
Abstract
Phosphodiesterase 4 (PDE4) belongs to a family of enzymes which catalyzes the breakdown of 3, 5′-adenosine cyclic monophosphate (cAMP) and is ubiquitously expressed in inflammatory cells. There is little evidence that inflammatory diseases are caused by increased expression of this isoenzyme, although human inflammatory cell activity can be suppressed by selective PDE4 inhibitors. Consequently, there is intense interest in the development of selective PDE4 inhibitors for the treatment of a range of inflammatory diseases, including asthma, chronic obstructive pulmonary disease (COPD), inflammatory bowel disease, and psoriasis. Recent clinical trials with roflumilast in COPD have confirmed the therapeutic potential of targeting PDE4 and recently roflumilast has been approved for marketing in Europe and the USA, although side effects such as gastrointestinal disturbances, particularly nausea and emesis as well as headache and weight loss, may limit the use of this drug class, at least when administered by the oral route. However, a number of strategies are currently being pursued in attempts to improve clinical efficacy and reduce side effects of PDE4 inhibitors, including delivery via the inhaled route, development of nonemetic PDE4 inhibitors, mixed PDE inhibitors, and/or antisense biologicals targeted toward PDE4.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Theophylline has been used in the treatment of asthma and chronic obstructive pulmonary disease (COPD) since the 1930s, although its popularity has declined due to the introduction of β2-adrenoceptor agonists and glucocorticosteroids. Several mechanisms have been proposed to explain the potential beneficial action of theophylline in respiratory disease. Theophylline has been shown to inhibit the activity of a cyclic 3′, 5′ nucleotide PDE with a K i of 100 μM (Butcher and Sutherland 1962), which has been suggested to contribute to its ability to promote suppressor cell activity in lymphocytes (Shore et al. 1978; Zocchi et al. 1985; O’Shaughnessy et al. 2007) and its beneficial actions in patients with asthma (Sullivan et al. 1994), COPD (Rennard 2004), or psoriasis (Papakostantinou et al. 2005). However, theophylline has been relegated to second- or third-line therapy for the treatment of respiratory disease and is not routinely used in the treatment of other inflammatory conditions such as rheumatoid arthritis, psoriasis, and inflammatory bowel disease, in part because it is not recognized as an anti-inflammatory drug, but more importantly because of the very significant drug interactions and narrow therapeutic window exhibited by theophylline; the side effect profile of theophylline includes nausea, emesis, gastrointestinal disturbances, and arrhythmias. These problems, in part, promoted an interest in understanding how the PDE family of enzymes regulated cAMP levels within cells and the potential for developing drugs inhibiting such enzymes, in a selective way as a way of improving the therapeutic window of theophylline.
There are presently 11 known families of PDEs and at least 21 isoforms with numerous splice variants that are characterized by differences in structure, substrate specificity, inhibitor selectivity, tissue and cell distribution, regulation by kinases, protein–protein interaction, and subcellular distribution (Conti and Beavo 2007; McCahill et al. 2008). However, targeting PDE4, an enzyme family that exclusively metabolizes cAMP (Houslay et al. 2005, 2007), has been the focus for the development of drugs that could prove beneficial in the treatment of depression, schizophrenia (Millar et al. 2007), respiratory diseases such as asthma and COPD (Barnes 2008), allergic skin diseases and psoriasis (Baumer et al. 2007), and inflammatory bowel disease (Keshavarzian et al. 2007).
It is therefore of interest that plasma levels achieved with a dose of theophylline that demonstrated significant anti-inflammatory activity (Sullivan et al. 1994) are well below the K i for PDE inhibition and suggested that PDE4 inhibition alone does not completely explain the clinical effectiveness of this drug (Barnes et al. 2005). Nevertheless, highly potent and selective PDE4 inhibitors have been developed to target a range of inflammatory diseases. However, targeting PDE enzymes is not unique to inflammatory diseases as exemplified with the development and clinical success of a number of PDE5 inhibitors for the treatment of erectile dysfunction as exemplified by sildenafil (Boolell et al. 1996).
2 Phosphodiesterase 4
PDE4 is highly selective for catalyzing the hydrolysis of cAMP, which terminates the downstream signalling of this second messenger (Houslay et al. 2007; Houslay 2010). There are 4 gene families (A, B, C, and D), where alternative mRNA splicing and the use of distinct promoters generates more than 20 splice variants (Houslay et al. 2007). While hydrolysis of cAMP is a common feature of this family, it is clear that these isoforms can be targeted to different domains and signalling complexes within the intracellular compartment, where they play a pivotal role in underpinning compartmentalized cAMP signaling (Houslay 2010). Furthermore, their activities can be differentially regulated by phosphorylation by various protein kinases, suggesting that these isoforms have specific functions in the control of cellular activity (McCahill et al. 2008).
X-ray crystallography has resolved the catalytic domain of these enzymes, which comprise three important domains: a bivalent metal binding pocket (Zn2+, Mg2+), which is thought to form a complex with the phosphate moiety of cAMP, a pocket containing an invariant glutamine (Q pocket), which forms hydrogen bonds with the nucleotide (purine) moiety of cAMP, and a solvent pocket. PDE4 inhibitors occupy this active site through a number of important interactions and prevent cAMP metabolism. These include indirect binding to the metal ions via the formation of hydrogen bonding to water while hydrophobic interactions between the planar ring structure of these inhibitors and hydrophobic amino acid residues such as phenylalanine and isoleucine serve to “clamp” the inhibitor within the active site. There are also hydrogen bond interactions between the aromatic ring structure of these inhibitors and the invariant glutamine residue in the Q pocket, the site which is normally occupied by the nucleotide moiety of cAMP (Xu et al. 2000; Card et al. 2004; Wang et al. 2007).
There are considerable challenges to the synthesis of subtype selective inhibitors due to the high degree of sequence and structural homology within the catalytic domains of the PDE4 subtypes (Xu et al. 2000; Card et al. 2004; Houslay et al. 2005; Wang et al. 2007). One possibility might be to exploit subtle differences between the interaction of these inhibitors with the catalytic active site. Alternatively, compounds that inhibit catalysis by targeting regions outside the catalytic site may be possible. Compounds that selectively interact with the N terminal regions of the respective PDE4, which contain phosphorylation sites and/or protein binding sequences, might indirectly interfere with PDE4 activity (Houslay et al. 2005). Moreover, inhibitors may act through a combined interaction with residues in regions that fold over or near the catalytic site, e.g., UCR2 which provides a regulatory region unique to PDE4 enzymes as well as some residues in the catalytic domain (Kranz et al. 2009; Burgin et al. 2010).
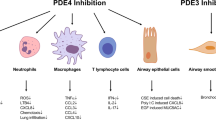
PDE4 is expressed in a number of cell types that are considered important in the pathogenesis of inflammatory disease (Table 1). It might reasonably be argued that targeting the inhibition of PDE4 could potentially suppress the function of numerous cell types. However, it is well known that other PDE enzymes are also expressed in these cells and the contribution of other PDEs to cell function (e.g., PDE3, PDE7) in the context of regulating inflammatory cell function is also being explored (Smith et al. 2004; Boswell-Smith et al. 2006a; Banner and Press 2009). Although it would seem prudent to develop subtype selective PDE4 inhibitors in attempts to maximize therapeutic benefit at the expense of adverse effects, there is also the possibility that nonselective PDE inhibitors might offer a better approach in targeting multiple target cells in the disease process. Indeed, it has been suggested that clozapine is a better antipsychotic than newer generation atypical antipsychotics because this drug targets numerous receptors and as such, has been described as a “magic shotgun” for the treatment of schizophrenia (Roth et al. 2004).
3 Asthma and PDE4
The underlying pathophysiological features of asthma are a consequence of the contribution of numerous cells including the epithelium, dendritic cells, T-lymphocytes, eosinophils, mast cells, and airway smooth muscle. Thus, there is a complex web of interconnecting interactions between different cell types and inflammatory mediators. Whilst glucocorticosteroids remain the mainstay treatment of this disease, there still remains a group of asthmatics with poor symptom control despite treatment with these drugs (Barnes 2008). It is therefore of interest that many of the cell types illustrated in Table 1 express PDE4 and inhibition of this enzyme family can suppress the function of these cells.
A number of studies have investigated whether the pathogenesis of asthma might be a consequence of increased PDE4 protein expression and activity. This hypothesis had some currency in view of studies in the field of dermatology, which purported to demonstrate the expression of a novel and distinct monocyte-derived cAMP-PDE obtained from individuals with atopic dermatitis. Monocytes from these individuals had increased functionality and this corresponded with the presence of protein with increased PDE activity. This was thought to be the underlying basis for the pathology associated with atopic dermatitis (Chan et al. 1993). However, soluble PDE4 activity was not increased in a range of peripheral blood leukocytes from atopic subjects of either mild or severe severity (Gantner et al. 1997b). Similarly, while increased total PDE catalytic activity was observed in peripheral blood monocytes from individuals with mild asthma, this was associated, paradoxically, with reduced PDE4 activity (Landells et al. 2001), and the expression of PDE4A, B, and D was not increased in peripheral blood CD4 positive T-lymphocytes in patients with mild asthma (Jones et al. 2007). Together these studies indicate that the underlying pathogenesis of mild asthma cannot be attributed to enhanced PDE4 expression or activity. A genome-wide association search has demonstrated single nucleotide polymorphisms on chromosome 5q12 for PDE4D (Himes et al. 2009).
