorylation is also reduced in cancer, although the specific kinase involved in this modification has not been identified yet (Asano et al. 2016). Other phosphorylation with repressive functions reduced in cancer is Ser121 involved in Hsp90 binding, mediated by MAPK-activated protein kinase 2 (MK2) and the 5′-AMP-activated protein kinase (AMPK). By contrast, during metabolic stress, AMPK-mediated phosphorylation of Ser121 increases and dictates HSF1 nuclear localization and stability (Asano et al. 2016; Dai et al. 2015; Guettouche et al. 2005). While several studies have revealed essential PTMs that contribute to HSF1 activity and stability in cancer, our knowledge about the different PTMs that regulate the role of HSF1 in other different diseases is very limited. These studies are necessary to fully understand the mechanisms that regulate HSF1 function under pathological conditions and may identify novel therapeutic targets.
Intriguingly, phosphorylation also serves as a platform for additional PTMs like sumoylation. GSK3β-induced phosphorylation at Ser303 is a prerequisite for Lys298 sumoylation, conjugation of SUMO-2/3, which results in inhibition of the transactivating capacity of HSF1 (Hietakangas et al. 2003). This sumoylation is mediated by the E1 SUMO-activating enzymes SAE1/2 and E2 SUMO-conjugating enzyme UBC9. The authors demonstrated the existence of a phosphorylation-dependent sumoylation motif within HSF1 (ΨKxExxSP, where Ψ is a branched hydrophobic amino acid and x is any amino acid), which resembles a consensus SUMO site (ΨKxE), that is utilized to prime proteins as a SUMO substrate. In general, sumoylation is facilitated by the aid of an E3 ligase, which increases sumoylation efficiency either by accelerating SUMO transfer from UBC9 to the substrate or merely by providing scaffolding support (Brunet Simioni et al. 2009). Site-specific mapping of the human SUMO proteome has revealed co-modifications with phosphorylation and the presence of several sumoylated-Lys on HSF1 (Hendriks et al. 2017). However, their roles in HSF1 regulation under physiological conditions are still unknown.
Another modification influencing HSF1’s function is Lys acetylation. Considering the studies reported, acetylation within the DBD has adverse effects on HSF1-DNA interaction. Acetylation at Lys80 in the DBD by p300 shortens the time of HSF1 on DNA, reducing HSF1 activation (Westerheide et al. 2009). In agreement with this, deacetylation at Lys80 by NAD + -dependent sirtuin 1 (SIRT1) accelerates HSF1 activity by increasing HSF1 DNA occupancy. Interestingly, the recognition of SIRT1 is as a nutrient sensor and longevity factor. The gradual loss of SIRT1 during aging correlates with the dissipation of HSF1 and reduced HSR and protein homeostasis in aging (Akerfelt et al. 2010). Zelin et al. demonstrated that general control non-repressed protein 5 (GCN5) histone acetyltransferases target HSF1 Lys80 in the presence of p23 chaperone, which disrupts HSF1 DNA binding (Zelin et al. 2012). An additional study identified that overexpression of histone deacetylases HDAC 7, HDAC9, and SIRT1 stimulated heat-triggered HSF1 DNA binding (Zelin and Freeman 2015). On the other hand, acetylation of Lys residues within the RD contributes to preventing HSF1 degradation. A study conducted in HeLa cells showed that acetylation at Lys208 and Lys298 by p300 prevents HSF1 from proteasomal degradation, maintaining HSF1 stability (Raychaudhuri et al. 2014). Also, HDAC6 has been implicated in the activation of HSF1 by repressing the Hsp90-HSF1 complex and promoting HSP gene expression (Boyault et al. 2007; Pernet et al. 2014). Future studies are warranted to demonstrate the relevance of these modifications in pathological stages in which there is a dysregulation in HSF1. A summary of the most relevant regulatory proteins based on the HSF1-PTMs they are responsible for, and their role in the regulation of HSF1, is shown in Fig. 4.

HSF1-regulatory enzymes and their contribution in HSF1 activation cycle. Upon proteotoxic stress, misfolded proteins titrate away the Hsp repressive complex, allowing HSF1 trimerization and nuclear accumulation. The HSF1 trimer binds to HSE in the promoter region of HSF1 target genes (old model of HSF-DNA binding: see Fig. 2). HSF1 activation is modified by several PTMs (see Fig. 1). Enzymes responsible for controlling nuclear translocation and activation include MEK and AMPK (with opposite functions). PGC-1α and different co-activators modulate HSF1 transcriptional capacity. Enzymes like HDAC7, HDAC9, and SIRT1 prolong HSF1 binding to the DNA, while acetylation by p300/CBP has an opposite effect and mediate DNA dissociation. Ubiquitin proteasome-dependent HSF1 degradation occurs by E3 ligases FBXW7 and NEDD4. FBXW7 is recruited by phosphorylation of HSF1 mediated by GSK3β, ERK, and CK2α’, whereas NEDD4 is accessed by p300/CBP-mediated acetylation (Figure reprinted from “Rethinking HSF1 in Stress, Development, and Organismal Health” by Li et al. 2017, with copyright permission from Elsevier. Figure legend has been modified accordingly for this publication)
4 Regulation of HSF1 and the HSR by Non-coding RNAs
Emerging evidence have shown that non-coding RNAs (ncRNAs) are actively involved in the regulation of HSF1 and the HSR (Place and Noonan 2014). A large class of small ncRNAs known as microRNAs (miRNAs) can regulate many biological processes by acting as post-transcriptional regulators of gene expression. Different miRNAs can bind in the 3’UTR of HSF1 altering its expression. miR-378 directly targets and represses the expression of HSF1 in cardiomyocytes (Jie Yuan et al. 2010) therefore affecting the induction of downstream HSPs in the heart while miR-608 operates as an HSF1 positive regulator in human breast cancer (Huang et al. 2012). Levels of HSF2 are also influenced by miRNAs (Björk et al. 2010; Cai et al. 2015). Other key determinant in the regulation of HSFs in human cells is the long non-coding RNAs (lncRNA). The lncRNA HSR1 (heat shock RNA-1), is upregulated during the HSR and plays an essential role in HSF1 trimerization and subsequent DNA binding activity forming a complex with the eukaryotic elongation factor 1A (eEF1A) (Shamovsky et al. 2006; Shamovsky and Nudler 2009). HSR1 is a foreign lncRNA derived from a bacterial genome, functioning as an exogenous auxiliary factor required for mammalian HSF1 activation upon stress conditions (Choi et al. 2015; Kim et al. 2010a, b).
HSF1 also regulates the expression of different non-coding RNAs (ncRNAs) involved in global suppression of transcription, translational processes, and protein aggregation (Lindquist 1986). Upon heat shock, HSF1 induces the expression of a class of lncRNAs known as Satellite III transcripts (Sat3) that accumulate at the site of transcription to form nuclear stress bodies (nSBs) (Jolly et al. 2004; Rizzi et al. 2004; Sengupta et al. 2009) and are known to co-localize with several RNA binding proteins and transcription factors such as HSF1 (Cotto et al. 1997; Jolly et al. 2004; Metz et al. 2004). Although knockdown of Sat3 transcripts does not affect HSF1 recruitment to the nSB-like structures, it has been recently demonstrated that Sat3 transcripts are essential for the recruitment of additional transcription regulators to the nSBs contributing to the heat-induced transcriptional silencing (Goenka et al. 2016). Transcription of the lncRNA satellite 2 (Sat2) is also strongly upregulated in the presence of heat shock in a HSF1-dependent manner (Tilman et al. 2012), and it has been involved in tumor progression (Tilman et al. 2012). Genome-wide studies revealed that HSF1 has also the ability to bind HSE present upstream of different miRNAs and regulate their expression under thermal stress (Srijit Das 2015). Interestingly, some of those HSF1 regulated miRNAs have shown inhibitory effects on HTT protein, and they are significantly depleted in HD, therefore contributing to increased mHTT expression and aggregation (Das and Bhattacharyya 2015). These studies demonstrate an integrated model of ncRNAs and HSF activity in the regulation of the HSR under physiological conditions and human diseases.
5 Role of HSF1 in Physiology and Disease
HSF1 is well known for its role in regulating the HSR, as discussed above. However, it has implications in many other processes, including aging, immune system maintenance, cancer, metabolic stress, neural development, and neurodegeneration. These functions can be accomplished by combining different protein-protein interactions and post-translational modifications that modulate HSF1 activity and stability and end up activating different transcriptional programs. Some of these functions are discussed below and summarized in Fig. 5.

Relationship between HSF1 and different physiological and pathological processes. Changes in activity and stability of HSF1 are responsible for regulating different cellular processes that are essential for life, including brain development, immune response, metabolism, and cell growth. Dysregulation of HSF1 contributes to different diseases like cancer and neurodegeneration. For each process, we included different proteins related to HSF1 and that contribute to the regulation of such a process. Proteins shown in a white box correspond to proteins that directly or indirectly regulate HSF1 activity or stability. In contrast, proteins shown in a blue box are proteins whose expression is influenced by HSF1
5.1 HSF1 in Aging and Inflammation
For every single living cell, there is constant exposure to environmental and physiological stresses during its lifespan. Therefore, proper mechanisms are necessary to execute an adequate response to ensure cell survival. However, during aging, there is a profound decline in the HSR that is accompanied by frail HSF1 activity and downregulation of its downstream target genes that compromise the survival of aged cells exposed to stressful conditions (Kregel 2002). Direct involvement of HSF1 in aging was reported by Garigan et al. showing that HSF1 knockdown speeded up the decline of tissue integrity and shortened lifespan in C. elegans (Garigan et al. 2002), while others showed that HSR significantly declines in this organism in early adulthood at reproductive maturity (reviewed in (Labbadia and Morimoto 2015)). These results have been supported by overexpression studies, in which HSF1 expression increased C. elegans lifespan by 20–40% (Hsu et al. 2003; Morley and Morimoto 2004). Examples in mammals include diminished HSF1-DNA binding capacity and Hsp accumulation in aged rats (21–26-month-old) upon heat shock and reduced HSF1-DNA binding in human lymphocytes and skin fibroblasts from old individuals (>70 years) when compared with those from a young age (20–40 years) (Fawcett et al. 1994; Gutsmann-Conrad et al. 1998; Jurivich et al. 1997; Locke and Tanguay 1996).
There is a coupling between longevity enhancing capacity of HSF1 with insulin/insulin-like signaling (ILS) pathway (Barna et al. 2012; Chiang et al. 2012). In C. elegans, stimulation of the insulin/insulin-like growth factor 1 receptor DAF-2 (IGF-1R in mammals) inactivates anti-aging and HSP genes via activation of protein kinase B (AKT) and phosphorylation of the longevity-related transcription factor DAF-16 (FOXO in mammals). Increased DAF-2 signaling inhibits HSF1 activity by promoting HSF1 to compete with negative regulators DAF16-dependent longevity-1 (DDL-1) and DDL-2, which accelerates aging (Chiang et al. 2012). Although it is unclear the integration of DAF-2 signaling to HSF1, both HSF1 and DAF-16 are needed for DAF2-mediated extension of lifespan (Cohen et al. 2006; Hsu et al. 2003; Morley and Morimoto 2004). Moreover, both HSF1 and DAF-16 activate the same small hsp genes, hsp-12 and hsp-16, which are essential to promote longevity (Hsu et al. 2003). The ILS-dependent lifespan extension is observed in mammals as well. Heterozygous Igf-1r knockout mice have shown to live approximately 26% longer than their wild-type littermates (Holzenberger et al. 2003). Also, sequence analysis of IGF-1R genes displayed over-representation of heterozygous mutations of the IGF1-R gene in female centenarians (Suh et al. 2008). While these studies did not explore the involvement of HSF1 on long-lived life, given that HSF1 plays a critical role in IGF-1R-dependent lifespan regulation, it is reasonable to speculate that HSF1 might be a controlling factor in the longevity of mammals as it is in invertebrates.
Human aging is also characterized by chronic low-grade inflammation. Several studies indicate that HSF1 modulates normal immune response and inflammation by controlling the expression of different cytokines. For example, HSF1 suppresses tumor necrosis factor-α (TNF-α) by binding to the HSE-like sequences within the TNF-α promoter, while it represses IL-1β through direct interaction with a regulator of IL-1β transcription, a nuclear factor for interleukin-6 (NF-IL6) (Xiao et al. 1999; Xie et al. 2002a). In the case of IL-6, HSF1 represses IL-6 by recruiting an IL-6 repressor activating transcription factor 3 (ATF3) into the open chromatin structure of IL-6 (Inouye et al. 2007). Interestingly, reports have linked HSF1 with the repression of HIV-induced inflammation by impairing the HIV-dependent expression of IL-6 (Inouye et al. 2007; Takii et al. 2010; Xie et al. 2002b), TNF-α (Muralidharan et al. 2014), and IL-1β (Xie et al. 2002a). Additionally, active HSF1 increases the expression of anti-inflammatory cytokine IL-10 (Zhang et al. 2012). Therefore, the decline of HSF1 activity during aging may contribute to increased inflammatory cytokines expression exacerbating aging.
5.2 Tumorigenesis and HSF1
Cancer cells are in a hostile environment enriched with stress, including hypoxia, acidity, ATP depletion, and lack of nutrients (Hanahan and Weinberg 2011). Thus, it can be assumed that under these conditions HSF1 may remain constitutively activated. In 2007, Dai et al. demonstrated that Hsf1−/− mice were far more resistant to skin-induced tumor formation than Hsf1+/+ mice (Dai et al. 2007). The authors showed that HSF1 steers cancer growth by modulating proliferation, signal transduction, protein translation, and glucose metabolism. Since then, there has been growing evidence that shows the increase of HSF1 in many cancer types, including breast cancer, colorectal cancer, gastric cancer, myeloma, non-small-cell lung cancer, oral squamous cell carcinoma, prostate cancer, and many other cancer types (Cui et al. 2015; Fok et al. 2018; Ishiwata et al. 2012; Li et al. 2018; Santagata et al. 2011; Tong et al. 2018). However, it is essential to clarify that increased HSF1 expression is not causative for tumorigenesis. The different roles of HSF1 in cell proliferation and cancer were extensively reviewed recently (Carpenter and Gokmen-Polar 2019; Gomez-Pastor et al. 2018; Jiang et al. 2015). Therefore, we will focus on the most recent and groundbreaking studies that have set up HSF1 as an essential target in cancer biology.
The mechanisms underlying increased HSF1 expression and activity in cancer are complex and vary between different types of tumors and cell types. Zhao et al. showed that the increased levels of HSF1 in ERBB2-overexpressing cancers are due to an elevation of HSF1 protein translation (Zhao et al. 2009). On the other hand, Kourtis et al. suggested that increased levels of HSF1 in melanoma are due to increased protein stability and decreased proteasomal degradation through decreased or mutated E3 ligase FBXW7 (see HSF1 PTMs section for further details) (Kourtis et al. 2015). As we previously mentioned in the above section, phosphorylation of Ser and Thr with repressive functions within HSF1 is significantly reduced in cancer cells, leading to increased HSF1 activity. In cells from human malignant peripheral nerve sheath tumor (MPNST) lacking neurofibromatosis type 1 (NF1), a tumor suppressor, there is an increase in RAS/MEK-mediated HSF1 Ser326 phosphorylation, which leads to HSF1 trimerization and nuclear translocation. Reports have indicated that this modification significantly contributes to cancer progression (Dai et al. 2012). Also, HSF1 acts in diverse signaling pathways driving cancer initiation, migration, invasion, and metastasis. For example, HSF1 enhances the initiation and progression of breast cancer by upregulating RNA-binding protein Hu-antigen R (HuR) and increasing hypoxia-inducible factor 1 (HIF-1) signaling (Gabai et al. 2012). Knockdown of HSF1 represses epithelial-mesenchymal transition (EMT), a facilitator of metastasis in cancer development, and reduces cell migration in ovarian cancer cells (Powell et al. 2016). Specifically, HSF1 is phosphorylated at Ser326 by AKT and binds to SLUG promoter, an EMT regulator, leading to the upregulation of SLUG in HER2-positive breast cancer cells (Carpenter et al. 2015). These findings suggest that the HSF1-SLUG axis is an essential pathway in cancer progression.
HSF1 exerts its effects on cancer progression, in part through the upregulation of Hsp. In MPNST, loss of NF1 increased HSF1 levels and Hsp90 expression, promoting carcinogenesis (Dai et al. 2012). HSF1 activation and Hsp90 expression are also essential in enhancing tumor growth in HER2-positive breast cancer (Schulz et al. 2014). Hsp70 also enhances tumorigenesis by acting as a survivor factor due to its anti-apoptotic effects. For example, Hsp70 and its co-chaperone BCL-2–associated athanogene 3 (BAG3) mediate apoptosis resistance in glioma cells by supporting cell survival through increasing the levels of pro-survival BCL-2 family members (Antonietti et al. 2017). In addition to the upregulation of Hsp90 and Hsp70, there is also an elevation in the expression of Hsp27 in a variety of different cancers, and it is involved in tumor progression and drug resistance (Fang et al. 2012; Vahid et al. 2016; Xu et al. 2006; Yu et al. 2014; Zhao et al. 2012). Interestingly, a study conducted in hepatocellular carcinoma showed that HSF1-mediated phosphorylation of Hsp27 rather than Hsp27 expression is necessary to promote migration and invasion of carcinoma cells (Fang et al. 2012).
Intriguingly, the upregulation of HSF1 in cancer is associated with the activity of tumor suppressor p53 (Toma-Jonik et al. 2019). Under stress conditions, HSF1 interacts with p53 in the nucleus and form a complex with DNA damage kinases to effect p53 phosphorylation in response to DNA damage (Logan et al. 2009). The presence of mutations in p53 protein is also connected to the activation of HSF1. Specifically, a gain-of-function p53 mutant variant (Arg280Lys) directly interacts with HSF1 phospho-Ser326, enhancing HSF1 transcriptional activity and Hsp90 expression in human breast cancer cells (Li et al. 2014a). Hence, the gain of function of p53 offers not only the drastic acceleration of oncogenic signaling but also the Hsp-induced survival capacity of cells through HSF1. On the other hand, DNA damage induces p53 activation and HSF1 downregulation, resulting in senescence (Kim et al. 2012). Moreover, HSF1 depletion causes growth inhibition of breast cancer cells by promoting p53-induced senescence (Meng et al. 2010), suggesting that lack or loss of p53 combined with HSF1 activation support tumorigenesis by regulating senescence and proliferation. These observations indicate that HSF1 may be a central mediator of the oncogenic function of different mutant variants of p53 observed in cancer cells.
Reports have shown that the transcriptional program triggered by HSF1 in malignant cells is different from those mediated by heat shock. The genes activated in this program drive oncogenic transformation by accelerating protein folding, translation, mitosis, invasion, metabolism, and metastasis and obstructing immune functions and apoptotic response (Mendillo et al. 2012). Since there is an elevation in the expression levels of HSF1 in many solid tumors and there is an association between its levels with low survival rate and metastasis (Ciocca et al. 2013; Mendillo et al. 2012), HSF1 has been proposed as a prognostic factor in cancer. In addition, there is a link between increased HSF1 and reduced survival in many different tumors like estrogen receptor-positive breast cancer, osteosarcoma, pancreatic cancer, melanoma, and esophageal squamous cell carcinoma (Kourtis et al. 2015; Liang et al. 2017; Liao et al. 2015; Santagata et al. 2011; Santagata et al. 2013; Tsukao et al. 2017; Zhou et al. 2017). In patients with hepatocellular carcinoma (HCC), expression of HSF1 was also found to be correlated with poor overall survival (Fang et al. 2012) and, specifically, the high levels of HSF1 phospho-Ser326 has been reported in HCC progression and invasion (Li et al. 2014b). Notably, the levels of HSF1 phospho-Ser326 have clinical significance in shorter overall survival in ovarian cancer patients (Yasuda et al. 2017). Collectively, the expression and activity of HSF1 can serve as a potential clinical biomarker for patients with cancers. Due to the high levels of HSF1 and its relevance in tumor growth, HSF1 has become a desirable target to treat cancer (Carpenter and Gokmen-Polar 2019; Dong et al. 2019). However, although HSF1 has been validated as a potent target in cancers by genetic knockdown studies, HSF1 inhibitors reported to date have lacked specificity and potency for clinical evaluation (Dong et al. 2019). Hence, it is necessary to make a more systematic design to develop more potent and specific HSF1 inhibitors in the future.
5.3 HSF1 in the Central Nervous System (CNS)
Ubiquitous expression of HSF1 in the developing brain implies a need for HSF1 during neurodevelopment (El Fatimy et al. 2014). It is now known that not only HSF1 but also HSF2 plays essential roles in brain function through modulation of neuronal migration, formation, and maintenance of neuronal synapses. Lack of HSF1 dramatically alters brain structure, neuronal differentiation, and synaptic formation (Chen et al. 2014; Hooper et al. 2016; Uchida et al. 2011). In the hippocampus, a brain area involved in learning and memory, the absence of HSF1 causes a decrease in the dendrite length of the dentate gyrus granule neurons and pyramidal neurons. It reduces dendritic spine density, resulting in reduced synapse formation (Uchida et al. 2011). Additional observations demonstrated that lack of HSF1 in the hippocampus causes low expression of polysialylated-neural cell adhesion molecule (PSA-NCAM), which is fundamental in synapse formation. Other synaptic proteins, such as synapsin and discs large MAGUK scaffold protein 4 (DLG4) involved in synaptogenesis, also depend on HSF1 (Chen et al. 2014). Due to the specific role of HSF1 in regulating synaptic proteins, alteration in the levels of HSF1 during embryogenesis results in alterations in synaptic fidelity and memory consolidation. In this line, HSF1 also regulates the expression of brain-derived neurotrophic factor (BDNF), an essential regulator of synaptogenesis and synaptic plasticity mechanisms underlying learning and memory (Chen et al. 2014; Cunha et al. 2010). Other functions attributed to HSF1 are the regulation of lipid raft formation, the subdomains of plasma essential for postsynaptic consolidation of memory receptors and long-term memory retention (Nagy et al. 2007; Suzuki and Yao 2014), and regulation of Ca2+ homeostasis through the activation of CALBINDIN expression in cerebellar Purkinje cells (Ingenwerth et al. 2016). The deficiency of HSF1 also results in loss of oligodendrocytes and severe demyelination, astrogliosis, increased activated microglia, and neuronal apoptosis across different brain regions (Homma et al. 2007).
HSF1 has critical protective roles in response to stresses during brain development. Research has shown that exposure of Hsf1-/- mice to stressful conditions during prenatal stages increases neuropsychiatric susceptibility like disorders in the newborns (Hashimoto-Torii et al. 2014). During exposure to stresses like alcohol or a maternal epileptic seizure, HSF1 is retained in the nucleus and activates Hsp70 expression. Intriguingly, under these conditions, HSF1 does not show a characteristic hyperphosphorylation pattern upon activation but rather presents reduced acetylation and sumoylation (El Fatimy et al. 2014). Interestingly, El Fatimy et al. demonstrated that prenatal alcohol exposure (fetal alcohol syndrome) causes brain structural abnormalities dependent on the formation of HSF1-HSF2 heterotrimers (El Fatimy et al. 2014). The group found that fetal alcohol exposure causes HSF1-HSF2 heterotrimers to bind to and downregulate doublecortin-like kinase 1 (Dclk1) expression, a gene that participates in neuronal migration and neurogenesis. The authors also demonstrated that lack of Hsf2 exerts a protective role during prenatal alcohol exposure facilitating HSF1 homo-trimerization and regulating neuronal migration genes. These results indicate the complexity of the different HSF1 regulatory mechanisms during physiological conditions and the importance of these transcription factors in every step of an organismal life.
5.4 Neurodegenerative Diseases and HSF1
As we described in the previous section (HSF1 in the CNS), HSF1 participates in the development of the CNS by controlling diverse processes such as neuronal migration, neurogenesis, glycogenesis, synapse formation, and neuronal survival (El Fatimy et al. 2014; Homma et al. 2007; Hooper et al. 2016; Ingenwerth et al. 2016; Uchida et al. 2011). Therefore, maintaining proper levels of HSF1 is essential to ensure the appropriate development and maintenance of the CNS. However, in the adult brain, HSF1 activity significantly declines, contributing to inflammation and neuronal death (Calderwood et al. 2009; Murshid et al. 2013). In line with these observations, there is a significant reduction in HSF1 in many age-related neurodegenerative diseases (NDs), like AD, PD, and amyotrophic lateral sclerosis (ALS), which are characterized by protein aggregation and reduced expression of the protein quality-control machinery (Homma et al. 2007; Santos and Saraiva 2004). Lowered HSF1 has also been reported in other heritable NDs, such as those related to polyglutamine expansions (discussed below and in the section Implication of HSF1 in HD). The causes of HSF1 depletion during neurodegeneration are not well understood and may differ between different NDs. Nevertheless, numerous studies conducted in different animal models of NDs imply that increasing the levels or activity of HSF1 has therapeutic potential (Neef et al. 2011). In this book chapter, we will briefly discuss the role of HSF1 in different NDs with particular emphasis on the advances made in HD.
AD is a common age-associated neurodegenerative disease most often caused by extracellular depositions of amyloid-β, a neurotoxic peptide produced by the amyloid-β precursor protein proteolysis, leading to neuronal death (Mavroudis et al. 2010). Increasing reports show that intracellular levels of HSF1 are essential to maintain neuronal survival in AD. A study conducted in plasma neural-derived exosomes from patients with AD showed a remarkable reduction in the levels of HSF1 compared with unaffected individuals (Goetzl et al. 2015). There was also a reduction in the levels of HSF1 in the cerebellum of an AD rat model (Jiang et al. 2013). Notably, several studies have shown that overexpression of HSF1 results in an increased number of Purkinje cells in the cerebellum, the numbers of which are reduced in patients with AD and mouse models, lowering amyloid-β levels and improving cognitive deficits (Jiang et al. 2013; Khalsa 2015; Kozuki et al. 2011; Lee et al. 2014; Pierce et al. 2013). In line with the beneficial effects of increased HSF1 levels on neuronal survival, boosting HSF1 activity provides synaptic protection in AD. Pharmacological activation of HSF1 by using Hsp90 inhibitors led to the upregulation of Hsp70 and Hsp25 and improved synaptic integrity and memory consolidation in AD mouse models (Chen et al. 2014; Wang et al. 2017). Despite the lack of evidence as to whether the decrease of HSF1 in AD is due to a pathological degradation, it is essential to note that increased CK2 has been reported in AD mouse models and patients with AD, contributing to the inflammatory phenotype characteristic of AD (Masliah et al. 1992; Perez et al. 2011; Rosenberger et al. 2016). However, the connection between CK2, HSF1, and inflammation in AD has yet to be explored.
