
Abstract
The cyclic nucleotides cAMP and cGMP are well-characterized second messenger molecules regulating many important intracellular processes, such as differentiation, proliferation, and apoptosis. The latter is a highly regulated process of programmed cell death wherein several regulatory proteins, like those belonging to the Bcl-2 family, are involved. The initiation of apoptosis is regulated by three different pathways: the intrinsic or mitochondrial, the extrinsic, and the ER stress pathway. Recently, it has been published that the pyrimidine cyclic nucleotides cCMP and cUMP also function as second messenger molecules, and additionally have an effect on apoptosis signaling pathways. cCMP induced PKA-independent apoptosis via the intrinsic and ER-stress pathway in S49 mouse lymphoma cells, and cCMP as well as cUMP induced apoptosis in human HEL cells via the intrinsic pathway. However, in human K-562 cells, which are known to be multidrug-resistant, cCMP and cUMP had no effect. Summarized in this chapter are the initiation of apoptosis by cCMP and cUMP regarding the various apoptotic pathways, the enzymes involved in apoptosis, as well as the most relevant methods for the detection and examination of apoptosis and the corresponding signaling pathways.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Apoptosis is a cellular mechanism of programmed cell death that is characterized by membrane blebbing, condensation of the cytoplasm and nucleus, DNA fragmentation, and cell shrinkage. DNA fragmentation results in various DNA strand breaks and can be analyzed with the TUNEL (TdT-mediated dUTP-biotin nick end labeling) assay. This assay was formerly considered as the gold standard for the identification of apoptotic cells (Darzynkiewicz et al. 2008). Understanding the cellular apoptosis mechanisms can provide the opportunity to treat diseases or to develop drugs by targeting apoptotic genes and pathways.
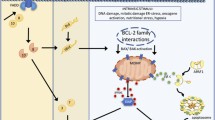
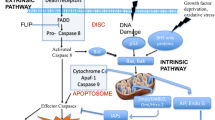
The central enzymes involved in apoptosis are caspases (Hengartner 2000). Generally, three different pathways of apoptosis induction can be distinguished (Fig. 1):
-
(1)
The extrinsic pathway is activated by death ligands like TNFα or FasL (Fas ligand) which bind to specific receptors. Afterwards, a so-called death-inducing signaling complex (DISC) is formed, the initiator caspase 8 is activated and triggers apoptosis by cleaving other downstream executioner caspases like caspase 3 and caspase 7, which can be analyzed by western blotting.
-
(2)
The intrinsic mitochondrial pathway is initiated within the cell by irreparable genetic damage, severe oxidative stress, hypoxia, or deprivation of survival factors increasing the mitochondrial permeability caused by the loss of mitochondrial membrane potential. This leads to the release of pro-apoptotic factors, such as cytochrome c, from the mitochondrium into the cytoplasm. The release of these pro-apoptotic factors can be analyzed by flow cytometry after intracellular staining with an appropriate antibody, e.g., cytochrome c-FITC antibody. Cytochrome c triggers the formation of a complex known as apoptosome which is composed of cytochrome c, APAF-1 (apoptotic protease activating factor 1), and caspase 9. The apoptosome is able to activate caspase 3. Several proteins of the Bcl-2 family are important for the regulation of this pathway. These proteins have a pro- or anti-apoptotic function by promoting or blocking the mitochondrial release of pro-apoptotic factors (Martinou and Youle 2011). RNA or protein expression of the Bcl-2 family members can be analyzed by real-time PCR or western blotting, respectively.
-
(3)
The third pathway is the intrinsic endoplasmatic reticulum (ER) pathway which leads to activation of caspase 12. This pathway is triggered by ER stress, e.g., by misfolded proteins (Szegezdi et al. 2003). Stimulation with brefeldin A (BFA), an inhibitor of intracellular protein transport, also induces ER stress. Thus, incubation of cells with BFA leads to a blockade of protein transport from the ER to the Golgi complex and an accumulation of proteins in the ER (Kaufman 1999). A potential cross-talk between the ER-induced apoptosis and the intrinsic pathway, controlled by Bcl-2 proteins, is under discussion (Häcki et al. 2000), and a function of several Bcl-2 proteins in ER stress has been reported as well (Siddiqui et al. 2015).

