
Abstract
Molybdenum and tungsten are, respectively, the only second and third transition metal ions with well-defined functions in living organisms and with a single exception are found in association with a novel pyranopterin dithiolene cofactor called molybdopterin. This review focusses on the catalytic mechanisms of the molybdenum and tungsten enzymes, with an emphasis on the molybdenum and tungsten sites. Most, but not all, of the enzymes catalyze oxygen atom transferase redox chemistry, with the metal cycling between M(VI) and M(IV) formal oxidation states during the catalytic cycle. We discuss the range of reactions and what is known of mechanism for both oxo-transferase and non-oxo-transferase molybdenum and tungsten enzymes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
This review will discuss the catalytic mechanisms of the molybdopterin-based molybdenum and tungsten enzymes. Molybdenum and tungsten are unique as the only second and third transition elements whose compounds play catalytic roles in biological systems [1]. We note in passing that cadmium plays roles in specific carbonic anhydrases from marine algae [2] but because Cd2+ is 4d10, like 3d10 Zn2+, it is not formally a transition metal ion and we therefore exclude it. Tungsten is also distinguished by being the heaviest element with a known biological role. Apart from nitrogenase, which is distinct from all other Mo-containing enzymes [3, 4], all Mo and W enzymes contain either one or two pyranopterin dithiolene cofactors, known as molybdopterin, which chelates the metal through the dithiolene group. In order to distinguish the enzymes from nitrogenase, the enzymes have often been referred to as the mononuclear Mo or W enzymes [5]. The structure of the molybdenum cofactor, molybdopterin bound to molybdate, is shown in Fig. 1 in the fully reduced tetrahydro form. The molybdopterin cofactor can be found in ring-closed tricyclic or ring-open bicyclic tautomers (Fig. 2), and for E.coli dissimilatory nitrate reductase, two different crystal structures have been reported, one clearly showing a tricyclic molybdopterin [6] and the other a bicyclic ring-open form [7], and the ring-open form has also been observed for ethylbenzene dehydrogenase [8]. Recent model compound studies have observed both ring-open and ring-closed forms [9]. Simple density functional theory calculations reveal that in the absence of protein, for the dihydro oxidation state, the ring-open form of molybdopterin is expected to be thermodynamically preferred, whereas for the tetrahydro form, the ring-closed form is more stable [10].

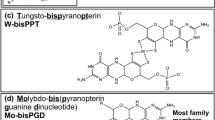
Schematic structure of the molybdenum cofactor, molybdopterin. One of the structures postulated for the cofactor prior to insertion into the target enzyme is shown, with the pterin ring in the tetrahydro redox state. In the protein the phosphate can be free as shown (molybdopterin or MPT) or attached to guanosine (molybdopterin guanine dinucleotide or MGD) or cytosine (molybdopterin cytidine dinucleotide or MCD)

Structure of ring-closed tricycle and ring-open bicycle molybdopterin, shown for the dihydro molybdopterin oxidation state
The field of molybdenum and tungsten enzymes has developed very rapidly; in the early 1970s, a total of six molybdenum enzymes were known [11], and while one tungsten enzyme had been reported [12]; it was not until 1992 that the tungsten enzymes were recognized as closely related to the mononuclear molybdenum enzymes [13]. Since this early work, a very large number of both molybdenum and tungsten enzymes have been discovered and characterized, and much is now known about their evolution, their biochemistry, and their catalytic mechanisms.
With some notable exceptions (Sect. 6), the mononuclear Mo and W enzymes catalyze two-electron redox reactions in which the metal cycles between formally M4+ and M6+ oxidation states with an oxygen atom from water being transferred either from or to substrate via the metal atom, according to the following stoichiometry:
Thus, the vast majority of the enzymes either form or break bonds between oxygen and carbon, with, respectively, the source or destination of the oxygen being water. Many of the enzymes possess multiple redox-active groups that appear to form an electron transfer chain within the protein, linking the oxidative and reductive half-reactions. An example is shown in Fig. 3, that of xanthine oxidase. The Mo site converts the substrate; in this case xanthine is oxidized to uric acid via reduction of the Mo site by two electrons, from Mo(VI) to Mo(IV). Reducing electrons then equilibrate through intramolecular electron transfer to the different acceptor sites within the protein [14] (Figs. 3 and 4). The redox behavior of such multi-redox site enzymes can be complex, for example, each xanthine oxidase monomer can accept up to 6 electrons, 2 at Mo, 1 at each of the 2 FeS clusters, and 2 at the flavin site, giving a total of 36 possible redox states ranging from fully oxidized to six-electron-reduced enzyme [14]. In xanthine oxidase the electrons from the oxidation of xanthine are finally accepted through two one-electron reductions of dioxygen to form the superoxide radical anion, O2·−. In enzymes that have two molybdopterin dithiolenes coordinated to molybdenum or tungsten, in most cases, one of the molybdopterins is positioned approximately between the molybdenum/tungsten site and an adjacent iron-sulfur cluster, and this is referred to as the proximal or sometimes the P molybdopterin, while the other is called the distal or the Q molybdopterin (Fig. 4). As has been previously discussed, the redox state of the pterin probably cannot be distinguished by crystallography, at least at poorer crystallographic resolutions [1], although the ring-open and ring-closed forms appear unambiguous [1, 6,7,8]. The pterin in the oxidized dihydro redox state, which has a more planar structure than the other redox states, has been suggested to direct electron transfer from the Mo site to the next acceptor in the protein, rather like an electrical wire [15].

Structure and large-scale catalytic mechanism of xanthine oxidase. At top is shown the crystal structure (pdb code 1F1Q) with the redox-active components depicted with CPK models. Below is shown both the oxidative half-reaction in which xanthine is converted to uric acid and the reductive half-reaction in which oxygen is converted to superoxide

Distal and proximal molybdopterin in Escherichia coli nitrate reductase. The proximal molybdopterin lies between the Mo and the first iron-sulfur cluster, FS0. Iron atoms are shown in green, sulfur in yellow, oxygen in red, carbon in gray, and nitrogen in blue. The string of five irons sulfur clusters is thought to act as an electron conduit to the Mo site
This review will focus primarily on the mechanisms that are thought to operate at the molybdenum and tungsten sites themselves, such as the reductive half-reaction of xanthine oxidase (Fig. 3). We will discuss some aspects of electron transfer but will neglect the parts of the catalytic processes that do not directly involve the molybdenum or tungsten.
The Mo/W enzymes are a very widespread group, playing important roles in all kingdoms of life [1], and are likely very ancient [1, 16]. The enzymes fulfill diverse roles, ranging from microbial respiration (e.g., the dissimilatory nitrate reductases), through the uptake of nitrogen in green plants (e.g., the assimilatory nitrate reductases), to important roles in human health (e.g., sulfite oxidases) and drug metabolism (e.g., aldehyde oxidase). At the time of writing, only five examples of Mo/W enzymes are known to occur in eukaryotes, and all are molybdenum enzymes: sulfite oxidase, nitrate reductase, xanthine oxidase/dehydrogenase (counted as a single enzyme), aldehyde oxidase, and the recently discovered mitochondrial amidoxime reducing component (mARC). In contrast the prokaryotes present a vast array of both Mo and W enzymes, including homologues of the eukaryotic enzymes mentioned above, and a great many others besides. The enzymes are divided into three broad families according to their oxidized Mo(VI) active site structures and are named for the best-studied members of each family [16]: the sulfite oxidase family, the xanthine oxidase family, and the dimethyl sulfoxide (DMSO) reductase family. Within each family there is considerable diversity, both in terms of the active site structure and in the details of the catalytic mechanisms. Our knowledge of the active sites and mechanisms of the enzymes derives from ground-breaking crystallographic work, plus numerous spectroscopic studies of the metal sites of the proteins [17].
The oxidized Mo(VI) active site structures of the prototypical members of each family are shown in Fig. 5:
-
The sulfite oxidase family includes, in addition to the sulfite oxidases themselves [18, 19], the nitrate reductases of fungi and green plants [20], a number of prokaryotic sulfite dehydrogenases [21, 22], and the mitochondrial amidoxime reducing component (mARC) [23]. Members of the family have a single molybdopterin dithiolene and possess a cysteine thiolate coordination of molybdenum, with a cis-dioxo Mo(VI) making up the total five-coordinate metal site (Fig. 5).
-
The xanthine oxidase family [24, 25] includes the aldehyde oxidases [26] plus a number of exotic members, in particular carbon monoxide dehydrogenases which contains a novel Mo/Cu binuclear active site [27, 28] and nicotinate dehydrogenases which contain Mo = Se coordination in the oxidized enzyme [29, 30]. Like sulfite oxidase, members of the xanthine oxidase family are five-coordinate in the oxidized form. The metal lacks a coordinating amino acid and has a structure that is either cis-dioxo-thio Mo(=O)2 = S at high pH or mono-oxo-thio Mo(OH)(=O) = S at low pH [24] (Fig. 5).
-
The DMSO reductase family is by far the most diverse and abundant family, containing a wide range of molybdenum enzymes, and all of the tungsten enzymes thus far discovered. All DMSO reductase family members contain two molybdopterins associated with the metal, with many but not all family members possessing a terminal oxo ligand to the metal and a coordinating amino acid. The DMSO family is thus further split into three main active site structural categories based on the coordinating amino acid. Type 1 possesses metal coordinated to either a cysteine or selenocysteine, Type 2 to an aspartate, and Type 3 to a serine. Figure 5 shows the active site of Rhodobacter sphaeroides DMSO reductase [31, 32] which is a Type 3 member. Many DMSO reductase family members might more properly be assigned to families of their own, because examples exist where no amino acid is bound to Mo or W [33], or the oxidized active site is apparently a des-oxo species.

