
Abstract
Polycyclic aromatic hydrocarbons (PAHs) are a class of hazardous organic contaminants that are widely distributed in nature, and many of them are potentially toxic to humans and other living organisms. Biodegradation is the major route of detoxification and removal of PAHs from the environment. Aerobic biodegradation of PAHs has been the subject of extensive research; however, reports on anaerobic biodegradation of PAHs are so far limited. Microbial degradation of PAHs under anaerobic conditions is difficult because of the slow growth rate of anaerobes and low energy yield in the metabolic processes. Despite the limitations, some anaerobic bacteria degrade PAHs under nitrate-reducing, sulfate-reducing, iron-reducing, and methanogenic conditions. Anaerobic biodegradation, though relatively slow, is a significant process of natural attenuation of PAHs from the impacted anoxic environments such as sediments, subsurface soils, and aquifers. This review is intended to provide comprehensive details on microbial degradation of PAHs under various reducing conditions, to describe the degradation mechanisms, and to identify the areas that should receive due attention in further investigations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- 2-Naphthoic acid
- Aerobic biodegradation
- Anaerobic biodegradation
- Anoxia
- Aromatic ring reduction
- Benzo(a)pyrene
- Biodegradation
- Bioremediation
- Fate of PAHs
- Genetics
- Iron-reducing bacteria
- Mechanism of anaerobic biodegradation
- Metabolite profiling
- Methanogenic bacteria
- Microbial degradation
- Naphthalene
- Naphthalene carboxylase
- Nitrate-reducing bacteria
- PAH sources
- Pathways of biodegradation
- Phenanthrene
- Polycyclic aromatic hydrocarbons
- Pyrene
- Sulfate-reducing bacteria
- Thermodynamics
1 Introduction
Polycyclic aromatic hydrocarbons (PAHs) are a group of hundreds of related organic aromatic compounds consisting of at least two (e.g., naphthalene) or more (e.g., anthracene) fused benzene rings arranged in linear (e.g., naphthalene, anthracene), angular [e.g., dibenzo(a,h)anthracene], or cluster (e.g., pyrene) fashion. PAHs that are composed of only fused benzene rings are classified as alternant PAHs (Smith and March 2007). In addition to the regular hexagonal benzene ring, non-alternant PAHs contain an additional annealed cyclic structure, for example, a tetragonal or a pentagonal ring. Thus, naphthalene, phenanthrene, and pyrene are alternant PAHs, while fluorene, fluoranthene, cyclopenta(d,e,j)phenanthrene, 7H-benzanthrene, and indeno(1,2,3-c,d)pyrene are non-alternant PAHs (Blumer 1976; Maliszewska-Kordybach 1999; Neilson 2013; Abdel-Shafy and Mansour 2016). Two- and three-ring PAHs are generally regarded as low-molecular-weight (LMW) PAHs, whereas those having four or more rings are considered as high-molecular-weight (HMW) PAHs. Incomplete combustion of carbonaceous materials and crude oil spills are the two major sources of PAHs in the environment (Maliszewska-Kordybach 1999; Lundstedt et al. 2007; Ohura 2007). They are ubiquitous environmental pollutants; many of them show toxic, mutagenic, and carcinogenic properties (White and Claxton 2004). Their complex and rigid aromatic structure, high resonance energy, and limited bioavailability make them chemically stable as well as resistant to microbial degradation. Due to the ubiquity, persistence, bioaccumulative tendency, and acute toxicity to biota, PAHs are regarded as a class of hazardous organic contaminants.
The United States Environmental Protection Agency (US EPA) announced 16 PAHs as “priority pollutants” in aquatic and terrestrial ecosystems (US EPA 1982). The US EPA (2008) extended the list of hazardous PAHs in January 2008 (see Fig. 1). Although PAHs are removed from the environment by physicochemical processes, biodegradation of the pollutants by bacteria, fungi, algae, and plants is regarded as the principal mechanism of detoxification and removal (Samanta et al. 2002). Aerobic degradation of PAHs is well studied, and associated biochemical mechanisms have already been elucidated. Aerobic bacterial degradation of PAHs initiates with the dioxygenase enzyme-catalyzed introduction of both atoms of molecular oxygen into the aromatic nucleus of PAHs (Cerniglia 1992; Kanaly and Harayama 2000; Haritash and Kaushik 2009). PAHs can dissipate from their sources to many environmental compartments where the oxygen level is too low or zero. Such an anaerobic environment exists in many habitats such as subsurface soil, groundwater, aquifer sediment, freshwater sediment, marine sediment, sewage sludge, anaerobic wastewater treatment plant, etc. Biodegradation of PAHs in an anaerobic environment is challenging because molecular oxygen that is involved in the first step of the degradation pathway is not available to serve as the terminal electron acceptor during aerobic respiration and as a substrate for dioxygenases. For many years, PAHs were thought to be refractory to anaerobic microbial degradation; unavailability of oxygen and lower energy yield in the anaerobic metabolism were believed to be the critical restraining factors. However, many facultative and strictly anaerobic bacteria and archaea are now known to degrade PAHs using alternative electron acceptors such as nitrate, iron(III), and sulfate. Furthermore, pathways of anaerobic naphthalene and 2-methylnaphthalene degradation in sulfate-reducing bacteria have been elucidated.

Chemical structure of some PAHs of environmental concern
The understanding in the field of anaerobic biodegradation of PAHs is expanding consistently. Therefore, a critical appraisal of the field will help researchers to keep abreast of trends and state of the art of the knowledge. Only a few reviews in this field are available; most of them discussed PAHs degradation in association with monoaromatic compounds like benzene and toluene (Meckenstock et al. 2004, 2016; Foght 2008; Meckenstock and Mouttaki 2011). That too, many of the reviews focused primarily on degradation and metabolism of PAHs by sulfate-reducing bacteria (SRB). As such, only limited information is available on the facultative anaerobic degradation of PAHs by nitrate reducers, iron reducers, and methanogens. Therefore, the present review aims at providing a comprehensive critique of the various aspects of anaerobic PAHs biodegradation. Initial few sections deal with the physicochemical properties, toxicity, sources, and possible fates of PAHs in the environment. The main discussion begins with an explanation of the process for development of anaerobic conditions and theoretical prediction of the thermodynamic feasibility of PAHs degradation under different reducing conditions. Later, PAHs degradation under various reducing conditions, their effects on anaerobic microbial community, and factors affecting anaerobic biodegradation are discussed. Furthermore, metabolic pathways of anaerobic PAHs degradation under various redox conditions and progress in the area of metabolic biomarker-based in situ degradation monitoring techniques are summarized. Finally, we identified some critical research gaps and suggested bioremediation approaches for mitigating PAHs contamination in anaerobic environments.
2 Physicochemical Properties of PAHs
The behavior, transport, and fate of PAHs in the environment largely depend on their physical and chemical properties. Three main aspects of PAHs as pollutants – bioavailability, persistence and bioaccumulation tendency – are strongly influenced by these physicochemical properties (Skupinska et al. 2004; Wiktorska et al. 2004; Abdel-Shafy and Mansour 2016). With an increase in molecular weight and number of benzene rings in the structure, their aqueous solubility decreases, resistance to oxidation and reduction increases, and vapor pressure drops. Common physical, chemical, and toxicological properties of some selected PAHs, those are listed in US EPA Toxic Release Inventory for polycyclic aromatic compounds, are listed in Table 1. Pure PAHs are white to pale yellow color solids. They are nonpolar, hydrophobic, and lipophilic. PAHs are slightly soluble in water, and, in general, their solubility in water decreases as the molecular weight increases (Table 1). They are soluble in many organic solvents and have a strong tendency to sorb to and accumulate in organic molecules of soil and sediments or in fat (Subashchandrabose et al. 2014). Water-soluble PAHs, such as naphthalene, have higher aqueous solubility and hence are more available for microbial degradation. HMW PAHs are less water soluble and thus less accessible for microbial attack and, therefore, remain persistent in the environment.
PAHs exist as a complex mixture in nature. The composition of a PAHs pool emitted from a combusted source depends on the properties of the combusting carbonaceous material and combustion temperature. Likewise, crude oil, petroleum fuels, coal tar, creosote, and asphalt contain different combinations of PAHs and their derivatives (Blumer 1976). Vapor pressure is an essential determinant of dispersion, transport, and fate of PAHs. LMW PAHs having higher vapor pressure are mostly emitted as gas phase in ambient air. HMW PAHs have lower vapor pressure, and they are released predominantly as particle form. Most of the time, the particles tend to be associated with the airborne particulates such as soot, dust, and fly ash. The octanol-water partition coefficient (logKOW) is also a crucial determinant of PAHs compartmentalization in the environment. It is a measure that expresses the extent of a substance to partition itself between an organic phase (n-octanol) and an aqueous phase. It is used for predicting the distribution of a substance in various environmental compartments, i.e., water, soil, sediment, and biota. A logKOW value greater than four indicates that a chemical is likely to be sorbed strongly to minerals and organic matters (Karickhoff et al. 1979; Means et al. 1980). The logKOW values for almost all the PAHs exceed four (Table 1). The values increase proportionally with the increase in molecular weight (Miller et al. 1985). Thus, benzo(a)pyrene (BaP) with a logKOW = 6.13 is expected to bind strongly to organic fraction of soil and sediment and to lipids of organisms. PAHs with high logKOW may disappear quickly from aqueous mixture but remain persistent in soil and sediment. The propensity of PAHs to sorb to lipids indicates the potential of bioaccumulation in living organisms. Moreover, PAHs exhibit some notable characteristics like photosensitivity, conductivity, heat resistance, and corrosion resistance (Miller and Olejnik 2001; Northrop et al. 1956; Stein and Fahr 1985). Although PAHs are relatively stable compounds, they are prone to several chemical alteration processes in the natural environment. They may be subjected to photooxidation, chemical oxidation with oxidizing agents, hydroxylation, nitration, emulsification, as well as a range of microbial degradation processes. Some of the breakdown products are less toxic than the parental PAHs, while some nitro-, oxy-, amino-, and hydroxy-derivatives pose even greater toxicity (Nielsen et al. 1983; Neilson et al. 1998; Yu 2002; Kim et al. 2013; Neilson 2013; Andersson and Achten 2015).
3 Sources of PAHs
PAHs may not be abundant only in our planet, they are proposed to be widely distributed in the universe and constitute up to one fifth of all the carbon present in the galaxy (Allamandola et al. 1989; Cohen and Barlow 2005; Tielens 2005). They might have formed just after a couple of billion years after the Big Bang. The presence of anthracene and pyrene in Red Triangle nebula has been suggested based on the spectral signature analysis (Mulas et al. 2006). The new “PAHs world” hypothesis argues that the primordial soup might contain PAHs that in eons of time underwent several difficult changes and eventually transformed into the starting materials such as purines and pyrimidines for the origin of life (Peeters 2011). If the theory were proven right, it would shed some light on explaining the ubiquity of PAHs on Earth. PAHs are present in every sphere of Earth: atmosphere, hydrosphere, lithosphere, and biosphere. They can enter the environment in several ways. Based on their origin and mode of distribution, the sources of PAHs can be categorized into three major groups: pyrogenic, petrogenic, and diagenetic and biogenic (Fig. 2).

Classification of PAHs sources based on their origin and mode of distribution
3.1 Pyrogenic Sources
Pyrolysis or thermal cracking is an irreversible thermochemical process in which organic matter decomposes at high temperature in the absence of oxygen. Incomplete combustion of fossil fuel and biomass during pyrolysis release a significant amount of hazardous substances including PAHs (Ross et al. 2002). Pyrogenic PAHs are typically formed at elevated temperature (350–1,200°C), although they can also be emitted from a low temperature (100–150°C) combustion process. Pyrogenic sources can be natural or anthropogenic. Many natural events, for instance, forest burning, bushfire, and volcanic eruption, release PAHs into the environment (Menzie et al. 1992; Zhang and Tao 2009). Anthropogenic emission sources can be divided further into four major subclasses based on the sources: domestic, industrial, automotive exhaust, and agricultural. Domestic emission results from cooking and heating activities. The burning of coal, oil, gas, garbage, wood, and other organic substances emit PAHs. Cigarette smoke, fireplace, and backyard barbecues also contribute to the emission.
Pyrogenic process is defined as high-temperature low-oxygen combustion process, domestic open burning of coal, peat, wood, straw, cow-dung-cake, rice husk briquettes, and garbage at a temperature as low as 150–200°C contributing to the emission of PAHs (Tsibart et al. 2014). Since the industrial revolution, the major portion of PAHs in the environment, especially in the ambient air, has been added directly or indirectly from industrial activities. Destructive distillation of coal to coke and coal tar, thermal cracking of petroleum residue, burning of fuels, metallurgical process such as aluminum smelting, rubber tire industry, cement manufacturing industry, waste incineration, bitumen and asphalt production, wood preservation, commercial heating plant, as well as manufactured gas plant sites (MGP) are some of the conventional sources of pyrogenic PAHs. Automotive emission sources include exhaust gases from automobile vehicles, railway, ship, aircraft, jet engine, and other motor vehicles. Some agricultural practices, for example, intentional burning of bushland and forest, straw and stubble, and moorland heather, also contribute to the buildup of PAHs concentration in the environment (Stogiannidis and Laane 2015; Abdel-Shafy and Mansour 2016). Pyrogenic emission contains both LMW and HMW PAHs. Due to their higher vapor pressure, LMW constituents are released in gaseous form, whereas HMW PAHs are abundant in particle-sorbed form. Generally, HMW PAHs share the significant part of emitted PAHs from a pyrogenic origin (Ou et al. 2004; Page et al. 2006; Boll et al. 2008). Pyrene, fluoranthene, BaP, chrysene, and, to a lesser extent, phenanthrene are found commonly in pyrogenic emission. The predominance of these parent PAHs over their alkylated homologues is used as an indication of pyrogenic origin (Blumer and Youngblood 1975; Laflamme and Hites 1978; Sporstol et al. 1983; Wang et al. 2001).
3.2 Petrogenic Sources
Petrogenic PAHs are constituents of petroleum products including crude oil, engine oils, lubricant, and their derivatives. Petroleum fuel has become the inevitable energy source since the dawn of the industrial revolution in the late eighteenth century. Dependency on fossil petroleum fuels leads to an extensive exploration and transportation of petrochemicals. Crude oil and refined petroleum fuels are rich in parental PAHs, alkyl-PAHs, azaarenes, and thiophenes (Grimmer et al. 1983). Petrogenic PAHs enter the environment through maturation, drilling, transportation, storage, use of crude oil, and related petrochemicals. Petrogenic sources can be natural or anthropogenic. Petroleum seeps from natural crude oil reservoir and erosion of sedimentary rock release PAHs into the environment. The release of petrogenic PAHs from anthropogenic activities is a significant route of PAHs contamination. Fuel-based industrial dependency has boosted economic growth; however, indiscriminate use of fossil fuels also engenders severe environmental pollution as a result of accidental as well as the intentional release of crude oil and refined products. Some important sources of petrogenic PAHs of anthropogenic origin are oceanic and freshwater oil spills, underground or aboveground storage tank leak, oil refinery waste, and leakage of crude and refined oil during transportation (Stogiannidis and Laane 2015). Since the last few decades, marine oil spills have become a recurring disaster. Amoco Cadiz (1978), Ixtoc I (1979), Atlantic Empress (1979), Exxon Valdez (1989), Kuwaiti Oil Lakes (1991), Kuwaiti Oil Fires (1991), Gulf War (1991), and Deepwater Horizon (2010) oil spills released massive amounts of crude and refined fuel rich in PAHs in aquatic environment (Hayakawa 2018).
Although the PAHs constituents of petrogenic sources vary greatly depending on the origins, LMW PAHs are the dominant representatives. Furthermore, most of the petrogenic release contains 16 US EPA priority pollutants and alkylated forms of 5 parental PAHs (alkylated naphthalene, phenanthrene, dibenzothiophene, fluorene, and chrysene). The parental compounds and “alkylated five” are used often as signature molecules for indicating petrogenic PAHs contamination of sediments (Laflamme and Hites 1978; Wang et al. 2001; Boll et al. 2008; Stogiannidis and Laane 2015). Coal tar and creosote are other important sources of PAHs. Creosote is a mixture of several hundreds of compounds; PAHs may constitute 90% of creosote. It is used widely as a wood preservative and waterproofing agent. Creosote enters soil and water mainly through wood preservation industry wastewater. Seeping and leakage of creosotes from treated timber may add PAHs in soil. A minor fraction (1–2%) of creosote is released in air through volatilization (Nestler 1974).
3.3 Diagenetic and Biogenic Sources
Not as pronounced and pernicious as pyrogenic and petrogenic sources, the biogenic and diagenetic process contribute to the environmental PAHs load. Crude oil, coal, and gases are formed from sedimentary algae, diatoms, phytoplankton, and bacteria through a process called diagenesis. Biogenic PAHs are derived from biosynthesis of the compounds in plant, phytoplankton, and microorganisms. Some endophytic fungi produce naphthalene (Daisy et al. 2002; Ezra et al. 2004). Naphthalene is also a major chemical component of Magnolia flower (Azuma et al. 1996). A microalga, Chlorella vulgaris, was found to synthesize several PAHs, including BaP, while growing in acetate-containing medium (Borneff et al. 1968).
In addition to the sources mentioned above, unburnt hard coal (bituminous coal) has recently been reported as a PAHs source (Achten and Hofmann 2009). Rochman et al. (2013) reported the sorption of several unsubstituted PAHs and their nitro- and methyl-substitutes on virgin polystyrene and polystyrene debris in marine environment suggesting that the polymers are a potential secondary source. Petrogenic sources are generally accidental and acute. They do not contribute to continuous contamination. In contrast, pyrogenic sources, especially incomplete combustion processes, are considered as the prominent and chronic sources of PAHs entering the environment (Duran and Cravo-Laureau 2016). Irrespective of sources, most of the released PAHs ultimately find their way to surface water, topsoil, the bottom surface of lakes, estuaries or rivers, and sediments either via airshed (dry and wet deposition) or watershed (e.g., urban runoff, rainfall, snow/ice fall, etc.).
4 Fate of PAHs in Aerobic Environments
The distribution, partition, transport, and fate of PAHs in the environment are intimately dependent on their sources, physicochemical properties, some environmental parameters, and biodegradation mechanisms. As their origin can be diverse, assigning a single source to a pool of PAHs in each environment is difficult (Blumer 1976). Moreover, post-emission alteration of PAHs gives rise to substitutes. As a result, the atmosphere contains a variable concentration of unsubstituted and substituted PAHs. A simplified overview of the possible fates of PAHs in the environment is presented in Fig. 3.

The possible fates of PAHs (in black dots) in the environment. Among the several possibilities, the most common ways of origin, transfer, and removal are illustrated
The fate of PAHs in air depends largely on temperature, humidity, precipitation, sunlight, and presence of atmospheric gases and acids. Temperature and humidity are the two main factors that govern the ratio of gas-to-particulate PAHs in the air (Maliszewska-Kordybach 1999). PAHs transformation in the atmosphere occurs mainly by chemical reactions and physical deposition. Reaction with ozone, nitrate and hydroxyl radicals, and acids derived from NOx and SOx and photolysis are accounted for the loss of gas-phase PAHs (Valerio et al. 1984). Alkyl-substituted PAHs are common constituents of crude and refined oil. Petroleum products are formed in sediment over a long period under pressure and temperature (150–200°C). Such comparatively mild temperature range favors the formation of alkylated derivatives so vigorously that the relative amounts of alkyl-PAHs may far exceed their parent compounds. Alkyl-PAHs have been found as the major pollutant in environments contaminated with crude and petroleum oil. Several methylated PAHs have been identified in urban air, street dust, and sediment. Alkyl-PAH also enter the environment through petrogenic, pyrogenic, and industrial sources (Miki et al. 2014; Tuyen et al. 2014; Wei et al. 2015). Nitro-PAHs are formed by nitration of parent PAHs during incomplete combustion or atmospheric gas-phase reaction. They enter the environment from automobile exhaust, waste incinerator, and domestic wood burning (Lima et al. 2005; Karavalakis et al. 2010; Shen et al. 2012; Bandowe and Meusel 2017). Oxygenated PAHs (oxy-PAHs) have one or more oxygen atom(s) attached to the aromatic structure of parent PAHs. Incomplete combustion is one of the major sources of oxy-PAHs. Parent PAHs may also transform to oxy-PAHs through light-induced reaction and chemical oxidation with singlet oxygen, peroxides, peroxyl radical, and hydroxyl radicals (Lundstedt et al. 2007). Particle-phase PAHs are also subjected to similar reactions. Most of the atmospheric PAHs deposit near their sources; however, PAHs with higher vapor pressure are transported to far away from their sources and are distributed worldwide. PAHs were detected in remote areas like Antarctic snow (Kukučka et al. 2010) and the Antarctic and Southern sea atmosphere (Cabrerizo et al. 2014). Physical removal of PAHs from the atmosphere generally occurs through dry deposition and wet deposition. In a dry deposition, PAHs are adsorbed on particulates and settle down slowly due to gravitational pull. The process depends on the size/mass of the particle and some environmental factors such as seasonal temperature, wind speed, turbulence, etc. Due to comparatively high aqueous solubility, atmospheric vapor-phase LMW PAHs may dissolve in cloud and raindrop that upon condensation of clouds settle down on Earth’s surface during precipitation. Particle-sorbed PAHs also settle on the surface through wet deposition. Transport of gas-phase PAHs from temperate or tropical warm regions of Earth to high-latitude cold regions is governed mainly by atmospheric temperature and vapor pressure of PAHs. The phenomenon can be explained by the global distillation effect theory. It predicts that atmospheric gas PAHs are transported to colder regions and condense. The deposited compounds may undergo several volatilization-transport-deposition cycles. This multiple hopping from low latitude to high latitude is known as the grasshopper effect. Consequently, PAHs would reach to the polar regions where low ambient temperature prevents their further transport; the effect is termed as “cold trap” or “cold finger” (Wania and Mackay 1993, 1995, 1996; Fernández and Grimalt 2003). Although not all atmospheric PAHs are removed, a significant portion finds their way to soil, water surface, and vegetation (Maliszewska-Kordybach 1999).
Soil can be contaminated with PAHs through different ways. Notable routes include dry and wet atmospheric deposition, automobile exhaust, sewage sludge, industrial effluent, seeping and leakage from coal tar creosote impregnated timbers, unburnt bituminous coal, roadway asphalts, accidental release of crude or refined oil during transportation and handling. PAHs in the soil can have different fates depending on the physical and chemical properties of PAHs, soil texture, soil organic matter (SOM) content, environmental conditions, and associated removal mechanisms. Sorption, sequestration, evaporation, photolysis, leaching, as well as biodegradation are the primary routes of PAHs processing in the soil. Sorption and sequestration processes play a significant role in PAHs accumulation in soil. Due to the strong sorption capacity of SOM and minerals, PAHs tend to be fixed with these substances (Means et al. 1980). The content, composition, and structure of SOM influence the sorption process. Also, soil particle size, the presence of clay minerals, and soil pH determine the extent of PAHs sorption to soil. Rhizosphere soil facilitates sorption of PAHs as root exudates increase soil structure (Wilcke 2000; Okere and Semple 2012). Evaporation or volatilization largely depends on daily and seasonal temperature. At elevated temperature, PAHs, mostly from topsoil, may evaporate quickly. Air current disperses the evaporated PAHs to a lower temperature region where they settle down through wet deposition (Sims and Overcash 1983; Wild and Jones 1995). Unlike atmospheric PAHs, very little soil PAHs are transformed through light energy. If any photo-destruction occurs, it remains confined to few millimeters of topsoil only. Therefore, photooxidation is not considered as a significant way of PAHs processing in the soil (Sims and Overcash 1983). Due to lower aqueous solubility and decreased mobility, leaching of unsubstituted PAHs is limited in the soil. However, semipolar derivatives (nitro-, oxy-, hydroxy-PAHs) show increased mobility in soil and hence dissipate to the soil column through micropores (Sims and Overcash 1983). Lipophilic nature of both PAHs and plant cuticle facilitate the accumulation of significant amounts of particle-bound PAHs in leaves, trunk, needles, and bark. Accumulated PAHs can enter the soil through plant litters during or at the end of vegetation period, precipitation, and near-stem runoff. Plant root system can uptake PAHs, and gas-phase PAHs may be accumulated by plant stroma and subsequently transported through vascular system or by diffusion; the cycle is completed at the end of vegetation (Wilcke 2000).