Apoptotic pathways: extrinsic, intrinsic and ER stress-activated pathways
Besides apoptosis, other cell death mechanisms exist, namely autophagy and necrosis. Autophagy is a catabolic degradative process and is often associated with anti-proliferative mechanisms and physiological processes like differentiation, development, and cancer (Reggiori and Klionsky 2002). During autophagy, cytoplasmatic components are delivered to lysosomes and eliminated. Important enzymes involved in this process are autophagy-related genes (ATG) and LC3 (light chain 3) A/B proteins which therefore function as autophagy marker proteins. On the contrary, cells undergoing programmed necrosis show an increase in cell volume, swelling of organelles, and disruption of the cell membrane. Intracellular components are released and an inflammatory response is thereby often induced (Ouyang et al. 2012).
2 Cyclic Nucleotides and Induction of Apoptotic Pathways via Membrane-Permeant cNMP-Analogs
cNMPs with a free amino group at the nucleobase like cAMP, cGMP, and cCMP can be dibutyrylated (db-cNMPs). Such analogs are membrane-permeable. Other cNMPs like cUMP and also cIMP lack a free amino group and dibutyrylated analogs are therefore not available. Db-cNMPs are intracellularly cleaved by unspecific esterases or amidases into two metabolites and butyrate (Schwede et al. 2000). The most prevalent metabolite is the monobutyrylated form of the cNMP with a remaining butyryl residue at the nucleobase, the other metabolite with the butyryl residue at the ribose is formed to a much lower extent. Due to these issues, another group of membrane-permeant analogs has been developed: cNMP analogs with an acetoxymethyl ester residue at the ribose (cNMP-AMs). Similar to db-cNMPs, the acetoxymethyl ester analogs penetrate the membrane, and their ester residue is intracellularly cleaved by unspecific esterases. But in contrast to the cleavage products of the db-cNMPs, the only cleavage product of the cNMP-AMs is the unmodified cNMP (Beckert et al. 2014; Schultz 2003).
3 Function of PKA, cAMP, cGMP, and cUMP in Apoptosis
3.1 PKA in Apoptosis
The best known and extensively investigated cAMP target in eukaryotic cells is cAMP-dependent protein kinase (PKA) (Francis and Corbin 1999; Taylor et al. 2008). PKA is a tetrameric holoenzyme consisting of two identical regulatory subunits, each containing two binding sites for cAMP and two catalytic domains (Taylor et al. 2004). Without binding of cAMP, the enzyme is catalytically inactive. After activation, the catalytic subunits phosphorylate serine and threonine residues of specific target proteins, e.g., the transcription factor cyclic cAMP response element-binding protein (CREB) or the cAMP-responsive element modulator (CREM), thereby altering gene expression (Mayr and Montminy 2001).
3.2 cAMP in Apoptosis
cAMP functions both as pro- and anti-apoptotic factor, depending on the condition of the cells (Insel et al. 2012). In S49 lymphoma cells, cAMP acts as pro-apoptotic stimulus and operates via PKA to induce a cell cycle arrest in the G1 phase and apoptosis (Yan et al. 2000). S49 cells are of thymic origin; the cells were derived from lymphoma of oil-treated BALB/c mice and established by van Daalen Wetters and Coffino (1987). In S49 kin− cells, mRNA for the catalytic subunits of PKA is expressed at a normal level, but the catalytic subunit protein is degraded rapidly. Therefore, the catalytic subunit is not detectable in S49 kin− cells at the protein level and, subsequently, the cells have no PKA activity (Orellana and McKnight 1990). As a consequence, S49 kin− cells are resistant to cAMP-induced apoptosis. Therefore, S49 cells are a useful tool to study the role of cAMP in apoptosis (Yan et al. 2000). Under basal conditions, cAMP and cCMP occur but neither cGMP nor cUMP are detectable in S49 cells (Hartwig et al. 2014).