The significance of these findings remains to be established given that this isoform is purportedly linked with emesis and targeting of PDE4A and PDE4B is seen as a rationale approach to develop nonemetic anti-inflammatory PDE4 inhibitors (Manning et al. 1999; Kranz et al. 2009). If this assertion is correct, then the challenge remains to develop a PDE4 subtype selective inhibitor that differs by at least three orders of magnitude, but this has yet to be achieved after more than a decade of work in this field. However, this hypothesis has been questioned following the discovery of PDE4D selective allosteric inhibitors, which promote anti-inflammatory activity and are largely devoid of emesis in animal models (Burgin et al. 2010). The implication of these novel findings is that selectivity for different PDE4 subtypes is not a critical determinant in avoiding side effects such as nausea, but whether partial inhibition can be achieved, as in this example using allosteric inhibitors which interact with upstream conserved regions (UCRs), and the catalytic domain of PDE4D (Burgin et al. 2010).
Numerous preclinical studies in models of allergic pulmonary inflammation have repeatedly documented the ability of PDE4 inhibitors to inhibit two important characteristic features of asthma, namely, the recruitment of eosinophils to the airways and bronchial hyperresponsiveness (Spina 2003). One disadvantage of these studies is the inability to ascertain the role of PDE4 isoforms because of the nonselective nature of the PDE4 inhibitors currently under development. However, the use of genetically modified mice has revealed some interesting findings. Airway inflammation characterized by recruitment of eosinophils to the airways of mice deficient in PDE4D was not different from that of wild-type controls (Hansen et al. 2000). This indicated that other PDE4 subtypes contribute in the metabolism of intracellular cAMP since cell recruitment to the airways was inhibited when animals were treated with nonselective PDE4 inhibitors (Kung et al. 2000; Kanehiro et al. 2001). However, airway obstruction caused by methacholine was enhanced in wild-type allergic mice, but abolished in PDE4D gene-deficient mice. These mice were hyporesponsive to methacholine, even in the absence of allergic sensitization, and this feature appeared to be related to an increase in the production of a dilator prostaglandin in the airways of these PDE4D gene-deficient mice (Hansen et al. 2000; Mehats et al. 2003). However, this effect was specific for methacholine because the enhanced airway obstruction in response to serotonin was unaffected by the removal of PDE4D (Hansen et al. 2000). This highlights the potential complementary role of PDE4 isoforms in regulating allergic airway inflammation, and the need to target more than one PDE4 isoform since the inflammatory response, bronchial hyperresponsiveness, and airway remodeling in allergic wild-type mice could be inhibited by nonselective PDE4 inhibitors such as rolipram or roflumilast (Kung et al. 2000; Kanehiro et al. 2001; Kumar et al. 2003). Whilst the use of gene knockout studies can offer insights into the function of proteins, it is likely that compensatory mechanisms at birth and the mixed background of the generated knockouts could confound attempts to define the biological role of a particular protein. The use of conditional PDE4 knockouts or a biological approach (e.g., siRNA) may be required to understand the biological role(s) of PDE4 subtypes completely (Huston et al. 2008).
The numerous preclinical studies reporting the anti-inflammatory potential of PDE4 inhibitors in models of allergic inflammation and in human cells in vitro have to some degree been corroborated in clinical trials in asthmatic subjects. Twice-daily treatment for 9.5 days with the PDE4 inhibitor CDP840 inhibited the development of the late phase response in asthmatic subjects by 30% (Harbinson et al. 1997). A similar degree of inhibition of the late phase response was observed following once daily treatment for 7- to 10-day with roflumilast (van Schalkwyk et al. 2005), while 2-week treatment with MK-0359 improved baseline FEV1 and reduced symptom scores in chronic asthmatic subjects (Lu et al. 2009). This late phase response is used by clinicians to model the inflammatory component following an allergic insult to the airways. In both allergen challenge studies, the effect of drug treatment on the acute allergen bronchoconstriction was modest and consistent with the lack of demonstrable action of PDE4 inhibition on both mast cell and airway smooth muscle function. This highlights the role of other PDE enzymes, namely PDE3 in the regulation of the function of these cell types in the airway (Table 1). Bronchial hyperresponsiveness was not reduced by these drugs, with only one study purporting to show modest protection against allergen-induced bronchial hyperresponsiveness (Louw et al. 2007). This suggests that PDE4 may not be a suitable target for this particular phenomenon, or that higher doses are required to provide complementary and persistent inhibition of the enzyme and hence attenuation of bronchial hyperresponsiveness. It is of interest that roflumilast has a plasma half-life of 16 h following a single oral administration and is metabolized by CYP3A4 to the active N-oxide metabolite, which has considerably greater bioavailability with a half-life of 20 h that would favor prolonged enzyme exposure (David et al. 2004). This favorable pharmacokinetic profile would be anticipated to produce longer periods of PDE4 inhibition. However, whilst there was a significant reduction in the activity of circulating monocytes in subjects maintained on roflumilast for 4 weeks, the magnitude of this change was small, resulting in only an approximately 1.3-fold reduction in TNFalpha production by monocytes in response to endotoxin challenge in vitro (Timmer et al. 2002). One could argue that only a partial inhibition of PDE4 activity was achieved at the dose employed in clinical studies and consequently suppression of inflammatory cell function within the airway tissue compartment is not maximal, a hypothesis borne out in several clinical studies (Gamble et al. 2003; Grootendorst et al. 2007).
Unfortunately, the doses that can be administered clinically with most orally active PDE4 inhibitors are limited by side effects, the most commonly reported being headache, nausea, and diarrhea of a mild-to-moderate severity (van Schalkwyk et al. 2005; Lu et al. 2009). The availability of other nonemetic drugs such as CDP840 (Harbinson et al. 1997) might provide hope that such PDE4 inhibitors can be developed for the treatment of asthma, a worthy goal to pursue in light of at least one clinical study reporting comparable clinical efficacy between roflumilast and the inhaled glucocorticosteroids beclomethasone diproprionate in persistent asthma (Bousquet et al. 2006). Such systemic side effects may also be eliminated by using the inhaled route such that relatively higher doses of PDE4 inhibitor can be delivered to the intrapulmonary compartment whilst reducing systemic bioavailability as recently reported with GSK256066 (Singh et al. 2010). However, another inhaled PDE inhibitor, UK 50,000 failed to show any clinical benefit (Phillips et al. 2007) and an early mixed PDE 3/4 inhibitor zardaverine actively elicited frank emesis at inhaled doses that induced bronchodilation (Brunnee et al. 1992). Mesenteric arteritis, characterized by inflammation of the arterioles is observed in rodents and primates following doses of some PDE4 inhibitors far exceeding those that would be used clinically; however, this condition has not been observed in subjects in clinical trials with roflumilast and therefore is unlikely to be a major issue and even less so if inhaled formulations of this drug class are developed (Spina 2008).
4 COPD and PDE4
Unlike asthma, COPD is primarily caused by cigarette smoking, although in developing countries smoke derived from burning biomass fuels is also a predisposing factor (Salvi and Barnes 2009). COPD is also an inflammatory disease but the nature of the inflammatory response is distinct from asthma. The inflammatory response in COPD is characterized by the activation of macrophages and airway epithelial cells, which in turn, secrete a range of chemokines and lipid mediators resulting in the recruitment of neutrophils and CD8+ T-lymphocytes to the lung. The secretion of a range of proteases from neutrophils (elastase, MMP9, cathepsins) and macrophages (MMP12) is thought to contribute toward fibrosis of the small airways and increased mucus secretion and destruction of the alveolar wall (Barnes 2008) These pathological changes give rise to the symptoms of cough, mucus secretion, dyspnea, and emphysema. Many of the cell types implicated in the COPD inflammatory response express PDE4 (Table 1).
The expression of PDE4A-D mRNA transcripts in peripheral blood neutrophils and CD8 T cells is not altered in subjects with mild COPD (Jones et al. 2007). However, the expression of PDE4A4 mRNA transcript was significantly increased in macrophages purified from bronchoalveolar lavage fluid from subjects with mild-to-moderate COPD compared with healthy subjects or smokers who did not present with COPD (Barber et al. 2004). Of the 12 PDE4 variants analyzed, only the activity of PDE4A4 was increased and suggested that local events/processes within the lung of subjects with COPD specifically upregulated this variant (Barber et al. 2004). However, the functional consequence of this change remains to be established in light of findings showing that PDE4 inhibition has only a modest effect in suppressing TNFα production from human macrophages derived from cultured monocytes. Moreover, the contribution of other PDEs (e.g., PDE3, PDE7) and other PDE4 isoforms in regulating function in this cell type clearly need to be explored (Gantner et al. 1997a; Smith et al. 2004). Genotyping studies have also revealed the presence of single nucleotide polymorphisms within the PDE4D gene in COPD subjects compared with smokers within the Japanese population that was in linkage disequilibrium with the IL-13 gene. However, the significance of these findings remains to be established since this relationship was absent in an Egyptian population (Homma et al. 2006).