Structures of the active sites in the oxidized Mo(VI) form of the prototypical members of the three families of enzymes. The structures shown are from crystallographic results (pdb codes 1SOX, 1F1Q and 1EU1, for sulfite oxidase, xanthine oxidase, and DMSO reductase, respectively), modified based on XAS data [17] to account for photoreduction, when present. For 1EU1 only the conformer model corresponding to the active enzyme is shown
The tungsten enzymes are alternatively allocated to a fourth family, the AOR family, named for Pyrococcus furiosus aldehyde oxidoreductase [13, 33]. This enzyme is still, however, incompletely characterized [17], and as there is greater structural variation in the accepted members of the DMSO reductase family, we choose to include the tungsten enzymes in that group. The tungsten enzymes are still relatively under-studied, so our review will focus on the molybdenum enzymes and where relevant point out possible parallels and examples from the tungsten enzymes.
2 Oxygen Atom Transferase Chemistry
Oxygen atom transferase activity is by far the most common activity among the molybdenum and tungsten enzymes. The exceptions are the molybdenum- and tungsten-containing formate dehydrogenases (Sect. 6.1), the molybdenum-containing pyrogallol transhydroxylases (Sect. 6.3), and the tungsten-containing acetylene hydratases (Sect. 6.2) and benzoyl co-A dehydrogenases (Sect. 6.4). These enzymes are discrete from the mainstream, are novel in their own right, and will be discussed in Sect. 6.
2.1 Spectator Oxo Effects
The enzymes that possess a cis-dioxo oxidized Mo(VI) active site may take advantage of what has been called the “spectator oxo effect” [34] to help drive the catalytic reaction. Here, the spectator oxo is an additional terminal oxygen atom bound to molybdenum that is not directly involved in oxygen atom transfer activity and remains after catalytic oxygen atom transfer. In cis-dioxo Mo(VI) species, both of the oxygens form Mo = O double bonds, with each comprised of a σ bond and a π-bond, and a non-bonding lone pair remaining on the oxygen (Fig. 6). In the monooxo Mo(IV) species, the oxygen forms a Mo ≡ O triple bond involving two electrons in 4d orbitals from the metal and four from the oxygen to give a σ bond and two π-bonds (Fig. 6). Such triple bonds cannot form in cis-dioxo Mo(VI) species because the use of two π-orbitals to make a triple bond would deprive the other oxygen of a d-orbital for its d-p π bond (Fig. 6). The bond strength of this Mo ≡ O triple bond with the spectator oxo is thought to act as a thermodynamic driver for oxygen atom transferase chemistry [34]. Numerous examples of cis-dioxo Mo(VI) and W(VI) enzymes are known, and specific examples of these will be discussed below.

Molecular orbitals for two simple examples, following [34]. (a) shows molecular orbitals for the hypothetical Mo(VI) system [MoO2Cl2] showing σ and π, and the oxygen lone pair (l.p.), (b) shows molecular orbitals for the Mo(IV) species [MoOCl4]2−, a σ bond and two d-p π bonds, constituting the Mo ≡ O triple bond
2.2 Non-spectator Oxygen Atom Transferase
Spectator oxo effects are undoubtedly part of the overall picture but only make sense as thermodynamic drivers for oxygen transfer from metal to substrate and not the reverse. It is noteworthy that both molybdenum and tungsten show remarkable flexibility in their coordination chemistry. Thus, searching the Cambridge structure database [35] gives a total of 374 sulfur-coordinated Mo(VI) compounds, of which 95 are des-oxo Mo(VI) species, 61 are monooxo species, 215 are cis-dioxo, and 3 are cis-trioxo Mo(VI) species. Tungsten shows similar flexibility in its coordination chemistry, with a total of 225 entries with sulfur-coordinated W(VI), comprised of 141 des-oxo, 54 monooxo, 27 cis-dioxo, and 3 cis-trioxo. At the time of writing, trioxo active sites are only known for mutant enzymes [36,37,38], but there are many examples of all the other categories of oxygen coordination. For now, we note that enzymes engaging in oxo atom transferase activity are expected to possess at least one terminal oxygen in the high-valent state, and the exceptions appear to be those enzymes that do not engage in such chemistry.
3 The Roles of Molybdopterin
Since the first structural information on molybdopterin began to emerge in 1982 [39], there have been suggestions of possible roles for molybdopterin beyond anchoring the active site within the protein [40]. In this section we will discuss current thinking on the roles that molybdopterin might play in the catalytic mechanism of the enzymes.
3.1 Molybdopterin in Electron Transfer
The first evidence that molybdopterin might function as an electron conduit for intramolecular electron transfer came from the initial crystal structures of molybdenum and tungsten enzymes [33, 41]; in particular this was suggested for the aldehyde oxidoreductase from Desulfovibrio gigas, a member of the xanthine oxidase family [41]. The notion that enzymes with two molybdopterins might show differential functions for the two cofactors, one acting as an electron conduit and the other to fine-tune the active site properties, was first suggested by Johnson and co-workers [42]. This work was subsequently expanded upon by Rothery et al. [15], with the suggestion that the molybdopterin acting as an electron conduit might be in the dihydro oxidation state and the other in the tetrahydro oxidation state. These workers have argued that the crystallographic results can be used to readily distinguish the different molybdopterin oxidation states. This was based on an alignment of DFT geometry-optimized structures at the relatively rigid aminopyrimidinone end of the cofactor, which showed large relative displacements of the other end (the pyranodithiolene) for the different oxidation states [15]. However, if the structures are aligned based on the entire molybdopterin structure, and not just one end, then the differences do not seem as marked. As we have previously noted [1], the resolution of many of the protein crystal structures may be insufficient to distinguish the structural differences of the different molybdopterin redox states (Fig. 7), and at present there is reason for cautious skepticism about these deductions.

Pterin redox state vs. fit to electron density. A difference map contoured at 3σ calculated using the modified coordinates (molybdopterin removed) and structure factors of Pichia angusta (now called Ogataea angusta) nitrate reductase (pdb code 2bih). The three redox isoforms of molybdopterin (shown in red, green, and blue) can all plausibly fit the electron density
Kirk and co-workers have discussed the case of dithiolene coordination in enzymes with a single Mo ≡ O [43]; this group will serve to define the axial direction and orient the Mo 4dxy redox-active orbital for maximal interaction with in-plane dithiolene sulfur p-orbitals (Fig. 8), yielding effective regeneration of the reduced enzyme following oxygen atom transfer. This role of a terminal oxo in facilitating electron transfer via the molybdopterin dithiolene has been referred to as the oxo-gate hypothesis [43].

Calculated molecular orbitals for an asymmetrically bound hypothetical Mo(V) dithiolene complex, as discussed by Kirk and co-workers [43], depicting the pseudo-σ interaction between Mo 4dxy and in-plane S 3p orbitals that may facilitate electron transfer
We now turn to the effects of protein upon the molybdopterin and the effect that this might have on the metal site and, in turn, upon mechanisms of catalysis.
3.2 The Pterin Twist Hypothesis
As discussed above and in previous work [1, 17, 44], the bulky molybdopterin enfolded in the protein is expected to restrict the motions of the ligands around the metal site, and neglect of such effects is a weakness of some early computational chemistry [44, 45]. Figure 9 shows the bis-molybdopterin (molybdopterin guanine dinucleotide, MGD) enfolded within the polypeptide of DMSO reductase, clearly illustrating that the forces exerted by the protein must impact the coordination environment around molybdenum. Recent work on arsenite oxidase illustrates this point nicely, with a hypothesis called the pterin twist hypothesis [46]. The basic idea is that the polypeptide acts to distort the active site to a conformation that approaches the transition state. We will discuss this enzyme more fully below, but, in brief, the observed structure of the oxidized Mo(VI) active site appears to be distorted by the protein exerting a twist on the dithiolene by up to 55 kJ mol−1 above the low-energy conformation of an isolated active site [46] (Fig. 10). The proposed distortion can be expressed as a function of a single variable (a twist angle φ; Fig. 10) and is a variant of the Rây-Dutt twist of classical coordination chemistry which is used to understand racemization of six-coordinate tris-chelate complexes [47, 48]. To date, none of the crystal structures determined for molybdenum enzymes which conduct oxo-transferase chemistry show simple octahedral-type geometry, suggesting that all show some tuning of the active site by the protein and molybdopterin. We note that for systems that have ring-open molybdopterin, these mechanisms will be inactive, because without the furan ring the pterin moiety can rotate relative to the dithiolene about the C4–C4a bond (see ring numbering Figs. 1 and 2).

Structure of DMSO reductase [pdb code 1EU1] showing the bis-molybdopterin MGD coordinated Mo site in red CPK (only the active conformer model is shown), with the polypeptide chain shown in yellow. The cofactor can be seen to be effectively enfolded by the protein, which is expected to restrict coordination geometry of the ligands at the metal site