Incubation of S49 wild-type (wt) cells with db-cAMP or other cAMP-increasing compounds induces a cell cycle arrest in the G1 phase, followed by delayed cytolysis which finally leads to cell death (Yan et al. 2000). But after incubation with cAMP analogs or cAMP-increasing stimuli, S49 kin− cells show neither G1 phase cell cycle arrest, nor mitochondria-dependent apoptosis, nor induction of PDE or other apoptosis-relevant proteins (Yan et al. 2000).
Surprisingly, apoptosis in S49 wt cells after treatment with cAMP-AM could not be detected (Wolter et al. 2015). This might be due to fast degradation of cAMP by phosphodiesterases (PDEs), since co-incubation with inhibitors of phosphodiesterases, like IBMX or rolipram, increased apoptosis in these cells. Moreover, activation of the PKA pathway and induction of apoptosis after treatment with PDE-resistant and cell-permeable cAMPS-AM analogs (Sp-cAMPS-AM and Sp-8-Br-cAMPS-AM) occurred in S49 wt cells as well (Wolter et al. 2015).
3.3 cGMP in Apoptosis
The best examined target of cGMP is cGMP-dependent protein kinase (PKG) (Hofmann 2005). cGMP acts as a pro-apoptotic factor in human breast cancer cell lines and in human cancer cells in general (Fallahian et al. 2011). However, stimulation with cGMP and cGMP-AM did not induce apoptosis in S49 wt or S49 kin− cells (Wolter et al. 2015). This could be due to rapid degradation of cGMP by PDEs or effective export via multidrug resistance proteins (MRPs).
Rat neuroblastoma B103 cells were used for a combination treatment for 48 h with high-concentrated cGMP-AM and cUMP-AM (both 200 μM). Prior to this treatment, cells were cultivated for 24 h in resting medium (RM), which had previously been used for studies on cCMP and cUMP generators (Hasan et al. 2014). After treatment, the cells became necrotic, whereas the individual substances had no effect and apoptosis could not be detected in B103 cells. However, based on the detection of necrotic cells in the described study, it could be possible that cUMP as well as cGMP induce apoptosis in other cell lines or primary cells.
4 Hitherto Known Targets of cCMP and cUMP
cCMP is a cyclic pyrimidine nucleotide which binds to and activates cAMP-dependent protein kinase (PKA) and cGMP-dependent protein kinase (PKG) (Desch et al. 2010; Hammerschmidt et al. 2012; Wolter et al. 2011). cCMP regulates the function of HCN2 and HCN4 channels (DeBerg et al. 2016; Zong et al. 2012). So far, only phosphodiesterase (PDE) 7A1 was identified as cCMP-degrading enzyme (Monzel et al. 2014). Another mechanism for cCMP decrease in cells is the export via MRP 5 (Laue et al. 2014).
cUMP also activates PKA and PKG (Wolter et al. 2011) and regulates HCN2 and HCN4 channels. cUMP is degraded by PDE3A, 3B, and 9A (Reinecke et al. 2011), and is exported via MRP4 and MRP5 (Laue et al. 2014). Thus, cCMP and cUMP fulfill prerequisites for a second messenger (Seifert 2015).
The MRP inhibitor probenecid (Feller et al. 1995) can be used to investigate the role of MRPs in the apoptosis resistance of cancer cells. In HEL cells, which were established from a patient with erythroleukemia (Martin and Papayannopoulou 1982), the apoptosis-inducing effect of cUMP-AM could be enhanced after pretreatment with probenecid (Fig. 2). In K-562 cells, which were obtained from a patient with chronic myelogenous leukemia (Lozzio and Lozzio 1975), pretreatment with probenecid resulted in apoptosis induction by cCMP-AM and cUMP-AM, whereas apoptosis was not induced without probenecid (Fig. 2) (Dittmar et al. 2016).