PD is characterized by α-synuclein aggregation and an age-dependent loss of dopaminergic neurons in the midbrain (Jellinger 2014). In PD, reports have shown low levels of HSF1 in both mouse models and patients (Kim et al. 2016). Kim et al. showed that α-synuclein accumulation enhanced HSF1 poly-ubiquitination and degradation by elevated ubiquitin E3 ligase neuronal precursor cell-expressed developmentally downregulated 4 (NEDD4) expression. Notably, the authors found that acetylation at Lys80 within the DBD of HSF1 makes it more accessible for NEDD4-mediated ubiquitination (Kim et al. 2016). It is unknown whether phosphorylation contributes to NEDD4-mediated HSF1 degradation in PD. However, several kinases induce HSF1 phosphorylation, such as CK2 in PD (Lee et al. 2004a; Mavroudis et al. 2010). Therefore, it is tentative to speculate that phosphorylation events may control HSF1 degradation in PD, as it happens in HD (Gomez-Pastor et al. 2017; Soncin et al. 2003). While the degradation mechanism of HSF1 in PD is not fully characterized, its therapeutic potential has been demonstrated by studies in which a constitutive active form of HSF1 (caHSF1) is expressed in a human cellular model of PD (Liangliang et al. 2010). In this study, caHSF1 reduced α-synuclein aggregation and toxicity by inducing the expression of Hsp70. As proof of concept, direct overexpression of Hsp70 in a Drosophila model of PD inhibited α-synuclein induced neurodegeneration (Auluck et al. 2002). Recently, a new pharmacological activator of HSF1 (echinochrome derivative U-133) administered to a rat model of PD showed increased levels of Hsp70 and neuroprotective effects, including decreased microglia activation, a-synuclein aggregation, and improved motor behavior (Ekimova et al. 2018).
ALS is characterized by progressive dysfunction and death of motor neurons in the brain and spinal cord. Mutations in several genes can cause familial ALS and contribute to the development of sporadic ALS. Mutations in the chromosome 9 open reading frame 72 (C9orf72) gene account for approximately 40% of familial ALS; superoxide dismutase (SOD1) gene mutations cause approximately 20% of familial ALS. TAR DNA binding protein (TARDBP) (also known as TDP-43) and the RNA binding protein fused in sarcoma/translocated in liposarcoma (FUS/TLS) mutations each account for about 5% of cases. Increased microtubule-associated protein Tau has also been linked to ALS (Schreiber et al. 2018). The depletion of HSF1 in ALS significantly contributes to increased oxidative stress and aggregation of TDP-43 (Batulan et al. 2003; Chen et al. 2016). In the cell model of ALS, HSF1-induced TDP-43 clearance is partly mediated by Hsp70 and its co-chaperone DNAJB2a (Jung et al. 2013). HSF1 also regulates taurine transporter (TauT) levels under oxidative stress conditions, acting as an antioxidant to protect motor neurons (Jung et al. 2013). These studies suggest that HSF1 partially protects motor neurons by compensating for constitutive oxidative stress, which is thought to be a key mechanism contributing to ALS’s pathogenesis.
Polyglutamine (polyQ) diseases are hereditary degenerative disorders characterized by the aberrant expansion of a CAG repeat in a specific gene, resulting in misfolding and aggregation of the disease-causing protein (La Spada and Taylor 2010). Multiple lines of evidence demonstrated that HSF1 and HSF1-dependent pathways strongly influence the pathogenesis of polyQ diseases. These studies are discussed in detail in the section Implication of HSF1 in HD. Understanding the specific roles of HSF1 in HD and other NDs may help develop new therapeutic strategies to increase the levels of HSF1 and prevent neurodegeneration across multiple NDs.
6 Protein Aggregation in HD
HD is a heritable neurodegenerative disease caused by a CAG trinucleotide (code for glutamine) repeat expansion within exon 1 of the HTT gene (MacDonald et al. 1993). The disease arises when the polyQ tract exceeds approximately 37 CAG repeats (Bates et al. 2015; Novak and Tabrizi 2010). The disease occurs in all populations but is most common in individuals of European ancestry, and it has an overall prevalence estimated to be 1 in every 10,000 individuals (Evans et al. 2013; Fisher and Hayden 2014; McColgan and Tabrizi 2018). Characteristic symptoms include progressive motor dysfunction (chorea, dystonia, bradykinesia, and incoordination), psychiatric disturbances (depression, obsessive-compulsive disorders, and anxiety) and cognitive decline (distractibility, impulsivity, and difficulty in multitasking) (Group 1996; Kieburtz et al. 2001; Novak and Tabrizi 2010). Other symptoms that accompany those mentioned above are weight loss and muscle wasting, metabolic dysfunction, and endocrine disturbance (Novak and Tabrizi 2010; Ross and Tabrizi 2011). Research has shown that such clinical aspects come from progressive and massive loss of neurons, predominantly GABAergic medium-sized spiny neurons (MSNs) located in the striatum. A brain region that controls movement and some forms of cognition (Albin et al. 1992; Deng et al. 2004; Ferrante et al. 1991; Ross and Tabrizi 2011; Rubinsztein 2003; Waldvogel et al. 2015). However, neurodegeneration in other brain regions, including cortex, thalamus, and hippocampus, is also observed as the disease progresses (Cepeda et al. 2007; Puigdellivol et al. 2016; Sieradzan and Mann 2001). The disease’s symptoms usually begin in the adult-onset (around age 34–40 years with 15–20 years of progression). However, when the length of the CAG expansion exceeds about >60, it manifests before age 20 years (Juvenile HD) and accounts for 5–10% of all HD cases (Chen et al. 2001; Gusella and MacDonald 2000; Morley et al. 2002; Novak and Tabrizi 2010; Rubinsztein 2003). Despite numerous studies in the past two decades that have been addressing the importance of mutant HTT (mHTT) protein aggregation and MSN degeneration in HD pathology, very little is known about the exact molecular mechanisms by which mHTT induces neuronal death. There are several proposed pathophysiological pathways in this regard, many of which are associated with protein quality control impairment. In this chapter, we will discuss those pathways in which HSF1 may play a key role.
6.1 Structure and Function of HTT
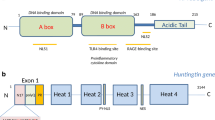
The HTT gene encodes a large ~350 kDa widely expressed protein composed of multiple domains (Fig. 6) that control conformation, protein localization, and function (reviewed in (Jimenez-Sanchez et al. 2017)). PolyQ expansion induces conformational changes in HTT, affecting structural aspects of different regions or the protein proteolytic cleavage, unfolding, and aggregation (Almeida et al. 2013). While HTT is considered a cytoplasmic protein, it can partially accumulate in the nucleus, which is enhanced by the polyQ expansion and is believed to aggravate the disease. The highly conserved domain N17 (17-amino acid-long N-terminus) can modulate nuclear localization and mHTT toxicity (Desmond et al. 2012). N17 interacts with the nuclear pore protein called translocated promoter region (TPR) that is involved in nuclear export, facilitating the shuttling of HTT from the nucleus to the cytoplasm (Cornett et al. 2005). PolyQ expansions decrease this interaction and increase the nuclear accumulation of HTT (Cornett et al. 2005). The N17 is also subject to several PTMs, including sumoylation (Steffan et al. 2004), phosphorylation (Aiken et al. 2009; Thompson et al. 2009), acetylation (Thompson et al. 2009), and ubiquitination that control interactions between N17 and other proteins (Atwal et al. 2007; Graham et al. 2006; Kalchman et al. 1996; Maiuri et al. 2013; Steffan et al. 2004; Thompson et al. 2009; Wellington et al. 2002). The polyQ tract starts right after the N17, and it is composed of a series of CAG repeats interrupted by a CAA codon (also codes for glutamine) as follows (CAG)n-CAA-CAG (where n ≤ 36 repeats in unaffected individuals). There is an inverse correlation between the polyQ tract length and the age of onset (AOO) (Langbehn et al. 2010). However, there is a lot of variability in the AOO that could not be explained by considering the number of polyQ. Two recent studies have shown that the number of the CAG uninterrupted track rather than the number of polyQ determines HD onset timing (Wright et al. 2019). Following the polyQ tract is a polyproline (polyP)-rich region that consists of 10–11 proline and functions in stabilizing polyQ conformation by forming proline-proline helixes (Bhattacharyya et al. 2006; Kim et al. 2009). These domains are followed by tandem arrays of a repeat called HEAT, which is named for the following four functionally characterized proteins: HTT, elongation factor 3 (EF3), the regulatory A subunit (65 kDa) of protein phosphatase 2A (PP2A), and target of rapamycin 1 (TORl). They form rod-like helical structures and serve as a scaffold for many protein interactions (Andrade and Bork 1995; Saudou and Humbert 2016). Recently, a nuclear localization sequence (NLS) has been described between amino acids 174 and 207, preceding the first HEAT domain, allowing the interaction with KARYOPHERIN β2. This protein mediates the nuclear import of proteins (Desmond et al. 2012). The authors showed that polyQ expansion decreased the interaction between the NLS containing region and karyopherin β2, increasing mHTT nuclear accumulation. A nuclear export signal (NES) can also be found in the C-terminus of HTT (Xia et al. 2003). Truant and colleagues have demonstrated that the C-terminus region’s proteolytic cleavage occurs during HD and proposes that lack of the C-terminus containing NES facilitates the nuclear accumulation of mHTT and pathogenesis (Xia et al. 2003).

Diagram of human wild-type HTT relative to its known domains and post-translational modifications. HTT is composed of different domains; N17 (containing the first 17 amino acids of the protein), polyQ tract (whose expansion is responsible for HD), polyP repeats, and five helical clusters of HEAT repeats. Several PTMs (e.g., acetylation [Ac, pink], phosphorylation [P, orange], sumoylation [S, green], and ubiquitination [Ub, blue]) alter HTT’s cell biology and toxic properties. Within the N17 region, we find (P1) P-Ser13 and P-Ser16, (Ac1) Ac-Lys9, S-Lys6, 9 and 13, and (S1) S-Lys9. Other PTMs can also be detected along with the protein (P2) P-Ser116, (P3) Ser-419, 421, 434, 533, 535, and 536, (P4) P-Ser1181 and 1,201, (P5) P-Ser2076, P(6) P-Ser2653 and 2,657, (Ac2) Ac-Lys178 and 236, (Ac3) Ac-Lys 345, (Ac4) Ac-Lys444, and (S2) S-Lys444. For a detailed list of all the PTMs identified in vivo and in vitro, see (Arbez et al. 2017). The corresponding amino acid sequence from exon 1 is shown in blue for the N17, in purple for the polyQ repeat, and in green for the polyP-rich region. Sequences were obtained from the NCBI protein database (Human: NP_002102.4)
Another factor that contributes to HD pathogenesis is CAG repeat somatic instability. Meaning that the CAG track undergoes progressive length increases over time in somatic tissues, particularly in HD’s most affected brain regions. Prior research demonstrated that CAG somatic instability could account for the increased susceptibility of MSNs to mHTT due to cell-type-specific transcriptional programs (Gonitel et al. 2008; Goula et al. 2012; Kennedy et al. 2003; Lee et al. 2011; Shelbourne et al. 2007). It is known that CAG length variation is tissue-dependent, and it is enhanced in the striatum of HD mouse models and HD patients. A hypothesis for this phenomenon is the tissue-specific positioning of the RNA pol II at the HTT locus, which is enhanced in the striatum (Goula et al. 2012). Pharmacological suppression of CAG instability in the HdhQ150 mouse improved neuropathology and demonstrated the role of somatic instability is pathogenesis (Budworth et al. 2015; Kovtun et al. 2007; Massey and Jones 2018). Also, several mismatch repair genes, including MSH3, influence somatic instability of CAG repeats and are considered critical genetic modifiers for the disease (Flower et al. 2019). Recently, the Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium demonstrated that the uninterrupted CAG repeat is a propensity for length instability leading to its somatic expansion (Lee et al. 2019). Other aspects contributing to the HD pathology include HTT transcriptional dysregulation and HTT mRNA abnormal splicing, two topics recently discussed in (Thomson and Leavitt 2018).
HTT is an essential protein required for normal development since the ablation of Htt in mice resulted in embryonic death (Zeitlin et al. 1995). However, the exact function of HTT is not known yet. There is an expression of HTT throughout the body, but it is highly active in the CNS, suggesting a potential role in brain physiology. Gain-of-function effects of mHTT have been proposed as the primary driver for neurodegeneration in HD. However, recent studies proposed an alternative hypothesis suggesting that loss-of-function may be at the forefront of the pathogenesis (reviewed in (Cattaneo 2003)). Studies using conditional deletion of HTT in mice’s forebrain resulted in neurodegeneration, demonstrating that HTT is necessary for neuronal function and survival. A loss-of-function mechanism may contribute to HD pathogenesis (Dragatsis et al. 2000). In support of this study, McKinstry and colleagues demonstrated that silencing HTT in the developing mouse cortex was viable but resulted in alterations in cortical and striatal synaptic connectivity similarly to those deficits observed in mHTT expressing mice (McKinstry et al. 2014). Other studies have also shown that HTT plays a crucial role in neurogenesis by maintaining the lineage potential of primitive neural stem cells during neural induction and synapse formation (Nguyen et al. 2013; Sun et al. 2001). Additionally, HTT has been related to protein scaffolding, vesicular and organelle trafficking, and transcription regulation (Benn et al. 2008; Caviston et al. 2007; Gunawardena et al. 2003; Lee et al. 2004b; Li et al. 2001; Orr et al. 2008; Parker et al. 2001; Rong et al. 2006; Zuccato et al. 2001). While it is still unclear the direct role of HTT in all these processes, it seems evident that alteration in the structure and conformation of HTT can alter many different pathways that complicate the study of HD and difficult the development of effective therapeutic strategies.
Accumulation and aggregation of mHTT are positively associated with mitochondrial dysfunction, which is critical in promoting MSNs degeneration and death in HD. The mHTT disrupts mitochondria by interacting with several mitochondrial proteins and regulators. Consequently, the outcome of mitochondrial perturbation is ATP deficiency and increased reactive oxygen species (ROS) production. Both factors contribute to the exacerbation of mitochondrial damage and mHTT aggregation. Research has shown increased mitochondrial fragmentation in cellular and mouse models of HD and the brain of patients with HD (Cherubini and Gines 2017). Mitochondrial fragmentation is related to the abnormal interaction between mHTT with the central regulator of protein fission and dynamin-related protein 1 (DRP-1), resulting in DRP-1 dysfunction (Cherubini and Gines 2017). The mHTT also disrupts retrograde and anterograde mitochondrial trafficking along axons, resulting in a reduced transport of mitochondria to synapses with high energy demands and contributing to synaptic dysfunction (Orr et al. 2008). Also, mHTT interacts with TIM23, a component of the mitochondrial inner membrane translocase, altering mitochondrial protein import, and leading to respiratory dysfunction and neuronal cell death (Yano et al. 2014). Mitochondria dysfunction is also caused by decreased transcription of nuclear-coded mitochondrial genes such as peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), a transcription coactivator that stimulates energy metabolism (Costa and Scorrano 2012; Cui et al. 2006; McConoughey et al. 2010; Weydt et al. 2006). One of the main pathways involved in the downregulation of PGC-1α in HD is the CREB/TAF4 signaling pathway, which is altered in the presence of mHTT (Cui et al. 2006). Very recently, Intihar et al. demonstrated that HSF1 also regulates the expression of PGC-1α in striatal like-cells, and deficits in HSF1 contribute to the downregulation of PGC-1α expression observed in HD (Intihar et al. 2019). The contribution of HSF1 to mitochondrial dysfunction in HD is discussed in the section HSF1 as a Potential Regulator of PolyQ-Dependent Mitochondrial Dysfunction.
6.2 HSR and Other Protein Quality Control Systems in HD
Full-length mHTT and proteolytic cleaved fragments have the propensity to misfold and aggregate, forming insoluble inclusion bodies, a hallmark of HD (Bates 2003; Davies et al. 1997; DiFiglia et al. 1997). Aggregates arise elsewhere in the cell, including cytoplasm, nucleus, dendrites, and axon terminals (DiFiglia et al. 2007; Vonsattel 2008). The insoluble aggregates have been observed in in vitro cell models of HD, mouse models of HD, and HD patients (Davies et al. 1997; DiFiglia et al. 1997; Gray et al. 2008; Sahl et al. 2012). However, it remains under debate whether the aggregates are a byproduct of a cellular attempt to protect neurons from misfolded mHTT protein or are instead the cause of pathology. Unfortunately, discriminating between these two hypotheses and correlating the presence of aggregates with the onset of a phenotype is technically tricky since in many cases quantifying small polyQ oligomers is challenging. When successful, it does not inform about all the different structural conformers present in the system. Studies conducted by Yang and colleagues demonstrated that preformed polyQ aggregates are highly toxic when directed to the nucleus, establishing proof that aggregates represent species with toxic properties (Yang et al. 2002). The pharmacological intervention aimed at inhibiting aggregate formation has shown beneficial effects in a mouse model of HD (Sánchez et al. 2003; Tanaka et al. 2004). Therefore, studying the regulatory mechanisms that lead to enhanced aggregation in HD may lead to more effective therapeutic strategies that can ameliorate neuronal death.
Neurons are post-mitotic cells in constant need of protein quality control to ensure protein homeostasis and keep cells functional. The HSR is a mechanism to cope with proteotoxic stress by inducing the expression of molecular chaperones and other Hsp, and it constitutes the first line of defense against aggregation. Due to the stress-inducible nature of Hsp, it would be expected to observe increased levels of Hsp in response to the accumulation and aggregation of mHTT. However, several studies demonstrated that Hsp shows a progressive decrease in cellular and mouse models of HD and that depletion of Hsp contributes to disease pathogenesis (Chafekar and Duennwald 2012; Hay et al. 2004). Research has shown decreased Hsp expression in postmortem brain tissue from patients with HD (Gomez-Pastor et al. 2017; Hodges et al. 2006). Multiple proposed hypotheses aim to explain the depletion of Hsp in HD. Hay et al. suggested that changes in the levels of Hsp are due to their sequestration into aggregates, a hypothesis supported by other studies (Park et al. 2013; Seidel et al. 2016; Yamanaka et al. 2008), while Labbadia et al. showed Hsp expression impairment in HD due to altered chromatin structure of the Hsp promoters (Labbadia et al. 2011). Finally, Gomez-Pastor et al. demonstrated that abnormal degradation of HSF1 during HD pathogenesis likely contributes to the downregulation of Hsp (Gomez-Pastor et al. 2017). Recently, a fascinating study conducted by Neueder and colleagues exposing wild-type and HD mice (HdhQ150 and R6/2) to a heat shock treatment demonstrated that HD mice are unable to induce Hsp in affected tissues, revealing a disruption in the HSR in HD mice (Neueder et al. 2017). Interestingly, recent global chaperone gene expression analysis in the adult mouse brain identified that the striatum shows intrinsically lower levels of Hsp compared to the cortex (Tebbenkamp and Borchelt 2010), which may contribute to the explanation of enhanced abrogation in the HSR in the striatum and the increased susceptibility of MSNs in the presence of mHTT.
The ubiquitin-proteasome system (UPS), an intracellular pathway for degrading unfolded and unnecessary proteins, also constitutes an essential defense mechanism against protein aggregation. The prominent presence of ubiquitin characterizes inclusion bodies in HD. Recent work using the CAG140Q knock-in mice demonstrated that the UPS activity is lower in the striatum than in other brain regions, which correlates with decreased ubiquitin-activating enzyme (UBE1) levels (Wade et al. 2014). Decreased UPS activity, together with the observation that mutations in the UPS components give rise to some neurodegenerative diseases, suggests that UPS impairment may contribute to HD (reviewed in (Ortega and Lucas 2014; Schipper-Krom et al. 2012)). Another route for degradation of dysfunctional or aggregated proteins is via autophagy, a lysosome-mediated degradation pathway, whose role in HD has been widely studied (reviewed in (Croce and Yamamoto 2019)). One example that demonstrates impairment in the autophagy pathway is the decreased expression of high-temperature requirement protein A2 (HTRA2/OMI), a positive regulator of autophagy (Li et al. 2010), in cultured striatal neurons and the striatum of patients with HD (Inagaki et al. 2008). Inagaki et al. proposed that HTRA2/OMI is relevant to the selective vulnerability of striatal neurons in HD. Also, Yamamoto and colleagues identified a new protein, autophagy linked FYVE (ALFY), that mediates selective macroautophagy of aggregated proteins and whose expression is essential for the clearance of HTT aggregates (Yamamoto and Simonsen 2011). Recent studies indicate that the macroautophagic machinery, comprised of a core group of autophagy-related proteins, such as ATG5, ATG7, and LC3, is compromised in HD (Croce and Yamamoto 2019; Filimonenko et al. 2010; Martinez-Vicente et al. 2010). These findings demonstrate that degradative systems responsible for HTT clearance are affected and contribute to the age-dependent accumulation of neuronal aggregates. Very recently, a study led by Li and colleagues implemented the use of a small-molecule-microarray-based screening to identify specific linkers between the autophagy protein LC3 and mHTT that promote cargo recognition and uptake of mHTT into the macroautophagy pathway for degradation (Li et al. 2019). Administration of these linkers to HD mouse models decreased HTT aggregates’ load and ameliorated motor deficits associated with HD (Li et al. 2019). This study establishes a start point for developing new therapies that exploit the degradative properties of different protein quality control systems as a strategy to remove mHTT aggregates and alleviate neurodegeneration.
7 Implication of HSF1 in HD
7.1 HSF1 and Hsp Function in HD
As discussed earlier, the HSR facilitates the folding of proteins and maintains protein homeostasis by inducing the expression of a set of molecular chaperones and Hsp (Bjork and Sistonen 2010; Lindquist 1986; Morimoto 1998). Hsp is a family of proteins named by their molecular weight whose functions require ATP (Fink 1999; Wang and Spector 2001). Some of the most critical Hsp players in the HSR include Hsp40, Hsp60, Hsp70, Hsp90, Hsp100, and the small Hsp (De Maio and Vazquez 2013; Kampinga et al. 2009). Among them, Hsp70 and its co-chaperone Hsp40 are central Hsp in the HSR. They are involved in folding proteins correctly and preserving polypeptides in a soluble conformation by binding to misfolded disease proteins, thereby preventing them from generating toxic protein structures (Hartl and Hayer-Hartl 2002). Early studies conducted in yeast expressing polyQ showed that Hsp70 and Hsp40 could alter the formation of detergent-insoluble mHTT aggregates to detergent-soluble amorphous structures (Muchowski et al. 2000). Additionally, immunolabeling and fluorescence resonance energy transfer (FRET) experiments showed that Hsp70 and Hsp40 directly bind to the N-terminus region of HTT within the inclusion bodies and interfere with the intramolecular rearrangement of HTT by modulating the interaction between HTT and other proteins (Hay et al. 2004; Jana et al. 2000; Schaffar et al. 2004). Atomic force microscopy has also revealed that Hsp70 and Hsp40 reduce the formation of spherical and annular structures of mHTT fragments, resulting in the partition of monomeric conformations of mHTT and acceleration of fibrillization (Wacker et al. 2004). Consequently, deletion of Hsp70 in the R6/2 HD mouse model increased mHTT inclusion bodies’ size and exacerbated HD-related physiological and neuropathological features (Wacker et al. 2009). Furthermore, single overexpression of different DNAJ proteins (Hsp40 family members) has shown protective properties in different HD animal models (Bason et al. 2019; Gillis et al. 2013; Kakkar et al. 2016). These studies demonstrated the fundamental role of Hsp70/Hsp40 in preventing polyQ aggregation.
In yeast, Hsp70 also assists Hsp104, a member of the Hsp100 family with disaggregase activity, by binding to protein aggregates and recruiting Hsp104 (Acebron et al. 2009; Okuda et al. 2015; Winkler et al. 2012). After Hsp104 is bound to protein aggregates, Hsp70 stimulates ATP-driven substrate-threading activity of Hsp104, leading to the extraction of single unfolded polypeptides from aggregates (Lum et al. 2004; Schlieker et al. 2004; Tessarz et al. 2008; Weibezahn et al. 2003; Zietkiewicz et al. 2004). Hsp110, the mammalian homolog of Hsp104, also interacts with and disassembles protein aggregates through its ATP-dependent activity (Gao et al. 2015; Garcia et al. 2017; Rampelt et al. 2012; Shorter 2011). In vitro and in vivo assays have recently shown that a trimeric chaperone complex formed by HSC70 (a member of the Hsp70 family), Hsp110, and DNAJB1 (Hsp40) completely suppresses fibrillization of HTT exon1 Q48, demonstrating the effective co-operation between the different Hsp (Scior et al. 2018). Hsp90 also plays a vital role in the triage of HTT. However, contrary to what would be expected, Hsp90 functions in promoting polyQ aggregation. Reports have shown that N-terminal fragments of HTT associate with Hsp90, and inhibition of Hsp90 increases ubiquitination and proteasomal degradation of mHTT by disrupting the Hsp90-mHTT complex (Baldo et al. 2012; He et al. 2017). Hsp90 diminishes the removal of mHTT aggregates by promoting the recruitment of a deubiquitinating enzyme (ubiquitin-specific protease 19 (USP19)) (He et al. 2017).
