
Abstract
Metastatic melanoma is associated with poor outcome and is largely refractory to the historic standard of care. In recent years, the development of targeted small-molecule inhibitors and immunotherapy has revolutionised the care and improved the overall survival of these patients. Therapies targeting BRAF and MEK to block the mitogen-activated protein kinase (MAPK) pathway were the first to show unprecedented clinical responses. Following these encouraging results, antibodies targeting immune checkpoint inhibition molecules cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), programmed cell death (PD)-1, and PD-ligand1(PD-L1) demonstrated sustained tumour regression in a significant subset of patients by enabling an anti-tumour immunologic response. Despite these landmark changes in practice, the majority of patients are either intrinsically resistant or rapidly acquire resistance to MAPK pathway inhibitors and immune checkpoint blockade treatment. The lack of response can be driven by mutations and non-mutational events in tumour cells, as well as by changes in the surrounding tumour microenvironment. Common resistance mechanisms bypass the dependence of tumour cells on initial MAPK pathway driver mutations during targeted therapy, and permit evasion of the host immune system to allow melanoma growth and survival following immunotherapy. This highlights the requirement for personalised treatment regimens that take into account patient-specific genetic and immunologic characteristics. Here we review the mechanisms by which melanomas display intrinsic resistance or acquire resistance to targeted therapy and immunotherapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Acquired resistance
- BRAF inhibitor
- Checkpoint inhibitor
- Immunotherapy
- Intrinsic resistance
- MAPK pathway
- MEK inhibitor
- Melanoma
- Targeted therapy
1 Melanoma Incidence and Clinical Subtypes
Malignant melanoma accounts for >80% of skin cancer-related deaths despite representing <1% of cases (NICE 2015). The current incidence and mortality of melanoma in Europe is over 100,000 new cases and over 22,000 deaths each year (http://www.IARC.fr). In most European countries rates are doubling every decade and in many countries melanoma will soon be the fourth most common cancer (Ferlay et al. 2013). The yearly incidence in the UK is 12,000 cases, which is rising by ~2% per year, and approximately 2,000 patients die from advanced disease. Approximately one third of melanoma patients are under 50 years of age, so this cancer disproportionately affects the elderly population (NICE 2015).
Melanoma stems from the malignant transformation of melanocytes, which are neural crest-derived, and migrate during development to colonise the skin, eye, and in scarce numbers, other tissues of the body. The most common type of melanoma arises from melanocytes in the skin and predominantly affects the population of European descent (Whiteman and Green 1999). The only known environmental risk factor for cutaneous melanoma is ultraviolet radiation (UVR) and informing the population at risk remains a public health challenge (Gilchrest et al. 1999). Previous efforts to use environmental factors to classify cutaneous melanoma include proposals for a divergent pathway model (Whiteman and Green 1999), where UVR exposure pattern, host susceptibility, and the site of the primary lesion are used as criteria to identify epidemiologically uniform subsets of patients. Indeed, cutaneous melanoma arising at sites chronically exposed to UVR (head and neck) is more likely to occur in adults with a high lifetime accumulation of UVR exposure, whereas melanomas arising at sites that are only intermittently exposed to UVR (trunk) appear in younger patients who received a lower lifetime exposure. This group of patients were more likely to report intermittent UVR exposure on the trunk through recreational activities (Whiteman et al. 2003; Curtin et al. 2005; Chang et al. 2009).
The most common founder mutation in melanomas arising in patients younger than 55 years of age is V600EBRAF (Davies et al. 2002; Cancer Genome Atlas Network 2015). In contrast, melanomas arising in the elderly are more likely to be driven by mutations in NRAS, NF1 and BRAF mutations that do not affect the codon V600 (Cancer Genome Atlas Network 2015; Krauthammer et al. 2015; Shain and Bastian 2016). Thus, the protein kinase BRAF is mutated in almost half of all cutaneous melanomas, and BRAF mutations are more frequently found to drive melanomas arising over skin that has been only intermittently exposed to sun such as the trunk and upper limbs (Cancer Genome Atlas Network 2015; Shain and Bastian 2016; Shain et al. 2015).
2 Melanoma Treatment Overview
Until the last decade, the standard of care for advanced disease included surgical resection, chemotherapy with the alkylating agent dacarbazine (DTIC), and high-dose interleukin 2 (IL-2), which only led to durable responses in a few patients with metastatic disease. The only approved adjuvant treatment for patients at high risk of progression was interferon-α-2b. Despite these treatments, metastatic melanoma remained associated with extremely poor prognosis and only curable if detected at an early, pre-metastatic stage (Balch et al. 2009). More recently, insights into the molecular mechanisms driving melanomagenesis have led to the development of targeted therapies, and the advances in immune checkpoint inhibitor therapies have also contributed to a revolution in melanoma care.
3 The Rationale for Targeted Therapies in Melanoma
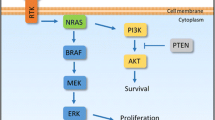
One of the principal signalling pathways in melanoma is the MAPK pathway, which signals via RAS (Young et al. 2009; Stephen et al. 2014) and RAF (Davies et al. 2002; Wan et al. 2004; Marais et al. 1997). The signalling cascade is triggered by stimulatory input from extracellular growth factors, cytokines and hormones that bind to the receptor tyrosine kinases (RTK) on the cell membrane (Schlessinger 2000). Once activated, the RTKs form stable dimers that undergo phosphorylation at multiple tyrosine residues that are in the cytosolic component of the protein. This chemical change drives activation of RAS proteins, and downstream proteins BRAF and CRAF, leading to activation and phosphorylation of the dual-specificity kinases MEK1 and MEK2; which in turn phosphorylate ERK1 and ERK2 (Holderfield et al. 2014). ERK proteins then phosphorylate approximately 50 cytoplasmic and nuclear proteins to control cell proliferation, differentiation, adhesion and migration (Roberts and Der 2007). This powerful signalling pathway presents negative regulators that limit ERK effects supplying feedback mechanism systems (Lito et al. 2013). In addition to the MAPK pathway, however, extracellular stimuli may affect multiple other pathways. Of particular significance to melanoma is the PI3K/AKT/mTOR pathway, which is upregulated across many cancer types (Fruman and Rommel 2014). Critically, these signalling pathways not only drive signals in a single direction from the extracellular space to the nucleus, but also interconnect at different levels creating a complex network (Fey et al. 2012). Thus, targeted therapies aim to not only interfere with a single signalling axis but also aim to target secondary signalling streams that the tumour may use to circumvent the target of a single node within the network.
