
Abstract
Alcohol use disorder (AUD) is a chronic relapsing brain disease that currently afflicts over 15 million adults in the United States. Despite its prevalence, there are only three FDA-approved medications for AUD treatment, all of which show limited efficacy. Because of their ability to alter expression of a large number of genes, often with great cell-type and brain-region specificity, transcription factors and epigenetic modifiers serve as promising new targets for the development of AUD treatments aimed at the neural circuitry that underlies chronic alcohol abuse. In this chapter, we will discuss transcriptional regulators that can be targeted pharmacologically and have shown some efficacy in attenuating alcohol consumption when targeted. Specifically, the transcription factors cyclic AMP-responsive element binding protein (CREB), peroxisome proliferator-activated receptors (PPARs), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and glucocorticoid receptor (GR), as well as the epigenetic enzymes, the DNA methyltransferases (DNMTs) and histone deacetylases (HDACs), will be discussed.
The original version of this chapter was revised. A correction to this chapter is available at https://doi.org/10.1007/164_2018_194.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- CREB
- DNA methylation
- DNA methyltransferase
- DNMT
- Glucocorticoid receptor
- HDAC
- Histone acetylation
- Histone deacetylase
- Nuclear factor kappa B
- PPAR
1 Introduction
Alcohol (ethanol) induces both rapid changes in receptor signaling and the longer-acting second messenger signal transduction cascades in the brain that culminate in chromatin remodeling and changes in gene expression. While acute alcohol can lead to transient changes in these signaling pathways, chronic alcohol use leads to persistent genome-wide epigenetic modifications and associated changes in gene expression that alter the neuronal circuitry involved in alcohol reward, craving, and the negative affective state that develops during ethanol withdrawal. Transcription factors and epigenetic modifiers therefore represent excellent targets for attenuating or reversing the pathological effects of chronic alcohol use on neuronal circuitry and ameliorating alcohol use disorder (AUD). In this chapter, we will discuss the role of transcription factors and chromatin-modifying enzymes in alcohol consumption and behaviors related to problematic alcohol use. Many pharmacological agents targeting transcriptional regulators and epigenetic enzymes have been developed that have shown efficacy in preclinical models of AUD.
2 Transcription Factors
2.1 Cyclic AMP-Responsive Element Binding Protein
Cyclic AMP-responsive element binding protein (CREB) is a transcription factor that is widely expressed in the nervous system and is critically involved in neuronal development, plasticity, and learning and memory (Silva et al. 1998). Activity of CREB is modulated by phosphorylation by a number of kinases and phosphatases, including protein kinase A (PKA, Fig. 1a) and calcium/calmodulin-dependent protein kinases (Soderling 1999; Mayr and Montminy 2001). Phosphorylated CREB (pCREB) binds to its coactivator CREB binding protein (CBP), a histone acetyltransferase (HAT) that acts to open chromatin and activate transcription (see Sect. 3), and this complex then binds to cAMP-response elements (CREs) in the DNA. As such, CREB activity is tightly regulated and can rapidly change to adapt to different stimuli.

Simplified diagram of transcriptional pathways and targets for intervention for alcohol use disorder (AUD) treatment. (a) The cAMP-responsive element binding protein (CREB) pathway. Adenylyl cyclase (AC) produces cAMP from AMP, activating protein kinase A (PKA). CREB is phosphorylated (pCREB) by several kinases, one of which is PKA. Once phosphorylated, CREB translocates to the nucleus and binds to cAMP-responsive elements (CRE) in the DNA to activate transcription of genes associated with AUD such as Bdnf and Npy. One method to activate CREB is to use compounds that inhibit the phosphodiesterases (PDEs) that hydrolyze cAMP, thus increasing cAMP levels and activating PKA. PDE inhibitors reduce alcohol consumption in animal models of AUD. (b) The peroxisome proliferator-activated receptor (PPAR) signaling pathway. PPARs are activated by their endogenous ligands, fatty acids (FA), or by synthetic agonists such as the thiazolidinediones and fibrates. Upon ligand binding, PPARs translocate to the nucleus and interact with retinoid X receptor (RXR) at peroxisome proliferator response elements (PPREs) to regulate gene transcription. PPAR agonists reduce alcohol consumption in animal models of AUD. (c) The nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. NF-κB exists as a dimer of different subunits and is complexed with an inhibitory molecule, inhibitor κB (IκB) in the cytosol. Activation of various receptors leads to activation of IκB kinase (IKKβ) and phosphorylation of IκB. This event targets IκB for degradation, releasing NF-κB for translocation to the nucleus to regulate gene expression at κB elements. IKKβ inhibitors reduce alcohol consumption in mice. (d) Glucocorticoid receptor (GR) pathway. GR is held in the cytosol by chaperone proteins. Once bound to its ligand, cortisol (in humans/nonhuman primates) or corticosterone (in rodents) (CORT), GR translocates to the nucleus and binds to glucocorticoid response elements (GREs) to regulate gene transcription. The GR antagonist mifepristone has shown efficacy in reducing alcohol consumption in rodents and humans
Polymorphisms in both the CREB1 gene (rs35349697) and the CBP gene CREBBP (rs3025684) are associated with alcohol addiction (Pal et al. 2014; Kumar et al. 2011). Two key transcriptional target genes of CREB are brain derived neurotrophic factor (BDNF) and neuropeptide Y (NPY) (Tao et al. 1998; Pandey et al. 2004). Polymorphisms in NPY have been associated with alcohol consumption, and a large body of literature has demonstrated that manipulation of the NPY system in rodents alters ethanol consumption (reviewed in Robinson and Thiele 2017). BDNF modulates neuronal development, differentiation, and survival and has been implicated in most psychiatric disorders, including addiction (Moonat et al. 2011; Greenwald et al. 2013; Lobo et al. 2010; Logrip et al. 2015). A single nucleotide polymorphism (SNP) of BDNF (Val66Met) has been associated with alcohol dependence. Minor allele carriers exhibit resistance to relapse (Wojnar et al. 2009) and decreased brain activation in networks associated with more severe dependence symptoms (Chen et al. 2015). Admittedly, this association has not always been found (Nedic et al. 2013; Forero et al. 2015), and discrepancies in study results may be partly attributable to differences in the frequency of the BDNF Val66Met allele across ethnic populations (Shimizu et al. 2004; Pivac et al. 2009).
Alcohol regulates CREB activity by modulating its phosphorylation. Acute ethanol treatment increases, while chronic ethanol attenuates, pCREB (Yang et al. 1998; Pandey et al. 2004). Similarly, withdrawal from ethanol after chronic exposure is characterized by decreased pCREB, without changes in total CREB (Pandey et al. 2001). In vitro, acute ethanol induces an increase in gene expression that is dependent on CREB phosphorylation, an effect that can be blocked by inhibiting PKA activity (Asher et al. 2002). It is possible that the decreased pCREB that results from chronic alcohol exposure is a direct result of reduced PKA activity. Chronic intermittent alcohol exposure in rats results in increased expression of protein kinase A inhibitor alpha (PKI-alpha), a member of a family of proteins implicated in reducing PKA activity (Repunte-Canonigo et al. 2007).
Changes in pCREB may mediate behaviors at each phase of the alcohol addiction cycle. The development of tolerance to the sedative effect of alcohol is associated with increased pCREB (Yang et al. 2003). Additionally, rats selectively bred for high alcohol consumption (alcohol-preferring, or P rats) have decreased CREB expression, pCREB, and CRE-DNA binding in the amygdala compared to their alcohol non-preferring counterparts (Pandey et al. 1999). Withdrawal from chronic alcohol exposure leads to decreased pCREB in the amygdala (Pandey et al. 2001). This reduction of pCREB in the amygdala, a brain region critical for anxiety-like behavior, is associated with both high anxiety and increased ethanol preference (Pandey et al. 2003). Similarly, mice that are deficient in CREB display more anxiety-like behavior relative to wild-type mice and show an attenuation of ethanol-induced anxiolysis (Pandey et al. 2004). Importantly, restoring CREB function in the amygdala of P rats can reduce both alcohol intake and anxiety-like behavior (Pandey et al. 2005).
Although there are no pharmacological agents that directly interact with CREB, CREB activity can be increased indirectly by elevating cAMP levels and activating PKA, which is achieved by inhibition of the phosphodiesterases (PDEs) that hydrolyze cAMP (Fig. 1a). Two recent review articles have discussed the effects of PDE inhibitors on alcohol consumption (Logrip 2015; Olsen and Liu 2016), so the results will only be briefly summarized here (Table 1). Rolipram, a phosphodiesterase-4 (PDE4) inhibitor, increases pCREB in the brain (Hu et al. 2016) and reduces alcohol intake in rats (Franklin et al. 2015; Wen et al. 2012) and in mice (Hu et al. 2011; Blednov et al. 2014; Liu et al. 2017). Other PDE4 inhibitors that reduce ethanol consumption in rodents are roflumilast, Ro 20-1724, mesopram, CDP 840, and piclamilast (Table 1). In addition, the PDE10 inhibitor TP-10 reduces ethanol self-administration by rats, and the nonselective PDE inhibitor ibudilast reduces ethanol consumption by high-drinking rats and ethanol-dependent mice (Bell et al. 2015; Logrip 2015). Most of the aforementioned studies observed a selective reduction in ethanol intake with no change in water or saccharin intake, but others have shown reductions in saccharin or sucrose intake with administration of TP-10 and rolipram (Logrip 2015; Franklin et al. 2015). This initial anhedonic behavior is likely a short-term side effect of drug administration. Rolipram, for instance, initially reduced sucrose intake in P rats, but intake normalized after 5 days of exposure, while suppression of ethanol intake continued (Franklin et al. 2015).
Ibudilast was recently tested in human subjects with mild to severe AUD and was found to improve mood after exposure to an alcohol cue or stress, and reduced craving, but did not change the subjective effects of alcohol (Ray et al. 2017). Ibudilast is currently approved in Japan for the treatment of asthma, multiple sclerosis, and cerebrovascular disease and is generally considered safe. However, ibudilast has gastrointestinal side effects that include nausea, vomiting, and abdominal pain. Roflumilast is a selective PDE4 inhibitor that is FDA-approved for the treatment of chronic obstructive pulmonary disease (COPD) and also has gastrointestinal side effects. Another PDE inhibitor that is currently being used clinically for the treatment of psoriasis is apremilast, a selective PDE4 inhibitor that may have fewer side effects, but animal and human studies need to be completed to evaluate whether it can reduce ethanol consumption. PDE inhibitors may, in fact, reduce drinking through their anti-inflammatory actions (Page and Spina 2011). PDE inhibitors reduce inflammatory neuroimmune responses, which are induced after chronic alcohol exposure and have emerged as important promoters of excessive alcohol intake (Crews et al. 2017; Titus et al. 2015). Thus, the ability of PDE inhibitors to reduce ethanol consumption may not necessarily be solely related to the enhancement of CREB signaling.