The marine environments including estuaries, coastal areas, ocean surface, and deep-sea shelter are diverse ecosystems. The fate of PAHs in the marine environment is determined mainly by the mode of PAHs entrance. Also, physical and chemical properties of PAHs, the presence of co-contaminants, sediment composition, environmental conditions, and hydrologic dynamics also influence PAHs fate in the marine environment (Latimer and Zheng 2003). Pyrogenic PAHs from combustion sources enter through urban runoff and atmospheric deposition. The accidental oil spill has become a significant means of PAHs entrance into the marine environment. Crude oil release from natural oil seep is the other notable source of PAHs in the environment. In the marine environment, petrogenic PAHs are more bioavailable because of the abundance of LMW PAHs, while pyrogenic PAHs are more recalcitrant as they remain sorbed to organic particulate matters. PAHs in the marine environment are transformed in abiotic processes such as volatilization, photooxidation, and chemical alteration. A significant portion of PAHs sinks vertically to marine sediment where microbial degradation becomes the primary fate of the pollutant. However, due to the hydrophobic nature of PAHs, they tend to become sorbed to sediment organic matters and mineral particles and thus become less bioavailable, hence persist in the marine environment (Acosta-González and Marqués 2016; Duran and Cravo-Laureau 2016).
Biodegradation is the principal mechanism for removing PAHs from the soil. The uptake and degradation of PAHs by microorganisms depend largely on soil temperature and other physicochemical properties of PAHs and the nature of the organisms. Bacteria, algae, and fungi can degrade many PAHs and their derivatives. Plants can also extract, sequester, and detoxify PAHs from the environment. In general, bacteria utilize PAHs as a carbon and energy source. Fungal degradation process, in contrast, leads to detoxification rather than mineralization (Cerniglia 1992; Samanta et al. 2002; Haritash and Kaushik 2009; Cerniglia and Sutherland 2010).
A growing body of literature deals with the bacterial degradation of PAHs from soil, water, and sediment under aerobic, microaerobic, and anaerobic conditions. Aerobic degradation of PAHs has been outlined in some excellent reviews (Cerniglia 1992; Juhasz and Naidu 2000; Kanaly and Harayama 2000; Peng et al. 2008; Haritash and Kaushik 2009). Aerobic bacterial degradation of PAHs, especially 2–5 ring PAHs, has been investigated well. Members of the genera, Pseudomonas, Sphingomonas, Mycobacteria, Burkholderia, Rhodococcus, Flavobacterium, Acinetobacter, and Klebsiella, have been frequently isolated from contaminated sites. Aerobic degradation of PAHs by bacteria involves the introduction of both atoms of oxygen to the aromatic structure producing cis-dihydrodiols. The enzyme dioxygenase is a multicomponent protein consisting of ferredoxin, ferredoxin reductase, and an iron-sulfur protein (Habe and Omori 2003). The resulting cis-dihydrodiols are then rearomatized to dihydroxylated intermediates by the action of dehydrogenases. Ring cleavage of the intermediates produce TCA cycle intermediates and finally mineralized to CO2 and H2O with the production of energy. In addition, identification of trans-dihydrodiol metabolites during PAHs degradation by certain strains of Mycobacterium and Streptomyces suggests cytochrome P450 oxygenases-mediated transformation also accounts for PAHs metabolism in bacteria (Sutherland et al. 1990; Tongpim and Pickard 1999).
Many fungi have been reported to degrade PAHs. In most of the cases, fungal degradation of PAHs is cometabolic. However, some fungi can utilize PAHs as the sole sources of carbon. For example, Fusarium solani was able to germinate on and mineralize BaP (Rafin et al. 2000). PAHs-degrading fungi generally belong to two major groups: (a) ligninolytic fungi that produce extracellular enzymes to degrade wood derived lignin and (b) non-ligninolytic fungi that do not possess lignin-degrading enzyme system. Lignin is a class of complex HMW compounds found in the vascular tissue of plants and some algae. Lignin bears structural resemblance to PAHs. Due to structural irregularity, lignin-decomposing enzymes show low substrate specificities; these enzymes can catalyze the transformation of several organic pollutants including PAHs. Among the wood-decaying and lignin-decomposing fungi, “white-rot fungi” has been studied extensively. Notable members of this group are Phanerochaete chrysosporium, Trametes versicolor, and Pleurotus ostreatus. The ligninolytic enzyme system involved in PAHs degradation comprises one or more of two glycosylated heme-containing peroxidases, lignin peroxidase (LiP), manganese-dependent peroxidase (MnP), and a copper-containing phenoloxidase, laccase. LiP oxidizes PAHs in the presence of H2O2, MnP oxidizes PAHs using Mn-dependent peroxidation of unsaturated lipids, whereas laccase oxidizes PAHs in the presence of phenol, aniline, 4-hydroxybenzoic acid, methionine, cysteine, or reduced glutathione as mediator. PAHs biodegradation by white-rot fungi initiates with the generation of hydroxyl free radical by the donation of one electron, which oxidizes the PAHs ring. The reaction products include PAH quinone and acids, which may be further metabolized to nontoxic intermediates or end products via ring fission (Cerniglia and Sutherland 2010).
Many non-ligninolytic hyphomycetes, zygomycetes, and ascomycetes can metabolize PAHs. Several species of Aspergillus, Penicillium, Fusarium, and Cunninghamella have been reported to transform and sometime mineralize PAHs. Many of these fungi utilize intracellular cytochrome 450 monooxygenases system that initiates PAHs metabolism through ring epoxidation reaction producing epoxide and water. The unstable epoxide is hydrated by an epoxide hydrolase to form trans-dihydrodiol or rearranged to phenol derivatives by nonenzymatic action. The reaction products, PAH trans-hydrodiol and phenols, are then methylated or form conjugates with sulfate, xylose, glucuronic acid, or glucose. Ligninolytic fungi may also involve in PAHs metabolism through the production of intracellular cytochrome P450 and epoxide hydrolase (Cerniglia and Sutherland 2010). Unfortunately, some fungal metabolites are more toxic than the substrate PAHs. Vázquez-Duhalt et al. (2001) reported the conversion of PAHs to mono-, di-, and tri-chlorinated compounds by the chloroperoxidase enzyme of Caldariomyces fumago in the presence of H2O2 and chloride ion. Some of these chlorinated compounds were more mutagenic than their parent PAHs.
PAHs are toxic to many aquatic animals and plants (Landrum et al. 1986; Yu 2002). Nevertheless, algal biotransformation of PAHs along with bacterial and fungal degradation is an important determinant of the fate of PAHs in the aquatic environment. Both fresh and marine water algae can degrade PAHs. A cyanobacterial strain, Agmenellum quadruplicatum PR-6, and a microalga, Oscillatoria sp. JCM, can oxidize naphthalene to 1-naphthol (Cerniglia et al. 1979, 1980). The green algae, Selenastrum capricornutum, Scenedesmus acutus, and Ankistrodesmus braunii, metabolize BaP through dioxygenase pathway and produce dihydrodiols and quinones. The degradation extent and metabolites were found to depend on light intensities, algal species, and dose (Schoeny et al. 1988; Warshawsky et al. 1995). Two diatoms, Skeletonema costatum and Nitzschia sp., isolated from mangrove aquatic ecosystem, were reported to accumulate and degrade phenanthrene and fluoranthene (Hong et al. 2008). Interesting enough, dead cells of algae retain PAHs removal capability. Lei et al. (2002) reported no significant differences in the removal of pyrene by live and dead cells of Chlamydomonas sp., Chlorella miniata, Chlorella vulgaris, Scenedesmus platydiscus, Scenedesmus quadricauda, S. capricornutum, and Synechocystis sp. This study suggested both biosorption (by dead cells) and bioaccumulation (inside live cells) as PAHs removal mechanisms by the microalgal cultures. Similarly, dead cells of S. capricornutum exhibited removal of several HMW PAHs including benz(a)anthracene, BaP, dibenzo(a,h)anthracene, indeno(1,2,3-cd)pyrene, and benzo(g,h,i)perylene (Luo et al. 2014). The same research team established that photocatalytic transformation of BaP is catalyzed by chlorophyll of dead algal cells through the formation of a high level of reactive singlet oxygen species (Luo et al. 2015). Soil microalgae have also been reported to degrade PAHs. A soil microalga, Chlorella sp. MM3, has recently been reported to degrade pyrene from both liquid media and soil slurry (Subashchandrabose et al. 2017). Algal transformation of PAHs is species-specific (Kirso and Irha 1998), and bacterial-algal consortia have been considered better suited than monoculture in the removal of PAHs from the environment (Warshawsky et al. 2007).
In addition to microbial degradation, several plants including grasses have been reported to play a role in the removal of PAHs from the environment. As with microbial bioremediation, phytoremediation of PAHs has been gaining recognition as an efficient pollutant-remediation technique (Sivaram et al. 2018). Plant-mediated transformation of PAHs involves the uptake of the pollutants from contaminated soil to the plant system through the root, translocation within the plant tissues, enzymatic breakdown or modification, conjugate formation, sequestration of conjugates within plant compartment, and further processing of the conjugates (Arthur et al. 2005). Moreover, plants facilitate immobilization of PAHs in soil and promote microbial degradation. Plant-microbe association, as in rhizosphere and mycorrhiza, is another means in determining the fate of PAHs in the environment (Ma et al. 2010).
As such, very little is known about the fate of PAHs in the anaerobic environment. Fresh and marine water sediments, sewage, subsurface aquifer sediment, and groundwater contamination with PAHs occur from anthropogenic activities such as shipping, boating, fishing, oil spill, leakage of coal tar, creosote and petroleum fuel from the surface storage tank, and gas production. The absence of molecular oxygen and sorption of PAHs to sediment organic matter are probably the most critical factors that govern persistence of PAHs in such marine environment.
5 Onset of Anoxia and Nature of Anoxic Environment
Unavailability of gaseous and dissolved oxygen in an environment renders it anoxic. The atmosphere, as we know it now, consists of the essential gaseous mixture for supporting life. The strong oxidizing gas, oxygen, is an absolute requirement for aerobic respiration. It acts as the terminal electron acceptor (TEA) in the aerobic cellular respiration process and participates in many biological reactions as a co-substrate. During the formation of Earth, it experienced extended anoxia until molecular oxygen began accumulating in the atmosphere when ancient microbial life forms breathed out the gas. After millions of years of accumulation, we are now breathing in an atmosphere consisting of ~21% oxygen (Margulis and Sagan 1997; Planavsky et al. 2014).
In well-structured and drained soil, gaseous oxygen penetrates through the cracks and pores. As a result, the topsoil layer becomes sufficiently oxygenated and supports aerobic microbial metabolism and root respiration (Drew 1990). Hydrosphere can be saturated with atmospheric oxygen to the extent that the maximum dissolved oxygen (DO) level can reach as much as 9–10 mg L−1 (McNeely et al. 1979). Often oxygen level becomes limited below few millimeters/centimeters along soil/sediment profile. Wet or waterlogged soil, sediment overlaid by stagnant or constant-depth water column, and subsurface groundwater aquifers generally contain very limited DO that, in many cases, reaches a zero value. Moreover, specific habitats like marine sediment remain in permanent anoxia (Kaiho 1994). Contrary to the complete aerobic and anaerobic environment, DO level in a hypoxic environment ranges between 0 and 4.5 mg L−1 (Wu 2002). Many habitats such as wetlands and swamps exhibit hypoxic condition. The DO level of an environment is highly influenced by temperature, salinity, and microbial activity (Brune et al. 2000). Sediment top layer receives considerable input of organic biomass from terrestrial and aquatic algae, plants, and animals. During the decomposition of organic compounds, aerobic microorganisms use available oxygen. As a result, DO level decreases with time. Once the oxygen demand of a habitat exceeds the oxygen dissolution rate, anoxia begins to develop. Moreover, continuous input of a large amount of natural and synthetic organic compounds in water, soil, or sediment environment exacerbates the DO level and creates an oxygen-depleted anaerobic condition (Burdige 2007).
A redox gradient along its depth characterizes an anoxic environment (Fig. 4). Extensive use of oxygen during microbial decomposition of organic matters fosters the formation of a redox gradient that is characterized by rapid decrease in DO level and redox potential across the gradient and the variable availability of alternate electron acceptors for microbial respiration (Fig. 4) (Brune et al. 2000; Li et al. 2009). Depending on the characteristics of the site, nitrate, manganese, iron, and sulfate become the dominant electron acceptors. The gradient starts at a transition zone where nitrate, manganese(IV), and iron(III) are used preferentially as a TEA. Down to the transition zone, sulfur reduction process turns out to be the prominent anaerobic respiration regime. The methanogenic zone may be developed further below a sulfidic regime where methane production often occurs through interspecies syntrophic metabolism (Cappenberg 1974; Acosta-González and Marqués 2016).

Schematic of a typical sediment column characterized by decreasing redox gradient below the oxidized zone. In the transition zone, microbial respiration processes use NO3−, NO2−, Mn4+, Fe3+, and SO42− as terminal electron acceptors. With increasing depth, redox potential drops dramatically, and sulfur reduction becomes predominant in the high negative redox potential zone. Further down to this sulfidic zone, methanogenic activity by strict anaerobic bacteria and archaea may be present
In the anoxic environment, anaerobes are the key players in geochemical cycling. Cellular respiration in oxygen-depleted conditions is challenging because oxygen is no longer available or “died out.” The Gibbs free energy (ΔG°′) change in the oxidation of NADH (E0′ = −320 mV) coupled with oxygen reduction to water (E0′ = +818 mV) is −220 KJ mol−1. When the same reaction coupled with NO3− reduction to NO2− (E0′ = +433 mV) and CO2 reduction to CH4 (E0′ = −244 mV), the free energy changes decrease by −56.2 KJ mol−1 and −157.2 KJ mol−1 for the respective processes (Thauer et al. 1977). Microorganisms being the first forms of life on Earth show adaptation to a range of oxygen environments. On the contrary, some of them are fitted out at surviving in the total absence of oxygen. It comes with a surprise to think that the first life form, no matter whether it emerged in an ancient “pond of soup” or in a hydrothermal vent (Nisbet and Sleep 2001; Weiss et al. 2016), might be capable of thriving under anoxic conditions. Still, we know very little about the physiology and metabolism of anaerobic microorganisms. Facultative anaerobes are the significant occupants in the transition zones, while strict anaerobes govern biogeochemical cycling at sulfate-reducing and methanogenic zones (Lovley 2001). Down to the gradient, the reduction potential drops abruptly, and the net energy yield during oxidation, per molecule, of organic matter decreases strongly. Anaerobic lifestyle, therefore, should be parsimonious enough to allow efficient survival, maintenance, and cell growth with a limited amount of available energy. Many anaerobic metabolisms even run close to the thermodynamic limits (Fuchs et al. 2011). Although several growth-limiting constraints exist, anaerobes contribute crucially to the global biogeochemical cycle.
6 Persistence of PAHs Under Anaerobic Conditions
In an anaerobic environment, PAHs are susceptible to microbial enzymatic degradation if they are bioavailable, enough appropriate inorganic electron acceptors are present, and the native microflora possess genetic setup for encoding necessary degradative enzymes. The absence of one or more of the prerequisites may affect PAHs degradation process that consequently would lead to recalcitrance and accumulation of the pollutants in the environment. Additionally, PAHs often occur with other mixed contaminants in real impacted sites. The presence of other toxic substances such as heavy metals, cyanides, and organic compounds can impede PAHs degradation (Kuppusamy et al. 2017).
Under anaerobic condition, PAHs may resist microbial degradation. Sharak Genthner et al. (1997) reported the persistence of many PAHs tested in their study under various redox conditions. Only little degradation of naphthalene, 1-methylnaphthalene (MN), and 2-MN occurred under methanogenic conditions. In a methanogenic sediment column, benzene as well as naphthalene remained persistent for 20–40 days even after the addition of simpler co-substrates such as acetate, benzoate, lactate, and phenol (Langenhoff et al. 1996). Bauer and Capone (1985) observed that although anthracene and phenanthrene were degraded relatively well under aerobic conditions, they remained persistent under anaerobic conditions. In another study, HMW PAHs in contaminated arctic soils remained refractory to biodegradation under nitrate-reducing conditions at both low (7°C) and moderate (20°C) temperature. Under aerobic conditions, however, the HMW PAHs were degraded well (Eriksson et al. 2003). Recently, Folwell et al. (2016) reported persistence of pyrene and naphthenic acids in oil-sand-water from tailing pond. Failure to establish PAHs-degrading enrichment culture in laboratory microcosm study may be attributed to the absence of appropriate electron acceptor, reducing conditions and essential nutrients in the culture media. In addition, very low or total absence of requisite microbial population and their slow adaptation to the contaminants may also lead to the failure in developing enrichments. Inhibition of degradation by co-occurring contaminants may also contribute to the apparent persistence of PAHs in the environment or a laboratory microcosm.
7 Feasibility of Anaerobic Biodegradation of PAHs
The stabilizing resonance energy of the aromatic compounds is the major hindrance to microbial degradation. Moreover, unavailability of oxygen in an anaerobic environment presents another critical challenge to microorganisms that require to use aromatic compounds as growth substrates (Fuchs et al. 2011). In the absence of oxygen, where it is no longer available to accept electrons during respiratory electron transport, anaerobes use several inorganic ions or compounds as TEA (Fig. 5). Nitrate is one of the first components in anaerobic nitrate respiration process; facultative nitrate reducers can harness energy by reducing nitrate to different nitrogen oxides and molecular dinitrogen. From a thermodynamic point of view, standard Gibbs free energy change in nitrate reduction process is close to that in aerobic respiration process (−220 KJ mol−1 vs −163.2 KJ mol−1) (Thauer et al. 1977). Thus, nitrate reduction is a widespread process often associated with degradation of POPs. Nitrate-reducing microorganisms mainly belong to the facultative anaerobic group of bacteria. If available, oxides of manganese and iron(III) can also act as TEA in anaerobic metabolism. Contaminants often move down to the gradient to the utterly anoxic zone. SRB often degrade organic compounds in strictly anaerobic conditions. Fortunately, sulfate is abundant in many anaerobic systems such as marine sediment. SRB are considered the oldest life forms that can be dated back to 3.5 billion years. Soon after the formation of Earth, SRB have been contributing to the biogeochemical cycling (Barton and Fauque 2009).

Conceptual representation of anaerobic degradation of PAHs under various reducing conditions. The standard Gibbs free energy changes (ΔG°′) for the redox couples under standard conditions and pH 7.0 in reduction processes of electron acceptor are obtained from Thauer et al. (1977)
The primary challenge in organic pollutant degradation through sulfur reduction process is low changes in Gibbs free energy. Despite the limitation, many SRB are known to degrade many POPs including PAHs. For a long time, anaerobic degradation of hydrocarbon compounds under methanogenic conditions was considered as thermodynamically improbable. However, the evidence is accumulating in support of the methanogenic degradation of crude oil in the deep subsurface environment (Aitken et al. 2004). Dolfing et al. (2009) calculated free energy change during the oxidation of PAHs under methanogenic conditions. The ΔG°′ values for methanogenic naphthalene, phenanthrene, anthracene, pyrene, and chrysene degradation ranged from −208.8 to −331.4 kJ mol−1, and the energy yield per mole CH4 generation was in the range of −27.1 to −34.8 kJ mol−1. Although the reaction is exergonic, sharing of energy among the associated interdependent microbial members of an anaerobic syntrophic metabolism makes the process challenging. The authors predicted that oxidation to H2/CO2 or conversion to acetate is energetically more favorable for PAHs degradation under methanogenic conditions. The calculation of free energy changes under standard conditions (25°C, atmospheric pressure) during anaerobic oxidation of four model PAHs indicates that the anaerobic oxidation processes under nitrate-reducing, sulfate-reducing, and methanogenic conditions are exergonic (Table 2). The Gibbs free energy change and the ATP produced per mole of substrate oxidation for any of the PAHs are the highest under denitrifying process and lowest under methanogenic conditions. With increasing molecular weight, energy yield also increases. However, the thermodynamic calculation is based on differences between the formation energy of reactants and products. Therefore, calculations of free energy changes under standard conditions are not directly applicable to environmental conditions. In a real environment, the biodegradability of PAHs depends on some factors that are not considered in a free energy change calculation. Such factors include molecular weight and conformation-related properties such as solubility, logKOW, affinity to organic matter, and bioavailability. Nevertheless, Table 2 indicates the feasibility of anaerobic degradation of PAHs such as naphthalene, phenanthrene, pyrene, and BaP.
8 PAHs Biodegradation Under Nitrate-Reducing Conditions
8.1 PAHs Degradation by Nitrate-Reducing Bacteria
Nitrate reduction is a crucial microbial respiration process that is often adopted by facultative anaerobes in many organic-rich and oxygen-depleted environments. The process produces enough energy that is comparable to aerobic respiration as the reduction potential of nitrate is close to oxygen. Denitrification process leads to the conversion of nitrate to dinitrogen via various oxides of nitrogen (Kuypers et al. 2018). Nitrate-reducing bacteria are versatile aromatic hydrocarbon degraders. Benzene degradation under the nitrate-reducing condition is a well-documented process (Majora et al. 1988; Nales et al. 1998; Burland and Edwards 1999; Coates et al. 2001; Folwell et al. 2016). Mihelcic and Luthy (1988a) were the first to demonstrate PAHs degradation under nitrate-reducing conditions. In this study, aqueous-phase concentration of spiked naphthalene and acenaphthene did not change during anaerobic incubation without an external electron acceptor. When nitrate was added to the culture medium, complete degradation of naphthalene and acenaphthene was observed despite a lag of about 2 weeks. Since then, PAHs degradation by several nitrate-reducing enrichments and pure cultures has been reported so far. Table 3 summarizes many of the available reports on PAHs degradation under nitrate-reducing conditions.
PAHs degradation under nitrate-reducing conditions is widespread in nature: from pristine to contaminated samples, temperate to arctic soils, freshwater to marine sediment, petrochemical to sewage sludge, etc. (Mihelcic and Luthy 1988b; Al-Bashir et al. 1990; Leduc et al. 1992; Murphy et al. 1995; MacRae and Hall 1998; McNally et al. 1998; Rockne and Strand 1998, 2001; Rockne et al. 2000; Chang et al. 2003; Eriksson et al. 2003; Ambrosoli et al. 2005; Dou et al. 2009; Lu et al. 2011; Yang et al. 2013; Liang et al. 2014; Qin et al. 2017, 2018). Most of these studies used classical ecology approach, i.e., microcosm incubation, to investigate PAHs degradation using contaminated or uncontaminated samples, non-reduced mineral salt, nitrate as the TEA and PAHs as the electron donor. Rockne and Strand (1998) adopted a fluidized bed reactor (FBR) approach for enriching PAHs-degrading nitrate-respiring bacteria (NRB). Langenhoff et al. (1996) studied naphthalene degradation in a soil percolation column. Also, PAHs degradation by pure bacterial cultures has also been established (McNally et al. 1998; Rockne et al. 2000; Yang et al. 2013; Qin et al. 2017, 2018).
