
Abstract
This review discusses the current mechanistic understanding of a group of mononuclear non-heme iron-dependent enzymes that catalyze four-electron oxidation of their organic substrates without the use of any cofactors or cosubstrates. One set of enzymes acts on α-ketoacid-containing substrates, coupling decarboxylation to oxygen activation. This group includes 4-hydroxyphenylpyruvate dioxygenase, 4-hydroxymandelate synthase, and CloR involved in clorobiocin biosynthesis. A second set of enzymes acts on substrates containing a thiol group that coordinates to the iron. This group is comprised of isopenicillin N synthase, thiol dioxygenases, and enzymes involved in the biosynthesis of ergothioneine and ovothiol. The final group of enzymes includes HEPD and MPnS that both carry out the oxidative cleavage of the carbon–carbon bond of 2-hydroxyethylphosphonate but generate different products. Commonalities amongst many of these enzymes are discussed and include the initial substrate oxidation by a ferric-superoxo-intermediate and a second oxidation by a ferryl species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mononuclear non-heme iron-dependent (NHI) enzymes catalyze an array of chemical transformations including hydroxylation, chlorination, and epimerization as well as both cyclization and ring cleavage of organic substrates [1, 2]. These transformations occur in a wide variety of biological processes in primary metabolism such as amino acid biosynthesis and degradation, and in secondary metabolism such as the biosynthesis of antibiotics and herbicides. NHI enzymes can generally be classified along at least two mechanistic axes. The first distinction is between those that incorporate atoms derived from O2 into their enzymatic products (oxygenases) and those that activate and transfer electrons to O2 without incorporating atoms derived from O2 into their enzymatic products (oxidases). A second defining feature within this class of enzymes is whether the protein requires a cosubstrate or an organic cofactor to facilitate activation of O2. Among the most common cosubstrates is α-ketoglutarate, which in the presence of O2 and substrate undergoes oxidative decarboxylation to generate an Fe(IV)=O species. This intermediate has been spectroscopically characterized in a number of non-heme iron enzymes, including those that effect hydroxylation [3–6], chlorination [7], and epimerization [8]. In these instances, catalysis consumes one molecule of cosubstrate per molecule of product generated. By contrast, cofactors such as tetrahydrobiopterin in tyrosine hydroxylase function as two-electron reservoirs to facilitate oxygen activation [9]. After turnover, the cofactor must be reduced by an exogenous source of electrons to restore the resting state of the enzyme. These mechanistic strategies limit these enzymes to two-electron oxidations of their substrates for each O2 consumed. Some examples of consecutive two-electron oxidations utilizing multiple O2 molecules have also been reported [10].
A remarkable second subset of NHI enzymes activates O2 without requiring any additional cosubstrates or cofactors. These enzymes, including both oxygenases and oxidases, extract all four electrons from their organic substrates for complete reduction of oxygen. This review will focus on three classes of such enzymes that all utilize a ferric-superoxo intermediate to initiate catalysis and that may involve a subsequent ferryl intermediate for a second step of oxidation [11]. The first group encompasses enzymes that use substrates containing an α-ketoacid moiety as a “high-energy” substrate capable of generating an Fe(IV)=O species. The second group comprises enzymes that oxidize substrates that bind to the active site Fe via a thiolate and facilitate oxygen activation via this highly covalent Fe–S bond. The final class is a pair of unusual enzymes that catalyze carbon–carbon bond scission of electron-deficient phosphonates. This review will not cover several other classes of enzymes that carry out four-electron oxidations of their substrates such as the ring-cleaving extradiol dioxygenases that are discussed in a different review in this issue, dinuclear enzymes like myo-inositol oxygenase [12], NHI dioxygenases such as Dke1 that cleave diketones [13–15], and members of the cofactor-independent oxidases/oxygenases [16].
Spring-loaded substrates: HPPD, HMS, and CloR
Thus far, three NHI enzymes have been discovered that use a substrate containing an α-ketoacid moiety, resulting in substrate decarboxylation and subsequent elaboration. The best studied of these enzymes is 4-hydroxyphenylpyruvate dioxygenase (HPPD) [17]. HPPD catalyzes the second step of tyrosine catabolism by transforming 4-hydroxyphenylpyruvate (HPP) to homogentisate in an O2- and Fe(II)-dependent manner (Fig. 1a). Homogentisate feeds into a number of important pathways in plants, including biosynthesis of plastoquinones and tocopherols [18]. Consequently, both natural inhibitors have evolved and synthetic herbicides have been developed to target HPPD [19]. In humans, abrogation of HPPD activity leads to tyrosinemia, with downstream effects ranging from impaired cognitive abilities to death [20].

Overview of HPPD, HMS, and CloR. a The reactions catalyzed by HPPD, HMS, and CloR. The stereochemistry of the initial CloR hydroxylation has not been defined. b Overlays of the crystal structures of Fe-HPPD (carbons in green, PDB ID: 1CJX) aligned with Co-HMS bound to 4-hydroxymandelate (carbons in aqua, PDB ID: 2R5 V). Given the sequence divergence of CloR from structurally characterized proteins, CloR did not yield high-confidence homology models from I-TASSER [125] and is, therefore, omitted. c A proposed mechanism for catalysis by HPPD, HMS, and the first oxidative decarboxylation by CloR. For details and discussion, see the text. d A mechanistic proposal for the second oxidative decarboxylation by CloR. A concerted β-scission mechanism is shown in red. ET electron transfer
An enzyme with distant homology to HPPD is 4-hydroxymandelate synthase (HMS). HMS was discovered during investigations into the biosynthesis of chloroeremomycin [21] and related glycopeptide antibiotics, including vancomycin. Feeding studies had shown that the (R)-4-hydroxyphenylglycine residues in these glycopeptides were derived from l-tyrosine. Because the gene clusters encoded epimerase domains, it was suspected that l-tyrosine was converted to (S)-4-hydroxphenylglycine, followed by epimerization and incorporation into the antibiotics. A protein with distant homology to HPPD was found in the chloroeremomycin gene cluster (34% identity), hinting that this protein might convert HPP not to homogentisate but to 4-hydroxymandelate, which could then be oxidized and transaminated to yield 4-hydroxyphenylglycine (Fig. 1a) [21]. Initial characterization of HMS demonstrated that it was an authentic dioxygenase, with both incipient oxygen atoms in the 4-hydroxymandelate product originating from 18O2 [21].
A stark difference between HPPD and HMS is the outcome of catalysis. Similar to many NHI hydroxylases, HMS hydroxylates the benzylic position of HPP, whereas HPPD hydroxylates the aromatic ring and triggers an unusual alkyl migration reminiscent of the 1,2-NIH shift (discussed below) [22]. These distinct reactivities with the same substrate spurred efforts to convert HPPD into a functional HMS and vice versa. These endeavors were empowered by the crystal structures of HPPD from Pseudomonas fluorescens [23], Streptomyces avermitilis [24], and Arabidopsis thaliana [25, 26]. Initial attempts based on rational design [27] and site-saturation mutagenesis [28] endowed HPPD with only weak activity HMS activity; no combination of active site mutants ever conferred HPPD-like activity to HMS, implying that ring hydroxylation and the ensuing alkyl shift require a more precisely tuned active site. A possible explanation for these difficulties was uncovered with the elucidation of the HMS crystal structure [29]. Comparing the two enzymes, the active site residues aligned by primary sequence were offset by ~1–1.5 Å (Fig. 1b), suggesting that simply converting residues from those in HPPD to those in HMS would be insufficient for properly remodeling the active site cavity. Yet another difference was that the HMS active site was much smaller than the HPPD active site (30 vs 63 Å3, respectively), indicating that more flexibility is required for the alkyl migration in HPPD [29].