In vivo models of COPD are very limited. However, the recruitment of neutrophils to the airways can be readily induced using the bacterial wall component, endotoxin, although it is widely appreciated that this stimulus can only model neutrophil recruitment to the airways (Leclerc et al. 2006). Neutrophil recruitment into the airways of wild-type mice was inhibited by around 50% in PDE4B and PDE4D-deficient mice and a greater degree of inhibition was observed when wild type mice were treated with rolipram (Ariga et al. 2004). This once again highlights the complementary roles of PDE4 isoforms in regulating neutrophil recruitment to the airways. Similarly, smoking-induced neutrophil recruitment to the airways, release of chemokines, and emphysematous changes to the lung were attenuated by PDE4 inhibitors (Martorana et al. 2005; Leclerc et al. 2006). Together, these studies highlight the utility of inhibiting PDE4 in cell types implicated in COPD.
A number of phase III clinical trials have assessed the potential utility of PDE4 inhibitors in the treatment of COPD (Rabe et al. 2005; Rennard et al. 2006; Calverley et al. 2007). All three studies report modest, but nevertheless significant improvements in spirometry over placebo, quality of life scores, and reduction in the number of exacerbations in the severest group of COPD subjects. Indeed, recent phase III clinical trials evaluating the effect of roflumilast in symptomatic moderate-to-severe asthmatics have reported more promising findings (Fabbri et al. 2009; Calverley et al. 2009). Roflumilast caused a significant 48-ml improvement in FEV1 compared with placebo and reduced exacerbation rates of moderate-to-severe intensity by 17% over a 52-week period (Calverley et al. 2009). Similarly, roflumilast consistently improved baseline FEV1 in moderate-to-severe COPD patients already taking either a long-acting β2-agonist (salmeterol) or a long-acting muscarinic antagonist (tiotropium bromide) by 49 and 80 ml, respectively, during 24 weeks of treatment (Fabbri et al. 2009).
The mechanism of the improvement in spirometry is unlikely to be due to relaxation of airway smooth muscle because this drug class has weak bronchodilator activity. It is possible that this improvement is due to an anti-inflammatory action of the drugs (Table 1), although no biomarker of inflammation was measured in these studies. However, separate studies have addressed whether PDE4 inhibitors are anti-inflammatory in patients with COPD. Both roflumilast (Grootendorst et al. 2007) and cilomilast (Gamble et al. 2003) reduced both the number of inflammatory cells, such as, neutrophils and lymphocytes recruited to the airways and the levels of two biochemical markers of this disease, namely IL-8 and neutrophil elastase. The magnitude of the change in the number of these inflammatory cells and concentration of mediators was between 30 and 50%, which might contribute to the beneficial action of these two PDE4 selective in the clinic trials. However, to date the substantial inhibition of PDE4 with an orally active inhibitor has not been achieved because of dose-limiting side effects, which may also explain the modest clinical benefit shown with these drugs. As discussed for asthma, it is plausible that other PDE species (e.g., PDE3, 7; see Table 1) may require targeting for a full anti-inflammatory therapeutic action to be attained in treating this disease (Banner and Press 2009).
The most common side effect reported with roflumilast included diarrhea (9%), headache (5%), and nausea (5%) (Rabe et al. 2005; Calverley et al. 2007, 2009; Fabbri et al. 2009). This was of the same order of magnitude as reported with cilomilast, although abdominal pain and vomiting were also reported for this drug (Rennard et al. 2006). The adverse effects seemed to disappear with continued use; nonetheless, these side effects were a major reason why patients did not continue with the study during the first 3–4 weeks of treatment. No cardiovascular liabilities were noted. One unanticipated side effect that was noted following prolonged treatment with roflumilast was a significant reduction in body weight in subjects. This might be attributed to gastrointestinal discomfort in susceptible patients, although it was interesting to note that weight loss was also reported in patients not complaining of gastrointestinal side effects and might be an effect worthy of further study (Fabbri et al. 2009; Calverley et al. 2009).
One might anticipate that the risk/benefit ratio would be improved by inhaled delivery of a PDE4 inhibitor directly to the lung. Unfortunately, to date only one study has evaluated the effect of an inhaled PDE4 nonselective subtype inhibitor. This was used to treat moderate-to-severe COPD and no significant improvement in baseline FEV1 was observed after 6 weeks of treatment (Vestbo et al. 2009). As no inflammatory biomarker was measured in this study, it is unclear whether a sufficient level of sustained PDE4 inhibition was achieved. This seems likely because adverse events such as headache and improvements in lung function that were noted at 2 weeks were absent after 6 weeks of treatment with the high dose. Whether inhaled PDE4 inhibitors will be of utility in COPD remains to be determined but there may be a cause for optimism in view of the amelioration of the allergen-induced late phase response in asthma with the inhaled PDE4 inhibitor GSK256066 (Singh et al. 2010). This demonstrates that in principle one can achieve inhibition of PDE4 following topical delivery to the lung that could lead to clinical benefit, but the relevant clinical trials need to be undertaken with GSK250666 to ascertain whether this drug will be of any clinical benefit to patients with COPD.
5 Other Inflammatory Diseases and PDE4
Numerous inflammatory conditions such as psoriasis (Nestle et al. 2009), rheumatoid arthritis (Taylor and Feldmann 2009), and inflammatory bowel disease (Abraham and Cho 2009) are characterized by a complex interplay between cells resulting in their activation and recruitment, release of proinflammatory cytokines, and tissue damage. Biologicals directed against cytokines are one of the most important therapies to emerge in the past decade and have revolutionized the treatment of inflammatory diseases as exemplified by those targeting TNFα (Nestle et al. 2009; Taylor and Feldmann 2009; Abraham and Cho 2009). It is therefore of interest that PDE4 inhibitors can suppress TNFα production by various inflammatory cell types, and inhibit the activity of immune cells so critical in these diseases (Table 1). It is therefore no surprise that PDE4 inhibitors are also being considered for the treatment of a number of nonrespiratory inflammatory diseases.
Earlier observations suggested the targeting of cAMP PDE might prove beneficial in the treatment of skin inflammatory disease. For example, potential deficits in adenylyl cyclase signaling in psoriasis (Wright et al. 1973) and expression of a novel monocyte-derived cAMP PDE in atopic dermatitis (Chan et al. 1993) were reported. Consequently, several selective PDE4 inhibitors have been evaluated in clinical trials. Patients with psoriasis and treated with apremilast, an orally active PDE4 inhibitor demonstrated an improvement in clinical scores and reduced T-cell number in the dermis in an open-label clinical trial (Gottlieb et al. 2008). Similarly, the systemic administration of apremilast reduced pathological features of psoriatic human skin transplanted onto immunocompromised mice (Schafer et al. 2010). One of the advantages of inflammatory dermatoses is that topical administration could limit the systemic liability associated with orally administered PDE4 inhibitors and this strategy has been adopted for the development of AN2728 for the treatment of psoriasis and atopic dermatitis with promising results in phase II clinical trials (Nazarian and Weinberg 2009; Akama et al. 2009).
Inflammatory bowel diseases might also be amenable to treatment with PDE4 inhibitors since Th1 and Th17 cells are implicated in this disease and the activity of these cell types can be suppressed by PDE4 inhibitors (Table 1). The PDE4 inhibitor, tetomilast, appeared to reduce various indices of inflammation in IL10-deficient mice that spontaneously develop chronic gastrointestinal inflammation characterized by mucus secretion, rectal prolapse, and diarrhea (Ichikawa et al. 2008). Tetomilast has also been evaluated in a phase II clinical trial in active ulcerative colitis (Schreiber et al. 2007). Improvement in disease as defined by a disease activity index of ≥3 was achieved in 53% (25 mg dose) and 39% (50 mg dose) of patients after 8 weeks of treatment, although this was not statistically different from the placebo group. However, post-hoc analysis revealed that patients with more severe disease showed significant improvement when treated with tetomilast. One of the potential issues with this study is the reporting of nausea and emesis, particularly with the higher dose of tetomilast which will be of concern when treating patients with gastrointestinal inflammation.
Rheumatoid arthritis is amenable to anti-TNFα monoclonal antibodies, which highlights an important role this cytokine plays in disease pathology. Similarly, T cells (e.g., Th1, Th17), macrophages, chondrocytes, and osteoblasts all play a role in this disease (Nestle et al. 2009). All these cell types express PDE4 and inhibition of this enzyme can lead to a reduction in cell activity that might be conducive to a beneficial action in this disease (Table 1).