In the natural environment, nitrate-reducing facultative anaerobes degrade both LMW and HMW PAHs. Several nitrate-reducing microcosm studies demonstrated naphthalene degradation using soil, sediment, and sludge samples. Acenaphthene, anthracene, fluorene, fluoranthene, phenanthrene, pyrene, and BaP degradation have also been reported. Al-Bashir et al. (1990) described naphthalene mineralization in both pristine and oil-contaminated soil slurry under denitrifying conditions. Naphthalene degradation was observed in a nitrate-amended sediment column only after the addition of benzoate (Langenhoff et al. 1996). McNally et al. (1998) isolated three nitrate-reducing facultative anaerobic pure bacterial cultures that could degrade acenaphthene, phenanthrene, and pyrene, both aerobically and anaerobically. A FBR enrichment culture that was developed from coal tar creosote-contaminated sediment could degrade naphthalene and phenanthrene (Rockne and Strand 1998). Subsequently, subculture was obtained through the transfer of FBR cell mass and biocarrier to PAHs amended media. The subculture showed nitrate-dependent mineralization of naphthalene and phenanthrene (Rockne and Strand 2001). Pure cultures isolated from the FBR enrichment showed higher degradation ability although at a low rate compared to the original FBR enrichments (Rockne et al. 2000). Degradation of acenaphthene, anthracene, phenanthrene, fluorene, and pyrene under nitrate-reducing conditions was also demonstrated in soil, sediment, and sludge (Chang et al. 2002, 2003; Yuan and Chang 2007). Eriksson et al. (2003) investigated anaerobic biodegradation potential of contaminated arctic soil at low temperature under nitrate-reducing conditions. Only naphthalene and 2-MN were entirely degraded by the enriched culture; fluorene and phenanthrene were also degraded to a lesser extent. HMW PAHs used in this study remained persistent at both 7 and 20°C temperature. Dou et al. (2009) demonstrated naphthalene degradation by the nitrate-reducing mixed culture at different doses. Among the 16 priority PAHs, 2- and 3-ring members have been shown to be degraded more efficiently by sediment enrichment culture under nitrate-reducing conditions than sulfate-reducing conditions (Lu et al. 2012). Experimental evidence of HMW PAHs degradation under the nitrate-reducing conditions is scarce. Only recently, some pure bacterial cultures capable of degrading HMW PAHs have been obtained. Yang et al. (2013) isolated Pseudomonas sp. JP1 form river sediment that can degrade BaP, fluoranthene, and phenanthrene. Liang et al. (2014) isolated a pyrene-degrading bacterium, Paracoccus denitrificans, from river sediment. Cellulosimicrobium cellulans CWS2 that has been isolated from coking plant soil could degrade BaP (Qin et al. 2018). Qin et al. (2017) isolated a BaP-degrading Microbacterium sp. strain from contaminated soil. It is worth noting here that some habitats such as a continental shelf, shallow lake, and wetland experience fluctuations in oxygen level on a daily or seasonal basis. Facultative anaerobes, especially NRB, might have a potential role in PAHs removal from these habitats. However, to the best of our knowledge, no study so far has investigated the role of nitrate reducers in PAHs removal from an environment that experiences fluctuating oxygen regime.
8.2 PAHs Biodegradation and Nitrate Consumption
PAHs degradation under nitrate-reducing conditions depends on the availability of nitrate to support respiration (Mihelcic and Luthy 1988a, b; Al-Bashir et al. 1990; Rockne and Strand 1998, 2001; Rockne et al. 2000). In a soil-water system, limiting nitrate concentration did not allow naphthalene and acenaphthene degradation. Only when excess nitrate was provided, degradation of the substrates commenced (Mihelcic and Luthy 1988b). Al-Bashir et al. (1990) demonstrated a linear relationship between naphthalene mineralization and nitrate depletion. Stoichiometric depletion of nitrate and degradation of PAHs was also observed by Rockne and Strand (1998). Recently, Qin et al. (2017) reported that BaP degradation by Microbacterium sp. was affected by C:N ratio and BaP:nitrate ratio of 1:33 resulting in 84.2% degradation in 10 days.
Nitrate demand for anaerobic oxidation (per mole of PAHs) depends on the reduction chemistry. Theoretically, one mole of naphthalene degradation requires 9.6 mol of nitrate assuming complete denitrification (Table 2) and 24 mol assuming partial reduction to nitrite (Dou et al. 2009). However, experimental values (10.71–12.02 mol) obtained by Dou et al. (2009) were in between the theoretical values. Additionally, the observed disproportion between nitrate consumption and nitrite accumulation suggested that only a fraction of nitrite is converted to dinitrogen rather than complete denitrification (Dou et al. 2009). A similar relationship between nitrate and PAHs depletion was also observed by Rockne and Strand (1998). The reaction stoichiometry of PAHs degradation and nitrate reduction is crucial for determining the extent of nitrate amendment for stimulation of biodegradation and, at the same time, avoiding adverse effects of nitrate, nitrite, and nitrogen oxides.
8.3 Enhanced PAHs Biodegradation by Nitrate Amendment
Available nitrate that initially supports the microbial degradation of organic compounds would be depleted in an environment with high contaminant load. It should be noted that total organic carbon load is also important in the context of nitrate availability in a given habitat. In such a situation, replenishing nitrate by external amendment could help to resume biodegradation. Naphthalene and phenanthrene degradation were ceased when nitrate was depleted from the media. Refeeding of the culture with nitrate re-established degradation of PAHs (Rockne and Strand 2001). Nitrate was injected into PAHs-contaminated Hamilton sediment, Canada, to enhance biodegradation. Among the 16 priority PAHs, 15 of them were degraded in the sediment (Murphy et al. 1995). Tang et al. (2005) demonstrated that slow release of nitrate from nitrocellulose in an anaerobic marine sediment increased phenanthrene degradation by 2–3 orders of magnitude.
The above findings suggest that nitrate addition to the anoxic contaminated environment may be useful for enhancing bioremediation. Nitrate amendment in PAHs-contaminated soil enhanced the abundance and activity of denitrifying bacteria and induced a shift in microbial community structure (Zhou et al. 2017). To examine the effect of nitrate addition to contaminated sediment, Xu et al. (2014) injected Ca(NO3)2 solution into the sediment of a field-scale in situ bioremediation site. They observed changes, induced by nitrate addition, in functional diversity, composition, structure, and dynamics of sediment microbial communities using GeoChip 4.0 gene array technology (Tu et al. 2014). Functional genes involved in C, N, P, and S cycling were enriched in metabolically versatile microbial members of the community. Reduced total organic carbon (TOC) as well as polybrominated diphenyl ethers and PAHs level after injection indicated that the nitrate amendment was effective in increasing potential of the sediment microflora in PAHs bioremediation. Xu et al. (2015) also reported enrichment of several aerobic PAHs-degrading genes in the nitrate-amended sediment. However, differential enrichment of genes involved in anaerobic PAHs degradation after nitrate amendment in a real contaminated environment has not been documented yet. It should be noted that the fate of supplied nitrate depends on soil chemistry, C:N ratio, total carbon load, temperature, and concentrations of nitrate-nitrite and sulfide (Tiedje et al. 1983; Akunna et al. 1993; Kraft et al. 2014). Excess of nitrate and nitrite affects biodiversity and ecosystem (Sutton et al. 2011). Therefore, the dosage of nitrate amendment to a contaminated sediment should be carefully determined to ensure considerable degradation and avoidance of excess nitrogen toxicity.
9 PAHs Biodegradation Under Iron-Reducing Conditions
Iron constitutes approximately 80% of the inner and outer cores of Earth. It is the fourth most abundant element in Earth’s crust (Frey and Reed 2012). Iron-reducing bacteria participate in the anaerobic degradation of organic matter (Canfield et al. 1993), BTEX compounds (Edwards et al. 1992; Jahn et al. 2005), phenols, and p-cresol (Lovley and Lonergan 1990). However, very few reports on PAHs degradation under iron-reducing conditions are available. Anderson and Lovley (1999) demonstrated anaerobic naphthalene oxidation to CO2 in petroleum-contaminated aquifer sediment where the iron reduction was the terminal electron-accepting process. An iron-reducing enrichment culture, N49, degraded naphthalene. It was enriched from a sediment sample of monitoring well set at a former MGP site (Kleemann and Meckenstock 2011). The culture is composed mainly of one bacterial member that is closely related to the significant organism in the iron-reducing, benzene-degrading enrichment culture, BF, as revealed by T-RFLP pattern and 16S rRNA gene sequences. Apart from naphthalene, N49 can also grow on 1-MN, 2-MN, 1-naphthoic acid (1-NA), or 2-NA. Hydrogenophaga sp. PYR1, an iron-reducing facultative anaerobe, has been recently isolated from PAHs-contaminated river sediment (Yan et al. 2017). This biosurfactant-producing bacterium degraded both pyrene and BaP under both aerobic and iron-reducing conditions. Marozava et al. (2018) enriched a 1-MN-degrading culture from contaminated soil at a former coal gasification site using Fe(III) as the TEA. The enrichment culture consisted of two bacteria related to uncultured Gram-positive Thermoanaerobacteraceae and uncultured Gram-negative Desulfobulbaceae. The culture could also grow on naphthalene and 2-MN.
10 PAHs Biodegradation Under Sulfate-Reducing Conditions
10.1 PAHs Degradation by Sulfate-Reducing Bacteria
Sulfur is one of the most abundant elements on Earth. Sulfate ion significantly influences microbial activities in anaerobic environments (Capone and Kiene 1988). Dissimilatory sulfate reduction by anaerobic bacteria and archaea is a crucial and perhaps one of the earliest biochemical processes on Earth. SRB play a crucial role in global sulfur cycling (Muyzer and Stams 2008). SRB belong to ~23 bacterial genera representing only 7 phylogenetic lineages, 5 within bacteria (Deltaproteobacteria, Gram-positive Clostridia, Nitrospirae, Thermodesulfobacteria, and Thermodesulfobiceae) and 2 within archaea (Euryarchaeota and Crenarchaeota) (Muyzer and Stams 2008). During anaerobic degradation of organic matter, SRB use sulfate as the TEA and produce hydrogen sulfide in this process. SRB are widespread in freshwater and marine sediment, aquifer materials, hydrothermal vent, volcanic mud, and anaerobic sludge (Widdel and Bak 1992; Muyzer and Stams 2008). Earth’s ocean is a main sink of sulfate; hence, sulfate is not a limiting nutrient in the marine environment. Thus, anaerobic degradation of organic matter in marine sediments by SRB becomes a major element cycling mechanism. Some SRB can degrade organic pollutants such as BTEX compounds (Edwards et al. 1992; Lovley et al. 1995; Phelps et al. 1996; Meckenstock et al. 2016). To date, PAHs degradation coupled with sulfate reduction has been demonstrated in many enrichments and pure cultures. Table 4 summarizes most of the available reports on PAHs degradation under sulfate-reducing conditions.
Among the PAHs, the processes of naphthalene and 2-MN degradation by SRB are better explored. Most of the information on genetics and biochemistry of anaerobic degradation of PAHs has been obtained from naphthalene and 2-MN-degrading SRB enrichments and pure cultures. SRB are abundant in sediment; so, it is not surprising that the majority of PAHs-degrading SRB cultures are obtained from freshwater and marine sediments. Coates et al. (1997) obtained naphthalene- and phenanthrene-degrading enrichment cultures from contaminated marine harbor sediments. PAHs oxidation rate was higher in the heavily contaminated sediment than that in less contaminated sediment. The former sediment enrichment could also degrade methylnaphthalene, fluorene, and fluoranthene under sulfate-reducing conditions. Bedessem et al. (1997) established several sulfate-reducing naphthalene-degrading enrichment cultures from creosote-contaminated aquifer sediment and maintained them throughout for 3 years. After repeated feeding with naphthalene, the duration of initial lag (1–20 weeks) was reduced to a minimum, and the adapted enrichment could mineralize 66% of added 14C-naphthalene to 14CO2 in 13 days. Zhang and Young (1997) enriched naphthalene- and phenanthrene-degrading culture from contaminated harbor sediment under strict sulfate-reducing conditions. The cultures could degrade 150–200 μM naphthalene and phenanthrene within 150 days. N47, which is one of the thoroughly investigated enrichment cultures, was derived from the soil of a contaminated aquifer near Stuttgart, Germany (Meckenstock et al. 2000). The culture utilized naphthalene and 2-MN without a significant lag (Annweiler et al. 2000; Meckenstock et al. 2000). Thus, N47 is one of the few anaerobic PAHs-degrading cultures that can consistently degrade naphthalene and 2-MN upon repeated transfer. Terminal fragment length analysis and 16S rRNA gene sequencing of N47 revealed that the culture is composed of an unidentified member of Deltaproteobacteria in association with 7% of Spirochaetes members. In addition to naphthalene and 2-MN, N47 can also co-metabolically degrade different poly- and heterocyclic aromatic hydrocarbon compounds (Safinowski et al. 2006). Rothermich et al. (2002) demonstrated mineralization of 14C-naphthalene and 14C-phenanthrene in sulfidogenic contaminated harbor sediment.
Moreover, degradation of in situ PAHs pool (naphthalene, 1-MN, 2-MN, acenaphthene, fluorene, phenanthrene, anthracene, fluoranthene, pyrene, benzo(a)anthracene, chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene, and BaP) was demonstrated in sediment microcosm (Safinowski et al. 2006). In this study, LMW PAHs except naphthalene were degraded more rapidly than HMW congeners. Davidova et al. (2007) enriched a phenanthrene-degrading culture from hydrocarbon-contaminated marine sediment. The culture is mainly composed of members of Deltaproteobacteria that are like other known hydrocarbon degraders and uncultured clones obtained from hydrocarbon-degrading communities. An SRB enrichment culture was obtained from swine sewage sludge that can degrade fluorene and phenanthrene (Tsai et al. 2009). The enrichment degraded 88% fluorene and 65% phenanthrene (initially 5 mg L−1 each) after 21 days of operation. However, when the substrate mixture was provided as a carbon source, the degradation rate decreased, indicating that an enrichment or a pure culture may degrade a single compound more efficiently than mixed substrates. Environmental contaminants exist as conglomeration and degradation efficiency of culture in microcosm might not necessarily be the same as in real contaminated site. An SRB pure culture, NaphS2, isolated from North Sea harbor sediment, degraded naphthalene and 2-MN under sulfate reduction conditions (Galushko et al. 1999). Two other pure strains, NaphS3 and NaphS6, were isolated from Mediterranean lagoon sediment using naphthalene as the substrate (Musat et al. 2009). Both strains also utilized 2-MN as the sole carbon source. All the pure bacterial strains (NaphS2, NaphS3, and NaphS6) are affiliated with δ-subclass of Proteobacteria and are closely related to SRB that degrade some other aromatic compounds. Five naphthalene-degrading SRB enrichment cultures (SobN1, MicN1, GölN1, EgN1, and EgN2) were enriched from contaminated groundwater and aquifer sediment (Kummel et al. 2015). These highly enriched cultures degraded naphthalene at a much higher concentration (7.8 mM) with appreciable degradation rates (7–13.3 μM day−1). Acenaphthene and phenanthrene degradation ability of the enrichment cultures was tested, but none of the cultures could utilize the PAHs. Very recently, Himmelberg et al. (2018) enriched phenanthrene-degrading and sulfate-reducing culture, namely, TRIP1, from muddy soil mixture sample in which a member of Desulfobacteraceae that was very closely related to naphthalene-degrading strain, NaphS2, dominated in the enrichment.
The coupling of sulfate reduction with PAHs degradation has been demonstrated in many sulfate-reducing enrichment cultures. Partial or complete inhibition of PAHs degradation upon the addition of sulfate reduction inhibitor is a hallmark of direct involvement of SRB in PAHs degradation process. Several studies used sodium molybdate (usually 20 mM) as sulfate reduction inhibitor to demonstrate the role of SRB in PAHs degradation (Coates et al. 1996; Bedessem et al. 1997; Annweiler et al. 2000; Meckenstock et al. 2000; Rothermich et al. 2002; Davidova et al. 2007). Besides, stoichiometric sulfate loss from the media assuming complete mineralization of substrate PAHs was also demonstrated (Davidova et al. 2007). Furthermore, the reduction of 35S-sulfate to 35S-sulfide during naphthalene degradation by a sulfate-reducing enrichment microcosm was demonstrated by Bedessem et al. (1997). Although anaerobic degradation of some PAHs, exceptionally low-molecular-weight congeners, under sulfate reduction condition is better investigated and biochemical mechanisms of naphthalene and 2-MN degradation have been elucidated, similar investigations on HMW PAHs such as pyrene, BaP, or chrysene are rarely reported. Pyrene degradation under sulfate-reducing conditions has been reported in some instances. However, no culture capable of consistently degrading pyrene has been identified.
10.2 Enhanced PAHs Biodegradation by Sulfate Amendment
Although sulfate is abundant in seawater, availability of sulfate in anoxic sediment depends on diffusion of seawater sulfate to sediment. Moreover, utilization of sulfate during anaerobic oxidation of organic substances may exceed sulfate supply from overlaying sulfate-rich water (Martens and Val Klump 1984). On the other hand, freshwater sediments are generally low in sulfate content compared to marine sediments. In general, sulfate reduction will predominate over methanogenesis if sulfate supply is enough to sustain the process (Muyzer and Stams 2008). In both fresh and marine water systems, when sedimentary sulfate concentration decreases because of the extensive activity of SRB or other physicochemical reasons, methanogenic degradation becomes the primary means of organic compound decomposition. Methanogenesis, however, is a less efficient energy-generating process than sulfate reduction. In sulfate-depleted sediment, the external amendment could help to restore SRB-mediated bioremediation. Sulfate addition has been shown to be effective in stimulating anaerobic benzene (Weiner et al. 1998; Anderson and Lovley 2000) and BTEX (Cunningham et al. 2001) degradation.
The addition of sulfate in the form of soluble sodium sulfate and less soluble gypsum to a sulfate-depleted methanogenic sediment stimulated naphthalene and 2-MN degradation (Rothermich et al. 2002). Similarly, the controlled release of sulfate in marine sediment significantly enhanced anaerobic degradation of phenanthrene (Tang et al. 2005). Sulfate addition in more soluble form may be practicable for enhancing bioremediation potential of contaminated groundwater and aquifer sediment, whereas addition of less soluble form (e.g., gypsum) may be useful in alleviating sulfate concentration in the marine system (Rothermich et al. 2002). Nonetheless, Bach et al. (2005) did not find any stimulatory effect of sulfate addition in a sulfate-deficient PAHs-contaminated estuarine sediment. To date, the effect of sulfate amendment on shaping sediment microbial community structure and function and in situ demonstrations of the stimulatory effect of sulfate in PAHs degradation have not been reported.
11 PAHs Biodegradation Under Methanogenic Conditions
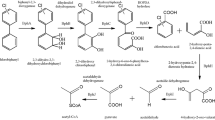
PAHs degradation under methanogenic conditions is a thermodynamically challenging process, as the net gain of ATP during the biodegradation process is extremely low (Dolfing et al. 2009). Complex organic compounds such as polysaccharides, halogenated organic compounds, alkanes, as well as PAHs can be transformed via syntrophic metabolism which involves cross-feeding between microbial species (Schink 1997). In anaerobic syntrophic metabolism, substrates are first hydrolyzed to acetate, longer-chain fatty acids, propionate, alcohols, CO2, formate, and H2 by fermentative bacteria. Subsequently, the other crucial participant of a syntrophic consortium, methanogenic bacteria, utilizes some of the products of the preceding fermentative metabolism to reduce CO2 to CH4. The conversion of the most oxidized state of carbon, i.e., CO2, to the most reduced form, i.e., CH4, generates adequate energy that makes the overall anaerobic transformation process thermodynamically feasible (McInerney et al. 2009). Christensen et al. (2004) inferred from a thermodynamic calculation that methanogenic degradation of naphthalene is feasible only in the presence of hydrogen-utilizing methanogens. PAHs are widespread in fuel-contaminated methanogenic sediments, oil reservoirs, and groundwater aquifers. Methanogenic degradation of PAHs can be a major route of PAHs detoxification in methane-rich environments. So far, degradation of some LMW PAHs under methanogenic conditions has been reported in contaminated soils, sewage and petrochemical sludge, and sediments (Table 5). However, the degradability of HMW PAHs under methanogenic conditions remains elusive due to the dearth of experimental evidence. Methanogenic degradation of PAHs with two or more rings was demonstrated in contaminated sewage sludge (Trably et al. 2003; Christensen et al. 2004; Cea-Barcia et al. 2013). Zhang et al. (2012b) demonstrated incorporation of labeled carbon from 13C6-anthracene in the microbial community of landfill leachate-contaminated subsurface aquifer sediment under methanogenic conditions. Production of methane relative to the sterile control in this study also suggested methanogenic degradation of anthracene. In another study, naphthalene- and phenanthrene-degrading methanogenic enrichment cultures were developed with Baltimore Harbor sediments without the addition of any external electron acceptors (Chang et al. 2001). As PAHs are natural components of crude oil, microorganisms that can degrade alkane compounds may also adapt to and degrade aromatic hydrocarbons. Berdugo-Clavijo et al. (2012) examined the ability of a methanogenic enrichment culture in biodegradation of naphthalene and methylated naphthalene substitutes. The methanogenic enrichment was obtained from aquifer sediment that was contaminated with natural gas compensate. The culture was able to degrade alkane fraction of crude oil with concomitant production of methane (Townsend et al. 2003; Gieg et al. 2008). Under methanogenic conditions, 2-MN- and 2,6-dimethylnaphthalene-amended enrichments produced methane gas compared to unamended controls. However, naphthalene- and 1-MN-amended culture did not show substantial amount of methane production. Interestingly, Toth et al. (2018) demonstrated development of naphthalene degradation ability of the methanogenic culture after long adaptation period. The findings suggested that methanogenic microorganisms in crude oil-contaminated sediment could adapt to PAHs and, over time, developed considerable degradation ability.
The involvement of methanogens in PAHs degradation has been demonstrated experimentally. In PAHs-degrading methanogenic enrichment cultures, the addition of methanogenesis inhibitor, bromoethane sulfonic acid (BESA), and eubacterial inhibitor, vancomycin, caused a significant reduction in degradation rates indicating the role of both methanogens and syntrophic bacteria (Chang et al. 2003). Similarly, the addition of BESA to a naphthalene- and phenanthrene-degrading consortia partially inhibited degradation and eliminated archaeal members from the consortia suggesting the involvement of methanogens in the degradation process (Chang et al. 2006). Fluorescence in situ hybridization analysis of naphthalene- and 1-MN-degrading enriched methanogenic consortia revealed that both bacteria and archaea were involved in the degradation process. Moreover, the presence of the members of Methanobacteriales in the consortia indicated the involvement of syntrophic obligate hydrogen- or formate-utilizing archaea (Christensen et al. 2004).