After the putative Fe(IV)=O is generated in HMS by chemistry similar to that in α-ketoglutarate-dependent enzymes, a standard hydrogen atom abstraction-rebound mechanism is believed to occur (Fig. 1c) [30]. Experimental attempts to elucidate the HPPD mechanism after generation of the putative Fe(IV)=O species have relied on indirect methods, including mutants of HPPD that uncouple oxidative decarboxylation from native product formation to instead make both the native product and shunt products. These mutated proteins were then reacted with deuterated substrates and the ratio of shunt to native products was determined to infer kinetic isotope effects (KIEs) on isotopically sensitive branching steps [31, 32]. These experiments led to the conclusion that a homolytic biradical mechanism was responsible for the alkyl migration, consistent with early DFT calculations [33]. Subsequent structure elucidation enabled more detailed quantum mechanics/molecular mechanics (QM/MM) calculations, which led to a revised hypothesis, in which the migration takes place via initial attack at C1 of the aromatic ring by the ferryl species (more accurately described as an Fe(III)-oxyl radical in the transition state [34]), followed by electron transfer to oxidize the ring to a resonance stabilized cationic species, and finally a heterolytic alkyl shift [35] (Fig. 1c). The latter is not unlike alkyl migration during a pinacol rearrangement. Alternative mechanisms (epoxidation of the aryl ring or ferryl attack at C2 or C6; not shown in Fig. 1c) were all more energetically demanding [35]. The revised proposal also recapitulated the experimentally observed large, inverse KIE when using substrate that was perdeuterated on the hydroxyphenyl ring [35].
The final member of this class of enzymes is CloR, which catalyzes two sequential oxidative decarboxylations in converting 3-dimethylallyl-4-hydroxyphenylpyruvate (3DMA-HPP) to 3-dimethylallyl-4-hydroxybenzoate (3DMA-HB) in the biosynthesis of the aminocoumarin antibiotic clorobiocin. The first decarboxylation converts 3DMA-HPP to 3-dimethylallyl-4-hydroxymandelate (3DMA-HMA) and the second produces 3DMA-HB (Fig. 1a) [36]. As 3DMA-HMA is an isolable intermediate, the enzyme is likely distributive. 18O2 labeling experiments demonstrated incorporation of two atoms from O2 in 3DMA-HMA and an additional atom derived from O2 in 3DMA-HB, confirming that CloR is an oxygenase. Though the first reaction is nearly identical to that catalyzed by HMS, CloR exhibits no primary sequence similarity to HMS. Despite this difference, it is quite likely that CloR follows an HMS-like mechanism to effect its first oxidation. The subsequent oxidative decarboxylation of 3DMA-HMA is intriguing because it features an α-hydroxyacid rather than the canonical α-ketoacid moiety. On the basis of studies with Fe(II)-HMA model complexes (R=H, Fig. 1a) [37–39], the mechanism shown in Fig. 1d has been proposed for the second oxidation to produce 3DMA-HB. In this proposal, a ferric-superoxo species abstracts a hydrogen atom to form an oxyl-substrate radical. Decarboxylation would generate a ketyl radical that can transfer an electron to the iron center to yield an aldehyde adjacent to a ferrous hydroperoxo (decarboxylation and electron transfer could also happen in a concerted process via β-scission). This latter species could effect a two-electron oxidation of the aldehyde to the acid, thereby incorporating one oxygen derived from O2 into the product (possibly via an Fe(IV)=O species attacking a hydrated aldehyde). Additional studies will be necessary to determine whether this mechanistic proposal based on model complexes is operative during enzymatic catalysis.
Oxidative ring closure: isopenicillin N synthase (IPNS)
It is likely not an overstatement to say that the discovery of penicillin fundamentally altered the course of medicine and human society in the twentieth century. Penicillins are a family of compounds that incorporate a β-lactam moiety, which covalently inhibits proteins critical to bacterial cell wall biosynthesis [40]. Early investigations into the biosynthesis of penicillin led to the formulation of a hypothesis in which the linear peptide δ-(l-α-aminoadipyl)-l-cysteinyl-d-valine (ACV) was desaturatively cyclized to install the two rings found in penicillins (Fig. 2a) [41]. Unique among the enzymes discussed in this review, the protein responsible for this transformation, isopenicillin N synthase (IPNS), is an oxidase rather than an oxygenase. Early experiments demonstrated that the activity of IPNS was maximized upon inclusion of Fe2+ and ascorbate [42]. Isolation of the enzyme from the native producer Cephalosporium acremonium [42, 43] enabled the determination of its N-terminal sequence and ultimately the identification of the gene encoding the protein [44]. Analysis of the purified protein by Mössbauer, EPR, and UV–Vis spectroscopy [45, 46] in tandem with EXAFS [47] demonstrated that under anaerobic conditions, ACV binds to the active site Fe(II) via its thiolate and renders the Fe(II) coordination environment more covalent. The O2-mimic NO was also shown to coordinate to the active site metal by Mössbauer spectroscopy, both in the presence or absence of substrate [46]. However, the overall architecture of the protein and the identities of any reactive intermediates during the catalytic cycle remained unknown.

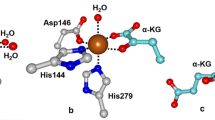
Overview of the chemistry effected by IPNS. a The native substrate of IPNS catalysis (left) and a substrate analog that does not undergo a second cyclization (right). b The crystal structure of the ternary complex of Fe(II)-IPNS-ACV-NO (PDB ID: 1BLZ). The distance between the oxygen atom of NO to the β carbon of cysteine in ACV is 3.3 Å (black dashes). c The structure of the product of the in crystallo reaction of Fe(II)-IPNS with ACmC reveals formation of the thiazolidine ring and sulfoxidation of the methylcysteine moiety (PDB ID: 1QJF). d A proposed mechanism for catalysis by IPNS. Instead of a formal ET in the second step after substrate binding, the two structures could also be considered resonance forms. For details, see the text
An elegant series of crystallographic studies provided key insights. The crystal structure of the Mn(II)-substituted protein showed that the active site was buried within a hydrophobic cavity of a jelly-roll motif, and that the active site metal was coordinated by four residues: 2 His and 1 Asp on one face of a pseudooctrahedron as well as a Gln [48]. The structure of the Fe(II)-containing enzyme in complex with ACV was subsequently solved under anaerobic conditions [49, 50]. The metal in the IPNS-Fe(II)-ACV cocrystal structure was ligated by the two His and the Asp observed in the Mn(II) cocrystal, and the ACV substrate displaced the Gln to bind to the Fe(II) through its thiolate, thereby verifying previous spectroscopic assignments [45–47]. A ternary complex of IPNS-Fe(II)-ACV-NO was also crystallized. NO bound cis to the thiolate with the oxygen atom in close proximity to the cysteinyl-β-carbon (3.3 Å), in prime position for the distal oxygen of a ferric-superoxo species to abstract a hydrogen atom and lead to β-lactam formation (black dashes in Fig. 2b). This positioning was congruent with the observation of a KIE on k cat/K m with ACV containing [3,3-2H2]-Cys but the absence of a KIE on k cat/K m with ACV containing [3-2H1]-Val, suggesting that the β-lactam ring forms first and is the first irreversible step involving the ACV substrate [51]. Formation of the second ring could then be initiated by the potent Fe(IV)-oxo intermediate formed in the first cyclization (Fig. 2d) performing an energetically more difficult hydrogen atom abstraction from C3 of valine. The IPNS-Fe(II)-ACV-NO complex has also been investigated extensively by spectroscopic and DFT studies, resulting in a detailed description of the bonding interactions [52]. These studies revealed that thiolate coordination is critical for rendering O2 binding more energetically favorable. The order of events was corroborated by an in crystallo reaction of IPNS-Fe(II) with O2 and a substrate analog [53]. As crystals of IPNS-Fe(II)-ACV pressurized with O2 had yielded only cocrystals of the substrate (ACV) or the product (isopenicillin N) but not any trapped intermediates, Baldwin and coworkers turned to a substrate analog (δ-(l-α-aminoadipyl)-l-cysteinyl-l-S-methyl cysteine, ACmC; Fig. 2a). Using this compound, the authors observed a monocyclic product with a sulfoxide coordinated to the active site Fe, consistent with, but not proving, the hypothesis that the second ring is generated by a ferryl intermediate (Fig. 2).