Nearly half of all cutaneous melanomas present a gain-of-function (GOF) mutation in the BRAF gene at the codon V600, which constitutively drives MAPK pathway activation via MEK1/2 and ERK1/2 (Davies et al. 2002; Wan et al. 2004). A single activating mutation in the V600 codon of BRAF, which usually leads to substitution of a valine residue to glutamic acid residue (V600E) (Dhomen et al. 2009), renders BRAF constitutively active but is not enough to drive tumour initiation (Pollock et al. 2003). Additional hits in tumour suppressors PTEN (Dankort et al. 2009), TP53 (Goel et al. 2009) or CDKN2A (Sharpless et al. 2003) are required to progress melanomagenesis. Critically, in physiological conditions where BRAF is wild type, RAF proteins dimerise upon activation (Wan et al. 2004; Luo et al. 1996; Farrar et al. 1996). By contrast, V600EBRAF is able to dimerise with CRAF (Wan et al. 2004), a RAF family protein, and function as a monomer vulnerable to inhibition (Poulikakos et al. 2011).
The second most common mutation is a GOF at codons G12/13 (approximately 5% of melanomas) or Q61 (17% of melanomas) of NRAS (Sekiya et al. 1984; Albino et al. 1989; van't Veer et al. 1989). BRAF and NRAS mutations occur in a mutually exclusive pattern in pre-treatment samples (Cancer Genome Atlas Network 2015). NRAS presents GTPase activity and when wild type, following the phosphorylation of RTKs, is activated by a guanine nucleotide exchange factor (GEF), which permits the exchange of GDP for GTP (Young et al. 2009). When RAS is mutated, it remains in GTP-bound state (Katz and McCormick 1997). RAS signals downstream by a variety of pathways, including both the MAPK and PI3K/Akt/mTOR pathways, and in human melanoma and mouse models of NRAS melanoma, like in BRAF, there is synergy to drive tumour progression when RAS alterations are combined with additional genetic hits in CDKN2A, TP53 and PTEN (Feng et al. 2013; Conde-Perez and Larue 2014). Critically, there are currently no successful drugs in clinic targeting oncogenic RAS.
In the tumours that are wild type for both BRAF and NRAS mutations, the more frequent identified drivers are NF1 loss-of-function (LOF) mutations, as well as alterations in KIT, TERT and CCND1 (Cancer Genome Atlas Network 2015; Krauthammer et al. 2015; Shain et al. 2015; Hodis et al. 2012).
4 RAF Inhibition in Melanoma
The discovery of the BRAF oncogene and MAPK pathway signalling led to the development of targeted BRAF inhibition therapy (vemurafenib (Chapman et al. 2011; Sosman et al. 2012; McArthur et al. 2014) and dabrafenib (Hauschild et al. 2012)) and targeted MEK kinase inhibition (trametinib (Flaherty et al. 2012)), although MEK inhibitor trials have shown less impressive responses than BRAF inhibitors. More recently, combination therapies have improved initial monotherapy results (Long et al. 2014; Ascierto et al. 2016). These new drugs achieve improved progression-free and overall survival, and about 90% of patients show some improvement and tumour regression, with up to approximately 50% achieving partial or complete responses. Although these responses are encouraging, the response to these drugs is limited, with only a small subset of patients developing long-term, durable responses. Unfortunately, approximately 50% of patients that present an initial improvement generally relapse within 7 months of treatment (acquired resistance) (Chapman et al. 2011; Hauschild et al. 2012). Additionally, there are about 10% of patients who do not respond to the targeted inhibition at all, and these are termed intrinsic or primary resistant to targeted therapy. Importantly, BRAF inhibitors lead to characteristic side effects, including photosensitivity, which can limit treatment, and the rapid development of cutaneous squamous cell carcinoma (cuSCC) (Su et al. 2012). Keratinocytic secondary neoplasia are thought to arise due to the paradoxical activation of the MAPK pathway in keratinocytes that are wild type for BRAF but present upstream RAS activation in chronically damaged skin (Su et al. 2012). In this cellular context, the BRAF inhibitor leads to increased downstream ERK signalling in BRAF wild type keratinocytes that give rise to cuSCC (Heidorn et al. 2010). Thus, combining BRAF and ERK inhibitors was predicted to decrease this side effect, and indeed, this combination treatment not only was demonstrably linked to improved progression free and overall survival compared to BRAF inhibitor in monotherapy (Larkin et al. 2014; Robert et al. 2015) but also decreased development of cuSCC. However, as in BRAF inhibitor monotherapy, resistance develops in most melanoma patients, and great efforts have been invested to understand how and when tumours resist or stop responding.