2.2 Peroxisome Proliferator-Activated Receptors
Peroxisome proliferator-activated receptors (PPARs) are a group of transcription factors belonging to the nuclear hormone receptor superfamily. PPARs are distributed throughout the body and contribute to a range of biological processes (for a review, see Berger and Moller 2002). Although primarily known for their role in regulating lipid metabolism, PPARs have also been shown to play a role in neuroprotection through repression of pro-inflammatory genes, including the inducible nitric oxide synthase gene (Pascual et al. 2005). PPARs function by translocating to the nucleus upon ligand binding (Fig. 1b). Saturated and unsaturated fatty acids, and their derivatives, are the endogenous ligands of PPARs, although a number of synthetic ligands have also been developed (Berger and Moller 2002). Once in the nucleus, PPARs form heterodimers with retinoid X receptors (RXRs) and bind to PPAR response elements (PPREs) in the promoter region of target genes. Coactivator proteins, such as steroid receptor coactivator-1 (SRC-1), then bind to the transcriptional complex to help initiate transcription (Zhu et al. 1996). The efficiency of coactivator proteins to aid in transcription depends upon which ligand is bound to the PPAR complex, allowing for dynamic control of PPAR target gene expression (Berger and Moller 2002).
Three isoforms of PPARs exist: PPARα, PPARγ, and PPARβ/δ. While most regions of the brain express all three isoforms, PPARβ/δ has the most widespread distribution and dense expression in the rat brain, with PPARγ showing the most restricted expression pattern (Moreno et al. 2004). All three isoforms may work in a coordinated fashion, with PPARβ/δ regulating the activity of the other two PPAR types (Aleshin et al. 2013). Importantly, PPARs are expressed in regions of the brain critical to addiction (i.e., the nucleus accumbens, ventral tegmental area, and amygdala) (Warden et al. 2016) and have recently been implicated in the addiction cycle (Flores-Bastias and Karahanian 2018). Data from a genome-wide association study (GWAS) of the genetics of alcoholism, the Collaborative Study on the Genetics of Alcoholism (COGA), supported an association with the genes encoding PPARγ and PPARα with alcohol withdrawal (Blednov et al. 2015). Intriguingly, while no genetic association was found in that GWAS study for PPARβ/δ, individuals with an AUD were shown to have altered expression of PPARβ/δ and PPARG coactivator 1 alpha (PGC-1alpha) protein in the amygdala and cortical regions of the brain (Ponomarev et al. 2012). Alterations in expression of PPARβ/δ in the brain may then be a consequence of chronic alcohol use. Alternatively, discrepancies in the results of these two studies could suggest that changes in epigenetic regulation of PPARβ/δ may increase risk for the development of an AUD.
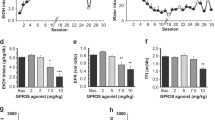
Several agonists of PPARs have proven efficacious in regulating alcohol intake in animal models (Table 2). Agonists of PPARγ and PPARα (pioglitazone, fenofibrate, and tesaglitazar), but not PPARβ/δ (GWO742), decreased alcohol intake and preference in C57BL/6J mice (Blednov et al. 2015; Ferguson et al. 2014). Furthermore, fenofibrate and tesaglitazar suppressed ethanol intake in wild-type mice but had no effect on PPARα null mice, supporting a direct role of PPARα in regulating drinking (Blednov et al. 2016). Interestingly, PPARα may be acting in a sex-dependent manner to regulate ethanol intake; while male mice showed reductions in ethanol intake with fenofibrate and tesaglitazar, female mice showed no response to fenofibrate and an attenuated response to tesaglitazar relative to male mice (Blednov et al. 2016).
Ferguson and colleagues collected amygdala and prefrontal cortex in mice that were given agonists to PPARα (fenofibrate), PPARα/γ (tesaglitazar), or PPARα/γ/β (bezafibrate) and, by using gene expression microarrays and a weighted gene co-expression network analysis (WGCNA), were able to identify gene expression networks that were associated with reduced drinking (Ferguson et al. 2014). Cell-type enrichment analysis showed that both fenofibrate and tesaglitazar targeted amygdala GABAergic interneurons in a coordinated manner while the nonselective PPAR agonist bezafibrate did not. Interestingly, fenofibrate and tesaglitazar both upregulated neuropeptide and dopaminergic signaling genes in the amygdala (including Avp [encoding vasopressin], Npy, and Pdyn [encoding dynorphin]), suggesting that these drugs may be acting in a manner independent of their anti-inflammatory effects to regulate drinking.
The PPARγ agonist pioglitazone has also been shown to be effective at reducing alcohol relapse in rats induced by the pharmacological stressor yohimbine (Stopponi et al. 2013). Interestingly, the opioid antagonist naltrexone (which is FDA-approved for AUD) reduces cue-induced reinstatement of alcohol seeking but has no effect on stress-induced reinstatement of alcohol seeking. When naltrexone and pioglitazone are given together, however, both relapse behaviors are reduced (Stopponi et al. 2013), suggesting that these drugs may work in independent, complementary manners to reduce alcohol relapse risk. In addition to regulating drinking behaviors, pioglitazone has also been shown to be protective against alcohol neurotoxicity (Kane et al. 2011; Tajuddin et al. 2014), suggesting that PPAR agonists may be effective treatments for both AUD and fetal alcohol spectrum disorder (FASD). Indeed, pioglitazone treatment in neonatal C57BL/6J mice blocked ethanol-induced neuroinflammatory cytokine and chemokine expression and microglial activation (Drew et al. 2015). Both fenofibrate and pioglitazone are FDA-approved and are currently being used clinically to improve metabolism and decrease inflammation for a range of conditions, including insulin resistance (Shah and Mudaliar 2010) and cardiovascular disease (Rosenson et al. 2012). Patients with AUD may need to be closely monitored on these drugs, given the rare, but serious, potential side effect of liver disease.
2.3 Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factors are activated by various immunological stimuli and work to amplify inflammatory responses. Since their discovery in the immune system, NF-κB factors have been found in a range of cell types (including neurons and glial cells). NF-κB consists of a family of five subunit proteins (p50, p65, p52, RelB, and c-Rel) that function as dimers. The dimers formed by these subunits are specific to cell type and developmental stage, and lend great specificity to downstream targets and function (Perkins 1997). Generally speaking, the p65/p50 heterodimer activates gene transcription (Li et al. 1994) and is the major NF-κB complex in the adult rodent brain (Yakovleva et al. 2011), while the p50 homodimer represses transcription (Guan et al. 2005). These dimers can be found in the cytoplasm under basal conditions bound to inhibitor κB (IκB) proteins. Upon immune activation, IκB is phosphorylated by the IκB kinase (IKKβ) and targeted for degradation, allowing NF-κB to translocate to the nucleus and regulate gene transcription (Fig. 1c). Although NF-κB is classically activated by immunological stimuli, it can also be activated by glutamate (Guerrini et al. 1995). Activation of NF-κB by synaptic transmission is dependent, at least partially, on calcium/calmodulin-dependent protein kinase II (CAMKII) activation (Meffert et al. 2003). NF-κB activity can also be regulated by additional neurotransmitter systems implicated in addiction. Stimulation of dopamine D2 receptors increases, while stimulation of D1 receptors decreases, NF-κB activity (Takeuchi and Fukunaga 2003). Opioid receptors have also been shown to activate NF-κB. Acute and long-term administration of a μ-opioid receptor agonist in primary cultures of cortical neurons increased NF-κB activity, an effect that was abolished by concurrent treatment with naloxone (Hou et al. 1996).
NF-κB activation has also been associated with alcohol dependence. Polymorphisms in the p50 protein precursor gene NFKB1 are correlated with an increased risk for developing an AUD, especially in individuals with an early onset of alcoholism (Edenberg et al. 2008). The brains of chronic alcoholics exhibit dysregulation in the NF-κB system, with reduced expression of the p50 homodimer and the p65 subunit in the dorsal prefrontal cortex (Okvist et al. 2007). Because the p50 homodimer is largely responsible for inhibiting transcription, its downregulation is associated with increased transcription of over 50 of its target genes in alcoholics (Okvist et al. 2007). The timing and dose of alcohol exposure plays a large role in determining what effect it will have on the NF-κB system. Acute alcohol exposure in C57BL/6J mice results in an upregulation of NF-κB activity, while chronic treatment downregulates its activity (Rulten et al. 2006). As an amplifier of inflammatory responses, NF-κB has been shown to regulate alcohol-induced neurotoxicity. Binge alcohol exposure activates microglia, increases NF-κB binding to DNA, and results in neurotoxicity in Sprague-Dawley rats (Crews et al. 2006). Furthermore, alcohol-induced neurotoxicity can be attributed to the activation of a number of pro-inflammatory genes by NF-κB (Zou and Crews 2010).
Anti-inflammatory compounds may be a promising strategy for manipulating the NF-κB system. Resveratrol, a natural polyphenol, prevents the acute ethanol-induced upregulation of NF-κB, decreases ethanol-induced pro-inflammatory gene transcription, and increases cognitive performance in rodents (Tiwari and Chopra 2013b). Similar therapeutic effects were found upon treatment with curcumin (Tiwari and Chopra 2013a), a biomolecule found in turmeric with well-defined anti-inflammatory pathways associated with inhibition of NF-κB (Shakibaei et al. 2007; Singh and Aggarwal 1995). Administration of the antioxidant butylated hydroxytoluene (BHT) can prevent NF-κB activation, neural damage, and pro-inflammatory gene induction that occur with ethanol exposure (Zou and Crews 2010). Specifically targeting the NF-κB pathway, the IKKβ inhibitors TPCA-1 and sulfasalazine (which prevent NF-κB from translocating to the nucleus) were able to reduce ethanol drinking in mice (Truitt et al. 2016) (Table 2). Sulfasalazine is an FDA-approved anti-inflammatory agent that is commonly used for the management of rheumatoid arthritis (Meier et al. 2013). Delayed liver toxicity may be a serious, albeit reversible, side effect of sulfasalazine in patients with AUD (Masood et al. 2016). The occurrence of such liver toxicity is rare (<1%) in the general population and is associated with a slowed acetylation of sulfasalazine metabolites (Tanigawara et al. 2002). Currently, it is not known whether patients with AUD would be at an increased risk for sulfasalazine-induced liver toxicity.
2.4 Glucocorticoid Receptors
The connection between stress and alcohol use has long been recognized. Stress has been shown to escalate drinking in nondependent and dependent populations (Nash and Maickel 1985; Russell et al. 2017; Spanagel et al. 2014), individuals with a family history of alcohol dependence exhibit increased stress responsivity (Uhart et al. 2006), and vulnerability to stress is a reliable indicator of relapse in alcohol-dependent individuals (Brown et al. 1995; Witkiewitz 2011). There are many targets in the physiological stress pathway that may contribute to stress-induced drinking (including corticotropin releasing factor), yet the importance of glucocorticoids has increasingly been recognized (Nash and Maickel 1988; Fahlke et al. 2000).
The hypothalamic–pituitary–adrenal (HPA) axis is the body’s primary stress response pathway and is responsible for the release of cortisol (in humans) or corticosterone (in rodents), herein referred to as CORT, from the adrenal glands. CORT binds to two receptors: the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). CORT has a tenfold higher affinity for MRs than for GRs, which allows MR occupancy to occur under basal conditions, and therefore restrict CORT levels at baseline via a negative feedback loop (Rupprecht et al. 1993). In contrast, GR occupancy occurs under conditions of high CORT release and is therefore largely responsible for facilitating recovery after a stressor, via negative feedback in the hypothalamus. Additionally, GRs help to promote memories of stressful events by increasing AMPA receptor expression, and thereby strengthening glutamatergic signaling, in the hippocampus and prefrontal cortex (Joels et al. 2012). The ratio of MR:GR functionality may confer resilience (when high) or vulnerability (when low) to a host of psychiatric conditions (ter Heegde et al. 2015). Under basal conditions, GRs are bound to chaperone proteins in the cytoplasm. Upon ligand binding, GR translocates to the nucleus and binds to glucocorticoid response elements (GREs) on the DNA that are often distal to the promoter region of target genes (Reddy et al. 2009) (Fig. 1d). GRs can also bind to noncanonical binding sites on DNA, interact indirectly with DNA via a tethered mechanism with other transcription factors, and interact synergistically with neighboring transcriptional regulatory proteins at combinatorial binding sites on the DNA (Ratman et al. 2013). These varied mechanisms allow for incredible complexity in the downstream transcriptional effects of GRs.