Both DFT [54] and QM/MM calculations [55] are in accord with a ferric-superoxo species initiating formation of the β-lactam ring and the more powerful iron(IV)-oxo species catalyzing formation of the thiazolidine ring via abstraction of a hydrogen atom from an unactivated C–H bond, with discrepancies mainly about the origin of protons at each step. Very recently, both of these reactive intermediates were detected by rapid-freeze-quench Mössbauer spectroscopy and UV–Vis absorption spectroscopy [56]. The ferric-superoxo species was barely detectable when IPNS was reacted with O2 and ACV, but it accumulated to higher levels (14% total 57Fe) in the reaction with ACV containing [3-2H2]-Cys, indicating that it decays by abstracting a H-atom from C3 of Cys. The iron(IV)-oxo species accumulated when IPNS was reacted with ACV and O2; when IPNS was instead reacted with ACV containing [2H8]-Val, the ferryl accumulated to a greater extent and decayed at a much slower rate (2H KIE > 30), revealing that the ferryl cleaves the C–H bond in Val that initiates thiazolidine formation. IPNS is only the third known instance in which a mononuclear ferric-superoxo intermediate has been trapped and spectroscopically characterized [56–58].
Thiol dioxygenases: CDO, ADO, and MDO
The thiol dioxygenases (TDOs) are a class of enzymes that oxidizes the sulfhydryl group of various molecules to sulfinates [59]. Activities within this family include the oxidation of cysteamine (2-aminoethanethiol) to hypotaurine by 2-aminoethanethiol dioxygenase (ADO) in mammalian livers [60, 61] and the oxidation of 3-mercaptopropionate to 3-sulfinopropionate by the bacterial enzyme 3-mercaptopropionate dioxygenase (MDO) (Fig. 3a) [62]. Another enzyme in this family is cysteine dioxygenase (CDO), which oxidizes cysteine to cysteine sulfinic acid (CSA) as the first step in cysteine catabolism en route to taurine synthesis [63]. As CDO was the first discovered TDO and dysfunction of CDO has been associated with both neurological [64] and autoimmune diseases [65, 66], CDO has been the best studied of the thiol dioxygenases. Consequently, this section of the review will focus primarily on CDO.

Overview of the current knowledge of the thiol dioxygenases. a Reactions catalyzed by ADO, MDO, and CDO. b The crystal structure of a eukaryotic Ni-CDO (carbons in green, PDB ID: 2ATF) overlaid with a prokaryotic Fe-CDO (carbons in aqua, PDB ID: 3EQE) demonstrates the preservation of the 3-His coordination sphere in each active site. The Cys-Tyr cross-link is observed only in the eukaryotic version; in the prokaryotic version, the position of the Cys is occupied by a Gly and the Cys is instead one residue removed and unable to form a cross-link. c Mechanistic proposal for catalysis by CDO. The majority of mechanistic research on the TDOs has focused on CDO, but a similar mechanism may be operative in ADO and MDO catalysis. The attack on the sulfur by the ferric-superoxo may be enhanced by an Fe(II)-thiyl radical cation resonance form in which the sulfur has donated an electron to the metal center. d The persulfenate intermediate that was observed crystallographically [80]
Initial work with CDO showed that both oxygen atoms in CSA derived from O2 and that activity was stimulated by Fe(II) [63, 67]. In vitro studies with rat CDO demonstrated that only these two components (Fe(II) and O2) were required for activity [68]. Despite these early advances, substantial difficulties in isolating the enzyme meant that only recently have mammalian CDOs become available in sufficient quantities for thorough in vitro investigations. The crystal structure of mouse CDO (100% identical sequence to rat CDO) illustrated that, rather unexpectedly, the active site metal was coordinated by a 3-His facial triad [69] (ADO and MDO also contain the iron-binding histidines in a multiple sequence alignment) [70] (Fig. 3b). The other unusual structural feature of this enzyme was a cross-link between a cysteine and a proximal tyrosine in the active site of mouse CDO [69], similar to enzymes such as galactose oxidase [71]. The cysteine residue forming this cross-link is not conserved among prokaryotic CDOs nor in ADO or MDO homologs (Fig. 3b). The cross-link is generated slowly during the course of hundreds of turnovers by the eukaryotic version of the protein [72], and thus recombinantly expressed CDO is a mixture of protein with and without the cross-link. The slow maturation of the enzymes means that isolation of a version entirely without the cross-link might be feasible under anaerobic expression/purification conditions. By comparing enzyme mixtures with ~50% cross-linked (as isolated) protein to those that had been fully matured (~100% cross-linked), it was shown that the mature eukaryotic form exhibits a catalytic efficiency 10- to 20-fold higher than that of the version without the cross-link; the prokaryotic version (naturally without the cross-link) has a catalytic efficiency that rivals that of the mature eukaryotic form [72]. These observations call into question how the cross-link enhances catalysis. Replacement of the active site cysteine with an alanine generated a eukaryotic CDO variant that cannot form the thioether bridge. In this variant, O2 consumption was less tightly coupled to CSA production (~45%) compared to WT CDO (~80%) [73]. Both QM/MM calculations and EPR spectroscopy (with Fe(III) and CN as a surrogate for Fe(II) and O2) have been used to contrast WT CDO with the Cys → Ala variant [73]. These studies demonstrated that the cross-linked residues are key to anchoring the cysteine substrate to the active site metal (in particular reducing the lability of the coordination of the amino group of the substrate to the active site metal).