Several transcriptomic and proteomic studies conducted in different mouse models of HD and patients with HD have demonstrated that several Hsp are in low abundance in HD affected tissues (Ament et al. 2017; Hodges et al. 2006; Kumar et al. 2016; Langfelder et al. 2016). The thought is that this defect contributes to polyQ aggregation and neurodegeneration. The importance of restoring the levels of Hsp has been widely demonstrated in cell cultures, yeast, fly, and mouse models of HD in which overexpression of different Hsp including Hsp70, different members of the Hsp40 family as well as different small Hsp decreased mHTT aggregation and neuropathological features of HD (Bason et al. 2019; Carmichael et al. 2000; Fuchs et al. 2009; Gillis et al. 2013; Gunawardena et al. 2003; Jana et al. 2000; Kuo et al. 2013; Wacker et al. 2004, 1999a). There is an association between the decreased expression of Hsp and impairment in the HSR (Chafekar and Duennwald 2012; Cowan et al. 2003; Duennwald and Lindquist 2008; Maheshwari et al. 2014; Neueder et al. 2017; Riva et al. 2012), and different hypothesis have been proposed in this regard (discussed in HSR and Other Protein Quality Control Systems in HD section). Different studies suggested that Hsp depletion is caused by their sequestration into aggregates (Park et al. 2013; Seidel et al. 2016; Yamanaka et al. 2008), while Labbadia et al. proposed that impaired Hsp expression in HD occurs at the level of transcription due to hypoacetylation of histone H4 at Hsp promoters altering nucleosome landscape and chromatin architecture (Labbadia et al. 2011). However, these studies did not fully explain how Hsp promoters are preferentially hypoacetylated over other HD gene promoters.
The transcriptional regulation of the HSR is primarily controlled by HSF1 (Parsell and Lindquist 1993; Wu 1995). The importance of HSF1 is demonstrated by the high susceptibility to heat-induced apoptosis in Hsf1-null fibroblasts (McMillan et al. 1998) and the failure to respond to thermal insults in Hsf1-null mice (Neueder et al. 2017; Xiao et al. 1999), whose defects are attributed to the improper induction of HSF1-induced Hsp. The fundamental role of HSF1 in HD was demonstrated by Hayashida and colleagues when they showed increased polyQ aggregation and shortened lifespan in a transgenic mouse model of HD (R6/2) lacking Hsf1 (Hayashida et al. 2010). Additionally, overexpression of a caHSF1 in the same mouse model enhanced Hsp expression, inhibited polyQ aggregation, and ameliorated HD-like symptoms (Fujimoto et al. 2005; Rimoldi et al. 2001). Other authors obtained similar results in C. elegans, Drosophila, and mammalian cells expressing polyQ (Homma et al. 2007; Hsu et al. 2003; Morley and Morimoto 2004), supporting the importance of HSF1 in HD. acknoHSF2 also plays an important role in polyQ aggregation since lack of HSF2 enhanced mHTT aggregation and shortened the lifespan of R6/2 mice (Shinkawa et al. 2011). Although the exact role of HSF2 in HD has not been fully characterized, it is possible that the defects observed in Hsf2 null mice are associated with an alteration in the ratio of HSF1-HSF2 heterotrimers and the impact on HSF1 activation.
Early studies conducted by Chafekar and Duennwald in immortalized striatal cells derived from wild-type mice expressing full-length HTT-Q7 (STHdhQ7) and knock-in HD mice expressing HTT-Q111 (STHdhQ111; HD cells) demonstrated a significant reduction in HSF1 in HD cells. HSF1 depletion leads to severe impairments when cells are exposed to heat shock conditions (Chafekar and Duennwald 2012). In the study, the authors demonstrated that both nuclear translocation and trimerization of HSF1 were significantly lower in STHdhQ111 cells than STHdhQ7, particularly after heat shock, presumably due to the lowered levels of HSF1 observed in the HD cells. Such deficits correlated with reduced expression of several Hsp upon HS in HD, a phenomenon widely observed in other studies (Chafekar and Duennwald 2012; Duennwald and Lindquist 2008; Maheshwari et al. 2014; Neueder et al. 2017). The authors also demonstrated a significant reduction of HSF1 levels, both mRNA and protein, in the striatum of STHdhQ111 knock-in mice compared to wild-type mice. As with the study above, studies conducted in R6/2 and zQ175 HD mice showed an age-dependent decrease in HSF1 levels in differentially affected brain regions (Gomez-Pastor et al. 2017; Maheshwari et al. 2014). HSF1 ChIP-seq in STHdhQ111 and STHdhQ7 cells under control (33 °C) or HS (42 °C) also showed a marked reduction in HSF1 DNA binding to its target genes in STHdhQ111 cells in both 33 and 42 °C compared to STHdhQ7 cells (Riva et al. 2012). Recently, studies conducted by Neueder et al. in which both R6/2 and CAG-140 HD mice were exposed to HS in vivo showed a significant impairment in the HSR, in which many affected genes were HSF1-dependent (Neueder et al. 2017). Further comparisons between HD and Hsf1 null mice transcriptomes in the absence of HS will be necessary to establish the extent to which HSF1 degradation contributes to large transcriptional changes in HD.
Very recently, Gomez-Pastor et al. demonstrated the existence of a pathological HSF1 degradation pathway in the presence of polyQ and suggested this process as a potential driving force for the HSR impairment in HD (Gomez-Pastor et al. 2017). The authors demonstrated that the presence of polyQ enhanced the expression of two proteins, protein kinase CK2α’ and the E3 ligase FBXW7, which sequentially phosphorylated and ubiquitylated HSF1 and signaled the protein for proteasomal degradation. The authors showed that CK2α’ mediated phosphorylation of Ser303/ 307 was a prerequisite for FBXW7-dependent ubiquitylation and degradation. Abrogation of HSF1 phosphorylation in vitro by either genetic mutation of the Ser303 into Ala and genetic and pharmacological inhibition of CK2α’ resulted in increased levels of HSF1 and Hsp70 and Hsp25 expression and decreased polyQ aggregation. As proof of concept, the authors generated a zQ175 HD mouse lacking one allele of the CK2α’ kinase and demonstrated increased levels of HSF1 and Hsp, decreased polyQ aggregation in the striatum, and improved HD-like phenotypes such as amelioration of synaptic deficits and decreased weight loss (Gomez-Pastor et al. 2017). These studies proposed CK2α’ as a potential therapeutic strategy to prevent HSF1 degradation in HD, enhance Hsp expression, and ameliorate protein aggregation. These and other studies have developed different therapeutic strategies aimed at activating HSF1 to enhance the levels of multiple Hsp simultaneously and ameliorate protein aggregation and neurodegeneration (Neef et al. 2011) and discussed in HSF1 as a Therapeutic Target in HD and Other PolyQ Diseases section.
7.2 Crosstalk Between HSF1 and the UPS: Implications in HD
When there is a failure in the correct folding of abnormal proteins, they can be targeted by the UPS for degradation or the lysosome-mediated autophagy pathway. In conjunction with the HSR, these systems ensure protein homeostasis and prevent the accumulation of toxic peptides (Hipp et al. 2012, 2019). However, eukaryotic proteasomes cannot digest polyQ sequences longer than 25Q residues (Venkatraman et al. 2004). Typically, polyQ diseases like HD possess polyQ sequences from 37-300Qs, exceeding standard proteasome products’ length and interfering with proteasome function. Inclusion bodies in HD contain ubiquitin and components of the proteasome (20S core proteasome, 19S regulatory complex, and 11S proteasome-activating complex) whose levels increase in an age-dependent manner (Davies et al. 1997; DiFiglia et al. 1997; Waelter et al. 2001). These observations suggest an age-dependent failure of the UPS to degrade polyQ (Sherman and Goldberg 2001). Also, the continued accumulation of these polyQ inclusions may impact the folding of other proteins exceeding the proteasome’s capacity and eventually reduce the cell’s ability to degrade other proteins (Bence et al. 2001).
Many investigators have connected dysregulation of the UPS and mHTT-induced toxicity in cellular models of HD (Bence et al. 2001; Bennett et al. 2005; Duennwald and Lindquist 2008; Hunter et al. 2007; Maynard et al. 2009), mouse models of HD (Bennett et al. 2007; Wang et al. 2008), and tissues from patients with HD (Bennett et al. 2007; Seo et al. 2004). The HSR and UPS pathways are closely interconnected. When there is a block in proteasome function by inhibitors such as MG132, abnormal proteins accumulate, and the expression of Hsp is enhanced (Bush et al. 1997; Lecomte et al. 2010; Mathew et al. 1998; Pirkkala et al. 2000). Also, damaged UPS is a potent stressor that stimulates HSR (Bush et al. 1997). Various studies indicated that the UPS network’s impairment activates HSF1 and HSF2, which in turn elevates Hsp expression (Lecomte et al. 2010; Mathew et al. 1998; Pirkkala et al. 2000). UPS-mediated induction of Hsp70 facilitates the E3 ubiquitin ligase C-terminus of HSC70-interacting protein (CHIP)-dependent ubiquitylation and degradation of protein aggregates (Lackie et al. 2017). The role of CHIP in promoting mHTT ubiquitylation and suppression of mHTT aggregation and toxicity has been extensively studied in cells, flies, zebrafish, and mouse models of HD (Al-Ramahi et al. 2006; Jana et al. 2005; Miller et al. 2005). CHIP is confined to the cytoplasm in unstressed conditions, but it translocates to the nucleus in response to cellular stress (Dai et al. 2003). CHIP promotes HSF1 trimerization and transcriptional activation of HSF1 and is a requirement for protection against stress-induced apoptosis in murine fibroblasts. Lastly, CHIP leads to stable interactions between Hsp70 and activated forms of HSF1. The authors detected CHIP in DNA-bound HSF1-containing complexes that were transcriptionally activated by heat shock (Dai et al. 2003). CHIP exerts a central and unique role in tuning the response to stress at multiple levels by regulating protein quality control and transcriptional activation of stress response signaling via HSF1.
There is a broad acceptance that the UPS function is abrogated in HD, although the causes of such a problem are unclear. It is still under debate whether deficits in the UPS activity cause mHTT accumulation or rather are the consequence of polyQ aggregation. Considering the intricate relationship between HSF1, Hsp70, and the UPS and the fact that HSF1 degradation and Hsp70 expression are impaired during HD, we speculate that the HSR impairment may cause UPS dysfunction in HD. However, further studies are necessary to unravel the role of the UPS in HD since alternative studies have shown no impairment in this system (Bett et al. 2006; Bowman et al. 2005; Diaz-Hernandez et al. 2003; Maynard et al. 2009; Seo et al. 2008).
Recent studies have demonstrated the existence of a mammalian membrane-associated proteasome complex, specifically expressed in the nervous system, that modulates neuronal function by degrading intracellular proteins into extracellular peptides, which can stimulate neuronal signaling (Ramachandran and Margolis 2017). This study challenged the notion that proteasomes function primarily to maintain protein homeostasis. In a follow-up study, the authors indicated that this neuronal-specific 20S membrane proteasome complex (NMP) exclusively degrades a large fraction of ribosome-associated nascent polypeptides that are newly synthesized during neuronal stimulation (Ramachandran et al. 2018). Interestingly, this NMP-mediated degradation is independent of canonical ubiquitylation pathways. However, several aspects of these NMPs remain unknown, including their potential role in the UPS in neurons, whether there is an alteration in their function during HD, and the extent HSF1 influences the regulation of NMPs.
7.3 PolyQ-Induced Oxidative and Metabolic Stress: Convergence into HSF1
Cells possess antioxidant mechanisms responsible for the elimination of harmful byproducts generated during biological processes. However, the failure of these systems often results in oxidative stress and cellular cytotoxicity. Oxidative stress is the imbalance between ROS/reactive nitrogen species (RNS) generation and the biological antioxidant defense system. Accumulation of ROS/RNS leads to damage of proteins, DNA, and lipids and further damages tissues and organs. Many studies in both HD patients and experimental mouse models of HD have documented that oxidative stress is a critical player in HD neuropathology (Johri and Beal 2012; Paul and Snyder 2019). Studies conducted in the striatum of different mouse models of HD (R6/1 and R6/2) revealed the accumulation of ROS, alteration in nitric oxide synthase (NOS) and superoxide dismutase (SOD) activities (Deckel et al. 2002; Perez-Severiano et al. 2002; Santamaria et al. 2001; Tabrizi et al. 2000), and a decline of antioxidant molecules such as ascorbate and antioxidant vitamins (Rebec et al. 2002). Moreover, increased levels of 8-hydroxydeoxyguanosine (8-OHDG), reports have shown that a product of oxidative stress-derived DNA damage is present in the urine, plasma, samples from striatal microdialysis, and brain of R6/2 and BACHD mice (Browne and Beal 2006; Gray et al. 2008; Stack et al. 2008). Studies conducted in plasma and postmortem tissues from patients with HD have also shown increased global oxidative stress manifested by increased lipid peroxidation products (malondialdehyde and lipofuscin), reduction in the antioxidant molecules, and increased DNA damage and fragmentation (Browne et al. 1999; Butterworth et al. 1998; Chen et al. 2007; Hersch et al. 2006; Tunez et al. 2011). The latter correlated with CAG repeat length and instability (Massey and Jones 2018) (see Structure and function of HTT section for details). Biochemical and proteomic studies conducted in the cerebral cortex and the striatum of HD patients also revealed decreased glutathione (GSH), a potent intracellular redox buffer, and a substantial elevation of glutathione peroxidases (GPX1 and 6), peroxiredoxins (PRDX1,2, and 6), and manganese-containing SOD (MnSOD/SOD2) (Beal et al. 1992; Sorolla et al. 2008). Endogenous ROS mainly originates from mitochondria during the synthesis of ATP. There is a large amount of evidence showing that mitochondrial dysfunction indeed plays a crucial role in polyQ-mediated oxidative stress and the pathogenesis of HD (Carmo et al. 2018b; Costa and Scorrano 2012; Damiano et al. 2010; Guedes-Dias et al. 2016; Lin and Beal 2006; Quintanilla and Johnson 2009). This topic is discussed in detail in the section HSF1 as a Potential Regulator of PolyQ-Dependent Mitochondrial Dysfunction.
HSF1 can sense and respond to oxidative stress by inducing the expression of several Hsp and other anti-apoptotic genes (Ahn and Thiele 2003; Kim et al. 2013; Raitt et al. 2000). Studies conducted by Ahn and Thiele using purified recombinant mammalian HSF1 and H2O2 demonstrated that HSF1 directly senses oxidative stress by forming a disulfide bond between two cysteine residues (C35 and C105) localized within the HSF1 DNA-binding domain (Ahn and Thiele 2003). C35-C105 disulfide bond formation is critical for HSF1 trimerization, nuclear accumulation, DNA binding, and target gene activation in the presence of H2O2 (Ahn and Thiele 2003). This process seems to be reversible since the addition of reducing agents such as DTT to an H2O2 activated-HSF1 trimer decreased binding to an Hsp70 HSE sequence. At the same time, removal of DTT by dialysis and re-exposure to H2O2 recovered HSF1 binding. Additional studies conducted in yeast showed that Hsf1 coordinates the expression of Hsp under oxidative stress conditions by interacting with the yeast oxidative stress response regulator Skn7 (Raitt et al. 2000). Interestingly, Skn7 contains a small polyQ at its C-terminus (Gutiérrez et al. 2017), which implies that polyQ dependent aggregation mechanisms may play a role in regulating the Hsf1-Skn7 mediated response to oxidative stress. While there are no human homologs described for Skn7, it seems to share sequence homology and function with the RHOA effector protein ROCK-I (Alberts et al. 1998), a modulator of dendritic spine morphogenesis upon Ca2+ signaling (Murakoshi et al. 2011). RHOA and ROCK-I mRNA and the induction of protein levels in HD human blood leukocytes and postmortem brain and R6/2 HD mouse brain tissue respond to oxidative stress conditions (Narayanan et al. 2016). Interestingly, studies conducted in cardiomyocytes showed that activation of RHOA negatively regulates the HSR via attenuation of the HSF1-HSE binding and significantly suppressed the proteotoxic stress-induced HSR (Meijering et al. 2015).
Upregulation of Hsp during oxidative stress is key to ensure proper protein folding and prevents further oxidative damage (Janowska et al. 2019). Rubinsztein and colleagues showed that Hsp27, an ATP-independent chaperone, has antioxidant effects on cells expressing mHTT and its overexpression decreased ROS and polyQ-mediated cell death. Surprisingly, the authors also showed that there was not an attribution of benefits exerted by Hsp27 to the suppression of polyQ aggregation but rather the amelioration of polyQ-mediated oxidative stress (Wyttenbach et al. 2002). Reports have shown that Hsp27 protects against oxidative stress through its ability to increase GSH levels and glucose-6-phosphate dehydrogenase activity, preventing the mitochondrial cytochrome-c release and maintaining optimal cellular detoxifying machinery (Paul et al. 2002; Preville et al. 1999). HSF1 plays a crucial role in regulating Hsp27 expression in HD (Gomez-Pastor et al. 2017). Both HSF1 depletion and subsequent downregulation of Hsp27 in HD can contribute to the damages caused by polyQ-induced oxidative stress (Gomez-Pastor et al. 2017). Interestingly, Hsp27 also modulates HSF1 transactivation activity by promoting conjugation of SUMO-2/3 at Lys298 on HSF1 (Brunet Simioni et al. 2009). These studies demonstrate how Hsp27 exerts a feedback inhibition of HSF1 transactivation and enlighten the strictly regulated interplay between HSF1, Hsp, and oxidative stress.
A critical transcription factor that regulates antioxidant proteins’ expression and protects against oxidative damage is the nuclear factor E2-related factor 2 (NRF2). NRF2 activation mitigates multiple pathogenic processes involved in HD and other neurodegenerative disorders through upregulation of antioxidant defenses, inhibition of inflammation, improvement of mitochondrial function, and maintenance of protein homeostasis (Dinkova-Kostova et al. 2018). NRF2 has become an attractive therapeutic target in neurodegeneration, and several small molecules activating NRF2 have shown protective effects in numerous models of neurodegenerative diseases. NRF2 have several common target genes with HSF1, including heme oxygenase 1 (HMOX1, also known as HSP32) (Maines and Ewing 1996; Prestera et al. 1995), HSP70 gene (Almeida et al. 2010), autophagy cargo protein sequestosome 1 (p62/SQSTM1) (Komatsu et al. 2010; Samarasinghe et al. 2014), and ATF3 (Dziunycz et al. 2014; Hoetzenecker et al. 2011). Recent studies conducted in MCF7 breast cancer cells have shown that NRF2 transcriptionally activates HSF1 under oxidative stress conditions, connecting these two critical regulators of cytoprotective mechanisms (Paul et al. 2018). While no studies have yet explored these two transcription factors’ dual activities in HD, there is evidence that alteration of their expression levels and activities may affect HD pathogenesis (Dinkova-Kostova et al. 2018; Gomez-Pastor et al. 2017; Quinti et al. 2017).
Oxidative and metabolic stresses and mitochondrial dysfunction are interconnected HD processes (Lou et al. 2016). Proteomic analysis conducted in yeast revealed that cells challenged to menadione, a ROS generator, upregulated antioxidant enzymes, several Hsp, and the expression of metabolic enzymes in a Hsf1-dependent fashion (Kim et al. 2013). Additionally, overexpression of alphaB-crystallin (αB-crys) in astrocytes reversed HD related phenotypes and neuronal cell loss in BACHD mice by restoring antioxidant protection and glucose uptake in astrocytes (Oliveira et al. 2016). On the other hand, AMPK is another important sensor for cell survival by modulating energy homeostasis. Ju and colleagues showed that AMPK is activated in the brains of mice and patients with HD, suggesting that this abnormal activation may contribute to neuronal degeneration in HD (Ju et al. 2014). The authors showed that mHTT-induced ROS contributes to the activation of AMPK-α1 and subsequently facilitates neurotoxicity in STHdhQ109 cells and the striatum of R6/2 mice while AMPK inhibition reduced the level of oxidative stress (Ju et al. 2014). However, conflicting reports have shown the opposite indicating that activation of AMPK in HD protects from neuronal dysfunction and vulnerability (Vazquez-Manrique et al. 2016). Therefore, it is unclear to what extent AMPK activation can be considered a detrimental response. However, the fact that AMPK negatively regulates HSF1 activation via phosphorylation at Ser121 (Dai et al. 2015) points out the possibility that activation of AMPK-α1 could contribute to the impairment of the HSR in HD and therefore negatively contribute to pathology.
7.4 HSF1 as a Potential Regulator of PolyQ-Dependent Mitochondrial Dysfunction
7.4.1 Mitochondrial Dysfunction and Excitotoxicity
Mitochondria are distinct cellular organelles that control vital physiological processes such as energy production, lipid metabolism, and Ca2+ signaling. There is a strong association between mitochondrial function defects with different NDs, including HD (Wang et al. 2019). The overall manifestation of mitochondrial impairment in HD appears in the abnormal mitochondrial morphology (Jin et al. 2013a; Kim et al. 2010a, b; Panov et al. 2002; Squitieri et al. 2006), alteration in the components of the electron transport chain (Browne et al. 1997; Gu et al. 1996; Guidetti et al. 2001; Sawa et al. 1999), reduction in cellular glucose uptake and enzymes in the oxidative metabolism (Antonini et al. 1996; Butterworth et al. 1985; Dubinsky 2017; Feigin et al. 2001; Sorolla et al. 2008; Tabrizi et al. 1999), and increased mitochondrial DNA damage (Acevedo-Torres et al. 2009; Horton et al. 1995; Liu et al. 2008). Reports have shown that all these defects play an important role in promoting MSN degeneration (Carmo et al. 2018a; Costa and Scorrano 2012; Lin and Beal 2006; Quintanilla and Johnson 2009). Indeed, it has been proposed that mHTT-mediated mitochondrial abnormalities underlie the specific vulnerability of MSNs in HD since this type of neurons require a higher-energy demand compared to other cell types in the brain (Ferrante et al. 1991; Mitchell and Griffiths 2003; Pickrell et al. 2011). This hypothesis is supported by additional studies in which administration of mitochondrial inhibitors in rodents (Beal et al. 1993; Borlongan et al. 1995; Brouillet et al. 1993) and non-human primates (Brouillet et al. 1995) produced selective striatal degeneration and behavioral defects evocative of HD (Brouillet et al. 1999; Brouillet et al. 2005).
The primary function of mitochondria is the generation of energy in the form of ATP. ATP is generated via oxidative phosphorylation through the action of the electron transport chain (ETC) in conjunction with the ATP synthase (Lenaz and Genova 2010). Several components of the ETC present altered levels in postmortem striatum and cortex of patients with HD and cell and mouse models of HD (Browne et al. 1997; Gu et al. 1996; Majumder et al. 2007; Quintanilla and Johnson 2009; Tabrizi et al. 1999; Tabrizi et al. 2000). Interestingly, lymphoblasts from patients with HD, but not from patients with ataxia type-1 (another polyQ disease), treated with toxins targeting different complexes of the ETC, showed increased mitochondrial depolarization and caspase 3-dependent apoptosis (Sawa et al. 1999). This study demonstrated the increased susceptibility of HD cells to mitochondrial dysfunction and mitochondrial-induced cell death. However, analyses conducted in pre-symptomatic patients with HD have not shown ETC abnormalities (Guidetti et al. 2001), suggesting that mitochondrial defects may be secondary to neurodegeneration.
The exact mechanism by which mHTT induces mitochondrial dysfunction is unclear, but several studies have linked these defects to alterations in Ca2+ buffering. The maintenance of mitochondrial Ca2+ levels is by the mitochondrial permeability transition pore (mPTP). When mitochondrial Ca2+ buffering capacity is overloaded, the mPTP opens and causes a decrease in mitochondrial membrane potential (MMP), uncoupling the oxidative phosphorylation and leading to cell death (Choo et al. 2004; Halestrap 2006; Krieger and Duchen 2002; Rasola et al. 2010). Alterations in Ca2+ buffering capacity and decreased threshold for mPTP opening are defects widely reported in HD (Brennan Jr. et al. 1985; Choo et al. 2004; Ciammola et al. 2006; Milakovic and Johnson 2005; Milakovic et al. 2006; Oliveira et al. 2006; Panov et al. 2002; Rockabrand et al. 2007; Squitieri et al. 2006). These defects are more significant in the striatum than in the rest of the brain (Gellerich et al. 2008). They are delayed in cyclosporin A and mPTP inhibitor (Milakovic and Johnson 2005). Additional studies conducted in mitochondria isolated from skeletal muscle from R6/2 mice have shown that reduced Ca2+ accumulation capacity and mPTP opening threshold are also responsible for energetic depression and muscle atrophy (Gizatullina et al. 2006). Further, immunocytochemistry and electron microscopy studies conducted in brain mitochondria obtained from YAC72 HD mice revealed a direct interaction between the N-terminal fragment of mHTT and the outer mitochondrial membrane (Choo et al. 2004; Jin et al. 2013b; Kim et al. 2010a, b; Panov et al. 2002; Rockabrand et al. 2007; Squitieri et al. 2006), suggesting a direct role of mHTT in the alteration of mPTP function.
Yan et al. initially reported that HSF1 plays a significant role in regulating mitochondrial activity (Yan et al. 2002). The authors showed that the heart of Hsf1-/- mice presented increased oxidative damage on a structural component of the mPTP (ANT1 protein), which resulted in decreased ANT1 catalytic activity and increased mPTP opening. Additional studies in oocytes and hepatocytes of Hsf1−/−mice showed mitochondrial functional deficits, ultrastructural abnormalities, and increased caspase-3 activation (Bierkamp et al. 2010; Canto 2017). Recent studies have connected HSF1 with increased mitochondrial membrane depolarization and decreased MMP observed in HD (Intihar et al. 2019). Intihar et al. showed that silencing Hsf1 in the wild-type striatal cell model STHdhQ7 resulted in decreased MMP compared to scramble conditions (Chafekar and Duennwald 2012; Cui et al. 2006; Gomez-Pastor et al. 2017; Intihar et al. 2019). More importantly, these HSF1-induced defects in STHhdQ7 cells recapitulated the MMP decrease observed in untreated STHhdQ111 HD cells (Chafekar and Duennwald 2012; Cui et al. 2006; Gomez-Pastor et al. 2017; Intihar et al. 2019). Due to the fact that HSF1 seems to regulate MMP and dramatically reduce its levels in HD (Gomez-Pastor et al. 2017), it is reasonable to hypothesize that HSF1 dysfunction in MSNs could be at the forefront of the increased MSN mitochondrial susceptibility to polyQ.