Preclinical studies have shown that PDE4 inhibitors can suppress pannus-like inflammation in animals injected with methylated bovine serum albumin. Furthermore, PDE4 inhibitors suppressed cytokine production from peritoneal macrophages and inhibited synovial fibroblast proliferation in culture (Kobayashi et al. 2007). The recent finding that PDE4 inhibitors suppressed IL-17 production from human peripheral blood mononuclear cells and CD4+ T lymphocytes and inhibited proliferation of human memory Th17 (Ma et al. 2008) could explain the preclinical findings. Indeed, the expression of IL-23 from human peripheral blood mononuclear cells, a cytokine which supports the expansion of Th17 cells was suppressed by PDE4 inhibition (Schafer et al. 2010). Similarly, roflumilast partially attenuated nitric oxide release from human chondrocytes in culture, these cells are involved in articular cartilage destruction in rheumatoid arthritis (Tenor et al. 2002). Loss of bone is also a feature of rheumatoid arthritis, and PDE3 and PDE4 inhibitors have been shown to augment the ability of prostaglandin E2 to promote murine osteoclast differentiation in co-cultures of calvarial osteoclasts and bone marrow cells (Noh et al. 2009). However, it is well established that PGE2 promotes bone formation in vivo, and this anabolic effect is also observed with PDE4 inhibitors in ovariectomized rats (Yao et al. 2007). PDE4 inhibitors appear to suppress inflammation, cartilage destruction, and bone loss in rheumatoid arthritis, but it remains to be established whether this drug class will be significantly better than current biological agents.
6 Unwanted Side Effects
Nausea and other gastrointestinal side effects are commonly reported side effects associated with many PDE4 inhibitors and the mechanism responsible for these side effects has been investigated in an attempt to discover nonemetic PDE4 inhibitors. The direct recording of neuronal activity within the area postrema of dogs conclusively demonstrated that substances known to cause nausea (e.g., apomorphine) caused the excitation of neurons within this anatomical location (Carpenter et al. 1988). Neuronal activity within the area postrema was also increased following the systemic administration of 8-bromo cAMP or following elevation of endogenous levels of cAMP within neurones by forskolin, an activator of adenylyl cyclase (Carpenter et al. 1988). Elevated levels of cAMP within the area postrema enhanced the emetogenic response. Dogs treated with theophylline and the PDE4 selective inhibitor, 4-(3-Butoxy-4-methoxyphenyl)methyl-2-imidazolidone (Ro 20-1724) reduced the emetic threshold of the D2 agonist, apomorphine (Carpenter et al. 1988). One consequence of elevated levels of cAMP is the transcriptional activation of the early response gene c-fos, following upstream activation of the transcription factor, cAMP response element binding (CREB) by protein kinase A. This method was employed to demonstrate increased c-fos immunoreactivity in neurons within area postrema and nucleus tractus solitarius following systemic administration of a PDE4 inhibitor and providing conclusive proof that this drug class can lead to the activation of neurons within the emetic centers of the central nervous system (CNS) (Bureau et al. 2006). Direct application of a highly potent PDE4 inhibitor via intracerebroventricular administration in order to limit systemic bioavailability provide a means of directly activating neurons within the CNS, consequently resulting in emesis in the ferret (Robichaud et al. 1999). This ability of PDE4 inhibitors to induce emesis in the ferret was inhibited by the alpha2-selective agonist, clonidine (Robichaud et al. 2001) and suggested that raising cAMP within central noradrenergic terminals by PDE4 inhibitors promoted emesis, and this could be attenuated via alpha2-adrenoceptor mediated inhibition of adenylyl cyclase.
The emetic response to systemically administered PDE4 inhibitors is reduced by anti-emetic agents including the 5HT3-antagonist, ondansetron, and the NK1 antagonist, (+)-(2S,3S)-3-(2-[11C]Methoxybenzylamino)-2-phenylpiperidine (CP-99,994) (Robichaud et al. 1999, 2001). Similarly, the increased expression of c-fos within the emetic centers of the brain was also reduced following treatment with the NK1 antagonist, RP67580, thus implicating substance P in this response (Bureau et al. 2006).
Many studies have documented the expression of PDE4D within the area postrema, nucleus tractus solitaris, and nodose ganglion neurons in various species including man and implicated this isoform in nausea and vomiting (Cherry and Davis 1999; Takahashi et al. 1999; Perez-Torres et al. 2000; Lamontagne et al. 2001). However, it should also be recognized that detectable transcripts for PDE4B were also found within the nucleus tractus solitaris and area postrema in humans and rodents, respectively, and could just as well be involved in the emetic response (Perez-Torres et al. 2000). Consequently, there is a general consensus that inhibition of PDE4D is responsible for side effects such as nausea and emesis. Since rodents lack an emetic reflex, it is not possible to directly investigate the role of different isoforms of PDE4 in emesis in this model. However, a surrogate biological response, which measures the reversal of anesthesia induced by alpha2-adrenoceptor agonists (e.g., clonidine, xylazine), has been used to study the role of PDE4 subtypes in emesis (Robichaud et al. 2001, 2002a). Deletion of PDE4D and not PDE4B reduced the duration of anesthesia induced by xylazine, compared with wild-type mice, and the ability of PDE4 inhibitors to shorten xylazine-induced anesthesia was impaired in PDE4D but not PDE4B knockout mice (Robichaud et al. 2002b). Together these studies suggested that PDE4 inhibitors with low affinity for PDE4D should have reduced emetic potential.
However, the validity of this hypothesis has been questioned in light of the development of PDE4 selective inhibitors which preferentially distribute to the brain and are relatively free from emesis in a range of animal models (Burgin et al. 2010). Furthermore, there are examples of nonselective PDE4 inhibitors that have in vivo anti-inflammatory activity but are not emetogenic (Aoki et al. 2000, 2001; Gale et al. 2002). Similarly, the emetic profile of various PDE4 inhibitors (PMNPQ > R-rolipram > CT-2450) could not be explained by a preferred selectivity for PDE4D over PDE4A or PDE4B (Robichaud et al. 1999, 2002b). Some PDE4 inhibitors may preferentially partition within the area postrema in the CNS and thereby enhancing the inhibition of PDE4D in area postrema neurons and explaining the differences in the emetic potential of these drugs (Aoki et al. 2001; Robichaud et al. 2002a). Indeed, brain penetration by PMNPQ was 46-fold greater than CT-2450; however, the concentration of the “low emetic” PDE4 inhibitor, CT-2450, within the CNS was still 475-fold greater than the PDE4D inhibitory potency for CT-2450, and presumably still in sufficient concentrations to inhibit this enzyme. Moreover, the area postrema is not completely behind the blood–brain barrier and therefore accessible to free drug within the circulation (Gross et al. 1990). Whether differential partitioning of these inhibitors within the area postrema accounts for why some PDE4 inhibitors have a reduced emetic profile remains to be established. Alternatively, partial inhibition of PDE4D within the emetic center of the brain may not to be sufficient to promote an emetogenic signal, although this degree of inhibition within inflammatory cells is sufficient enough to exert an in vitro anti-inflammatory effect (Burgin et al. 2010).
A number of preclinical studies have highlighted a number of other potential disadvantages to targeting PDE4, and these include the development of mesenteric vasculitis (Spina 2004), immunosuppression (Spina 2004), heart failure, and arrhythmia (Lehnart et al. 2005). However, none of these events appear to be realized in phase II and phase III clinical trials undertaken to date, at least with cilomilast and roflumilast. Similarly, slow release theophylline has been used for decades in the treatment of asthma and COPD and has not been associated with a number of these potentially adverse events despite being shown to cause mesenteric vasculitis preclinically in some models (Nyska et al. 1998; Ohta et al. 2004). It has also been suggested that PDE4 inhibitors may have pro-inflammatory properties, which is based on the finding that at very high doses, roflumilast (100 mg/kg) promoted the recruitment of neutrophils to the airways and this correlated with the release of IL-8 from cultured endothelial cells in vitro (McCluskie et al. 2006). This mechanism might explain why animals that have been chronically treated with high doses of PDE4 inhibitors document vasculitis. However, the concentrations required to achieve these untoward effects are at least 1,000 times greater than the ED50 and EC50 values reported for roflumilast against several in vivo biomarkers of inflammation and cell function in vitro, respectively (Bundschuh et al. 2001; Hatzelmann and Schudt 2001). It is unlikely that the plasma concentrations required to produce this purported pro-inflammatory effect could be achieved even with chronic dosing. Similarly, another study has shown that PDE4 inhibitors, at concentrations that are pharmacologically relevant, delay apoptosis of neutrophils and eosinophils, an effect that increased when combined with β2-agonists (Parkkonen et al. 2007). However, the extent to which these findings translate into the clinic is unclear, particularly as the beneficial effect of roflumilast in moderate-to-severe COPD subjects was not compromised in patients taking long-acting β2-agonists (Fabbri et al. 2009). The clinical evidence suggests that PDE4 inhibitors suppress rather than exacerbate inflammation in the airways (Gamble et al. 2003; Grootendorst et al. 2007). Furthermore, there was no evidence of an increased incidence of pneumonia in patients with COPD clinically treated with roflumilast, which might be anticipated if PDE4 inhibitors interfere with host defense (Fabbri et al. 2009; Calverley et al. 2009). Interestingly, weight loss was an unexpected effect in subjects independent of any reports of gastrointestinal discomfort (Fabbri et al. 2009; Calverley et al. 2009) and could be a result of the well-recognized effect of elevating cAMP in adipocytes in promoting lipolysis since it has been previously reported that β2-agonists have a well-known lipolytic action. Mice deficient in PDE4D but not PDE4B have abnormal growth development as demonstrated by reduced fertility and litter size by female mice, reduced growth rates, and body weight in both sexes, the latter a consequence of a significant reduction in the weight of bone, muscle, and body organs (Jin et al. 1999; Jin and Conti 2002). Although the effect of these gene deficiencies were not studied on lipolysis per se, both PDE4B and PDE4D are present in rat adipocytes and inhibition of these enzymes promotes basal lipolysis (Wang and Edens 2007).