The majority of experimental investigations into CDO have addressed how CDO binds and activates O2. EPR spectroscopic studies with Fe(II)-CDO-NO [74] and Fe(III)-CDO-CN [73] as Fe(II)-CDO-O2 mimics have demonstrated an obligate ordered binding in which binding of L-Cys to CDO primes binding of the O2 mimic. This same order of binding is presumably also followed by Fe(II)-CDO-O2 and would be congruent with thiolate ligation triggering O2 binding as suggested for other enzymes including IPNS as discussed above. Interestingly, the conversion of cysteine to CSA can be accomplished by adding superoxide (generated by xanthine/xanthine oxidase) to Fe(III)-CDO [75]. Crucially, the superoxide does not reduce the Fe(III) to Fe(II), demonstrating that the superoxide is not merely acting as a one-electron donor to generate Fe(II)-CDO that could then react normally with cysteine and O2. Additionally, this study reported the trapping and characterization of a ferric-superoxide species. The decay of this species was kinetically matched to the appearance of CSA, but the rate of CSA formation was ~200-fold slower than the k cat of CDO, suggesting that an alternative reaction manifold is traversed.
Though this artificially generated Fe(III)-superoxo species may not be relevant to the native catalytic cycle, multiple in silico studies have also predicted an Fe(III)-superoxo in either the quintet [76–78] or singlet [79] state as the initial reactive intermediate. Following O2 binding, the sulfur atom might be activated by a minor resonance form as the radical cation, leading to attack of the distal oxygen atom of the superoxo on the sulfur and formation of a four-membered ring (Fig. 3c) [52, 77, 79]. Heterolytic scission of the oxygen–oxygen bond would generate an iron(IV)-oxo species that, after rotation of the sulfenate or dissociation and recoordination (possibly through oxygen; not drawn), could perform oxygen atom transfer to generate the product. Notably, this proposed catalytic cycle does not invoke a persulfenate intermediate that was observed in a rat CDO crystal structure and was proposed to catalyze sulfinate formation via an isomerization (Fig. 3d) [80]. Computational calculations found that the activation energy required for product formation via a persulfenate was prohibitively high and, therefore, this intermediate could be a crystallographic artifact [79]. To test its catalytic competence, the CDO-bound persulfenate crystal was generated again, and LC–MS used to analyze whether cysteine persulfenate or CSA (which are isobaric) were present in the crystal and/or the crystallization drop [81]. This analysis detected cysteine persulfenate in the crystal (CSA was not observed crystallographically) but neither compound in the drop, suggesting that the cysteine persulfenate does not turnover.
Recently, pre-steady-state single turnover studies of CDO reacted with cysteine and O2 enabled the detection of a transient intermediate that absorbed maximally at 500 and 640 nm [82]. Higher concentrations of Fe(II)-CDO and O2 led to increased formation of the intermediate, and the rate of decay of the intermediate was independent of the O2 concentration. Use of rapid-freeze-quench Mössbauer spectroscopy did not succeed in elucidating the nature of the intermediate, but DFT calculations suggest that the absorption spectrum is consistent with the singly oxygenated bicyclic structure I shown in Fig. 3c [82]. Further work will be required to unambiguously define the structure of this intermediate. Collectively, the observations detailed in this section suggest the mechanism depicted in Fig. 3c.
Sulfoxide synthases: EgtB and OvoA
Ergothioneine and the ovothiols are structurally similar thiohistidines (Fig. 4a, b). The redox potential of the ovothiols (−0.09 V vs SHE) permits them to function as protective radical scavengers, as is the case during oxidative maturation of sea urchin eggs [83]. By contrast, the biological role of ergothioneine is less clear in both the producing organisms (fungi and mycobacteria) as well as higher organisms exposed to ergothioneine. Despite this uncertainty, microbially produced ergothioneine is specifically imported by a transporter in mammals and accumulates in certain areas of the human body including the central nervous system, liver, and kidneys [84].

Overview of catalysis by the sulfoxide-inserting enzymes EgtB and OvoA. a The biosynthetic pathway to ergothioneine. b The ovothiol A biosynthetic pathway. c The site selectivity of OvoA catalysis is dependent on the number of methyl groups on Nα of L-His. d An overlay of the Mn–EgtB–dimethylhistidine–γ-glutamyl cysteine complex (carbons in aqua, PDB ID: 4X8D) with the apo OvoA homology model (carbons in green), generated for this review using I-TASSER [125]. Key active site residues are preserved in the OvoA homology model, including the three histidines that would ligate the active site metal (one is partially obscured by the substrates in the EgtB structure). The homology model lacks an active site metal, explaining why the histidine side chains do not overlay with the histidine side chains in the EgtB structure. e A mechanistic proposal for catalysis by EgtB and OvoA. As with CDO, a minor resonance form with an Fe(II)-thiyl radical cation may activate O2 for attack on the sulfur atom. The ferrous-peroxysulfur species initially implicated based on DFT calculations (bottom) has instead been proposed to lead to non-productive dioxygenation to generate CSA as in CDO (Fig. 3c)
After initial isolation from ergot in 1909 [85], the gene cluster responsible for the biosynthesis of ergothioneine remained elusive until recently. Cell-free extracts from Neurospora crassa were reported to produce ergothioneine from the amino acids histidine, methionine, and cysteine via (at least) two intermediates: N α-trimethyl histidine (hercynine) and hercynylcysteine sulfoxide [86, 87] (Fig. 4a). In 2010, these reports were leveraged to locate candidate methyltransferases with homologs present in strains producing ergothioneine such as Mycobacterium avium but absent in non-producers like Escherichia coli and Bacillus subtilis [88]. One such methyltransferase was encoded adjacent to a gene for a PLP-binding protein. This candidate protein methylated L-His in the presence of SAM to generate hercynine. A nearby protein contained a domain reminiscent of those in formylglycine-generating enzymes. This protein, EgtB, effected the Fe(II)-dependent addition of γ-glutamyl cysteine (furnished by EgtA) and subsequent oxidation to yield a sulfoxide product. Sequential amide bond hydrolysis by EgtC and sulfur elimination by the lyase EgtE yields ergothioneine. The intermediacy of the sulfoxide intermediate was surprising, as it had not previously been detected in cell-free experiments. Homologs of EgtB contain a strongly conserved HX3HXE motif, implying that it is a member of the facial triad enzyme family with the Fe(II) site ligated by 2-His-1-Glu [88].
Given the similar structure of ergothioneine and ovothiol, it was hypothesized that ovothiol biosynthesis would employ a distant homolog of EgtB as a 5-histidylcysteine sulfoxide synthase. Genome mining of the producer Erwinia tasmaniensis yielded a candidate cluster with an EgtB homolog (E < 10−7), named OvoA [89]. In contrast to sulfoxide incorporation at C2 by EgtB, OvoA modified the C5 position of the imidazole ring of L-His. The site selectivity of the transformation was confirmed upon retention of the deuterium label in the product when using C2-2H1-His as substrate. Though the observed rate of formation was low (1.9 ± 0.2 min−1), a better pair of substrates was not found. No KIE was detected during a competition assay between L-His and L-C2,C5,Cα-2H3-His, indicating that hydrogen removal does not occur in a rate-determining step or that the enzyme has a high commitment to catalysis. Additionally, mutation of any of the residues in the HX3HXE motif in OvoA resulted in a >100-fold attenuation of activity, implying that these residues are catalytically important (i.e., likely bind iron).