Disrupted intracellular Ca2+ levels in neurons can be provoked by excessive glutamatergic signaling. Glutamate is the primary excitatory neurotransmitter in the CNS and has critical functions in controlling perception, reward circuitry, and cognition. In HD, glutamate release changes, glutamate uptake, and postsynaptic signaling converge into Ca2+ buffering dysregulation and promote mitochondrial energy failure and cell death. This phenomenon is known as excitotoxicity (Dong et al. 2009; Zhou et al. 2013). Different neuronal receptors respond to the extracellular levels of glutamate. However, ionotropic receptors such as N-methyl-D-aspartic acid receptor (NMDAR) and α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPAR) are the major players (Danysz and Parsons 2003). Reports have shown that mHTT disrupts glutamatergic transmission by reducing the levels of glial glutamate transporter 1 (GLT1) and therefore reducing astrocytic glutamate reuptake (Behrens et al. 2002; Estrada-Sanchez et al. 2009; Li et al. 2000; Lievens et al. 2001; Shin et al. 2005). Decreased GLT1 expression in HD leads to increased extracellular glutamate concentration, and excessive Ca2+ influx through the NMDARs, and increased NMDAR-mediated currents in the striatum from HD mice (Fan et al. 2007; Laforet et al. 2001; Milnerwood et al. 2010; Okamoto et al. 2009; Qiu et al. 2013; Song et al. 2003; Sun et al. 2001; Zeron et al. 2002). Interestingly, in silico analyses have revealed several putative HSE in the promoter region of both human and mouse GLT1 (Liu et al. 2011). Liu et al. showed an increase in GLT1 expression in NG108–15 cells (a hybrid mouse neuroblastoma-rat glioma cell line) exposed to heat shock. The authors also showed that administration of the neuroprotective drug riluzole increased the amount of HSF1 in NG108–15 cells by slowing HSF1 turnover and increasing the levels of GLT1 (Liu et al. 2011). Also, cells exposed to glutamate excitotoxic stress survived better in the presence of riluzole, indicating that increased HSF1 and GLT1 levels protect cells from excitotoxicity. Despite all the evidence discussed, it is still unknown whether depletion of HSF1 in the striatum of HD mice is responsible for GLT1 downregulation and glutamate excitotoxicity in HD (Chafekar and Duennwald 2012; Maheshwari et al. 2014).
7.5 Crosstalk Between HSF1 and Mitochondrial-Mediated Apoptotic Pathways
Mitochondria are a reservoir for pro-apoptotic factors and play a fundamental role in regulating cell death (Dumollard et al. 2009; Suzuki et al. 1999). Dysregulation of two major transcription factors, tumor suppressor p53 and PGC-1α, has been connected to mitochondrial-mediated apoptosis in HD. p53 is responsible for regulating pro-apoptotic genes such as BCL2-associated X (BAX) and p53 upregulated modulator of apoptosis (PUMA), and its activation triggers mitochondrial-dependent intrinsic apoptosis. There is an increase in p53 levels in cell and mouse models of HD and the striatum and cortex of patients with HD (Bae et al. 2005; Reynolds et al. 2018). Also, transcriptomic analysis in the striatum of an allelic series of HD knock-in mice demonstrated CAG length-dependent activation of p53 signaling pathways (Langfelder et al. 2016). Bae et al. also showed that p53 interacts with HTT, and there is an enhancement in such interaction in the presence of polyQ. It is still unknown how mHTT increases p53, although the authors hypothesized a dysregulation in p53 turnover. They also showed that genetic and pharmacological inhibition of p53 provided neuroprotection by altering transcription and improving mitochondrial function (Bae et al. 2005). However, the mechanism by which p53 inhibition mediated such beneficial effects in HD is unknown. Additional studies showed that mHTT increased phosphorylation of p53 on Ser46, a key PTMs involved in decoupling p53 from the apoptosis inhibitor iASPP, thereby inducing the expression of apoptotic target genes (Bae et al. 2005; Grison et al. 2011; Yu et al. 2001). Alternative studies suggested p53 induces mitochondrial damage and necrosis by directly binding to DRP-1, a primary mitochondrial fission protein (Guo et al. 2013, 2014).
Reciprocal regulation between p53 and HSF1 has been reported in different contexts such as DNA damage and cancer (Jin et al. 2009; Logan et al. 2009; Oda et al. 2018), suggesting that the abnormal degradation of HSF1 may accelerate the harmful effects of p53 on mitochondrial dysfunction in HD. The primary mechanism responsible for keeping p53 levels under control depends on an E3 ligase MDM2, a gene regulated by p53 that promotes p53 ubiquitylation and degradation (Barak et al. 1993; Haupt et al. 1997; Momand et al. 1992). When MDM2 is inefficient or fails, an alternative degradation mechanism involving the small chaperone αB-crys and the E3 ligase FBX4 is activated (Jin et al. 2009). The αB-crys interacts with p53 and facilitates the recruitment of FBX4 (Watanabe et al. 2009). The accumulation of p53 in αB-crys−/− cells is due to the inability of p53 to interact with FBX4 (Jin et al. 2009). The transcriptional regulation of αB-crys expression is made by HSF1 (Gomez-Pastor et al. 2017), and its expression is downregulated in HD (Gomez-Pastor et al. 2017; Hodges et al. 2006). Consistent with this observation, Hsf1 deficient cells express reduced levels of αB-crys and accumulate p53 (Jin et al. 2009). Interestingly, p53 positively regulates the expression of FBXW7 (Kimura et al. 2003; Mao et al. 2004), an E3 ligase involved in the degradation of HSF1 and whose levels are increased in HD (Gomez-Pastor et al. 2017; Kourtis et al. 2015). Therefore, p53 and HSF1 may operate on a unified pathological pathway that controls mitochondrial function and neuronal integrity in HD (Fig. 7).

Model for p53-HSF1-PGC-1α integrated responses in HD. Crosstalk between the transcription factors p53, HSF1, and PGC-1α in regulating transcription, protein homeostasis, mitochondrial function, and apoptosis. There are alterations in different pathways (CREB/TAF4, CK2α’/FBXW7, and MDM2) in the presence of mHTT, which independently leads to the deregulation of the levels and functions of all three transcription factors. However, HSF1 becomes a key player in the subsequent regulation of the levels of p53 and PGC-1α by directly regulating the transcription of PGC-1α and controlling p53 protein stability in HD. The potential role of p53 in regulating the HSF1 degradation pathway in HD would add a positive feedback loop into the p53-HSF1-PGC-1α axis, which triggers mitochondrial dysfunction and neuronal death. Reprinted and modified from “Mitochondrial Dysfunction in Huntington’s Disease; Interplay Between HSF1, p53 and PGC-1α Transcription Factors” by Intihar et al. 2019, with permission from the original authors
PGC-1α governs an additional mechanism essential for the regulation of mitochondrial function. PGC-1α is a transcription factor that regulates the expression of nuclear-encoded mitochondrial genes and participates in the regulation of mitochondrial biogenesis, ROS detoxification, and oxidative phosphorylation (Johri et al. 2013; Puigserver and Spiegelman 2003; Wu et al. 1999). Downregulation of PGC-1α has been reported in cell and mouse models of HD as well as in postmortem brain, muscle biopsies, and myoblast cultures from HD patients (Chafekar and Duennwald 2012; Cui et al. 2006; Gomez-Pastor et al. 2017; Intihar et al. 2019; Turner and Schapira 2010; Weydt et al. 2006). Along with a decrease in the expression of PGC-1α, there is also a decrease in the expression of several PGC-1α-dependent targets and MSN markers (Lucas et al. 2012; Weydt et al. 2006). The role of PGC-1α in HD is supported by several studies conducted in different mouse models. Ppargc1a (a gene encoding PGC-1α) null mice displayed similar defects to those observed in HD such as mitochondrial dysfunction, myelination deficits, degeneration of striatal neurons, white matter atrophy, and motor alterations (Cui et al. 2006; Leone et al. 2005; Lin et al. 2004; Lucas et al. 2012; Weydt et al. 2006; Xiang et al. 2011). Recent studies where PGC-1α was knocked out in MSNs showed that PGC-1α is necessary for MSN transcriptional homeostasis and function (McMeekin et al. 2018). Although the loss of PGC-1α in MSNs does not replicate an HD-like phenocopy, its target genes are altered in a CAG-length and age-dependent fashion, suggesting a potential role of PGC-1α in the selective vulnerability of MSNs in HD (McMeekin et al. 2018). In contrast, overexpression of Ppargc1a rescued HD neurological phenotypes and neurodegeneration and decreased mHTT aggregation (La Spada 2012; St-Pierre et al. 2006). Similarly, pharmacological activation of PGC-1α ameliorated both neuropathological features and HD phenotype in different mouse models of HD (Chandra et al. 2016; Chiang et al. 2010; Jin et al. 2013a; Johri et al. 2013).
Despite all the studies showing the importance of PGC-1α in HD, the mechanism responsible for PGC-1α downregulation is still unclear. Different studies have suggested that an impairment in the CREB/TAF4 signaling pathway in HD (Cui et al. 2006; Gines et al. 2003; Weydt et al. 2006) controls the expression of PGC-1α by binding to cAMP-response elements (CRE) present in the PGC-1α promoter (Fig. 7). Other studies indicated that increased extrasynaptic NMDAR activity causes PGC-1α depletion (Dickey et al. 2016). More recently, different studies showed that HSF1 directly binds to a non-canonical HSE present in the PGC-1α promoter and regulates its expression (Chafekar and Duennwald 2012; Cui et al. 2006; Gomez-Pastor et al. 2017; Intihar et al. 2019; Ma et al. 2015). Intihar et al. documented a reduction in HSF1 binding to PGC-1α promoter in STHdhQ111 cells compared to STHdhQ7 cells and that overexpression of HSF1 in STHdhQ111 cells restored PGC-1α expression and improved cell viability (Chafekar and Duennwald 2012; Cui et al. 2006; Gomez-Pastor et al. 2017; Intihar et al. 2019). Consistent with this view, increased HSF1 levels in the striatum of zQ175; CK2α’ +/−mice enhanced the expression of PGC-1α and its target genes (Gomez-Pastor et al. 2017) and demonstrated an interconnection between HSF1 and PGC-1α in HD. An additional regulatory mechanism that can potentially explain PGC-1α downregulation in HD involves p53, which has been shown to bind to PGC-1α promoter in neuroblastoma cells and regulates its expression (Aquilano et al. 2013). These studies indicate that HSF1, p53, and PGC-1α conform to an intricate pathway that can operate together in controlling mitochondrial function (Fig. 7). Future efforts should focus on elucidating the molecular mechanisms that directly link the interplay between HSF1, p53, and PGC-1α in HD.
7.6 Excitatory Synapse Regulation in HD; Connection Between HSF1 and Mitochondrial Dysfunction
The striatum receives inputs from both the cortex and thalamus and coordinates these inputs to regulate movement and cognition. Among the different chemical synapses that control striatum function, glutamatergic excitatory synapses play a fundamental role. Several lines of evidence suggested that early clinical manifestations of HD may arise from excitatory synaptic dysfunction, including progressive MSNs axonal degeneration (Albin et al. 1992), alteration of neurite outgrowth and maintenance (Trushina et al. 2004), disruption of synaptic gene expression (Luthi-Carter et al. 2003; Manczak and Reddy 2015; Rozas et al. 2010; Smith et al. 2014), collapse in axonal transport of organelles and neurotrophic factors (Gunawardena et al. 2003; McGuire et al. 2006; Trushina et al. 2004), and an imbalance between excitatory and inhibitory systems (Benn et al. 2007; Twelvetrees et al. 2010). Although the exact function of HTT is not known, McKinstry et al. demonstrated the requirement of HTT for normal excitatory synapse development in both cortical and striatal circuits (McKinstry et al. 2014). Several additional studies have shown the specific deterioration in the cortico-striatal circuitry in HD and proposed that these defects may be responsible for striatum degeneration (Bunner and Rebec 2016; Cepeda et al. 2007; Dogan et al. 2015; Rebec 2018). However, other studies in YAC128 and zQ175 HD mouse models have suggested an early dysfunction in the thalamo-striatal circuit, which occurs before an overt HD phenotype (Gomez-Pastor et al. 2017; Kolodziejczyk and Raymond 2016). Interestingly, Gomez-Pastor et al. showed that manipulation of the HSF1 degradation pathway in the zQ175 mice by removing one allele of CK2α’ resulted in increased HSF1 levels and restoration of the thalamo-striatal synapse connectivity (Gomez-Pastor et al. 2017). Unfortunately, the mechanism by which HSF1 regulates this specific circuitry in HD is still unknown.
Previous studies have demonstrated that HSF1 plays a vital role in the regulation of synapse function and formation. Hsf1 deficient mice showed a significant decrease in the number of dendritic spines and dendrite length in the hippocampus, downregulation of polysialylated-neural cell adhesion molecule (PSA-NCAM) required for synapse formation. They decreased expression of the postsynaptic protein PSD-95 needed to anchor NMDARs and AMPARs to the postsynaptic membrane (Uchida et al. 2011). Also, the authors showed that overexpression of a caHSF1 in Hsf1−/−mice rescued PSD-95 expression and improved synapse function (Uchida et al. 2011). Other studies that support the role of HSF1 in the regulation of synaptic genes were conducted in cells and mouse models of AD treated with the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) (Chen et al. 2014). In this study, the authors showed that 17-AAG treatment enhanced HSF1 activity and increased the expression of synapsin I and synaptophysin (presynaptic proteins regulating vesicle cycling) and PSD-95, leading to the prevention of amyloid-β-induced memory loss (Chen et al. 2014). Also, the identity of several HSE in different synaptic genes, including DLG4 (encoding PSD-95) and DLG1 (encoding SAP97) (Chen et al. 2014; Ting et al. 2011). These studies suggest that HSF1 plays a direct role in the structural and functional integrity of synaptic connections underpinning learning and memory. However, since the role of HSF1 in synaptic gene expression impairment in HD has not been established yet, other hypotheses may be possible (Fig. 8).

Working model for the connection between HSF1, mitochondrial impairment, and synapse dysfunction in HD. mHTT alters excitatory synaptic transmission and mitochondrial function by altering Ca2+ buffering capacity and PGC-1α and p53 levels. mHTT is also responsible for promoting HSF1 degradation, which results in downregulation of Hsp and synaptic proteins. The effects caused by HSF1 depletion then further disrupt glutamate release and uptake in neurons, resulting in excitotoxicity. (Reprinted from “Excitatory synapse impairment and mitochondrial dysfunction in Huntington’s disease: heat shock factor 1 (HSF1) converging mechanisms” by (Zarate and Gomez-Pastor 2020), with permission from Neural Regeneration Research)
Reports have shown that chaperones exert protective functions at the synapse level, and the sole overexpression of Hsp70 in cortical cells provides protection against glutamatergic excitotoxicity and improves glutamatergic synaptic transmission (Mokrushin et al. 2005; Song et al. 2016). Therefore, upregulation of Hsp by HSF1 might impact excitatory synapses in HD by improving protein folding and scaffolding at the synapse zone. An additional mechanism that can explain the restoration of excitatory synapses is an increase in HSF1 levels in the correction of mitochondrial function. Mitochondria are essential in synaptic transmission through ATP production, Ca2+ homeostasis, synthesis of glutamate, synaptic vesicle recycling, and functional maintenance of dendritic spines (Smith et al. 2016; Zeron et al. 2002). As discussed above, HSF1 regulates mitochondrial function and MMP by controlling the expression of PGC-1α in HD cells (Gomez-Pastor et al. 2017; Zarate and Gomez-Pastor 2020). Therefore, HSF1 could modulate excitatory synapses in HD throughout its action on PGC-1α. However, further studies are warranted to ascertain how these mechanisms are selective to thalamo-striatal excitatory synapses, whether HSF1-dependent PGC-1α expression constitutes a causal role in MSNs degeneration in HD, and how HSF1 maintains synapse integrity in the adult brain.
7.7 Role of HSF1 in Other PolyQ-Related Diseases
HD belongs to a family of polyQ-related diseases that include spinal and bulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy (DRPLA), and several spinocerebellar ataxias (SCAs) (Williams and Paulson 2008). As in HD, the cause of all these diseases is a CAG repeat expansion in the mutated gene. Although it occurs in entirely unrelated genes that affect different cell types and brain regions, all these polyQ diseases share several features. The characterization of PolyQ diseases is nuclear localization of the toxic protein (Orr 2012; Orr and Zoghbi 2007), the propensity for the mutant proteins to form insoluble aggregates in neurons (Gusella and MacDonald 2000), instability in the CAG repeat number with a tendency for further elongations (Schols et al. 2004), and an inverse correlation between the CAG repeat length and the age of onset (Zoghbi and Orr 2000). Similar to HD, depletion of HSF1 as well as impairment of Hsp expression has been documented in many polyQ diseases and demonstrated that failure in the HSF1-Hsp axis enhances pathogenesis (Evert et al. 2018; Katsuno et al. 2005; Kondo et al. 2013; Tsai et al. 2005). Different studies have also reported the neuroprotective effects of activating HSF1 in many cellular and rodent models of polyQ diseases (Fujimoto et al. 2005; Rimoldi et al. 2001), indicating that failure in the HSR is a common and underlying mechanism in several polyQ-related diseases.
In SBMA, the CAG expansion occurs in the first exon of the androgen receptor (AR) gene (Katsuno et al. 2012; La Spada et al. 1991) and affects males exclusively (Chen et al. 2018; Grunseich et al. 2014). Nuclear accumulation of the polyQ-AR occurs in both neuronal and non-neuronal cells but preferentially affects motor neurons in the brainstem and spinal cord (Adachi et al. 2005; Katsuno et al. 2002). Kondo et al. showed a significant decrease in HSF1 levels in different brain regions of the SBMA mouse model AR-97Q and autopsy specimens from patients with SBMA. The authors also observed that pathogenic polyQ-AR accumulation was greater in tissues where HSF1 levels usually are lower (brain and pancreas). In contrast, tissues with relatively higher HSF1 levels (liver and testis) did not show polyQ-AR accumulation. Interestingly, Hsf1 depletion expanded the distribution of polyQ-AR accumulation to those tissues that are generally not affected, aggravating the SBMA phenotype. On the contrary, lentiviral-mediated overexpression of HSF1 decreased the accumulation of polyQ-AR and neuronal atrophy in the brain of AR-97Q mice (Kondo et al. 2013). Pharmacological activation of HSF1 has also shown beneficial effects on reducing polyQ-AR aggregation and amelioration of SBMA-like phenotypes (Bott et al. 2016; Hargitai et al. 2003). Treatment with arimoclomol, a molecule known to prolong the activation of HSF1, induced chaperones expression and enhanced motor neuron survival, and delayed disease progression in SBMA mice (Bott et al. 2016); (Hargitai et al. 2003).
DRPLA is a rare autosomal dominant polyQ disease and the most HD-like disease among the polyQ disorders. It is clinically characterized by cerebellar ataxia, chorea, myoclonus, epilepsy, dementia, and seizures (Tsuji 2012). The CAG repeat expansion occurs in ATROPHIN-1 (Atro), a transcriptional corepressor (Zhang et al. 2002). Although there are not many studies evaluating the role of HSF1 in this disease, Nisoli and colleagues demonstrated that overexpression of Hsp40 significantly suppressed polyQ-Atro toxicity in a Drosophila model of DRPLA (Nisoli et al. 2010). Additional studies in HeLa cells transfected with an expression vector containing 81 CAG repeats of DRPLA cDNA and expressing a dominant active form of HSF1 (Ad-HSF1-ΔRDT) showed decreased polyQ aggregates compared to cells expressing wild-type HSF1 (Fujimoto et al. 2005; Rimoldi et al. 2001). The authors also demonstrated that the effect of Ad-HSF1-ΔRDT on polyQ aggregates suppression was more efficient than overexpression of any of the significant Hsp (Hsp27, Hsp40, Hsp70, and Hsp110). These pieces of evidence suggest a beneficial role of activating HSF1 in DRPLA. However, additional in vivo studies will be necessary to determine the implications of HSF1 in this disease fully.
SCAs are a heterogeneous group of autosomal dominant inherited ataxias that are characterized by degeneration of Purkinje cells in the cerebellum and occasional degeneration in the brainstem, spinal cord, and basal ganglia (Schols et al. 2004; Zoghbi 2000). To date, at least 43 subtypes of SCA have been classified depending on their genetic locus (Klockgether et al. 2019; Sun et al. 2016). Among them, six SCA subtypes, SCA 1, 2, 3, 6, 7, and 17, are the most common ones, and the genes affected are ATAXIN-1 (Banfi et al. 1994), ATAXIN-2 (Satterfield and Pallanck 2006), ATAXIN-3 (Kawaguchi et al. 1994), α1A subunit of the voltage-dependent calcium channel Cav2.1 (CACNA1A) (Orr 2012), ATAXIN-7 (David et al. 1997), and TATA-box binding protein (TBP) (Koide et al. 1999), respectively. The signs and symptoms may vary between different SCAs, but they also share some phenotypes like an uncoordinated walk (gait), poor hand-eye coordination, and abnormal speech (dysarthria). Interestingly, Ingenwerth and colleagues reported that mice lacking Hsf1 showed gait and other motor symptoms reminiscent of cerebellar ataxia. Also, they showed that CALBINDIN, a calcium-binding protein, was reduced in the cerebellum of Hsf1−/−mice and suggested a role of HSF1 in the regulation of Purkinje cell calcium homeostasis (Ingenwerth et al. 2016). A study conducted in a cell model of SCA6 demonstrated that the levels of HSF1 and Hsp70 were significantly downregulated (Li et al. 2009). Similarly, Tsai et al. showed decreased expression of Hsp27 and Hsp70 in transformed lymphoblastoid cells from patients with SCA7 (Tsai et al. 2005). Conversely, activation of HSF1 by a small molecule HSF1A inhibits the interaction between HSF1 and the chaperonin complex TRiC (Neef et al. 2014), suppressed polyQ aggregation, and ameliorated neurotoxicity in a fly model of SCA3 (Neef et al. 2010). These studies led to pharmacological and genetic manipulations of HSF1 in different SCAs to explore the potential role of HSF1 as a therapeutic target and showed successful induction of Hsp expression and amelioration of polyQ aggregation (Adachi et al. 2003; Chan et al. 2000; Chang et al. 2013; Chen et al. 2010; Cummings et al. 2001; Fujikake et al. 2008; Ghosh and Feany 2004; Gong and Golic 2006; Helmlinger et al. 2004; Katsuno et al. 2005; Kobayashi et al. 2000; Malik et al. 2013; Wang et al. 2013; Warrick et al. 1999b).
Rimoldi et al. utilized a non-neuronal cell system of SCA1 expressing ATAXIN-1 with polyQ tracts of different lengths and determined the effect of de-repressed HSF1 mutant variants on polyQ aggregation (Rimoldi et al. 2001). They showed that HSF1-S303G and S307G, variants unable to be phosphorylated, significantly induced Hsp70 and Hsp40 expression and abolished nuclear polyQ-ATAXIN-1 accumulation. Other pharmacological approaches have used Hsp90 inhibition to activate HSF1. Ding et al. showed that treatment of a neuronal cell model of SCA1 with BIIB021, a synthetic Hsp90 inhibitor, increased the transactivation capacity of HSF1, leading to decreased aggregation and toxicity induced by mutant ATAXIN-1 (Ding et al. 2016). Administration of 17-AAG, another known Hsp90 inhibitor, suppressed inclusion body formation, and eye degeneration in a Drosophila model of SCA3 in an HSF1-dependent manner (Fujikake et al. 2008). Alternative treatments with herbal extracts with HSF1 activation properties have also reduced toxic aggregates formation in cells expressing mutant ATAXIN-3 (Chang et al. 2013). The effects of Hsp90 inhibitors in cellular and Drosophila SCA models are promising; however further studies are required to determine the effects of these inhibitors in more relevant in vivo models of SCA. This strategy has already been tested in the R6/2 HD mice and showed that pharmacological activation of HSF1 using the Hsp90 inhibitor NVP-Hsp990 failed to maintain long-term benefits in vivo, a problem that may be due to progressive loss of HSF1 protein in HD (Gomez-Pastor et al. 2017). Since previous reports have shown HSF1 depletion in SCAs, further experiments will be necessary to uncover the causes that mediate HSF1-Hsp depletion in the different polyQ diseases and whether this may impact future pharmacological studies.
8 HSF1 as a Therapeutic Target in HD and Other PolyQ Diseases
Over the past decade, there has been a profound advancement in understanding the molecular mechanisms responsible for neuronal death in HD and other polyQ diseases, emphasizing those pathways that lead to protein misfolding and aggregation. This knowledge has resulted in development of several molecules with therapeutic potential (Neef et al. 2011). However, current advances in clinical trials are focused on using antisense oligonucleotides against HTT to lower HTT expression (Imbert et al. 2019; Lemprière 2019; Tabrizi et al. 2019). This section will discuss the strategies focused on the activation of the HSR as a potential future venue to ameliorate protein aggregation and neurodegeneration in HD and other polyQ diseases.