Importantly, alterations in GR expression and activity have been linked to alcohol abuse risk. Genetic polymorphisms in the GR gene (NR3C1) are associated with age of onset of alcohol use and abuse, a phenotype strongly correlated with risk of developing an AUD (Desrivieres et al. 2011). Individuals with alcohol dependence also exhibit a delayed and/or blunted hormonal response to a pharmacological stressor, suggesting dysregulation of the HPA axis with heavy alcohol use (Wand and Dobs 1991). Alcohol can alter CORT and GR expression in such a way as to promote intake. Chronic intermittent ethanol exposure in rats increased peak CORT levels, transiently decreased GR signaling in the medial prefrontal cortex (mPFC) during early withdrawal, and then increased GR signaling during protracted abstinence, an effect accompanied by reinstatement of ethanol seeking (Somkuwar et al. 2017). Similarly, acute alcohol withdrawal in rats produced decreases in GR expression in other regions of the brain critical to stress/alcohol pathways, including the nucleus accumbens and bed nucleus of the stria terminalis, whereas protracted abstinence led to increased GR expression in these brain areas and escalated compulsive alcohol intake (Vendruscolo et al. 2012). CORT can also contribute to alcohol-induced neurodegeneration. Chronic alcohol exposure in adrenalectomized rats given high levels of CORT showed exacerbation of neurodegeneration, while low-dose CORT (commensurate with basal CORT levels) did not exacerbate alcohol neurotoxicity (Cippitelli et al. 2014). CORT acutely suppresses the immune system, but repeated exposures to CORT have been shown to activate microglia in mice, a response driven by GR activation (Nair and Bonneau 2006). In this way, alcohol may increase pro-inflammatory responses in the brain via direct mechanisms involving NF-κB signaling (as mentioned earlier in this chapter) as well as via GR activation.
Inhibition of GRs can reverse many of these alcohol phenotypes. Chronic treatment with mifepristone, a nonselective GR antagonist, prevented dependence-induced escalations in drinking and compulsive responding for alcohol exhibited during protracted abstinence in rats (Vendruscolo et al. 2012) (Table 2). Interestingly, mifepristone reduced alcohol intake in dependent, but not nondependent rats, suggesting that the GR dysregulation that occurs with chronic alcohol exposure is a unique risk factor for escalated use (Vendruscolo et al. 2015). Mifepristone may also prove to aid in the treatment of symptoms associated with ethanol withdrawal. Rats treated with mifepristone showed a dose-dependent reduction in several withdrawal-related behaviors, including tremor and tail rigidity (Sharrett-Field et al. 2013), and a single treatment of mifepristone in mice reduced the cognitive deficits observed during withdrawal (Jacquot et al. 2008). Daily doses of mifepristone have also been shown to attenuate alcohol-induced hippocampal neurodegeneration in rats in a dose-dependent manner (Cippitelli et al. 2014). Although most work with mifepristone has been conducted in animals, preliminary research in humans has shown promising results. Just 1 week of mifepristone treatment in alcohol dependent human subjects reduced both alcohol craving and consumption (Vendruscolo et al. 2015).
Mifepristone shows great promise as a treatment for AUD, but there are limitations to its use. In addition to blocking GRs, mifepristone is also a potent antagonist of the progesterone receptor and is most commonly used clinically to terminate pregnancies. As such, female patients receiving mifepristone can experience vaginal bleeding due to endometrial thickening. Other side effect profiles are low, even with chronic treatment. Long-term, low-dose mifepristone used to treat uterine fibroids resulted in no significant side effects (Kapur et al. 2016). At much higher doses, mifepristone has been used to treat Cushing’s syndrome, which is characterized by chronic, excessive exposure to glucocorticoids. At these high doses, mifepristone can cause more serious side effects, including hypertension, hypokalemia, and edema (Cuevas-Ramos et al. 2016).
Taken together, these results suggest that GR activation is critical for both the development and maintenance of AUD, and disrupting GR signaling using mifepristone may be promising for preventing relapse and treating withdrawal symptoms in alcohol-dependent individuals.
3 Epigenetic Modifiers
3.1 DNA Methyltransferases
In mammals, DNA methylation is catalyzed by three DNA methyltransferases (DNMTs): DNMT1, DNMT3A, and DNMT3B (Day et al. 2015). These enzymes add a methyl group to the fifth carbon position of the cytosine (5mC) found adjacent to guanine (as cytosine-phosphate-guanine dinucleotides, or CpGs) (Zovkic et al. 2013), using the methyl donor S-adenosyl methionine (SAM). The genome contains regions that are rich in CpGs, known as CpG islands, which are often found in gene regulatory or promoter regions. The methyl-binding domain proteins (e.g., MeCP2) directly interact with 5mC, that then recruit chromatin-modifying proteins and transcriptional repressor complexes to the DNA (Zovkic et al. 2013). Thus, DNAm is normally associated with repression of gene expression (Fig. 2).

Simplified diagram of epigenetic modifications and epigenetic enzyme targets for intervention for AUD. DNA (gray line) is wrapped around histone octamers to form the nucleosome (shown in blue), the basic unit of chromatin. Top panel: DNA methylation is catalyzed by DNA methyltransferases (DNMTs) and is associated with condensed chromatin and repression of gene expression. DNMT inhibitors reduce alcohol consumption in animal models of AUD. Bottom panel: histone acetylation is catalyzed by the histone acetyltransferases (HATs) and is generally associated with open chromatin, increased transcription factor availability, and activation of gene expression. Removal of acetyl groups from histones is achieved by the histone deacetylases (HDACs). HDAC inhibitors reduce alcohol consumption in animal models of AUD
DNAm in the brain plays an important role in learning and memory (Zovkic et al. 2013), and evidence is accumulating that DNAm is also important in AUD (Tulisiak et al. 2017). Increased expression of DNMT1 protein and DNMT3a and 3b genes in rodent brains, and decreased expression of DNMT3A and 3B genes in human blood samples, have been observed after chronic ethanol exposure (Barbier et al. 2015; Bonsch et al. 2006; Warnault et al. 2013; Qiao et al. 2017). The difference in DNMT3A and 3B expression between brain and blood samples may be due to species differences (rat vs. human), different tissue-specific responses to ethanol, or duration or timing of alcohol exposure. Nonetheless, changes in DNMTs after chronic ethanol exposure indicate that DNAm might be altered by alcohol.
Additional circumstantial evidence that DNAm is altered by alcohol exposure has come from an analysis of postmortem cerebellum from human subjects with AUD compared with that of control subjects. In this study, the authors measured increased transcript levels of enzymes involved in the one-carbon metabolism pathway, which generates the methyl donor SAM (Gatta et al. 2017). Correlated with this was an increase in the ratio of SAM to s-adenosylhomocysteine (SAH) in the cerebellum, which would increase the activity of DNMTs (Auta et al. 2017; Gatta et al. 2017). Similar changes in the SAM/SAH ratio occurred in rat cerebellum after chronic ethanol drinking, indicating that the changes are induced by alcohol (Auta et al. 2017). Finally, mRNA levels of an enzyme (tet methylcytosine dioxygenase 1, TET1) that removes methyl groups from cytosines was decreased in the cerebellum of subjects with AUD (Gatta et al. 2017). Together, these results indicate that changes in enzymes that regulate DNAm are associated with chronic alcohol exposure and suggest that there might be increased total DNAm in the brain. However, more detailed studies described below, examining differentially methylated regions after chronic alcohol use, suggest that specific genomic locations associated with excessive drinking can be either hypo- or hypermethylated.
More explicit evidence that changes in DNAm are associated with alcohol use was first provided by analysis of genomic DNA from blood cells of alcoholic patients, with increased total DNAm associated with alcoholism (Bonsch et al. 2004). Another study found decreased DNAm at repetitive DNA elements (Alu) associated with alcohol use in lymphocytes from healthy individuals (Zhu et al. 2012). Candidate gene approaches in blood cells have demonstrated associations between increased DNAm and AUD at the promoters for alpha synuclein (SNCA), homocysteine-induced endoplasmic reticulum protein (HERPUD1), serotonin transporter (SLC6A4), monoamine oxidase A (MAOA), prodynorphin (PDYN), and aldehyde dehydrogenase 2 (ALDH2) (Bleich et al. 2006; Bonsch et al. 2005; Philibert et al. 2008a, b; D’Addario et al. 2017; Pathak et al. 2017). More recently, genome-wide profiling of methylated regions in whole blood cells or human lymphoblast cell lines has demonstrated several significant differentially methylated regions or differentially methylated cytosines associated with high levels of alcohol drinking or dependence (Philibert et al. 2012; Clark et al. 2015; Liu et al. 2016; Zhang et al. 2013; Zhao et al. 2013). In another study by Philibert et al. (2014), differentially methylated cytosines were identified in peripheral mononuclear cell DNA from subjects with heavy alcohol use compared with controls. Genome-wide DNAm was measured in the AUD subjects as they entered a treatment facility and ~25 days later, during abstinence. Many of the differentially methylated cytosines identified between controls and alcohol-dependent subjects were reversed during abstinence to levels similar to controls. Similarly, Bruckmann et al. (2017) found that some of the differentially methylated cytosines identified in CD3+ T cells between healthy controls and alcohol-dependent patients reverted back to control levels after abstinence, demonstrating a potential causal role for alcohol in changing DNAm in blood cells.
Studies in blood, lymphoblast cell lines, or other peripheral tissues may not represent the DNAm patterns in the brain associated with alcohol use. A few studies have found similar changes occurring in postmortem brain and peripheral tissues. Notably, DNAm in the promoter for the gene encoding the delta subunit of the GABA-A receptor (GABRD) was increased in the cerebellum of human subjects with AUD compared with controls, similar to what has been observed in lymphocytes (Gatta et al. 2017; Liu et al. 2016). The increase in DNAm in the promoter of GABRD in the cerebellum was associated with decreased GABRD expression (Gatta et al. 2017). In the precuneus brain region from AUD subjects, 244 hypomethylated and 188 hypermethylated regions were associated with alcohol dependence (Hagerty et al. 2016). These differentially methylated cytosines overlapped with those found in buccal (cheek) cells collected from the same subjects (Hagerty et al. 2016). Together, these studies indicate that alterations in DNAm in the brain occur with chronic alcohol use and that some of these changes are similar to those occurring in peripheral tissues, suggesting that DNAm changes at particular genetic loci could be used as a diagnostic measure for AUD and possibly treatment response.
Additional genome-wide analysis of DNAm in different brain regions in humans, and monkeys has been performed in order to identify both region-specific changes in DNAm and potential new candidate genes for AUD. Analysis of prefrontal cortex tissue from AUD and control subjects found 1,812 differentially methylated cytosines mapping to 1,099 genes that were significantly associated with AUD (Wang et al. 2016). In rhesus macaques, differentially methylated regions in the nucleus accumbens discriminated abstinent monkeys from low/binge drinkers and heavy/very-heavy drinkers and were located in genes encoding synaptic, cell signaling, and receptor trafficking mediators (Cervera-Juanes et al. 2017a, b) that could, in theory, be targets for pharmacological intervention.