7 PDE4 Inhibition and the Future
Strategies at improving the risk/benefit ratio for PDE4 inhibition will be important if this drug class is to be widely used in the treatment of inflammatory diseases. The therapeutic window between the anti-inflammatory action of these drugs and side effects such as nausea and emesis is probably not wide enough for cilomilast, and may limit the use of roflumilast. It is of interest that there are a number of PDE4 inhibitors currently in development that appear to lack significant emetic action; these include oglemilast and IPL512602 (Boswell-Smith et al. 2006b), apremilast (Schafer et al. 2010), and the recently reported compounds produced by the Biotech company Decode (Burgin et al. 2010), but the molecular basis for this lack of emesis has not been published.
Most PDE4 inhibitors under development are designed for oral administration; however, the inhaled route would deliver a PDE4 inhibitor directly to target cells within the lung and thereby minimize systemic absorption and of course this is a widely accepted route of administration in pulmonary medicine as a way of minimizing systemic side effects with other drug classes. Both AWD 12-281 (N-(3,5-dichloropyrid-4-yl)-[1-(4-fluorobenzyl)-5-hydroxy-indole-3-yl]-glyoxylic acid amide) (Kuss et al. 2003) and UK-500,001 (Phillips et al. 2007; Vestbo et al. 2009) are examples of inhaled PDE4 inhibitors. Indeed, a 7-day treatment with the inhaled PDE4 inhibitor GSK256066 produced modest attenuation of both the early and the late asthmatic response to antigen challenge (Singh et al. 2010). It clearly should be possible to obtain anti-inflammatory activity by direct delivery of this drug class to the lung, although this does not always mean a complete loss of emesis and nausea, as exemplified by the mixed PDE3/4 inhibitor zardaverine, which although inducing bronchodilation clinically also induced nausea (Brunnee et al. 1992).
Another approach might be the use of antisense oligodeoxynucleotides targeting PDE4, which could be delivered by the inhaled route. The positive results obtained in the successful targeting of the adenosine A1 receptor in a rabbit model of allergic inflammation (Nyce and Metzger 1997) illustrates the potential of this approach. A preclinical study has demonstrated that antisense oligonucleotides against mRNA for PDE4B/4D and PDE7 delivered topically to the lung by the endotracheal route of administration suppressed inflammation following 2 weeks of cigarette smoke exposure in mice (Fortin et al. 2009). The advantage of this technique is that side effects such as nausea and emesis are likely to be avoided. Alternatively, the recently identified allosteric modulators of PDE4 are proposed to confer lower emetic potential because despite only partially inhibiting PDE4 activity, the effect is nonetheless sufficient to inhibit inflammatory cell activity completely (Burgin et al. 2010). This is an important possibility that could be exploited in the discovery of nonemetic PDE4 inhibitors for the treatment of inflammatory diseases.
Another reason that targeting PDE4 alone may not fully resolve airway inflammation is the fact that there are other PDE types exist in structural and inflammatory cells in the lung (Table 1). Therefore, targeting multiple PDEs may be required for optimal anti-inflammatory action. For example, the macrophage is viewed as a critical cell type in the pathogenesis of COPD (Barnes 2008); however, the ability of these cells to release TNFα in response to endotoxin was only inhibited to a small degree by PDE4 inhibitors (Hatzelmann and Schudt 2001) and the potential functional involvement of PDE3 and PDE7 in these cells cannot be completely ignored. The inhibitory action of PDE4 inhibitors on the cellular activity of CD8+ T-lymphocytes and macrophages was significantly increased in the presence of PDE7 selective inhibitors (Smith et al. 2004). Similarly, combined PDE3 and PDE4 inhibitor in a single molecule offers the advantage of delivering a bronchodilator and anti-inflammatory substance. Moreover, it is likely that retention of the inhibitor within the lung may be required in order to maintain anti-inflammatory activity with the airways (Boswell-Smith et al. 2006a).
8 Conclusion
A number of clinical trials assessing the efficacy of PDE4 inhibitors for the treatment of inflammatory disease including asthma, COPD, atopic dermatitis, and psoriasis have demonstrated moderate success. However, the dose-limiting side effects such as nausea, emesis, and headache potentially limit the utility of these drugs. Importantly, there are examples of PDE4 inhibitors that have low emetogenic potential, although the molecular basis of this phenomenon remains to be established. Other strategies include topical delivery (e.g., inhalation for administration to the lung; direct application to the skin), development of subtype selective PDE4 inhibitors, use of mixed PDE inhibitors, interference with PDE4 activation, targeting proteins that are involved in locating PDE4 to specific microcellular domains and finally the potential for use of antisense oligonucleotides may offer another solution to the problem of targeting PDE4 in inflammation is a cause for optimism.
Conflict of Interest.
The authors are consultants for Veronapharma plc who are developing RPL554 as a novel inhaled PDE3/4 inhibitor for the treatment of inflammatory airways disease. CP is also a founder and has equity in Veronapharma plc.
Abbreviations
- COPD:
-
Chronic obstructive pulmonary disease
- PDE:
-
Phosphodiesterase
References
Abraham C, Cho JH (2009) Inflammatory bowel disease. N Engl J Med 361:2066–2078
Akama T, Baker SJ, Zhang YK, Hernandez V, Zhou H, Sanders V, Freund Y, Kimura R, Maples KR, Plattner JJ (2009) Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg Med Chem Lett 19:2129–2132
Aoki M, Kobayashi M, Ishikawa J, Saita Y, Terai Y, Takayama K, Miyata K, Yamada T (2000) A novel phosphodiesterase type 4 inhibitor, YM976 (4-(3-chlorophenyl)-1, 7-diethylpyrido[2, 3-d]pyrimidin-2(1H)-one), with little emetogenic activity. J Pharmacol Exp Ther 295:255–260
Aoki M, Fukunaga M, Sugimoto T, Hirano Y, Kobayashi M, Honda K, Yamada T (2001) Studies on mechanisms of low emetogenicity of YM976, a novel phosphodiesterase type 4 inhibitor. J Pharmacol Exp Ther 298:1142–1149
Ariga M, Neitzert B, Nakae S, Mottin G, Bertrand C, Pruniaux MP, Jin SL, Conti M (2004) Nonredundant function of phosphodiesterases 4D and 4B in neutrophil recruitment to the site of inflammation. J Immunol 173:7531–7538
Banner KH, Press NJ (2009) Dual PDE3/4 inhibitors as therapeutic agents for chronic obstructive pulmonary disease. Br J Pharmacol 157:892–906
Barber R, Baillie GS, Bergmann R, Shepherd MC, Sepper R, Houslay MD, Heeke GV (2004) Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokers. Am J Physiol Lung Cell Mol Physiol 287:L332–L343
Barnes PJ (2008) The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest 118:3546–3556
Barnes PJ, Adcock IM, Ito K (2005) Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J 25:552–563
Baumer W, Hoppmann J, Rundfeldt C, Kietzmann M (2007) Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm Allergy Drug Targets 6:17–26
Boolell M, Gepi-Attee S, Gingell JC, Allen MJ (1996) Sildenafil, a novel effective oral therapy for male erectile dysfunction. Br J Urol 78:257–261
Boswell-Smith V, Spina D, Oxford AW, Comer MB, Seeds EA, Page CP (2006a) The pharmacology of two novel long-acting phosphodiesterase 3/4 inhibitors, RPL554 [9, 10-dimethoxy-2(2, 4, 6-trimethylphenylimino)-3-(n-carbamoyl-2-aminoethyl) -3, 4, 6, 7-tetrahydro-2H-pyrimido[6, 1-a]isoquinolin-4-one] and RPL565 [6, 7-dihydro-2-(2, 6-diisopropylphenoxy)-9, 10-dimethoxy-4H-pyrimido[6, 1-a]i soquinolin-4-one]. J Pharmacol Exp Ther 318:840–848
Boswell-Smith V, Spina D, Page CP (2006b) Phosphodiesterase inhibitors. Br J Pharmacol 147(Suppl 1):S252–S257