Apoptosis induction after pretreatment with probenecid (PROB) in HEL and K-562 cells. 1 × 105 cells were treated with 150 μM cCMP-AM, 50 μM PO4-AM3, and 5 μM BFA for 24 h and 48 h, respectively. Apoptosis was measured by flow cytometry with a MACSQuant Analyzer. Shown are data as means ± SD. Data were analyzed by one-way ANOVA followed by Dunnett’s test (****/####/$$$$, p < 0.0001; n.s., not significant, p > 0.05; n = 4). Data were adapted from Dittmar et al. (2016)
5 cCMP in Cell Proliferation and Apoptosis
First hints for a possible role of cCMP in the mechanism of cell proliferation arose from early experiments with mouse lymphocytic leukemia cells derived from ascetic fluid (L-1210 cells). In these experiments, performed by Bloch in 1974, cCMP enhanced cell proliferation in a dose-specific manner. A significant increase in cell proliferation was already obtained at 10 μM cCMP (Bloch 1974; Bloch et al. 1974). To determine the cell number, Bloch used the trypan blue exclusion method, which is a relatively simple method. Another method to determine cell proliferation is the incorporation of 3H- or 14C-thymidine (Adams 1980) or BrdU (Rothaeusler and Baumgarth 2007).
In contrast to the observations by Bloch et al. (Bloch 1974; Bloch et al. 1974), cCMP had no pro-proliferative effect, but inhibited cell proliferation and induced apoptosis in S49 wt and S49 kin− cells (Wolter et al. 2015). Using S49 cells as a model system, hitherto known target proteins of cCMP like PKA, PKG, Epac, and HCN channels can be excluded in the apoptosis-inducing mechanism. These proteins could be excluded by real-time PCR, since mRNA of PKG, HCN2, and HCN4 channels was not expressed in S49 wt and S49 kin− cells. Furthermore, stimulation of S49 cells with Epac-activating compounds did not alter cell proliferation (Wolter et al. 2015).
In S49 cells, cCMP induced apoptosis via the intrinsic apoptotic pathway and also via the ER stress pathway, whereas the extrinsic pathway was not involved. Induction of apoptosis was specific for cCMP-AM, because cAMP-AM, cGMP-AM, and cUMP-AM failed to activate apoptotic pathways in both S49 cell lines. Moreover, unmodified cCMP or other tested unmodified cNMPs (cAMP, cGMP, and cUMP) failed to induce apoptosis in S49 cells as well. These results indicate that extracellular effects like binding of cCMP to specific membrane receptors are not involved in the apoptosis-inducing mechanism of cCMP in S49 cells (Wolter et al. 2015).
In HEL cells, cCMP induced apoptosis also via the intrinsic apoptotic pathway. In contrast to S49 cells, induction of apoptosis was not specific for cCMP-AM, since cAMP-AM, cGMP-AM, and cUMP-AM activated the apoptotic pathway as well. Similar to S49 cells, unmodified cNMPs failed to induce apoptosis in HEL cells (Dittmar et al. 2016).
Recently, activation of the p44/p42 MAPK pathway by db-cCMP in mouse tissue lysates has been reported (Wolfertstetter et al. 2015). In contrast, activation of this pathway by cCMP-AM or db-cCMP in S49 cells was not detected. These differences could point to a cell type-specific activation pattern for cCMP, comparable to cAMP (Insel et al. 2012).
cCMP-induced apoptosis in S49 and HEL cells is caspase-dependent because the apoptotic mechanism was partly inhibited by the pan-caspase inhibitor Z-VAD-FMK (Dittmar et al. 2016; Wolter et al. 2015). Since caspases are the relevant enzymes in apoptosis, the various mechanisms can be distinguished by their characteristic activated and cleaved caspases (Hengartner 2000). Caspases activated by cCMP were identified by western blot analysis for S49 cells (Fig. 3) (Wolter et al. 2015) and by a fluorometric caspase 3 activation assay for HEL cells (Fig. 4).