One of the most exploited strategies to induce the HSR and ameliorate protein aggregation is Hsp90 inhibitors. As discussed earlier, Hsp90 suppresses HSF1 multimerization and transactivation (Bharadwaj et al. 1999; Zou et al. 1998), and therefore pharmacological inhibition of Hsp90 leads to HSF1 activation and Hsp induction. Well-studied Hsp90 inhibitors are geldanamycin, 17-AAG, 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), celastrol, radicicol, and Hsp990. Geldanamycin is a benzoquinone ansamycin antitumor antibiotic, and Sittler and colleagues reported its first therapeutic effect in a cell culture model of HD (Sittler et al. 2001). The authors showed that geldanamycin induces the expression of Hsp40 and Hsp70 and suppresses aggregation of mHTT. Its anti-aggregation activity was subsequently tested in hippocampal slice cultures derived from HD mice (Hay et al. 2004). However, recent reports have shown that geldanamycin has low solubility, inadequate blood-brain-barrier permeability, and toxicity (Bose and Cho 2017). Less toxic derivatives of geldanamycin are 17-AAG and 17-DMAG. Treatment of 17-AAG strikingly decreased degeneration of photoreceptor neurons in Drosophila models of both HD and SCA3 (Fujikake et al. 2008). Importantly, 17-AAG benefits were abolished when HSF1 was knocked down (Fujikake et al. 2008). Research has shown that 17-DMAG inhibits the formation of mHTT aggregates with higher efficiency than geldanamycin and 17-AAG (Herbst and Wanker 2007). Celastrol constitutes a new class of Hsp90 inhibitors that, unlike the classical Hsp90 inhibitors (geldanamycin and 17-AAG), does not block ATP binding to Hsp90. Instead, Celastrol inhibits Hsp90 activity by binding to its C-terminus domain and blocking Hsp90 oligomerization (Zhang et al. 2008, 2009). Celastrol contributes to the activation of HSF1 and Hsp induction (Westerheide et al. 2004) and suppresses polyQ-induced aggregation in vitro (Wang et al. 2005; Zhang and Sarge 2007). However, celastrol has shown activity against numerous targets besides HSF1 (Lee et al. 2006; Sreeramulu et al. 2009; Yang et al. 2006; Zhang and Sarge 2007) and therefore, the HSF1-dependency in the effects mediated by celastrol is unclear.
The use of 17-AAG has been explored in mouse models of SBMA and animal models from other proteinopathies such as AD and frontotemporal dementia and demonstrated its benefits in improving synaptic connectivity and behavior (Chen et al. 2014; Ho et al. 2013; Katsuno et al. 2005; Waza et al. 2005). Unfortunately, the effects of 17-AAG and other Hsp90 inhibitors on HSF1 activation in HD progression in the mammalian brain have remained unknown due to their low efficiency in crossing the blood-brain barrier (Ebrahimi-Fakhari et al. 2013; Egorin et al. 2001; Porter et al. 2010). A new Hsp90 inhibitor developed by Novartis and with brain-penetrant properties is Hsp990 (Jackrel and Shorter 2011; Menezes et al. 2012). Oral administration of Hsp990 increased Hsp, reduced aggregate load in the brain, and improved motor symptoms at early stages in R6/2 HD mice (Labbadia et al. 2011). Unfortunately, the use of Hsp990 failed to provide long-term benefits in the R6/2 mice and resulted in the attenuation of the HSR. The authors attributed this defect to an alteration in the chromatin structure on the Hsp promoters that resulted in chronic Hsp downregulation. However, more recent studies suggested that failure to provide long-term benefits upon Hsp90 inhibition in vivo may be due to the pathological degradation of HSF1 reported in HD (Gomez-Pastor et al. 2017). In support of this study, Chafekar and Duennwald reported that STHdhQ111 cells treated with radicicol, another Hsp90 inhibitor, showed very modest or absent effects on the levels of Hsp70 and Hsp27 as well as increased cell sensitivity (Chafekar and Duennwald 2012). They hypothesized that lack of Hsp induction might be due to the low levels of HSF1 observed in those cells compared with wild-type STHdhQ7 cells (Chafekar and Duennwald 2012). Therefore, these studies suggest that activation of HSF1 via Hsp90 inhibition in HD may not be a successful therapeutic strategy after all.
Due to the toxicity issues of many Hsp90 inhibitors reported in HD, other strategies have investigated alternative ways of inducing the HSR (Calamini et al. 2011). Calamini and colleagues developed a high-throughput screening using HeLa cells stably transfected with a heat shock-inducible reporter containing the proximal human HSPA2 (a gene encoding Hsp70.1) promoter sequence upstream of a luciferase reporter gene and measured the activation of the HSR in more than 900,000 molecules. The authors identified a series of small molecules (PR, proteostasis regulators) that induced HSF1-dependent chaperone expression, did not inhibit Hsp90 activity, and restored protein folding in PC12 cells expressing HTTQ74-GFP and in C. elegans expressing YFP-tagged Q35 protein. Besides the activation of HSF1, the study revealed several molecules provided proteome stability by activating other factors, including DAF-16/FOXO and SKN-1/NRF-2, and suggested an activation in HSR via alternative mechanisms. An additional screening conducted by Neef and colleagues used a humanized yeast-based high-throughput screen insensitive to proteotoxic stress and Hsp90 inhibition and identified a small molecule activator of human HSF1, so-called HSF1A (Neef et al. 2010). The study reported that HSF1A repressed aggregation and cytotoxicity of mHTT in a cell culture model of HD and ameliorated polyQ-induced cytotoxicity in a Drosophila model of SCA3. Further studies revealed that HSF1A inhibits the repressive interaction between HSF1 and the chaperonin complex TRiC/CCT, consequently increasing HSF1 activity and Hsp expression (Neef et al. 2014). However, the effects of the PRs or HSF1A in mouse models of HD or other polyQ diseases are still unknown.
Instead of disrupting the interaction between HSF1 and its negative regulators, other strategies have searched for molecules that directly enhance HSF1 activity. Bimoclomol and its derivative arimoclomol are non-toxic hydroxylamine derivatives and are co-inducers of the HSR under stress conditions (Kalmar et al. 2002; Vigh et al. 1997). Reports have suggested that bimoclomol directly binds to HSF1, increasing the time and duration of HSF1 binding to the respective target genes and therefore prolonging the activation of HSF1 (Hargitai et al. 2003). The direct binding was analyzed by measuring binding of radiolabeled [3H]bimoclomol to purified proteins by equilibrium dialysis where [3H]bimoclomol showed binding to both recombinant and native HSF1 but not Hsp70 or Hsp90. However, other studies suggested that bimoclomol enhances HSF1 activity indirectly through altering plasma membrane fluidity (Torok et al. 2003). The mechanism of action for prolonged HSF1 activity by bimoclomol or arimoclomol remains unclear. Despite the controversy in the mechanism of action of these molecules, both bimoclomol and arimoclomol have shown cytoprotective value in mitigating motor neuron degeneration in SBMA (Malik et al. 2013) as well as in an array of different diseases such as ALS (Kalmar et al. 2008; Kieran et al. 2004), diabetes (Biro et al. 1997; Erdo and Erdo 1998), cardiac dysfunction (Jednakovits et al. 2000; Lubbers et al. 2002; Polakowski et al. 2002), and cerebrovascular disorders (Melville et al. 1997). Unfortunately, arimoclomol did not improve motor function or rescue of striatal pathology in YAC128 HD mice (Pouladi et al. 2010). An additional therapeutic strategy pursuing prolonged HSF1 DNA binding is the use of SIRT1 agonists. SIRT1 is a deacetylase responsible for deacetylating HSF1-Lys80 in the DBD domain and promoting DNA binding release (Westerheide et al. 2009). Studies in different animal models provided convincing evidence that SIRT1 and different SIRT1 agonists like resveratrol protect neurons in cell and mouse models of HD and C. elegans, although there were controversial results reported in a fly model (Duan 2013; Parker et al. 2005). The therapeutic potential of resveratrol is currently being tested in clinical trials for HD (ClinicalTrials.gov identifier: NCT02336633). The study has revealed a reduction in neurodegeneration among HD patients treated with resveratrol by showing an improvement in brain energy profiles measured by 31P-magnetic resonance spectroscopy.
It is essential to remember that HSF1 undergoes a progressive and pathological degradation in HD, and therefore strategies aimed at stimulating HSF1 activity may not be effective in the long-term. Thus, other strategies are investigating the stabilization of HSF1 protein levels rather than activity stimulation as a more attractive strategy to ameliorate long-term protein aggregation in HD. Riluzole, an FDA approved anti-glutamatergic agent for ALS treatment, has shown the ability to increase HSF1 protein levels by disrupting the association between HSF1 and the chaperone-mediated autophagy pathway (Yang et al. 2008). Moreover, riluzole increased the expression of the glutamate transporter GLT1 and abolished NMDA-induced excitotoxicity in an HSF1-dependent manner (Liu et al. 2011). Experiments using animal models of HD and other proteinopathies have determined the neuroprotective effects of riluzole (Douhou et al. 2002; Scherfler et al. 2005; Schiefer et al. 2002). More importantly, riluzole alleviated clinical symptoms by preserving brain structure and increasing neurotrophin production in patients with HD (Bonelli and Hofmann 2007; Squitieri et al. 2009). Although this molecule failed in phase III HD clinical trials in 2007 (ClinicalTrials.gov Identifier: NCT00277602), its efficacy is currently being tested to treat other polyQ disorders such as SCA2 (ClinicalTrials.gov Identifier: NCT03347344). The use of protein kinase CK2 inhibitors (TID43, Emodin, and CX4945) has also shown the ability to increase HSF1 protein levels by preventing Ser303/307-phosphorylation-dependent degradation. CK2 inhibitors increased Hsp expression and decreased mHTT load and cell death in different cellular models of HD. Inhibition of CK2 has been recently proposed as a potential target to treat different neurological and psychiatric disorders due to its ability to modulate HSF1 levels and inflammation (Castello et al. 2017; Gomez-Pastor et al. 2017). Further studies will be necessary to address these inhibitors’ potential benefits in animal models of polyQ-related diseases.
Many of the molecules discussed in this section, whether synthetic or natural, have shown some forms of neuroprotection against HD or other polyQ diseases in different cellular models. However, many of them have failed when tested in more relevant and physiological animal models. Many of the studies have also explored the single use of one molecule or another but never a combination of them. Due to the complexity of the HSR, it may be necessary to combine different strategies to obtain a synergistic effect on the activation and stabilization of HSF1 and Hsp expression and, therefore, a more successful strategy towards preventing long-term aggregation and neurodegeneration.
9 Conclusion
Since the discovery of HSF1 in 1984, hundreds of studies have contributed to improving our understanding of this critical transcription factor’s protective roles. While HSF1 was initially ascribed to modulate the Hsp in response to heat shock conditions, we know now that HSF1 controls very diverse physiological processes, including cell growth and differentiation, apoptosis, oxidative stress, aging, immune and inflammatory responses, and neuronal development through the regulation of numerous target genes, mainly in a temperature-independent manner. It is not difficult to predict that defects in HSF1 activation at any stage of an organismal life may lead to devastating consequences. The transcriptional program governed by HSF1 allows cells to survive under adverse conditions induced by proteotoxic stress in diverse diseases. Cumulative evidence presented in this book chapter has demonstrated the critical role of HSF1 in different neurological disorders, specifically in HD. Various groundbreaking studies on HSF1 have helped to elucidate the mechanisms by which neurons fail to fight aggregation and succumb to death, contributing to the design and exploration of potential therapeutic strategies to aid HSF1 in the course of HD and other pathologies. While we have made tremendous advancements in understanding HSF1 role in physiology and disease, many questions remain unanswered. To design effective and successful therapies in the future to ameliorate the negative effects of aging and neurodegeneration, it will be necessary to fully characterize the differential roles of HSF1 in the young vs. adult brain, assess the spatiotemporal effects of altering HSF1 levels in the adult brain, and characterize all the factors that contribute to the HSR impairment in the aging mammalian brain.
Abbreviations
- 17-AAG:
-
17-allylamino-17-demethoxygeldanamycin
- 17-DMAG:
-
17-dimethylaminoethylamino-17-demethoxygeldanamycin
- 8-OHDG:
-
8-hydroxydeoxyguanosine
- AD:
-
Alzheimer’s disease
- AKT:
-
Protein kinase B
- ALFY:
-
Autophagy-linked FYVE
- ALS:
-
Amyotrophic lateral sclerosis
- AMPAR:
-
α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor
- AMPK:
-
5′-AMP-activated protein kinase
- AOO:
-
Age of onset
- AR:
-
Androgen receptor
- ATF3:
-
Activating transcription factor 3
- Atro:
-
ATROPHIN-1
- BAG3:
-
BCL-2–associated athanogene 3
- BAX:
-
BCL2-associated X
- BDNF:
-
Brain-derived neurotrophic factor
- CACNA1A:
-
α1A subunit of the voltage-dependent calcium channel Cav2.1
- caHSF1:
-
Constitutive active form of HSF1
- CCT:
-
Chaperonin containing TCP-1
- CHIP:
-
C-terminus of HSC70-interacting protein
- ChIP-seq:
-
Chromatin immunoprecipitation followed by sequencing
- CK2:
-
Casein kinase holoenzyme
- CK2α’:
-
CK2 α prime
- CRE:
-
cAMP-response elements
- DBD:
-
DNA binding domain
- Dclk1:
-
Doublecortin-like kinase 1
- DDL-1:
-
DAF16-dependent longevity-1
- DRP-1:
-
Dynamin-related protein 1
- DRPLA:
-
Dentatorubral-pallidoluysian atrophy
- EF3:
-
Elongation factor 3
- EMT:
-
Epithelial-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- ETC:
-
Electron transport chain
- FBXW7:
-
F-box and WD repeat domain containing 7
- FILIP-1 L:
-
Filamin A interacting protein 1-like
- FRET:
-
Fluorescence resonance energy transfer
- FUS/TLS:
-
Fused in sarcoma/translocated in liposarcoma
- GCN5:
-
General control non-repressed protein 5
- GLT1:
-
Glutamate transporter 1
- GPX:
-
Glutathione peroxidases
- GSH:
-
Glutathione
- GSK3β:
-
Glycogen synthase kinase 3β
- HD:
-
Huntington’s disease
- HDAC6:
-
Histone deacetylase 6
- HIF-1:
-
Hypoxia-inducible factor 1
- HMOX1:
-
Heme oxygenase 1
- HR-A/B:
-
Heptad repeat-A/B
- HSE:
-
Heat shock element
- HSFs:
-
Heat shock transcription factors
- Hsp:
-
Heat shock protein(s)
- HSR:
-
Heat shock response
- HTRA2/OMI:
-
High-temperature requirement protein A2
- HTT:
-
Huntingtin
- HuR:
-
Hu-antigen R
- IGF-1R:
-
Insulin/insulin-like growth factor 1 receptor
- IL-6:
-
Interleukin-6
- ILS:
-
Insulin/insulin-like signaling
- LZ:
-
Leucine zipper domains
- MEK:
-
Mitogen-activated protein kinase kinase
- MK2:
-
MAPK-activated protein kinase 2
- MMP:
-
Mitochondrial membrane potential
- MnSOD:
-
Manganese-containing SOD
- MPNST:
-
Malignant peripheral nerve sheath tumor
- mPTP:
-
Mitochondrial permeability transition pore
- MSNs:
-
Medium-sized spiny neurons
- N17:
-
17-amino acid-long N-terminus
- NDs:
-
Neurodegenerative diseases
- NEDD4:
-
Neuronal precursor cell-expressed developmentally downregulated 4
- NES:
-
Nuclear export signal
- NF1:
-
Neurofibromatosis type 1
- NF-IL6:
-
Nuclear factor for interleukin-6
- NLS:
-
Nuclear localization sequence
- NMDAR:
-
N-methyl-D-aspartic acid receptor
- NMP:
-
Neuronal-specific 20S membrane proteasome complex
- NOS:
-
Nitric oxide synthase
- NRF2:
-
Nuclear factor E2-related factor 2
- OD:
-
Oligomerization domains
- PD:
-
Parkinson’s disease
- PGC-1α:
-
Peroxisome proliferator-activated receptor-γ coactivator-1α
- PIM2:
-
Proviral integrations of Moloney virus 2
- PLK1:
-
Polo-like kinase 1
- polyP:
-
Polyproline
- polyQ:
-
Polyglutamine
- PP2A:
-
Protein phosphatase 2A
- PR:
-
Proteostasis regulators
- PRDX:
-
Peroxiredoxins
- PSA-NCAM:
-
Polysialylated-neural cell adhesion molecule
- PTMs:
-
Post-translational modifications
- PUMA:
-
p53 upregulated modulator of apoptosis
- RD:
-
Regulatory domain
- RNS:
-
ROS/reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SAE:
-
SUMO-activating enzyme
- SBMA:
-
Spinal and bulbar muscular atrophy
- SCAs:
-
Spinocerebellar ataxias
- SIRT1:
-
Sirtuin 1
- SOD:
-
Superoxide dismutase
- SQSTM1:
-
Sequestosome 1
- TAD:
-
Transactivation domain
- TARDBP:
-
TAR DNA binding protein
- TauT:
-
Taurine transporter
- TBP:
-
TATA-box binding protein
- TNF-α:
-
Tumor necrosis factor-α
- TOR1:
-
Target of rapamycin 1
- TPR:
-
Translocated promoter region
- TRiC:
-
T-complex protein-1 ring complex
- UPS:
-
Ubiquitin proteasome system
- USP19:
-
Ubiquitin-specific protease 19
- VCP:
-
Valosin-containing protein
- αB-crys:
-
AlphaB-crystallin
References
Acebron SP, Martin I, del Castillo U, Moro F, Muga A (2009) DnaK-mediated association of ClpB to protein aggregates. A bichaperone network at the aggregate surface. FEBS Lett 583:2991–2996
Acevedo-Torres K, Berrios L, Rosario N et al (2009) Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington’s disease. DNA Repair (Amst) 8:126–136
Adachi H, Katsuno M, Minamiyama M et al (2003) Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci 23:2203–2211
Adachi H, Katsuno M, Minamiyama M et al (2005) Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 128:659–670
Ahn SG, Thiele DJ (2003) Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev 17:516–528
Aiken CT, Steffan JS, Guerrero CM et al (2009) Phosphorylation of threonine 3: implications for huntingtin aggregation and neurotoxicity. J Biol Chem 284:29427–29436
Akerfelt M, Trouillet D, Mezger V, Sistonen L (2007) Heat shock factors at a crossroad between stress and development. Ann N Y Acad Sci 1113:15–27
Akerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11:545–555
Alberts AS, Bouquin N, Johnston LH, Treisman R (1998) Analysis of RhoA-binding proteins reveals an interaction domain conserved in heterotrimeric G protein beta subunits and the yeast response regulator protein Skn7. J Biol Chem 273:8616–8622
Albin RL, Reiner A, Anderson KD et al (1992) Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann Neurol 31:425–430
Almeida DV, da Silva Nornberg BF, Geracitano LA, Barros DM, Monserrat JM, Marins LF (2010) Induction of phase II enzymes and hsp70 genes by copper sulfate through the electrophile-responsive element (EpRE): insights obtained from a transgenic zebrafish model carrying an orthologous EpRE sequence of mammalian origin. Fish Physiol Biochem 36:347–353
Almeida B, Fernandes S, Abreu IA, Macedo-Ribeiro S (2013) Trinucleotide repeats: a structural perspective. Front Neurol 4:76
Al-Ramahi I, Lam YC, Chen HK et al (2006) CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem 281:26714–26724
Ament SA, Pearl JR, Grindeland A et al (2017) High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet 26:913–922
Amin J, Ananthan J, Voellmy R (1988) Key features of heat shock regulatory elements. Mol Cell Biol 8:3761–3769
Anckar J, Sistonen L (2011) Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem 80:1089–1115
Andrade MA, Bork P (1995) HEAT repeats in the Huntington’s disease protein. Nat Genet 11:115–116
Antonietti P, Linder B, Hehlgans S et al (2017) Interference with the HSF1/HSP70/BAG3 pathway primes glioma cells to matrix detachment and BH3 mimetic-induced apoptosis. Mol Cancer Ther 16:156–168
Antonini A, Leenders KL, Spiegel R et al (1996) Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain 119(Pt 6):2085–2095
Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR (2013) p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal 18:386–399
Arbez N, Ratovitski T, Roby E et al (2017) Posttranslational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J Biol Chem 292:19238
Asano Y, Kawase T, Okabe A et al (2016) IER5 generates a novel hypo-phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci Rep 6:19174
Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet 16:2600–2615
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a drosophila model for Parkinson’s disease. Science 295:865–868
Bae BI, Xu H, Igarashi S et al (2005) p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47:29–41
Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K (2012) A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J Biol Chem 287:1406–1414
Banfi S, Servadio A, Chung MY et al (1994) Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat Genet 7:513–520
Barak Y, Juven T, Haffner R, Oren M (1993) mdm2 expression is induced by wild type p53 activity. EMBO J 12:461–468
Barna J, Princz A, Kosztelnik M, Hargitai B, Takacs-Vellai K, Vellai T (2012) Heat shock factor-1 intertwines insulin/IGF-1, TGF-beta and cGMP signaling to control development and aging. BMC Dev Biol 12:32
Bason M, Meister-Broekema M, Alberts N et al (2019) Astrocytic expression of the chaperone DNAJB6 results in non-cell autonomous protection in Huntington’s disease. Neurobiol Dis 124:108–117
Bates G (2003) Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 361:1642–1644
Bates GP, Dorsey R, Gusella JF et al (2015) Huntington disease. Nat Rev Dis Primers 1:15005
Batulan Z, Shinder GA, Minotti S et al (2003) High threshold for induction of the stress response in motor neurons is associated with failure to activate HSF1. J Neurosci 23:5789–5798
Beal MF, Matson WR, Storey E et al (1992) Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J Neurol Sci 108:80–87
Beal MF, Brouillet E, Jenkins BG et al (1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13:4181–4192
Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain 125:1908–1922
Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292:1552–1555
Benn CL, Slow EJ, Farrell LA et al (2007) Glutamate receptor abnormalities in the YAC128 transgenic mouse model of Huntington’s disease. Neuroscience 147:354–372
Benn CL, Sun T, Sadri-Vakili G et al (2008) Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci 28:10720–10733
Bennett EJ, Bence NF, Jayakumar R, Kopito RR (2005) Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell 17:351–365
Bennett EJ, Shaler TA, Woodman B et al (2007) Global changes to the ubiquitin system in Huntington’s disease. Nature 448:704–708
Bett JS, Goellner GM, Woodman B, Pratt G, Rechsteiner M, Bates GP (2006) Proteasome impairment does not contribute to pathogenesis in R6/2 Huntington’s disease mice: exclusion of proteasome activator REGgamma as a therapeutic target. Hum Mol Genet 15:33–44
Bharadwaj S, Ali A, Ovsenek N (1999) Multiple components of the HSP90 chaperone complex function in regulation of heat shock factor 1 In vivo. Mol Cell Biol 19:8033–8041
Bhattacharyya A, Thakur AK, Chellgren VM et al (2006) Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol 355:524–535
Bierkamp C, Luxey M, Metchat A, Audouard C, Dumollard R, Christians E (2010) Lack of maternal Heat Shock Factor 1 results in multiple cellular and developmental defects, including mitochondrial damage and altered redox homeostasis, and leads to reduced survival of mammalian oocytes and embryos. Dev Biol 339:338–353
Biro K, Jednakovits A, Kukorelli T, Hegedus E, Koranyi L (1997) Bimoclomol (BRLP-42) ameliorates peripheral neuropathy in streptozotocin-induced diabetic rats. Brain Res Bull 44:259–263
Bjork JK, Sistonen L (2010) Regulation of the members of the mammalian heat shock factor family. FEBS J 277:4126–4139
Björk JK, Sandqvist A, Elsing AN, Kotaja N, Sistonen L (2010) miR-18, a member of Oncomir-1, targets heat shock transcription factor 2 in spermatogenesis. Development 137:3177–3184
Bonelli RM, Hofmann P (2007) A systematic review of the treatment studies in Huntington’s disease since 1990. Expert Opin Pharmacother 8:141–153
Borlongan CV, Koutouzis TK, Freeman TB, Cahill DW, Sanberg PR (1995) Behavioral pathology induced by repeated systemic injections of 3-nitropropionic acid mimics the motoric symptoms of Huntington’s disease. Brain Res 697:254–257
Bose S, Cho J (2017) Targeting chaperones, heat shock factor-1, and unfolded protein response: promising therapeutic approaches for neurodegenerative disorders. Ageing Res Rev 35:155–175
Bott LC, Badders NM, Chen KL et al (2016) A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum Mol Genet 25:1979–1989
Bowman AB, Yoo SY, Dantuma NP, Zoghbi HY (2005) Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin-proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum Mol Genet 14:679–691
Boyault C, Zhang Y, Fritah S et al (2007) HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 21:2172–2181
Brennan WA Jr, Bird ED, Aprille JR (1985) Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem 44:1948–1950
Brouillet E, Jenkins BG, Hyman BT et al (1993) Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem 60:356–359
Brouillet E, Hantraye P, Ferrante RJ et al (1995) Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci U S A 92:7105–7109
Brouillet E, Conde F, Beal MF, Hantraye P (1999) Replicating Huntington’s disease phenotype in experimental animals. Prog Neurobiol 59:427–468
Brouillet E, Jacquard C, Bizat N, Blum D (2005) 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem 95:1521–1540
Browne SE, Beal MF (2006) Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal 8:2061–2073
Browne SE, Bowling AC, MacGarvey U et al (1997) Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 41:646–653
Browne SE, Ferrante RJ, Beal MF (1999) Oxidative stress in Huntington’s disease. Brain Pathol 9:147–163
Brunet Simioni M, De Thonel A, Hammann A et al (2009) Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene 28:3332–3344
Budworth H, Harris FR, Williams P et al (2015) Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet 11:e1005267
Bunner KD, Rebec GV (2016) Corticostriatal dysfunction in Huntington’s disease: the basics. Front Hum Neurosci 10:317