Three pharmacological agents have been used to inhibit DNMTs and determine the effect on behaviors related to AUD in animal models: the nucleoside analogs 5-azacytidine (azacytidine), 5-aza-2′deoxycytidine (decitabine), and RG108, a non-nucleoside DNMT inhibitor (Table 3). Mice treated with azacytidine or decitabine reduced their ethanol intake in intermittent access procedures that model binge-like drinking (Warnault et al. 2013; Ponomarev et al. 2017). In alcohol-dependent rats, infusion of RG108 into the cerebral ventricles resulted in decreased alcohol intake after a three-week period of forced abstinence when compared with vehicle-treated rats (Barbier et al. 2015). Similarly, infusion of decitabine into the mPFC of chronically drinking rats decreased ethanol intake but increased anxiety-like behavior (Qiao et al. 2017). The timing of administration appears to be important for the ability of DNMT inhibitors to reduce ethanol drinking. When ethanol-dependent mice were given intracerebroventricular azacytidine during the induction of dependence (immediately before ethanol vapor exposure), they subsequently consumed more ethanol in a 2BC test (Qiang et al. 2014). However, generally the evidence indicates that DNMT inhibitors reduce ethanol intake in rodent models of binge and dependence-induced drinking.
Azacytidine and decitabine are FDA-approved for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Both of these drugs have high toxicity (Gnyszka et al. 2013) and serious side effects of these drugs include increased bruising, bleeding, and infection. Azacytidine is contraindicated for individuals with liver tumors, and those with liver and kidney disease should be monitored carefully. As a result, these drugs should not be used for those individuals with alcohol-associated liver disease. A newer nucleoside analog, zebularine, has less toxicity (Gnyszka et al. 2013) but has not yet been tested in animal models of AUD. In summary, DNMT inhibitors represent a promising pharmacotherapeutic approach to treat AUD, but newer generation compounds, such as zebularine and RG-108, which are not yet FDA-approved, require further investigation.
3.2 Histone Deacetylases and Histone Acetyltransferases
DNA wrapped around a histone octamer forms the nucleosome, an integral building block of chromatin structure. Changes in the acetylation state of histone tails are intimately involved in chromatin remodeling and transcriptional alterations. HATs are enzymes that add acetyl groups to lysine residues on histone proteins, while histone deacetylases (HDACs) remove these acetyl groups (Elvir et al. 2017; Haberland et al. 2009) (Fig. 2). Generally, HATs activate transcription while HDACs repress it, although there are exceptions (Haberland et al. 2009; Sacconnay et al. 2016). The mammalian genome encodes 11 “classical,” zinc-dependent HDACs that are categorized into four families (class I: HDAC1, 2, 3, and 8; class IIa: HDAC4, 5, 7, and 9; class IIb: HDAC6 and 10; and class IV: HDAC11). A separate family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases, called sirtuins (or class III HDACs), comprises 7 members (SIRT1-7) (Sacconnay et al. 2016). Recent evidence shows alterations in histone acetylation/deacetylation and chromatin structure in several psychiatric disorders, suggesting that these processes may underlie motivated behaviors, including drug addiction (Elvir et al. 2017; Pena et al. 2014).
Ethanol changes the acetylation state of histones after acute and chronic exposure and during withdrawal. Acute ethanol exposure led to changes in histone H3 and H4 acetylation in the amygdala, hippocampus, and cortex (D’Addario et al. 2013; Finegersh and Homanics 2014; Pandey et al. 2008; Sakharkar et al. 2012). Chronic ethanol exposure and/or withdrawal altered acetylation of histones H3 and H4 in the amygdala, ventral tegmental area, cortex, nucleus accumbens, dorsal striatum, and hippocampus (Arora et al. 2013; Bohnsack et al. 2017; Botia et al. 2012; D’Addario et al. 2013; Dominguez et al. 2016; Finegersh et al. 2015; Qiang et al. 2011; Shibasaki et al. 2011; Simon-O’Brien et al. 2015; You et al. 2014; Pandey et al. 2008). In general, the changes in total histone H3 and H4 acetylation induced by acute alcohol exposure appear to be the opposite to those of withdrawal from chronic alcohol exposure. However, this depends on the brain region. Alcohol-induced changes in acetylation in the promoter regions of specific genes such as GABRA1 (Arora et al. 2013; Bohnsack et al. 2017), PDYN (D’Addario et al. 2013), PNOC (D’Addario et al. 2013), BDNF (You et al. 2014), ARC (You et al. 2014), NPY (Pandey et al. 2008; Sakharkar et al. 2012), and GRIN2B (NR2B) (Qiang et al. 2011, 2014) were associated with changes in gene expression, which has important consequences for behaviors such as anxiety during withdrawal (Pandey et al. 2008) and ethanol consumption (Qiang et al. 2014).
Treatment with HDAC inhibitors is effective in reducing ethanol intake in multiple models of AUD (Table 3). Mice treated with Trichostatin A (TSA) or Vorinostat (SAHA) (inhibitors of class I, II, and IV HDACs) consumed less ethanol in a limited-access binge-drinking test, and rats treated with SAHA also self-administered less ethanol in an operant task and exhibited reduced alcohol-seeking behavior (Warnault et al. 2013). Treatment with the HDAC1/2 inhibitor valproic acid (VPA) decreased 2BC ethanol consumption and preference by rats and also blocked ethanol reward, as measured in the conditioned place preference test (Al Ameri et al. 2014). In the Warnault and Al Ameri studies, the effect of the HDAC inhibitors on histone acetylation in the brain was not measured, but Warnault et al. did observe a decrease in total histone H4 acetylation in the nucleus accumbens after binge drinking by mice and ethanol self-administration by rats (Warnault et al. 2013). In summary, these studies indicate that TSA, SAHA, and VPA decrease binge-like drinking or alcohol reward/reinforcement-related behavioral measures.
Heavy-drinking rats treated with the HDAC1/3 inhibitor Entinostat (MS-275) also self-administered less ethanol and exhibited reduced relapse (reinstatement) to ethanol seeking after a period of abstinence, an effect that was associated with increased histone H4 acetylation in the nucleus accumbens and dorsolateral striatum (Jeanblanc et al. 2015). Treatment with the nonselective HDAC class I and IIa inhibitor sodium butyrate (NaB), or MS-275, reduced operant ethanol self-administration by ethanol-dependent rats, but these compounds did not affect responding for ethanol in nondependent animals (Simon-O’Brien et al. 2015). In addition, NaB treatment reduced ethanol drinking by rats in an intermittent access 2BC drinking experiment and prevented the escalation of ethanol intake that occurs after alcohol deprivation (Simon-O’Brien et al. 2015). In this study, histone H3 lysine 9 acetylation varied between ethanol-dependent and nondependent rats depending on the brain region, and NaB treatment did not uniformly increase histone acetylation in all brain regions as might be predicted of an HDAC inhibitor. For instance, in the prefrontal cortex of ethanol-dependent rats, NaB decreased histone H3 acetylation (Simon-O’Brien et al. 2015). This demonstrates that there are complicated region-specific alterations in total histone acetylation after alcohol exposure. In the studies described above in which HDAC inhibitors were tested for their role in ethanol consumption, histone acetylation at specific gene promoters was not examined. Identifying these genes and the brain regions in which they act to regulate alcohol drinking is a clear area for future research. Nonetheless, these studies demonstrate that NaB and MS-275 treatment can prevent relapse to alcohol drinking in animals that are alcohol-dependent.
Ethanol withdrawal causes anxiety, promotes relapse to drinking, and is associated with several changes in the amygdala: increased nuclear HDAC activity, decreased acetylated histones, decreased expression of the HAT CBP (CREB binding protein), decreased expression of Npy, Bdnf, and Arc, and decreased dendritic spine density (Pandey et al. 2008; You et al. 2014). Treatment of rats with TSA reversed ethanol withdrawal-induced anxiety and the epigenetic, gene expression, and structural changes observed in the amygdala (Pandey et al. 2008; You et al. 2014). HDAC inhibitors are also effective in a genetic model of AUD, the alcohol-preferring P rat. P rats have higher anxiety and ethanol intake compared with alcohol non-preferring NP rats, higher levels of nuclear HDAC activity, more HDAC2 protein, decreased acetylated histones, and decreased NPY protein in the amygdala (Moonat et al. 2013; Sakharkar et al. 2014). Treatment of P rats with TSA, or with HDAC2 siRNA in the amygdala, reduced anxiety and ethanol intake and normalized the associated epigenetic alterations and NPY levels (Moonat et al. 2013; Sakharkar et al. 2014). Since anxiety is associated with an increased risk of relapse in alcohol-dependent individuals (Schellekens et al. 2015), preventing the development of withdrawal-induced anxiety through the use of HDAC inhibitors may be a promising method for encouraging abstinence in recovering alcoholics.
Changes in histone acetylation can persist long after the initial exposure to alcohol. Adolescence is a period of brain development in which synaptic structural modifications and changes in neural plasticity are occurring. Exposure of animals to alcohol during adolescence leads to long-lasting alterations in histone acetylation that persist into adulthood and these changes are associated with increased anxiety-like behavior and high levels of ethanol consumption (Kokare et al. 2017; Pandey et al. 2015; Pascual et al. 2009, 2012; Sakharkar et al. 2016). Treatment of adult rats with TSA after they had been exposed to ethanol during adolescence normalized the high levels of anxiety, ethanol intake, and alcohol-induced histone acetylation and gene expression changes (Pandey et al. 2015; Sakharkar et al. 2016). These studies suggest that the persistent histone acetylation changes associated with alcohol exposure during adolescence can be reversed by treatment with HDAC inhibitors in adulthood and attenuate pathological anxiety and excessive drinking.
Taken together, HDAC inhibitors have been found generally to decrease excessive ethanol-drinking and ethanol-seeking behavior in rodents. However, there are a few exceptions. Wolstenholme et al. found that treatment of mice with TSA increased voluntary 2BC ethanol intake, and Qiang et al. demonstrated that treatment of mice with TSA during exposure to ethanol vapor subsequently led to increased ethanol drinking (Qiang et al. 2014; Wolstenholme et al. 2011). In addition, Ponomarev et al. found that SAHA had no effect on either a binge-drinking test or in a chronic intermittent drinking protocol (Ponomarev et al. 2017). These results indicate that the timing of administration of HDAC inhibitors may be important when considering them for AUD treatment.
SAHA is FDA-approved for the treatment of cutaneous T-cell lymphoma. Serious side effects include increased risk of developing blood clots, increased bruising, bleeding, or susceptibility to infection, and increased blood sugar levels. There are no known interactions with light alcohol drinking. SAHA might be a viable option to move forward in clinical studies. VPA has long been prescribed for the treatment of seizures and bipolar disorder. Serious side effects of VPA include blistering, peeling, or red skin rash, confusion, memory problems, suicidal thoughts, and depression. VPA can also cause liver problems, pancreatitis, and thrombocytopenia. As also noted for the DNMT inhibitors (azacytidine and decitabine), liver disease may preclude the use of VPA in alcohol-dependent patients. Finally, it should be mentioned that systemic administration of DNMT and HDAC inhibitors could have potentially deleterious effects on gene expression in several tissues because they block the activity of enzymes expressed throughout the body.