Activation of the executioner caspase 3 after cCMP-AM treatment in S49 wt and S49 kin− cells. 1 × 106 cells were treated with 150 μM cCMP-AM and 50 μM PO4-AM3 for 20 h. Expressions of caspase 3 and cleaved caspase 3 were analyzed using western blotting. Data were adapted from Wolter et al. (2015)

Activation of the executioner caspase 3 after cCMP-AM and cUMP-AM treatment in HEL cells. 8 × 105 cells were treated with 150 μM cCMP-AM, 50 μM PO4-AM3, and 5 μM BFA for 20 h. Caspase 3 activity was measured at 400 nm excitation and 505 nm emission in a fluorometer. Shown are data as means ± SD. Data were analyzed by one-way ANOVA followed by Dunnett’s test (***p < 0.001; ****p < 0.0001; n = 5). Data were adapted from Dittmar et al. (2016)
Furthermore, intracellular cCMP also activated caspases 9 and 12 in S49 cells (Wolter et al. 2015). Caspase 12 is involved in the ER stress apoptotic pathway (Nakagawa et al. 2000). The extrinsic apoptotic pathway is not involved in cCMP-promoted apoptosis, since activation of caspase 8 was not detected after cCMP-AM treatment. BFA activates the ER stress apoptotic pathway and was therefore used as a negative control in the caspase 8 activation assay (Kaufman 1999), whereas a combination of cycloheximide and tumor necrosis factor (TNF) α was used as positive control (Fig. 5).

No activation of caspase 8 after cCMP-AM treatment in S49 wt and S49 kin− cells. 1 × 106 cells were treated with 150 μM cCMP-AM, 50 μM PO4-AM3, and 4 μM BFA for 20 h. As a positive control, cells were also incubated with a combination of 100 ng/ml cyclohexemide and 1 ng/ml TNFα μM for 4 h. Caspase 8 activity was measured at 400 nm excitation and 505 nm emission in a fluorometer. Shown are data as means ± SD. Data were analyzed by one-way ANOVA followed by Dunnett’s test (***p < 0.001; n = 5). Data were adapted from Wolter et al. (2015)
The involvement of the mitochondria-dependent apoptosis pathway was investigated by the depolarization of the mitochondrial membrane potential (Δψ) in HEL cells after treatment with cCMP-AM (Fig. 6) (Dittmar et al. 2016).

Loss of mitochondrial membrane potential after cCMP-AM treatment in HEL cells. 6 × 105 cells were treated with 150 μM cCMP-AM, 50 μM PO4-AM3, and 5 μM BFA for 24 h. Mitochondrial membrane potential was analyzed by flow cytometry. Shown are data as means ± SD. Data were analyzed by one-way ANOVA followed by Dunnett’s test (***p < 0.001; ****p < 0.0001; n = 3). Data were adapted from Dittmar et al. (2016)
Caspase 9 is an initiator caspase of the intrinsic apoptotic pathway (Elmore 2007). The importance of this pathway for cAMP-induced apoptosis in S49 wt cells and the release of cytochrome c were shown by Zhang et al. (2008). cCMP also induced cytochrome c release in these cells, as shown by flow cytometry analysis with a specific antibody (Fig. 7). Surprisingly, cytochrome c release was also detected in S49 kin− cells, but to a lesser extent (Fig. 7). PKA activity is not absolutely required for cytochrome c release, but could be responsible for the stronger release of cytochrome c by cCMP in S49 wt cells, since S49 kin− cells lack PKA (Orellana and McKnight 1990).

Cytochrome c release after cCMP-AM treatment. 5 × 105 cells were treated with 50 μM PO4-AM3, 150 μM cCMP-AM and 0.05 μM staurosporine for 24 h. After fixation, the S49 wt and S49 kin− cells were stained with an anti-cytochrome c-FITC antibody and analyzed by flow cytometry. Shown are the SSC-A (side scatter-area) and the FITC signal of representative dot blots. Data were adapted from Wolter et al. (2015)
Additional cell lines from human, mouse, rat, and insect species were treated with cCMP-AM and analyzed by flow cytometry. PO4-AM3 was used as a control for the acetoxymethyl residue. Thus, cell lines that are resistant to cCMP-AM treatment and cell lines that become apoptotic due to treatment with cCMP-AM were identified (summarized in Tables 1 and 2).
Comparative analysis of these results could help to identify hitherto unknown cCMP target protein molecules. In addition, highly active PDEs and/or MRPs could account for the apoptosis resistance of certain cell lines.