Bousquet J, Aubier M, Sastre J, Izquierdo JL, Adler LM, Hofbauer P, Rost KD, Harnest U, Kroemer B, Albrecht A, Bredenbroker D (2006) Comparison of roflumilast, an oral anti-inflammatory, with beclomethasone dipropionate in the treatment of persistent asthma. Allergy 61:72–78
Brunnee T, Engelstatter R, Steinijans VW, Kunkel G (1992) Bronchodilatory effect of inhaled zardaverine, a phosphodiesterase III and IV inhibitor, in patients with asthma. Eur Respir J 5:982–985
Bundschuh DS, Eltze M, Barsig J, Wollin L, Hatzelmann A, Beume R (2001) In vivo efficacy in airway disease models of roflumilast, a novel orally active PDE4 inhibitor. J Pharmacol Exp Ther 297:280–290
Bureau Y, Handa M, Zhu Y, Laliberte F, Moore CS, Liu S, Huang Z, Macdonald D, Xu DG, Robertson GS (2006) Neuroanatomical and pharmacological assessment of Fos expression induced in the rat brain by the phosphodiesterase-4 inhibitor 6-(4-pyridylmethyl)-8-(3-nitrophenyl) quinoline. Neuropharmacology 51:974–985
Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, Thorsteinsdottir M, Hrafnsdottir S, Hagen T, Kiselyov AS, Stewart LJ, Gurney ME (2010) Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol 28:63–70
Butcher RW, Sutherland EW (1962) Adenosine 3′, 5′-phosphate in biological materials. I. Purification and properties of cyclic 3′, 5′-nucleotide phosphodiesterase and use of this enzyme to characterize adenosine 3′, 5′-phosphate in human urine. J Biol Chem 237:1244–1250
Calverley PM, Sanchez-Toril F, McIvor A, Teichmann P, Bredenbroeker D, Fabbri LM (2007) Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 176:154–161
Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ (2009) Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet 374:685–694
Card GL, England BP, Suzuki Y, Fong D, Powell B, Lee B, Luu C, Tabrizizad M, Gillette S, Ibrahim PN, Artis DR, Bollag G, Milburn MV, Kim SH, Schlessinger J, Zhang KY (2004) Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 12:2233–2247
Carpenter DO, Briggs DB, Knox AP, Strominger N (1988) Excitation of area postrema neurones by transmitters, peptides and cyclic nucleotides. J Neurophysiol 59:358–369
Chan SC, Reifsnyder D, Beavo JA, Hanifin JM (1993) Immunochemical characterization of the distinct monocyte cyclic AMP-phosphodiesterase from patients with atopic dermatitis. J Allergy Clin Immunol 91:1179–1188
Cherry JA, Davis RL (1999) Cyclic AMP phosphodiesterases are localized in regions of the mouse brain associated with reinforcement, movement and affect. J Comp Neurol 407:287–301
Conti M, Beavo J (2007) Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem 76:481–511
David M, Zech K, Seiberling M, Weimar C, Roflumilast BTD (2004) a novel, oral, selective PDE4 inhibitor, shows high absolute bioavailability. J Allergy Clin Immunol 113:S220–S221, Ref Type: Abstract
Dunkern TR, Feurstein D, Rossi GA, Sabatini F, Hatzelmann A (2007) Inhibition of TGF-beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol 572:12–22
Essayan DM, Kagey-Sobotka A, Lichtenstein LM, Huang S-K (1997) Differential regulation of human antigen-specific Th1 and Th2 lymphocyte responses by isozyme selective cyclic nucleotide phosphodiesterase inhibitors. J Pharmacol Exp Ther 282:505–512
Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, Rabe KF (2009) Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet 374:695–703
Fortin M, D’Anjou H, Higgins ME, Gougeon J, Aube P, Moktefi K, Mouissi S, Seguin S, Seguin R, Renzi PM, Paquet L, Ferrari N (2009) A multi-target antisense approach against PDE4 and PDE7 reduces smoke-induced lung inflammation in mice. Respir Res 10:39
Fuhrmann M, Jahn H-U, Seybold J, Neurohr C, Barnes PJ, Hippenstiel S, Kraemer HJ, Suttorp N (1999) Identification and function of cyclic nucleotide phosphodiesterase isoenzymes in airway epithelial cells. Am J Respir Cell Mol Biol 20:292–302
Gale DD, Landells LJ, Spina D, Miller AJ, Smith K, Nichols T, Rotshteyn Y, Tonelli A, Lacouture P, Burch RM, Page CP, O’Connor BJ (2002) Pharmacokinetic and pharmacodynamic profile following oral administration of the phosphodiesterase (PDE)4 inhibitor V11294A in healthy volunteers. Br J Clin Pharmacol 54:478–484
Gamble E, Grootendorst DC, Brightling CE, Troy S, Qiu Y, Zhu J, Parker D, Matin D, Majumdar S, Vignola AM, Kroegel C, Morell F, Hansel TT, Rennard SI, Compton C, Amit O, Tat T, Edelson J, Pavord ID, Rabe KF, Barnes NC, Jeffery PK (2003) Antiinflammatory effects of the phosphodiesterase-4 inhibitor cilomilast (Ariflo) in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 168:976–982
Gantner F, Kupferschmidt R, Schudt C, Wendel A, Hatzelmann A (1997a) In vitro differentiation of human monocytes to macrophages: Change of PDE profile and its relationship to suppression of tumour necrosis factor-alpha release by PDE inhibitors. Br J Pharmacol 121:221–231
Gantner F, Tenor H, Gekeler V, Schudt C, Wendel A, Hatzelmann A (1997b) Phosphodiesterase profiles of highly purified human peripheral blood leukocyte populations from normal and atopic individuals: a comparative study. J Allergy Clin Immunol 100:527–535
Gantner F, Gotz C, Gekeler V, Schudt C, Wendel A, Hatzelmann A (1998) Phosphodiesterase profile of human B lymphocytes from normal and atopic donors and the effects of PDE inhibition on B cell proliferation. Br J Pharmacol 123:1031–1038
Gottlieb AB, Strober B, Krueger JG, Rohane P, Zeldis JB, Hu CC, Kipnis C (2008) An open-label, single-arm pilot study in patients with severe plaque-type psoriasis treated with an oral anti-inflammatory agent, apremilast. Curr Med Res Opin 24:1529–1538
Grootendorst DC, Gauw SA, Verhoosel RM, Sterk PJ, Hospers JJ, Bredenbroker D, Bethke TD, Hiemstra PS, Rabe KF (2007) Reduction in sputum neutrophil and eosinophil numbers by the PDE4 inhibitor roflumilast in patients with COPD. Thorax 62:1081–1087
Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS (1990) Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol 259:R1131–R1138
Haddad JJ, Land SC, Tarnow-Mordi WO, Zembala M, Kowalczyk D, Lauterbach R (2002) Immunopharmacological potential of selective phosphodiesterase inhibition. I. Differential regulation of lipopolysaccharide-mediated proinflammatory cytokine (interleukin-6 and tumor necrosis factor-alpha) biosynthesis in alveolar epithelial cells. J Pharmacol Exp Ther 300:559–566
Hansen G, Jin S, Umetsu DT, Conti M (2000) Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc Natl Acad Sci USA 97:6751–6756
Harbinson PL, MacLeod D, Hawksworth R, O’Toole S, Sullivan PJ, Heath P, Kilfeather S, Page CP, Costello J, Holgate ST, Lee TH (1997) The effect of a novel orally active selective PDE4 isoenzyme inhibitor (CDP840) on allergen-induced responses in asthmatic subjects. Eur Respir J 10:1008–1014
Hatzelmann A, Schudt C (2001) Anti-inflammatory and immunomodulatory potential of the novel PDE4 inhibitor roflumilast in vitro. J Pharmacol Exp Ther 297:267–279
Heystek HC, Thierry AC, Soulard P, Moulon C (2003) Phosphodiesterase 4 inhibitors reduce human dendritic cell inflammatory cytokine production and Th1-polarizing capacity. Int Immunol 15:827–835
Himes BE, Hunninghake GM, Baurley JW, Rafaels NM, Sleiman P, Strachan DP, Wilk JB, Willis-Owen SA, Klanderman B, Lasky-Su J, Lazarus R, Murphy AJ, Soto-Quiros ME, Avila L, Beaty T, Mathias RA, Ruczinski I, Barnes KC, Celedon JC, Cookson WO, Gauderman WJ, Gilliland FD, Hakonarson H, Lange C, Moffatt MF, O’Connor GT, Raby BA, Silverman EK, Weiss ST (2009) Genome-wide association analysis identifies PDE4D as an asthma-susceptibility gene. Am J Hum Genet 84:581–593
Homma S, Sakamoto T, Hegab AE, Saitoh W, Nomura A, Ishii Y, Morishima Y, Iizuka T, Kiwamoto T, Matsuno Y, Massoud HH, Massoud HM, Hassanein KM, Sekizawa K (2006) Association of phosphodiesterase 4D gene polymorphisms with chronic obstructive pulmonary disease: relationship to interleukin 13 gene polymorphism. Int J Mol Med 18:933–939