In terms of treating AUD patients with compounds targeting epigenetic modifiers, the aforementioned animal studies may provide insight into compounds that may work best for different subtypes of alcoholism or at different phases of the addiction cycle. For instance, DNMT inhibitors might be useful in decreasing binge-like drinking, and a non-nucleoside DNMT inhibitor such as RG-108 could also be effective in treating alcohol-dependent individuals during abstinence to prevent relapse. SAHA may be effective in reducing binge-like drinking and also anxiety during alcohol withdrawal. Finally, the HDAC1/3 inhibitor MS-275 appears to have limited toxicity (Subramanian et al. 2010), is currently in clinical trials for cancer treatment, and may be another option for treating alcohol-dependent patients during abstinence to prevent relapse.
4 Conclusions and Future Directions
Several small molecule compounds have been developed that target transcriptional regulators and epigenetic enzymes that have shown effectiveness in reducing alcohol drinking in several rodent models of AUD. One of these compounds, mifepristone, has shown promising results in a human laboratory study and is already FDA-approved for other conditions. Future studies should focus on translating the findings of other compounds to clinical studies to determine if they can reduce excessive alcohol drinking in human subjects with AUD. Several new promising candidates exist, including PDE4 inhibitors, PPARα/γ agonists, HDAC inhibitors, and DNMT inhibitors. A focus on repurposing those compounds that are already FDA-approved for other conditions may be an efficient mechanism to getting these into clinical use for those suffering from AUD.
Change history
29 January 2019
Figure 1 was published incorrectly in this chapter. The original chapter was corrected.
References
Al Ameri M, Al Mansouri S, Al Maamari A, Bahi A (2014) The histone deacetylase (HDAC) inhibitor valproic acid reduces ethanol consumption and ethanol-conditioned place preference in rats. Brain Res 1583:122–131. https://doi.org/10.1016/j.brainres.2014.07.051
Aleshin S, Strokin M, Sergeeva M, Reiser G (2013) Peroxisome proliferator-activated receptor (PPAR)beta/delta, a possible nexus of PPARalpha- and PPARgamma-dependent molecular pathways in neurodegenerative diseases: review and novel hypotheses. Neurochem Int 63(4):322–330. https://doi.org/10.1016/j.neuint.2013.06.012
Arora DS, Nimitvilai S, Teppen TL, McElvain MA, Sakharkar AJ, You C, Pandey SC, Brodie MS (2013) Hyposensitivity to gamma-aminobutyric acid in the ventral tegmental area during alcohol withdrawal: reversal by histone deacetylase inhibitors. Neuropsychopharmacology 38(9):1674–1684. https://doi.org/10.1038/npp.2013.65
Asher O, Cunningham TD, Yao L, Gordon AS, Diamond I (2002) Ethanol stimulates cAMP-responsive element (CRE)-mediated transcription via CRE-binding protein and cAMP-dependent protein kinase. J Pharmacol Exp Ther 301(1):66–70
Auta J, Zhang H, Pandey SC, Guidotti A (2017) Chronic alcohol exposure differentially alters one-carbon metabolism in rat liver and brain. Alcohol Clin Exp Res 41(6):1105–1111. https://doi.org/10.1111/acer.13382
Barbier E, Tapocik JD, Juergens N, Pitcairn C, Borich A, Schank JR, Sun H, Schuebel K, Zhou Z, Yuan Q, Vendruscolo LF, Goldman D, Heilig M (2015) DNA methylation in the medial prefrontal cortex regulates alcohol-induced behavior and plasticity. J Neurosci 35(15):6153–6164. https://doi.org/10.1523/JNEUROSCI.4571-14.2015
Barson JR, Karatayev O, Chang GQ, Johnson DF, Bocarsly ME, Hoebel BG, Leibowitz SF (2009) Positive relationship between dietary fat, ethanol intake, triglycerides, and hypothalamic peptides: counteraction by lipid-lowering drugs. Alcohol 43(6):433–441. https://doi.org/10.1016/j.alcohol.2009.07.003
Bell RL, Lopez MF, Cui C, Egli M, Johnson KW, Franklin KM, Becker HC (2015) Ibudilast reduces alcohol drinking in multiple animal models of alcohol dependence. Addict Biol 20(1):38–42. https://doi.org/10.1111/adb.12106
Berger J, Moller DE (2002) The mechanisms of action of PPARs. Annu Rev Med 53:409–435. https://doi.org/10.1146/annurev.med.53.082901.104018
Blednov YA, Benavidez JM, Black M, Harris RA (2014) Inhibition of phosphodiesterase 4 reduces ethanol intake and preference in C57BL/6J mice. Front Neurosci 8:129. https://doi.org/10.3389/fnins.2014.00129
Blednov YA, Benavidez JM, Black M, Ferguson LB, Schoenhard GL, Goate AM, Edenberg HJ, Wetherill L, Hesselbrock V, Foroud T, Harris RA (2015) Peroxisome proliferator-activated receptors alpha and gamma are linked with alcohol consumption in mice and withdrawal and dependence in humans. Alcohol Clin Exp Res 39(1):136–145. https://doi.org/10.1111/acer.12610
Blednov YA, Black M, Benavidez JM, Stamatakis EE, Harris RA (2016) PPAR agonists: I. Role of receptor subunits in alcohol consumption in male and female mice. Alcohol Clin Exp Res 40(3):553–562. https://doi.org/10.1111/acer.12976
Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, Bonsch D (2006) Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin Exp Res 30(4):587–591. https://doi.org/10.1111/j.1530-0277.2006.00068.x
Bohnsack JP, Patel VK, Morrow AL (2017) Ethanol exposure regulates Gabra1 expression via histone deacetylation at the promoter in cultured cortical neurons. J Pharmacol Exp Ther 363(1):1–11. https://doi.org/10.1124/jpet.117.242446
Bonsch D, Lenz B, Reulbach U, Kornhuber J, Bleich S (2004) Homocysteine associated genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm (Vienna) 111(12):1611–1616. https://doi.org/10.1007/s00702-004-0232-x
Bonsch D, Lenz B, Kornhuber J, Bleich S (2005) DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 16(2):167–170
Bonsch D, Lenz B, Fiszer R, Frieling H, Kornhuber J, Bleich S (2006) Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm (Vienna) 113(9):1299–1304. https://doi.org/10.1007/s00702-005-0413-2
Botia B, Legastelois R, Alaux-Cantin S, Naassila M (2012) Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in mice. PLoS One 7(10):e47527. https://doi.org/10.1371/journal.pone.0047527
Brown SA, Vik PW, Patterson TL, Grant I, Schuckit MA (1995) Stress, vulnerability and adult alcohol relapse. J Stud Alcohol 56(5):538–545
Bruckmann C, Islam SA, MacIsaac JL, Morin AM, Karle KN, Di Santo A, Wust R, Lang I, Batra A, Kobor MS, Nieratschker V (2017) DNA methylation signatures of chronic alcohol dependence in purified CD3(+) T-cells of patients undergoing alcohol treatment. Sci Rep 7(1):6605. https://doi.org/10.1038/s41598-017-06847-z
Cervera-Juanes R, Wilhelm LJ, Park B, Grant KA, Ferguson B (2017a) Alcohol-dose-dependent DNA methylation and expression in the nucleus accumbens identifies coordinated regulation of synaptic genes. Transl Psychiatry 7(1):e994. https://doi.org/10.1038/tp.2016.266
Cervera-Juanes R, Wilhelm LJ, Park B, Grant KA, Ferguson B (2017b) Genome-wide analysis of the nucleus accumbens identifies DNA methylation signals differentiating low/binge from heavy alcohol drinking. Alcohol 60:103–113. https://doi.org/10.1016/j.alcohol.2016.11.003
Chen J, Hutchison KE, Calhoun VD, Claus ED, Turner JA, Sui J, Liu J (2015) CREB-BDNF pathway influences alcohol cue-elicited activation in drinkers. Hum Brain Mapp 36(8):3007–3019. https://doi.org/10.1002/hbm.22824
Cippitelli A, Damadzic R, Hamelink C, Brunnquell M, Thorsell A, Heilig M, Eskay RL (2014) Binge-like ethanol consumption increases corticosterone levels and neurodegeneration whereas occupancy of type II glucocorticoid receptors with mifepristone is neuroprotective. Addict Biol 19(1):27–36. https://doi.org/10.1111/j.1369-1600.2012.00451.x
Clark SL, Aberg KA, Nerella S, Kumar G, McClay JL, Chen W, Xie LY, Harada A, Shabalin AA, Gao G, Bergen SE, Hultman CM, Magnusson PK, Sullivan PF, van den Oord EJ (2015) Combined whole methylome and genomewide association study implicates CNTN4 in alcohol use. Alcohol Clin Exp Res 39(8):1396–1405. https://doi.org/10.1111/acer.12790
Crews F, Nixon K, Kim D, Joseph J, Shukitt-Hale B, Qin L, Zou J (2006) BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol Clin Exp Res 30(11):1938–1949. https://doi.org/10.1111/j.1530-0277.2006.00239.x
Crews FT, Lawrimore CJ, Walter TJ, Coleman LG Jr (2017) The role of neuroimmune signaling in alcoholism. Neuropharmacology 122:56–73. https://doi.org/10.1016/j.neuropharm.2017.01.031
Cuevas-Ramos D, Lim DST, Fleseriu M (2016) Update on medical treatment for Cushing’s disease. Clin Diabetes Endocrinol 2:16. https://doi.org/10.1186/s40842-016-0033-9
D’Addario C, Caputi FF, Ekstrom TJ, Di Benedetto M, Maccarrone M, Romualdi P, Candeletti S (2013) Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J Mol Neurosci 49(2):312–319. https://doi.org/10.1007/s12031-012-9829-y
D’Addario C, Shchetynsky K, Pucci M, Cifani C, Gunnar A, Vukojevic V, Padyukov L, Terenius L (2017) Genetic variation and epigenetic modification of the prodynorphin gene in peripheral blood cells in alcoholism. Prog Neuropsychopharmacol Biol Psychiatry 76:195–203. https://doi.org/10.1016/j.pnpbp.2017.03.012
Day JJ, Kennedy AJ, Sweatt JD (2015) DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu Rev Pharmacol Toxicol 55:591–611. https://doi.org/10.1146/annurev-pharmtox-010814-124527
Desrivieres S, Lourdusamy A, Muller C, Ducci F, Wong CP, Kaakinen M, Pouta A, Hartikainen AL, Isohanni M, Charoen P, Peltonen L, Freimer N, Elliott P, Jarvelin MR, Schumann G (2011) Glucocorticoid receptor (NR3C1) gene polymorphisms and onset of alcohol abuse in adolescents. Addict Biol 16(3):510–513. https://doi.org/10.1111/j.1369-1600.2010.00239.x
Dominguez G, Dagnas M, Decorte L, Vandesquille M, Belzung C, Beracochea D, Mons N (2016) Rescuing prefrontal cAMP-CREB pathway reverses working memory deficits during withdrawal from prolonged alcohol exposure. Brain Struct Funct 221(2):865–877. https://doi.org/10.1007/s00429-014-0941-3
Drew PD, Johnson JW, Douglas JC, Phelan KD, Kane CJ (2015) Pioglitazone blocks ethanol induction of microglial activation and immune responses in the hippocampus, cerebellum, and cerebral cortex in a mouse model of fetal alcohol spectrum disorders. Alcohol Clin Exp Res 39(3):445–454. https://doi.org/10.1111/acer.12639