A lingering question from these initial reports was why Nature selected an apparent four-electron oxidation to effect the two-electron oxidation of thiol insertion on imidazole, as the sulfoxide is no longer present in either ergothioneine or ovothiol, and the mechanism of oxygen removal has only begun to be studied [90]. DFT calculations regarding possible reactive intermediates in the catalytic mechanism of EgtB and OvoA provided one explanation. Because neither the crystal structure of EgtB or OvoA was known at the time of these calculations, the authors instead calculated the gas-phase thermodynamic free energies of possible reactive intermediates [91]. From these calculations, the authors proposed that neither a ferric-superoxo nor a ferryl-oxo intermediate was competent to oxidize the imidazole substrate. Instead, a four-membered ferrous-peroxysulfur species (Fe-OOS) reminiscent of that proposed in CDO catalysis [78, 79] was proposed as the oxidant. In the computational studies, this intermediate led to formation of the sulfoxide en route to sulfur insertion, though the authors cautioned that in the absence of any structural information it was impossible to draw definitive conclusions.
Despite the dearth of structural information, substantial in-roads were made exploring the substrate specificity and the site selectivity of EgtB and OvoA [92, 93]. Although EgtB did not accept the substrates of OvoA (l-cysteine and L-His) (Fig. 4b), OvoA exhibited relaxed substrate specificity and oxidatively added the non-native dipeptide γ-glutamyl cysteine to the expected C5 imidazole carbon of His (i.e., R 2 = γ-glutamyl-Cys; Fig. 4b) [92]. Interestingly, when OvoA was incubated with cysteine and N α-mono-, -di-, or –trimethylated histidine (hycernine), modification at the C5 imidazole carbon went from 60% to trace (<5%) to 0%, respectively, with a corresponding increase in functionalization at the C2 position (Fig. 4c). In addition to L-His derivatives, OvoA also coupled cysteine with D-His to yield a mixture of isomers [3:2 C5 (native): C2 (non-native)] [93]. Collectively, these results implicate different binding modes of substrate leading to site-selective outcomes. Even in the absence of structural information for OvoA, this hypothesis could potentially be tested by determining the substrate K m leading to each product [94], as OvoA might have a different affinity for the substrate in the different binding modes.
Surprisingly, when the crystal structure of EgtB was determined, the active site iron was coordinated by a 3-His facial triad rather than the anticipated 2-His-1-Glu ligand set [95]. As mentioned previously, the 3-His facial triad coordination sphere has been observed in only a handful of enzymes, including Dke1 [96] and CDO [69], further underscoring mechanistic similarities between these enzymes and the TDOs. The conservation of the third His in OvoA and a homology model generated for this review strongly suggests that OvoA is also a 3-His facial triad enzyme (Fig. 4d). Hints of mechanistic similarities between EgtB/OvoA and CDO existed prior to the report of the crystal structure, such as the observation that OvoA incubated with hercynine (the normal substrate for EgtB oxidation) and cysteine produces primarily CSA, the product of CDO-type chemistry [97]. Thus, it seems that the reactive oxygen species is poised to effect dioxygenation of sulfur in OvoA if the sulfur is not precisely oriented for addition to the imidazole ring. More careful reexamination revealed that, even when OvoA was incubated with its native substrates (L-Cys and L-His), approximately 10% of the cysteine is converted to CSA [97]. Very recently, the structural insights gleaned from the EgtB crystal structure were used to assess the importance of various active site residues [98]. A single point mutation in EgtB, Y377F, completely uncoupled substrate consumption from sulfoxide synthase activity with the native substrates (hercynine and γ-glutamyl cysteine), with EgtB exclusively oxidizing γ-glutamyl cysteine to the sulfinic acid with a catalytic efficiency that approached (~40%) that of WT murine CDO. In the structure, Tyr377 is hydrogen bonded to a water molecule that coordinates to the iron. A hypothesis to explain this diversion of activity is shown in Fig. 4e [93]. The initially formed superoxo species can be drawn in several formal resonance forms (we use the sulfur radical cation notation for consistency with the CDO studies). Proton transfer in the WT enzyme from Tyr377 generates a peroxo species, and the sulfur with radical character attacks the imidazole (again the positions of the electrons are drawn to provide a nitrogen-based radical but alternative resonance structures are possible). Deprotonation, by the tyrosinate at position 377, and sulfur oxidation via either the peroxo species or via a ferryl would generate the product. In the absence of Tyr377, CDO-like chemistry ensues.
With structural information now available, a number of experiments are feasible including QM/MM calculations to test various enzymatic mechanisms. Additionally, co-crystallization/soaking experiments with substrates could definitively resolve whether alternate binding conformations are accessible that lead to isomeric products. As OvoA exhibits relaxed substrate specificity, obtaining a crystal structure of this enzyme would also facilitate these experiments. Given the similarities between EgtB/OvoA and CDO, it would be interesting to apply experiments used to interrogate CDO to EgtB and OvoA. Doing so might help tease out whether these proteins indeed rely on similar intermediates.
Carbon–carbon bond cleavage: HEPD and MPnS
Phosphonates and phosphinates are reduced phosphorus compounds with one and two carbon–phosphorus bonds, respectively. These molecules often exhibit potent antibiotic or herbicidal activities [99–102]. Phosphinothricin (PT, glufosinate), for example, is the active ingredient in commercial herbicides such as Liberty and Basta (Fig. 5a). During the course of investigating the phosphinothricin tripeptide (PTT) biosynthetic pathway, knockout experiments with the native producer, Streptomyces viridochromogenes, in combination with heterologous expression of proteins from the biosynthetic pathway demonstrated that the protein encoded by phpD converted 2-hydroxyethylphosphonate (HEP) to hydroxymethylphosphonate (HMP) [103]. Because these were in vivo experiments, the precise requirements for catalysis by PhpD at the time were unknown. Many of these details were elucidated by in vitro reconstitution of activity and determination of the crystal structure of Cd2+-substituted PhpD in complex with the HEP substrate [104]. The active site metal was coordinated by 2-His-1-Glu on one face of a pseudooctrahedron, indicating that despite limited sequence homology to characterized proteins, it was a member of the facial triad family. As PhpD required only Fe(II) and O2 for catalysis and incorporated both oxygen atoms into the products, HMP and formate (Fig. 5a), it was renamed 2-hydroxyethylphosphonate dioxygenase (HEPD). In comparison to the other enzymes discussed in this review, HEPD oxidizes a relatively unactivated substrate that cannot easily facilitate O2 activation. HEP does not contain a thiol group that upon binding to the iron can activate it for catalysis, nor does it contain an α-keto acid functionality.

Overview of catalysis by HEPD and MPnS. a The biosynthetic pathways in which HEPD and MPnS are operative. b An overlay of the Cd-HEPD-HEP crystal structure (carbons in green, PDB ID: 3GBF) and an apo MPnS homology model (carbons in aqua) generated by I-TASSER [125]. An isoleucine is predicted to occupy the space of the carboxylate ligand in HEPD (E176), but a Gln residue in MPnS that aligns with the Fe-binding Glu of hydroxypropylphosphonate epoxidase is also predicted to be nearby. The identity of the third iron-binding ligand in MPnS remains unclear. c Scheme detailing the results of labeling experiments with MPnS, HEPD, and HEPD-E176H. d A mechanistic proposal invoking a consensus mechanism for HEPD and MPnS. Product identity is governed by whether the MPn radical combines with the ferric hydroxide (HEPD) or attacks formate (MPnS). For details, see the text
Many phosphonate natural products are bioactive, but some instead play structural roles, particularly as alternate polar headgroups for lipids and exopolysaccharides [105, 106]. One such example is methylphosphonate (MPn), which is an alternate headgroup for exopolysaccharides that decorate the surface of Nitrosopumilus maritimus, a ubiquitous marine archaeon [107]. MPn is produced from HEP by methylphosphonate synthase (MPnS), which has distant sequence homology to HEPD and is likewise reliant on only Fe(II) and O2 for catalysis. A sequence alignment of HEPD and MPnS (as well as the distantly related peroxidase hydroxypropylphosphonate epoxidase, HppE [108, 109]) indicated that MPnS contains iron-binding residues at only the two histidines, raising the specter that either MPnS is a 2-His only enzyme (like the halogenase SyrB2 [110]) or that the third ligand is not well conserved in the alignment.