Several members of methanogenic PAHs-degrading community have been identified. Microbial community analysis revealed the dominance of archaeal members affiliated with Methanosaeta and Methanoculleus species and bacterial members related to the Clostridiaceae in aquifer sediment enrichments that degrade 2-MN and 2,6-dimethylnaphthalene (Berdugo-Clavijo et al. 2012). The cultures were previously reported incapable of degrading naphthalene under methanogenic conditions. However, after repeated transfer and tuning of the culture conditions, an enrichment culture capable of mineralizing naphthalene to methane has been obtained recently (Toth et al. 2018). Using next-generation sequencing and DNA-stable isotope probing techniques, the authors identified an unclassified Clostridiaceae species as a putative naphthalene degrader along with a Desulfuromonadales phylotype (function unknown). In another study, clone library analysis of phenanthrene-degrading leachate-contaminated sediment microcosm identified a community consisting of γ-Proteobacteria dominated by members of Citrobacter and Pseudomonas and archaea represented by members of Methanosarcina, Methanobacterium, and Thermogymnomonas (Zhang et al. 2012a). Using 13C6-anthracene and DNA-based stable isotope probing technique, Zhang et al. (2012b) demonstrated incorporation of radiolabel into three Proteobacteria phylotypes represented by the genera Methylibium and Legionella and an unclassified Rhizobiales. Methanogenic PAHs degradation and biogas/methane production may be correlated. Degradation of PAHs in laboratory-scale stirred-tank sewage sludge digester resulted in decreased biogas production but increased PAHs removal (Trably et al. 2003). In contrast, near stoichiometric production of methane was reported in methanogenic enrichment culture amended with 2-MN and 2, 6-dimethylnaphthalene (Berdugo-Clavijo et al. 2012).
12 Anaerobic PAHs Degradation by Pure Cultures of Bacteria
Pure cultures are invaluable especially in investigating the molecular mechanism of biodegradation. Several pure cultures of bacteria capable of degrading PAHs anaerobically have been isolated (Table 6). Among them, only three are strictly anaerobic sulfate-reducing bacterial strains, whereas most of the isolates are facultative anaerobic nitrate reducers. The scarcity of strictly anaerobic PAHs-degrading pure isolates may be due to (a) complex nutrient requirement, (b) slow growth, (c) failure to provide in situ-like incubation environment, and (d) obligatory mutualistic dependence among members of degrading consortium. Attempts to isolate Deltaproteobacterium sp. from naphthalene-degrading enrichment culture, N47, failed even though most of the genes/proteins involved in degradation are associated with this bacterium (Safinowski and Meckenstock 2006; Selesi et al. 2010; Bergmann et al. 2011b). In another case, Martirani-Von Abercron et al. (2016) isolated several naphthalene-degrading and nitrate-reducing bacterial strains. However, nitrate-respiring and naphthalene-degrading properties of the pure isolates could not be reproduced in liquid culture. A naphthalene-degrading pure culture, NaphS2, was isolated from the anoxic black sediment of North Sea (Galushko et al. 1999). Two other naphthalene-degrading SRB cultures, NaphS3 and NaphS6, were obtained from anoxic Mediterranean lagoon sediment (Musat et al. 2009). Much information on anaerobic naphthalene degradation has been obtained from Deltaproteobacteria NaphS2. Rockne et al. (2000) isolated three nitrate-reducing naphthalene-degrading facultative anaerobic bacteria. Stain NAP-3-1 and NAP-4 are phylogenetically close to Pseudomonas stutzeri and Vibrio pelagius, respectively. McNally et al. (1998) demonstrated anthracene, phenanthrene, and pyrene degradation by nitrate-reducing facultative anaerobic bacteria, Pseudomonas stutzeri SAG-R, P. fluorescens W-2, and P. putida KBM-1. Recently, Microbacterium sp. (Qin et al. 2017) and Cellulosimicrobium cellulans CWS2 (Qin et al. 2018) have been shown to degrade BaP under nitrate-reducing conditions. The efficiency of pure cultures over enrichment cultures in anaerobic PAHs degradation is not fully understood. Pure strains, NAP-3-1 and NAP-4, were less efficient in degrading naphthalene compared to their efficiency in the consortia; better degradation in co-culture may be due to the consortium synergy (Rockne et al. 2000). Pure cultures may not necessarily reflect the bioremediation potency of the environment from which they are obtained. Nevertheless, the value of obtaining pure bacterial strains for the elucidation of the molecular mechanism of degradation remains crucial.
13 Effects of PAHs Contamination on Anaerobic Microbial Community
PAHs input in an environment changes the total organic matter content. Depending on solubility and bioavailability of contaminants, an alteration in dissolved and particulate organic matter content is expected. The altered ratio, together with the toxicity of PAHs, would have a consequence on the natural microbial community. Knowledge on PAHs-induced changes in the microbial community is almost vague, as only a few studies have been conducted so far. Thus, PAHs-induced selection pressure on the microbial community is evident from the available few instances of evidence. Chang et al. (2005) investigated the effect of naphthalene and phenanthrene on methanogenic microbial community in harbor sediment. They found that SRB dominated the baseline community. Addition of naphthalene or phenanthrene triggered a marked shift toward the enrichment of methanogenic community in that sediment.
Comparative analysis of 16S rRNA genes indicated that naphthalene- and phenanthrene-degrading communities were different. Wan et al. (2012) reported an increased abundance of archaeal community in anthracene-treated methanogenic sediment. Alejandro et al. (2013) investigated the impact of Prestige oil spill on the microbial community in contaminated subtidal sediment after 18 and 53 months of the spill. The spilled oil contained naphthalene and its methylated derivatives. Along the depth, aerobic hydrocarbon degraders were abundant in the upper zone, NRB were present in higher number in the oxidized zone, and their number decreased with depth while SRB count reached a maximum at depth of 12–15 cm. The deep anoxic sediment was dominated by strictly anaerobic SRB. Although the spill caused a decrease in bacterial count, aromatic-oxidizing cultivable populations increased with time. The time-dependent difference in community distribution was also evident. Gammaproteobacteria and Deltaproteobacteria were the dominant phyla in the contaminated sediment as indicated by 16S rRNA gene analysis and fluorescent in situ hybridization analysis. Within the Deltaproteobacteria, Desulfobacteraceae was the most abundant, and Desulfarculales constituted half of the specific sequences. This study indicates that introduction of crude oil PAHs in anaerobic sediment exerts some toxic effect to the microbial community; however, resistant bacterial community will eventually develop and participate in PAHs cleaning from the impacted environment. Recently, Martirani-Von Abercron et al. (2016) investigated the effect of PAHs on different environmental samples under nitrate-reducing conditions. The samples were collected from the diverse environment: rice paddy fields, activated sludge, compost pile, lagoon sediment, and marine sediment. MPN enumeration indicated the presence of nitrate reducers and naphthalene, 2-MN, 2-NA, and anthracene degraders in all samples. Bacterial community analysis of the non-spiked control samples indicated that the samples, with one exception, were dominated by Proteobacteria followed by a different proportion of Bacteroidetes, Chloroflexi, and Actinobacteria. After enriching the samples in naphthalene and 2-MN, significant changes in the bacterial community occurred. Groups of uncultured and poorly characterized Acidobacteria, Firmicutes, and Verrucomicrobia could be enriched over Proteobacteria.
A recent study showed rapid shaping of the methanogenic microbiome in full-scale anaerobic digester reactors amended with naphthalene, fluorene, anthracene, phenanthrene, and fluoranthene (Oko et al. 2017). The adaptation was more rapid with oil and gas processing wastewater treatment reactor sludge (OG) than with municipal solid waste reactor sludge (MS). Over time, PAHs, feed, and nutrient-dependent succession in the bacterial and archaeal community was observed. After 14 days of incubation with PAHs, the relative abundance of the Euryarchaeota group increased by 35 and 90% in MS and OG communities, respectively, while the total abundance of bacteria decreased. In contrast, Ribeiro et al. (2018) argued that the microbial community structure in sediment microcosms amended with naphthalene and fluoranthene was mainly shaped by sample type and incubation time rather than the PAHs. In addition, Zhou et al. (2017) observed that pyrene amendment did not affect the activity and abundance of soil denitrifiers as well as microbial respiration. Also, pyrene addition to soil did not cause any change in microbial community.
14 Factors Affecting Anaerobic Biodegradation of PAHs
Many factors affect bioavailability and degradability of PAHs in the environment. In addition to the physicochemical properties of PAHs, some characteristics of the contaminated sites such as organic matter content, soil/sediment texture, clay minerals, pH, temperature, salinity, nutrient availability, and redox potential affect biodegradation of PAHs (Wilson and Jones 1993). Apart from these factors, constituents of a contaminating PAHs mixture and presence of co-contaminants such as heavy metals, cyanides, and other organic compounds also affect the microbial degradation of PAHs (Kuppusamy et al. 2017). As described in Sects. 8.3 and 10.2, the availability of TEAs such as nitrate and sulfate is a prominent factor affecting anaerobic degradation. The field of anaerobic PAHs degradation is nascent; very few studies so far have investigated the role of factors in determining anaerobic degradation of PAHs.
14.1 Influence of Structural Complexity of PAHs on Biodegradability
In general, structurally simple compounds are better biodegradable than complex compounds. Xenobiotic compounds are comparatively persistent to biodegradation. Several rules for predicting biodegradability of natural and synthetic compounds based on structural and chemical properties have been proposed, but numerous exceptions make them overly generalized (Kobayashi and Rittmann 1982). For example, benzene is structurally simpler and show more water solubility than naphthalene. Even so, it is chemically more stable than naphthalene. Benzene remained persistent in anaerobic sediment columns while naphthalene was degraded (Langenhoff et al. 1996). Hypothetically, LMW PAHs, as they are less complex in structure and more soluble in water, should be more readily degradable than their HMW congeners. Indeed, this premise is supported by many studies. For instance, Rothermich et al. (2002) observed quicker removal of 2–3 ring PAHs than 4–5 ring congeners. In a nitrate-reducing marine sediment enrichment, LMW PAHs (acenaphthene, phenanthrene, and fluorine) were degraded more efficiently than heavier fluoranthene and pyrene; the more complex PAHs like chrysene and benz(a)anthracene were degraded very slowly, and BaP remained persistent (MacRae and Hall 1998). Naphthalene was better degraded than phenanthrene by a denitrifying fluidized bed enrichment culture (Rockne and Strand 1998). Ambrosoli et al. (2005) reported a trend of three PAHs in the following order: fluorene > phenanthrene > pyrene. In a contaminated arctic soil, naphthalene and 2-methylnaphthalene were degraded under nitrate-reducing conditions, but HMW PAHs showed persistence for 90 days (Eriksson et al. 2003). However, complexity and solubility are not the only factors that determine degradability of PAHs. In a sediment enrichment, naphthalene degradation was slow compared to other 2–5 ring PAHs tested, including BaP (Rothermich et al. 2002). Relatively complex peri-fused pyrene was better degraded than cata-condensed anthracene by denitrifying strain KBM-1 (McNally et al. 1998). Three-ring phenanthrene was degraded slowly compared to five-ring BaP by a facultative anaerobe, Pseudomonas sp. JP1 (Liang et al. 2014). Interestingly, Murphy et al. (1995) demonstrated equal degradation rate of 15 priority PAHs in real contaminated sediment upon nitrate supplement. It is thus apparent that a simple correlation between structural complexity and biodegradability during anaerobic PAHs degradation cannot be drawn conclusively.
It may seem that increasing concentration of PAHs in an environment could impede their degradability. Available evidence is contrasting enough that does not allow to arrive at a general conclusion. Al-Bashir et al. (1990) reported that denitrifying soil slurry enrichment culture could mineralize naphthalene at aqueous-phase saturation level (50 ppm) and beyond (200 and 500 ppm) at the same rate. However, at concentration beyond the saturation level, degradation rate plummet after substrate concentration decreased to aqueous-phase solubility limit. The authors suggested that desorption of substrate rather than concentration determines the degradability. Dou et al. (2009) determined the degradation rates of naphthalene at a different initial concentration by mixed bacterial culture. Degradation rate increased with the increase in naphthalene concentration in 5–30 mg L−1 range. A BaP-degrading pure culture, Cellulosimicrobium cellulans CWS2, degraded 10 mg L−1 of BaP more efficiently than 5 mg L−1 concentration (Qin et al. 2018). Zhou et al. (2017) reported that increase in concentration of pyrene from 30 to 60 mg L−1 cause an increase in pyrene removal efficiency in denitrifying soil microcosms. Nevertheless, it can be inferred that apart from structural complexity and concentration of the contaminants, some yet unknown factors also affect degradability of PAHs in anaerobic environment.
14.2 Effect of Prior Exposure to PAHs
Experimental evidence does not allow reaching a generalized assumption that a site with previous contamination history possesses higher bioremediation potency than a pristine one. It is tempting to assume that microorganisms in a long-term contaminated site are better adapted to the pollutant(s); hence more efficient in degrading contaminants. Indeed, PAHs-degrading enrichment and pure cultures have been frequently isolated from contaminated samples. In such habitats, metabolically versatile microorganisms that can survive and utilize otherwise toxic pollutants confront selection pressure and might get competitive advantages over other community members. However, bacterial degradation of PAHs in pristine environments has also been reported in many studies. One possible explanation for the scenario is the possession of uninduced genetic machinery within the microbial members of an uncontaminated environment. Those unexpressed metabolic “toolboxes” are possibly induced after exposure to pollutants. Additionally, not all community members may have the ability to initialize attacking the pollutants. In the natural environment, many compounds including toxic contaminants are degraded co-metabolically (Hazen 2010).
Comparison between anaerobic PAHs removal efficiency of contaminated and uncontaminated (or less contaminated) indicates that samples with previous exposure history sometimes have greater bioremediation potential. Naphthalene and phenanthrene degradation in two sulfidogenic marine sediments that differ in the degree of contamination history was investigated (Coates et al. 1996). Both the substrates were readily degraded in one sediment that was severely contaminated with PAHs (33 mg of PAHs kg−1 of sediment). In contrast, the less contaminated sediment (4 mg PAHs kg−1) showed very minimal degradation. Interestingly, naphthalene degradation ability of the less contaminated sediment was stimulated when it was incubated with some of the heavily contaminated sediment (Coates et al. 1997). Hayes et al. (1999) observed a similar correspondence between the exposure level of a sample and its PAHs removal potential. In both studies, long-term exposure of the pristine-like samples to PAHs also resulted in the development of better degradation capacity. Eriksson et al. (2003) observed a better degradation of naphthalene, 2-MN, fluorene, and phenanthrene in a nitrate-reducing arctic soil contaminated with fuel.
Previous exposure, however, does not always lead to the development of better remediation capacity. Al-Bashir et al. (1990) observed that prior exposure did not help in improving PAHs degradation rates and reduction of the lag phase. The denitrifying Pseudomonas stutzeri strain SAG-R was isolated from a soil of creosote-contaminated hazardous waste site. It could degrade anthracene, phenanthrene, and pyrene more efficiently than the other two bacteria tested (McNally et al. 1998). Surprisingly, one of the strains, Pseudomonas putida KBM-1, which was isolated from almost pristine soil, could also degrade all the three PAHs at the rates comparable to those of the strain SAG-R. Sometimes, prolonged exposure to PAHs promotes the abundance of specialized degrading microbial community that is reflected in enhanced biodegradation rate, but such contamination history is not a prerequisite in all cases.
14.3 Effect of Soil Amendments and Heavy Metals
The effects of nitrate and sulfate amendment in enhancing natural attenuation of PAHs in an anaerobic environment and changes in abundance, activity, and community structure following the addition of inorganic electron acceptors have already been discussed. Li et al. (2015) investigated the effect of bicarbonate addition on anaerobic degradation of fluorene, phenanthrene, fluoranthene, and pyrene. Addition of 20 mM bicarbonate did not cause any significant change in PAHs removal rate. A similar result was also reported by Bach et al. (2005). Li et al. (2011) observed that addition of 1,160 mg L−1 of Mn(IV) to a PAHs degrading enriched bacterial consortium decreased the degradation rates of fluorene, phenanthrene, fluoranthene, and pyrene by 31–70%. The effect of bio-stimulating agents such as fertilizers, labile carbon sources in the form of organic acids, and surfactants was studied by Agarry and Owabor (2011). When added individually, all the amendments such as Tween 80, silicone oil, pig dung, and NPK fertilizer stimulated the degradation of naphthalene and anthracene in marine sediment. Pig dung and a mixture of pig dung with Tween 80 were the best in PAHs removal when tested individually or in combination. Organic acids such as acetate, lactate, and pyruvate were also found to be stimulatory in PAHs degradation (Chang et al. 2002, 2008; Bach et al. 2005). Langenhoff et al. (1996) reported that naphthalene degradation in a nitrate-reducing sediment column was only commenced after the addition of benzoate.
Very recently, Qin et al. (2018) investigated the effect of Fe2+, Zn2+, Cu(II), Mn2+, Hg2+, Co2+, Pb2+, and Cd2+ on BaP degradation by a nitrate-reducing bacterium, Cellulosimicrobium cellulans CWS2. Among the metal ions, Cd2+ inhibited the degradation; Fe2+, Zn2+, Hg2+, and Pb2+ did not show any toxic effect, while other metals slightly decreased the degradation rate.
14.4 Biosurfactants
Low solubility of PAHs in water is a major degradation rate-limiting factor. Biosurfactants are amphiphilic detergent-like molecules that promote release of sorbed PAHs and increase their aqueous concentrations by emulsification or solubilization process (Deziel et al. 1996; Ron and Rosenberg 2002; Johnsen et al. 2005; Mulligan 2005). The role of biosurfactants in aerobic PAHs degradation was better investigated. However, reports on biosurfactant production and role of biosurfactants in anaerobic degradation of hydrocarbon compounds are rare. Yan et al. (2017) reported biosurfactant production by iron-reducing facultative anaerobic strain, Hydrogenophaga sp. PYR1, that could degrade pyrene and BaP. Iron(III) stimulated the biosurfactant production, and the best production was observed with ferric citrate. Biosurfactant production by PAHs-degrading anaerobic bacteria and mechanism of enhanced anaerobic biodegradation by biosurfactants remain unknown.
15 Genetics and Biochemistry of Anaerobic PAHs Biodegradation
Our current understanding of the molecular mechanism of PAHs degradation is limited. Molecular mechanisms of anaerobic degradation of naphthalene and 2-MN in SRB are well studied. Difficulties in maintaining anaerobic cultures and their enzymes and extremely slow growth rate of anaerobic PAHs-degrading bacteria are the two main factors that hinder in-depth molecular investigation of anaerobic PAHs metabolism (Foght 2008; Meckenstock et al. 2016). Most of the information related to anaerobic degradation of naphthalene and 2-MN has been garnered from sulfate-reducing pure culture, NaphS2, and a freshwater enrichment culture, N47 (Meckenstock et al. 2016). The pathways for anaerobic BaP degradation by nitrate-reducing bacteria, Cellulosimicrobium cellulans CWS2 (Qin et al. 2018), Microbacterium sp. (Qin et al. 2017), and Pseudomonas sp. JP1 (Liang et al. 2014), and iron-reducing bacterial strain, Hydrogenophaga sp. PYR1 (Yan et al. 2017), have been proposed. Anaerobic degradation pathways of naphthalene and 2-MN in NaphS2 and N47 under sulfate-reducing conditions are analogous to anaerobic benzene and toluene degradation pathways, respectively (Foght 2008; Meckenstock et al. 2016). In contrast, the proposed pathways for anaerobic degradation of HMW PAHs, for example, BaP, in several nitrate- and iron-reducing pure cultures indicate that they are entirely different from anaerobic naphthalene and 2-MN degradation pathways. This variation could be specific for PAHs (LMW vs HMW) or reducing conditions (facultative anaerobic vs strict anaerobic or nitrate-reducing vs sulfate-reducing conditions) or organism-specific. In view of the currently available information, anaerobic metabolism of naphthalene and 2-MN is summarized in detail, and metabolism of phenanthrene and BaP has been outlined briefly in the following subsections.
15.1 Mechanism of Naphthalene Biodegradation
15.1.1 Initial Activation of Naphthalene
At least three different activation mechanisms, i.e., hydroxylation, carboxylation, and methylation, of naphthalene under entirely anaerobic conditions have been proposed (Fig. 6). Bedessem et al. (1997) detected an unresolved isomer naphthalenol (naphthol) as a principal intermediate during naphthalene degradation under sulfate-reducing conditions. Therefore, hydroxylation was proposed as the initial activation mechanism. Several lines of supporting evidence bake this plausible hypothesis. Indeed, benzene could be hydroxylated to phenol under methanogenic and sulfate- and iron-reducing conditions (Vogel and Grbic-Galic 1986; Caldwell and Suflita 2000). Moreover, phenol formation from benzene has recently been reported in an anaerobic benzene-oxidizing bacterium, Geobacter metallireducens (Zhang et al. 2013). Ideally, an activated intermediate of a recalcitrant compound should be readily utilizable by the degrading bacteria. Expectedly, Mihelcic and Luthy (1988a) observed that naphthol was more readily degraded than naphthalene by nitrate-reducing enrichment culture. On the contrary, the inability of several sulfate-reducing and naphthalene-degrading bacteria in using naphthol as substrate and absence of the compound in cellular metabolites exclude it as an intermediate of the metabolic pathways (Zhang and Young 1997; Meckenstock et al. 2000; Musat et al. 2009; Kleemann and Meckenstock 2011).

The reactions of the upper pathway of anaerobic naphthalene and 2-methylnaphthalene degradation. Solid arrows direct to the formation of 2-NA-CoA ester following initial activation of naphthalene by carboxylation reaction and 2-MN activation by fumarate addition reaction. Dotted arrows indicate the alternative pathways
Most of the available literature dealing with anaerobic naphthalene degradation support carboxylation as the first activation step (Fig. 6). Zhang and Young (1997) reported the accumulation of 2-NA in the supernatant of a naphthalene-degrading and sulfate-reducing enrichment culture. The culture could also use 2-NA for growth without any lag indicating the ready degradability of the compound as a typically activated compound. To confirm the origin of the carboxyl group, the enrichment culture was incubated in the presence of naphthalene and 14C-labeled bicarbonate. Results indicated the incorporation of 14CO2 to the most negative carbon atom (C2) of the naphthalene ring.
Interestingly, carboxylation is not exclusive for naphthalene; phenanthrenecarboxylic acid was also detected in phenanthrene-amended sulfate-reducing enrichment culture (Zhang and Young 1997). The position of the carboxylation in phenanthrene ring remained unknown for many years. Using deuterated phenanthrene and 13C-labeled bicarbonate, Davidova et al. (2007) unequivocally presented the evidence that phenanthrene is also carboxylated at the C2 position, and the reaction produces 2-phenanthrenecarboxylic acid. Furthermore, carboxylation is not unique to SRB; 2-NA has also been detected as a primary metabolite during naphthalene degradation by iron-reducing bacteria (Kleemann and Meckenstock 2011). Notably, several in situ metabolic profiling investigations identified carboxylated PAHs including 2-NA in PAHs-contaminated sites (Gieg and Toth 2017). Carboxylated PAHs are neither synthesized commercially nor occur as by-products of any known biological reactions; so the presence of these compounds in contaminated sites indicates that carboxylation may be a common activation reaction for many, if not all, PAHs. However, carboxylated intermediates are absent in cellular metabolites of phenanthrene- and BaP-degrading cultures (Tsai et al. 2009; Liang et al. 2014; Qin et al. 2017, 2018; Yan et al. 2017). Likewise, Toth et al. (2018) did not find any carboxylated, methylated, or fumarate substituted naphthalene metabolites in a methanogenic naphthalene-degrading enrichment culture. Therefore, a generalized activating step for all PAHs under anaerobic condition perhaps does not exist.