Houslay MD (2010) Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci 35:91–100
Houslay MD, Schafer P, Zhang KY (2005) Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today 10:1503–1519
Houslay MD, Baillie GS, Maurice DH (2007) cAMP-Specific phosphodiesterase-4 enzymes in the cardiovascular system: a molecular toolbox for generating compartmentalized cAMP signaling. Circ Res 100:950–966
Huston E, Lynch MJ, Mohamed A, Collins DM, Hill EV, MacLeod R, Krause E, Baillie GS, Houslay MD (2008) EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci USA 105:12791–12796
Ichikawa H, Okamoto S, Kamada N, Nagamoto H, Kitazume MT, Kobayashi T, Chinen H, Hisamatsu T, Hibi T (2008) Tetomilast suppressed production of proinflammatory cytokines from human monocytes and ameliorated chronic colitis in IL-10-deficient mice. Inflamm Bowel Dis 14:1483–1490
Jin SL, Conti M (2002) Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci USA 99:7628–7633
Jin SL, Richard FJ, Kuo WP, D’Ercole AJ, Conti M (1999) Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc Natl Acad Sci USA 96:11998–12003
Jones NA, Boswell-Smith V, Lever R, Page CP (2005) The effect of selective phosphodiesterase isoenzyme inhibition on neutrophil function in vitro. Pulm Pharmacol Ther 18:93–101
Jones NA, Leport M, Holand T, Vos T, Morgan M, Fink M, Pruniaux MP, Berthelier C, O’Connor BJ, Bertrand C, Page CP (2007) Phosphodiesterase (PDE) 7 in inflammatory cells from patients with asthma and COPD. Pulm Pharmacol Ther 20:60–68
Kanehiro A, Ikemura T, Makela MJ, Lahn M, Joetham A, Dakhama A, Gelfand EW (2001) Inhibition of phosphodiesterase 4 attenuates airway hyperresponsiveness and airway inflammation in a model of secondary allergen challenge. Am J Respir Crit Care Med 163:173–184
Keshavarzian A, Mutlu E, Guzman JP, Forsyth C, Banan A (2007) Phosphodiesterase 4 inhibitors and inflammatory bowel disease: emerging therapies in inflammatory bowel disease. Expert Opin Investig Drugs 16:1489–1506
Kobayashi K, Suda T, Manabe H, Miki I (2007) Administration of PDE4 inhibitors suppressed the pannus-like inflammation by inhibition of cytokine production by macrophages and synovial fibroblast proliferation. Mediat Inflamm 2007:58901
Kohyama T, Liu X, Zhu YK, Wen FQ, Wang HJ, Fang Q, Kobayashi T, Rennard SI (2002) Phosphodiesterase 4 inhibitor cilomilast inhibits fibroblast-mediated collagen gel degradation induced by tumor necrosis factor-alpha and neutrophil elastase. Am J Respir Cell Mol Biol 27:487–494
Kranz M, Wall M, Evans B, Miah A, Ballantine S, Delves C, Dombroski B, Gross J, Schneck J, Villa JP, Neu M, Somers DO (2009) Identification of PDE4B Over 4D subtype-selective inhibitors revealing an unprecedented binding mode. Bioorg Med Chem 17:5336–5341
Kumar RK, Herbert C, Thomas PS, Wollin L, Beume R, Yang M, Webb DC, Foster PS (2003) Inhibition of inflammation and remodeling by roflumilast and dexamethasone in murine chronic asthma. J Pharmacol Exp Ther 307:349–355
Kung TT, Crawley Y, Luo B, Young S, Kreutner W, Chapman RW (2000) Inhibition of pulmonary eosinophilia and airway hyperresponsiveness in allergic mice by rolipram: involvement of endogenously released corticosterone and catecholamines. Br J Pharmacol 130:457–463
Kuss H, Hoefgen N, Johanssen S, Kronbach T, Rundfeldt C (2003) In vivo efficacy in airway disease models of N-(3, 5-dichloropyrid-4-yl)-[1-(4-fluorobenzyl)-5-hydroxy-indole-3-yl]-glyo xylic acid amide (AWD 12–281), a selective phosphodiesterase 4 inhibitor for inhaled administration. J Pharmacol Exp Ther 307:373–385
Lamontagne S, Meadows E, Luk P, Normandin D, Muise E, Boulet L, Pon DJ, Robichaud A, Robertson GS, Metters KM, Nantel F (2001) Localization of phosphodiesterase-4 isoforms in the medulla and nodose ganglion of the squirrel monkey. Brain Res 920:84–96
Landells LJ, Szilagy CM, Jones NA, Banner KH, Allen JM, Doherty A, O’Connor BJ, Spina D, Page CP (2001) Identification and quantification of phosphodiesterase 4 subtypes in CD4 and CD8 lymphocytes from healthy and asthmatic subjects. Br J Pharmacol 133:722–729
Leclerc O, Lagente V, Planquois JM, Berthelier C, Artola M, Eichholtz T, Bertrand CP, Schmidlin F (2006) Involvement of MMP-12 and phosphodiesterase type 4 in cigarette smoke-induced inflammation in mice. Eur Respir J 27:1102–1109
Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, Richter W, Jin SL, Conti M, Marks AR (2005) Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123:25–35
Louw C, Williams Z, Venter L, Leichtl S, Schmid-Wirlitsch C, Bredenbroker D, Bardin PG (2007) Roflumilast, a phosphodiesterase 4 inhibitor, reduces airway hyperresponsiveness after allergen challenge. Respiration 74:411–417
Lu S, Liu N, Dass SB, Reiss TF, Knorr BA (2009) Randomized, placebo-controlled study of a selective PDE4 inhibitor in the treatment of asthma. Respir Med 103:342–347
Ma R, Yang BY, Wu CY (2008) A selective phosphodiesterase 4 (PDE4) inhibitor Zl-n-91 suppresses IL-17 production by human memory Th17 cells. Int Immunopharmacol 8:1408–1417
Manning CD, Burman M, Christensen SB, Cieslinski LB, Essayan DM, Grous M, Torphy TJ, Barnette MS (1999) Suppression of human inflammatory cell function by subtype- selective PDE4 inhibitors correlates with inhibition of PDE4A and PDE4B. Br J Pharmacol 128:1393–1398
Martin-Chouly CA, Astier A, Jacob C, Pruniaux MP, Bertrand C, Lagente V (2004) Modulation of matrix metalloproteinase production from human lung fibroblasts by type 4 phosphodiesterase inhibitors. Life Sci 75:823–840
Martorana PA, Beume R, Lucattelli M, Wollin L, Lungarella G (2005) Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am J Respir Crit Care Med 172:848–853
McCahill AC, Huston E, Li X, Houslay MD (2008) PDE4 associates with different scaffolding proteins: modulating interactions as treatment for certain diseases. Handb Exp Pharmacol (186); 125-166
McCluskie K, Klein U, Linnevers C, Ji YH, Yang A, Husfeld C, Thomas GR (2006) Phosphodiesterase type 4 inhibitors cause proinflammatory effects in vivo. J Pharmacol Exp Ther 319:468–476
Mehats C, Jin SL, Wahlstrom J, Law E, Umetsu DT, Conti M (2003) PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J 17:1831–1841
Millar JK, Mackie S, Clapcote SJ, Murdoch H, Pickard BS, Christie S, Muir WJ, Blackwood DH, Roder JC, Houslay MD, Porteous DJ (2007) Disrupted in schizophrenia 1 and phosphodiesterase 4B: towards an understanding of psychiatric illness. J Physiol 584:401–405
Nazarian R, Weinberg JM (2009) AN-2728, a PDE4 inhibitor for the potential topical treatment of psoriasis and atopic dermatitis. Curr Opin Investig Drugs 10:1236–1242
Nestle FO, Kaplan DH, Barker J (2009) Psoriasis. N Engl J Med 361:496–509
Noh AL, Yang M, Lee JM, Park H, Lee DS, Yim M (2009) Phosphodiesterase 3 and 4 negatively regulate receptor activator of nuclear factor-kappaB ligand-mediated osteoclast formation by prostaglandin E2. Biol Pharm Bull 32:1844–1848
Nyce JW, Metzger WJ (1997) DNA antisense therapy for asthma in an animal model. Nature 385:721–725
Nyska A, Herbert RA, Chan PC, Haseman JK, Hailey JR (1998) Theophylline-induced mesenteric periarteritis in F344/N rats. Arch Toxicol 72:731–737
O’Shaughnessy MJ, Chen ZM, Gramaglia I, Taylor PA, Panoskaltsis-Mortari A, Vogtenhuber C, Palmer E, Grader-Beck T, Boussiotis VA, Blazar BR (2007) Elevation of intracellular cyclic AMP in alloreactive CD4(+) T Cells induces alloantigen-specific tolerance that can prevent GVHD lethality in vivo. Biol Blood Marrow Transplant 13:530–542
Ohta K, Fukuchi Y, Grouse L, Mizutani R, Rabe KF, Rennard SI, Zhong NS (2004) A prospective clinical study of theophylline safety in 3810 elderly with asthma or COPD. Respir Med 98:1016–1024
Papakostantinou E, Xenos K, Markantonis SL, Druska S, Stratigos A, Katsambas A (2005) Efficacy of 2 weeks’ application of theophylline ointment in psoriasis vulgaris. J Dermatol Treat 16:169–170