The sequence homology between HEPD and MPnS combined with identical requirements for catalysis (HEP, Fe(II) and O2) suggests a consensus mechanism in which product identity is determined by branching at an intermediate in the catalytic cycle. To address whether the same early intermediate is used in both enzymes to activate substrate, competitive 18O KIE experiments were performed with natural abundance O2. In both proteins, discrimination against 18O increased when the proteins were reacted with HEP dideuterated at C2 [111]. This outcome signifies that for both enzymes the initial abstraction at C2 must occur prior to or during the first irreversible step involving O2 [112, 113]. This result implies that abstraction cannot take place via a ferryl species, which would be formed after the first irreversible step (O–O cleavage), rendering the 18O KIE insensitive to substrate deuteration. Additional evidence for a consensus mechanism arose from the unusual observation that the pro-(R) hydrogen at C2 of HEP is quantitatively incorporated by MPnS into MPn [114]. This result was intriguing because during catalysis by HEPD, the same pro-(R) hydrogen at C2 is quantitatively incorporated into formate [115] (Fig. 5c). These findings suggested that an MPn radical might exist in catalysis that could either recombine with a ferric hydroxide to make HMP during HEPD catalysis or abstract a hydrogen atom from formate in MPnS catalysis (Fig. 5d). An alternative that cannot be completely ruled out involves oxidation of the MPn radical to the cation by the ferric hydroxide. In that case, the resulting MPn cation would either be attacked by the hydroxide bound to the iron(II) site (HEPD) or by a hydride from formate (MPnS). Regardless of whether the branch point for the two outcomes occurs at an MPn radical or MPn cation, one would predict loss of the stereochemical integrity at the original C1 position of the substrate, and this has indeed been experimentally verified by stereospecific deuterium labeling [115].
Support that the MPn radical (or cation) is the branch point of catalysis comes from an HEPD variant that produces both HMP and MPn in near equal quantities [116]. In this variant, HEPD-E176H, the iron-binding Glu was replaced by a His; unexpectedly, the latter did not bind the active site metal in the crystal structure of the mutant. Reacting HEPD-E176H with (R)-[2-2H1]-HEP substantially biased product formation away from MPn and towards HMP with a calculated KIE of ~10, consistent with the branch point involving transfer of this hydrogen atom (Fig. 5d). Importantly, in the traces of MPn produced with this labeled substrate, deuterium was still incorporated into the product.
The weight of evidence suggests that HEPD and MPnS diverge very late in their enzymatic mechanisms. However, the intermediates after initial hydrogen atom abstraction but before the MPn radical remain unknown. Several computational efforts have been made to identify the species at the heart of HEPD catalysis [117–120]. These studies have generally been congruent with one another in suggesting that homolytic β-scission from a gem-diol radical would yield the MPn radical that would then recombine with the ferric hydroxide to hydroxylate MPn (Fig. 5d); in MPnS catalysis, this MPn radical could instead abstract a hydrogen atom from formate. Discrepancies are primarily over whether the initial ketyl radical transfers an electron to the active site iron followed by hydroperoxylation and homolytic cleavage of the hydroperoxo species [117, 118] or whether an Fe(IV)-oxo species is responsible for generating the gem-diol radical via H-atom abstraction from an O–H bond [119, 120]. This proposed cleavage is unusual because Fe(IV)=O species in enzymes have only been observed to cleave C–H bonds, though H-atom abstraction from heteroatoms has been proposed before [121, 122], and could be involved in the CloR reaction discussed above. Structural information for MPnS would help elucidate the determinants for product identity in the two enzymes and possibly empower the complete interconversion of HEPD and MPnS reactivity. Spectroscopic experiments of trapped intermediates will also be required to tease apart the catalytic cycles of these proteins.
Outlook
It has been over 60 years since Hayaishi and Mason first demonstrated the existence of oxygenases by showing incorporation of molecular O2 into the products of enzymatic catalysis [123, 124]. Although the pace of discovery was initially slow, improved experimental techniques and the advent of cheap genome sequencing have dramatically accelerated the speed at which enzymes with novel reactivity have been discovered in recent years. Investigating the mechanisms of non-heme iron enzymes holds the prospect of rationally tuning the reactivity of O2 towards different substrates, explaining the fascination with interconverting the activities of similar enzymes. Though enzymes requiring cosubstrates such as α-ketoglutarate or organic cofactors catalyze a dizzying array of reactions, from an atom economy and engineering standpoint it would be ideal to develop catalysts that can carry out reactions without requiring cosubstrates or expensive cofactors such as NADH/NADPH. Extending oxygen activation towards substrates that are not electron rich would be particularly attractive. From the knowledge gained thus far, four-electron oxidations in most cases require specialized substrates that activate a ferric-superoxide for subsequent chemistry such as a thiol or α-keto acid group. But in principle, such a thiolate ligand could also be supplied by the protein. If oxidation of the proteinaceous thiolate ligand could be prevented, enzymes might be sufficiently activated to harvest the oxidizing potential of first a ferric-superoxo and subsequently a ferryl species. The type of substrate activation that can be initiated by a ferric-superoxo will likely still be restricted, however. Although in HEPD and MPnS, a thiol ligand does not appear necessary, the C–H bond in the substrate that is abstracted is still relatively weak because it is adjacent to an alkoxide group. The step in which a ferryl is formed also has specific commonalities in several of the enzymes covered. A ferric hydroperoxo species is thought to be reduced to the ferrous hydroperoxo form by electron transfer from an intermediate formed from substrate. This intrinsic reactivity may be difficult to build into a substrate and will likely always limit the ability to achieve a four-electron oxidation of substrate in engineered systems. However, if past experience is a good predictor of future findings, then further investigations into this class of enzymes may uncover new design rules.