Computed effects of a pterin twist, φ, between extremes defined by point group symmetry; C 2v for φ = 0° and C 2 for φ = 33°. The geometry in the enzyme [46] is deduced to be close to φ = 0° which elevates the energy above the minimum by ~55 kJ mol−1. The centrally placed superimposed structures show the transformation from φ = 0° to φ = 33°
4 Reliability of Structural Information
Before we turn to considering the enzymes themselves, we will briefly consider some problems with the structural methods that we rely on for our insights into mechanism. Essentially the sources of all quantitative structural information can be divided into spectroscopy and crystallography. For spectroscopy a detailed review has been recently reported [17]. Methods such as electron paramagnetic resonance (EPR) and its derivatives and vibrational spectroscopies such as resonance Raman and Fourier transform infrared spectroscopy can provide insights into metal ion coordination, and a major problem with all of these relates to confidence that the bulk of the sample is being investigated. For example, with EPR, only the paramagnetic Mo(V) form is being probed, and EPR gives no direct information at all upon the oxidized Mo(VI) or the fully reduced Mo(IV) forms. X-ray absorption spectroscopy provides access to all oxidation states, but analysis and in particular over-interpretation of data can mislead researchers, as we have discussed previously [17]. For crystallography, multiple occupancies of active sites within a near-identical polypeptide fold have misled researchers [17] (see Sect. 5.3), and the identity of some atoms has posed challenges in other studies, for example, with copper being mistaken for selenium [49], with consequent incorrect conclusions about the mechanism [14]. A major problem with almost all of the crystal structures of molybdenum and tungsten enzymes is photoreduction [50,51,52]. This occurs when X-ray-induced photochemical scission of water, or other components of the sample, produces energetic free radical species, such as hydrated electrons and hydroxyl radicals. The former are strong reductants and the latter are potent oxidants, and while most photochemical products recombine to form starting materials and heat, a proportion can react with other components of the sample. The photoreduction of redox-active metals is substantially exacerbated by crystallographic cryoprotectants such as polyethylene glycols or glycerol [52]. This is because these species are very effective hydroxyl radical scavengers, leaving an excess of highly reducing hydrated electrons that can act upon the redox-active metal sites [52]. The same compounds that are added as cryoprotectants in crystallography are also commonly used as glassing agents for X-ray absorption spectroscopy to prevent artifacts due to crystalline ice diffraction [53]. While X-ray absorption spectroscopy is also susceptible to photoreduction, the sensitivity of the near-edge region to electronic structure means that this is usually detected [51, 52]. Moreover, the high energy of the X-ray beam at the Mo K-edge means that X-ray diffraction from any ice occurs at sufficiently low Bragg angles that ice diffraction artifacts are not registered by the detector; hence there is no need to add glassing agents, and the tendency for photoreduction is therefore much reduced [52]. While photoreduction of active sites was not recognized as such in some of the early crystallographic work on the Mo and W enzymes, crystallographers are increasingly aware of these issues, and a photoreduced active site is unlikely to be viewed as oxidized in any modern work.
5 The Enzymes
5.1 The Sulfite Oxidase Family
5.1.1 The Sulfite Oxidases
These enzymes catalyze the oxidation of sulfite to sulfate:
As discussed above, these enzymes possess cis-dioxo Mo(VI) sites. Like most molybdenum and tungsten enzymes, nearly all structures of sulfite oxidases and sulfite dehydrogenases show clear signs that the enzyme has been photoreduced in the X-ray beam. The only exceptions are enzymes with site-directed mutations [36] that render the molybdenum site very difficult to reduce [37, 38]. Our understanding of the active sites and the catalytic mechanism has been derived from a combination of crystallographic and spectroscopic methods [17, 54]. The active site of sulfite oxidases from animals and plants, and the bacterial sulfite dehydrogenases have been studied by a variety of methods, including X-ray absorption spectroscopy, X-ray crystallography, and, with the 4d1 Mo(V) form, electron paramagnetic resonance and its derivative methods such as electron nuclear double resonance and electron spin echo envelope modulation [17].
Sulfite oxidases are found in both plants and animals. In mammals the enzyme is essential for health as it is part of the pathway for cysteine catabolism, and individuals possessing mutant sulfite oxidase with partial activity show buildup of abnormal sulfur metabolites, very severe neurological impairment, and usually death in utero or in infancy [55,56,57]. Residing in the mitochondrial inner-membrane space, the enzyme is dimeric with a subunit mass of about 52,000 with each monomer possessing molybdenum associated with one molybdopterin and a cytochrome b 5 iron-heme. Plant sulfite oxidases show considerable similarity at the molybdenum site, but lack the heme, and thus are the simplest of the eukaryotic molybdenum enzymes. The crystal structures of sulfite oxidases reveal a number of highly relevant details of the active site structure. The first sulfite oxidase crystal structure reported was that of the chicken enzyme [18], which was crystallized in the presence of sulfate. The structure showed two basic arginine-rich anion-binding pockets near the molybdenum site, at approximately 5 Å and 10 Å from Mo, both with sulfate bound, which serve to channel and locate substrate within the active site [18]. The electron density observed crystallographically at the 5 Å pocket was most consistent with mixed sulfite/sulfate occupancy in the pocket [18], with the three sulfite oxygen atoms oriented away from Mo so that the sulfur lone pair would point in the direction of the equatorial Mo = O oxygen, orienting the substrate correctly for catalytic oxygen atom transfer. The attack of the sulfite lone pair on this equatorial oxo ligand has been suggested to result in population of a Mo–O dxy-pπ antibonding orbital, which in turn has been hypothesized to labilize the equatorial oxygen atom for transfer to substrate [58]. It has also been suggested that other aspects of the active site might fine-tune properties, specifically that the Ooxo–Mo–S–C(Cys) torsion angle is important in controlling overlap between a cysteine sulfur p-orbital and one of the two Mo = O π* orbitals, with consequent activation of the oxo for transfer to substrate [59]. Figure 11 summarizes current views of the sulfite oxidase catalytic mechanism at the molybdenum site.

Simplified sulfite oxidase oxidative half-reaction catalytic mechanism. The initial Mo(VI) enzyme is reduced to the Mo(IV) by substrate, with water subsequently liberating product. Mo(V) EPR signals corresponding to the product complex have been observed with the competitive inhibitors phosphate and arsenate (see Ref. [17])
5.1.2 The Nitrate Reductases
The assimilatory plant and fungal nitrate reductases are very closely related to the sulfite oxidases and quite discrete from the dissimilatory nitrate reductases of prokaryotes. They catalyze the conversion of nitrate to nitrite, which is a key part of the nitrogen cycle:
This reaction is known as the reductive half-reaction of nitrate reductase. Like the animal sulfite oxidases, plant and fungal nitrate reductases possess molybdenum and cytochrome b 5 and share structural features [60], with very similar oxidized Mo(VI) and reduced Mo(IV) active site structures [17, 61]. In addition the enzymes contain a flavin adenine dinucleotide (FAD) cofactor that serves to provide electrons to the cytochrome b 5 and molybdenum sites. The source of reduction of FAD is either NADH or NADPH; NADH-specific forms exist in higher plants, whereas NADPH-specific enzymes are found in some fungi. Bi-specific forms that can accept electrons from either NADPH or NADH are found in various plant and fungal species [62]. The structure of the nitrate reductase from the methylotrophic yeast Pichia angusta (now called Ogataea angusta) has been reported [60] and shows a basic substrate pocket ~5 Å from molybdenum homologous to that found in sulfite oxidase, but, with one fewer arginine, consistent with the lower charge on both substrate and product [60]: [NO3]− vs. [SO3]2−. Yeast nitrate reductase and chicken sulfite oxidase (both alas with photoreduced Mo sites) are compared in Fig. 12. The nitrate reductase possesses two rather than three arginine residues in the substrate binding pocket of the active site, plus a highly conserved methionine (Met427) that is needed for activity. Other differences include an asparagine residue in place of a tyrosine (Fig. 12). In an elegant demonstration of the close relationship between the two enzymes, a triple site-directed sulfite oxidase mutant (Tyr322 → Asn, Arg450 → Met, Val452 → Met) has been constructed to successfully convert sulfite oxidase into a nitrate reductase [63], with all three substitutions required to maximize activity [63]. As shown in Fig. 12, the structures of the molybdenum sites themselves are very similar indeed, with no substantive difference between the aforementioned Ooxo–Mo–S–C(Cys) torsion angle [59]. Given that one enzyme is an oxidase and the other a reductase, it seems unlikely that the postulated tuning using the cysteine torsion angle is important in the catalysis of either.

Comparison of active sites of chicken sulfite oxidase (pdb code 1SOX) and yeast nitrate reductase (pdb code 2BII), together with a superposition showing sulfite oxidase (red) vs. nitrate reductase (green)
5.1.3 Mitochondrial Amidoxime Reducing Component
Quite recently a previously unknown eukaryotic molybdenum enzyme was reported [64], which is now thought to be part of the sulfite oxidase family [65]. This enzyme was named mARC (mitochondrial amidoxime reducing component) for its ability to reduce N-hydroxylated species, such as some amidoxime prodrugs into their active amino form drugs. At the time of writing, mARC is only the fifth eukaryotic molybdenum enzyme known and perhaps the simplest as it possesses only a molybdenum site. Both plant and animal mARC enzymes have been reported, and all known mammalian genomes contain two different mARC genes: MARC1 and MARC2 with the two enzymes known as mARC-1 and mARC-2, respectively. In general mARCs have broad substrate specificity, and the biochemical roles they play are still emerging [65, 66]. In addition to the amidoxime reductase activity already mentioned, the mARC enzymes are able to reduce a range of N-hydroxylated compounds, such as 6-N-hydroxylaminopurine to adenine, as well as nitrite to the cardiovascular-signaling molecule nitric oxide (NO). While the study of these enzymes is still in its infancy, and the active sites remain poorly characterized with only preliminary XAS and magnetic resonance reported to date [67, 68], there is no doubt that more work will follow [69, 70] to give important details about these intriguing enzymes.
We now turn to a quite different aspect of the sulfite oxidase mechanism. Although our primary focus in this review are the mechanisms specific to the molybdenum or tungsten active sites, the electron transfer between molybdenum and heme in sulfite oxidase proved something of a conundrum. Since it involves Mo active site components, we will discuss it briefly here. The first sulfite oxidase crystal structure [18] showed a long-range Mo···Fe distance of 32 Å, which is surprisingly large given the reported rapid rates of electron transfer between these two sites [71]. Moreover, the molybdopterin was observed to point away from the cytochrome b 5 heme, the least favorable orientation if this heterocycle was involved in electron transfer [18]. It was later suggested that the cytochrome domain, connected to the molybdenum by a flexible peptide, was mobile in solution and would reorient, docking with the Mo domain for effective electron transfer [71], with the observed conformation in the crystal structure being due to crystal packing forces. Subsequent molecular mechanics indicated that such domain motion was possible, although this study, which had no imposed constraints or restraints, failed to show the specific docking that had been postulated [72]. Figure 13 shows schematic diagrams of the different domain arrangements. Subsequent steered molecular mechanics calculations did suggest a docked structure suitable for electron transfer [73], but a later electron-electron double resonance study of solution samples containing both Mo(V) and Fe(III) again indicated a Mo···Fe separation of 32 Å [74], strikingly the identical distance that had been determined by crystallography [18]. Added to this were results indicating that some of the amino acid residues of the Mo site were important for efficient electron transfer between iron and molybdenum [75]. Of particular note is the effect of the clinical mutation Arg160 → Gln, corresponding to Arg138 → Gln in chicken sulfite oxidase (Fig. 12). This mutation gave a three order of magnitude increase in the K m for sulfite [18] and substantially poorer intramolecular rates of electron transfer [76]. Recently structural information on the bacterial sulfite dehydrogenase from Sinorhizobium meliloti has been reported [77]. This enzyme has discrete proteins that act together containing the molybdenum site and the cytochrome electron acceptor, called SorT and SorU, respectively. The crystal structures of the SorT and SorU proteins have been solved, both alone and in complex [77], and with the analogue of the clinical mutation Arg78 → Gln plus a number of other site-directed mutants [78]. The structure of wild-type SorT in complex with SorU showed that Arg78 of SorT hydrogen bonds to the propionate of the SorU heme and the Arg78 mutants all showed substantially impaired ability to transfer electrons to SorU. This work compellingly showed that SorU docks close to the Mo site of SorT in a position analogous to that proposed for sulfite oxidase (Fig. 13b) and that the aforementioned active site arginine Arg78 (Arg138 chicken) plays a pivotal role in both active site structure and in facilitating electron transfer to the heme. The reason why this conformation has not yet been actually observed in sulfite oxidase is as yet unclear, and the answer to this must await further studies.

Sulfite oxidase domain motion (a) shows the crystallographically observed homodimer with cytochrome domains (red) and Mo domains (green) oriented unfavorably for electron transfer between Mo and Fe. The postulated “docked” relocation of the cytochrome domains is shown in (b) with the detail of the hypothetical situation of Mo and Fe sites in (c) (corresponding to the orange box in b)
5.2 The Xanthine Oxidase Family
5.2.1 Xanthine Oxidase and Aldehyde Oxidase
Xanthine oxidase and dehydrogenase are two different forms of the same enzyme, encoded by the same gene, the prototypical member of the xanthine oxidase family. From the molybdenum perspective, the oxidase and the dehydrogenase are essentially identical and can be interconverted either by proteolysis or by formation of a disulfide at the flavin site [79]. Xanthine oxidase is linked to gout, a painful condition in which crystals of monosodium urate monohydrate form in joint capsules [80, 81], in particular that at the base of the big toe. Gout can be prevented by administering the xanthine oxidase/dehydrogenase inhibitor allopurinol, which is oxidized in vivo by the enzyme to the active form that is known as alloxanthine or oxypurinol (Fig. 14) and which associates covalently with Mo in the inhibitory complex, forming a Mo–N bond [82, 83]. Human xanthine oxidase/dehydrogenase deficiency is known as xanthinuria, and, to the embarrassment of xanthine oxidase researchers, some individuals suffering from this can be in excellent health and completely asymptomatic, while others develop various problems associated with buildup of xanthine or hypoxanthine [84, 85].