5 Tumour Cell Intrinsic or Autonomous Resistance to RAF Inhibition in Melanoma due to Mutational or Genetic Events
Resistance to BRAF inhibitors is predominantly linked to reactivation of the MAPK pathway (Lito et al. 2013; Nazarian et al. 2010; Maertens et al. 2013; Whittaker et al. 2013). As monomeric V600EBRAF is the relevant target of RAF inhibitors, all changes that increase the proportion of RAF dimerisation likely decrease sensitivity to BRAF inhibitors (Poulikakos et al. 2011). Following the rationale that increasing RAS signalling to drive RAF heterodimer formation should decrease therapy response, we find that upstream, acquired GOF mutations in RAS (Nazarian et al. 2010), LOF mutations in NF1 (Maertens et al. 2013) and several GOF mutations in RTKs (Whittaker et al. 2013) have all shown to drive tumour resistance. Apart from these mechanisms that reactivate RAS, there are structural changes in oncogenic BRAF due to aberrant splicing that can lead to resistance (Poulikakos et al. 2011; Solit and Rosen 2011). For example, p61-V600EBRAF splice variants retain an active kinase activity but are unable to bind RAS. They dimerise regardless of RAS status and drive constitutive signalling to ERK, uncoupled from upstream regulation (Poulikakos et al. 2011). More recently, a screen for resistance mechanisms using patient derived xenografts (PDX) found that duplications of the BRAF kinase domain led to resistance in approximately 10% of PDX (Kemper et al. 2016). This was found to hold true in a validation cohort of resistant patient samples. Additional drivers of resistance are the increased expression of CRAF or copy number increase of BRAF, which possibly also drive dimer formation and increase signalling throughput.
There are also mechanisms of resistance that arise downstream of the inhibition site BRAF within the MAPK cascade, that bypass the effect of the drug. Importantly, the specific mutations in MEK1 (MAP2K1) coding amino acid changes P124L and Q56P are able to decrease response to BRAF inhibition (Wagle et al. 2011), but not all MEK1 somatic mutations lead to equivalent reactivation of the pathway downstream of MEK1 (Shi et al. 2012). Additionally, the increased expression of the MAPK kinase kinase COT (MAP3K8) is thought to lead to direct activation of MEK1, circumventing RAF inhibition (Johannessen et al. 2010). More recently, the role of ERK reactivation driving resistance has been further highlighted by massively parallel sequencing efforts revealing mutations in genes encoding for proteins in the cohesion complex that participate in the organisation of chromatids, STAG2 and STAG3 (Shen et al. 2016). Mutant proteins are shown to drive the reactivation of ERK signalling to drive resistance by inhibiting the expression of the phosphatase DUSP6, an inhibitor and regulator of ERK activity in melanoma. Other implicated negative regulators include the LOF of DUSP4 (Shen et al. 2016).
Further molecular alternatives driving resistance are the activation of parallel signalling, such as the activation of PI3K/AKT/mTOR pathway by deletion or inactivating mutations of the negative pathway regulator and tumour suppressor PTEN (Xing et al. 2012), and the inactivation of the tumour suppressor RB1 (Xing et al. 2012) to decrease the requirement for BRAF/MEK signalling. The discovery of these contributing signalling pathways provides new rationales of second-line therapies, as demonstrated by the success of combinations of MAPK and PI3K/AKT/mTOR pathway inhibitors to overcome acquired resistance to monotherapy targeting BRAF alone (Greger et al. 2012).
To add further complexity, patients who present disease progression following initial therapeutic response to BRAF inhibition are more likely to progress presenting metastasis at previously uninvolved sites, and there is a selection for more aggressive clones (Paraiso et al. 2015). Importantly, tumour heterogeneity and heterogeneous mechanisms of resistance are present in tumours and at different metastatic sites simultaneously (Shi et al. 2014). Recent efforts to dissect the degree of heterogeneity and its clinical implications have led to transcriptional studies targeting thousands of single tumour cells, and show that at the cellular level in all tumours, there are distinct transcriptional patterns within each tumour that display varying degrees of predicted responsiveness to BRAF inhibition (Tirosh et al. 2016). Thus, there are subclones of cells, in varying proportions within each tumour, expressing molecular programmes that make them less likely to respond to therapy and vulnerable to selection during disease progression (Tirosh et al. 2016).
6 Tumour Cell Intrinsic or Autonomous Resistance to RAF Inhibition in Melanoma due to Non-mutational Events
As described above, additional genetic damage to PTEN or RB1 leads to activation of alternative signalling pathways that will lead to therapeutic failure (Xing et al. 2012). However, we already observe the activation of parallel signalling pathways, leading to progressive “dampening” (adaptation) of response to BRAF inhibitors during therapy in the absence of additional genetic hits. This occurs because there is a progressive switch of cells to rely on other signalling pathways such as PI3K/AKT as a natural consequence of the new pressures exerted by BRAF inhibition (Lito et al. 2012). This rewiring or switch happens because BRAF inhibitors will initially shut down transcription of key targets of proliferation, leading to response, via suppression of ERK function, but will also decrease the expression of negative pathway regulators downstream of ERK such as DUSP6 (negative regulator of ERK activity) and Sprouty (SPRY (Tsavachidou et al. 2004); inhibitor of GRB2, an adaptor protein transducing signals between RTKs and RAS) (Lito et al. 2013; Lito et al. 2012). Thus, inherent tumour cell adaptation to targeted therapy, in the absence of additional genetic events, per se provides an explanation for progressive decline in therapeutic response.
Another way tumour cells co-opt established biological cellular signalling pathways to advance tumour progression in the absence of genetic mutations is by up-regulation or activation of RTKs (Whittaker et al. 2013). Tumour cells express multiple RTKs that integrate extracellular signals to modulate intracellular events feeding multiple cascades that will affect the fundamental MAPK and PI3K/AKT pathways, and influence proliferation and survival. Therefore, up-regulation of RTKs and increases in soluble RTK ligands and growth factors are all described mechanisms to reduce innate drug sensitivity and drive acquired resistance. Critical RTKs that are implicated in reducing response to treatment due to activation or up-regulation include MET, IFG-1R (Villanueva et al. 2010), EGFR and PDGFRbeta (Wilson et al. 2012). In melanoma, the expression of the RTK ligand HGF is a well-known driver of poor response (Straussman et al. 2012).