References
Kovaleva EG, Lipscomb JD (2008) Nat Chem Biol 4:186–193
Loenarz C, Schofield CJ (2008) Nat Chem Biol 4:152–156
Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM Jr (2003) J Am Chem Soc 125:13008–13009
Price JC, Barr EW, Tirupati B, Bollinger JM Jr, Krebs C (2003) Biochemistry 42:7497–7508
Hoffart LM, Barr EW, Guyer RB, Bollinger JM Jr, Krebs C (2006) Proc Natl Acad Sci USA 103:14738–14743
Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM, Krebs C, Fitzpatrick PF (2007) J Am Chem Soc 129:11334–11335
Galonić DP, Barr EW, Walsh CT, Bollinger JM Jr, Krebs C (2007) Nat Chem Biol 3:113–116
W-c Chang, Y Guo, C Wang, SE Butch, Rosenzweig AC, Boal AK, Krebs C, Bollinger JM Jr (2014) Science 343:1140–1144
Fitzpatrick PF (2003) Biochemistry 42:14083–14091
Cochrane RVK, Vederas JC (2014) Acc Chem Res 47:3148–3161
van der Donk WA, Krebs C, Bollinger JM Jr (2010) Curr Opin Struct Biol 20:673–683
Bollinger JM Jr, Diao Y, Matthews ML, Xing G, Krebs C (2009) Dalton Trans 38:905–914
Straganz GD, Glieder A, Brecker L, Ribbons DW, Steiner W (2003) Biochem J 369:573–581
Straganz GD, Nidetzky B (2005) J Am Chem Soc 127:12306–12314
Diebold AR, Straganz GD, Solomon EI (2011) J Am Chem Soc 133:15979–15991
Fetzner S, Steiner RA (2010) Appl Microbiol Biotechnol 86:791–804
Hager SE, Gregerman RI, Knox WE (1957) J Biol Chem 225:935–948
Norris SR, Shen X, Della Penna D (1998) Plant Physiol 117:1317–1323
Schulz A, Ort O, Beyer P, Kleinig H (1993) FEBS Lett 318:162–166
Tomoeda K, Awata H, Matsuura T, Matsuda I, Ploechl E, Milovac T, Boneh A, Scott CR, Danks DM, Endo F (2000) Mol Gen Metab 71:506–510
Choroba OW, Williams DH, Spencer JB (2000) J Am Chem Soc 122:5389–5390
Guroff G, Renson J, Udenfriend S, Daly JW, Jerina DM, Witkop B (1967) Science 157:1524–1530
Serre L, Sailland A, Sy D, Boudec P, Rolland A, Pebay-Peyroula E, Cohen-Addad C (1999) Structure 7:977–988
Brownlee JM, Johnson-Winters K, Harrison DHT, Moran GR (2004) Biochemistry 43:6370–6377
Yang C, Pflugrath JW, Camper DL, Foster ML, Pernich DJ, Walsh TA (2004) Biochemistry 43:10414–10423
Fritze IM, Linden L, Freigang J, Auerbach G, Huber R, Steinbacher S (2004) Plant Physiol 134:1388–1400
Gunsior M, Ravel J, Challis GL, Townsend CA (2004) Biochemistry 43:663–674
O’Hare HM, Huang F, Holding A, Choroba OW, Spencer JB (2006) FEBS Lett 580:3445–3450
Brownlee J, He P, Moran GR, Harrison DHT (2008) Biochemistry 47:2002–2013
Krebs C, Galonic Fujimori D, Walsh CT, Bollinger JM Jr (2007) Acc Chem Res 40:484–492
Shah DD, Conrad JA, Heinz B, Brownlee JM, Moran GR (2011) Biochemistry 50:7694–7704
Shah DD, Conrad JA, Moran GR (2013) Biochemistry 52:6097–6107
Borowski T, Bassan A, Siegbahn PEM (2004) Biochemistry 43:12331–12342
Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, Spencer JB, Solomon EI (2006) Proc Natl Acad Sci USA 103:12966–12973
Wójcik A, Broclawik E, Siegbahn PEM, Lundberg M, Moran G, Borowski T (2014) J Am Chem Soc 136:14472–14485
Pojer F, Kahlich R, Kammerer B, Li SM, Heide L (2003) J Biol Chem 278:30661–30668
Paine TK, Paria S, Que L Jr (2010) Chem Commun 46:1830–1832
Paria S, Chatterjee S, Paine TK (2014) Inorg Chem 53:2810–2821
Sheet D, Bhattacharya S, Paine TK (2015) Chem Commun 51:7681–7684
Waxman DJ, Strominger JL (1983) Annu Rev Biochem 52:825–869
Arnstein HRV, Clubb ME (1958) Biochem J 68:528–535
Pang CP, Chakravarti B, Adlington RM, Ting HH, White RL, Jayatilake GS, Baldwin JE, Abraham EP (1984) Biochem J 222:789–795
Hollander IJ, Shen YQ, Heim J, Demain AL, Wolfe S (1984) Science 224:610–612
Samson SM, Belagaje R, Blankenship DT, Chapman JL, Perry D, Skatrud PL, VanFrank RM, Abraham EP, Baldwin JE, Queener SW, Ingolia TD (1985) Nature 318:191–194
Chen VJ, Orville AM, Harpel MR, Frolik CA, Surerus KK, Münck E, Lipscomb JD (1989) J Biol Chem 264:21677–21681
Orville AM, Chen VJ, Kriauciunas A, Harpel MR, Fox BG, Münck E, Lipscomb JD (1992) Biochemistry 31:4602–4612
Scott RA, Wang S, Eidsness MK, Kriauciunas A, Frolik CA, Chen VJ (1992) Biochemistry 31:4596–4601
Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, Baldwin JE (1995) Nature 375:700–704
Roach PL, Clifton IJ, Hensgens CMH, Shibata N, Long AJ, Strange RW, Hasnain SS, Schofield CJ, Baldwin JE, Hajdu J (1996) Eur J Biochem 242:736–740
Roach PL, Clifton IJ, Hensgens CM, Shibata N, Schofield CJ, Hajdu J, Baldwin JE (1997) Nature 387:827–830
Baldwin JE, Adlington RM, Moroney SE, Field LD, Ting H-H (1984) J Chem Soc Chem Commun 15:984–986. doi:10.1039/C39840000984
Brown CD, Neidig ML, Neibergall MB, Lipscomb JD, Solomon EI (2007) J Am Chem Soc 129:7427–7438
Burzlaff NI, Rutledge PJ, Clifton IJ, Hensgens CM, Pickford M, Adlington RM, Roach PL, Baldwin JE (1999) Nature 401:721–724
Lundberg M, Siegbahn PEM, Morokuma K (2008) Biochemistry 47:1031–1042
Lundberg M, Kawatsu T, Vreven T, Frisch MJ, Morokuma K (2009) J Chem Theory Comput 5:222–234
Tamanaha E, Zhang B, Guo Y, Chang W-C, Barr EW, Xing G, St. Clair J, Ye S, Neese F, Bollinger JM, Krebs C (2016) J Am Chem Soc 138:8862–8874
Mbughuni MM, Chakrabarti M, Hayden JA, Bominaar EL, Hendrich MP, Münck E, Lipscomb JD (2010) Proc Natl Acad Sci USA 107:16788–16793
Chiang C-W, Kleespies ST, Stout HD, Meier KK, Li P-Y, Bominaar EL, Que L, Münck E, Lee W-Z (2014) J Am Chem Soc 136:10846–10849
Stipanuk MH, Simmons CR, Andrew Karplus P, Dominy JE (2011) Amino Acids 41:91–102
Cavallini D, Scandurra R, De Marco C (1963) J Biol Chem 238:2999–3005
Dominy JE, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, Stipanuk MH (2007) J Biol Chem 282:25189–25198