Upper panel: Purine oxidation reactions catalyzed by xanthine oxidase/dehydrogenase. In the figure the more stable keto tautomers are shown, although oxidations by the enzyme are thought to be initially to the enol form (C–OH). Lower panel: the xanthine oxidase/dehydrogenase inhibitors allopurinol and oxypurinol; as with the upper panel, the more stable tautomers are shown
The xanthine oxidase family also includes a range of other eukaryotic enzymes, as well as a substantial number of prokaryotic enzymes. Treating the oxidized active enzymes with cyanide converts the active site to an inactive “desulfo” form, with liberation of thiocyanate and reduction to Mo(IV). The cyanide-labile sulfur is now known to be a Mo = S group in the oxidized enzyme, and its presence together with molybdopterin and oxygen coordination of the molybdenum defines the xanthine oxidase family. When oxidized, the inactive desulfo form of the enzyme contains a Mo = O group in place of the Mo = S. Xanthine oxidase has already been shown in Fig. 3 as an example of a multi-redox center molybdenum enzyme; the enzyme is a homodimer, with each subunit containing one Mo, two different [Fe2S2] clusters, and a flavin [86,87,88]. The physiological role of xanthine oxidase is thought to be purine oxidation, successively converting purine to hypoxanthine to xanthine to uric acid (Fig. 14). The enzyme has very broad substrate specificity and is capable of oxidation of a wide range of aldehydes [88]. It is also very closely related to the aldehyde oxidases [26], which also have broad substrate specificity, catalyzing oxidation of aldehydes to carboxylic acids (Eq. 4). These enzymes are of pharmaceutical importance through their role in clearing a number of aldehyde drugs and nitrogen heterocycles such as nicotine [89].
Protein crystallography and spectroscopy have provided detailed structural information and potential insights about the catalytic mechanism. Early views on mechanism were advanced by Bray and co-workers, who used EPR spectroscopy in conjunction with substitution with stable isotopes. Labeling with 2H showed that the proton from the substrate carbon undergoing oxygen atom transfer to form product is transferred to the molybdenum as a Mo(V)–SH (see [17] and refs. therein) and that the oxygen transferred to substrate originates from oxygen coordinated to Mo (see [17] and refs. therein). Early proposed mechanisms were formulated in terms of a molybdenum-bound hydride acceptor and molybdenum-bound oxygen acting as a nucleophile. A number of xanthine oxidase family members have Mo = Se in place of Mo = S; these include purine hydroxylase and xanthine dehydrogenases which have been purified and characterized from Clostridium purinolyticum [90] and nicotinate dehydrogenase from Eubacterium barkeri, which has been structurally characterized [91]. Moreover, early work indicated that desulfo turkey xanthine dehydrogenase could, at least in part, be reactivated by treatment with selenide, suggesting that the Mo = Se form of this enzyme could show activity [92]. As Mo = Se must play a similar role to Mo = S, and because Mo = Se is an outstanding nucleophile and a poor electrophile, these observations support a nucleophilic role for Mo = S/Se in the catalytic mechanism [93]. Some aspects of the catalytic mechanism are summarized in Fig. 15; the Mo = S abstracts a proton from the carbon undergoing oxo transfer, followed by formation of the Mo–O–C bond with concomitant reduction of the metal to Mo(IV). Importantly, pH-dependent changes of the active site structure correspond with one molybdenum-oxygen bond shortening from 1.97 Å at pH 6 to 1.75 Å at pH 10, consistent with the active oxygen deprotonating from Mo–OH at low pH to Mo = O at high pH [24].

Proposed catalytic mechanism for xanthine oxidase, involving the highly conserved active site residue Glu 1,261. Colors indicate the fate of atoms in the reaction as probed by stable isotopes starting with 1; see [17] for a comprehensive review of the literature. The Mo(V) species 3 is the much-studied Very Rapid intermediate in which product is covalently bound to Mo [17]
5.2.2 Carbon Monoxide Dehydrogenase
The most unusual members of the xanthine oxidase family are undoubtedly the bacterial molybdenum-containing carbon monoxide dehydrogenases, owing to the presence of a Cu/Mo binuclear site in these systems. In essence, these enzymes possess a xanthine oxidase active site modified with an additional two-coordinate cuprous ion bound to a cysteine residue and to what in other more conventional enzymes would be Mo = S to form a Cu/Mo binuclear active site, as shown in Fig. 16, a structure deduced from a combination of crystallography [94] and XAS [95]. The enzymes catalyze oxidation of carbon monoxide to carbon dioxide, Eq. (5):

Proposed catalytic mechanism for carbon monoxide dehydrogenase involving binding of CO to Cu(I) in the active site; for a comprehensive review, see [17]
The substrate CO is thought to bind to Cu, positioning it adjacent to Mo in an ideal location for oxygen atom transfer from Mo to form CO2, as shown in Fig. 16. The enzyme produces a novel Mo(V) EPR signal, with coupling from both I = 3/2 63,65Cu and I = 5/2 95,97Mo [94, 96, 97] with evidence of I = 3/2 13C hyperfine when 13CO is used to develop the signal. This most novel member of the xanthine oxidase family thus provides a striking example of the flexibility of molybdenum enzyme active sites. How this system evolved, with ancient molybdenum alongside relatively recent copper, is at present unresolved.
5.3 The DMSO Reductase Family
The DMSO reductase family constitutes by far the most varied and numerous of the three families of enzymes. With two molybdopterin associated with the metal, this family is found in both archaea and bacteria, but not in eukaryotes. It includes all known tungsten enzymes, a wide range of molybdenum enzymes in various metabolic and respiratory roles, and the few known molybdenum/tungsten enzymes that are not oxo-transferases. The best studied are the Rhodobacter DMSO reductases. The R. capsulatus and R. sphaeroides enzymes have both been extensively studied, and as there appear to be only trivial differences between them, we will not distinguish them here. The DMSO reductases catalyze the following reaction:
The group of very similar enzymes includes trimethylamine N-oxide reductase [98] dimethyl sulfide dehydrogenase [99], which catalyzes the reverse reaction of Eq. (6), and the biotin sulfoxide reductases [100]. Together these enzymes comprise the Type 3 DMSO reductase family members.
5.3.1 Rhodobacter DMSO Reductase
The active site structures of this enzyme were a source of early controversy, and because the reader may come across some of this early work, we will discuss this topic very briefly here. The first characterization of the active site was by XAS [101], and notably, given what was to later unfold, this work discussed the possibility of multiple structures at the active site [101]. Subsequent crystallography [102] gave very surprising conclusions, with unlikely molybdenum coordination environments, and was distinctly at odds with the XAS [101]. More crystallography followed by two different research groups [103,104,105], plus another XAS study which seemed to confirm one of the crystal structures [106]. Meanwhile, additional studies by resonance Raman spectroscopy disagreed with the crystallography and instead favored the conclusions of the original XAS study [42]. Subsequent to all of this work, the XAS was revisited with substantially the same result as the initial study [107]. This later study also pointed out that the structures that were suggested from analysis of the X-ray crystallography were chemically implausible, with some showing supposedly non-bonded atoms with overlapping van der Waals radii, and reinforced the earlier suggestion [101] that multiple forms of the active site might exist [107]. Finally, the crystallography was reexamined at higher resolution [108] and interpreted in terms of two different active site structures enfolded within near-identical proteins. Still later work explored the culprits; in part the choice of buffer (HEPES) and exposure to oxygen had modified the enzyme active site [109], a process which could be reversed by reduction and catalytic turnover [107].
The true structure of the oxidized active enzyme is now agreed to have two molybdopterin dithiolenes bound to give a total of four sulfur donors to Mo, with a serine oxygen coordinated and a single Mo = O, as shown in Fig. 5 and schematically in Fig. 17. The spectator oxo effect (Sect. 2.1) is unimportant in DMSO reductase and its siblings, because they possess only a single Mo = O ligand in the oxidized Mo(VI) form. As discussed above, the terminal oxo group may facilitate electron transfer [43] via one of the two molybdopterins, which have been suggested to function differently in catalysis [15, 42].

Proposed catalytic mechanism for DMSO reductase; for a comprehensive review of spectroscopic evidence, see [17]. The DMSO binds to reduced enzyme 2 to produce a DMSO bound form 3 that has been studied both by crystallography and spectroscopy. The oxygen is transferred to Mo to form 4, and release of dimethyl sulfide regenerates oxidized enzyme 1
Various product- and product analogue-bound forms of DMSO reductase have been characterized spectroscopically [107, 110]. Most notably dimethyl sulfide binds to the oxidized enzyme to make a catalytically relevant DMSO complex (Fig. 17), and dimethyl selenide forms a similar complex [110]; moreover, trimethylarsine reacts with oxidized Mo(VI) enzyme to make a trimethylarsine-N-oxide bound to a reduced Mo(IV) site with stoichiometric Mo and As, which can be characterized by XAS from both the Mo and the As perspective [110] as coordinated through a Mo–O = As bond, suggesting a structure for a substrate complex. A schematic diagram of the catalytic process most probably employed in DMSO reductase is shown in Fig. 17. The enzyme has been extensively studied by electron paramagnetic resonance spectroscopy [17], and the Mo(V) form has been examined by XAS and found to be five-coordinate, with four sulfurs from two molybdopterins plus one Mo–OH ligand but lacking the serine oxygen ligand to Mo [45]. How this coordination fits into the proposed catalytic cycle is at present unclear, but it illustrates that the active sites of these enzymes can by dynamic and change their coordination chemistry under different conditions.
5.3.2 Prokaryotic Nitrate Reductases
A widespread and well-studied group of Mo enzymes are the prokaryotic nitrate reductases [111, 112]. These can be divided into three distinct categories: the periplasmic nitrate reductase (Nap), the respiratory nitrate reductases (Nar), and the assimilatory nitrate reductase (Nas). All are DMSO reductase family members, although the Nas enzymes are not as completely studied. Structures for Nap indicate that these are Type 1 with a cysteine donor to Mo [113] plus another ligand which may be a sulfido [114], while the Nar enzymes are Type 2 DMSO reductase family members, with aspartate coordination to the metal [6, 7]. Both active site structures and possible mechanisms have recently been reviewed for the Nap enzymes [112, 114] which suggest that the cysteine may dissociate to allow nitrate to bind, in a postulated mechanism related to that suggested for formate dehydrogenase, which we will discuss below (Sect. 6.1). The Nar nitrate reductases have also been extensively studied using both crystallography [6, 7] and a variety of spectroscopic methods [17, 115]. The active site of Nar has two molybdopterin dithiolene ligands with an aspartate (Asp222) providing oxygen coordination to Mo. The enzyme shows a curious reductive activation [116] that we can speculate may be related to the ring-open/ring-closed forms of molybdopterin discussed above [6, 7] (Fig. 2). Possibly, the reductive activation is due to conversion of inactive and ring-open dihydro form, to an active and ring-closed tetrahydro form [116], and these are the forms that have been observed crystallographically [6, 7]. Insofar as the catalytic mechanism is concerned, nitrate is thought to bind to the Mo(IV) enzyme in a manner analogous to the nitrate complex that has been observed by EPR for the Mo(V) oxidation state [117], and a simplified mechanism is shown in Fig. 18.