Edenberg HJ, Xuei X, Wetherill LF, Bierut L, Bucholz K, Dick DM, Hesselbrock V, Kuperman S, Porjesz B, Schuckit MA, Tischfield JA, Almasy LA, Nurnberger JI Jr, Foroud T (2008) Association of NFKB1, which encodes a subunit of the transcription factor NF-kappaB, with alcohol dependence. Hum Mol Genet 17(7):963–970. https://doi.org/10.1093/hmg/ddm368
Elvir L, Duclot F, Wang Z, Kabbaj M (2017) Epigenetic regulation of motivated behaviors by histone deacetylase inhibitors. Neurosci Biobehav Rev. https://doi.org/10.1016/j.neubiorev.2017.09.030
Fahlke C, Lorenz JG, Long J, Champoux M, Suomi SJ, Higley JD (2000) Rearing experiences and stress-induced plasma cortisol as early risk factors for excessive alcohol consumption in nonhuman primates. Alcohol Clin Exp Res 24(5):644–650
Ferguson LB, Most D, Blednov YA, Harris RA (2014) PPAR agonists regulate brain gene expression: relationship to their effects on ethanol consumption. Neuropharmacology 86:397–407. https://doi.org/10.1016/j.neuropharm.2014.06.024
Finegersh A, Homanics GE (2014) Acute ethanol alters multiple histone modifications at model gene promoters in the cerebral cortex. Alcohol Clin Exp Res 38(7):1865–1873. https://doi.org/10.1111/acer.12465
Finegersh A, Ferguson C, Maxwell S, Mazariegos D, Farrell D, Homanics GE (2015) Repeated vapor ethanol exposure induces transient histone modifications in the brain that are modified by genotype and brain region. Front Mol Neurosci 8:39. https://doi.org/10.3389/fnmol.2015.00039
Flores-Bastias O, Karahanian E (2018) Neuroinflammation produced by heavy alcohol intake is due to loops of interactions between Toll-like 4 and TNF receptors, peroxisome proliferator-activated receptors and the central melanocortin system: a novel hypothesis and new therapeutic avenues. Neuropharmacology 128:401–407. https://doi.org/10.1016/j.neuropharm.2017.11.003
Forero DA, Lopez-Leon S, Shin HD, Park BL, Kim DJ (2015) Meta-analysis of six genes (BDNF, DRD1, DRD3, DRD4, GRIN2B and MAOA) involved in neuroplasticity and the risk for alcohol dependence. Drug Alcohol Depend 149:259–263. https://doi.org/10.1016/j.drugalcdep.2015.01.017
Franklin KM, Hauser SR, Lasek AW, McClintick J, Ding ZM, McBride WJ, Bell RL (2015) Reduction of alcohol drinking of alcohol-preferring (P) and high-alcohol drinking (HAD1) rats by targeting phosphodiesterase-4 (PDE4). Psychopharmacology (Berl) 232(13):2251–2262. https://doi.org/10.1007/s00213-014-3852-3
Gatta E, Auta J, Gavin DP, Bhaumik DK, Grayson DR, Pandey SC, Guidotti A (2017) Emerging role of one-carbon metabolism and DNA methylation enrichment on delta-containing GABAA receptor expression in the cerebellum of subjects with alcohol use disorders (AUD). Int J Neuropsychopharmacol. https://doi.org/10.1093/ijnp/pyx075
Gnyszka A, Jastrzebski Z, Flis S (2013) DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res 33(8):2989–2996
Greenwald MK, Steinmiller CL, Sliwerska E, Lundahl L, Burmeister M (2013) BDNF Val(66)Met genotype is associated with drug-seeking phenotypes in heroin-dependent individuals: a pilot study. Addict Biol 18(5):836–845. https://doi.org/10.1111/j.1369-1600.2011.00431.x
Guan H, Hou S, Ricciardi RP (2005) DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J Biol Chem 280(11):9957–9962. https://doi.org/10.1074/jbc.M412180200
Guerrini L, Blasi F, Denis-Donini S (1995) Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci U S A 92(20):9077–9081
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10(1):32–42. https://doi.org/10.1038/nrg2485
Hagerty SL, Bidwell LC, Harlaar N, Hutchison KE (2016) An exploratory association study of alcohol use disorder and DNA methylation. Alcohol Clin Exp Res 40(8):1633–1640. https://doi.org/10.1111/acer.13138
Hou YN, Vlaskovska M, Cebers G, Kasakov L, Liljequist S, Terenius L (1996) A mu-receptor opioid agonist induces AP-1 and NF-kappa B transcription factor activity in primary cultures of rat cortical neurons. Neurosci Lett 212(3):159–162
Hu W, Lu T, Chen A, Huang Y, Hansen R, Chandler LJ, Zhang HT (2011) Inhibition of phosphodiesterase-4 decreases ethanol intake in mice. Psychopharmacology (Berl) 218(2):331–339. https://doi.org/10.1007/s00213-011-2290-8
Hu S, Cao Q, Xu P, Ji W, Wang G, Zhang Y (2016) Rolipram stimulates angiogenesis and attenuates neuronal apoptosis through the cAMP/cAMP-responsive element binding protein pathway following ischemic stroke in rats. Exp Ther Med 11(3):1005–1010. https://doi.org/10.3892/etm.2015.2958
Jacquot C, Croft AP, Prendergast MA, Mulholland P, Shaw SG, Little HJ (2008) Effects of the glucocorticoid antagonist, mifepristone, on the consequences of withdrawal from long term alcohol consumption. Alcohol Clin Exp Res 32(12):2107–2116. https://doi.org/10.1111/j.1530-0277.2008.00799.x
Jeanblanc J, Lemoine S, Jeanblanc V, Alaux-Cantin S, Naassila M (2015) The class I-specific HDAC inhibitor MS-275 decreases motivation to consume alcohol and relapse in heavy drinking rats. Int J Neuropsychopharmacol 18(9). https://doi.org/10.1093/ijnp/pyv029
Joels M, Sarabdjitsingh RA, Karst H (2012) Unraveling the time domains of corticosteroid hormone influences on brain activity: rapid, slow, and chronic modes. Pharmacol Rev 64(4):901–938. https://doi.org/10.1124/pr.112.005892
Kane CJ, Phelan KD, Han L, Smith RR, Xie J, Douglas JC, Drew PD (2011) Protection of neurons and microglia against ethanol in a mouse model of fetal alcohol spectrum disorders by peroxisome proliferator-activated receptor-gamma agonists. Brain Behav Immun 25(Suppl 1):S137–S145. https://doi.org/10.1016/j.bbi.2011.02.016
Kapur A, Angomchanu R, Dey M (2016) Efficacy of use of long-term, low-dose mifepristone for the treatment of fibroids. J Obstet Gynaecol India 66(Suppl 1):494–498. https://doi.org/10.1007/s13224-016-0861-7
Karahanian E, Quintanilla ME, Fernandez K, Israel Y (2014) Fenofibrate – a lipid-lowering drug – reduces voluntary alcohol drinking in rats. Alcohol 48(7):665–670. https://doi.org/10.1016/j.alcohol.2014.08.004
Kokare DM, Kyzar EJ, Zhang H, Sakharkar AJ, Pandey SC (2017) Adolescent alcohol exposure-induced changes in alpha-melanocyte stimulating hormone and neuropeptide Y pathways via histone acetylation in the brain during adulthood. Int J Neuropsychopharmacol 20(9):758–768. https://doi.org/10.1093/ijnp/pyx041
Kumar D, Deb I, Chakraborty J, Mukhopadhyay S, Das S (2011) A polymorphism of the CREB binding protein (CREBBP) gene is a risk factor for addiction. Brain Res 1406:59–64. https://doi.org/10.1016/j.brainres.2011.05.048
Li CC, Dai RM, Chen E, Longo DL (1994) Phosphorylation of NF-KB1-p50 is involved in NF-kappa B activation and stable DNA binding. J Biol Chem 269(48):30089–30092
Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, Just AC, Duan Q, Boer CG, Tanaka T, Elks CE, Aslibekyan S, Brody JA, Kuhnel B, Herder C, Almli LM, Zhi D, Wang Y, Huan T, Yao C, Mendelson MM, Joehanes R, Liang L, Love SA, Guan W, Shah S, McRae AF, Kretschmer A, Prokisch H, Strauch K, Peters A, Visscher PM, Wray NR, Guo X, Wiggins KL, Smith AK, Binder EB, Ressler KJ, Irvin MR, Absher DM, Hernandez D, Ferrucci L, Bandinelli S, Lohman K, Ding J, Trevisi L, Gustafsson S, Sandling JH, Stolk L, Uitterlinden AG, Yet I, Castillo-Fernandez JE, Spector TD, Schwartz JD, Vokonas P, Lind L, Li Y, Fornage M, Arnett DK, Wareham NJ, Sotoodehnia N, Ong KK, van Meurs JB, Conneely KN, Baccarelli AA, Deary IJ, Bell JT, North KE, Liu Y, Waldenberger M, London SJ, Ingelsson E, Levy D (2016) A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. https://doi.org/10.1038/mp.2016.192
Liu X, Hao PD, Yang MF, Sun JY, Mao LL, Fan CD, Zhang ZY, Li DW, Yang XY, Sun BL, Zhang HT (2017) The phosphodiesterase-4 inhibitor roflumilast decreases ethanol consumption in C57BL/6J mice. Psychopharmacology (Berl) 234(16):2409–2419. https://doi.org/10.1007/s00213-017-4631-8
Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ (2010) Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330(6002):385–390. https://doi.org/10.1126/science.1188472
Logrip ML (2015) Phosphodiesterase regulation of alcohol drinking in rodents. Alcohol 49(8):795–802. https://doi.org/10.1016/j.alcohol.2015.03.007
Logrip ML, Vendruscolo LF, Schlosburg JE, Koob GF, Zorrilla EP (2014) Phosphodiesterase 10A regulates alcohol and saccharin self-administration in rats. Neuropsychopharmacology 39(7):1722–1731. https://doi.org/10.1038/npp.2014.20
Logrip ML, Barak S, Warnault V, Ron D (2015) Corticostriatal BDNF and alcohol addiction. Brain Res 1628(Pt A):60–67. https://doi.org/10.1016/j.brainres.2015.03.025
Masood U, Sharma A, Nijjar S, Krenzer B (2016) Unusual case of an alcoholic with liver injury from sulfasalazine use. J Basic Clin Pharm 8(1):38–39. https://doi.org/10.4103/0976-0105.195126
Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2(8):599–609. https://doi.org/10.1038/35085068
Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (2003) NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci 6(10):1072–1078. https://doi.org/10.1038/nn1110
Meier FM, Frerix M, Hermann W, Muller-Ladner U (2013) Current immunotherapy in rheumatoid arthritis. Immunotherapy 5(9):955–974. https://doi.org/10.2217/imt.13.94
Moonat S, Sakharkar AJ, Zhang H, Pandey SC (2011) The role of amygdaloid brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein and dendritic spines in anxiety and alcoholism. Addict Biol 16(2):238–250. https://doi.org/10.1111/j.1369-1600.2010.00275.x
Moonat S, Sakharkar AJ, Zhang H, Tang L, Pandey SC (2013) Aberrant histone deacetylase2-mediated histone modifications and synaptic plasticity in the amygdala predisposes to anxiety and alcoholism. Biol Psychiatry 73(8):763–773. https://doi.org/10.1016/j.biopsych.2013.01.012
Moreno S, Farioli-Vecchioli S, Ceru MP (2004) Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience 123(1):131–145
Nair A, Bonneau RH (2006) Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol 171(1–2):72–85. https://doi.org/10.1016/j.jneuroim.2005.09.012