In sulfate-reducing enrichment culture N47, naphthalene is carboxylated by the enzyme naphthalene carboxylase. The enzyme activity was demonstrated in a crude cell extract of N47 (Mouttaki et al. 2012). In the reaction mixture of a crude cell extract of N47, naphthalene and 13C-bicarbonate, this enzyme produced 13C-labeled 2-NA at a rate of 0.12 nmol min−1 mg−1 of protein. Interestingly, the enzyme activity was ATP-independent. Divalent cations (Mn2+ and Mg2+) and chelating agent (EDTA) do not affect the activity. Moreover, the enzyme is biotin-independent, susceptible to oxygen exposure, and affected by strong reducing agents like sodium dithionite and Ti(III)citrate. Many of the characteristics of the naphthalene carboxylase enzyme suggest that it belongs to UbiD-like carboxylase enzyme family (Meckenstock et al. 2016). A recent proteogenomic study has revealed the presence of an alpha-subunit of the putative naphthalene carboxylase in N47 (Bergmann et al. 2011b). The ORF of the putative carboxylase shows 48 and 45% sequence similarity to alpha-subunit of phenylphosphate carboxylase (PpcA) of Aromatoleum aromaticum EbN1 (Rabus et al. 2005) and putative anaerobic benzene carboxylase of the iron-reducing and benzene-degrading culture, BF (Abu Laban et al. 2010), respectively. Also, molecular mass (53.23 kDa) and peptide chain length (481 amino acids) of putative naphthalene carboxylase are almost analogous to those of the EbN1 PpcA (53.99 kDa and 485 amino acids, respectively). Despite the similarities, naphthalene carboxylase is different from PpcA as the latter requires ATP and phosphorylated substrate.
A completely different mechanism of naphthalene activation was also proposed for sulfate-reducing Deltaproteobacterial culture, N47 (Safinowski and Meckenstock 2006). According to the proposal, naphthalene is first methylated to 2-MN and then subsequently transformed to the central metabolite 2-NA (Fig. 6). This pathway is analogous to the anaerobic conversion of benzene to toluene (Coates et al. 2002). The methylation theory is based on the detection of naphthyl-2-methylsuccinic acid (NMS) and naphthyl-2-methylenesuccinic acid (NeMS) in naphthalene-supplemented N47 culture. These two metabolites are highly specific for anaerobic 2-MN degradation; Safinowski and Meckenstock (2006) therefore reasoned that the metabolites were produced from 2-MN which, in turn, was derived from naphthalene by a methylation reaction.
Additionally, when naphthalene-degrading N47 culture was transferred to 2-MN-amended medium, rapid onset of degradation occurred without a pronounced lag (Safinowski and Meckenstock 2006). However, in vice versa scenario, i.e., when 2-MN-adapted culture was transferred to naphthalene-amended medium, degradation commenced after a long period of ~100 days. The authors suggested that naphthalene-grown cells contained all necessary enzyme(s) required for 2-MN degradation, while the enzyme(s) were not induced in 2-MN-grown cells that caused such a long adaptation period. While it came to the origin of the methyl group, the authors argued that it could be generated from bicarbonate via a reverse carbon monoxide (CO) dehydrogenase pathway. CO dehydrogenase activity of about 0.0974 μmol min−1 mg−1 of protein was found in culture N47 using methyl viologen as the TEA. Although methylation is not regarded as a primary mechanism of benzene activation during anaerobic degradation, Ulrich et al. (2005) detected toluene in benzene-degrading and nitrate-reducing enrichment culture. Like benzene, naphthalene could also be methylated, as it is more reactive than benzene in electrophilic substitution and addition reaction. According to quantum mechanical calculations, the net loss in stabilization energy for the first step in electrophilic substitution or addition is higher for benzene than that for naphthalene. Therefore, it is expected that electrophilic substitution will occur for naphthalene if it reacts with strong methyl group-donating compounds. However, later studies on N47 failed to detect methylated derivative in culture incubated with naphthalene and a methyl group donor such as methyltetrahydrofolate, S-adenosyl-l-methionine, and methylcobalamin (Mouttaki et al. 2012).
Convincing evidence that cancels out methylation as an initial activation mechanism has been provided by Musat et al. (2009). When naphthalene-grown cells of three sulfate-reducing pure bacterial strains, NaphS2, NaphS3, and NaphS6, were transferred to 2-MN, substrate utilization did not commence before a long adaptation period. The second evidence was obtained by growing NaphS2 in deuterated(d8) naphthalene and unlabeled 2-MN. Most of the d8 label was found in the carboxylation product 2-NA, while succinic acid adduct 2-naphthylmethylsuccinate was found unlabeled. In addition, gel electrophoresis of 2-MN-grown cell extracts revealed high-molecular-mass co-migrating protein bands. These protein bands were only specific for 2-MN-grown cells, indicating that the bacteria use different activation mechanisms for naphthalene and 2-MN. Nevertheless, traces of 2-MN-specific metabolites were also detected in naphthalene-grown cultures of NaphS2, NaphS3, and NaphS6 (Musat et al. 2009). The authors argued that these metabolites were probably produced via back reaction starting from 2-naphthoyl-CoA and succinyl-CoA. Recently, enzymes specific for 2-MN degradation have been found in several naphthalene-degrading and sulfate-reducing cultures of Desulfobacteraceae (Kummel et al. 2015). One possible explanation for this scenario may be the co-induction of 2-MN-specific gene clusters by naphthalene.
15.1.2 Initial Activation of 2-Methylnaphthalene
In many senses, anaerobic degradation 2-MN and toluene show striking similarities. Most of the information on anaerobic toluene degradation has been obtained from Azoarcus sp. strain T (Beller and Spormann 1997) and Thauera aromatica (Biegert et al. 1996). Anaerobic toluene degradation in these organisms initiates with the conversion of toluene to benzylsuccinic acid by the enzyme benzylsuccinate synthase (Bss) that catalyzes the addition of fumarate to the methyl side chain of toluene. Likewise, the initial activation of 2-MN involves similar fumarate addition step that leads to the production of naphthyl-2-methylsuccinic acid by the catalytic action of 2-naphthylmethylsuccinate synthase (Nms) (Fig. 6). The enzyme reaction was demonstrated in a dense cell suspension of culture N47 with fumarate (Safinowski and Meckenstock 2004). Musat et al. (2009) observed 2-MN-specific co-migrating protein band in gel electrophoresis of cell extract from sulfate-reducing cultures, NaphS2, NaphS3, and NaphS6. Peptide sequencing of the putative large subunit of Nms (NmsA) revealed that the protein shares sequence similarity to the large catalytic subunit of Bss (BssA). Whole genome and proteome sequencing of the culture N47 revealed Nms genes, nmsA, nmsB, and nmsC, corresponding to the three subunits of the Nms protein. The α (95.9 kDa), β (7.9 kDa), and γ (7.8 kDa) subunits share sequence similarities to the corresponding subunits of Bss enzyme. Notably, the NmsA sequences of N47 show more similarity (92%) to those of NaphS6 compared to any BssA sequences. In addition to the gene products of nmsABC, NmsD, a putative Nms-activating enzyme that shares sequence similarity to putative 1-methyl alkyl-succinate synthase activase (MasG) of Azoarcus sp. strain HxN1, was also identified in N47 (Selesi et al. 2010).
During 2-MN degradation by a sulfate-reducing consortium that was enriched from marine harbor estuarine sediment, Sullivan et al. (2001) found very different degradation metabolites. They argued that 2-NA is not a product of direct carboxylation or fumarate addition. Instead 2-NA was proposed to be generated via the oxidation of the methyl side chain. Also, several carboxylated-2-MN metabolites and their reduced derivatives were detected. As in the case of naphthalene, several alternative activation mechanisms of 2-MN activation, i.e., oxidation and carboxylation, may exist in nature.
15.1.3 Conversion of Naphthyl-2-Methylsuccinic Acid to 2-NA
Despite the difference in naphthalene and 2-MN activation mechanisms, both the substrates channeled to the central metabolic pathway via the common intermediate 2-NA. Unlike one-step naphthalene activation reaction (naphthalene to 2-NA), conversion of 2-MN to 2-NA via naphthyl-2-methylsuccinic acid (NMS) is a multistep process (Fig. 6). As in toluene degradation, the reactions following NMS generation proceed via the formation of corresponding CoA ester, NMS-CoA. In a subsequent reaction, NMS-CoA is oxidized to naphthyl-2-methylenesuccinyl-CoA (NMeS-CoA) by naphthyl-2-methylsuccinyl-CoA dehydrogenase activity. The activity of the succinyl-CoA:naphthyl-2-methylsuccinate CoA-transferase enzyme, which converts NMS to the corresponding CoA ester, has been measured in crude cell extract of culture N47. This CoA-transferase enzyme shares significant similarity to succinyl-CoA:(R)-benzylsuccinate CoA-transferase, and both belong to the family(III) of CoA-transferases. In addition, naphthyl-2-methylsuccinyl-CoA dehydrogenase activity has also been measured in crude extract of N47 (Safinowski and Meckenstock 2004). The rest of the steps leads to the conversion of NMeS-CoA to 2-NA. The reaction series should proceed via the formation of oxidized intermediates as in benzoyl-CoA pathway of anaerobic toluene degradation. However, the predicted intermediates, naphthyl-2-hydroxymethyl-succinyl-CoA and naphthyl-2-oxomethyl-succinyl-CoA, are not yet detected in any 2-MN-degrading culture extracts. Nevertheless, recent proteogenomic studies on culture N47 (Selesi et al. 2010; Bergmann et al. 2011a) and NaphS2 (Didonato et al. 2010) provide interesting clues suggesting the presence of all the necessary genes and enzymes for the putative oxidation reactions. The bns (beta-oxidation of naphthyl-2-methylsuccinate) operon in N47 consists of eight genes, bnsABCDEFGH. Functions of the gene products were predicted from orthologous sequences of toluene-degrading Aromatoleum aromaticum EbN1, Thauera aromatica, and Azoarcus sp. strain T (Meckenstock et al. 2016).
15.1.4 Formation of 2-Naphthoyl-CoA from 2-NA by CoA Ligase
Anaerobic metabolism of aromatic acid proceeds via the formation of a thioester with coenzyme A (CoA) by CoA ligase or CoA-transferase enzyme (Fuchs et al. 2011). Observing the formation of 2-NA and reduced 2-NA derivatives, Zhang et al. (2000) speculated that 2-NA might be thioesterified with CoA-SH so that the subsequent reactions would proceed through the formation of ring reduction products in a manner that is similar to the anaerobic benzoyl-CoA pathway (Harwood et al. 1998). Indeed, the –CO-S-CoA group facilitates further electron transfer and ring reduction by lowering the midpoint potential of the first electron transfer process (Johann and Georg 1997). A phototrophic bacterium, Rhodopseudomonas palustris, produces a 4-hydroxybenzoate-coenzyme A ligase that can catalyze the MgATP-requiring thioesterification reaction with 4-hydroxybenzoate (or benzoate) and coenzyme A. Benzoate-CoA ligase of a facultative anaerobe, T. aromatica, can also perform benzoyl-CoA-forming catalysis in the presence of benzoate, MgATP, and coenzyme A (Schühle et al. 2003). An indication for the presence of similar CoA ligase enzyme in NaphS2 and N47 has been obtained from proteogenomic investigations (Didonato et al. 2010; Bergmann et al. 2011b). Deltaproteobacterium NaphS2 genome contains a gene (NPH_5477) that encodes a putative 2-NA-CoA ligase with 50% similarity to E. coli phenylacetate-CoA ligase. During differential growth on benzene and naphthalene, NPH_5477 was upregulated (Didonato et al. 2010). Sequence comparison between 2-NA-CoA ligase in N47 and benzoate-CoA ligase of R. palustris revealed the presence of several ORFs. Among the most related ORFs, N47_B20660 was found to be expressed exclusively in naphthalene- and 2-NA-grown cultures (Bergmann et al. 2011b). However, experimental evidence confirming the presence and activity of 2-NA-CoA ligase has not yet been provided.
15.1.5 Ring Reduction of 2-NA
Following the formation of common 2-NA intermediate, both naphthalene and 2-MN degradation proceeds through identical sequential ring reduction steps that ultimately pave the way of ring cleavage (Fig. 7). Early GC-MS analysis of culture extract from the culture N47 provided clues for the formation of several metabolites by ring reduction (Meckenstock et al. 2000). Two tetrahydro-derivatives, 1,2,3,4-tetrahydro-2-naphthoic acid (1,2,3,4-THNA) and 5,6,7,8-tetrahydro-2-naphthoic acid (5,6,7,8-THNA), in addition to octahydro-2-NA and decahydro-2-NA, in naphthalene-fed culture were detected (Meckenstock et al. 2000). A similar analysis on another naphthalene-degrading and sulfate-reducing enrichment culture extracts also revealed the formation of five-ring reduction products: dihydro-; 5,6,7,8-tetrahydro-; hexahydro-; octahydro-; and decahydro-2-NA (Zhang et al. 2000). The formation of these metabolites has suggested that 2-NA is sequentially reduced through five successive steps toward the production of decahydro-2-NA. However, decahydro-2-NA was proposed as a dead-end metabolite (Annweiler et al. 2002).

Formation of the lower pathway central metabolite pimeloyl-CoA from 2-naphthoyl-CoA via sequential ring reduction and two ring-opening reactions in the anaerobic degradation of naphthalene and 2-MN
Detection of only deuterated 5,6,7,8-THNA (but not 1,2,3,4-THNA) in the study by Zhang et al. (2000) has provided an important clue that the ring reduction might start in the non-substituted ring (ring II) rather than the carboxyl-substituted ring (ring I). On the other hand, accumulation of 1,2,3,4-THNA, even in small amounts, together with 5,6,7,8-THNA in the study of Meckenstock et al. (2000) could not deduce where the first reduction was initiated from the metabolites’ profile. If the ring reduction starts at the ring I, then 1,2,3,4-THNA should have been the major metabolite. To resolve the apparent discrepancy, Annweiler et al. (2002) analyzed the metabolites formed while growing the culture N47 on 1,2,3,4-tetrahydronaphthalene (tetralin). Only 5,6,7,8-THNA was detected in culture extract indicating that the addition of C1 unit requires an aromatic ring and the ring reduction starts from the unsubstituted ring II. However, recent metabolite profiling studies have reported the presence of 1,2,3,4-THNA in PAHs-contaminated samples (Aitken et al. 2004; Griebler et al. 2004; Wawrik et al. 2012). Therefore, ring reduction may initiate in either of the rings. Conditions that favor a reduction process are still unknown.
High resonance energy of mono- or polyaromatic compounds makes a ring cleavage reaction very challenging in a biological system. Before cleavage of the ring structure, anaerobic bacteria adopt a strategy that involves the dearomatization of the ring. Reductive dearomatization of the central metabolite benzoyl-CoA is a well-known process in anaerobic degradation of monoaromatic BTEX compounds (Boll 2005; Fuchs et al. 2011). The dearomatization process that leads to the production of 1,5dienoyl-CoA (cyclohex-1,5-diene-1-carboxy-CoA) is catalyzed by benzoyl-CoA reductase (Bcr) enzyme (Boll and Fuchs 1995; Fuchs et al. 2011). Two classes of Bcr have been reported so far. They catalyze the formation of the same product but differ in some properties; ATP dependence is one of the most striking features of the enzyme. Bcr I activity has been reported so far in Thauera aromatica (Breese et al. 1998), Rhodopseudomonas palustris (Egland et al. 1997), and Azoarcus evansii (Harwood et al. 1998). Oxygen-sensitive Bcr I catalyzes two-electron transfer from reduced ferredoxin to the substrate with stoichiometric hydrolysis of two molecules of ATP to ADP and PPi. On the other hand, class II Bcr activity in obligate anaerobic Geobacter metallireducens does not require ATP (Kung et al. 2009). N47 genome contains genes that are similar to the genes encoding the four subunits of class I Bcr in Azoarcus sp. However, the gene similar to BamB that codes for the active site of Bcr II is absent in N47. In contrast, NaphS2 genome harbors gene analogous to both classes of Bcr (Didonato et al. 2010). The activity of the putative naphthoyl-CoA reductase (Ncr) has been shown in the crude cell extracts of N47 which catalyzes sodium dithionite-dependent four-electron transfer reaction, converting NCoA to 5,6,7,8-THN-CoA. Although sequences of ncrABCD and bcrABCD are similar, unlike Bcr I protein, Ncr activity in N47 is independent of ATP and insensitive to oxygen indicating the novelty of Ncr within the family of reductases (Eberlein et al. 2013b). Ncr has been purified and characterized from N47 culture. The enzyme is a 150 kDa dimeric protein consisting of two 72 kDa subunits. It contains FMN and FAD cofactors and [4Fe-4S] clusters. It is classified as a member of old yellow enzyme (OYE) family based on the presence of flavin cofactors and iron-sulfur cluster as well as sequence similarity to cyclohexa-1,5-diene-1-carboxyl-CoA oxidase from T. aromatica (Eberlein et al. 2013a).
Notably, no dihydro derivative formation was observed during NCoA reduction. However, when ncr gene of N47 (N47_G38220) was heterologously expressed in Escherichia coli, the extract of recombinant cells catalyzed the conversion of NCoA to dihydro-2-naphthoyl-CoA (DHN-CoA) rather than 5,6,7,8-THN-CoA (Eberlein et al. 2013a). Further, Estelmann et al. (2015) investigated the expression of three putative ncr (N47_G38220 from N47 and NPH_5475 and NPH_1753 from NaphS2) in E. coli host. All ncr gene products could convert NCoA to a two-electron reduced metabolite 5,6-dihydro-2-naphthoyl-CoA (5,6-DHN-CoA). However, none of the enzymes showed a four-electron reduction of NCoA to THN-COA. Genes encoding putative 5,6-DHN-CoA reductases from both N47 (N47-G38210) and NaphS2 (NPH_5476) were expressed in E. coli. The gene products catalyzed the formation of 5,6,7,8-THN-CoA from 5,6-DHN-CoA (Estelmann et al. 2015). Subsequent reduction of 5,6,7,8-THN-CoA to hexahydro-2-naphthoyl-CoA (HHNCoA) in N47 involves another two-electron reduction step that is catalyzed by an ATP-dependent and oxygen-sensitive THN-CoA reductase enzyme (Eberlein et al. 2013b). Therefore, reduction of NCoA to HHNCoA involves three enzymatic reduction steps. The first two enzymes in this series belong to the OYE family, and the other, HHNCoA, is identical to class I Bcr family. In both N47 and NaphS2, genes encoding four putative subunits of THN-CoA reductase have been identified as a part of a gene cluster. In N47, the gene cluster forms thn operon. Protein prediction from the operon indicates that the operon contains genes that may encode several putative enzymes in addition to THN-CoA reductase. The predicted enzymes include an oxidoreductase, enoyl-CoA hydratases/hydrolases, acyl-CoA dehydrogenases, 3-hydroxyacyl-CoA dehydrogenases, and acetyl-CoA thiolases/transferases (Meckenstock et al. 2016). By comparing the putative functions of the enzymes with the functions of analogous enzymes in benzoyl-CoA pathway, Meckenstock et al. (2016) hypothesized that downstream degradation of hexahydronaphthoyl-CoA could proceed via β-oxidation-like reactions, and the first ring cleavage would occur through a thiolytic cleavage of acetyl-CoA producing a cyclohexanoic acid-CoA ester derivative.
15.1.6 Ring Cleavage of Reduced 2-NA Products
The downstream ring cleavage pathways in anaerobic naphthalene and 2-MN degradation proceed through cyclohexanoic acid rather than monoaromatic compound (Annweiler et al. 2002; Weyrauch et al. 2017). In naphthalene-degrading N47 culture extracts, GC-MS analysis revealed the presence of two ring cleavage products (Annweiler et al. 2002). The first product consisted of a cyclohexane ring with two carboxylic acid side chains with C11H16O4-diacid. The exact constituents of the diacid side chains are not known yet. The second ring cleavage product was detected as a cis-2-carboxycyclohexylacetic acid that was assumed to be a β-oxidation product of the diacid. Recently, metabolism of cis-2-carboxycyclohexylacetic acid-CoA ester (2-(carboxymethyl)cyclohexane-1-carboxylic acid-CoA ester) by N47 culture extracts has been demonstrated by Weyrauch et al. (2017). When cis-2-carboxycyclohexylacetic acid-CoA ester (m/z = 936) was incubated with a cell-free extract of the culture N47 and NaphS2 in the presence of ferrocenium hexafluorophosphate as the artificial electron acceptor, two new compounds were detected in GC-MS representing m/z values of 934 and 952, respectively. This indicates that metabolism of cis-2-carboxycyclohexylacetic acid proceeds via α-, β-desaturation and a subsequent water addition at the β-position. The dehydrogenase and hydratase enzymes catalyzing the two reactions show similarity in reaction to corresponding enzymes of branched acyl-CoA ester metabolism. The acyl-CoA dehydrogenase introduces a double bond to cis-2-carboxycyclohexylacetic acid producing 2-carboxycyclohexylideneacetyl-CoA that is converted into 1-hydroxy-2-carboxycyclohexylacetyl CoA in the hydratase-catalyzed reaction. The remaining cyclohexane ring opening and central acetyl-CoA metabolism substrates production proceed via pimeloyl-CoA. Before converting to CoA ester of dicarboxylic C7 pimelic acid, the ring structure of 1-hydroxy-2-carboxycyclohexylacetic acid-CoA ester is opened by a novel class of ring-cleaving lyase. In the subsequent thiolytic cleavage reaction, the ring cleavage reaction product, 3-oxononanedioyl-CoA, is converted to pimeloyl-CoA by thiolase (Weyrauch et al. 2017).
Further degradation of pimeloyl-CoA to glutaryl-CoA proceeds via β-oxidation (Fig. 8). Pimeloyl-CoA is converted sequentially to 2,3-dehydropimeloyl-CoA, 3-hydroxypimeloyl-CoA, and 3-oxopimeloyl-CoA probably by acyl-CoA dehydrogenase, acyl-CoA hydratase, and β-hydroxyacyl-CoA dehydrogenase, respectively. Although no peak representing glutaryl-CoA was detected during pimeloyl-CoA conversion, cell-free extracts of both N47 and NaphS2 strains can utilize glutaryl-CoA (Weyrauch et al. 2017). In the glutaryl-CoA assay, four major peaks appeared in LC-MS chromatogram representing glutaconyl-CoA, crotonyl-CoA, 3-hydroxybutyryl-CoA, and acetyl-CoA. A similar conversion process of glutaryl-CoA to glutaconyl-CoA and glutaconyl-CoA to crotonyl-CoA has been previously reported in strict anaerobes (Schöcke and Schink 1999; Müller and Schink 2000; Wischgoll et al. 2009). Unlike facultative anaerobes that exploit decarboxylating glutaryl-CoA dehydrogenase only for the direct conversion of glutaryl-CoA to crotonyl-CoA, strict anaerobes use non-decarboxylating glutaryl-CoA dehydrogenase and glutaconyl-CoA decarboxylase to produce crotonyl-CoA from glutaryl-CoA via glutaconyl-CoA. By the formation of TCA cycle intermediate, acetyl-CoA, the pathway merges to the central respiration pathway.