Postulated catalytic cycle of Nar nitrate reductase
Another relative of this family is the selenate reductase from Thauera selenatis [118], which has a monooxo Mo(VI) site and a des-oxo Mo(IV) site with close to four Mo–S ligands in both. The enzyme also possesses a mysterious selenium-containing group bound to a metal ion, most probably Fe [118].
5.3.3 Ethylbenzene Dehydrogenase
This enzyme catalyzes the anaerobic hydroxylation of ethylbenzene to (S )-1-phenylethanol in a highly stereospecific reaction [119], which is the first step in the anaerobic degradation of ethylbenzene [120]. The enzyme can hydroxylate a series of other compounds with methylene carbons adjacent to a benzene or other heterocyclic ring [121], and in most cases the reaction is strongly enantioselective [121]. The crystal structure of the enzyme shows a type 2 DMSO reductase family member [119], with the molybdenum coordinated by two molybdopterin dithiolenes and an aspartate side chain and one terminal oxygen group [119]. Similarly to one of the two Nar structures [7], one of the two molybdopterins has a bicycle ring-open structure, while the other is a tricyle furan (Fig. 2). Current views of the catalytic mechanism [122] involve abstraction of one hydrogen from the methylene by the Mo = O group of the oxidized Mo(VI) enzyme to form a free radical intermediate with a Mo(V) site with hydrogen from an adjacent histidine residue stabilizing the hydroxyl ligand. This is followed by a second one-electron transfer with the radical becoming a carbocation Mo(IV) species which then associates with Mo to form a bridging OH. Displacement of this by water then releases product and subsequent reoxidation through intramolecular electron transfer to regenerate the oxidized active site (Fig. 19). When 1,2-diethylbenzene is used as a substrate, the product is nearly racemic, arguing for the proposed reaction mechanism [123].

Postulated reaction cycle of ethylbenzene dehydrogenase. We note that alternative mechanisms have been suggested for this enzyme for hydroxylation of 4-ethylphenol involving a nearby aspartate and a quinone methide intermediate [120]
5.3.4 Arsenite Oxidase and Arsenate Reductase
These enzymes catalyze the oxidation of toxic arsenite to less toxic arsenate (arsenite oxidase) or the reverse (arsenate reductase), as shown in Eq. (7)
Arsenite oxidase has been characterized from two different organisms by both crystallography [124, 125] and XAS [46, 126], with consistent results for each technique. As is often the case, and as we have discussed above, the crystal structures are clearly of photoreduced enzymes and show differences from the structure suggested from XAS [46, 126]. In the oxidized enzyme the active site possesses a cis-dioxo Mo(VI) with four nearly equivalent sulfur donors from the two molybdopterin dithiolenes, consistent with a square-based prismatic type geometry. The proposed catalytic mechanism is shown in Fig. 20 [46]. The pterin twist mechanism, discussed above, was postulated to distort the active site away from the low-energy octahedral-type geometry toward the transition state [46], energetically lowering the barrier between the initial substrate complex (Fig. 20, 1) by adopting a geometry more typical of reduced Mo(IV) (Fig. 20). The reaction is assisted by the spectator oxo effect [46], which is also discussed above. The fully reduced product (Fig. 20) resembles the structures observed by X-ray crystallography. The enzyme is also remarkable in having fully crossed-over Mo(VI)/Mo(V) and Mo(V)/Mo(IV) redox potentials, so that the Mo(V) oxidation state cannot be observed [127] and the enzyme works only in the Mo(VI) and Mo(IV) formal oxidation states (Fig. 20).

Proposed catalytic mechanism for arsenite oxidase. The oxidized enzyme 1, distorted by the protein through a pterin twist, reacts with arsenite via a transition state 2 from which is liberated arsenate
The reduction of arsenate to arsenite is not as biochemically challenging as the reverse reaction from a thermodynamic perspective [46]. The arsenate reductases that concern us here are molybdenum-containing enzymes, but we note in passing that bacteria and yeasts possess unrelated non-Mo-containing arsenate reductases [128]. Although these are themselves unrelated, both prokaryotic and eukaryotic enzymes employ glutaredoxin with reduced glutathione as the source of electrons [128]. There is also a human enzyme about which little is known, which may be related to these enzymes as exogenous thiols are needed for activity [129]. These systems are not directly related evolutionarily or biochemically to the molybdenum-containing enzyme that is our focus, and we will not discuss them further.
The Mo-containing arsenate reductase from Shewanella sp. ANA-3 has been studied crystallographically [1130], and, as is often observed, the initially oxidized enzyme appears to have been photoreduced during data collection [130]. The XAS of this enzyme has not yet been reported, but the detailed crystal structures suggest a catalytic mechanism that is quite distinct from that of arsenite oxidase [130] and an active site that belongs to the Type 1 DMSO reductase family, with cysteine coordination of Mo. The proposed mechanism is shown in Fig. 21, and in many ways resembles that of DMSO reductase, and is as expected for an oxo-transferase acting in a reductive capacity. The active site structure of the enzyme is unusual in having near-octahedral-type coordination around molybdenum.

Proposed catalytic mechanism for arsenate reductase
6 Non-oxo-transferase Molybdenum and Tungsten Enzymes
As we have discussed above, the vast majority of the molybdenum and tungsten enzymes are oxo-transferases, but there are a few noteworthy exceptions as we now discuss.
6.1 Formate Dehydrogenase and Formylmethanofuran Dehydrogenase
Both tungsten and molybdenum formate dehydrogenases are known; the first tungsten enzyme to be described was the formate dehydrogenase from the thermophile Clostridium thermoaceticum [12]. Formate dehydrogenases are noteworthy in that they are one category of molybdenum/tungsten enzymes that do not employ oxygen atom transferase chemistry nor do they use water as a source of oxygen. They catalyze the conversion of formate to carbon dioxide or carbonate, as shown in Eq. (8).
Both molybdenum and tungsten formate dehydrogenases contain two molybdopterins bound to the metal, and are found with selenocysteine associated with the metal [131], although cysteine coordinated analogues are also known [131]. E. coli has two different formate dehydrogenases (FDH), and much work has been done on the catalytic component of one of these, called FDHH. We will focus on discussion of this enzyme here, although the other E. coli enzyme appears to be similar. There has been debate about the structure of the active site of FDHH, and the original crystallography [132] has been reinterpreted [133, 134]. Our discussion here is based upon recent work on the mechanism [135] while noting that there is still contention [136], taking into account the definitive identification by XAS of a selenium sulfide covalent bond [137, 138]. The mechanism is guided by elegant experiments using stable isotopes; when 13C-labeled formate in 18O-enriched water is used, the enzyme produced 13CO2 containing no 18O [139], conclusively establishing that the enzyme does not catalyze incorporation of oxygen from water into product. Moreover, deuterated formate 2HCO2 − was used to show that the proton of formate is associated with the molybdenum after catalytic turnover [139]. A summary of a possible mechanism for formate dehydrogenase is shown in Fig. 22.

Effectively the reverse reaction to that catalyzed by formate dehydrogenase is catalyzed by formylmethanofuran dehydrogenase [151, 152], which fixes CO2 (Fig. 23) and is the first step in the process of methane generation by some Archaea. The most prominent mechanism of biological CO2 fixation is photosynthesis, but many others exist [140] of which archaeal methanogenesis is thought to be among the most ancient. While this mechanism was much more prevalent in the pre-photosynthetic world, it is still widespread in the environment, being responsible for such phenomena as marsh gas, deep-sea methane clathrates [141], and gastro-intestinal flatulence in mammals [142]. As we have discussed above, both molybdenum and tungsten enzymes are known, and both have two molybdopterins and sulfide and cysteine sulfur donors forming an active site that is analogous to that of the cysteine-containing formate dehydrogenases and the selenocysteine enzymes discussed above. It seems likely that its catalytic mechanism is related to that of formate dehydrogenase [151, 152]. Aside from the chemistry that occurs at the tungsten site, the overall structure of the enzyme deserves comment.

Reaction catalyzed by formylmethanofuran dehydrogenase
The enzyme exists as two linked dodecamers (12), forming an overall tetracosamer (24) of some 800 kDa, containing a total of 46 iron-sulfur clusters. The surprising complexity of the enzyme seems to almost invite superlatives. The tungsten sites (4 per tetracosamer) are buried deep within the protein, and to reach them the substrate CO2 must diffuse along a 40 Å hydrophobic internal channel before it is reduced to formate or formic acid. The product must then traverse another predominantly hydrophilic internal channel, this time some 43 Å-long, to a binuclear zinc site where it reacts with methanofuran to form formylmethanofuran [151]. The remarkable arrangement of iron-sulfur clusters in this novel enzyme is shown in Fig. 24, and while these clearly serve as an electrical connection, coupling the four tungsten sites over some 206 Å, why such complex arrangements are needed and the exact function of the [4Fe-4S] cluster arrays is as yet unclear [151].