Both the expression and loss of MITF, the critical melanoma lineage survival factor, are additional mechanisms of resistance to BRAF pathway inhibition. MITF plays a central role in ensuring melanocyte survival, and regulates multiple critical survival and antiapoptotic genes (Levy et al. 2006).
Different MITF expression levels have been linked to distinct melanoma tumour and cell behaviour, where high levels mediate differentiation, moderate levels promote proliferation, and low/absent levels drive a more invasive phenotype (Levy et al. 2006). Therefore, the multifaceted role of MITF as a consequence of varying expression levels, with seemingly opposing effects, is further mirrored during the development of resistance. Specifically, overexpression of MITF reduces the therapeutic effect of BRAF inhibitors (Johannessen et al. 2013; Smith et al. 2016; Haq et al. 2013; Van Allen et al. 2014), MEK inhibitors (Smith et al. 2013) and combination treatments (Shi et al. 2014) via increased cAMP pathway signalling (Johannessen et al. 2013).
However, at the other end of the expression spectrum, loss of MITF is also a common occurrence in acquired resistance (Muller et al. 2014). Critically, in cell clones down-regulating MITF, there is an inversely correlated up-regulation of the RTK AXL that enhances tumour resistance. The expression programme in MITF low/AXL high tumours is implicated in driving the resistant phenotype and has now been found to exist in a continuum in subsets of tumour cells that may otherwise express a predominating “AXL low” expression programme that would predict response. A recent study has found the expression programme observed in “MITF low/AXL high” resistant samples can already be detected at the single cell level in treatment-naïve samples, and these transcriptomic features are subsequently increased in resistant, progressive disease (Muller et al. 2014; Konieczkowski et al. 2014). Additionally, the neighbouring stromal cells also influence the expression programme at the single cell level (Tirosh et al. 2016).
A landmark 2015 study performed transcriptome and methylome analysis of matched melanoma samples taken before treatment and during disease progression. One of the critical findings in this paper is that resistant samples displayed a more homogeneous pattern of expression that was in contrast to heterogeneity shown at the DNA level. The acquired resistance transcriptome correlated with YAP1 enrichment, MET high and LEF1 low expression levels. Moreover, MAPK pathway inhibition with targeted therapies led to direct methylation changes that were therapy time-dependent, affecting key regulatory genes and the transcriptome across the resistant samples. One of the other striking findings in the paper is that in resistant samples, there is a predominance of NF-Kβ inflammation that correlates with monocyte expression and M2 macrophages (tumour associated macrophages), suggesting MAPK pathway inhibition affects the inflammatory context of the tumour. Indeed, as a consequence of the M2 switch in macrophage phenotype, the researchers observed that as MAPK pathway inhibitor resistance arose, there was a parallel increase in CD8+ T cell deficiency, exhaustion of the cell population and down-regulation of the antigen presentation molecules in about half the resistant samples (Hugo et al. 2015). Thus, this shows that the study of resistance must extend beyond DNA analysis to incorporate non-genomic approaches with a focus on the consequences on the tumour as well as on the immune tumour ecosystem. Unravelling these changes impacts the therapeutic options of patients, as this work shows that when patients progress on targeted therapies they likely respond less to immunotherapy. Providing further evidence of how a deep understanding of the immune context underpins therapeutic choice, there are data to support the up-regulation of PD-L1 and the tumour-infiltrating immune populations in metastatic melanoma before MAPK inhibition are associated with progression and survival (Hugo et al. 2017; Massi et al. 2015).
7 Tumour Cell Extrinsic or Microenvironmental Resistance to RAF Inhibition in Melanoma
Research focus into the mechanisms of drug resistance in melanoma has predominantly centred on properties intrinsic to tumour cells, but disease progression and resistance to targeted therapies is no longer considered an exclusive function of genomic and non-genomic modifications of tumour cells. The importance of the tumour microenvironment in supporting resistance to MAPK inhibition is slowly unravelling, and a more complex, comprehensive interpretation must incorporate knowledge of cross talk between discrete cellular compartments including the tumour and supporting stroma (Tape et al. 2016). As we have already alluded to, recent studies delineate a clear contribution of macrophages and fibroblast-derived factors that are well known to confer resistance to MAPK pathway inhibitors.
Fibroblasts are well-known facilitators of melanoma progression, and there is a bidirectional communication through direct cell-to-cell contact and via the secretion of soluble factors to promote melanoma invasion, survival and growth (Li et al. 2003). In melanoma, an extrinsic, non-cell autonomous signal leading to HGF secretion by the stromal cells may lead to resistance (Straussman et al. 2012). HGF can bind to RTKs that will increase intracellular signalling to drive up-regulation of RAS, and ultimately, reactivation of MAPK pathway (Straussman et al. 2012). Furthermore, HGF is also known to contribute to resistance in human cell lines treated with a BRAF inhibitor by down-regulating the pro-apoptotic response. A more recent paper investigated how genetically unharmed stroma evolves under treatment with targeted therapy. Importantly, they demonstrated that in areas of high stromal density, fibroblasts present a hyperactivation of MAPK pathway that elicits a qualitative change in the tumour matrix via integrin β1 and FAK to induce ERK, providing an early subset of melanoma cells with the capacity to rapidly tolerate treatment (Hirata et al. 2015). Moreover, the effect of fibroblasts may vary in patients who are elderly, as melanoma cells in contact with aged fibroblasts are more invasive (Kaur et al. 2016). A recent study has shown that aged fibroblasts increase the secretion of sFRP2, a β-catenin inhibitor that decreases MITF expression, leading to downregulation of the redox regulator APE1, thus rendering melanoma cells more sensitive to oxidative stress and secondarily driving resistance to BRAF inhibition (Kaur et al. 2016). Together, all these studies demonstrate that melanoma cells and fibroblasts are both under pressure to adapt to targeted inhibitors. Thus, fibroblasts change their matrix providing melanoma cells with a new microenvironment that will enable adjacent tumour cells to evade therapy at a very early stage.