Lower pathway of anaerobic naphthalene and 2-methylnaphthalene degradation. Pimeloyl-CoA is converted to the TCA cycle intermediate acetyl-CoA via glutaryl-CoA. Pimeloyl-CoA is derived from 2-naphthoyl-CoA that is in turn generated during the anaerobic oxidation of the parent substrates
15.2 Mechanism of Phenanthrene and BaP Biodegradation
As mentioned above, phenanthrene degradation in a sulfate-reducing enrichment culture starts with the carboxylation at the C2 position (Davidova et al. 2007). Recently, Himmelberg et al. (2018) detected 2-phenanthroic acid as the primary metabolite in phenanthrene-degrading and sulfate-reducing enrichment culture. Detection of carboxylated phenanthrene also suggests that the anaerobic phenanthrene metabolism might follow analogous steps as in naphthalene biodegradation. The study of Himmelberg et al. (2018) also provided some clues that indicate possibility for the existence of a similar metabolic route. Like 2-NA, 2-phenanthroic acid is also converted to corresponding CoA ester by the enzyme 2-phenanthroate-CoA ligase. Moreover, several ring-reduced products were also identified, indicating the occurrence of ring reduction steps that will make the ring cleavage possible at later stages. Tsai et al. (2009) detected phenol and p-cresol in phenanthrene-degrading and sulfate-reducing enrichment culture. The authors proposed that phenanthrene degradation proceed through a series of hydration and hydrolysis reactions and a decarboxylation reaction on p-cresol. Phenol was also detected in the fluorene-amended cultures.
Reports proposing anaerobic BaP degradation mechanism are extremely sporadic. BaP degradation pathways have been reported for facultative anaerobes, Pseudomonas sp. JP1 (Liang et al. 2014), Microbacterium sp. (Qin et al. 2017), and Cellulosimicrobium cellulans CWS2 (Qin et al. 2018), and biosurfactant-producing and iron-reducing Hydrogenophaga sp. PYR1 (Yan et al. 2017). The metabolites of BaP degradation identified so far are listed in Table 7. If BaP biodegradation is thought to proceed via a pathway that is comparable to naphthalene degradation pathway, carboxylated BaP should be a metabolite in upper degradation pathway. However, such a metabolite is not reported yet in any BaP-degrading culture. Available proposed BaP degradation pathway does not merge at a central metabolite like benzoic acid or naphthoic acid as in benzene and naphthalene degradation, respectively. In addition, a common activation reaction in BaP-degrading pathway cannot be expected, not only for the absence of carboxylated metabolites, but proposed pathways indicate several distinct first step metabolites even for the same strain. For instance, both 4,5-dihydrobenzo(a)pyrene and 7,8,9,10-tetrahydrobenzo(a)pyrene have been suggested as initial reduction step products for BaP-degrading and nitrate-reducing Microbacterium sp. (Qin et al. 2017). Similarly, Liang et al. (2014) suggested 1,12-dimethylbenz(a)anthracene, 7,8,9,10-tetrahydrobenzo(a)pyrene, and 5-ethylchrysene as products of alternative initial reactions. According to Qin et al. (2017), BaP degradation proceeds through initial reduction followed by formation of unsubstituted BaP congeners (pyrene and chrysene) and subsequently through ring opening that leads to the production of naphthalene and its hydroxyl derivatives. In contrast, the pathway proposed by Liang et al. (2014) involves methylated derivatives in addition to the reduced and congener derivatives.
At this moment, reaching a consensus about BaP biodegradation seems difficult. However, proposed pathways indicate the formation of some common metabolites during BaP degradation. Pyrene and phenanthrene have been detected as BaP metabolites in cultures of Pseudomonas sp. JP1, Microbacterium sp., Hydrogenophaga sp. PYR1, and Cellulosimicrobium cellulans CWS2 (Qin et al. 2017). Chrysene is the common BaP metabolite in Pseudomonas sp. JP1 and Microbacterium sp. The same 7,8,9,10-tetrahydrobenzo(a)pyrene has been detected in both Microbacterium sp. and Pseudomonas sp. JP1. Detection of similar compounds may be indicative of the existence of novel pathway(s) for BaP biodegradation. In contrast, detection of reduced derivatives, 4,5-dihydrobenzo(a)pyrene and 7,8,9,10-tetrahydrobenzo(a)pyrene, indicates the involvement of ring reduction steps as in naphthalene and 2-MN degradation.
16 Metabolite Profiling for Monitoring In Situ Anaerobic PAHs Biodegradation
Degradation metabolites of various environmental contaminants are used as diagnostic biomarkers for investigating the biotic fate of the compounds in the real environments (Callaghan 2013; Gieg and Toth 2017). Metabolite profiling or metabolome analysis provides unambiguous evidence that biodegradation is occurring or has already occurred since metabolites are the products of enzyme-catalyzed reactions. Suitability of a compound as a biomarker is determined by some essential criteria. The metabolite generated during the active biodegradation process must be specific to the parent compound monitored and is not a natural as well as xenobiotic compound and is biodegradable (Callaghan 2013). The selection criteria necessitate in-depth knowledge of the degradation pathway(s) and analytical competence. Availability and continuous improvement of efficient extraction methods and high precision hyphenated separation and detection techniques have made the task of detection of compound-specific biomarker(s) easier for both field and laboratory investigations. However, our current understanding of anaerobic PAHs metabolism is very limited. Insight into the naphthalene and 2-MN metabolism provided some specific metabolites that are used as biomarkers. These include naphthyl-2-methylsuccinic acid, naphthyl-2-methylenesuccinic acid, 2-NA, 5,6,7,8-tetrahydronaphthoic acid, and hexahydronaphthoic acids. Therefore, these metabolites have been used as metabolic markers for monitoring PAHs biodegradation mainly in groundwater samples obtained from PAHs-impacted sites (Table 8).
In addition to the detection of signature metabolites that provide evidence of ongoing or already occurred biodegradation, metabolite profiling provides a strong indication of the presence of yet to be discovered metabolites and even the existence of alternative novel degradation pathway(s). For example, methylnaphthoic acid, which might be a metabolite of either naphthalene or 2-MN or both, has been identified (Aitken et al. 2004). This metabolite is not reported in any PAHs-degrading enrichment or pure culture study. The same is true for 2,6-dimethylnapthoic acid. Detection of indanoic acid, acenaphthenoic acid, and acenaphthylenoic acid indicates that these metabolites result from carboxylation of the parent compounds. Drawing analogy to naphthalene and phenanthrene activation pathways, it can be inferred that the same carboxylating activation mechanism also works for other unsubstituted PAHs. Detection of 1-NA, 2-NA, 1-hydroxy-2-naphthoic acid, and 2-hydroxy-3-naphthoic acid is suggestive of the existence of yet undiscovered degradation pathways. To harness the maximum from a metabolite profiling, further studies in anaerobic degradation of HMW as well as carcinogenic PAHs such as BaP are required.
17 Anaerobic Bioremediation of PAHs
Integrated remediation approaches that combine physical, chemical, and biological treatment technologies have been shown to be more effective than a single treatment (Kuppusamy et al. 2017). However, remediation of anaerobic environments contaminated with PAHs is quite challenging. Anaerobic remediation options available are less compared to aerobic treatment processes and limited in terms of feasibility and applicability. Contamination of anaerobic subsurface soils, aquifers, sediments, and sludge with PAHs is a worldwide problem. Addition of oxygen to anoxic environments for stimulating in situ degradation of organic pollutants has very limited success, and the process encounters technical difficulties (Thomas and Ward 1989; Morgan and Watkinson 1992; Lovley et al. 1994). Several physical and chemical remediation technologies for the removal of the pollutants from anaerobic environments are available. Mechanical removal of contaminated sediment (dredging), removal of the overlying water body and subsequent physical removal of contaminated sediment (dry excavation), covering the contaminated sediment with fresh material (capping), and complete isolation of a contaminated area with containment barriers are associated with limited success (Perelo 2010). Chemical remediation options such as oxidation with H2O2, modified Fenton’s reagent, activated sodium persulfate, potassium permanganate, and their combinations were shown to be effective in the removal of PAHs from contaminated sediments (Ferrarese et al. 2008). The practicability of physical and chemical treatment technologies is questioned due to cost, limited efficiency, technical complexity, destruction of habitats, and increased exposure to site workers and native organisms.
Bioremediation approaches offer several advantages over physical and chemical remediation options. But, investigations on the feasibility of laboratory- and field-scale bioremediation of PAHs from anaerobic environments are limited. Some studies suggested biostimulation (enhanced removal of contaminants following the addition of electron acceptors and/or nutrients) as a successful option for remediation of PAHs. For instance, nitrate addition in nitrate-deficient contaminated sediments resulted in significant removal of PAHs (Murphy et al. 1995; Rockne and Strand 2001; Tang et al. 2005; Yang et al. 2013). Similarly, sulfate addition in contaminated sediments promoted biodegradation of naphthalene, 2-MN, and phenanthrene (Rothermich et al. 2002; Tang et al. 2005). On the other hand, the arbitrary addition of electron acceptors appeared to be futile. Johnson and Ghosh (1998) compared the effect of nitrate and sulfate addition to dredged contaminated sediments. The sediments were rich in sulfate and supported appreciable anaerobic degradation of PAHs without any amendment. Addition of sulfate further enhanced PAHs degradation; however, nitrate addition did not have any stimulatory effect. Likewise, induction of methanogenesis by the addition of dextrin did not promote PAHs degradation. Similarly, Mn(IV) addition to contaminated sediment, where sulfate was the dominant electron acceptor, inhibited PAHs biodegradation (Li et al. 2011). Determination of dominant electron-accepting process in contaminated anaerobic environments, demonstration of the presence of requisite microorganisms, and laboratory-scale investigation into the efficacy of electron acceptor(s) seem to be critical steps in the process of anaerobic remediation through biostimulation.
Although laboratory-scale demonstrations of anaerobic PAHs degradation under various reducing conditions by enrichment and pure cultures are available, evidence suggesting their role in the degradation of PAHs from contaminated environments is still lacking. Future research should focus on evaluating the efficacy of PAHs-degrading cultures from real contaminated samples. The direct application of microorganisms as bioaugmentation agents also require site characterization and understanding of the degradation processes. Further research must also be directed toward feasibility assessment to establish the chance of success, effectiveness, and applicability. Moreover, laboratory investigations should focus on establishing the ability of native microorganisms to degrade PAHs from the anaerobic environments, deciphering the degradation mechanisms, identification of influencing parameters, and demonstration of bioremediation in laboratory-scale bioreactors. Behavior of anaerobic PAHs-degrading bacteria in the presence of mixed contaminants, for example, PAHs + heavy metals + cyanides, should be investigated. As strict anaerobes are sensitive to atmospheric oxygen, inoculation of the contaminated sites with microorganisms would also require special techniques. In this direction, facultative anaerobes would offer more flexibility in terms of oxygen sensitivity and inoculation techniques.
18 Conclusion
Field- and laboratory-scale investigations have provided ample evidence that LMW PAHs are biodegradable under various anaerobic conditions. Besides, anaerobic degradation of 4–5 ring PAHs, for example, BaP, is also known for some facultative anaerobes. However, most of the carcinogenic PAHs are HMW compounds and have more compact ring system compared to the LMW congeners. Still, there is a dearth of evidence of biodegradability of HMW PAHs, especially in strictly anaerobic sulfate-reducing and methanogenic conditions. Most of the existing literature shows anaerobic biodegradation in enrichment cultures, and very few pure cultures have been isolated. As the biomarker-based in situ monitoring depends upon the knowledge on biochemical transformation pathways, anaerobic PAHs-degrading pure culture isolation should receive due attention from concerned researchers. Factors that affect anaerobic PAHs degradation are still poorly understood. In many real contaminated environments, PAHs exist with other co-contaminants such as heavy metals, BTEX compounds, cyanides, and phenolics. Effect of co-contaminants is almost neglected in anaerobic PAHs degradation studies.
Furthermore, interactions among degrading microorganisms in anaerobic environments should be investigated, as the natural environment is an interactive and interdependent system regarding niche and nutrient cycling. Biochemistry of naphthalene and 2-MN degradation in SRB is now known. The mechanisms may not necessarily be the same for the other reducing conditions. It is especially relevant for facultative anaerobes that can switch among aerobic, hypoxic, and anaerobic modes. Thus, future studies should focus on elucidating the mechanism of PAHs degradation in both facultative anaerobic and strictly anaerobic bacteria. Identification of HMW PAHs degradation metabolites will also guide to develop tools for contamination monitoring. Information on the suitability of bioremediation, for instance, nutrient amendment or bioaugmentation, in the restoration of PAHs-impacted anaerobic environments should come from field-scale investigations. The field of anaerobic degradation of PAHs is about to pass its infancy, and it’s now expanding reasonably well. Now it is our time to learn how to translate the ever-growing knowledge into remediation planning.
19 Summary
Many natural environments such as subsurface soil, groundwater, freshwater, and marine sediment and sludge are devoid of oxygen, i.e., anaerobic. Feasibility of degradation of hazardous polycyclic aromatic hydrocarbons (PAHs) in anaerobic environment was questioned for a long time. Thermodynamically, anaerobic biodegradation of PAHs under different reducing conditions is feasible despite lower energy yield compared to the aerobic process. So far, degradation of PAHs by facultative and strict anaerobic bacteria and archaea under nitrate-, sulfate-, and iron-reducing and methanogenic conditions have been reported. But, experimental evidence of high-molecular-weight (HMW) PAHs degradation is still lacking. Metabolic pathways for low-molecular-weight (LMW) naphthalene and 2-methylnaphthalene (2-MN) in SRB have been well investigated. In SRB, naphthalene is activated by carboxylation at C2 position, whereas 2-MN is activated by a fumarate addition reaction. Subsequently, anaerobic bacteria employ ring reductases system to overcome the resonance energy of PAHs. Striking dissimilarities between degradation pathways of LMW PAHs and HMW PAHs suggest that the anaerobic degradation mechanisms are either organism-specific, reducing condition-specific or substrate-specific, or all. Because of the limited understanding of anaerobic PAHs metabolism, in situ diagnosis of the impacted environment based on metabolite profiling is still underdeveloped. PAHs exert a selection pressure on the anaerobic microbial community that is often reflected in marked change in abundance, diversity, and function. Inorganic electron acceptor amendment could be a viable method for enhancing anaerobic biodegradation of PAHs. Further investigations on anaerobic degradation of PAHs, especially HMW members, under different redox conditions are crucial to understand the natural attenuation process and to develop approaches for remediation of anaerobic environments contaminated with PAHs.
Abbreviations
- ΔG°′:
-
Standard Gibbs free energy change
- 1,2,3,4-THNA:
-
1,2,3,4-Tetrahydro-2-naphthoic acid
- 1-MN:
-
1-Methylnaphthalene
- 1-NA:
-
1-Naphthoic acid
- 2-DMNA:
-
2-Dimethylnaphthalene
- 2-MN:
-
2-Methylnaphthalene
- 2-NA:
-
2-Naphthoic acid
- 5,6,7,8-THNA:
-
5,6,7,8-Tetrahydro-2-naphthoic acid
- ADP:
-
Adenosine diphosphate
- ATP:
-
Adenosine triphosphate
- BaP:
-
Benzo(a)pyrene
- Bcr:
-
Benzoyl-CoA reductase
- BESA:
-
Bromoethane sulfonic acid
- Bns:
-
Beta-oxidation of naphthyl-2-methylsuccinate
- Bss:
-
Benzylsuccinate synthase
- BTEX:
-
Benzene, toluene, ethylbenzene, and xylene
- CoA:
-
Coenzyme A
- DO:
-
Dissolved oxygen
- E0′:
-
Standard reduction potential
- FBR:
-
Fluidized bed reactor
- GC:
-
Gas chromatography
- H2O2:
-
Hydrogen peroxide
- HH-2-NA:
-
Hexahydro-2-naphthoic acid
- HMW:
-
High molecular weight
- kDa:
-
Kilodalton
- LC:
-
Liquid chromatography
- LC-ESI-MS-MS:
-
Liquid chromatography electrospray ionization tandem mass spectrometry
- LiP:
-
Lignin peroxidase
- LMW:
-
Low molecular weight
- logKOW:
-
Octanol-water partition coefficient
- MGP:
-
Manufactured gas plant sites
- MNA:
-
Methylnaphthoic acid
- MnP:
-
Manganese-dependent peroxidase
- MS:
-
Mass spectrometry
- Ncr:
-
Naphthoyl-CoA reductase
- NMeS:
-
Naphthyl-2-methylenesuccinic acid
- Nms:
-
2-Napthylmethylsuccinate synthase
- NMS:
-
Naphthyl-2-methylsuccinic acid
- NRB:
-
Nitrate-reducing bacteria
- OYE:
-
Old yellow enzyme
- PAHs:
-
Polycyclic aromatic hydrocarbons
- POP:
-
Persistent organic pollutants
- PpcA:
-
Phenylphosphate carboxylase
- Q-TOF-MS:
-
Quadrupole time-of-flight mass spectrometry
- rRNA:
-
Ribosomal RNA
- SOM:
-
Soil organic matter
- SRB:
-
Sulfate-reducing bacteria
- TCA:
-
Tricarboxylic acid
- TEA:
-
Terminal electron acceptor
- THNA:
-
Tetrahydronaphthoic acid
- TOC:
-
Total organic carbon
- T-RFLP:
-
Terminal restriction fragment length polymorphism
- UbiD:
-
3-Polyprenyl-4-hydroxybenzoate decarboxylase
- US EPA:
-
United States Environmental Protection Agency
References
Abdel-Shafy HI, Mansour MSM (2016) A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egypt J Pet 25:107–123
Abu Laban N, Selesi D, Rattei T, Tischler P, Meckenstock RU (2010) Identification of enzymes involved in anaerobic benzene degradation by a strictly anaerobic iron-reducing enrichment culture. Environ Microbiol 12:2783–2796
Achten C, Hofmann T (2009) Native polycyclic aromatic hydrocarbons (PAH) in coals – a hardly recognized source of environmental contamination. Sci Total Environ 407:2461–2473
Acosta-González A, Marqués S (2016) Bacterial diversity in oil-polluted marine coastal sediments. Curr Opin Biotechnol 38:24–32
Agarry SE, Owabor CN (2011) Anaerobic bioremediation of marine sediment artificially contaminated with anthracene and naphthalene. Environ Technol 32:1375–1381
Aitken CM, Jones DM, Larter S (2004) Anaerobic hydrocarbon biodegradation in deep subsurface oil reservoirs. Nature 431:291
Akunna JC, Bizeau C, Moletta R (1993) Nitrate and nitrite reductions with anaerobic sludge using various carbon sources: glucose, glycerol, acetic acid, lactic acid and methanol. Water Res 27:1303–1312
Al-Bashir B, Cseh T, Leduc R, Samson R (1990) Effect of soil/contaminant interactions on the biodegradation of naphthalene in flooded soil under denitrifying conditions. Appl Microbiol Biotechnol 34:414–419
Alejandro A-G, Ramon R-M, Silvia M (2013) Characterization of the anaerobic microbial community in oil-polluted subtidal sediments: aromatic biodegradation potential after the Prestige oil spill. Environ Microbiol 15:77–92
Allamandola L, Tielens A, Barker J (1989) Interstellar polycyclic aromatic hydrocarbons - the infrared emission bands, the excitation/emission mechanism, and the astrophysical implications. Astrophys J Suppl Ser 71:733–775
Ambrosoli R, Petruzzelli L, Minati JL, Marsan FA (2005) Anaerobic PAH degradation in soil by a mixed bacterial consortium under denitrifying conditions. Chemosphere 60:1231–1236
Anderson RT, Lovley DR (1999) Naphthalene and benzene degradation under Fe(III)-reducing conditions in petroleum-contaminated aquifers. Biorem J 3:121–135
Anderson RT, Lovley DR (2000) Anaerobic bioremediation of benzene under sulfate-reducing conditions in a petroleum-contaminated aquifer. Environ Sci Technol 34:2261–2266
Andersson JT, Achten C (2015) Time to say goodbye to the 16 EPA PAHs? Toward an up-to-date use of PACs for environmental purposes. Polycyc Aromat Compd 35:330–354
Annweiler E, Materna A, Safinowski M, Kappler A, Richnow HH, Michaelis W, Meckenstock RU (2000) Anaerobic degradation of 2-methylnaphthalene by a sulfate-reducing enrichment culture. Appl Environ Microbiol 66:5329–5333
Annweiler E, Michaelis W, Meckenstock RU (2002) Identical ring cleavage products during anaerobic degradation of naphthalene, 2-methylnaphthalene, and tetralin indicate a new metabolic pathway. Appl Environ Microbiol 68:852–858
Arthur EL, Rice PJ, Rice PJ, Anderson TA, Baladi SM, Henderson KLD, Coats JR (2005) Phytoremediation – an overview. Crit Rev Plant Sci 24:109–122
Azuma H, Toyota M, Asakawa Y, Kawano S (1996) Naphthalene – a constituent of Magnolia flowers. Phytochemistry 42:999–1004
Bach Q-D, Kim S-J, Choi S-C, Oh Y-S (2005) Enhancing the intrinsic bioremediation of PAH-contaminated anoxic estuarine sediments with biostimulating agents. J Microbiol 43:319–324
Bandowe BA, Meusel H (2017) Nitrated polycyclic aromatic hydrocarbons (nitro-PAHs) in the environment - a review. Sci Total Environ 581-582:237–257