Burchfiel ET, Vihervaara A, Guertin MJ, Gomez-Pastor R, Thiele DJ (2020) Comparative interactomes of HSF1 in stress and disease reveal a role for CTCF in HSF1-mediated gene regulation. J Biol Chem 296:100097
Bush KT, Goldberg AL, Nigam SK (1997) Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem 272:9086–9092
Butterworth J, Yates CM, Reynolds GP (1985) Distribution of phosphate-activated glutaminase, succinic dehydrogenase, pyruvate dehydrogenase and gamma-glutamyl transpeptidase in post-mortem brain from Huntington’s disease and agonal cases. J Neurol Sci 67:161–171
Butterworth NJ, Williams L, Bullock JY, Love DR, Faull RL, Dragunow M (1998) Trinucleotide (CAG) repeat length is positively correlated with the degree of DNA fragmentation in Huntington’s disease striatum. Neuroscience 87:49–53
Cai H, Xue Y, Wang P, Wang Z, Li Z, Hu Y, Shang X, Liu Y (2015) The long noncoding RNA TUG1 regulates blood-tumor barrier permeability by targeting miR-144. Oncotarget 6:19759–19779
Calamini B, Silva MC, Madoux F et al (2011) Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol 8:185–196
Calderwood SK, Murshid A, Prince T (2009) The shock of aging: molecular chaperones and the heat shock response in longevity and aging–a mini-review. Gerontology 55:550–558
Canto C (2017) The heat shock factor HSF1 juggles protein quality control and metabolic regulation. J Cell Biol 216:551–553
Carmichael J, Chatellier J, Woolfson A, Milstein C, Fersht AR, Rubinsztein DC (2000) Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington’s disease. Proc Natl Acad Sci U S A 97:9701–9705
Carmo C, Naia L, Lopes C, Rego AC (2018a) Mitochondrial dysfunction in Huntington’s disease. Adv Exp Med Biol 1049:59–83
Carmo C, Naia L, Lopes C, Rego AC (2018b) Mitochondrial dysfunction in Huntington’s disease. In: Polyglutamine disorders. Springer, pp 59–83
Carpenter RL, Gokmen-Polar Y (2019) HSF1 as a cancer biomarker and therapeutic target. Curr Cancer Drug Targets 19:515–524
Carpenter RL, Paw I, Dewhirst MW, Lo HW (2015) Akt phosphorylates and activates HSF-1 independent of heat shock, leading to Slug overexpression and epithelial-mesenchymal transition (EMT) of HER2-overexpressing breast cancer cells. Oncogene 34:546–557
Castello J, Ragnauth A, Friedman E, Rebholz H (2017) CK2-An emerging target for neurological and psychiatric disorders. Pharmaceuticals (Basel, Switzerland) 10:7
Cattaneo E (2003) Dysfunction of wild-type huntingtin in Huntington disease. News Physiol Sci 18:34–37
Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur EL (2007) Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A 104:10045–10050
Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS (2007) The corticostriatal pathway in Huntington’s disease. Prog Neurobiol 81:253–271
Chafekar SM, Duennwald ML (2012) Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS One 7:e37929–e37e29
Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM (2000) Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 9:2811–2820
Chandra A, Sharma A, Calingasan NY et al (2016) Enhanced mitochondrial biogenesis ameliorates disease phenotype in a full-length mouse model of Huntington’s disease. Hum Mol Genet 25:2269–2282
Chang KH, Chen WL, Lee LC et al (2013) Aqueous extract of Paeonia lactiflora and Paeoniflorin as aggregation reducers targeting chaperones in cell models of spinocerebellar ataxia 3. Evid Based Complement Alternat Med 2013:471659
Chen Y, Barlev NA, Westergaard O, Jakobsen BK (1993) Identification of the C-terminal activator domain in yeast heat shock factor: independent control of transient and sustained transcriptional activity. EMBO J 12:5007–5018
Chen S, Berthelier V, Yang W, Wetzel R (2001) Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 311:173–182
Chen CM, Wu YR, Cheng ML et al (2007) Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem Biophys Res Commun 359:335–340
Chen CM, Lee LC, Soong BW et al (2010) SCA17 repeat expansion: mildly expanded CAG/CAA repeat alleles in neurological disorders and the functional implications. Clin Chim Acta 411:375–380
Chen Y, Wang B, Liu D et al (2014) Hsp90 chaperone inhibitor 17-AAG attenuates Abeta-induced synaptic toxicity and memory impairment. J Neurosci 34:2464–2470
Chen HJ, Mitchell JC, Novoselov S et al (2016) The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain 139:1417–1432
Chen Z, Sequeiros J, Tang B, Jiang H (2018) Genetic modifiers of age-at-onset in polyglutamine diseases. Ageing Res Rev 48:99–108
Cherubini M, Gines S (2017) Mitochondrial fragmentation in neuronal degeneration: toward an understanding of HD striatal susceptibility. Biochem Biophys Res Commun 483:1063–1068
Chiang MC, Chen CM, Lee MR et al (2010) Modulation of energy deficiency in Huntington’s disease via activation of the peroxisome proliferator-activated receptor gamma. Hum Mol Genet 19:4043–4058
Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL (2012) HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148:322–334
Choi D, Oh HJ, Goh CJ, Lee K, Hahn Y (2015) Heat shock RNA 1, known as a eukaryotic temperature-sensing noncoding RNA, is of bacterial origin. J Microbiol Biotechnol 25:1234–1240
Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet 13:1407–1420
Ciammola A, Sassone J, Alberti L et al (2006) Increased apoptosis, huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington’s disease subjects. Cell Death Differ 13:2068–2078
Ciocca DR, Arrigo AP, Calderwood SK (2013) Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol 87:19–48
Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A (2006) Opposing activities protect against age-onset proteotoxicity. Science 313:1604–1610
Cornett J, Cao F, Wang CE et al (2005) Polyglutamine expansion of huntingtin impairs its nuclear export. Nat Genet 37:198–204
Costa V, Scorrano L (2012) Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J 31:1853–1864
Cotto J, Fox S, Morimoto R (1997) HSF1 granules: a novel stress-induced nuclear compartment of human cells. J Cell Sci 110(Pt 23):2925–2934
Cowan KJ, Diamond MI, Welch WJ (2003) Polyglutamine protein aggregation and toxicity are linked to the cellular stress response. Hum Mol Genet 12:1377–1391
Croce KR, Yamamoto A (2019) A role for autophagy in Huntington’s disease. Neurobiol Dis 122:16–22
Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D (2006) Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127:59–69
Cui J, Tian H, Chen G (2015) Upregulation of nuclear heat shock factor 1 contributes to tumor angiogenesis and poor survival in patients with non-small cell lung cancer. Ann Thorac Surg 100:465–472
Cummings CJ, Sun Y, Opal P et al (2001) Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet 10:1511–1518
Cunha C, Brambilla R, Thomas KL (2010) A simple role for BDNF in learning and memory? Front Mol Neurosci 3:1
Dai Q, Zhang C, Wu Y et al (2003) CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J 22:5446–5458
Dai C, Whitesell L, Rogers AB, Lindquist S (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130:1005–1018
Dai C, Santagata S, Tang Z et al (2012) Loss of tumor suppressor NF1 activates HSF1 to promote carcinogenesis. J Clin Invest 122:3742–3754
Dai S, Tang Z, Cao J et al (2015) Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J 34:275–293
Damiano M, Galvan L, Déglon N, Brouillet E (2010) Mitochondria in Huntington’s disease. Biochim Biophys Acta (BBA)-Mol Basis Dis 1802:52–61
Danysz W, Parsons CG (2003) The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: preclinical evidence. Int J Geriatr Psychiatry 18:S23–S32
Das S, Bhattacharyya NP (2015) Heat shock factor 1-regulated miRNAs can target huntingtin and suppress aggregates of mutant huntingtin. Microrna 4:185–193
David G, Abbas N, Stevanin G et al (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17:65–70
Davies SW, Turmaine M, Cozens BA et al (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90:537–548
De Maio A, Vazquez D (2013) Extracellular heat shock proteins: a new location, a new function. Shock 40:239–246
Deckel AW, Tang V, Nuttal D, Gary K, Elder R (2002) Altered neuronal nitric oxide synthase expression contributes to disease progression in Huntington’s disease transgenic mice. Brain Res 939:76–86
Deng YP, Albin RL, Penney JB, Young AB, Anderson KD, Reiner A (2004) Differential loss of striatal projection systems in Huntington’s disease: a quantitative immunohistochemical study. J Chem Neuroanat 27:143–164
Desmond CR, Atwal RS, Xia J, Truant R (2012) Identification of a karyopherin beta1/beta2 proline-tyrosine nuclear localization signal in huntingtin protein. J Biol Chem 287:39626–39633
Diaz-Hernandez M, Hernandez F, Martin-Aparicio E et al (2003) Neuronal induction of the immunoproteasome in Huntington’s disease. J Neurosci 23:11653–11661
Dickey AS, Pineda VV, Tsunemi T et al (2016) PPAR-δ is repressed in Huntington’s disease, is required for normal neuronal function and can be targeted therapeutically. Nat Med 22:37–45
DiFiglia M, Sapp E, Chase KO et al (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993
DiFiglia M, Sena-Esteves M, Chase K et al (2007) Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A 104:17204–17209
Ding Y, Adachi H, Katsuno M et al (2016) BIIB021, a synthetic Hsp90 inhibitor, induces mutant ataxin-1 degradation through the activation of heat shock factor 1. Neuroscience 327:20–31
Dinkova-Kostova AT, Kostov RV, Kazantsev AG (2018) The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J 285:3576–3590
Dogan I, Eickhoff CR, Fox PT et al (2015) Functional connectivity modeling of consistent cortico-striatal degeneration in Huntington’s disease. Neuroimage Clin 7:640–652
Dong XX, Wang Y, Qin ZH (2009) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30:379–387
Dong B, Jaeger AM, Thiele DJ (2019) Inhibiting heat shock factor 1 in cancer: a unique therapeutic opportunity. Trends Pharmacol Sci 40:986–1005
Douhou A, Debeir T, Murer MG et al (2002) Effect of chronic treatment with riluzole on the nigrostriatal dopaminergic system in weaver mutant mice. Exp Neurol 176:247–253
Dragatsis I, Levine MS, Zeitlin S (2000) Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet 26:300–306
Duan W (2013) Targeting sirtuin-1 in Huntington’s disease: rationale and current status. CNS Drugs 27:345–352
Dubinsky JM (2017) Towards an understanding of energy impairment in Huntington’s disease brain. J Huntingtons Dis 6:267–302
Duennwald ML, Lindquist S (2008) Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev 22:3308–3319
Dumollard R, Carroll J, Duchen MR, Campbell K, Swann K (2009) Mitochondrial function and redox state in mammalian embryos. Semin Cell Dev Biol 20:346–353
Dziunycz PJ, Lefort K, Wu X et al (2014) The oncogene ATF3 is potentiated by cyclosporine A and ultraviolet light A. J Invest Dermatol 134:1998–2004
Ebrahimi-Fakhari D, Saidi L-J, Wahlster L (2013) Molecular chaperones and protein folding as therapeutic targets in Parkinson’s disease and other synucleinopathies. Acta Neuropathol Commun 1:79
Egorin MJ, Zuhowski EG, Rosen DM, Sentz DL, Covey JM, Eiseman JL (2001) Plasma pharmacokinetics and tissue distribution of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) in CD2F1 mice1. Cancer Chemother Pharmacol 47:291–302
Ekimova IV, Plaksina DV, Pastukhov YF et al (2018) New HSF1 inducer as a therapeutic agent in a rodent model of Parkinson’s disease. Exp Neurol 306:199–208
El Fatimy R, Miozzo F, Le Mouël A et al (2014) Heat shock factor 2 is a stress-responsive mediator of neuronal migration defects in models of fetal alcohol syndrome. EMBO Mol Med 6:1043–1061
Ellis RJ (2007) Protein misassembly: macromolecular crowding and molecular chaperones. Adv Exp Med Biol 594:1–13
Erdo F, Erdo SL (1998) Bimoclomol protects against vascular consequences of experimental subarachnoid hemorrhage in rats. Brain Res Bull 45:163–166
Estrada-Sanchez AM, Montiel T, Segovia J, Massieu L (2009) Glutamate toxicity in the striatum of the R6/2 Huntington’s disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiol Dis 34:78–86
Evans SJ, Douglas I, Rawlins MD, Wexler NS, Tabrizi SJ, Smeeth L (2013) Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry 84:1156–1160
Evert BO, Nalavade R, Jungverdorben J et al (2018) Upregulation of miR-370 and miR-543 is associated with reduced expression of heat shock protein 40 in spinocerebellar ataxia type 3. PLoS One 13:e0201794
Fan MM, Fernandes HB, Zhang LY, Hayden MR, Raymond LA (2007) Altered NMDA receptor trafficking in a yeast artificial chromosome transgenic mouse model of Huntington’s disease. J Neurosci 27:3768–3779
Fang F, Chang R, Yang L (2012) Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer 118:1782–1794
Farkas T, Kutskova YA, Zimarino V (1998) Intramolecular repression of mouse heat shock factor 1. Mol Cell Biol 18:906–918
Fawcett TW, Sylvester SL, Sarge KD, Morimoto RI, Holbrook NJ (1994) Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J Biol Chem 269:32272–32278
Feigin A, Leenders KL, Moeller JR et al (2001) Metabolic network abnormalities in early Huntington’s disease: an [(18)F]FDG PET study. J Nucl Med 42:1591–1595
Ferrante RJ, Kowall NW, Richardson EP Jr (1991) Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci 11:3877–3887
Filimonenko M, Isakson P, Finley KD et al (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 38:265–279
Fink AL (1999) Chaperone-mediated protein folding. Physiol Rev 79:425–449
Fisher ER, Hayden MR (2014) Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord 29:105–114
Flower M, Lomeikaite V, Ciosi M et al (2019) MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain 142:1876–1886
Fok JHL, Hedayat S, Zhang L et al (2018) HSF1 is essential for myeloma cell survival and a promising therapeutic target. Clin Cancer Res 24:2395–2407
Fuchs M, Poirier DJ, Seguin SJ et al (2009) Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem J 425:245–255
Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T (2008) Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem 283:26188–26197
Fujimoto M, Takaki E, Hayashi T et al (2005) Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem 280:34908–34916
Gabai VL, Meng L, Kim G, Mills TA, Benjamin IJ, Sherman MY (2012) Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol Cell Biol 32:929–940
Gao X, Carroni M, Nussbaum-Krammer C et al (2015) Human Hsp70 Disaggregase reverses Parkinson’s-linked alpha-Synuclein amyloid fibrils. Mol Cell 59:781–793
Garcia VM, Nillegoda NB, Bukau B, Morano KA (2017) Substrate binding by the yeast Hsp110 nucleotide exchange factor and molecular chaperone Sse1 is not obligate for its biological activities. Mol Biol Cell 28:2066–2075
Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics 161:1101–1112
Gellerich FN, Gizatullina Z, Nguyen HP et al (2008) Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J Biol Chem 283:30715–30724
Ghosh S, Feany MB (2004) Comparison of pathways controlling toxicity in the eye and brain in drosophila models of human neurodegenerative diseases. Hum Mol Genet 13:2011–2018
Gillis J, Schipper-Krom S, Juenemann K et al (2013) The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J Biol Chem 288:17225–17237
Gines S, Seong IS, Fossale E et al (2003) Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet 12:497–508
Gizatullina ZZ, Lindenberg KS, Harjes P et al (2006) Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann Neurol 59:407–411
Goenka A, Sengupta S, Pandey R, Parihar R, Mohanta GC, Mukerji M, Ganesh S (2016) Human satellite-III non-coding RNAs modulate heat-shock-induced transcriptional repression. J Cell Sci 129:3541–3552
Goetzl EJ, Boxer A, Schwartz JB et al (2015) Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann Clin Transl Neurol 2:769–773
Gomez AV, Galleguillos D, Maass JC, Battaglioli E, Kukuljan M, Andres ME (2008) CoREST represses the heat shock response mediated by HSF1. Mol Cell 31:222–231
Gomez-Pastor R, Burchfiel ET, Neef DW et al (2017) Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat Commun 8:14405
Gomez-Pastor R, Burchfiel ET, Thiele DJ (2018) Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol 9:4–19
Gong WJ, Golic KG (2006) Loss of Hsp70 in Drosophila is pleiotropic, with effects on thermotolerance, recovery from heat shock and neurodegeneration. Genetics 172:275–286
Gonitel R, Moffitt H, Sathasivam K et al (2008) DNA instability in postmitotic neurons. Proc Natl Acad Sci U S A 105:3467–3472
Goula AV, Stys A, Chan JP, Trottier Y, Festenstein R, Merienne K (2012) Transcription elongation and tissue-specific somatic CAG instability. PLoS Genet 8:e1003051
Graham RK, Deng Y, Slow EJ et al (2006) Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 125:1179–1191
Gray M, Shirasaki DI, Cepeda C et al (2008) Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 28:6182–6195
Green M, Schuetz TJ, Sullivan EK, Kingston RE (1995) A heat shock-responsive domain of human HSF1 that regulates transcription activation domain function. Mol Cell Biol 15:3354–3362
Grison A, Mantovani F, Comel A et al (2011) Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc Natl Acad Sci U S A 108:17979–17984
Group H (1996) Unified Huntington’s disease rating scale: reliability and consistency. Mov Disord 11:136–142
Grunseich C, Rinaldi C, Fischbeck KH (2014) Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis 20:6–9
Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH (1996) Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol 39:385–389
Guedes-Dias P, Pinho BR, Soares TR, de Proença J, Duchen MR, Oliveira JM (2016) Mitochondrial dynamics and quality control in Huntington’s disease. Neurobiol Dis 90:51–57
Guertin MJ, Lis JT (2010) Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet 6:e1001114
Guettouche T, Boellmann F, Lane WS, Voellmy R (2005) Analysis of phosphorylation of human heat shock factor 1 in cells experiencing a stress. BMC Biochem 6:4
Guidetti P, Charles V, Chen EY et al (2001) Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol 169:340–350
Gunawardena S, Her LS, Brusch RG et al (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40:25–40
Guo Y, Guettouche T, Fenna M et al (2001) Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J Biol Chem 276:45791–45799
Guo X, Disatnik MH, Monbureau M, Shamloo M, Mochly-Rosen D, Qi X (2013) Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J Clin Invest 123:5371–5388
Guo X, Sesaki H, Qi X (2014) Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem J 461:137–146
Gusella JF, MacDonald ME (2000) Molecular genetics: unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci 1:109–115
Gutiérrez JI, Brittingham G, Wang X, Fenyö D, Holt LJ (2017) The largest SWI/SNF polyglutamine domain is a pH sensor. bioRxiv:165043
Gutsmann-Conrad A, Heydari AR, You S, Richardson A (1998) The expression of heat shock protein 70 decreases with cellular senescence in vitro and in cells derived from young and old human subjects. Exp Cell Res 241:404–413
Halestrap AP (2006) Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 34:232–237
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hargitai J, Lewis H, Boros I et al (2003) Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem Biophys Res Commun 307:689–695
Hartl FU, Hayer-Hartl M (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858
Hashimoto-Torii K, Torii M, Fujimoto M et al (2014) Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron 82:560–572
Haupt Y, Maya R, Kazaz A, Oren M (1997) Mdm2 promotes the rapid degradation of p53. Nature 387:296–299
Hay DG, Sathasivam K, Tobaben S et al (2004) Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet 13:1389–1405
Hayashida N, Fujimoto M, Tan K et al (2010) Heat shock factor 1 ameliorates proteotoxicity in cooperation with the transcription factor NFAT. EMBO J 29:3459–3469
He WT, Xue W, Gao YG et al (2017) HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci Rep 7:14797
Helmlinger D, Bonnet J, Mandel JL, Trottier Y, Devys D (2004) Hsp70 and Hsp40 chaperones do not modulate retinal phenotype in SCA7 mice. J Biol Chem 279:55969–55977
Hendriks IA, Lyon D, Young C, Jensen LJ, Vertegaal AC, Nielsen ML (2017) Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat Struct Mol Biol 24:325–336
Hentze N, Le Breton L, Wiesner J, Kempf G, Mayer MP (2016) Molecular mechanism of thermosensory function of human heat shock transcription factor Hsf1. elife 5:e11576
Herbst M, Wanker EE (2007) Small molecule inducers of heat-shock response reduce polyQ-mediated huntingtin aggregation. A possible therapeutic strategy. Neurodegener Dis 4:254–260
Hersch SM, Gevorkian S, Marder K et al (2006) Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 66:250–252
Hietakangas V, Ahlskog JK, Jakobsson AM et al (2003) Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol Cell Biol 23:2953–2968
Hipp MS, Patel CN, Bersuker K et al (2012) Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J Cell Biol 196:573–587
Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20:421–435
Ho SW, Tsui YTC, Wong TT et al (2013) Effects of 17-allylamino-17-demethoxygeldanamycin (17-AAG) in transgenic mouse models of frontotemporal lobar degeneration and Alzheimer’s disease. Transl Neurodegener 2:24–24
Hodges A, Strand AD, Aragaki AK et al (2006) Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 15:965–977
Hoetzenecker W, Echtenacher B, Guenova E et al (2011) ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med 18:128–134
Holzenberger M, Dupont J, Ducos B et al (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421:182–187
Homma S, Jin X, Wang G et al (2007) Demyelination, astrogliosis, and accumulation of ubiquitinated proteins, hallmarks of CNS disease in hsf1-deficient mice. J Neurosci 27:7974–7986
Hooper PL, Durham HD, Török Z, Crul T, Vígh L (2016) The central role of heat shock factor 1 in synaptic fidelity and memory consolidation. Cell Stress Chaperones 21:745–753
Horton TM, Graham BH, Corral-Debrinski M et al (1995) Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 45:1879–1883
Hsu A-L, Murphy CT, Kenyon C (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300:1142–1145
Hu Y, Mivechi NF (2011) Promotion of heat shock factor Hsf1 degradation via adaptor protein filamin A-interacting protein 1-like (FILIP-1L). J Biol Chem 286:31397–31408
Huang AJ, Yu KD, Li J, Fan L, Shao ZM (2012) Polymorphism rs4919510:C>G in mature sequence of human microRNA-608 contributes to the risk of HER2-positive breast cancer but not other subtypes. PLoS One 7:e35252
Hunter JM, Lesort M, Johnson GV (2007) Ubiquitin-proteasome system alterations in a striatal cell model of Huntington’s disease. J Neurosci Res 85:1774–1788
Imbert M, Blandel F, Leumann C, Garcia L, Goyenvalle A (2019) Lowering mutant huntingtin using Tricyclo-DNA antisense oligonucleotides as a therapeutic approach for Huntington’s disease. Nucl Acid Ther 29:256–265
Inagaki R, Tagawa K, Qi ML et al (2008) Omi/HtrA2 is relevant to the selective vulnerability of striatal neurons in Huntington’s disease. Eur J Neurosci 28:30–40
Ingenwerth M, Estrada V, Stahr A, Müller HW, von Gall C (2016) HSF1-deficiency affects gait coordination and cerebellar calbindin levels. Behav Brain Res 310:103–108
Inouye S, Fujimoto M, Nakamura T et al (2007) Heat shock transcription factor 1 opens chromatin structure of interleukin-6 promoter to facilitate binding of an activator or a repressor. J Biol Chem 282:33210–33217
Intihar TA, Martinez EA, Gomez-Pastor R (2019) Mitochondrial dysfunction in Huntington’s disease; interplay between HSF1, p53 and PGC-1α transcription factors. Front Cell Neurosci 13:103
Ishiwata J, Kasamatsu A, Sakuma K et al (2012) State of heat shock factor 1 expression as a putative diagnostic marker for oral squamous cell carcinoma. Int J Oncol 40:47–52
Jackrel ME, Shorter J (2011) Shock and awe: unleashing the heat shock response to treat Huntington disease. J Clin Invest 121:2972–2975
Jaeger AM, Makley LN, Gestwicki JE, Thiele DJ (2014) Genomic heat shock element sequences drive cooperative human heat shock factor 1 DNA binding and selectivity. J Biol Chem 289:30459–30469
Jaeger AM, Pemble CW, Sistonen L, Thiele DJ (2016) Structures of HSF2 reveal mechanisms for differential regulation of human heat-shock factors. Nat Struct Mol Biol 23:147–154
Jana NR, Tanaka M, Wang G, Nukina N (2000) Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet 9:2009–2018
Jana NR, Dikshit P, Goswami A et al (2005) Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem 280:11635–11640
Janowska MK, Baughman HER, Woods CN, Klevit RE (2019) Mechanisms of small heat shock proteins. Cold Spring Harb Perspect Biol 11:a034025
Jednakovits A, Kurucz I, Nanasi PP (2000) Effect of subchronic bimoclomol treatment on vascular responsiveness and heat shock protein production in spontaneously hypertensive rats. Life Sci 67:1791–1797
Jellinger KA (2014) The pathomechanisms underlying Parkinson’s disease. Expert Rev Neurother 14:199–215
Jiang YQ, Wang XL, Cao XH, Ye ZY, Li L, Cai WQ (2013) Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res 1519:105–111
Jiang S, Tu K, Fu Q et al (2015) Multifaceted roles of HSF1 in cancer. Tumour Biol 36:4923–4931
Jie Yuan HM, Gong H, Zhou N, Liang Y, Niu Y, Zou Y (2010) MicroRNA-378, a novel regulator of heat shock transcription factor-1, involves development of cardiac hypertrophy. Congress of Cardiology, Beijing
Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC (2017) Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med 7:a024240
Jin X, Moskophidis D, Hu Y, Phillips A, Mivechi NF (2009) Heat shock factor 1 deficiency via its downstream target gene alphaB-crystallin (Hspb5) impairs p53 degradation. J Cell Biochem 107:504–515
Jin J, Albertz J, Guo Z et al (2013a) Neuroprotective effects of PPAR-gamma agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J Neurochem 125:410–419
Jin YN, Yu YV, Gundemir S et al (2013b) Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS One 8:e57932
Jin X, Qiao A, Moskophidis D, Mivechi NF (2018) Modulation of heat shock factor 1 activity through silencing of Ser303/Ser307 phosphorylation supports a metabolic program leading to age-related obesity and insulin resistance. Mol Cell Biol 38:e00095-18
Johri A, Beal MF (2012) Antioxidants in Huntington’s disease. Biochim Biophys Acta 1822:664–674
Johri A, Chandra A, Beal MF (2013) PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic Biol Med 62:37–46
Jolly C, Metz A, Govin J, Vigneron M, Turner BM, Khochbin S, Vourc’h C (2004) Stress-induced transcription of satellite III repeats. J Cell Biol 164:25–33