Additional stromal cells that play a demonstrated role in the development of resistance are macrophages (Ruffell and Coussens 2015). Tumours from patients treated with MAPK pathway inhibitors present an increased density of tumour-associated macrophages. These tumour macrophages secrete the melanoma growth factor TNFα that drives, in an NF-kβ-dependent manner, the expression of MITF, leading to resistance (Smith et al. 2014). Moreover, studies show that combination of MAPK and NF-kβ inhibitors delay the appearance of resistance (Smith et al. 2014). TNFα is known to block apoptosis in cells where BRAF is inhibited, and additionally contributes to melanoma invasion and vascularisation of tumours (Gray-Schopfer et al. 2007). As TNFα and NF-kβ play context-specific roles within cancer progression, it will be critical to further investigate whether the signalling pathways described to promote therapy failure secondary to macrophage infiltration hold true across different cancer types and other targeted therapies. These studies, together with the recent discovery that MAPK pathway inhibition leads to CD8+ T cell depletion, highlight how the pressure exerted by novel MAPK inhibitor therapies not only impact tumour evolution by genomic and non-genomic events, but also shape the behaviour of the supporting connective tissue and immune cell populations and function.
8 Immunotherapies in Melanoma
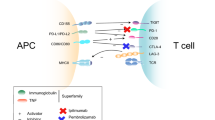
Another landmark change in melanoma therapy has been the development of antibodies to elicit an antitumor immunologic response (Brahmer et al. 2012; Hodi et al. 2010; Topalian et al. 2012; Kaufman et al. 2013; Mellman et al. 2011; Topalian et al. 2014; Wolchok et al. 2013). These novel therapies target the immune checkpoints that exist in physiological conditions to counterbalance immune activation. In cancer, these critical signalling barriers inhibit the activation of antitumoural immune responses. The inhibition of these checkpoints allows the immune system to target cancer cells and leads to long-term disease control. In melanoma, there is improved survival by targeting the cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) checkpoint molecule, the inhibitory T-cell receptor programmed death 1 (PD-1) receptor and the PD-1 ligand PD-L1. The mechanisms of action have been extensively reviewed. Other novel immunotherapies include adoptive T cell therapy, which is based on the isolation and ex vivo expansion of T cells that are tumour-specific (Restifo et al. 2012). This therapy allows the vast expansion of T cells, which are then infused into patients to target tumour cells. However, there is great variability of response to immunotherapies across different cancer types, and even within cancer types where immunotherapy is successful, such as melanoma, lung cancer and renal cell cancer, there is a proportion of patients who never respond. Similarly to targeted therapy patients, these are termed intrinsically resistant patients. There is also a subset of patients who initially respond but later progress, indicating the development of acquired resistance to immunotherapy. Thus, there are tumours that cannot be detected by the immune system (intrinsically resistant), and tumours where the cells adapt to immunotherapy and progressively outgrow the inhibition (adaptive immune resistance).
9 Intrinsic and Adaptive Resistance to Immunotherapies in Melanoma
Intrinsic resistance arises in cells that present genetic or non-genetic changes that afford the tumour natural resistance, as seen, for example, in tumours that express few molecular changes that are recognised as foreign by the immune system (Snyder et al. 2014; McGranahan et al. 2016). This affords one explanation as to why tumours with fewer mutation loads are less likely to respond to immunotherapy. During tumour evolution, there is a tendency for tumours to lose a proportion of their non-silent mutations, which can potentially lead to a lower ratio of antigenic epitopes, leading to immunoadaptation of tumours (Rooney et al. 2015). One of the necessary elements linked to response is the presence of pre-existing T cells within the tumour. The absence of effector T cells within a melanoma has been linked in humans and mice to melanoma-cell-intrinsic oncogenic activation of the WNT/β-catenin pathway (Spranger et al. 2015; Spranger and Gajewski 2016). Importantly, analysis of pre-anti-PD-1 treatment and post-treatment samples has shown a greater proportion of mutations in the DNA repair gene BRCA2 in responding patients (Hugo et al. 2017). By contrast, the samples that are intrinsically resistant to anti-PD-1 therapy have upregulation of genes involved in a non-melanocytic, more mesenchymal behaviour (AXL, ROR2, WNT5A, LOXL2, TWIST2, TAGLN, FAP), as well as genes involved in angiogenesis and wound healing (Hugo et al. 2017). Curiously, this expression profile significantly overlaps with the expression profile observed in samples that have developed resistance to MAPK pathway inhibition (Hugo et al. 2017), implying convergence of resistance-expression programmes.
Cooperating with the loss of antigenicity and the intrinsic characteristics in the tumour that render them more or less vulnerable to immunotherapy are additional mechanisms arising during immune response that directly inhibit tumour-targeting T cells. For example, resistances can be acquired by new resistance-driving mutations in genes involved in interferon-receptor signalling and in antigen presentation (Zaretsky et al. 2016). Additionally, during the initial phase of the interaction between a tumour cell and an antigen-specific T cell, the T cell produces interferon-γ (Ribas 2015; Peng et al. 2012). Interferon-γ signalling has a dual anti-tumour and immune inhibitor role in this interaction (Shankaran et al. 2001). It first serves as a mediator and amplifier of the immune effect, exerting a chemoattraction role to recruit further leukocytes, macrophages and natural killer T cells, but also has a second effect dampening the immune response to cancer cells by expressing factors aimed to reduce the immune anti-tumour effect (Rooney et al. 2015; Bald et al. 2014), such as indolamin 2.2 dioxygenase (IDO) (Peng et al. 2016). IDO is a critical enzyme that when expressed, interferes with appropriate T cell function. Most critically, following an interferon-γ stimulus, T cells express the ligand PD-L1 that binds to the PD1 receptor leading to inactivation of T cells (Spranger et al. 2013; Tumeh et al. 2014; Pardoll 2012). Other interferon-γ-dependent checkpoints that have also been described to mediate inhibitory immune loops are most prominently the carcinoembryonic antigen cell adhesion molecule-1 (CEACAM1) that forms heterodimers with TIM-3, an activation-induced inhibition molecule involved in immune tolerance and T-cell exhaustion, to inhibit T cell function (Huang et al. 2015). This interaction is an attractive target for additional immune checkpoint blockade in cancers expressing CEACAM1 and TIM-3 (Huang et al. 2015).