Bruland N, Wübbeler JH, Steinbüchel A (2009) J Biol Chem 284:660–672
Sörbo B, Ewetz L (1965) Biochem Biophys Res Commun 18:359–363
Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG (1990) Neurosci Lett 110:216–220
Gordon C, Emery P, Bradley H, Waring RH (1992) Lancet 339:25–26
Emery P, Bradley H, Arthur V, Tunn E, Waring R (1992) Rheumatology 31:449–451
Lombardini JB, Singer TP, Boyer PD (1969) J Biol Chem 244:1172–1175
Chai SC, Jerkins AA, Banik JJ, Shalev I, Pinkham JL, Uden PC, Maroney MJ (2005) J Biol Chem 280:9865–9869
McCoy JG, Bailey LJ, Bitto E, Bingman CA, Aceti DJ, Fox BG, Phillips GN (2006) Proc Natl Acad Sci USA 103:3084–3089
Straganz GD, Nidetzky B (2006) ChemBioChem 7:1536–1548
Ito N, Phillips SEV, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KDS, Knowles PF (1991) Nature 350:87–90
Dominy JE, Hwang J, Guo S, Hirschberger LL, Zhang S, Stipanuk MH (2008) J Biol Chem 283:12188–12201
Li W, Blaesi EJ, Pecore MD, Crowell JK, Pierce BS (2013) Biochemistry 52:9104–9119
Pierce BS, Gardner JD, Bailey LJ, Brunold TC, Fox BG (2007) Biochemistry 46:8569–8578
Crawford JA, Li W, Pierce BS (2011) Biochemistry 50:10241–10253
Blaesi EJ, Gardner JD, Fox BG, Brunold TC (2013) Biochemistry 52:6040–6051
Blaesi EJ, Fox BG, Brunold TC (2014) Biochemistry 53:5759–5770
de Visser SP, Straganz GD (2009) J Phys Chem A 113:1835–1846
Kumar D, Thiel W, de Visser SP (2011) J Am Chem Soc 133:3869–3882
Simmons CR, Krishnamoorthy K, Granett SL, Schuller DJ, Dominy JE, Begley TP, Stipanuk MH, Karplus PA (2008) Biochemistry 47:11390–11392
Souness RJ, Kleffmann T, Tchesnokov EP, Wilbanks SM, Jameson GB, Jameson GNL (2013) Biochemistry 52:7606–7617
Tchesnokov EP, Faponle AS, Davies CG, Quesne MG, Turner R, Fellner M, Souness RJ, Wilbanks SM, de Visser SP, Jameson GNL (2016) Chem Commun 52:8814–8817
Turner E, Hager LJ, Shapiro BM (1988) Science 242:939
Gründemann D, Harlfinger S, Golz S, Geerts A, Lazar A, Berkels R, Jung N, Rubbert A, Schömig E (2005) Proc Natl Acad Sci USA 102:5256–5261
Tanret C (1909) Compt Rend 149:222–224
Ishikawa Y, Melville DB (1970) J Biol Chem 245:5967–5973
Ishikawa Y, Israel SE, Melville DB (1974) J Biol Chem 249:4420–4427
Seebeck FP (2010) J Am Chem Soc 132:6632–6633
Braunshausen A, Seebeck FP (2011) J Am Chem Soc 133:1757–1759
Song H, Hu W, Naowarojna N, Her AS, Wang S, Desai R, Qin L, Chen X, Liu P (2015) Sci Rep 5:11870
Bushnell EAC, Fortowsky GB, Gauld JW (2012) Inorg Chem 51:13351–13356
Song H, Leninger M, Lee N, Liu P (2013) Org Lett 15:4854–4857
Mashabela GTM, Seebeck FP (2013) Chem Commun 49:7714–7716
Thuresson ED, Lakkides KM, Smith WL (2000) J Biol Chem 275:8501–8507
Goncharenko KV, Vit A, Blankenfeldt W, Seebeck FP (2015) Angew Chem Int Ed 54:2821–2824
Straganz GD, Diebold AR, Egger S, Nidetzky B, Solomon EI (2010) Biochemistry 49:996–1004
Song H, Her AS, Raso F, Zhen Z, Huo Y, Liu P (2014) Org Lett 16:2122–2125
Goncharenko KV, Seebeck FP (2016) Chem Commun 52:1945–1948
White AK, Metcalf WW (2007) Annu Rev Microbiol 61:379–400
Metcalf WW, van der Donk WA (2009) Annu Rev Biochem 78:65–94
McGrath JW, Chin JP, Quinn JP (2013) Nat Rev Microbiol 11:412–419
Chin JP, McGrath JW, Quinn JP (2016) Curr Opin Chem Biol 31:50–57
Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, Kelleher NL, Metcalf WW (2007) Nat Chem Biol 3:480–485
Cicchillo RM, Zhang H, Blodgett JAV, Whitteck JT, Li G, Nair SK, van der Donk WA, Metcalf WW (2009) Nature 459:871–874
Horiguchi M, Kandatstu M (1959) Nature 184:901–902
Hilderbrand RL (1983) The role of phosphonates in living systems. CRC Press, Boca Raton
Metcalf WW, Griffin BM, Cicchillo RM, Gao J, Janga SC, Cooke HA, Circello BT, Evans BS, Martens-Habbena W, Stahl DA, van der Donk WA (2012) Science 337:1104–1107
Higgins LJ, Yan F, Liu P, Liu HW, Drennan CL (2005) Nature 437:838–844
Wang C, Chang W-C, Guo Y, Huang H, Peck SC, Pandelia ME, Lin G-M, Liu H-w, Krebs C, Bollinger JM (2013) Science 342:991–995
Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL (2006) Nature 440:368–371
Zhu H, Peck SC, Bonnot F, van der Donk WA, Klinman JP (2015) J Am Chem Soc 137:10448–10451
Roth JP (2009) Acc Chem Res 42:399–408
Ashley DC, Brinkley DW, Roth JP (2010) Inorg Chem 49:3661–3675
Cooke HA, Peck SC, Evans BS, van der Donk WA (2012) J Am Chem Soc 134:15660–15663
Whitteck JT, Malova P, Peck SC, Cicchillo RM, Hammerschmidt F, van der Donk WA (2011) J Am Chem Soc 133:4236–4239
Peck SC, Chekan JR, Ulrich EC, Nair SK, van der Donk WA (2015) J Am Chem Soc 137:3217–3220
Hirao H, Morokuma K (2010) J Am Chem Soc 132:17901–17909
Hirao H, Morokuma K (2011) J Am Chem Soc 133:14550–14553
Du L, Gao J, Liu Y, Liu C (2012) J Phys Chem B 116:11837–11844
Du L, Gao J, Liu Y, Zhang D, Liu C (2012) Org Biomol Chem 10:1014–1024
Borowski T, de Marothy S, Broclawik E, Schofield CJ, Siegbahn PEM (2007) Biochemistry 46:3682–3691
Thrower J, Mirica LM, McCusker KP, Klinman JP (2006) Biochemistry 45:13108–13117
Hayaishi O, Katagiri M, Rothberg S (1955) J Am Chem Soc 77:5450–5451
Mason HS, Fowlks WL, Peterson E (1955) J Am Chem Soc 77:2914–2915
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) Nat Method 12:7–8
Acknowledgements
Our work on phosphonate biosynthesis is supported by the National Institutes of Health (P01 GM 077596).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Peck, S.C., van der Donk, W.A. Go it alone: four-electron oxidations by mononuclear non-heme iron enzymes. J Biol Inorg Chem 22, 381–394 (2017). https://doi.org/10.1007/s00775-016-1399-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-016-1399-y