Parkkonen J, Hasala H, Moilanen E, Giembycz MA, Kankaanranta H (2007) Phosphodiesterase 4 inhibitors delay human eosinophil and neutrophil apoptosis in the absence and presence of salbutamol. Pulm Pharmacol Ther 21:499–506
Perez-Torres S, Miro X, Palacios JM, Cortes R, Puigdomenech P, Mengod G (2000) Phosphodiesterase type 4 isozymes expression in human brain examined by in situ hybridization histochemistry and[3H]rolipram binding autoradiography. Comparison with monkey and rat brain. J Chem Neuroanat 20:349–374
Peter D, Jin SL, Conti M, Hatzelmann A, Zitt C (2007) Differential expression and function of phosphodiesterase 4 (PDE4) subtypes in human primary CD4+ T cells: predominant role of PDE4D. J Immunol 178:4820–4831
Phillips P, Bennetts M, Banner K, Ward J, Wessels D, Fuhr R (2007) The PDE4 inhibitor UK-500,001 does not significantly inhibit airway responses to allergen and histamine. Eur Resp J: 490s. Ref Type: Abstract
Rabe KF, Bateman ED, O’Donnell D, Witte S, Bredenbroker D, Bethke TD (2005) Roflumilast – an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 366:563–571
Rennard SI (2004) Treatment of stable chronic obstructive pulmonary disease. Lancet 364:791–802
Rennard SI, Schachter N, Strek M, Rickard K, Amit O (2006) Cilomilast for COPD: results of a 6-month, placebo-controlled study of a potent, selective inhibitor of phosphodiesterase 4. Chest 129:56–66
Robichaud A, Tattersall FD, Choudhury I, Rodger IW (1999) Emesis induced by inhibitors of type IV cyclic nucleotide phosphodiesterase (PDE IV) in the ferret. Neuropharmacology 38:289–297
Robichaud A, Savoie C, Stamatiou PB, Tattersall FD, Chan CC (2001) PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology 40:262–269
Robichaud A, Savoie C, Stamatiou PB, Lachance N, Jolicoeur P, Rasori R, Chan CC (2002a) Assessing the emetic potential of PDE4 inhibitors in rats. Br J Pharmacol 135:113–118
Robichaud A, Stamatiou PB, Jin SL, Lachance N, Macdonald D, Laliberte F, Liu S, Huang Z, Conti M, Chan CC (2002b) Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest 110:1045–1052
Roth BL, Sheffler DJ, Kroeze WK (2004) Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat Rev Drug Discov 3:353–359
Salvi SS, Barnes PJ (2009) Chronic obstructive pulmonary disease in non-smokers. Lancet 374:733–743
Sanz MJ, Cortijo J, Taha MA, Cerda-Nicolas M, Schatton E, Burgbacher B, Klar J, Tenor H, Schudt C, Issekutz AC, Hatzelmann A, Morcillo EJ (2007) Roflumilast inhibits leukocyte-endothelial cell interactions, expression of adhesion molecules and microvascular permeability. Br J Pharmacol 152:481–492
Schafer P, Parton A, Gandhi A, Capone L, Adams M, Wu L, Bartlett J, Loveland M, Gilhar A, Cheung YF, Baillie G, Houslay M, Man HW, Muller G, Stirling D (2010) Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol 159:842–855
Schreiber S, Keshavarzian A, Isaacs KL, Schollenberger J, Guzman JP, Orlandi C, Hanauer SB (2007) A randomized, placebo-controlled, phase II study of tetomilast in active ulcerative colitis. Gastroenterology 132:76–86
Shichijo M, Inagaki N, Nakai N, Kimata M, Nakahata T, Serizawa I, Iikura Y, Saito H, Nagai H (1998) The effects of anti-asthma drugs on mediator release from cultured human mast cells. Clin Exp Allergy 28:1228–1236
Shore A, Dosch H, Gelfand EW (1978) Induction and separation of antigen-dependent T helper and T suppressor cells in man. Nature 274:586–587
Singh D, Petavy F, Macdonald AJ, Lazaar AL, O’Connor BJ (2010) The inhaled phosphodiesterase 4 inhibitor GSK256066 reduces allergen challenge responses in asthma. Respir Res 11:26–35
Smith SJ, Brookes-Fazakerley S, Donnelly LE, Barnes PJ, Barnette MS, Giembycz MA (2003) Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am J Physiol Lung Cell Mol Physiol 284:L279–L289
Smith SJ, Cieslinski LB, Newton R, Donnelly LE, Fenwick PS, Nicholson AG, Barnes PJ, Barnette MS, Giembycz MA (2004) Discovery of BRL 50481 [3-(N, N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol Pharmacol 66:1679–1689
Spina D (2003) Phosphodiesterase-4 inhibitors in the treatment of inflammatory lung disease. Drugs 63:2575–2594
Spina D (2004) The potential of PDE4 inhibitors in respiratory disease. Curr Drug Targets Inflamm Allergy 3:231–236
Spina D (2008) PDE4 inhibitors: current status. Br J Pharmacol 155:308–315
Spina D, Harrison S, Page CP (1995) Regulation by phosphodiesterase isoenzymes of non-adrenergic non- cholinergic contraction in guinea-pig isolated main bronchus. Br J Pharmacol 116:2334–2340
Sullivan P, Bekir S, Jaffar Z, Page C, Jeffery P, Costello J (1994) Anti-inflammatory effects of low-dose oral theophylline in atopic asthma [published erratum appears in Lancet 1994 Jun 11; 343(8911):1512]. Lancet 343:1006–1008
Takahashi M, Terwilliger R, Lane C, Mezes PS, Conti M, Duman RS (1999) Chronic antidepressant administration increases the expression of cAMP-specific phosphodiesterase 4A and 4B isoforms. J Neurosci 19:610–618
Taylor PC, Feldmann M (2009) Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol 5:578–582
Tenor H, Hedbom E, Hauselmann HJ, Schudt C, Hatzelmann A (2002) Phosphodiesterase isoenzyme families in human osteoarthritis chondrocytes–functional importance of phosphodiesterase 4. Br J Pharmacol 135:609–618
Timmer W, Leclerc V, Birraux G, Neuhauser M, Hatzelmann A, Bethke T, Wurst W (2002) The new phosphodiesterase 4 inhibitor roflumilast is efficacious in exercise-induced asthma and leads to suppression of LPS-stimulated TNF-alpha ex vivo. J Clin Pharmacol 42:297–303
van Schalkwyk E, Strydom K, Williams Z, Venter L, Leichtl S, Schmid-Wirlitsch C, Bredenbroker D, Bardin PG (2005) Roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor, attenuates allergen-induced asthmatic reactions. J Allergy Clin Immunol 116:292–298
Vestbo J, Tan L, Atkinson G, Ward J (2009) A controlled trial of 6-weeks’ treatment with a novel inhaled phosphodiesterase type-4 inhibitor in COPD. Eur Respir J 33:1039–1044
Wang H, Edens NK (2007) mRNA expression and antilipolytic role of phosphodiesterase 4 in rat adipocytes in vitro. J Lipid Res 48:1099–1107
Wang H, Peng MS, Chen Y, Geng J, Robinson H, Houslay MD, Cai J, Ke H (2007) Structures of the four subfamilies of phosphodiesterase-4 provide insight into the selectivity of their inhibitors. Biochem J 408:193–201
Weston MC, Anderson N, Peachell PT (1997) Effects of phosphodiesterase inhibitors on human lung mast cell and basophil function. Br J Pharmacol 121:287–295
Wright RK, Mandy SH, Halprin KM, Hsia SL (1973) Defects and deficiency of adenyl cyclase in psoriatic skin. Arch Dermatol 107:47–53
Xu RX, Hassell AM, Vanderwall D, Lambert MH, Holmes WD, Luther MA, Rocque WJ, Milburn MV, Zhao Y, Ke H, Nolte RT (2000) Atomic structure of PDE4: insights into phosphodiesterase mechanism a specificity. Science 288:1822–1825
Yao W, Tian XY, Chen J, Setterberg RB, Lundy MW, Chmielzwski P, Froman CA, Jee WS (2007) Rolipram, a phosphodiesterase 4 inhibitor, prevented cancellous and cortical bone loss by inhibiting endosteal bone resorption and maintaining the elevated periosteal bone formation in adult ovariectomized rats. J Musculoskelet Neuronal Interact 7:119–130
Zocchi MR, Pardi R, Gromo G, Ferrero E, Ferrero ME, Besana C, Rugarli C (1985) Theophylline induced non specific suppressor activity in human peripheral blood lymphocytes. J Immunopharmacol 7:217–234
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Page, C.P., Spina, D. (2011). Phosphodiesterase Inhibitors in the Treatment of Inflammatory Diseases. In: Francis, S., Conti, M., Houslay, M. (eds) Phosphodiesterases as Drug Targets. Handbook of Experimental Pharmacology, vol 204. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-17969-3_17
Download citation
DOI: https://doi.org/10.1007/978-3-642-17969-3_17
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-17968-6
Online ISBN: 978-3-642-17969-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)