There are additional interferon-independent mechanisms of adaptation to immunotherapy. For example, in some tumours, mutations in cancer cells affecting major signalling pathways per se can also lead to expression of PD-L1 in tumours (Parsa et al. 2007; Mittendorf et al. 2014; Marzec et al. 2008; Atefi et al. 2014; Akbay et al. 2013; Shin et al. 2016). There is also a CD8+ T cell-dependent accumulation of regulatory T cells expressing FOXP3+ that exert an inhibitory influence over the immune response, which is mediated by chemokine release (Spranger et al. 2013). Regulatory T cells are immunosuppressive, downregulate the proliferation and induction of effector T cells and are critical to control autoimmunity or excess immunity. Additionally, tumour specific T cells mounting an immune response during the killing of cancer cells produce the major modulating inflammatory cytokine TNFα, which leads to a decrease in the expression of melanoma-specific genes, involved in melanocyte lineage differentiation, and an increase in the expression of genes that signal dedifferentiation and a biological profile more in keeping with neural crest-derived cells or immature melanocytes (Ribas and Tumeh 2012). In adoptive T cell transfer therapy, melanoma evolves to a less differentiated state that mediates resistance due to TNFα secretion (Landsberg et al. 2012). Thus, the loss of melanoma-specific antigens provides another mechanism of immunoevasion. Furthermore, a seminal recent study shows progressive selection of non-immunogenic clones, a process termed immunoediting, occurs during cancer progression via a T cell-dependent immunoselection process (Matsushita et al. 2012).
Taken together, research shows the fine regulatory relationships aimed to deliver accurate immune responses to infection and limit excess cytotoxicity in homeostasis are co-opted to the tumours’ advantage in cancer.
10 Conclusions and Future Avenues
There is a rapid changing landscape of therapies for advanced melanoma patients. Current efforts aim to enhance the efficacy of existing targeted and immunotherapies, by understanding the biology driving response and resistance to guide therapeutic care. One of the challenges is identifying the first line therapies or combinations of therapies that will lead to more prolonged clinical responses, taking into consideration the genetic and immunological differences in each patient. Moreover, in this rapidly changing landscape of novel therapies, new strategies to overcome resistances are currently underway. For example, promising new drugs block the reactivation of MAPK pathway in both BRAF and NRAS tumours, without driving the paradoxical activation of the MAPK pathway (Girotti et al. 2015). Another novel approach to overcome resistance includes the disruption of mitochondrial biogenesis using the small-molecule HSP90 inhibitor gamitrinib (Zhang et al. 2016). New drug discovery efforts have also found encouraging results using compounds triggering ER Stress in MAPK inhibitor resistant cells (Cerezo et al. 2016). For novel immunotherapies, understanding the activation levels of significant checkpoint molecules and the immunosuppressive context within each tumour sample will aid physicians select appropriate therapies or combinations of therapies to improve survival.
References
Akbay EA et al (2013) Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 3:1355–1363. doi:10.1158/2159-8290.CD-13-0310
Albino AP et al (1989) Analysis of Ras oncogenes in malignant melanoma and precursor lesions: correlation of point mutations with differentiation phenotype. Oncogene 4:1363–1374
Ascierto PA et al (2016) Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 17:1248–1260. doi:10.1016/S1470-2045(16)30122-X
Atefi M et al (2014) Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 20:3446–3457. doi:10.1158/1078-0432.CCR-13-2797
Balch CM et al (2009) Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 27:6199–6206. doi:10.1200/JCO.2009.23.4799
Bald T et al (2014) Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov 4:674–687. doi:10.1158/2159-8290.CD-13-0458
Brahmer JR et al (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465. doi:10.1056/NEJMoa1200694
Cancer Genome Atlas Network (2015) Genomic classification of cutaneous melanoma. Cell 161:1681–1696. doi:10.1016/j.cell.2015.05.044
Cerezo M et al (2016) Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell 30:183. doi:10.1016/j.ccell.2016.06.007
Chang YM et al (2009) Sun exposure and melanoma risk at different latitudes: a pooled analysis of 5700 cases and 7216 controls. Int J Epidemiol 38:814–830. doi:10.1093/ije/dyp166
Chapman PB et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516. doi:10.1056/NEJMoa1103782
Conde-Perez A, Larue L (2014) Human relevance of NRAS/BRAF mouse melanoma models. Eur J Cell Biol 93:82–86. doi:10.1016/j.ejcb.2013.10.010
Curtin JA et al (2005) Distinct sets of genetic alterations in melanoma. N Engl J Med 353:2135–2147. doi:10.1056/NEJMoa050092
Dankort D et al (2009) Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet 41:544–552. doi:10.1038/ng.356
Davies H et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. doi:10.1038/nature00766
Dhomen N et al (2009) Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 15:294–303. doi:10.1016/j.ccr.2009.02.022
Farrar MA, Alberol-Ila J, Perlmutter RM (1996) Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 383:178–181. doi:10.1038/383178a0
Feng Y et al (2013) Inhibition of melanoma development in the Nras((Q61K)) ::Ink4a(−/−) mouse model by the small molecule BI-69A11. Pigment Cell Melanoma Res 26:136–142. doi:10.1111/pcmr.12033
Ferlay J et al (2013) Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer 49:1374–1403. doi:10.1016/j.ejca.2012.12.027
Fey D, Croucher DR, Kolch W, Kholodenko BN (2012) Crosstalk and signaling switches in mitogen-activated protein kinase cascades. Front Physiol 3:355. doi:10.3389/fphys.2012.00355
Flaherty KT et al (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 367:107–114. doi:10.1056/NEJMoa1203421
Fruman DA, Rommel C (2014) PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 13:140–156. doi:10.1038/nrd4204
Gilchrest BA, Eller MS, Geller AC, Yaar M (1999) The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med 340:1341–1348. doi:10.1056/NEJM199904293401707