Crystal structure of the tetracosamer ~800 kDa formylmethanofuran dehydrogenase (pdb: 5t61), containing 46 [4Fe-4S] clusters (green/yellow) and four tungsten sites, two in each dodecamer. The overall hourglass shape of the two dodecamer subunit arrangements is clear, with two of the [4Fe-4S] clusters (one from each dodecamer) acting as a bridge between them. The locations of the tungsten sites in the top and bottom of the figure are shown (W), as are the bound methanofuran (MR). The four external [4Fe-4S] clusters (marked e−) are the postulated entry points of reductive electrons
6.2 Acetylene Hydratase
Acetylene hydratase is a second exception to the rule that molybdenum and tungsten enzymes are oxo-transferases. This enzyme is also unusual among molybdopterin-based enzyme in a second regard, in that the metal in the active site of the enzyme plays no apparent redox role, an anomaly shared only with the enzyme pyrogallol transhydroxylase, which we will discuss below (Sect. 6.3). The enzyme catalyzes the net hydration reaction converting acetylene to acetaldehyde, as shown in Eq. (9)
This reaction is exothermic by more than 110 kJ mol−1 and is of historical industrial interest, as the hydration of acetylene was the major source of acetaldehyde [143], and the analogous reaction with propyne (CH3C ≡ CH) was used to produce acetone. In those early industrial processes, Hg(II) compounds served as catalysts to activate the acetylene, with attack of water yielding an enol (vinyl alcohol from acetylene) which then underwent tautomerization to form the keto product (acetaldehyde). The tungsten of acetylene hydratase [144] seems to play a role that is in some ways analogous to the Hg(II) of industrial chemistry, binding acetylene via the C ≡ C triple bond with attack of water on one carbon to make vinyl alcohol via a multi-stage process involving a nearby aspartate residue [145].
6.3 Pyrogallol Transhydroxylase
The pyrogallol transhydroxylase from the anaerobe Pelobacter acidigallici is another molybdenum enzyme for which water is not the source of oxygen, and like acetylene hydratase it does not catalyze a redox reaction [146]. The enzyme catalyzes the conversion of pyrogallol (1,2,3-trihydroxybenzene) to phloroglucinol (1,3,5-trihydroxybenzene), effectively moving –OH from the two- to the five-ring position. The enzyme uses a co-substrate 1,2,3,5-tetrahydroxybenzene, which in fact is the primary source of the product phloroglucinol (Fig. 25). The enzyme binds pyrogallol to Mo via the C3–OH, and protons are abstracted from the 2 and 3 positions by nearby aspartate and histidine residues, respectively, stabilizing the diketo tautomer (1 hydroxy-2,3-benzoquinone). The co-substrate is located in the active site with its C2–OH adjacent to the pyrogallol C5 position, and its proton is picked up by a nearby tyrosine, forming an ether linkage between the substrate and co-substrate to make the intermediate 2,6-dihydroxy-4-(2,4,6-trihydroxyphenoxy)cyclohexa-2,5-dien-1-one. The catalytic cycle is completed by breaking the C2–O bond, forming phloroglucinol from the co-substrate and co-substrate from the substrate. The enzyme is clearly related to others in the DMSO reductase family [146] and even possesses iron-sulfur clusters that apparently serve no redox purpose. The sole purpose of the molybdenum appears to be to bind the substrate C3–OH, and the sole purpose of the two molybdopterins seems to be to appropriately locate the Mo in the active site.

Reaction catalyzed by pyrogallol transhydroxylase
6.4 Benzoyl-CoA Reductase
Our final example of a non-oxo-transferase is the tungsten-containing enzyme benzoyl-CoA reductase, which has been suggested to operate at the lowest redox potential of any biological reaction known to date [147]. It catalyzes the biological analogue (Fig. 26) of the Birch reduction, in which alkali metals (e.g., Na, Li) in solution in liquid ammonia are used to reduce aromatic rings [148]. Benzoyl Co-A dehydrogenase has tungsten coordinated by five sulfurs from two molybdopterin dithiolenes and a single cysteine residue. A sixth ligand is observed crystallographically but its identity is uncertain [147], although the crystal structure suggests that it is linear and diatomic, and enzyme activity increases upon addition of cyanide. The XAS is consistent with the sixth ligand as –CN, but FT-IR shows no evidence of such ligands. Tungsten does not appear to be in direct contact with the substrate, but rather it seems that the uncertain sixth ligand bridges to the aromatic ring of benzoyl-Co-A [147]. Cluka et al. [149] have employed computational chemistry to support water as the sixth ligand, but at the time of writing the identity of this pivotal ligand, and hence the exact nature of the catalytic mechanism of this intriguing enzyme, still seems uncertain.