Nash JF Jr, Maickel RP (1985) Stress-induced consumption of ethanol by rats. Life Sci 37(8):757–765
Nash JF Jr, Maickel RP (1988) The role of the hypothalamic-pituitary-adrenocortical axis in post-stress induced ethanol consumption by rats. Prog Neuropsychopharmacol Biol Psychiatry 12(5):653–671
Nedic G, Perkovic MN, Sviglin KN, Muck-Seler D, Borovecki F, Pivac N (2013) Brain-derived neurotrophic factor Val66Met polymorphism and alcohol-related phenotypes. Prog Neuropsychopharmacol Biol Psychiatry 40:193–198. https://doi.org/10.1016/j.pnpbp.2012.09.005
Okvist A, Johansson S, Kuzmin A, Bazov I, Merino-Martinez R, Ponomarev I, Mayfield RD, Harris RA, Sheedy D, Garrick T, Harper C, Hurd YL, Terenius L, Ekstrom TJ, Bakalkin G, Yakovleva T (2007) Neuroadaptations in human chronic alcoholics: dysregulation of the NF-kappaB system. PLoS One 2(9):e930. https://doi.org/10.1371/journal.pone.0000930
Olsen CM, Liu QS (2016) Phosphodiesterase 4 inhibitors and drugs of abuse: current knowledge and therapeutic opportunities. Front Biol (Beijing) 11(5):376–386. https://doi.org/10.1007/s11515-016-1424-0
Page CP, Spina D (2011) Phosphodiesterase inhibitors in the treatment of inflammatory diseases. Handb Exp Pharmacol 204:391–414. https://doi.org/10.1007/978-3-642-17969-3_17
Pal A, Chakraborty J, Das S (2014) Association of CREB1 gene polymorphism with drug seeking behaviour in eastern Indian addicts. Neurosci Lett 570:53–57. https://doi.org/10.1016/j.neulet.2014.03.064
Pandey SC, Mittal N, Lumeng L, Li TK (1999) Involvement of the cyclic AMP-responsive element binding protein gene transcription factor in genetic preference for alcohol drinking behavior. Alcohol Clin Exp Res 23(9):1425–1434
Pandey SC, Roy A, Mittal N (2001) Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the CREB gene transcription factor in rat cortex. J Pharmacol Exp Ther 296(3):857–868
Pandey SC, Roy A, Zhang H (2003) The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol Clin Exp Res 27(3):396–409. https://doi.org/10.1097/01.ALC.0000056616.81971.49
Pandey SC, Roy A, Zhang H, Xu T (2004) Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci 24(21):5022–5030. https://doi.org/10.1523/JNEUROSCI.5557-03.2004
Pandey SC, Zhang H, Roy A, Xu T (2005) Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest 115(10):2762–2773. https://doi.org/10.1172/JCI24381
Pandey SC, Ugale R, Zhang H, Tang L, Prakash A (2008) Brain chromatin remodeling: a novel mechanism of alcoholism. J Neurosci 28(14):3729–3737. https://doi.org/10.1523/JNEUROSCI.5731-07.2008
Pandey SC, Sakharkar AJ, Tang L, Zhang H (2015) Potential role of adolescent alcohol exposure-induced amygdaloid histone modifications in anxiety and alcohol intake during adulthood. Neurobiol Dis 82:607–619. https://doi.org/10.1016/j.nbd.2015.03.019
Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK (2005) A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature 437(7059):759–763. https://doi.org/10.1038/nature03988
Pascual M, Boix J, Felipo V, Guerri C (2009) Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J Neurochem 108(4):920–931. https://doi.org/10.1111/j.1471-4159.2008.05835.x
Pascual M, Do Couto BR, Alfonso-Loeches S, Aguilar MA, Rodriguez-Arias M, Guerri C (2012) Changes in histone acetylation in the prefrontal cortex of ethanol-exposed adolescent rats are associated with ethanol-induced place conditioning. Neuropharmacology 62(7):2309–2319. https://doi.org/10.1016/j.neuropharm.2012.01.011
Pathak H, Frieling H, Bleich S, Glahn A, Heberlein A, Haschemi Nassab M, Hillemacher T, Burkert A, Rhein M (2017) Promoter polymorphism rs886205 genotype interacts with DNA methylation of the ALDH2 regulatory region in alcohol dependence. Alcohol Alcohol 52(3):269–276. https://doi.org/10.1093/alcalc/agw106
Pena CJ, Bagot RC, Labonte B, Nestler EJ (2014) Epigenetic signaling in psychiatric disorders. J Mol Biol 426(20):3389–3412. https://doi.org/10.1016/j.jmb.2014.03.016
Perkins ND (1997) Achieving transcriptional specificity with NF-kappa B. Int J Biochem Cell Biol 29(12):1433–1448
Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A (2008a) MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet 147B(5):565–570. https://doi.org/10.1002/ajmg.b.30778
Philibert RA, Sandhu H, Hollenbeck N, Gunter T, Adams W, Madan A (2008b) The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am J Med Genet B Neuropsychiatr Genet 147B(5):543–549. https://doi.org/10.1002/ajmg.b.30657
Philibert RA, Plume JM, Gibbons FX, Brody GH, Beach SR (2012) The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genet 3:54. https://doi.org/10.3389/fgene.2012.00054
Philibert RA, Penaluna B, White T, Shires S, Gunter T, Liesveld J, Erwin C, Hollenbeck N, Osborn T (2014) A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics 9(9):1212–1219. https://doi.org/10.4161/epi.32252
Pivac N, Kim B, Nedic G, Joo YH, Kozaric-Kovacic D, Hong JP, Muck-Seler D (2009) Ethnic differences in brain-derived neurotrophic factor Val66Met polymorphism in Croatian and Korean healthy participants. Croat Med J 50(1):43–48
Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD (2012) Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci 32(5):1884–1897. https://doi.org/10.1523/JNEUROSCI.3136-11.2012
Ponomarev I, Stelly CE, Morikawa H, Blednov YA, Mayfield RD, Harris RA (2017) Mechanistic insights into epigenetic modulation of ethanol consumption. Alcohol 60:95–101. https://doi.org/10.1016/j.alcohol.2017.01.016
Qiang M, Denny A, Lieu M, Carreon S, Li J (2011) Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics 6(9):1095–1104. https://doi.org/10.4161/epi.6.9.16924
Qiang M, Li JG, Denny AD, Yao JM, Lieu M, Zhang K, Carreon S (2014) Epigenetic mechanisms are involved in the regulation of ethanol consumption in mice. Int J Neuropsychopharmacol 18(2). https://doi.org/10.1093/ijnp/pyu072
Qiao X, Yin F, Ji Y, Li Y, Yan P, Lai J (2017) 5-Aza-2′-deoxycytidine in the medial prefrontal cortex regulates alcohol-related behavior and Ntf3-TrkC expression in rats. PLoS One 12(6):e0179469. https://doi.org/10.1371/journal.pone.0179469
Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, De Bosscher K (2013) How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol 380(1–2):41–54. https://doi.org/10.1016/j.mce.2012.12.014
Ray LA, Bujarski S, Shoptaw S, Roche DJ, Heinzerling K, Miotto K (2017) Development of the neuroimmune modulator ibudilast for the treatment of alcoholism: a randomized, placebo-controlled, human laboratory trial. Neuropsychopharmacology 42(9):1776–1788. https://doi.org/10.1038/npp.2017.10
Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM (2009) Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res 19(12):2163–2171. https://doi.org/10.1101/gr.097022.109
Repunte-Canonigo V, Lutjens R, van der Stap LD, Sanna PP (2007) Increased expression of protein kinase A inhibitor alpha (PKI-alpha) and decreased PKA-regulated genes in chronic intermittent alcohol exposure. Brain Res 1138:48–56. https://doi.org/10.1016/j.brainres.2006.09.115
Robinson SL, Thiele TE (2017) The role of neuropeptide Y (NPY) in alcohol and drug abuse disorders. Int Rev Neurobiol 136:177–197. https://doi.org/10.1016/bs.irn.2017.06.005
Rosenson RS, Wright RS, Farkouh M, Plutzky J (2012) Modulating peroxisome proliferator-activated receptors for therapeutic benefit? Biology, clinical experience, and future prospects. Am Heart J 164(5):672–680. https://doi.org/10.1016/j.ahj.2012.06.023
Rulten SL, Ripley TL, Hunt CL, Stephens DN, Mayne LV (2006) Sp1 and NFkappaB pathways are regulated in brain in response to acute and chronic ethanol. Genes Brain Behav 5(3):257–273. https://doi.org/10.1111/j.1601-183X.2005.00157.x
Rupprecht R, Reul JM, van Steensel B, Spengler D, Soder M, Berning B, Holsboer F, Damm K (1993) Pharmacological and functional characterization of human mineralocorticoid and glucocorticoid receptor ligands. Eur J Pharmacol 247(2):145–154
Russell MA, Almeida DM, Maggs JL (2017) Stressor-related drinking and future alcohol problems among university students. Psychol Addict Behav 31(6):676–687. https://doi.org/10.1037/adb0000303
Sacconnay L, Carrupt PA, Nurisso A (2016) Human sirtuins: structures and flexibility. J Struct Biol 196(3):534–542. https://doi.org/10.1016/j.jsb.2016.10.008
Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC (2012) Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res 36(1):61–71. https://doi.org/10.1111/j.1530-0277.2011.01581.x
Sakharkar AJ, Zhang H, Tang L, Baxstrom K, Shi G, Moonat S, Pandey SC (2014) Effects of histone deacetylase inhibitors on amygdaloid histone acetylation and neuropeptide Y expression: a role in anxiety-like and alcohol-drinking behaviours. Int J Neuropsychopharmacol 17(8):1207–1220. https://doi.org/10.1017/S1461145714000054
Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT, Pandey SC (2016) A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Struct Funct 221(9):4691–4703. https://doi.org/10.1007/s00429-016-1196-y
Schellekens AF, de Jong CA, Buitelaar JK, Verkes RJ (2015) Co-morbid anxiety disorders predict early relapse after inpatient alcohol treatment. Eur Psychiatry 30(1):128–136. https://doi.org/10.1016/j.eurpsy.2013.08.006
Shah P, Mudaliar S (2010) Pioglitazone: side effect and safety profile. Expert Opin Drug Saf 9(2):347–354. https://doi.org/10.1517/14740331003623218
Shakibaei M, John T, Schulze-Tanzil G, Lehmann I, Mobasheri A (2007) Suppression of NF-kappaB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: implications for the treatment of osteoarthritis. Biochem Pharmacol 73(9):1434–1445. https://doi.org/10.1016/j.bcp.2007.01.005
Sharrett-Field L, Butler TR, Berry JN, Reynolds AR, Prendergast MA (2013) Mifepristone pretreatment reduces ethanol withdrawal severity in vivo. Alcohol Clin Exp Res 37(8):1417–1423. https://doi.org/10.1111/acer.12093
Shibasaki M, Mizuno K, Kurokawa K, Ohkuma S (2011) Enhancement of histone acetylation in midbrain of mice with ethanol physical dependence and its withdrawal. Synapse 65(11):1244–1250. https://doi.org/10.1002/syn.20947