Girotti MR et al (2015) Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 27:85–96. doi:10.1016/j.ccell.2014.11.006
Goel VK et al (2009) Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 28:2289–2298. doi:10.1038/onc.2009.95
Gray-Schopfer VC, Karasarides M, Hayward R, Marais R (2007) Tumor necrosis factor-alpha blocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer Res 67:122–129. doi:10.1158/0008-5472.CAN-06-1880
Greger JG et al (2012) Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther 11:909–920. doi:10.1158/1535-7163.MCT-11-0989
Haq R et al (2013) BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A 110:4321–4326. doi:10.1073/pnas.1205575110
Hauschild A et al (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380:358–365. doi:10.1016/S0140-6736(12)60868-X
Heidorn SJ et al (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140:209–221. doi:10.1016/j.cell.2009.12.040
Hirata E et al (2015) Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 27:574–588. doi:10.1016/j.ccell.2015.03.008
Hodi FS et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723. doi:10.1056/NEJMoa1003466
Hodis E et al (2012) A landscape of driver mutations in melanoma. Cell 150:251–263. doi:10.1016/j.cell.2012.06.024
Holderfield M, Deuker MM, McCormick F, McMahon M (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14:455–467. doi:10.1038/nrc3760
Huang YH et al (2015) CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517:386–390. doi:10.1038/nature13848
Hugo W et al (2015) Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 162:1271–1285. doi:10.1016/j.cell.2015.07.061
Hugo W et al (2017) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 168:542. doi:10.1016/j.cell.2017.01.010
Johannessen CM et al (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468:968–972. doi:10.1038/nature09627
Johannessen CM et al (2013) A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504:138–142. doi:10.1038/nature12688
Katz ME, McCormick F (1997) Signal transduction from multiple Ras effectors. Curr Opin Genet Dev 7:75–79
Kaufman HL et al (2013) The Society for Immunotherapy of Cancer consensus statement on tumour immunotherapy for the treatment of cutaneous melanoma. Nat Rev Clin Oncol 10:588–598. doi:10.1038/nrclinonc.2013.153
Kaur A et al (2016) sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 532:250–254. doi:10.1038/nature17392
Kemper K et al (2016) BRAF(V600E) kinase domain duplication identified in therapy-refractory melanoma patient-derived xenografts. Cell Rep 16:263–277. doi:10.1016/j.celrep.2016.05.064
Konieczkowski DJ et al (2014) A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 4:816–827. doi:10.1158/2159-8290.CD-13-0424
Krauthammer M et al (2015) Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet 47:996–1002. doi:10.1038/ng.3361
Landsberg J et al (2012) Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 490:412–416. doi:10.1038/nature11538
Larkin J et al (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 371:1867–1876. doi:10.1056/NEJMoa1408868
Levy C, Khaled M, Fisher DE (2006) MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med 12:406–414. doi:10.1016/j.molmed.2006.07.008
Li G et al (2003) Function and regulation of melanoma-stromal fibroblast interactions: when seeds meet soil. Oncogene 22:3162–3171. doi:10.1038/sj.onc.1206455
Lito P et al (2012) Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22:668–682. doi:10.1016/j.ccr.2012.10.009
Lito P, Rosen N, Solit DB (2013) Tumor adaptation and resistance to RAF inhibitors. Nat Med 19:1401–1409. doi:10.1038/nm.3392
Long GV et al (2014) Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N Engl J Med 371:1877–1888. doi:10.1056/NEJMoa1406037
Luo Z et al (1996) Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 383:181–185. doi:10.1038/383181a0
Maertens O et al (2013) Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov 3:338–349. doi:10.1158/2159-8290.CD-12-0313
Marais R, Light Y, Paterson HF, Mason CS, Marshall CJ (1997) Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J Biol Chem 272:4378–4383
Marzec M et al (2008) Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A 105:20852–20857. doi:10.1073/pnas.0810958105
Massi D et al (2015) The status of PD-L1 and tumor-infiltrating immune cells predict resistance and poor prognosis in BRAFi-treated melanoma patients harboring mutant BRAFV600. Ann Oncol 26:1980–1987. doi:10.1093/annonc/mdv255
Matsushita H et al (2012) Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482:400–404. doi:10.1038/nature10755
McArthur GA et al (2014) Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol 15:323–332. doi:10.1016/S1470-2045(14)70012-9
McGranahan N et al (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351:1463–1469. doi:10.1126/science.aaf1490
Mellman I, Coukos G, Dranoff G (2011) Cancer immunotherapy comes of age. Nature 480:480–489. doi:10.1038/nature10673
Mittendorf EA et al (2014) PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res 2:361–370. doi:10.1158/2326-6066.CIR-13-0127
Muller J et al (2014) Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 5:5712. doi:10.1038/ncomms6712
Nazarian R et al (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468:973–977. doi:10.1038/nature09626
NICE (2015) Melanoma: assessment and management. In: National Institute for Health and Care Excellence: clinical guidelines
Paraiso KH et al (2015) Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov 5:264–273. doi:10.1158/2159-8290.CD-14-0293
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264. doi:10.1038/nrc3239
Parsa AT et al (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13:84–88. doi:10.1038/nm1517
Peng W et al (2012) PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res 72:5209–5218. doi:10.1158/0008-5472.CAN-12-1187
Peng W et al (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6:202–216. doi:10.1158/2159-8290.CD-15-0283
Pollock PM et al (2003) High frequency of BRAF mutations in nevi. Nat Genet 33:19–20. doi:10.1038/ng1054
Poulikakos PI et al (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480:387–390. doi:10.1038/nature10662
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281. doi:10.1038/nri3191
Ribas A (2015) Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov 5:915–919. doi:10.1158/2159-8290.CD-15-0563