Reaction catalyzed by benzoyl-CoA reductase
7 Molybdenum Versus Tungsten
Holm, Solomon, and coworkers [150] have compared the chemistry of molybdenum and tungsten and its relation to the catalytic function of the molybdenum and tungsten enzymes. There are a few examples where organisms have both molybdenum and tungsten versions of similar enzymes, such as the formylmethanofuran dehydrogenases [151, 152]. There are also examples of native Mo enzymes where molybdenum has been successfully substituted by tungsten, including sulfite oxidase, where the tungsten enzyme essentially lacked any catalytic activity and was not reduced by sulfite [153] and DMSO reductase which showed substantially increased activity relative to the native molybdenum enzyme [154, 155].
We now turn to the question of why nature chose molybdenum and tungsten and why some organisms show preference for one over the other [1]. We have already mentioned that the enzymes are thought to be very ancient [1, 156]. Molybdenum and tungsten share somewhat non-metallic chemistry in that both form oxyanions [MO4]2− that are stable at lower redox potentials. Oxygen atom transferase chemistry would have been more problematic in the anoxic primordial earth than in our current oxic world [157], and the capability of these metals to carry out such chemistry at low redox potentials may have been essential for early life. The hyperthermophilic archaea are thought to have diverged from the rest of life earlier than many other organisms [158]. These organisms contain only tungsten enzymes, and thus it has been suggested that the tungsten enzymes were the precursors of the molybdenum enzymes [159]. Moreover, tungsten may have been more available than molybdenum in the primordial environment; modern seawater has much higher levels of molybdate than tungstate (100 nM and 60 pM, respectively) [160], and in the primordial oceans, this is likely to have been reversed because the anoxic sulfidic waters would precipitate insoluble MoS2 while tungsten compounds would remain soluble [161].
As discussed by Holm, Solomon, and co-workers [150], relativistic effects are much more important for tungsten than for molybdenum, with relativistic contraction of the core orbitals enhancing bond strengths and destabilizing metal centered d-orbitals, thereby lowering redox potentials. Tungsten complexes are also more sensitive to oxygen [150], and the choice of molybdenum rather than tungsten may therefore depend more upon cellular redox conditions than other factors. Overall, tungsten is chemically better suited for catalysis of lower-potential redox reactions under anaerobic conditions, and at higher operating temperatures [150], which is what is observed in nature.
8 Concluding Remarks
Interest in the molybdenum and tungsten enzymes will continue to be stimulated by fields as diverse as drug metabolism [162] and potential biofuels [163]. Progress in the future will continue to be driven by structural and spectroscopic studies and will be facilitated by site-directed mutagenesis and discovering diversity with the revolution in metagenomic sequencing. Despite the rich diversity in metal coordination environment and enzyme function, there are some rules that can be observed. Low-molecular weight Mo(VI) and W(VI) compounds in the Cambridge Structural Database [35] predominantly show octahedral-type coordination environments. But the enzymes are different; nearly all of the enzyme active sites that conduct oxo-transferase chemistry show non-octahedral-type coordination of the metal. Significantly, for three out of four of the enzymes currently known that do not conduct oxo-transferase chemistry, namely, acetylene hydratase, pyrogallol transhydroxylase, and benzoyl-CoA reductase, the active sites do have octahedral-type coordination geometry. This suggests that the oxo-transferases provide examples of entatic states in which the active site is distorted by the protein to approach a transition state [46]. As we have discussed, conclusions related to catalytic mechanism combine a number of factors. The information from crystallography is of pivotal importance, but the possibility of photoreduction and the limitations of resolution are important, as is the information from spectroscopy of various forms. In all cases not only the metal site but also the protein and molybdopterin must be considered. Despite the tremendous progress that has been made since the early days, and as we have discussed, there remain important unanswered questions related to the catalytic mechanisms. There is still confusion about the roles of various groups, and those that are excellent nucleophiles are probably acting as such. Among the most intriguing of these is the role of selenium in some of the enzymes, which is sometimes present as a metal-bound terminal selenide and sometimes as selenocysteine. Selenium is the essential element with by far the lowest crustal abundance [164], and it is incorporated into biological molecules at tremendous metabolic cost [165]. Hence, for enzymes such as the selenium-containing formate dehydrogenases (Sect. 6.1), it seems most unlikely that evolution would have incorporated selenium without a very good reason that is closely related to its chemistry. As we have previously discussed [166], selenium is probably the best nucleophile available to living organisms, and it therefore seems unlikely that its roles in catalysis would not involve nucleophilic attack. A number of proposed mechanisms have postulated ligand dissociation from molybdenum or tungsten (e.g., cysteine sulfur in the Nap nitrate reductases; Sect. 5.3), and while dissociation of one molybdopterin dithiolene has been observed in inactive forms of DMSO reductase family members [167], no catalytic role for such dissociation has, to our knowledge, yet been proposed in any system. Many other open questions also remain, such as the role played by ring-open and ring-closed forms of molybdopterin (Sect. 3) and the nature of the all-important mystery ligand in benzoyl-CoA reductase (Sect. 6.4). A different level of interest relates to the exact reasons behind the incredible complexity of formylmethanofuran dehydrogenase (Sect. 6.1). Finally, the roles played by molybdenum and tungsten in the origins of life on earth and in early organisms have been a topic of recent discussion [1, 156]. In particular the active sites of molybdenum and tungsten enzymes are members of only a handful of biological reagents capable of genuine n = 2 redox chemistry [127, 168], and such reactions may have played pivotal roles in life’s origins [169].
References
Pushie MJ, Cotelesage JJH, George GN (2014) Metallomics 6:15–24
Lane T, Saito MA, George GN, Pickering IJ, Prince RC, Morel FFM (2005) Nature 435:42
Hiller CJ, Rettberg LA, Lee CC, Stiebritz MT, Hu Y (2019) Current understanding of the biosynthesis of the unique nitrogenase cofactor core. Struct Bond. https://springerlink.bibliotecabuap.elogim.com/chapter/10.1007/430_2018_29
Wenke BB, Spatzal T (2019) Looking at nitrogenase: insights from modern structural approaches. Struct Bond. https://springerlink.bibliotecabuap.elogim.com/chapter/10.1007/430_2018_28
Hille R, Halt J, Basu P (2014) Chem Rev 114:3963–4038
Jormakka M, Richardson D, Byrne B, Iwata S (2004) Structure 12:95–104
Bertero MG, Rothery RA, Palak M, Hou C, Lim D, Blasco F, Weiner JH, Strynadka NCJ (2003) Nat Struct Biol 10:681–687
Kloer DP, Hagel C, Heider J, Schulz GE (2006) Structure 14:1377–1388
Gisewhite DR, Yang J, Williams BR, Esmail A, Stein BW, Kirk ML, Burgmayer SJN (2018) J Am Chem Soc 140:12808–12818
Cotelesage JJH, Crawford AM, Prince RC, George GN (unpublished)
Bray RC, Swann JC (1972) Struct Bond 11:107–144
Ljundahl LG, Andreesen JR (1975) FEBS Lett 54:279–282
George GN, Prince RC, Mukund S, Adams MWW (1992) J Am Chem Soc 114:3521–3523
Olson JS, Ballou DP, Palmer G, Massey V (1974) J Biol Chem 249:4363–4382
Rothery RA, Stein B, Solomonson M, Kirk ML, Weiner JH (2012) Proc Natl Acad Sci U S A 109:14773–14778
Stetter KO (2006) Philos Trans R Soc B 361:1837–1843
Pushie MJ, George GN (2011) Coord Chem Rev 255(9–10):1055–1084
Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC (1997) Cell 91:973–983
Schrader N, Fischer K, Theis K, Mendel RR, Schwarz G, Kisker C (2003) Structure 11:1251–1263
Fischer K, Barbier G, Hecht H-J, Mendel RR, Campbell WH, Schwarz G (2005) Plant Cell 17:1167–1179
Kappler U, Bailey S (2005) J Biol Chem 280:24999–25007
McGrath AP, Laming EL, Casas Garcia GP, Kvansakul M, Guss JM, Trewhella J, Calmes B, Bernhardt PV, Hanson GR, Kappler U, Maher MJ (2015) Elife 4:e09066–e09066
Plitzko B, Havemeyer A, Kunze T, Clement B (2015) J Biol Chem 290:10126–10135
Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF (2000) Proc Natl Acad Sci U S A 97:10723–10728
Doonan CJ, Stockert A, Hille R, George GN (2005) J Am Chem Soc 127:4518–4522
Coelho C, Foti A, Hartmann T, Santos-Silva T, Leimkuhler S, Romao MJ (2015) Nat Chem Biol 11:779–783
Dobbek H, Gremer L, Kiefersauer R, Huber R, Meyer O (2002) Proc Natl Acad Sci U S A 99:15971–15976
Gnida M, Ferner R, Gremer L, Meyer O, Meyer-Klaucke W (2003) Biochemistry 42:222–230
Gladyshev VN, Khangulov SV, Stadtman TC (1994) Proc Natl Acad Sci U S A 91:232–236
Wagener N, Pierik AJ, Ibdah A, Hille R, Dobbek H (2009) Proc Natl Acad Sci U S A 106:1055–11060
George GN, Hilton J, Temple C, Prince RC, Rajagopalan RC (1999) J Am Chem Soc 121:1256–1266
Li HK, Temple C, Rajagopalan KV, Schindelin H (2000) J Am Chem Soc 122:7673–7680
Chan MK, Mukund S, Kletzin A, Adams MWW, Rees DC (1995) Science 267:1463–1469
Rappé AK, Goddard WA (1982) J Am Chem Soc 104:448–456
Groom CR, Bruno IJ, Lightfoot MP, Ward SC (2016) Acta Cryst B72:171–179
Qiu JA, Wilson HL, Pushie MJ, Kisker C, George GN, Rajagopalan KV (2010) Biochemistry 49:3989–4000
George GN, Garrett RM, Prince RC, Rajagopalan KV (1996) J Am Chem Soc 118:8588–8592
George GN, Garrett RM, Prince RC, Rajagopalan KV (2004) Inorg Chem 43:8456–8460
Johnson JL, Rajagopalan KV (1982) Proc Natl Acad Sci U S A 79:6856–6860
Rajagopalan KV (1991) In: Meister A (ed) Advances in enzymology and related areas of molecular biology. Wiley, New York, pp 215–290
Romão MJ, Archer M, Moura I, Moura JJG, LeGall J, Engh R, Schneider M, Hof P, Huber R (1995) Science 270:1170–1176
Garton SD, Hilton J, Hiroyuki O, Crouse BR, Rajagopalan KV, Johnson MK (1997) J Am Chem Soc 119:12906–12916
McNaughton RL, Helton ME, Rubie ND, Kirk ML (2000) Inorg Chem 39:4386–4387
Cotelesage JJH, Pushie MJ, Grochulski P, Pickering IJ, George GN (2012) J Inorg Biochem 115:127–137
Pushie MJ, Cotelesage JJH, Lyashenko G, Hille R, George GN (2013) Inorg Chem 52:2830–2837
Warelow TP, Pushie MJ, Cotelesage JJH, Santini JM, George GN (2017) Sci Rep 7:1757/1–1757/7
Rây P, Dutt NK (1943) J Indian Chem Soc 20:81–92
Rzepa HS, Cass ME (2007) Inorg Chem 46:8024–8031
Dobbek H, Gremer L, Meyer O, Huber R (1999) Proc Natl Acad Sci U S A 96:8884–8889
George GN, Pickering IJ, Kisker C (1999) Inorg Chem 38:2539–2540
George GN, Pickering IJ, Pushie MJ, Nienaber K, Hackett MJ, Ascone I, Hedman B, Hodgson KO, Aitken JB, Levina A, Glover C, Lay PA (2012) J Synchrotron Radiat 19:875–886
Nienaber KH, Pushie MJ, Cotelesage JJH, Pickering IJ, George GN (2018) J Phys Chem Lett 9:540–544