Barton LL, Fauque GD (2009) Biochemistry, physiology and biotechnology of sulfate-reducing bacteria. Adv Appl Microbiol 68:41–98
Bauer JE, Capone DG (1985) Degradation and mineralization of the polycyclic aromatic hydrocarbons anthracene and naphthalene in intertidal marine sediments. Appl Environ Microbiol 50:81–90
Bedessem ME, Swoboda-Colberg NG, Colberg PJ (1997) Naphthalene mineralization coupled to sulfate reduction in aquifer-derived enrichments. FEMS Microbiol Lett 152:213–218
Beller HR, Spormann AM (1997) Anaerobic activation of toluene and o-xylene by addition to fumarate in denitrifying strain T. J Bacteriol 179:670–676
Berdugo-Clavijo C, Dong X, Soh J, Sensen CW, Gieg LM (2012) Methanogenic biodegradation of two-ringed polycyclic aromatic hydrocarbons. FEMS Microbiol Ecol 81:124–133
Bergmann F, Selesi D, Weinmaier T, Tischler P, Rattei T, Meckenstock RU (2011a) Genomic insights into the metabolic potential of the polycyclic aromatic hydrocarbon degrading sulfate-reducing Deltaproteobacterium N47. Environ Microbiol 13:1125–1137
Bergmann FD, Selesi D, Meckenstock RU (2011b) Identification of new enzymes potentially involved in anaerobic naphthalene degradation by the sulfate-reducing enrichment culture N47. Arch Microbiol 193:241–250
Biegert T, Fuchs G, Heider J (1996) Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur J Biochem 238:661–668
Blumer M (1976) Polycyclic aromatic compounds in nature. Sci Am 234:34–45
Blumer M, Youngblood WW (1975) Polycyclic aromatic hydrocarbons in soils and recent sediments. Science 188:53–55
Boll M (2005) Dearomatizing benzene ring reductases. J Mol Microbiol Biotechnol 10:132–142
Boll M, Fuchs G (1995) Benzoyl-coenzyme a reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism: ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur J Biochem 234:921–933
Boll ES, Christensen JH, Holm PE (2008) Quantification and source identification of polycyclic aromatic hydrocarbons in sediment, soil, and water spinach from Hanoi, Vietnam. J Environ Monit 10:261–269
Borneff J, Selenka F, Kunte H, Maximos A (1968) Experimental studies on the formation of polycyclic aromatic hydrocarbons in plants. Environ Res 2:22–29
Breese K, Boll M, Alt-Morbe J, Schagger H, Fuchs G (1998) Genes coding for the benzoyl-CoA pathway of anaerobic aromatic metabolism in the bacterium Thauera aromatica. Eur J Biochem 256:148–154
Brune A, Frenzel P, Cypionka H (2000) Life at the oxic–anoxic interface: microbial activities and adaptations. FEMS Microbiol Rev 24:691–710
Burdige DJ (2007) Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem Rev 107:467–485
Burland SM, Edwards EA (1999) Anaerobic benzene biodegradation linked to nitrate reduction. Appl Environ Microbiol 65:529–533
Cabrerizo A, Galbán-Malagón C, Del Vento S, Dachs J (2014) Sources and fate of polycyclic aromatic hydrocarbons in the Antarctic and Southern Ocean atmosphere. Glob Biogeochem Cycles 28:1424–1436
Caldwell ME, Suflita JM (2000) Detection of phenol and benzoate as intermediates of anaerobic benzene biodegradation under different terminal electron-accepting conditions. Environ Sci Technol 34:1216–1220
Callaghan AV (2013) Metabolomic investigations of anaerobic hydrocarbon-impacted environments. Curr Opin Biotechnol 24:506–515
Canfield DE, Thamdrup B, Hansen JW (1993) The anaerobic degradation of organic matter in Danish coastal sediments: iron reduction, manganese reduction, and sulfate reduction. Geochim Cosmochim Acta 57:3867–3883
Capone DG, Kiene RP (1988) Comparison of microbial dynamics in marine and freshwater sediments: contrasts in anaerobic carbon catabolism. Limnol Oceanogr 33:725–749
Cappenberg TE (1974) Interrelations between sulfate-reducing and methane-producing bacteria in bottom deposits of a fresh-water lake. I. Field observations. Antonie Van Leeuwenhoek 40:285–295
Cea-Barcia G, Carrère H, Steyer JP, Patureau D (2013) Evidence for PAH removal coupled to the first steps of anaerobic digestion in sewage sludge. Int J Chem Eng 2013:1–6. https://doi.org/10.1155/2013/450542
Cerniglia CE (1992) Biodegradation of polycyclic aromatic hydrocarbons. In: Rosenberg E (ed) Microorganisms to combat pollution. Springer, Dordrecht, pp 227–244. https://doi.org/10.1007/978-94-011-1672-5_16
Cerniglia C, Sutherland J (2010) Degradation of polycyclic aromatic hydrocarbons by fungi. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 2079–2110
Cerniglia CE, Gibson DT, Van Baalen C (1979) Algal oxidation of aromatic hydrocarbons: formation of 1-naphthol from naphthalene by Agmenellum quadruplicatum, strain PR-6. Biochem Biophys Res Commun 88:50–58
Cerniglia CE, Van Baalen C, Gibson DT (1980) Metabolism of naphthalene by the cyanobacterium Oscillatoria sp., strain JCM. Microbiology 116:485–494
Chang W, Jones TN, Holoman TRP (2001) Anaerobic polycyclic aromatic hydrocarbon(PAH)-degrading enrichment cultures under methanogenic conditions. In: Sixth international in situ and on site bioremediation symposium, pp 205–209
Chang B, Shiung L, Yuan S (2002) Anaerobic biodegradation of polycyclic aromatic hydrocarbon in soil. Chemosphere 48:717–724
Chang B, Chang S, Yuan S (2003) Anaerobic degradation of polycyclic aromatic hydrocarbons in sludge. Adv Environ Res 7:623–628
Chang W, Um Y, Hoffman B, Holoman TRP (2005) Molecular characterization of polycyclic aromatic hydrocarbon (PAH)-degrading methanogenic communities. Biotechnol Prog 21:682–688
Chang W, Um Y, Holoman TRP (2006) Polycyclic aromatic hydrocarbon (PAH) degradation coupled to methanogenesis. Biotechnol Lett 28:425–430
Chang B-V, Chang IT, Yuan SY (2008) Anaerobic degradation of phenanthrene and pyrene in mangrove sediment. Bull Environ Contam Toxicol 80:145–149
Christensen N, Batstone DJ, He Z, Angelidaki I, Schmidt J (2004) Removal of polycyclic aromatic hydrocarbons (PAHs) from sewage sludge by anaerobic degradation. Water Sci Technol 50:237–244
Coates JD, Anderson RT, Lovley DR (1996) Oxidation of polycyclic aromatic hydrocarbons under sulfate-reducing conditions. Appl Environ Microbiol 62:1099–1101
Coates JD, Woodward J, Allen J, Philp P, Lovley DR (1997) Anaerobic degradation of polycyclic aromatic hydrocarbons and alkanes in petroleum-contaminated marine harbor sediments. Appl Environ Microbiol 63:3589–3593
Coates JD, Chakraborty R, Lack JG, O’connor SM, Cole KA, Bender KS, Achenbach LA (2001) Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two strains of Dechloromonas. Nature 411:1039
Coates JD, Chakraborty R, McInerney MJ (2002) Anaerobic benzene biodegradation – a new era. Res Microbiol 153:621–628
Cohen M, Barlow MJ (2005) Polycyclic aromatic hydrocarbon emission bands in selected planetary nebulae: a study of the behaviour with gas phase C/O ratio. Mon Not R Astron Soc 362:1199–1207
Cunningham JA, Rahme H, Hopkins GD, Lebron C, Reinhard M (2001) Enhanced in situ bioremediation of BTEX-contaminated groundwater by combined injection of nitrate and sulfate. Environ Sci Technol 35:1663–1670
Daisy BH, Strobel GA, Castillo U, Ezra D, Sears J, Weaver DK, Runyon JB (2002) Naphthalene, an insect repellent, is produced by Muscodor vitigenus, a novel endophytic fungus. Microbiology 148:3737–3741
Davidova IA, Gieg LM, Duncan KE, Suflita JM (2007) Anaerobic phenanthrene mineralization by a carboxylating sulfate-reducing bacterial enrichment. ISME J 1:436–442
Deziel E, Paquette G, Villemur R, Lepine F, Bisaillon J (1996) Biosurfactant production by a soil Pseudomonas strain growing on polycyclic aromatic hydrocarbons. Appl Environ Microbiol 62:1908–1912
Didonato RJ Jr et al (2010) Genome sequence of the deltaproteobacterial strain Naphs2 and analysis of differential gene expression during anaerobic growth on naphthalene. PLoS One 5(11):e14072
Dolfing J, Xu A, Gray ND, Larter SR, Head IM (2009) The thermodynamic landscape of methanogenic PAH degradation. Microb Biotechnol 2:566–574
Dou J, Liu X, Ding A (2009) Anaerobic degradation of naphthalene by the mixed bacteria under nitrate reducing conditions. J Hazard Mater 165:325–331
Drew MC (1990) Sensing soil oxygen. Plant Cell Environ 13:681–693
Duran R, Cravo-Laureau C (2016) Role of environmental factors and microorganisms in determining the fate of polycyclic aromatic hydrocarbons in the marine environment. FEMS Microbiol Rev 40:814–830
Eberlein C, Estelmann S, Seifert J, Von Bergen M, Müller M, Meckenstock RU, Boll M (2013a) Identification and characterization of 2-naphthoyl-coenzyme A reductase, the prototype of a novel class of dearomatizing reductases. Mol Microbiol 88:1032–1039
Eberlein C, Johannes J, Mouttaki H, Sadeghi M, Golding BT, Boll M, Meckenstock RU (2013b) ATP-dependent/−independent enzymatic ring reductions involved in the anaerobic catabolism of naphthalene. Environ Microbiol 15:1832–1841
Edwards E, Wills L, Reinhard M, Grbić-Galić D (1992) Anaerobic degradation of toluene and xylene by aquifer microorganisms under sulfate-reducing conditions. Appl Environ Microbiol 58:794–800
Egland PG, Pelletier DA, Dispensa M, Gibson J, Harwood CS (1997) A cluster of bacterial genes for anaerobic benzene ring biodegradation. Proc Natl Acad Sci U S A 94:6484–6489
Eriksson M, Sodersten E, Yu Z, Dalhammar G, Mohn WW (2003) Degradation of polycyclic aromatic hydrocarbons at low temperature under aerobic and nitrate-reducing conditions in enrichment cultures from northern soils. Appl Environ Microbiol 69:275–284
Estelmann S, Blank I, Feldmann A, Boll M (2015) Two distinct old yellow enzymes are involved in naphthyl ring reduction during anaerobic naphthalene degradation. Mol Microbiol 95:162–172
Ezra D, Hess WM, Strobel GA (2004) New endophytic isolates of Muscodor albus, a volatile-antibiotic-producing fungus. Microbiology 150:4023–4031
Fernández P, Grimalt JO (2003) On the global distribution of persistent organic pollutants. Chimia 57:514–521
Ferrarese E, Andreottola G, Oprea IA (2008) Remediation of PAH-contaminated sediments by chemical oxidation. J Hazard Mater 152:128–139
Foght J (2008) Anaerobic biodegradation of aromatic hydrocarbons: pathways and prospects. J Mol Microbiol Biotechnol 15:93–120
Folwell BD, McGenity TJ, Price A, Johnson RJ, Whitby C (2016) Exploring the capacity for anaerobic biodegradation of polycyclic aromatic hydrocarbons and naphthenic acids by microbes from oil-sands-process-affected waters. Int Biodeterior Biodegrad 108:214–221
Frey PA, Reed GH (2012) The ubiquity of iron. ACS Chem Biol 7:1477–1481
Fuchs G, Boll M, Heider J (2011) Microbial degradation of aromatic compounds - from one strategy to four. Nat Rev Microbiol 9:803–816
Galushko A, Minz D, Schink B, Widdel F (1999) Anaerobic degradation of naphthalene by a pure culture of a novel type of marine sulphate-reducing bacterium. Environ Microbiol 1:415–420
Gieg LM, Toth CR (2017) Signature metabolite analysis to determine in situ anaerobic hydrocarbon biodegradation. In: Boll M (ed) Anaerobic utilization of hydrocarbons, oils, and lipids, Handbook of hydrocarbon and lipid microbiology. Springer, Cham, pp 1–30
Gieg LM, Duncan KE, Suflita JM (2008) Bioenergy production via microbial conversion of residual oil to natural gas. Appl Environ Microbiol 74:3022–3029
Griebler C, Safinowski M, Vieth A, Richnow HH, Meckenstock RU (2004) Combined application of stable carbon isotope analysis and specific metabolites determination for assessing in situ degradation of aromatic hydrocarbons in a tar oil-contaminated aquifer. Environ Sci Technol 38:617–631
Grimmer G, Jacob J, Naujack K-W (1983) Profile of the polycyclic aromatic compounds from crude oils. Fresenius Z Anal Chem 314:29–36
Habe H, Omori T (2003) Genetics of polycyclic aromatic hydrocarbon metabolism in diverse aerobic bacteria. Biosci Biotechnol Biochem 67:225–243
Haritash AK, Kaushik CP (2009) Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs): a review. J Hazard Mater 169:1–15
Harwood CS, Burchhardt G, Herrmann H, Fuchs G (1998) Anaerobic metabolism of aromatic compounds via the benzoyl-CoA pathway. FEMS Microbiol Rev 22:439–458
Hayakawa K (2018) Oil spills and polycyclic aromatic hydrocarbons. In: Hayakawa K (ed) Polycyclic aromatic hydrocarbons: environmental behavior and toxicity in East Asia. Springer, Singapore, pp 213–223. https://doi.org/10.1007/978-981-10-6775-4_16
Hayes LA, Nevin KP, Lovley DR (1999) Role of prior exposure on anaerobic degradation of naphthalene and phenanthrene in marine harbor sediments. Org Geochem 30:937–945
Haynes WM (2014) CRC handbook of chemistry and physics. CRC Press, Boca Raton
Hazen TC (2010) Cometabolic bioremediation. In: Timmis KN (ed) Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, pp 2505–2514. https://doi.org/10.1007/978-3-540-77587-4_185
Himmelberg AM, Brüls T, Farmani Z, Weyrauch P, Barthel G, Schrader W, Meckenstock RU (2018) Anaerobic degradation of phenanthrene by a sulfate-reducing enrichment culture. Environ Microbiol 20:3589–3600
Hong Y-W, Yuan D-X, Lin Q-M, Yang T-L (2008) Accumulation and biodegradation of phenanthrene and fluoranthene by the algae enriched from a mangrove aquatic ecosystem. Mar Pollut Bull 56:1400–1405
IARC (2010) Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr Eval Carcinog Risks Hum 92:1
Jahn MK, Haderlein SB, Meckenstock RU (2005) Anaerobic degradation of benzene, toluene, ethylbenzene, and o-xylene in sediment-free iron-reducing enrichment cultures. Appl Environ Microbiol 71:3355–3358
Joback KG, Reid RC (1987) Estimation of pure-component properties from group-contributions. Chem Eng Commun 57:233–243
Jobelius C, Ruth B, Griebler C, Meckenstock RU, Hollender J, Reineke A, Frimmel FH, Zwiener C (2010) Metabolites indicate hot spots of biodegradation and biogeochemical gradients in a high-resolution monitoring well. Environ Sci Technol 45:474–481
Johann H, Georg F (1997) Anaerobic metabolism of aromatic compounds. Eur J Biochem 243:577–596
Johnsen AR, Wick LY, Harms H (2005) Principles of microbial PAH-degradation in soil. Environ Pollut 133:71–84
Johnson K, Ghosh S (1998) Feasibility of anaerobic biodegradation of PAHs in dredged river sediments. Water Sci Technol 38:41–48
Juhasz AL, Naidu R (2000) Bioremediation of high molecular weight polycyclic aromatic hydrocarbons: a review of the microbial degradation of benzo[a]pyrene. Int Biodeterior Biodegrad 45:57–88
Kaiho K (1994) Benthic foraminiferal dissolved-oxygen index and dissolved-oxygen levels in the modern ocean. Geology 22:719–722
Kanaly RA, Harayama S (2000) Biodegradation of high-molecular-weight polycyclic aromatic hydrocarbons by bacteria. J Bacteriol 182:2059–2067
Karavalakis G, Deves G, Fontaras G, Stournas S, Samaras Z, Bakeas E (2010) The impact of soy-based biodiesel on PAH, nitro-PAH and oxy-PAH emissions from a passenger car operated over regulated and nonregulated driving cycles. Fuel 89:3876–3883
Karickhoff SW, Brown DS, Scott TA (1979) Sorption of hydrophobic pollutants on natural sediments. Water Res 13:241–248
Kim K-H, Jahan SA, Kabir E, Brown RJ (2013) A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ Int 60:71–80
Kirso U, Irha N (1998) Role of algae in fate of carcinogenic polycyclic aromatic hydrocarbons in the aquatic environment. Ecotoxicol Environ Saf 41:83–89
Kleemann R, Meckenstock RU (2011) Anaerobic naphthalene degradation by Gram-positive, iron-reducing bacteria. FEMS Microbiol Ecol 78:488–496
Kobayashi H, Rittmann BE (1982) Microbial removal of hazardous organic compounds. Environ Sci Technol 16:170A–183A
Kraft B, Tegetmeyer H, Sharma R, Klotz M, Ferdelman T, Hettich R, Geelhoed J, Strous M (2014) The environmental controls that govern the end product of bacterial nitrate respiration. Science 345:676–679
Kukučka P, Lammel G, Dvorská A, Klánová J, Möller A, Fries E (2010) Contamination of Antarctic snow by polycyclic aromatic hydrocarbons dominated by combustion sources in the polar region. Environ Chem 7:504–513
Kummel S et al (2015) Anaerobic naphthalene degradation by sulfate-reducing Desulfobacteraceae from various anoxic aquifers. FEMS Microbiol Ecol 91(3):fiv006
Kung JW et al (2009) Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc Natl Acad Sci U S A 106:17687–17692
Kuppusamy S, Thavamani P, Venkateswarlu K, Lee YB, Naidu R, Megharaj M (2017) Remediation approaches for polycyclic aromatic hydrocarbons (PAHs) contaminated soils: technological constraints, emerging trends and future directions. Chemosphere 168:944–968
Kuypers MMM, Marchant HK, Kartal B (2018) The microbial nitrogen-cycling network. Nat Rev Microbiol 16:263
Laflamme RE, Hites RA (1978) The global distribution of polycyclic aromatic hydrocarbons in recent sediments. Geochim Cosmochim Acta 42:289–303
Landrum P, Giesy J, Oris J, Allred P (1986) Photoinduced toxicity of polycyclic aromatic hydrocarbons to aquatic organisms. In: Vandermuelen J, Hrudey S (eds) Oil in freshwater: chemistry, biology, countermeasure technology. Pergamon Press, Elmsford, pp 304–318
Langenhoff AAM, Zehnder AJB, Schraa G (1996) Behaviour of toluene, benzene and naphthalene under anaerobic conditions in sediment columns. Biodegradation 7:267–274
Latimer J, Zheng J (2003) The sources, transport and fate of PAHs in the marine environment. In: Douben P (ed) PAHs: an ecotoxicological perspective. Wiley, West Sussex, pp 7–53
Leduc R, Samson R, Al-Bashir B, Al-Hawari J, Cseh T (1992) Biotic and abiotic disappearance of four pah compounds from flooded soil under various redox conditions. Water Sci Technol 26:51–60
Lei A, Wong Y, Tam N (2002) Removal of pyrene by different microalgal species. Water Sci Technol 46:195–201
Li C-H, Zhou H-W, Wong Y-S, Tam NF-Y (2009) Vertical distribution and anaerobic biodegradation of polycyclic aromatic hydrocarbons in mangrove sediments in Hong Kong, South China. Sci Total Environ 407:5772–5779
Li CH, Ye C, Wong YS, Tam NF (2011) Effect of Mn(IV) on the biodegradation of polycyclic aromatic hydrocarbons under low-oxygen condition in mangrove sediment slurry. J Hazard Mater 190:786–793
Li C, Wong Y, Wang H, Tam N (2015) Anaerobic biodegradation of PAHs in mangrove sediment with amendment of NaHCO3. J Environ Sci 30:148–156
Liang L, Song X, Kong J, Shen C, Huang T, Hu Z (2014) Anaerobic biodegradation of high-molecular-weight polycyclic aromatic hydrocarbons by a facultative anaerobe Pseudomonas sp. JP1. Biodegradation 25:825–833
Lima ALC, Farrington JW, Reddy CM (2005) Combustion-derived polycyclic aromatic hydrocarbons in the environment – a review. Environ Forensic 6:109–131
Lovley DR (2001) Anaerobes to the rescue. Science 293:1444–1446
Lovley DR, Lonergan DJ (1990) Anaerobic oxidation of toluene, phenol, and p-cresol by the dissimilatory iron-reducing organism, GS-15. Appl Environ Microbiol 56:1858–1864
Lovley DR, Woodward JC, Chapelle FH (1994) Stimulated anoxic biodegradation of aromatic hydrocarbons using Fe (III) ligands. Nature 370:128
Lovley DR, Coates JD, Woodward JC, Phillips E (1995) Benzene oxidation coupled to sulfate reduction. Appl Environ Microbiol 61:953–958
Lu X, Zhang T, Fang HH-P, Leung KM, Zhang G (2011) Biodegradation of naphthalene by enriched marine denitrifying bacteria. Int Biodeterior Biodegrad 65:204–211
Lu X-Y, Li B, Zhang T, Fang HH (2012) Enhanced anoxic bioremediation of PAHs-contaminated sediment. Bioresour Technol 104:51–58
Lundstedt S et al (2007) Sources, fate, and toxic hazards of oxygenated polycyclic aromatic hydrocarbons (PAHs) at PAH-contaminated sites. Ambio 36:475–485
Luo L, Wang P, Lin L, Luan T, Ke L, Tam NFY (2014) Removal and transformation of high molecular weight polycyclic aromatic hydrocarbons in water by live and dead microalgae. Process Biochem 49:1723–1732
Luo L, Lai X, Chen B, Lin L, Fang L, Tam NF, Luan T (2015) Chlorophyll catalyse the photo-transformation of carcinogenic benzo[a]pyrene in water. Sci Rep 5:12776
Ma B, He Y, Chen H-H, Xu J-M, Rengel Z (2010) Dissipation of polycyclic aromatic hydrocarbons (PAHs) in the rhizosphere: synthesis through meta-analysis. Environ Pollut 158:855–861
Ma C, Wang Y, Zhuang L, Huang D, Zhou S, Li F (2011) Anaerobic degradation of phenanthrene by a newly isolated humus-reducing bacterium, Pseudomonas aeruginosa strain PAH-1. J Soils Sed 11:923–929
MacRae JD, Hall KJ (1998) Biodegradation of polycyclic aromatic hydrocarbons (PAH) in marine sediment under denitrifying conditions. Water Sci Technol 38:177–185
Majora DW, Mayfielda CI, Barkerb JF (1988) Biotransformation of benzene by denitrification in aquifer sand. Groundwater 26:8–14
Maliszewska-Kordybach B (1999) Sources, concentrations, fate and effects of polycyclic aromatic hydrocarbons (PAHs) in the environment. Part A: PAHs in air. Pol J Environ Stud 8:131–136
Margulis L, Sagan D (1997) Microcosmos: four billion years of microbial evolution. University of California Press, New York
Marozava S, Mouttaki H, Muller H, Abu Laban N, Probst AJ, Meckenstock RU (2018) Anaerobic degradation of 1-methylnaphthalene by a member of the Thermoanaerobacteraceae contained in an iron-reducing enrichment culture. Biodegradation 29:23–39
Martens CS, Val Klump J (1984) Biogeochemical cycling in an organic-rich coastal marine basin 4. An organic carbon budget for sediments dominated by sulfate reduction and methanogenesis. Geochim Cosmochim Acta 48:1987–2004
Martirani-Von Abercron S-M, Pacheco D, Benito-Santano P, Marín P, Marqués S (2016) Polycyclic aromatic hydrocarbon-induced changes in bacterial community structure under anoxic nitrate reducing conditions. Front Microbiol 7:1775
McInerney MJ, Sieber JR, Gunsalus RP (2009) Syntrophy in anaerobic global carbon cycles. Curr Opin Biotechnol 20:623–632
McNally DL, Mihelcic JR, Lueking DR (1998) Biodegradation of three-and four-ring polycyclic aromatic hydrocarbons under aerobic and denitrifying conditions. Environ Sci Technol 32:2633–2639
McNeely RN, Neimanis VP, Dwyer L (1979) Water quality sourcebook: a guide to water quality parameters. Waters Directorate, Water Quality Branch, Ottawa
Means JC, Wood SG, Hassett JJ, Banwart WL (1980) Sorption of polynuclear aromatic hydrocarbons by sediments and soils. Environ Sci Technol 14:1524–1528