Ju TC, Chen HM, Chen YC, Chang CP, Chang C, Chern Y (2014) AMPK-alpha1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim Biophys Acta 1842:1668–1680
Jung MK, Kim KY, Lee NY et al (2013) Expression of taurine transporter (TauT) is modulated by heat shock factor 1 (HSF1) in motor neurons of ALS. Mol Neurobiol 47:699–710
Jurivich DA, Qiu L, Welk JF (1997) Attenuated stress responses in young and old human lymphocytes. Mech Ageing Dev 94:233–249
Kakkar V, Mansson C, de Mattos EP et al (2016) The S/T-rich motif in the DNAJB6 chaperone delays Polyglutamine aggregation and the onset of disease in a mouse model. Mol Cell 62:272–283
Kalchman MA, Graham RK, Xia G et al (1996) Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem 271:19385–19394
Kalmar B, Burnstock G, Vrbova G, Urbanics R, Csermely P, Greensmith L (2002) Upregulation of heat shock proteins rescues motoneurones from axotomy-induced cell death in neonatal rats. Exp Neurol 176:87–97
Kalmar B, Novoselov S, Gray A, Cheetham ME, Margulis B, Greensmith L (2008) Late stage treatment with arimoclomol delays disease progression and prevents protein aggregation in the SOD1 mouse model of ALS. J Neurochem 107:339–350
Kampinga HH, Hageman J, Vos MJ et al (2009) Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14:105–111
Katsuno M, Adachi H, Kume A et al (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35:843–854
Katsuno M, Sang C, Adachi H et al (2005) Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 102:16801–16806
Katsuno M, Tanaka F, Adachi H et al (2012) Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog Neurobiol 99:246–256
Kawaguchi Y, Okamoto T, Taniwaki M et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221–228
Kennedy L, Evans E, Chen CM et al (2003) Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet 12:3359–3367
Khalsa DS (2015) Stress, meditation, and Alzheimer’s disease prevention: where the evidence stands. J Alzheimers Dis 48:1–12
Kieburtz K, Penney JB, Corno P et al (2001) Unified Huntington’s disease rating scale: reliability and consistency. Neurology 11:136–142
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (2004) Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 10:402–405
Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I (2009) Secondary structure of huntingtin amino-terminal region. Structure 17:1205–1212
Kim DS, Lee Y, Hahn Y (2010a) Evidence for bacterial origin of heat shock RNA-1. RNA 16:274–279
Kim J, Moody JP, Edgerly CK et al (2010b) Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet 19:3919–3935
Kim G, Meriin AB, Gabai VL et al (2012) The heat shock transcription factor Hsf1 is downregulated in DNA damage-associated senescence, contributing to the maintenance of senescence phenotype. Aging Cell 11:617–627
Kim I-S, Kim H, Kim Y-S, Jin I, Yoon H-S (2013) HSF1-mediated oxidative stress response to menadione in Saccharomyces cerevisiae KNU5377Y3 by using proteomic approach. Adv Biosci Biotechnol 4:44
Kim E, Wang B, Sastry N et al (2016) NEDD4-mediated HSF1 degradation underlies alpha-synucleinopathy. Hum Mol Genet 25:211–222
Kim E, Sakata K, Liao F-F (2017) Bidirectional interplay of HSF1 degradation and UPR activation promotes tau hyperphosphorylation. PLoS Genet 13:e1006849
Kimura T, Gotoh M, Nakamura Y, Arakawa H (2003) hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci 94:431–436
Klockgether T, Mariotti C, Paulson HL (2019) Spinocerebellar ataxia. Nat Rev Dis Primers 5:24
Kobayashi Y, Kume A, Li M et al (2000) Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 275:8772–8778
Koide R, Kobayashi S, Shimohata T et al (1999) A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 8:2047–2053
Kolodziejczyk K, Raymond LA (2016) Differential changes in thalamic and cortical excitatory synapses onto striatal spiny projection neurons in a Huntington disease mouse model. Neurobiol Dis 86:62–74
Komatsu M, Kurokawa H, Waguri S et al (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223
Kondo N, Katsuno M, Adachi H et al (2013) Heat shock factor-1 influences pathological lesion distribution of polyglutamine-induced neurodegeneration. Nat Commun 4:1405
Korfanty J, Stokowy T, Widlak P et al (2014) Crosstalk between HSF1 and HSF2 during the heat shock response in mouse testes. Int J Biochem Cell Biol 57:76–83
Kourtis N, Moubarak RS, Aranda O et al (2015) FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol 17:322–332
Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT (2007) OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447:447–452
Kozuki M, Kurata T, Miyazaki K et al (2011) Atorvastatin and pitavastatin protect cerebellar Purkinje cells in AD model mice and preserve the cytokines MCP-1 and TNF-alpha. Brain Res 1388:32–38
Kregel KC (2002) Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J Appl Physiol (1985) 92:2177–2186
Krieger C, Duchen MR (2002) Mitochondria, Ca2+ and neurodegenerative disease. Eur J Pharmacol 447:177–188
Kumar A, Zhang J, Tallaksen-Greene S et al (2016) Allelic series of Huntington’s disease knock-in mice reveals expression discorrelates. Hum Mol Genet 25:1619–1636
Kuo Y, Ren S, Lao U, Edgar BA, Wang T (2013) Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis 4:e833
La Spada AR (2012) PPARGC1A/PGC-1alpha, TFEB and enhanced proteostasis in Huntington disease: defining regulatory linkages between energy production and protein-organelle quality control. Autophagy 8:1845–1847
La Spada AR, Taylor JP (2010) Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11:247–258
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352:77–79
Labbadia J, Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84:435–464
Labbadia J, Cunliffe H, Weiss A et al (2011) Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 121:3306–3319
Lackie RE, Maciejewski A, Ostapchenko VG et al (2017) The Hsp70/Hsp90 chaperone machinery in neurodegenerative diseases. Front Neurosci 11:254
Laforet GA, Sapp E, Chase K et al (2001) Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington’s disease. J Neurosci 21:9112–9123
Langbehn DR, Hayden MR, Paulsen JS (2010) CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet 153b:397–408
Langfelder P, Cantle JP, Chatzopoulou D et al (2016) Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci 19:623–633
Leach MD, Farrer RA, Tan K et al (2016) Hsf1 and Hsp90 orchestrate temperature-dependent global transcriptional remodelling and chromatin architecture in Candida albicans. Nat Commun 7:11704
Lecomte S, Desmots F, Le Masson F et al (2010) Roles of heat shock factor 1 and 2 in response to proteasome inhibition: consequence on p53 stability. Oncogene 29:4216–4224
Lee G, Tanaka M, Park K et al (2004a) Casein kinase II-mediated phosphorylation regulates alpha-synuclein/synphilin-1 interaction and inclusion body formation. J Biol Chem 279:6834–6839
Lee WC, Yoshihara M, Littleton JT (2004b) Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc Natl Acad Sci U S A 101:3224–3229
Lee JH, Koo TH, Yoon H et al (2006) Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol 72:1311–1321
Lee YJ, Kim EH, Lee JS et al (2008) HSF1 as a mitotic regulator: phosphorylation of HSF1 by Plk1 is essential for mitotic progression. Cancer Res 68:7550–7560
Lee JM, Pinto RM, Gillis T, St Claire JC, Wheeler VC (2011) Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One 6:e23647
Lee JM, Shin MS, Ji ES et al (2014) Treadmill exercise improves motor coordination through ameliorating Purkinje cell loss in amyloid beta23-35-induced Alzheimer’s disease rats. J Exerc Rehabil 10:258–264
Lee J-M, Correia K, Loupe J et al (2019) CAG repeat not Polyglutamine length determines timing of Huntington’s disease onset. Cell 178:887–900.e14
Lemprière S (2019) Antisense oligonucleotide reduces mutant protein in patients with HD. Nat Rev Neurol 15:435–435
Lenaz G, Genova ML (2010) Structure and organization of mitochondrial respiratory complexes: a new understanding of an old subject. Antioxid Redox Signal 12:961–1008
Leone TC, Lehman JJ, Finck BN et al (2005) PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol 3:e101
Li H, Li SH, Johnston H, Shelbourne PF, Li XJ (2000) Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet 25:385–389
Li H, Li SH, Yu ZX, Shelbourne P, Li XJ (2001) Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J Neurosci 21:8473–8481
Li L, Saegusa H, Tanabe T (2009) Deficit of heat shock transcription factor 1-heat shock 70 kDa protein 1A axis determines the cell death vulnerability in a model of spinocerebellar ataxia type 6. Genes Cells 14:1253–1269
Li B, Hu Q, Wang H et al (2010) Omi/HtrA2 is a positive regulator of autophagy that facilitates the degradation of mutant proteins involved in neurodegenerative diseases. Cell Death Differ 17:1773–1784
Li D, Yallowitz A, Ozog L, Marchenko N (2014a) A gain-of-function mutant p53-HSF1 feed forward circuit governs adaptation of cancer cells to proteotoxic stress. Cell Death Dis 5:e1194
Li S, Ma W, Fei T et al (2014b) Upregulation of heat shock factor 1 transcription activity is associated with hepatocellular carcinoma progression. Mol Med Rep 10:2313–2321
Li J, Labbadia L, Morimoto R (2017) Rethinking HSF1 in stress, development, and organismal health. Trends Cell Biol 27(12):895–905
Li J, Song P, Jiang T et al (2018) Heat shock factor 1 epigenetically stimulates Glutaminase-1-dependent mTOR activation to promote colorectal carcinogenesis. Mol Ther 26:1828–1839
Li Z, Wang C, Wang Z et al (2019) Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 575:203–209
Liang W, Liao Y, Zhang J et al (2017) Heat shock factor 1 inhibits the mitochondrial apoptosis pathway by regulating second mitochondria-derived activator of caspase to promote pancreatic tumorigenesis. J Exp Clin Cancer Res 36:64
Liangliang X, Yonghui H, Shunmei E, Shoufang G, Wei Z, Jiangying Z (2010) Dominant-positive HSF1 decreases alpha-synuclein level and alpha-synuclein-induced toxicity. Mol Biol Rep 37:1875–1881
Liao Y, Xue Y, Zhang L, Feng X, Liu W, Zhang G (2015) Higher heat shock factor 1 expression in tumor stroma predicts poor prognosis in esophageal squamous cell carcinoma patients. J Transl Med 13:338
Lievens JC, Woodman B, Mahal A et al (2001) Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis 8:807–821
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Lin J, Wu PH, Tarr PT et al (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 119:121–135
Lindquist S (1986) The heat-shock response. Annu Rev Biochem 55:1151–1191
Liu CS, Cheng WL, Kuo SJ, Li JY, Soong BW, Wei YH (2008) Depletion of mitochondrial DNA in leukocytes of patients with poly-Q diseases. J Neurol Sci 264:18–21
Liu AY, Mathur R, Mei N, Langhammer CG, Babiarz B, Firestein BL (2011) Neuroprotective drug riluzole amplifies the heat shock factor 1 (HSF1)- and glutamate transporter 1 (GLT1)-dependent cytoprotective mechanisms for neuronal survival. J Biol Chem 286:2785–2794
Locke M, Tanguay RM (1996) Diminished heat shock response in the aged myocardium. Cell Stress Chaperones 1:251–260
Logan IR, McNeill HV, Cook S et al (2009) Heat shock factor-1 modulates p53 activity in the transcriptional response to DNA damage. Nucleic Acids Res 37:2962–2973
Lou S, Lepak VC, Eberly LE et al (2016) Oxygen consumption deficit in Huntington disease mouse brain under metabolic stress. Hum Mol Genet 25:2813–2826
Lu M, Kim HE, Li CR et al (2008) Two distinct disulfide bonds formed in human heat shock transcription factor 1 act in opposition to regulate its DNA binding activity. Biochemistry 47:6007–6015
Lu M, Lee YJ, Park SM et al (2009) Aromatic-participant interactions are essential for disulfide-bond-based trimerization in human heat shock transcription factor 1. Biochemistry 48:3795–3797
Lubbers NL, Polakowski JS, Wegner CD et al (2002) Oral bimoclomol elevates heat shock protein 70 and reduces myocardial infarct size in rats. Eur J Pharmacol 435:79–83
Lucas EK, Dougherty SE, McMeekin LJ, Trinh AT, Reid CS, Cowell RM (2012) Developmental alterations in motor coordination and medium spiny neuron markers in mice lacking pgc-1α. PLoS One 7:e42878
Lum R, Tkach JM, Vierling E, Glover JR (2004) Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J Biol Chem 279:29139–29146
Luthi-Carter R, Apostol BL, Dunah AW et al (2003) Complex alteration of NMDA receptors in transgenic Huntington’s disease mouse brain: analysis of mRNA and protein expression, plasma membrane association, interacting proteins, and phosphorylation. Neurobiol Dis 14:624–636
Ma X, Xu L, Alberobello AT et al (2015) Celastrol protects against obesity and metabolic dysfunction through activation of a HSF1-PGC1alpha transcriptional Axis. Cell Metab 22:695–708
MacDonald ME, Ambrose CM, Duyao MP et al (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72:971–983
Mahat DB, Salamanca HH, Duarte FM, Danko CG, Lis JT (2016) Mammalian heat shock response and mechanisms underlying its genome-wide transcriptional regulation. Mol Cell 62:63–78
Maheshwari M, Bhutani S, Das A et al (2014) Dexamethasone induces heat shock response and slows down disease progression in mouse and fly models of Huntington’s disease. Hum Mol Genet 23:2737–2751
Maines MD, Ewing JF (1996) Stress response of the rat testis: in situ hydridization and immunohistochemical analysis of heme oxygenase-1 (HSP32) induction by hyperthermia. Biol Reprod 54:1070–1079
Maiuri T, Woloshansky T, Xia J, Truant R (2013) The huntingtin N17 domain is a multifunctional CRM1 and ran-dependent nuclear and cilial export signal. Hum Mol Genet 22:1383–1394
Majumder P, Raychaudhuri S, Chattopadhyay B, Bhattacharyya NP (2007) Increased caspase-2, calpain activations and decreased mitochondrial complex II activity in cells expressing exogenous huntingtin exon 1 containing CAG repeat in the pathogenic range. Cell Mol Neurobiol 27:1127–1145
Malik B, Nirmalananthan N, Gray AL, La Spada AR, Hanna MG, Greensmith L (2013) Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy. Brain 136:926–943
Manczak M, Reddy PH (2015) Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington’s disease. Hum Mol Genet 24:7308–7325
Mao JH, Perez-Losada J, Wu D et al (2004) Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432:775–779
Martinez-Vicente M, Talloczy Z, Wong E et al (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13:567–576
Masliah E, Iimoto DS, Mallory M, Albright T, Hansen L, Saitoh T (1992) Casein kinase II alteration precedes tau accumulation in tangle formation. Am J Pathol 140:263–268
Massey TH, Jones L (2018) The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech 11:dmm031930
Mathew A, Mathur SK, Morimoto RI (1998) Heat shock response and protein degradation: regulation of HSF2 by the ubiquitin-proteasome pathway. Mol Cell Biol 18:5091–5098
Mavroudis IA, Fotiou DF, Adipepe LF et al (2010) Morphological changes of the human purkinje cells and deposition of neuritic plaques and neurofibrillary tangles on the cerebellar cortex of Alzheimer’s disease. Am J Alzheimers Dis Other Dement 25:585–591
Maynard CJ, Bottcher C, Ortega Z et al (2009) Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc Natl Acad Sci U S A 106:13986–13991
McColgan P, Tabrizi SJ (2018) Huntington’s disease: a clinical review. Eur J Neurol 25:24–34
McConoughey SJ, Basso M, Niatsetskaya ZV et al (2010) Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med 2:349–370
McGuire JR, Rong J, Li SH, Li XJ (2006) Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem 281:3552–3559
McKinstry SU, Karadeniz YB, Worthington AK et al (2014) Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci 34:9455–9472
McMeekin LJ, Li Y, Fox SN et al (2018) Cell-specific deletion of PGC-1α from medium spiny neurons causes transcriptional alterations and age-related motor impairment. J Neurosci 38:3273–3286
McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ (1998) Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem 273:7523–7528
Meijering RA, Wiersma M, van Marion DM et al (2015) RhoA activation sensitizes cells to proteotoxic stimuli by abrogating the HSF1-dependent heat shock response. PLoS One 10:e0133553
Melville MW, Hansen WJ, Freeman BC, Welch WJ, Katze MG (1997) The molecular chaperone hsp40 regulates the activity of P58IPK, the cellular inhibitor of PKR. Proc Natl Acad Sci U S A 94:97–102
Mendillo ML, Santagata S, Koeva M et al (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150:549–562
Menezes DL, Taverna P, Jensen MR et al (2012) The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol Cancer Ther 11:730–739
Meng L, Gabai VL, Sherman MY (2010) Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 29:5204–5213
Metz A, Soret J, Vourc’h C, Tazi J, Jolly C (2004) A key role for stress-induced satellite III transcripts in the relocalization of splicing factors into nuclear stress granules. J Cell Sci 117:4551–4558
Milakovic T, Johnson GV (2005) Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem 280:30773–30782
Milakovic T, Quintanilla RA, Johnson GV (2006) Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences. J Biol Chem 281:34785–34795
Miller VM, Nelson RF, Gouvion CM et al (2005) CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci 25:9152–9161
Milnerwood AJ, Gladding CM, Pouladi MA et al (2010) Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 65:178–190
Mitchell IJ, Griffiths MR (2003) The differential susceptibility of specific neuronal populations: insights from Huntington’s disease. IUBMB Life 55:293–298
Mokrushin AA, Pavlinova LI, Plekhanov AY (2005) Heat shock protein HSP70 increases the resistance of cortical cells to glutamate excitotoxicity. Bull Exp Biol Med 140:1–5
Momand J, Zambetti GP, Olson DC, George D, Levine AJ (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245
Morano KA, Grant CM, Moye-Rowley WS (2012) The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 190:1157–1195
Morimoto RI (1998) Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 12:3788–3796
Morley JF, Morimoto RI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15:657–664
Morley JF, Brignull HR, Weyers JJ, Morimoto RI (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99:10417–10422
Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A 97:7841–7846
Murakoshi H, Wang H, Yasuda R (2011) Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature 472:100–104
Muralidharan S, Ambade A, Fulham MA, Deshpande J, Catalano D, Mandrekar P (2014) Moderate alcohol induces stress proteins HSF1 and hsp70 and inhibits proinflammatory cytokines resulting in endotoxin tolerance. J Immunol 193:1975–1987
Murshid A, Eguchi T, Calderwood SK (2013) Stress proteins in aging and life span. Int J Hyperth 29:442–447
Nagy E, Balogi Z, Gombos I et al (2007) Hyperfluidization-coupled membrane microdomain reorganization is linked to activation of the heat shock response in a murine melanoma cell line. Proc Natl Acad Sci U S A 104:7945–7950
Nakai A (2009) Heat shock transcription factors and sensory placode development. BMB Rep 42:631–635
Nakai A, Tanabe M, Kawazoe Y, Inazawa J, Morimoto RI, Nagata K (1997) HSF4, a new member of the human heat shock factor family which lacks properties of a transcriptional activator. Mol Cell Biol 17:469–481
Narayanan KL, Chopra V, Rosas HD, Malarick K, Hersch S (2016) Rho kinase pathway alterations in the brain and leukocytes in Huntington’s disease. Mol Neurobiol 53:2132–2140
Neef DW, Turski ML, Thiele DJ (2010) Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol 8:e1000291
Neef DW, Jaeger AM, Thiele DJ (2011) Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov 10:930–944
Neef DW, Jaeger AM, Gomez-Pastor R, Willmund F, Frydman J, Thiele DJ (2014) A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep 9:955–966
Neudegger T, Verghese J, Hayer-Hartl M, Hartl FU, Bracher A (2016) Structure of human heat-shock transcription factor 1 in complex with DNA. Nat Struct Mol Biol 23:140–146
Neueder A, Gipson TA, Batterton S et al (2017) HSF1-dependent and -independent regulation of the mammalian in vivo heat shock response and its impairment in Huntington’s disease mouse models. Sci Rep 7:12556
Newton EM, Knauf U, Green M, Kingston RE (1996) The regulatory domain of human heat shock factor 1 is sufficient to sense heat stress. Mol Cell Biol 16:839–846
Nguyen GD, Gokhan S, Molero AE, Mehler MF (2013) Selective roles of normal and mutant huntingtin in neural induction and early neurogenesis. PLoS One 8:e64368
Nisoli I, Chauvin JP, Napoletano F et al (2010) Neurodegeneration by polyglutamine Atrophin is not rescued by induction of autophagy. Cell Death Differ 17:1577–1587
Novak MJ, Tabrizi SJ (2010) Huntington’s disease. BMJ 340:c3109
Oda T, Sekimoto T, Kurashima K, Fujimoto M, Nakai A, Yamashita T (2018) Acute HSF1 depletion induces cellular senescence through the MDM2-p53-p21 pathway in human diploid fibroblasts. J Cell Sci 131:jcs210724
Okamoto S, Pouladi MA, Talantova M et al (2009) Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15:1407–1413
Okuda M, Niwa T, Taguchi H (2015) Single-molecule analyses of the dynamics of heat shock protein 104 (Hsp104) and protein aggregates. J Biol Chem 290:7833–7840
Oliveira JM, Chen S, Almeida S et al (2006) Mitochondrial-dependent Ca2+ handling in Huntington’s disease striatal cells: effect of histone deacetylase inhibitors. J Neurosci 26:11174–11186
Oliveira AO, Osmand A, Outeiro TF, Muchowski PJ, Finkbeiner S (2016) alphaB-Crystallin overexpression in astrocytes modulates the phenotype of the BACHD mouse model of Huntington’s disease. Hum Mol Genet 25:1677–1689
Orosz A, Wisniewski J, Wu C (1996) Regulation of Drosophila heat shock factor trimerization: global sequence requirements and independence of nuclear localization. Mol Cell Biol 16:7018–7030
Orr HT (2012) Cell biology of spinocerebellar ataxia. J Cell Biol 197:167–177
Orr HT, Zoghbi HY (2007) Trinucleotide Repeat Disorders. Annu Rev Neurosci 30:575–621
Orr AL, Li S, Wang CE et al (2008) N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci 28:2783–2792
Ortega Z, Lucas JJ (2014) Ubiquitin-proteasome system involvement in Huntington’s disease. Front Mol Neurosci 7:77
Page TJ, Sikder D, Yang L et al (2006) Genome-wide analysis of human HSF1 signaling reveals a transcriptional program linked to cellular adaptation and survival. Mol BioSyst 2:627–639
Panov AV, Gutekunst CA, Leavitt BR et al (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 5:731–736
Park SH, Kukushkin Y, Gupta R et al (2013) PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154:134–145
Parker JA, Connolly JB, Wellington C, Hayden M, Dausset J, Neri C (2001) Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci U S A 98:13318–13323
Parker JA, Arango M, Abderrahmane S et al (2005) Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet 37:349–350
Parsell DA, Lindquist S (1993) The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet 27:437–496
Paul BD, Snyder SH (2019) Impaired redox signaling in Huntington’s disease: therapeutic implications. Front Mol Neurosci 12:68
Paul C, Manero F, Gonin S, Kretz-Remy C, Virot S, Arrigo AP (2002) Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol 22:816–834
Paul S, Ghosh S, Mandal S, Sau S, Pal M (2018) NRF2 transcriptionally activates the heat shock factor 1 promoter under oxidative stress and affects survival and migration potential of MCF7 cells. J Biol Chem 293:19303–19316
Pelham HR (1982) A regulatory upstream promoter element in the Drosophila hsp 70 heat-shock gene. Cell 30:517–528
Perez DI, Gil C, Martinez A (2011) Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med Res Rev 31:924–954
Perez-Severiano F, Escalante B, Vergara P, Rios C, Segovia J (2002) Age-dependent changes in nitric oxide synthase activity and protein expression in striata of mice transgenic for the Huntington’s disease mutation. Brain Res 951:36–42
Pernet L, Faure V, Gilquin B, Dufour-Guerin S, Khochbin S, Vourc’h C (2014) HDAC6-ubiquitin interaction controls the duration of HSF1 activation after heat shock. Mol Biol Cell 25:4187–4194
Pickrell AM, Fukui H, Wang X, Pinto M, Moraes CT (2011) The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci 31:9895–9904
Pierce A, Podlutskaya N, Halloran JJ et al (2013) Over-expression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer’s-like deficits in mice modeling the disease. J Neurochem 124:880–893
Pincus D, Anandhakumar J, Thiru P, Guertin MJ, Erkine AM, Gross DS (2018) Genetic and epigenetic determinants establish a continuum of Hsf1 occupancy and activity across the yeast genome. Mol Biol Cell 29:3168–3182
Pirkkala L, Alastalo TP, Zuo X, Benjamin IJ, Sistonen L (2000) Disruption of heat shock factor 1 reveals an essential role in the ubiquitin proteolytic pathway. Mol Cell Biol 20:2670–2675
Place RF, Noonan EJ (2014) Non-coding RNAs turn up the heat: an emerging layer of novel regulators in the mammalian heat shock response. Cell Stress Chaperones 19:159–172
Polakowski JS, Wegner CD, Cox BF (2002) Bimoclomol elevates heat shock protein 70 and cytoprotects rat neonatal cardiomyocytes. Eur J Pharmacol 435:73–77
Porter JR, Fritz CC, Depew KM (2010) Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr Opin Chem Biol 14:412–420
Pouladi M, Carroll J, dar Santos R, Bertram L, Hayden M (2010) B12 treatment with arimoclomol does not lead to rescue of motor or striatal deficits in the YAC128 mouse model of Huntington’s disease. J Neurol Neurosurg Psychiatry 81:A14–A14
Powell CD, Paullin TR, Aoisa C, Menzie CJ, Ubaldini A, Westerheide SD (2016) The heat shock transcription factor HSF1 induces ovarian cancer epithelial-mesenchymal transition in a 3D spheroid growth model. PLoS One 11:e0168389
Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM (1995) Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE). Mol Med 1:827–837
Preville X, Salvemini F, Giraud S et al (1999) Mammalian small stress proteins protect against oxidative stress through their ability to increase glucose-6-phosphate dehydrogenase activity and by maintaining optimal cellular detoxifying machinery. Exp Cell Res 247:61–78
Prince TL, Lang BJ, Guerrero-Gimenez ME, Fernandez-Muñoz JM, Ackerman A, Calderwood SK (2020) HSF1: primary factor in molecular chaperone expression and a major contributor to cancer morbidity. Cell 9:1046
Puigdellivol M, Saavedra A, Perez-Navarro E (2016) Cognitive dysfunction in Huntington’s disease: mechanisms and therapeutic strategies beyond BDNF. Brain Pathol 26:752–771