Shimizu E, Hashimoto K, Iyo M (2004) Ethnic difference of the BDNF 196G/A (val66met) polymorphism frequencies: the possibility to explain ethnic mental traits. Am J Med Genet B Neuropsychiatr Genet 126B(1):122–123. https://doi.org/10.1002/ajmg.b.20118
Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21:127–148. https://doi.org/10.1146/annurev.neuro.21.1.127
Simon-O’Brien E, Alaux-Cantin S, Warnault V, Buttolo R, Naassila M, Vilpoux C (2015) The histone deacetylase inhibitor sodium butyrate decreases excessive ethanol intake in dependent animals. Addict Biol 20(4):676–689. https://doi.org/10.1111/adb.12161
Singh S, Aggarwal BB (1995) Activation of transcription factor NF-kappa B is suppressed by curcumin (diferuloylmethane) [corrected]. J Biol Chem 270(42):24995–25000
Soderling TR (1999) The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci 24(6):232–236
Somkuwar SS, Vendruscolo LF, Fannon MJ, Schmeichel BE, Nguyen TB, Guevara J, Sidhu H, Contet C, Zorrilla EP, Mandyam CD (2017) Abstinence from prolonged ethanol exposure affects plasma corticosterone, glucocorticoid receptor signaling and stress-related behaviors. Psychoneuroendocrinology 84:17–31. https://doi.org/10.1016/j.psyneuen.2017.06.006
Spanagel R, Noori HR, Heilig M (2014) Stress and alcohol interactions: animal studies and clinical significance. Trends Neurosci 37(4):219–227. https://doi.org/10.1016/j.tins.2014.02.006
Stopponi S, Somaini L, Cippitelli A, Cannella N, Braconi S, Kallupi M, Ruggeri B, Heilig M, Demopulos G, Gaitanaris G, Massi M, Ciccocioppo R (2011) Activation of nuclear PPARgamma receptors by the antidiabetic agent pioglitazone suppresses alcohol drinking and relapse to alcohol seeking. Biol Psychiatry 69(7):642–649. https://doi.org/10.1016/j.biopsych.2010.12.010
Stopponi S, de Guglielmo G, Somaini L, Cippitelli A, Cannella N, Kallupi M, Ubaldi M, Heilig M, Demopulos G, Gaitanaris G, Ciccocioppo R (2013) Activation of PPARgamma by pioglitazone potentiates the effects of naltrexone on alcohol drinking and relapse in msP rats. Alcohol Clin Exp Res 37(8):1351–1360. https://doi.org/10.1111/acer.12091
Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL (2010) Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (Basel) 3(9):2751–2767. https://doi.org/10.3390/ph3092751
Tajuddin N, Moon KH, Marshall SA, Nixon K, Neafsey EJ, Kim HY, Collins MA (2014) Neuroinflammation and neurodegeneration in adult rat brain from binge ethanol exposure: abrogation by docosahexaenoic acid. PLoS One 9(7):e101223. https://doi.org/10.1371/journal.pone.0101223
Takeuchi Y, Fukunaga K (2003) Differential regulation of NF-kappaB, SRE and CRE by dopamine D1 and D2 receptors in transfected NG108-15 cells. J Neurochem 85(3):729–739
Tanigawara Y, Kita T, Aoyama N, Gobara M, Komada F, Sakai T, Kasuga M, Hatanaka H, Sakaeda T, Okumura K (2002) N-acetyltransferase 2 genotype-related sulfapyridine acetylation and its adverse events. Biol Pharm Bull 25(8):1058–1062
Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20(4):709–726
ter Heegde F, De Rijk RH, Vinkers CH (2015) The brain mineralocorticoid receptor and stress resilience. Psychoneuroendocrinology 52:92–110. https://doi.org/10.1016/j.psyneuen.2014.10.022
Titus DJ, Oliva AA, Wilson NM, Atkins CM (2015) Phosphodiesterase inhibitors as therapeutics for traumatic brain injury. Curr Pharm Des 21(3):332–342
Tiwari V, Chopra K (2013a) Protective effect of curcumin against chronic alcohol-induced cognitive deficits and neuroinflammation in the adult rat brain. Neuroscience 244:147–158. https://doi.org/10.1016/j.neuroscience.2013.03.042
Tiwari V, Chopra K (2013b) Resveratrol abrogates alcohol-induced cognitive deficits by attenuating oxidative-nitrosative stress and inflammatory cascade in the adult rat brain. Neurochem Int 62(6):861–869. https://doi.org/10.1016/j.neuint.2013.02.012
Truitt JM, Blednov YA, Benavidez JM, Black M, Ponomareva O, Law J, Merriman M, Horani S, Jameson K, Lasek AW, Harris RA, Mayfield RD (2016) Inhibition of IKKbeta reduces ethanol consumption in C57BL/6J mice. eNeuro 3(5). https://doi.org/10.1523/ENEURO.0256-16.2016
Tulisiak CT, Harris RA, Ponomarev I (2017) DNA modifications in models of alcohol use disorders. Alcohol 60:19–30. https://doi.org/10.1016/j.alcohol.2016.11.004
Uhart M, Oswald L, McCaul ME, Chong R, Wand GS (2006) Hormonal responses to psychological stress and family history of alcoholism. Neuropsychopharmacology 31(10):2255–2263. https://doi.org/10.1038/sj.npp.1301063
Vendruscolo LF, Barbier E, Schlosburg JE, Misra KK, Whitfield TW Jr, Logrip ML, Rivier C, Repunte-Canonigo V, Zorrilla EP, Sanna PP, Heilig M, Koob GF (2012) Corticosteroid-dependent plasticity mediates compulsive alcohol drinking in rats. J Neurosci 32(22):7563–7571. https://doi.org/10.1523/JNEUROSCI.0069-12.2012
Vendruscolo LF, Estey D, Goodell V, Macshane LG, Logrip ML, Schlosburg JE, McGinn MA, Zamora-Martinez ER, Belanoff JK, Hunt HJ, Sanna PP, George O, Koob GF, Edwards S, Mason BJ (2015) Glucocorticoid receptor antagonism decreases alcohol seeking in alcohol-dependent individuals. J Clin Invest 125(8):3193–3197. https://doi.org/10.1172/JCI79828
Wand GS, Dobs AS (1991) Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrinol Metab 72(6):1290–1295. https://doi.org/10.1210/jcem-72-6-1290
Wang F, Xu H, Zhao H, Gelernter J, Zhang H (2016) DNA co-methylation modules in postmortem prefrontal cortex tissues of European Australians with alcohol use disorders. Sci Rep 6:19430. https://doi.org/10.1038/srep19430
Warden A, Truitt J, Merriman M, Ponomareva O, Jameson K, Ferguson LB, Mayfield RD, Harris RA (2016) Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep 6:27618. https://doi.org/10.1038/srep27618
Warnault V, Darcq E, Levine A, Barak S, Ron D (2013) Chromatin remodeling – a novel strategy to control excessive alcohol drinking. Transl Psychiatry 3:e231. https://doi.org/10.1038/tp.2013.4
Wen RT, Zhang M, Qin WJ, Liu Q, Wang WP, Lawrence AJ, Zhang HT, Liang JH (2012) The phosphodiesterase-4 (PDE4) inhibitor rolipram decreases ethanol seeking and consumption in alcohol-preferring Fawn-Hooded rats. Alcohol Clin Exp Res 36(12):2157–2167. https://doi.org/10.1111/j.1530-0277.2012.01845.x
Witkiewitz K (2011) Predictors of heavy drinking during and following treatment. Psychol Addict Behav 25(3):426–438. https://doi.org/10.1037/a0022889
Wojnar M, Brower KJ, Strobbe S, Ilgen M, Matsumoto H, Nowosad I, Sliwerska E, Burmeister M (2009) Association between Val66Met brain-derived neurotrophic factor (BDNF) gene polymorphism and post-treatment relapse in alcohol dependence. Alcohol Clin Exp Res 33(4):693–702. https://doi.org/10.1111/j.1530-0277.2008.00886.x
Wolstenholme JT, Warner JA, Capparuccini MI, Archer KJ, Shelton KL, Miles MF (2011) Genomic analysis of individual differences in ethanol drinking: evidence for non-genetic factors in C57BL/6 mice. PLoS One 6(6):e21100. https://doi.org/10.1371/journal.pone.0021100
Yakovleva T, Bazov I, Watanabe H, Hauser KF, Bakalkin G (2011) Transcriptional control of maladaptive and protective responses in alcoholics: a role of the NF-kappaB system. Brain Behav Immun 25(Suppl 1):S29–S38. https://doi.org/10.1016/j.bbi.2010.12.019
Yang X, Horn K, Baraban JM, Wand GS (1998) Chronic ethanol administration decreases phosphorylation of cyclic AMP response element-binding protein in granule cells of rat cerebellum. J Neurochem 70(1):224–232
Yang X, Oswald L, Wand G (2003) The cyclic AMP/protein kinase A signal transduction pathway modulates tolerance to sedative and hypothermic effects of ethanol. Alcohol Clin Exp Res 27(8):1220–1225. https://doi.org/10.1097/01.ALC.0000081626.02910.19
You C, Zhang H, Sakharkar AJ, Teppen T, Pandey SC (2014) Reversal of deficits in dendritic spines, BDNF and Arc expression in the amygdala during alcohol dependence by HDAC inhibitor treatment. Int J Neuropsychopharmacol 17(2):313–322. https://doi.org/10.1017/S1461145713001144
Zhang R, Miao Q, Wang C, Zhao R, Li W, Haile CN, Hao W, Zhang XY (2013) Genome-wide DNA methylation analysis in alcohol dependence. Addict Biol 18(2):392–403. https://doi.org/10.1111/adb.12037
Zhao R, Zhang R, Li W, Liao Y, Tang J, Miao Q, Hao W (2013) Genome-wide DNA methylation patterns in discordant sib pairs with alcohol dependence. Asia Pac Psychiatry 5(1):39–50. https://doi.org/10.1111/appy.12010
Zhu Y, Qi C, Calandra C, Rao MS, Reddy JK (1996) Cloning and identification of mouse steroid receptor coactivator-1 (mSRC-1), as a coactivator of peroxisome proliferator-activated receptor gamma. Gene Expr 6(3):185–195
Zhu ZZ, Hou L, Bollati V, Tarantini L, Marinelli B, Cantone L, Yang AS, Vokonas P, Lissowska J, Fustinoni S, Pesatori AC, Bonzini M, Apostoli P, Costa G, Bertazzi PA, Chow WH, Schwartz J, Baccarelli A (2012) Predictors of global methylation levels in blood DNA of healthy subjects: a combined analysis. Int J Epidemiol 41(1):126–139. https://doi.org/10.1093/ije/dyq154
Zou J, Crews F (2010) Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcohol Clin Exp Res 34(5):777–789. https://doi.org/10.1111/j.1530-0277.2010.01150.x
Zovkic IB, Guzman-Karlsson MC, Sweatt JD (2013) Epigenetic regulation of memory formation and maintenance. Learn Mem 20(2):61–74. https://doi.org/10.1101/lm.026575.112
Acknowledgements
A.W.L. is supported by the National Institute on Alcohol Abuse and Alcoholism (INIA U01 AA020912 and the Center for Alcohol Research in Epigenetics P50 AA022538).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Savarese, A.M., Lasek, A.W. (2018). Transcriptional Regulators as Targets for Alcohol Pharmacotherapies. In: Grant, K., Lovinger, D. (eds) The Neuropharmacology of Alcohol . Handbook of Experimental Pharmacology, vol 248. Springer, Cham. https://doi.org/10.1007/164_2018_101
Download citation
DOI: https://doi.org/10.1007/164_2018_101
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-96522-2
Online ISBN: 978-3-319-96523-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)