Ribas A, Tumeh PC (2012) Cancer therapy: tumours switch to resist. Nature 490:347–348. doi:10.1038/nature11489
Robert C et al (2015) Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 372:30–39. doi:10.1056/NEJMoa1412690
Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310. doi:10.1038/sj.onc.1210422
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160:48–61. doi:10.1016/j.cell.2014.12.033
Ruffell B, Coussens LM (2015) Macrophages and therapeutic resistance in cancer. Cancer Cell 27:462–472. doi:10.1016/j.ccell.2015.02.015
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Sekiya T et al (1984) Molecular cloning and the total nucleotide sequence of the human c-Ha-ras-1 gene activated in a melanoma from a Japanese patient. Proc Natl Acad Sci U S A 81:4771–4775
Shain AH, Bastian BC (2016) From melanocytes to melanomas. Nat Rev Cancer 16:345–358. doi:10.1038/nrc.2016.37
Shain AH et al (2015) Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet 47:1194–1199. doi:10.1038/ng.3382
Shankaran V et al (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410:1107–1111. doi:10.1038/35074122
Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L (2003) Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene 22:5055–5059. doi:10.1038/sj.onc.1206809
Shen CH et al (2016) Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat Med 22:1056–1061. doi:10.1038/nm.4155
Shi H et al (2012) Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov 2:414–424. doi:10.1158/2159-8290.CD-12-0022
Shi H et al (2014) Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4:80–93. doi:10.1158/2159-8290.CD-13-0642
Shin DS et al (2016) Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov 7:188–201. doi:10.1158/2159-8290.CD-16-1223
Smith MP et al (2013) Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J Natl Cancer Inst 105:33–46. doi:10.1093/jnci/djs471
Smith MP et al (2014) The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov 4:1214–1229. doi:10.1158/2159-8290.CD-13-1007
Smith MP et al (2016) Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 29:270–284. doi:10.1016/j.ccell.2016.02.003
Snyder A et al (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189–2199. doi:10.1056/NEJMoa1406498
Solit DB, Rosen N (2011) Resistance to BRAF inhibition in melanomas. N Engl J Med 364:772–774. doi:10.1056/NEJMcibr1013704
Sosman JA et al (2012) Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 366:707–714. doi:10.1056/NEJMoa1112302
Spranger S, Gajewski TF (2016) Tumor-intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 5:e1086862. doi:10.1080/2162402X.2015.1086862
Spranger S et al (2013) Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 5:200ra116. doi:10.1126/scitranslmed.3006504
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523:231–235. doi:10.1038/nature14404
Stephen AG, Esposito D, Bagni RK, McCormick F (2014) Dragging Ras back in the ring. Cancer Cell 25:272–281. doi:10.1016/j.ccr.2014.02.017
Straussman R et al (2012) Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487:500–504. doi:10.1038/nature11183
Su F et al (2012) RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366:207–215. doi:10.1056/NEJMoa1105358
Tape CJ et al (2016) Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell 165:910–920. doi:10.1016/j.cell.2016.03.029
Tirosh I et al (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352:189–196. doi:10.1126/science.aad0501
Topalian SL et al (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454. doi:10.1056/NEJMoa1200690
Topalian SL et al (2014) Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 32:1020–1030. doi:10.1200/JCO.2013.53.0105
Tsavachidou D et al (2004) SPRY2 is an inhibitor of the ras/extracellular signal-regulated kinase pathway in melanocytes and melanoma cells with wild-type BRAF but not with the V599E mutant. Cancer Res 64:5556–5559. doi:10.1158/0008-5472.CAN-04-1669
Tumeh PC et al (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571. doi:10.1038/nature13954
Van Allen EM et al (2014) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4:94–109. doi:10.1158/2159-8290.CD-13-0617
van't Veer LJ et al (1989) N-ras mutations in human cutaneous melanoma from sun-exposed body sites. Mol Cell Biol 9:3114–3116
Villanueva J et al (2010) Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18:683–695. doi:10.1016/j.ccr.2010.11.023
Wagle N et al (2011) Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 29:3085–3096. doi:10.1200/JCO.2010.33.2312
Wan PT et al (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867
Whiteman D, Green A (1999) The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med 341:766–767
Whiteman DC et al (2003) Melanocytic nevi, solar keratoses, and divergent pathways to cutaneous melanoma. J Natl Cancer Inst 95:806–812
Whittaker SR et al (2013) A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov 3:350–362. doi:10.1158/2159-8290.CD-12-0470
Wilson TR et al (2012) Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487:505–509. doi:10.1038/nature11249
Wolchok JD et al (2013) Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 369:122–133. doi:10.1056/NEJMoa1302369
Xing F et al (2012) Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene 31:446–457. doi:10.1038/onc.2011.250
Young A et al (2009) Ras signaling and therapies. Adv Cancer Res 102:1–17. doi:10.1016/S0065-230X(09)02001-6
Zaretsky JM et al (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819–829. doi:10.1056/NEJMoa1604958
Zhang G et al (2016) Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest 126:1834–1856. doi:10.1172/JCI82661
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Acknowledgements
Acknowledgements
MW is funded by Cancer Research UK and AV is funded by Wellcome Trust (110078/Z/15/Z).
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Winder, M., Virós, A. (2017). Mechanisms of Drug Resistance in Melanoma. In: Mandalà, M., Romano, E. (eds) Mechanisms of Drug Resistance in Cancer Therapy. Handbook of Experimental Pharmacology, vol 249. Springer, Cham. https://doi.org/10.1007/164_2017_17
Download citation
DOI: https://doi.org/10.1007/164_2017_17
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-10506-8
Online ISBN: 978-3-030-10507-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)