Puigserver P, Spiegelman BM (2003) Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 24:78–90
Qiu J, Tan YW, Hagenston AM et al (2013) Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun 4:2034
Qu Z, Titus A, Xuan Z, D’Mello SR (2018) Neuroprotection by heat shock Factor-1 (HSF1) and Trimerization-deficient mutant identifies novel alterations in gene expression. Sci Rep 8:17255
Quintanilla RA, Johnson GV (2009) Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull 80:242–247
Quinti L, Dayalan Naidu S, Trager U et al (2017) KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc Natl Acad Sci U S A 114:E4676–E4685
Rabindran SK, Haroun RI, Clos J, Wisniewski J, Wu C (1993) Regulation of heat shock factor trimer formation: role of a conserved leucine zipper. Science 259:230–234
Raitt DC, Johnson AL, Erkine AM et al (2000) The Skn7 response regulator of Saccharomyces cerevisiae interacts with Hsf1 in vivo and is required for the induction of heat shock genes by oxidative stress. Mol Biol Cell 11:2335–2347
Ramachandran KV, Margolis SS (2017) A mammalian nervous-system-specific plasma membrane proteasome complex that modulates neuronal function. Nat Struct Mol Biol 24:419–430
Ramachandran KV, Fu JM, Schaffer TB, Na CH, Delannoy M, Margolis SS (2018) Activity-dependent degradation of the nascentome by the neuronal membrane proteasome. Mol Cell 71:169–177.e6
Rampelt H, Kirstein-Miles J, Nillegoda NB et al (2012) Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J 31:4221–4235
Rasola A, Sciacovelli M, Pantic B, Bernardi P (2010) Signal transduction to the permeability transition pore. FEBS Lett 584:1989–1996
Raychaudhuri S, Loew C, Körner R et al (2014) Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell 156:975–985
Rebec GV (2018) Corticostriatal network dysfunction in Huntington’s disease: deficits in neural processing, glutamate transport, and ascorbate release. CNS Neurosci Ther 24:281–291
Rebec GV, Barton SJ, Ennis MD (2002) Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. J Neurosci 22:Rc202
Reynolds RH, Petersen MH, Willert CW et al (2018) Perturbations in the p53/miR-34a/SIRT1 pathway in the R6/2 Huntington’s disease model. Mol Cell Neurosci 88:118–129
Rimoldi M, Servadio A, Zimarino V (2001) Analysis of heat shock transcription factor for suppression of polyglutamine toxicity. Brain Res Bull 56:353–362
Riva L, Koeva M, Yildirim F et al (2012) Poly-glutamine expanded huntingtin dramatically alters the genome wide binding of HSF1. J Huntingtons Dis 1:33–45
Rizzi N, Denegri M, Chiodi I, Corioni M, Valgardsdottir R, Cobianchi F, Riva S, Biamonti G (2004) Transcriptional activation of a constitutive heterochromatic domain of the human genome in response to heat shock. Mol Biol Cell 15:543–551
Rockabrand E, Slepko N, Pantalone A et al (2007) The first 17 amino acids of huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet 16:61–77
Rong J, McGuire JR, Fang ZH et al (2006) Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J Neurosci 26:6019–6030
Rosenberger AF, Morrema TH, Gerritsen WH et al (2016) Increased occurrence of protein kinase CK2 in astrocytes in Alzheimer’s disease pathology. J Neuroinflammation 13:4
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98
Rozas JL, Gomez-Sanchez L, Tomas-Zapico C, Lucas JJ, Fernandez-Chacon R (2010) Presynaptic dysfunction in Huntington’s disease. Biochem Soc Trans 38:488–492
Rubinsztein DC (2003) The molecular pathology of Huntington’s disease (HD). Curr Med Chem Immunol Endocr Metab Agents 3:329–340
Sahl SJ, Weiss LE, Duim WC, Frydman J, Moerner WE (2012) Cellular inclusion bodies of mutant huntingtin exon 1 obscure small fibrillar aggregate species. Sci Rep 2:895
Samarasinghe B, Wales CT, Taylor FR, Jacobs AT (2014) Heat shock factor 1 confers resistance to Hsp90 inhibitors through p62/SQSTM1 expression and promotion of autophagic flux. Biochem Pharmacol 87:445–455
Sánchez I, Mahlke C, Yuan J (2003) Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 421:373–379
Sandqvist A, Björk JK, Akerfelt M et al (2009) Heterotrimerization of heat-shock factors 1 and 2 provides a transcriptional switch in response to distinct stimuli. Mol Biol Cell 20:1340–1347
Santagata S, Hu R, Lin NU et al (2011) High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A 108:18378–18383
Santagata S, Mendillo ML, Tang YC et al (2013) Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341:1238303
Santamaria A, Perez-Severiano F, Rodriguez-Martinez E et al (2001) Comparative analysis of superoxide dismutase activity between acute pharmacological models and a transgenic mouse model of Huntington’s disease. Neurochem Res 26:419–424
Santos SD, Saraiva MJ (2004) Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience 126:657–663
Satterfield TF, Pallanck LJ (2006) Ataxin-2 and its Drosophila homolog, ATX2, physically assemble with polyribosomes. Hum Mol Genet 15:2523–2532
Saudou F, Humbert S (2016) The biology of huntingtin. Neuron 89:910–926
Sawa A, Wiegand GW, Cooper J et al (1999) Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med 5:1194–1198
Schaffar G, Breuer P, Boteva R et al (2004) Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell 15:95–105
Scherfler C, Sather T, Diguet E et al (2005) Riluzole improves motor deficits and attenuates loss of striatal neurons in a sequential double lesion rat model of striatonigral degeneration (Parkinson variant of multiple system atrophy). J Neural Transm (Vienna) 112:1025–1033
Schiefer J, Landwehrmeyer GB, Luesse HG et al (2002) Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington’s disease. Mov Disord 17:748–757
Schipper-Krom S, Juenemann K, Reits EAJ (2012) The ubiquitin-proteasome system in Huntington’s disease: are proteasomes impaired, initiators of disease, or coming to the rescue? Biochem Res Int 2012:12
Schlieker C, Tews I, Bukau B, Mogk A (2004) Solubilization of aggregated proteins by ClpB/DnaK relies on the continuous extraction of unfolded polypeptides. FEBS Lett 578:351–356
Schols L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3:291–304
Schreiber S, Spotorno N, Schreiber F et al (2018) Significance of CSF NfL and tau in ALS. J Neurol 265:2633–2645
Schulz R, Streller F, Scheel AH et al (2014) HER2/ErbB2 activates HSF1 and thereby controls HSP90 clients including MIF in HER2-overexpressing breast cancer. Cell Death Dis 5:e980
Scior A, Buntru A, Arnsburg K et al (2018) Complete suppression of Htt fibrilization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J 37:282–299
Seidel K, Siswanto S, Fredrich M et al (2016) Polyglutamine aggregation in Huntington’s disease and spinocerebellar ataxia type 3: similar mechanisms in aggregate formation. Neuropathol Appl Neurobiol 42:153–166
Sengupta S, Parihar R, Ganesh S (2009) Satellite III non-coding RNAs show distinct and stress-specific patterns of induction. Biochem Biophys Res Commun 382:102–107
Seo H, Sonntag KC, Isacson O (2004) Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann Neurol 56:319–328
Seo H, Kim W, Isacson O (2008) Compensatory changes in the ubiquitin-proteasome system, brain-derived neurotrophic factor and mitochondrial complex II/III in YAC72 and R6/2 transgenic mice partially model Huntington’s disease patients. Hum Mol Genet 17:3144–3153
Shamovsky I, Nudler E (2009) Isolation and characterization of the heat shock RNA 1. Methods Mol Biol 540:265–279
Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E (2006) RNA-mediated response to heat shock in mammalian cells. Nature 440:556–560
Shelbourne PF, Keller-McGandy C, Bi WL et al (2007) Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet 16:1133–1142
Sherman MY, Goldberg AL (2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29:15–32
Shi Y, Mosser DD, Morimoto RI (1998) Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev 12:654–666
Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ (2005) Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 171:1001–1012
Shinkawa T, Tan K, Fujimoto M, Hayashida N, Yamamoto K, Takaki E, Takii R, Prakasam R, Inouye S, Mezger V, Nakai A (2011) Heat shock factor 2 is required for maintaining proteostasis against febrile-range thermal stress and polyglutamine aggregation. Mol Biol Cell 22(19):3571–3583
Shorter J (2011) The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS One 6:e26319
Sieradzan KA, Mann DM (2001) The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol Appl Neurobiol 27:1–21
Singh IS, Hasday JD (2013) Fever, hyperthermia and the heat shock response. Int J Hyperth 29:423–435
Sittler A, Lurz R, Lueder G et al (2001) Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet 10:1307–1315
Smith GA, Rocha EM, McLean JR et al (2014) Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum Mol Genet 23:4510–4527
Smith HL, Bourne JN, Cao G et al (2016) Mitochondrial support of persistent presynaptic vesicle mobilization with age-dependent synaptic growth after LTP. elife 5:e15275
Soncin F, Zhang X, Chu B et al (2003) Transcriptional activity and DNA binding of heat shock factor-1 involve phosphorylation on threonine 142 by CK2. Biochem Biophys Res Commun 303:700–706
Song C, Zhang Y, Parsons CG, Liu YF (2003) Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-D-aspartate receptors. J Biol Chem 278:33364–33369
Song H, Kim W, Kim SH, Kim KT (2016) VRK3-mediated nuclear localization of HSP70 prevents glutamate excitotoxicity-induced apoptosis and Abeta accumulation via enhancement of ERK phosphatase VHR activity. Sci Rep 6:38452
Sorger PK, Pelham HR (1988) Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54:855–864
Sorolla MA, Reverter-Branchat G, Tamarit J, Ferrer I, Ros J, Cabiscol E (2008) Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med 45:667–678
Squitieri F, Cannella M, Sgarbi G et al (2006) Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech Ageing Dev 127:217–220
Squitieri F, Orobello S, Cannella M et al (2009) Riluzole protects Huntington disease patients from brain glucose hypometabolism and grey matter volume loss and increases production of neurotrophins. Eur J Nucl Med Mol Imaging 36:1113–1120
Sreeramulu S, Gande SL, Gobel M, Schwalbe H (2009) Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl 48:5853–5855
Srijit Das NPB (2015) Widening HSF1 horizon: the microRNA connection [Research highlight]. RNA Dis 2:e596
Stack EC, Matson WR, Ferrante RJ (2008) Evidence of oxidant damage in Huntington’s disease: translational strategies using antioxidants. Ann N Y Acad Sci 1147:79–92
Steffan JS, Agrawal N, Pallos J et al (2004) SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304:100–104
St-Pierre J, Drori S, Uldry M et al (2006) Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127:397–408
Suh Y, Atzmon G, Cho MO et al (2008) Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A 105:3438–3442
Sun Y, Savanenin A, Reddy PH, Liu YF (2001) Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem 276:24713–24718
Sun YM, Lu C, Wu ZY (2016) Spinocerebellar ataxia: relationship between phenotype and genotype – a review. Clin Genet 90:305–314
Suzuki T, Yao W-D (2014) Molecular and structural bases for postsynaptic signal processing: interaction between postsynaptic density and postsynaptic membrane rafts. J Neurorestoratol 2:1–14
Suzuki A, Tsutomi Y, Yamamoto N, Shibutani T, Akahane K (1999) Mitochondrial regulation of cell death: mitochondria are essential for procaspase 3-p21 complex formation to resist Fas-mediated cell death. Mol Cell Biol 19:3842–3847
Tabrizi SJ, Cleeter MW, Xuereb J, Taanman JW, Cooper JM, Schapira AH (1999) Biochemical abnormalities and excitotoxicity in Huntington’s disease brain. Ann Neurol 45:25–32
Tabrizi SJ, Workman J, Hart PE et al (2000) Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol 47:80–86
Tabrizi SJ, Leavitt BR, Landwehrmeyer GB et al (2019) Targeting Huntingtin expression in patients with Huntington’s disease. N Engl J Med 380:2307–2316
Takii R, Inouye S, Fujimoto M et al (2010) Heat shock transcription factor 1 inhibits expression of IL-6 through activating transcription factor 3. J Immunol 184:1041–1048
Tanaka M, Machida Y, Niu S et al (2004) Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10:148–154
Tang Z, Dai S, He Y et al (2015) MEK guards proteome stability and inhibits tumor-suppressive amyloidogenesis via HSF1. Cell 160:729–744
Tebbenkamp AT, Borchelt DR (2010) Analysis of chaperone mRNA expression in the adult mouse brain by meta analysis of the Allen Brain Atlas. PLoS One 5:e13675
Tessarz P, Mogk A, Bukau B (2008) Substrate threading through the central pore of the Hsp104 chaperone as a common mechanism for protein disaggregation and prion propagation. Mol Microbiol 68:87–97
Thompson LM, Aiken CT, Kaltenbach LS et al (2009) IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol 187:1083–1099
Thomson SB, Leavitt BR (2018) Transcriptional regulation of the Huntingtin gene. J Huntingtons Dis 7:289–296
Tilman G, Arnoult N, Lenglez S, Van Beneden A, Loriot A, De Smet C, Decottignies A (2012) Cancer-linked satellite 2 DNA hypomethylation does not regulate Sat2 non-coding RNA expression and is initiated by heat shock pathway activation. Epigenetics 7:903–913
Ting YK, Morikawa K, Kurata Y et al (2011) Transcriptional activation of the anchoring protein SAP97 by heat shock factor (HSF)-1 stabilizes K(v) 1.5 channels in HL-1 cells. Br J Pharmacol 162:1832–1842
Toma-Jonik A, Vydra N, Janus P, Widlak W (2019) Interplay between HSF1 and p53 signaling pathways in cancer initiation and progression: non-oncogene and oncogene addiction. Cell Oncol (Dordr) 42:579–589
Tong Y, Li Y, Gu H et al (2018) HSF1, in association with MORC2, downregulates ArgBP2 via the PRC2 family in gastric cancer cells. Biochim Biophys Acta Mol basis Dis 1864:1104–1114
Torok Z, Tsvetkova NM, Balogh G et al (2003) Heat shock protein coinducers with no effect on protein denaturation specifically modulate the membrane lipid phase. Proc Natl Acad Sci U S A 100:3131–3136
Trushina E, Dyer RB, Badger JD 2nd et al (2004) Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 24:8195–8209
Tsai HF, Lin SJ, Li C, Hsieh M (2005) Decreased expression of Hsp27 and Hsp70 in transformed lymphoblastoid cells from patients with spinocerebellar ataxia type 7. Biochem Biophys Res Commun 334:1279–1286
Tsuji S (2012) Dentatorubral-pallidoluysian atrophy. Handb Clin Neurol 103:587–594
Tsukao Y, Yamasaki M, Miyazaki Y et al (2017) Overexpression of heat-shock factor 1 is associated with a poor prognosis in esophageal squamous cell carcinoma. Oncol Lett 13:1819–1825
Tunez I, Sanchez-Lopez F, Aguera E, Fernandez-Bolanos R, Sanchez FM, Tasset-Cuevas I (2011) Important role of oxidative stress biomarkers in Huntington’s disease. J Med Chem 54:5602–5606
Turner C, Schapira AH (2010) Mitochondrial matters of the brain: the role in Huntington’s disease. J Bioenerg Biomembr 42:193–198
Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL et al (2010) Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 65:53–65
Uchida S, Hara K, Kobayashi A et al (2011) Impaired hippocampal spinogenesis and neurogenesis and altered affective behavior in mice lacking heat shock factor 1. Proc Natl Acad Sci U S A 108:1681–1686
Vahid S, Thaper D, Gibson KF, Bishop JL, Zoubeidi A (2016) Molecular chaperone Hsp27 regulates the Hippo tumor suppressor pathway in cancer. Sci Rep 6:31842
Vazquez-Manrique RP, Farina F, Cambon K et al (2016) AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum Mol Genet 25:1043–1058
Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14:95–104
Verma P, Pfister JA, Mallick S, D’Mello SR (2014) HSF1 protects neurons through a novel trimerization- and HSP-independent mechanism. J Neurosci 34:1599–1612
Vigh L, Literati PN, Horvath I et al (1997) Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med 3:1150–1154
Vihervaara A, Sergelius C, Vasara J et al (2013) Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci U S A 110:E3388–E3397
Vihervaara A, Mahat DB, Guertin MJ, Chu T, Danko CG, Lis JT, Sistonen L (2017) Transcriptional response to stress is pre-wired by promoter and enhancer architecture. Nat Commun 8(1):255
Vonsattel JP (2008) Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol 115:55–69
Vydra N, Toma A, Widlak W (2014) Pleiotropic role of HSF1 in neoplastic transformation. Curr Cancer Drug Targets 14:144–155
Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ (2004) Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol 11:1215–1222
Wacker JL, Huang SY, Steele AD et al (2009) Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J Neurosci 29:9104–9114
Wade BE, Wang CE, Yan S et al (2014) Ubiquitin-activating enzyme activity contributes to differential accumulation of mutant huntingtin in brain and peripheral tissues. J Neurosci 34:8411–8422
Waelter S, Boeddrich A, Lurz R et al (2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12:1393–1407
Waldvogel HJ, Kim EH, Tippett LJ, Vonsattel JP, Faull RL (2015) The neuropathology of Huntington’s disease. Curr Top Behav Neurosci 22:33–80
Wang K, Spector A (2001) ATP causes small heat shock proteins to release denatured protein. Eur J Biochem 268:6335–6345
Wang J, Gines S, MacDonald ME, Gusella JF (2005) Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci 6:1
Wang J, Martin E, Gonzales V, Borchelt DR, Lee MK (2008) Differential regulation of small heat shock proteins in transgenic mouse models of neurodegenerative diseases. Neurobiol Aging 29:586–597
Wang AM, Miyata Y, Klinedinst S et al (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9:112–118
Wang B, Liu Y, Huang L et al (2017) A CNS-permeable Hsp90 inhibitor rescues synaptic dysfunction and memory loss in APP-overexpressing Alzheimer’s mouse model via an HSF1-mediated mechanism. Mol Psychiatry 22:990–1001
Wang Y, Xu E, Musich PR, Lin F (2019) Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther 25:816–824
Warrick JM, Chan HY, Gray B, Chai Y, Paulson HL, Bonini NM (1999a) Suppression of polyglutamine-mediated neurodegeneration in drosophila by the molecular chaperone HSP70. Nat Genet 23:425–428
Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM (1999b) Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 23:425–428
Watanabe G, Kato S, Nakata H, Ishida T, Ohuchi N, Ishioka C (2009) alphaB-crystallin: a novel p53-target gene required for p53-dependent apoptosis. Cancer Sci 100:2368–2375
Waza M, Adachi H, Katsuno M et al (2005) 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med 11:1088–1095
Weibezahn J, Schlieker C, Bukau B, Mogk A (2003) Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem 278:32608–32617
Wellington CL, Ellerby LM, Gutekunst CA et al (2002) Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington’s disease. J Neurosci 22:7862–7872
Westerheide SD, Bosman JD, Mbadugha BN et al (2004) Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem 279:56053–56060
Westerheide SD, Anckar J, Stevens SM, Sistonen L, Morimoto RI (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323:1063–1066
Weydt P, Pineda VV, Torrence AE et al (2006) Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab 4:349–362
Williams AJ, Paulson HL (2008) Polyglutamine neurodegeneration: protein misfolding revisited. Trends Neurosci 31:521–528
Winkler J, Tyedmers J, Bukau B, Mogk A (2012) Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol 198:387–404
Wright GEB, Collins JA, Kay C et al (2019) Length of uninterrupted CAG, independent of Polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet 104:1116–1126
Wu C (1995) Heat shock transcription factors: structure and regulation. Annu Rev Cell Dev Biol 11:441–469
Wu Z, Puigserver P, Andersson U et al (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124
Wyttenbach A, Sauvageot O, Carmichael J, Az-Latoud C, Arrigo AP, Rubinsztein DC (2002) Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet 11:1137–1151
Xia J, Lee DH, Taylor J, Vandelft M, Truant R (2003) Huntingtin contains a highly conserved nuclear export signal. Hum Mol Genet 12:1393–1403
Xiang Z, Valenza M, Cui L et al (2011) Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington’s disease. J Neurosci 31:9544–9553
Xiao X, Zuo X, Davis AA et al (1999) HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J 18:5943–5952
Xie Y, Chen C, Stevenson MA, Auron PE, Calderwood SK (2002a) Heat shock factor 1 represses transcription of the IL-1beta gene through physical interaction with the nuclear factor of interleukin 6. J Biol Chem 277:11802–11810
Xie Y, Chen C, Stevenson MA, Hume DA, Auron PE, Calderwood SK (2002b) NF-IL6 and HSF1 have mutually antagonistic effects on transcription in monocytic cells. Biochem Biophys Res Commun 291:1071–1080
Xu L, Chen S, Bergan RC (2006) MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 25:2987–2998
Yamamoto A, Simonsen A (2011) Alfy-dependent elimination of aggregated proteins by macroautophagy: can there be too much of a good thing? Autophagy 7:346–350
Yamamoto A, Mizukami Y, Sakurai H (2005) Identification of a novel class of target genes and a novel type of binding sequence of heat shock transcription factor in Saccharomyces cerevisiae. J Biol Chem 280:11911–11919
Yamanaka T, Miyazaki H, Oyama F et al (2008) Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J 27:827–839
Yan L-J, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ (2002) Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J 21:5164–5172
Yang W, Dunlap JR, Andrews RB, Wetzel R (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet 11:2905–2917
Yang H, Chen D, Cui QC, Yuan X, Dou QP (2006) Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 66:4758–4765
Yang J, Bridges K, Chen KY, Liu AY (2008) Riluzole increases the amount of latent HSF1 for an amplified heat shock response and cytoprotection. PLoS One 3:e2864
Yang T, Ren C, Lu C et al (2019) Phosphorylation of HSF1 by PIM2 induces PD-L1 expression and promotes tumor growth in breast cancer. Cancer Res 79:5233–5244
Yano H, Baranov SV, Baranova OV et al (2014) Inhibition of mitochondrial protein import by mutant huntingtin. Nat Neurosci 17:822–831
Yasuda K, Hirohashi Y, Mariya T et al (2017) Phosphorylation of HSF1 at serine 326 residue is related to the maintenance of gynecologic cancer stem cells through expression of HSP27. Oncotarget 8:31540–31553
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7:673–682
Yu L, Yuan X, Wang D, Barakat B, Williams ED, Hannigan GE (2014) Selective regulation of p38beta protein and signaling by integrin-linked kinase mediates bladder cancer cell migration. Oncogene 33:690–701
Zarate N, Gomez-Pastor R (2020) Excitatory synapse impairment and mitochondrial dysfunction in Huntington’s disease: heat shock factor 1 (HSF1) converging mechanisms. Neural Regen Res 15:69–70
Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A (1995) Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet 11:155–163
Zelin E, Freeman BC (2015) Lysine deacetylases regulate the heat shock response including the age-associated impairment of HSF1. J Mol Biol 427:1644–1654
Zelin E, Zhang Y, Toogun OA, Zhong S, Freeman BC (2012) The p23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol Cell 48:459–470
Zeron MM, Hansson O, Chen N et al (2002) Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 33:849–860
Zhang YQ, Sarge KD (2007) Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med (Berl) 85:1421–1428
Zhang S, Xu L, Lee J, Xu T (2002) Drosophila atrophin homolog functions as a transcriptional corepressor in multiple developmental processes. Cell 108:45–56
Zhang T, Hamza A, Cao X et al (2008) A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther 7:162–170
Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D (2009) Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem 284:35381–35389
Zhang H, Zhang L, Yu F et al (2012) HSF1 is a transcriptional activator of IL-10 gene expression in RAW264.7 macrophages. Inflammation 35:1558–1566
Zhao YH, Zhou M, Liu H et al (2009) Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 28:3689–3701
Zhao M, Shen F, Yin YX, Yang YY, Xiang DJ, Chen Q (2012) Increased expression of heat shock protein 27 correlates with peritoneal metastasis in epithelial ovarian cancer. Reprod Sci 19:748–753
Zheng X, Krakowiak J, Patel N et al (2016) Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. elife 5:e18638
Zhou X, Hollern D, Liao J, Andrechek E, Wang H (2013) NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis 4:e560
Zhou Z, Li Y, Jia Q et al (2017) Heat shock transcription factor 1 promotes the proliferation, migration and invasion of osteosarcoma cells. Cell Prolif 50:e12346
Zietkiewicz S, Krzewska J, Liberek K (2004) Successive and synergistic action of the Hsp70 and Hsp100 chaperones in protein disaggregation. J Biol Chem 279:44376–44383
Zoghbi HY (2000) Spinocerebellar ataxias. Neurobiol Dis 7:523–527
Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23:217–247
Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–480
Zuccato C, Ciammola A, Rigamonti D et al (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 293:493–498
Acknowledgments
We thank Nicole Zarate for her constructive comments during the preparation of this book chapter, Rachel Mansky for proofreading, and all the authors and journals that kindly gave us permit to use figures and materials for this chapter.
Disclosure of Interests
All authors declare they have no conflict of interest.
Ethical Approval for Studies Involving Humans
This article does not contain any studies with human participants performed by any of the authors.
Ethical Approval for Studies Involving Animals
This article does not contain any studies with animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
This work was funded by NIH R01 NS110694 (to R.G.P.) and the University of Minnesota Medical School’s Biomedical Research Awards for Interdisciplinary New Science (BRAINS) program (to R.G.P.).
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kim, H., Gomez-Pastor, R. (2022). HSF1 and Its Role in Huntington’s Disease Pathology. In: Turksen, K. (eds) Cell Biology and Translational Medicine, Volume 19. Advances in Experimental Medicine and Biology(), vol 1410. Springer, Cham. https://doi.org/10.1007/5584_2022_742
Download citation
DOI: https://doi.org/10.1007/5584_2022_742
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-28419-9
Online ISBN: 978-3-031-28420-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)