George GN (2016) X-ray absorption spectroscopy of molybdenum and tungsten enzymes. In: Kirk ML, Hille R, Schulzke C (eds) Molybdenum and tungsten enzymes: spectroscopic and theoretical investigations. Royal Society of Chemistry, Series on Meta, pp 121–167, ISBN 978-1-78262-878-1
Harris HH, George GN, Rajagopalan KV (2006) Inorg Chem 45:493–495
Shih VE, Abroms IF, Johnson JL, Carney M, Mandell R, Robb RM, Cloherty JP, Rajagopalan KV (1977) N Engl J Med 297:1022–1028
Johnson JL, Coyne KE, Garrett RM, Zabot M-T, Dorche C, Kisker C, Rajagopalan KV (2002) Hum Mutat 20:74
Karakas E, Wilson HL, Graf TN, Xiang S, Jaramillo-Busquets S, Rajagopalan KV, Kisker C (2005) J Biol Chem 280:33506–33515
Peariso K, McNaughton RL, Kirk ML (2002) J Am Chem Soc 124:9006–9007
Izumi Y, Glaser T, Rose K, McMaster J, Basu P, Enemark JH, Hedman B, Hodgson KO, Solomon EI (1999) J Am Chem Soc 121:10035–10046
Fischer K, Barbier G, Hecht H-J, Mendel RR, Campbell WH, Schwarz G (2005) Plant Cell 17:1167–1179
George GN, Mertens JA, Campbell WA (1999) J Am Chem Soc 121:9730–9731
Cambell WH, Kinghorn KR (1990) Trends Biochem Sci 15:315–319
Qiu JA, Wilson HL, Rajagopalan KV (2012) Biochemistry 51:1134–1147
Havemeyer A, Bittner F, Wollers S, Mendel R, Kunze T, Clement B (2006) J Biol Chem 281:34796–34802
Schneider J, Girreser U, Havemeyer A, Bittner F, Clement B (2018) Chem Res Toxicol 31:447–453
Llamas A, Chamizo-Ampudia A, Tejada-Jimenez M, Galvan A, Fernandez E (2017) Biofactors 43:486–494
Rajapakshe A, Astashkin AV, Klein EL, Reichmann D, Mendel RR, Bittner F, Enemark JH (2011) Biochemistry 50:8813–8822
Giles LJ, Ruppelt C, Yang J, Mendel RR, Bittner F, Kirk ML (2014) Inorg Chem 53:9460–9462
Yang J, Giles LJ, Ruppelt C, Mendel RR, Bittner F, Kirk ML (2015) J Am Chem Soc 137:5276–5279
Kubitza C, Ginsel C, Bittner F, Havemeyer A, Clement B, Scheidiga AJ (2018) Acta Cryst F74:337–344
Pacheco A, Hazzard JT, Tollin G, Enemark JH (1999) J Biol Inorg Chem 4:390–401
Pushie MJ, George GN (2010) J Phys Chem B 114:3266–3275
Utesch T, Mroginski MA (2010) J Phys Chem Lett 1:2159–2164
Atashkin AV, Rajapakshe A, Cornelison MJ, Johnson-Winters K, Enemark JH (2012) J Phys Chem B 116:1942–1950
Feng C, Wilson HL, Hurley JK, Hazzard JT, Tollin G, Rajagopalan KV, Enemark JH (2003) Biochemistry 42:12235–12242
Emesh S, Rapson TD, Rajapakshe SA, Kappler U, Bernhardt PV, Tollin G, Enemark JH (2009) Biochemistry 48:2156–2163
McGrath AP, Laming EL, Casus Garcia GPM, Guss JM, Trewella J, Calmes B, Bernhardt PV, Hanson GR, Kappler U, Maher MJ (2015) eLife 4:e09066/1–26
Hsiao J-C, McGrath AP, Kielmann L, Kalimuthu P, Darain F, Bernhardt PV, Harmer J, Lee M, Meyers K, Maher MJ, Kappler U (2018) BBA-Bioenergetics 1859:19–27
Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF (2000) Proc Natl Acad Sci U S A 97:10723–10728
Burt HM, Dutt YC (1989) J Cryst Growth 94:15–22
Pascual E, Addadi L, Andrés M, Sivera F (2015) Nat Rev Rheumatol 11:725–730
Williams JW, Bray RC (1981) Biochem J 195:753–760
Hawkes TR, George GN, Bray RC (1984) Biochem J 218:961–968
Dent CE, Philpot GR (1954) Lancet 266:182–185
Ichida K, Amaya Y, Nishino T, Hosoya T, Sakai O (1997) J Clin Invest 99:2391–2397
Bray RC (1975) In: Boyer PD (ed) The enzymes. Academic Press, New York, pp 299–419
Enroth C, Eger BT, Okamoto K, Nishino T, Nishino T, Pai EF (2000) Proc Natl Acad Sci U S A 97:10723–10728
Hille R (1996) Chem Rev 96:2757–2816
Kitamura S, Sugihara K, Ohta S (2006) Drug Metab Pharmacokinet 21:83–98
Self WT, Stadtman TC (2000) Proc Natl Acad Sci U S A 97:7208–7213
Wagener N, Pierik AJ, Ibdah A, Hille R, Dobbek H (2009) Proc Natl Acad Sci U S A 106:1055–11060
Cleere WF, Coughlan MP (1974) Biochem J 143:331–340
Kim JH, Ryan MG, Knaut H, Hille R (1996) J Biol Chem 271:6771–6780
Dobbek H, Gremer L, Kiefersauer R, Huber R, Meyer O (2002) Proc Natl Acad Sci U S A 99:15971–15976
Gnida M, Ferner R, Gremer L, Meyer O, Meyer-Klaucke W (2003) Biochemistry 42:222–230
Zhang B, Hemann CF, Hille R (2010) J Biol Chem 285:12571–12578
Gourlay C, Nielsen DJ, White JM, Knottenbelt SZ, Kirk ML, Young CG (2006) J Am Chem Soc 128:2164–2165
Zhang L, Johnson Nelson K, Rajagopalan KV, George GN (2008) Inorg Chem 47:1074–1078
Creevey NL, McEwan AG, Hanson GR, Bernhardt PV (2008) Biochemistry 47:3770–3776
Temple CA, George GN, Hilton J, George MJ, Prince RC, Barber MJ, Rajagopalan KV (2000) Biochemistry 39:4046–4052
George GN, Hilton J, Rajagopalan KV (1996) J Am Chem Soc 118:1113–1117
Schindelin H, Kisker C, Hilton J, Rajagppalan KV, Rees DC (1996) Science 272:1615–1621
Schneider F, Löwe J, Huber R, Schindelin H, Kisker C, Knäblein J (1996) J Mol Biol 263:53–69
McAlpine AS, McEwan AG, Shaw AL, Bailey S (1997) J Biol Inorg Chem 2:690–701
McAlpine AS, McEwan AG, Bailey SJ (1998) J Mol Biol 275:613–623
Baugh PE, Garner CD, Charnock JM, Collison D, Davies ES, McAlpine AS, Bailey S, Lane I, Hanson GR, McEwan AG (1997) J Biol Inorg Chem 2:634–643
George GN, Hilton J, Temple C, Prince RC, Rajagopalan KV (1999) J Am Chem Soc 121:1256–1266
Li HK, Temple C, Rajagopalan KV, Schindelin H (2000) J Am Chem Soc 122:7673–7680
Bray RC, Adams B, Smith AT, Bennett B, Bailey S (2000) Biochemistry 39:11258–11269
George GN, Nelson KJ, Harris HH, Doonan CJ, Rajagopalan KV (2007) Inorg Chem 46:3097–3104
Sparacino-Watkinsa C, Stolz JF, Basua P (2014) Chem Soc Rev 43:676–706
Coelho C, Romão MJ (2015) Protein Sci 24:1901–1911
Dias JM, Than ME, Humm A, Huber R, Bourenkov GP, Bartunik HD, Bursakov S, Calvete J, Calderia J, Carneiro C, Moura JJG, Moura I, Romão MJ (1999) Structure 7:65–79
Cerqueira NM, Pakhira B, Sarkar S (2015) J Biol Inorg Chem 30:323–335
Rendon J, Biaso F, Ceccaldi P, Toci R, Seduk F, Magalon A, Guigliarelli B, Grimaldi S (2017) Inorg Chem 56:4422–4434
Ceccaldi P, Rendon J, Léger C, Toci R, Guigliarelli B, Magalon A, Grimaldi S, Fourmond V (2015) Biochim Biophys Acta 1847:1055–1063
George GN, Bray RC, Morpeth FF, Boxer DH (1985) Biochem J 227:925–931
Maher MJ, Santini J, Pickering IJ, Prince RC, Macy JM, George GN (2004) Inorg Chem 43:402–404
Kloer DP, Hagel C, Heider J, Schulz GE (2006) Structure 14:1377–1388
Heider J, Szaleniec M, Sünwoldt K, Boll M (2016) J Mol Microbiol Biotechnol 26:45–62
Szaleniec M, Hagel C, Menke M, Nowak P, Witko M, Heider J (2007) Biochemistry 46:7637–7646
Szaleniec M, Borowski T, Schühle K, Nowak P, Witko M, Heider J (2010) J Am Chem Soc 132:6014–6024
Szaleniec M, Dudzik A, Kozik B, Borowski T, Heider J, Witko M (2014) J Inorg Biochem 139:9–20
Ellis PJ, Conrads T, Hille R, Kuhn P (2001) Structure 9:125–132
Warelow TP, Oke M, Schoepp-Cothenet B, Dahl JU, Bruselat N, Sivalingam GN, Leimkühler S, Thalassinos K, Kappler U, Naismith JH, Santini JM (2013) PLoS One 8:e72535
Conrads T, Hemann C, George GN, Pickering IJ, Prince RC, Hille R (2002) J Am Chem Soc 124:11276–11277
Hoke KR, Cobb N, Armstrong FA, Hille R (2004) Biochemistry 43:1667–1674
Mukhopadhyay R, Rosen BP (2002) Environ Health Perspect 110:745–748
Radabaugh TR, Aposhian HV (2000) Chem Res Toxicol 13:26–30
Glasser NR, Oyala PH, Osborne TH, Santini JM, Newman DK (2018) Proc Natl Acad Sci U S A 115:E8614–E8623
Grimaldi S, Schoepp-Cothenet B, Ceccaldi P, Guigliarelli B, Magalon A (2013) Biochim Biophys Acta Bioenerg 1827:1048–1085
Boyington JC, Gladyshev VN, Khangulov SV, Stadtman TC, Sun PD (1997) Science 275:1305–1308
Raaijmakers H, Macieira S, Dias JM, Teixeira S, Bursakov S, Huber R, Moura JJ, Moura I, Romão MJ (2002) Structure 10:1261–1273
Raaijmakers H, Romão MJ (2006) J Biol Inorg Chem 11:849–854
Mota CS, Rivas MG, Brondino CD, Moura I, Moura JJ, Gonzalez PJ, Cerqueira NM (2011) J Biol Inorg Chem 16:1255–1268
Robinson WE, Bassegoda A, Reisner E, Hirst J (2017) J Am Chem Soc 139:9927–9936
George GN, Colangelo CM, Dong J, Scott RA, Khangulov SV, Gladyshev VN, Stadtman TC (1998) J Am Chem Soc 120:1267–1273
George GN, Costa C, Moura JJG, Moura I (1999) J Am Chem Soc 121:2625–2626
Khangulov SV, Gladyshev VN, Dismukes CG, Stadtman TC (1998) Biochemistry 37:3518–3528
Berg IA, Kockelkorn D, Ramos-Vera WH, Say RF, Zarzycki J, Hügler M, Alber BE, Fuchs G (2010) Nat Rev Microbiol 8:447–459
Pancost RD, Sinninghe Damsté JS, de Lint S, van der Maarel MJEC, Gottschal JC (2000) Appl Environ Microbiol 66:1126–1132
Schwörer B, Thauer RK (1991) Arch Microbiol 155:459–465
Ponomarev DA, Shevchenko SM (2007) J Chem Educ 84:1725–1726
Seiffert GB, Ullmann GM, Messerschmidt A, Schink B, Kroneck PMH, Einsle O (2007) Proc Natl Acad Sci U S A 104:3073–3077
Liao R-Z, Yu J-G, Himo F (2010) Proc Natl Acad Sci U S A 107:22523–22527
Messerschmidt A, Niessen H, Abt D, Einsle O, Schink B, Kroneck PMH (2004) Proc Natl Acad Sci U S A 101:11571–11576
Weinert T, Huwiler SG, Kung JW, Weidenweber S, Hellwig P, Stärk HJ, Biskup T, Weber S, Cotelesage JJ, George GN, Ermler U, Boll M (2015) Nat Chem Biol:586–591
Boll M, Fuchs G (1995) Eur J Biochem:921–933
Culka M, Huwiler SG, Boll M, Ullmann GM (2017) J Am Chem Soc 139:14488–14500
Holm RH, Solomon EI, Majumdar A, Tenderholt A (2011) Coord Chem Rev 255:993–1015
Wagner T, Ermler U, Shima S (2016) Science 354:114–117
Niks D, Hille R (2018) Protein Sci. https://doi.org/10.1002/pro.3498
Johnson JL, Rajagopalan KV (1976) J Biol Chem 251:5505–5511
Stewart LJ, Bailey S, Bennett B, Charnock JM, Garner CD, McAlpine AS (2000) J Mol Biol 299:593–600
Garner CD, Stewart LJ (2002) Met Ions Biol Syst 39:699–726
Schoepp-Cothenet B, van Lis R, Philippot P, Magalon A, Russell MJ, Nitschke W (2012) Sci Rep 2:263/1–263/5
Williams RJP, Fraústo da Silva JJR (2002) Biochem Biophys Res Commun 292:293–299
Stetter KO (2006) Philos Trans R Soc B 361:1837–1843
Kletzin A, Adams MWW (1996) FEMS Microbiol Rev 18:5–63
Bruland KW, Lohan MC (2006) In: Elderfield H (ed) The oceans and marine geochemistry. Elsevier, North Holland, pp 23–47
Licht S (1988) J Electrochem Soc 135:2971–2975
Romão MJ, Coelho C, Santos-Silva T, Foti A, Terao M, Garattini E, Leimkühler S (2017) Curr Opin Chem Biol 37:39–47
Bassegoda A, Madden C, Wakerley DW, Reisner E, Hirst J (2014) J Am Chem Soc 136:15473–15476
Wedepohl KH (1995) Geochim Cosmochim Acta 59:1217–1232
Reich HJ, Hondal RJ (2016) ACS Chem Biol 11:821–841
Dolgova NV, Nehzati S, Choudhury S, Regnier N, Crawford AM, Ponomarenko O, George GN, Pickering IJ (2018) Biochim Biophys Acta 1862:2383–2392
Zhang L, Johnson Nelson K, Rajagopalan KV, George GN (2008) Inorg Chem 47:1074–1078
Nitschke W, Russell MJ (2009) J Mol Evol 69:484–496
Nitschke W, Russell MJ (2011) Bioessays 34:106–109
Acknowledgments
Research in the authors’ laboratory is funded by the Natural Sciences and Engineering Research Council of Canada, the Saskatchewan Health Research Foundation (SHRF), the University of Saskatchewan, the Canada Foundation for Innovation, Chevron Energy Research Co., and a Canada Research Chair award (to G.N.G). R.C.P. acknowledges support from the Diane Gunson benevolence fund.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Crawford, A.M., Cotelesage, J.J.H., Prince, R.C., George, G.N. (2018). The Catalytic Mechanisms of the Molybdenum and Tungsten Enzymes. In: Ribbe, M. (eds) Metallocofactors that Activate Small Molecules. Structure and Bonding, vol 179. Springer, Cham. https://doi.org/10.1007/430_2018_30
Download citation
DOI: https://doi.org/10.1007/430_2018_30
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-25896-2
Online ISBN: 978-3-030-25897-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)