Meckenstock RU, Mouttaki H (2011) Anaerobic degradation of non-substituted aromatic hydrocarbons. Curr Opin Biotechnol 22:406–414
Meckenstock RU, Annweiler E, Michaelis W, Richnow HH, Schink B (2000) Anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Appl Environ Microbiol 66:2743–2747
Meckenstock RU, Safinowski M, Griebler C (2004) Anaerobic degradation of polycyclic aromatic hydrocarbons. FEMS Microbiol Ecol 49:27–36
Meckenstock RU et al (2016) Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J Mol Microbiol Biotechnol 26:92–118
Menzie CA, Potocki BB, Santodonato J (1992) Exposure to carcinogenic PAHs in the environment. Environ Sci Technol 26:1278–1284
Mihelcic JR, Luthy RG (1988a) Degradation of polycyclic aromatic hydrocarbon compounds under various redox conditions in soil-water systems. Appl Environ Microbiol 54:1182–1187
Mihelcic JR, Luthy RG (1988b) Microbial degradation of acenaphthene and naphthalene under denitrification conditions in soil-water systems. Appl Environ Microbiol 54:1188–1198
Miki S, Uno S, Ito K, Koyama J, Tanaka H (2014) Distributions of polycyclic aromatic hydrocarbons and alkylated polycyclic aromatic hydrocarbons in Osaka Bay, Japan. Mar Pollut Bull 85:558–565
Miller JS, Olejnik D (2001) Photolysis of polycyclic aromatic hydrocarbons in water. Water Res 35:233–243
Miller MM, Wasik SP, Huang GL, Shiu WY, Mackay D (1985) Relationships between octanol-water partition coefficient and aqueous solubility. Environ Sci Technol 19:522–529
Morgan P, Watkinson RJ (1992) Factors limiting the supply and efficiency of nutrient and oxygen supplements for the in situ biotreatment of contaminated soil and groundwater. Water Res 26:73–78
Mouttaki H, Johannes J, Meckenstock RU (2012) Identification of naphthalene carboxylase as a prototype for the anaerobic activation of non-substituted aromatic hydrocarbons. Environ Microbiol 14:2770–2774
Mulas G, Malloci G, Joblin C, Toublanc D (2006) Estimated IR and phosphorescence emission fluxes for specific polycyclic aromatic hydrocarbons in the red rectangle. A&A 446:537–549
Müller JA, Schink B (2000) Initial steps in the fermentation of 3-hydroxybenzoate by Sporotomaculum hydroxybenzoicum. Arch Microbiol 173:288–295
Mulligan CN (2005) Environmental applications for biosurfactants. Environ Pollut 133:183–198
Murphy T, Moller A, Brouwer H (1995) In situ treatment of Hamilton Harbour sediment. Aquat Ecosyst Health Manag 4:195–203
Musat F et al (2009) Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ Microbiol 11:209–219
Muyzer G, Stams AJM (2008) The ecology and biotechnology of sulphate-reducing bacteria. Nat Rev Microbiol 6:441
Nales M, Butler BJ, Edwards EA (1998) Anaerobic benzene biodegradation: a microcosm survey. Biorem J 2:125–144
Neilson AN (2013) PAHs and related compounds: chemistry, vol 3. Springer, Berlin
Neilson A, Allard A, Remberger M (1998) The handbook of environmental chemistry. Part J. PAHs and related compounds. Springer, Berlin
Nestler FHM (1974) Characterization of wood-preserving coal-tar creosote by gas-liquid chromatography. Anal Chem 46:46–53
Nielsen T, Ramdahl T, Bjørseth A (1983) The fate of airborne polycyclic organic matter. Environ Health Perspect 47:103
Nisbet EG, Sleep NH (2001) The habitat and nature of early life. Nature 409:1083
Northrop D, Simpson O, Mott N (1956) Electronic properties of aromatic hydrocarbons I. Electrical conductivity. Proc R Soc Lond A 234:124–135
O’Neil MJ (2013) The Merck index: an encyclopedia of chemicals, drugs, and biologicals. RSC Publishing, London
Ohlenbusch G, Zwiener C, Meckenstock RU, Frimmel FH (2002) Identification and quantification of polar naphthalene derivatives in contaminated groundwater of a former gas plant site by liquid chromatography–electrospray ionization tandem mass spectrometry. J Chromatogr 967:201–207
Ohura T (2007) Environmental behavior, sources, and effects of chlorinated polycyclic aromatic hydrocarbons. Sci World J 7:372–380
Oka AR, Phelps CD, Zhu X, Saber DL, Young L (2011) Dual biomarkers of anaerobic hydrocarbon degradation in historically contaminated groundwater. Environ Sci Technol 45:3407–3414
Okere U, Semple K (2012) Biodegradation of PAHs in ‘pristine’soils from different climatic regions. J Bioremed Biodegr S1:006
Oko BJ, Tao Y, Stuckey DC (2017) Dynamics of two methanogenic microbiomes incubated in polycyclic aromatic hydrocarbons, naphthenic acids, and oil field produced water. Biotechnol Biofuels 10:1–13
Ou S, Zheng J, Zheng J, Richardson BJ, Lam PK (2004) Petroleum hydrocarbons and polycyclic aromatic hydrocarbons in the surficial sediments of Xiamen Harbour and Yuan Dan Lake, China. Chemosphere 56:107–112
Page DS, Brown JS, Boehm PD, Bence AE, Neff JM (2006) A hierarchical approach measures the aerial extent and concentration levels of PAH-contaminated shoreline sediments at historic industrial sites in Prince William Sound, Alaska. Mar Pollut Bull 52:367–379
Parisi VA, Brubaker GR, Zenker MJ, Prince RC, Gieg LM, Da Silva MLB, Alvarez PJJ, Suflita JM (2009) Field metabolomics and laboratory assessments of anaerobic intrinsic bioremediation of hydrocarbons at a petroleum-contaminated site. Microb Biotechnol 2:202–212
Peeters E (2011) The PAH hypothesis after 25 years. Proc Int Astron Union 7:149–161
Peng R-H et al (2008) Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbiol Rev 32:927–955
Perelo LW (2010) In situ and bioremediation of organic pollutants in aquatic sediments. J Hazard Mater 177:81–89
Phelps CD, Battistelli J, Young L (2002) Metabolic biomarkers for monitoring anaerobic naphthalene biodegradation in situ. Environ Microbiol 4:532–537
Phelps CD, Kazumi J, Young LY (1996) Anaerobic degradation of benzene in BTX mixtures dependent on sulfate reduction. FEMS Microbiol Lett 145:433–437
Planavsky NJ et al (2014) Evidence for oxygenic photosynthesis half a billion years before the Great Oxidation Event. Nat Geosci 7:283
Qin W, Zhu Y, Fan F, Wang Y, Liu X, Ding A, Dou J (2017) Biodegradation of benzo(a)pyrene by Microbacterium sp. strain under denitrification: degradation pathway and effects of limiting electron acceptors or carbon source. Biochem Eng J 121:131–138
Qin W, Fan F, Zhu Y, Huang X, Ding A, Liu X, Dou J (2018) Anaerobic biodegradation of benzo(a)pyrene by a novel Cellulosimicrobium cellulans CWS2 isolated from polycyclic aromatic hydrocarbon-contaminated soil. Braz J Microbiol 49:258–268
Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhardt R (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Rafin C, Potin O, Veignie E, Sahraoui L-HA, Sancholle M (2000) Degradation of Benzo[a]Pyrene as sole carbon source by a non white rot fungus, Fusarium solani. Polycyc Aromat Compd 21:311–329
Ribeiro H et al (2018) Potential of dissimilatory nitrate reduction pathways in polycyclic aromatic hydrocarbon degradation. Chemosphere 199:54–67
Rochman CM, Manzano C, Hentschel BT, Simonich SLM, Hoh E (2013) Polystyrene plastic: a source and sink for polycyclic aromatic hydrocarbons in the marine environment. Environ Sci Technol 47:13976–13984
Rockne KJ, Strand SE (1998) Biodegradation of bicyclic and polycyclic aromatic hydrocarbons in anaerobic enrichments. Environ Sci Technol 32:3962–3967
Rockne KJ, Strand SE (2001) Anaerobic biodegradation of naphthalene, phenanthrene, and biphenyl by a denitrifying enrichment culture. Water Res 35:291–299
Rockne KJ, Chee-Sanford JC, Sanford RA, Hedlund BP, Staley JT, Strand SE (2000) Anaerobic naphthalene degradation by microbial pure cultures under nitrate-reducing conditions. Appl Environ Microbiol 66:1595–1601
Ron EZ, Rosenberg E (2002) Biosurfactants and oil bioremediation. Curr Opin Biotechnol 13:249–252
Ross AB et al (2002) Measurement and prediction of the emission of pollutants from the combustion of coal and biomass in a fixed bed furnace. Fuel 81:571–582
Rothermich MM, Hayes LA, Lovley DR (2002) Anaerobic, sulfate-dependent degradation of polycyclic aromatic hydrocarbons in petroleum-contaminated harbor sediment. Environ Sci Technol 36:4811–4817
Safinowski M, Meckenstock RU (2004) Enzymatic reactions in anaerobic 2-methylnaphthalene degradation by the sulphate-reducing enrichment culture N47. FEMS Microbiol Lett 240:99–104
Safinowski M, Meckenstock RU (2006) Methylation is the initial reaction in anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Environ Microbiol 8:347–352
Safinowski M, Griebler C, Meckenstock RU (2006) Anaerobic cometabolic transformation of polycyclic and heterocyclic aromatic hydrocarbons: evidence from laboratory and field studies. Environ Sci Technol 40:4165–4173
Samanta SK, Singh OV, Jain RK (2002) Polycyclic aromatic hydrocarbons: environmental pollution and bioremediation. Trends Biotechnol 20:243–248
Schink B (1997) Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61:262–280
Schöcke L, Schink B (1999) Energetics and biochemistry of fermentative benzoate degradation by Syntrophus gentianae. Arch Microbiol 171:331–337
Schoeny R, Cody T, Warshawsky D, Radike M (1988) Metabolism of mutagenic polycyclic aromatic hydrocarbons by photosynthetic algal species. Mutat Res 197:289–302
Schühle K, Gescher J, Feil U, Paul M, Jahn M, Schägger H, Fuchs G (2003) Benzoate-coenzyme A ligase from Thauera aromatica: an enzyme acting in anaerobic and aerobic pathways. J Bacteriol 185:4920–4929
Selesi D et al (2010) Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J Bacteriol 192:295–306
Sharak Genthner BR, Townsend GT, Lantz SE, Mueller JG (1997) Persistence of polycyclic aromatic hydrocarbon components of creosote under anaerobic enrichment conditions. Arch Environ Contam Toxicol 32:99–105
Shen G, Tao S, Wei S, Zhang Y, Wang R, Wang B, Li W, Shen H, Huang Y, Chen Y, Chen H, Yang Y, Wang W, Wang X, Liu W, Simonich SLM (2012) Emissions of parent, nitro, and oxygenated polycyclic aromatic hydrocarbons from residential wood combustion in rural China. Environ Sci Technol 46:8123–8130
Sims RC, Overcash MR (1983) Fate of polynuclear aromatic compounds (PNAs) in soil-plant systems. Residue Rev 88:1–68
Sivaram AK, Logeshwaran P, Subashchandrabose SR, Lockington R, Naidu R, Megharaj M (2018) Comparison of plants with C3 and C4 carbon fixation pathways for remediation of polycyclic aromatic hydrocarbon contaminated soils. Sci Rep 8:2100
Skupinska K, Misiewicz I, Kasprzycka-Guttman T (2004) Polycyclic aromatic hydrocarbons: physicochemical properties, environmental appearance and impact on living organisms. Acta Pol Pharm 61:233–240
Smith MB, March J (2007) March’s advanced organic chemistry: reactions, mechanisms, and structure. Wiley, Hoboken
Sporstol S, Gjos N, Lichtenthaler RG, Gustavsen KO, Urdal K, Oreld F, Skei J (1983) Source identification of aromatic hydrocarbons in sediments using GC/MS. Environ Sci Technol 17:282–286
Stein SE, Fahr A (1985) High-temperature stabilities of hydrocarbons. J Phys Chem 89:3714–3725
Stogiannidis E, Laane R (2015) Source characterization of polycyclic aromatic hydrocarbons by using their molecular indices: an overview of possibilities. Rev Environ Contam Toxicol 234:49–133
Subashchandrabose SR, Krishnan K, Gratton E, Megharaj M, Naidu R (2014) Potential of fluorescence imaging techniques to monitor mutagenic PAH uptake by microalga. Environ Sci Technol 48:9152–9160
Subashchandrabose SR, Logeshwaran P, Venkateswarlu K, Naidu R, Megharaj M (2017) Pyrene degradation by Chlorella sp. MM3 in liquid medium and soil slurry: possible role of dihydrolipoamide acetyltransferase in pyrene biodegradation. Algal Res 23:223–232
Sullivan ER, Zhang X, Phelps C, Young L (2001) Anaerobic mineralization of stable-isotope-labeled 2-methylnaphthalene. Appl Environ Microbiol 67:4353–4357
Sutherland JB, Freeman JP, Selby AL, Fu PP, Miller DW, Cerniglia CE (1990) Stereoselective formation of a K-region dihydrodiol from phenanthrene by Streptomyces flavovirens. Arch Microbiol 154:260–266
Sutton MA, Oenema O, Erisman JW, Leip A, van Grinsven H, Winiwarter W (2011) Too much of a good thing. Nature 472:159
Tang YJ, Carpenter S, Deming J, Krieger-Brockett B (2005) Controlled release of nitrate and sulfate to enhance anaerobic bioremediation of phenanthrene in marine sediments. Environ Sci Technol 39:3368–3373
Thauer RK, Jungermann K, Decker K (1977) Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev 41:100–180
Thomas J, Ward C (1989) In situ biorestoration of organic contaminants in the subsurface. Environ Sci Technol 23:760–766
Tiedje JM, Sexstone AJ, Myrold DD, Robinson JA (1983) Denitrification: ecological niches, competition and survival. Antonie Van Leeuwenhoek 48:569–583
Tielens AG (2005) The physics and chemistry of the interstellar medium. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511819056
Tongpim S, Pickard MA (1999) Cometabolic oxidation of phenanthrene to phenanthrene trans-9, 10-dihydrodiol by Mycobacterium strain S1 growing on anthracene in the presence of phenanthrene. Can J Microbiol 45:369–376
Toth CRA, Berdugo-Clavijo C, O’Farrell CM, Jones GM, Sheremet A, Dunfield PF, Gieg LM (2018) Stable isotope and metagenomic profiling of a methanogenic naphthalene-degrading enrichment culture. Microorganisms 6(3):E65
Townsend GT, Prince RC, Suflita JM (2003) Anaerobic oxidation of crude oil hydrocarbons by the resident microorganisms of a contaminated anoxic aquifer. Environ Sci Technol 37:5213–5218
Trably E, Patureau D, Delgenes J (2003) Enhancement of polycyclic aromatic hydrocarbons removal during anaerobic treatment of urban sludge. Water Sci Technol 48:53–60
Tsai J-C, Kumar M, Lin J-G (2009) Anaerobic biotransformation of fluorene and phenanthrene by sulfate-reducing bacteria and identification of biotransformation pathway. J Hazard Mater 164:847–855
Tsibart A, Gennadiev A, Koshovskii T, Watts A (2014) Polycyclic aromatic hydrocarbons in post-fire soils of drained peatlands in western Meshchera (Moscow region, Russia). Solid Earth 5:1305–1317
Tu Q et al (2014) GeoChip 4: a functional gene-array-based high-throughput environmental technology for microbial community analysis. Mol Ecol Resour 14:914–928
Tuyen LH, Tue NM, Takahashi S, Suzuki G, Viet PH, Subramanian A, Bulbule KA, Parthasarathy P, Ramanathan A, Tanabe S (2014) Methylated and unsubstituted polycyclic aromatic hydrocarbons in street dust from Vietnam and India: occurrence, distribution and in vitro toxicity evaluation. Environ Pollut 194:272–280
Ulrich AC, Beller HR, Edwards EA (2005) Metabolites detected during biodegradation of 13C6-benzene in nitrate-reducing and methanogenic enrichment cultures. Environ Sci Technol 39:6681–6691
US EPA (1980) Ambient water quality criteria for polynuclear aromatic hydrocarbons. Office of Water Regulation and Standards, Washington. EPA 440/5–80–069
US EPA (1982) An exposure and risk assessment for benzo(a)pyrene and other polycyclic aromatic hydrocarbons. Vol. 1. Summary. United States Environmental Protection Agency, Washington. EPA-440/4-85-020-V1. 1982
US EPA (2008) Polycyclic aromatic hydrocarbons (PAHs). Office of the Solid Waste, United States Environmental Protection Agency, Washington. https://archive.epa.gov/epawaste/hazard/wastemin/web/pdf/pahs.pdf. Accessed 12 Nov 2018
Valerio F, Bottino P, Ugolini D, Cimberle MR, Tozzi GA, Frigerio A (1984) Chemical and photochemical degradation of polycyclic aromatic hydrocarbons in the atmosphere. Sci Total Environ 40:169–188
Vázquez-Duhalt R, Ayala M, Márquez-Rocha FJ (2001) Biocatalytic chlorination of aromatic hydrocarbons by chloroperoxidase of Caldariomyces fumago. Phytochemistry 58:929–933
Vogel TM, Grbic-Galic D (1986) Incorporation of oxygen from water into toluene and benzene during anaerobic fermentative transformation. Appl Environ Microbiol 52:200–202
Wan R, Zhang S, Xie S (2012) Microbial community changes in aquifer sediment microcosm for anaerobic anthracene biodegradation under methanogenic condition. J Environ Sci 24:1498–1503
Wang X-C, Zhang Y-X, Chen RF (2001) Distribution and partitioning of polycyclic aromatic hydrocarbons (PAHs) in different size fractions in sediments from Boston Harbor, United States. Mar Pollut Bull 42:1139–1149
Wania F, Mackay D (1993) Global fractionation and cold condensation of low volatility organochlorine compounds in polar regions. Ambio 22:10–18
Wania F, Mackay D (1995) A global distribution model for persistent organic chemicals. Sci Total Environ 160:211–232
Wania F, Mackay D (1996) Peer reviewed: tracking the distribution of persistent organic pollutants. Environ Sci Technol 30:390A–396A
Warshawsky D, Cody T, Radike M, Reilman R, Schumann B, LaDow K, Schneider J (1995) Biotransformation of benzo[a]pyrene and other polycyclic aromatic hydrocarbons and heterocyclic analogs by several green algae and other algal species under gold and white light. Chem Biol Interact 97:131–148
Warshawsky D, LaDow K, Schneider J (2007) Enhanced degradation of benzo[a]pyrene by Mycobacterium sp. in conjunction with green alga. Chemosphere 69:500–506
Wawrik B et al (2012) Field and laboratory studies on the bioconversion of coal to methane in the San Juan Basin. FEMS Microbiol Ecol 81:26–42
Wei C, Bandowe BAM, Han Y, Cao J, Zhan C, Wilcke W (2015) Polycyclic aromatic hydrocarbons (PAHs) and their derivatives (alkyl-PAHs, oxygenated-PAHs, nitrated-PAHs and azaarenes) in urban road dusts from Xi’an, Central China. Chemosphere 134:512–520
Weiner JM, Lauck TS, Lovley DR (1998) Enhanced anaerobic benzene degradation with the addition of sulfate. Biorem J 2:159–173
Weiss MC, Sousa FL, Mrnjavac N, Neukirchen S, Roettger M, Nelson-Sathi S, Martin WF (2016) The physiology and habitat of the last universal common ancestor. Nat Microbiol 1:16116
Weyrauch P, Zaytsev AV, Stephan S, Kocks L, Schmitz OJ, Golding BT, Meckenstock RU (2017) Conversion of cis-2-carboxycyclohexylacetyl-CoA in the downstream pathway of anaerobic naphthalene degradation. Environ Microbiol 19:2819–2830
White P, Claxton L (2004) Mutagens in contaminated soil: a review. Mutat Res 567:227–345
Widdel F, Bak F (1992) Gram-negative mesophilic sulfate-reducing bacteria. In: The prokaryotes. Springer, New York, pp 3352–3378
Wiktorska K, Misiewicz-Krzeminska I, Kasprzycka-Guttman T (2004) Polycyclic aromatic hydrocarbons: physicochemical properties, environmental appearance and impact on living organisms. Acta Pol Pharm 61(3):233–240
Wilcke W (2000) Synopsis polycyclic aromatic hydrocarbons (PAHs) in soil – a review. J Plant Nutr Soil Sci 163:229–248
Wild SR, Jones KC (1995) Polynuclear aromatic hydrocarbons in the United Kingdom environment: a preliminary source inventory and budget. Environ Pollut 88:91–108
Wilson SC, Jones KC (1993) Bioremediation of soil contaminated with polynuclear aromatic hydrocarbons (PAHs): a review. Environ Pollut 81:229–249
Wischgoll S, Taubert M, Peters F, Jehmlich N, von Bergen M, Boll M (2009) Decarboxylating and nondecarboxylating glutaryl-coenzyme a dehydrogenases in the aromatic metabolism of obligately anaerobic bacteria. J Bacteriol 191:4401–4409
Wu RS (2002) Hypoxia: from molecular responses to ecosystem responses. Mar Pollut Bull 45:35–45
Xu M et al (2014) Elevated nitrate enriches microbial functional genes for potential bioremediation of complexly contaminated sediments. ISME J 8:1932
Xu M, He Z, Zhang Q, Liu J, Guo J, Sun G, Zhou J (2015) Responses of aromatic-degrading microbial communities to elevated nitrate in sediments. Environ Sci Technol 49:12422–12431
Yan Z, Zhang Y, Wu H, Yang M, Zhang H, Hao Z, Jiang H (2017) Isolation and characterization of a bacterial strain Hydrogenophaga sp. PYR1 for anaerobic pyrene and benzo[a]pyrene biodegradation. RSC Adv 7:46690–46698
Yang X, Ye J, Lyu L, Wu Q, Zhang R (2013) Anaerobic biodegradation of pyrene by Paracoccus denitrificans under various nitrate/nitrite-reducing conditions. Water Air Soil Pollut 224:1578
Yu H (2002) Environmental carcinogenic polycyclic aromatic hydrocarbons: photochemistry and phototoxicity. J Environ Sci Health C 20:149–183
Yuan SY, Chang BV (2007) Anaerobic degradation of five polycyclic aromatic hydrocarbons from river sediment in Taiwan. J Environ Sci Health B 42:63–69
Zhang Y, Tao S (2009) Global atmospheric emission inventory of polycyclic aromatic hydrocarbons (PAHs) for 2004. Atmos Environ 43:812–819
Zhang X, Young LY (1997) Carboxylation as an initial reaction in the anaerobic metabolism of naphthalene and phenanthrene by sulfidogenic consortia. Appl Environ Microbiol 63:4759–4764
Zhang X, Sullivan ER, Young LY (2000) Evidence for aromatic ring reduction in the biodegradation pathway of carboxylated naphthalene by a sulfate reducing consortium. Biodegradation 11:117–124
Zhang S, Wang Q, Xie S (2012a) Molecular characterization of phenanthrene-degrading methanogenic communities in leachate-contaminated aquifer sediment. Int J Environ Sci Technol 9:705–712
Zhang S, Wang Q, Xie S (2012b) Stable isotope probing identifies anthracene degraders under methanogenic conditions. Biodegradation 23:221–230
Zhang T, Tremblay P-L, Chaurasia AK, Smith JA, Bain TS, Lovley DR (2013) Anaerobic benzene oxidation via phenol in Geobacter metallireducens. Appl Environ Microbiol 79:7800–7806
Zhou Z, Yao Y, Wang M, Zuo X (2017) Co-effects of pyrene and nitrate on the activity and abundance of soil denitrifiers under anaerobic condition. Arch Microbiol 199:1091–1101
Acknowledgment
Kartik Dhar is grateful to the University of Newcastle for UNIPRS and UNRS central scholarship and to the University of Chittagong, Chittagong 4331, Bangladesh, for granting study leave.
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Dhar, K., Subashchandrabose, S.R., Venkateswarlu, K., Krishnan, K., Megharaj, M. (2019). Anaerobic Microbial Degradation of Polycyclic Aromatic Hydrocarbons: A Comprehensive Review. In: de Voogt, P. (eds) Reviews of Environmental Contamination and Toxicology Volume 251. Reviews of Environmental Contamination and Toxicology, vol 251. Springer, Cham. https://doi.org/10.1007/398_2019_29
Download citation
DOI: https://doi.org/10.1007/398_2019_29
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27148-0
Online ISBN: 978-3-030-27149-7
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)