5.1 Relevant Genes in Apoptosis
During apoptosis, expression of a broad range of genes belonging to the Bcl-2 protein family, proteins related to p53, or cyclin-dependent kinases is altered (Dlamini et al. 2015). Their expression can be analyzed by real-time PCR. After RNA preparation, cDNA is reversely transcribed using moloney murine leukemia virus reverse transcriptase (MLV-RT) or another appropriate enzyme. Sybr Green and gene-specific primers for relevant genes in apoptosis and a housekeeping gene (e.g., Actb, Gus, or HPRT) or TaqMan probes can be used to analyze mRNA expression. Analysis of real-time PCR data and determination of gene expression alteration can be evaluated using the ΔΔCt equitation (Livak and Schmittgen 2001).
After different exposure times of S49 wt and S49 kin− cells with cCMP-AM and PO4-AM3, expression of Bim and other important apoptotic genes was analyzed by quantitative real-time PCR. The pro-apoptotic protein Bim belongs to the Bcl-2 protein family and was induced after cAMP stimulation in S49 wt cells (Zhang and Insel 2004). No significant induction of Bim or Bcl-2 was detectable in S49 wt or in S49 kin− cells after treatment with cCMP-AM, but other apoptotic relevant genes were induced in S49 wt cells, since mRNA expression of Gadd45α, cFos, and Nr4A1 was increased. On the contrary, no increase in mRNA expression of analyzed genes was detected in S49 kin− cells. Treatment with the control substance PO4-AM3 did not alter gene expression in both cell lines (Wolter et al. 2015).
5.2 Cell Cycle Analysis: G1 Arrest
Yan et al. showed that isoproterenol and also cAMP caused cell cycle arrest at G1 and increased the number of dead S49 wt cells (Yan et al. 2000). Cell viability was reduced after 36 h of treatment and the majority of S49 wt cells were dead after 72 h. cCMP-AM induced apoptosis in S49 wt and S49 kin− cells much faster, but no G1 arrest could be determined by cell cycle analysis. Forskolin (Fsk) was used as a positive control for cell cycle analysis and increased the subG1 and G0/1 population in S49 wt cells, but not in S49 kin− cells as expected (Fig. 8a, b, unpublished data).

(a) Representative histograms of original flow cytometric data. S49 wt and S49 kin− cells were incubated with 30 μM forskolin (Fsk), 100 μM cCMP-AM, and 33 μM PO4-AM3 for 24 h. Cells were fixed with ethanol, digested with RNase A, and stained with PI. Cell cycle analysis was performed using flow cytometry where results are displayed as a histogram. (b) Cell cycle analysis of S49 wt and S49 kin− cells. Intervals for the different phases in the histograms were used to calculate statistics for subG1 and G0/1. Shown are data as means ± SD. Data were analyzed by one-way ANOVA followed by Bonferroni’s post-test (*p < 0.05; ****p < 0.0001; n = 3)
Cell cycle analysis was also performed with higher concentrations (up to 200 μM cCMP-AM) and for longer time periods of treatment. The results were similar: G1 arrest was evident neither in S49 wt nor in S49 kin− cells. These differences after treatment with forskolin and cCMP-AM for cell cycle analysis are additional indications for a specific mechanism. Hitherto known cCMP targets were already excluded as targets for induction of apoptosis by cCMP, indicating the existence of other targets and mechanisms which should be investigated using S49 kin− cells as model system.
However, cell cycle analysis of HEL cells after treatment with cCMP-AM and cUMP-AM showed different results: SubG1 population was increased by cCMP-AM while G2/M population was simultaneously decreased (Fig. 9), whereas G0/1 and S populations were not affected, suggesting a cell cycle block at the G2/M checkpoint (Dittmar et al. 2016).

Cell cycle analysis of HEL cells. 5 × 105 cells were treated with 150 μM cCMP-AM, 50 μM PO4-AM3, and 5 μM BFA for 24 h. Samples were analyzed by flow cytometry with a MACSQuant Analyzer. Shown are data as means ± SD. Data were analyzed by one-way ANOVA followed by Dunnett’s test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; n = 3). Data were adapted from Dittmar et al. (2016)
5.3 Interaction of cCMP and cUMP with Bcl-2 Family Proteins and Induction of Apoptosis
Proteins belonging to the Bcl2-family play a very important role in apoptosis. Direct binding between cCMP or cUMP and Bcl-2 proteins can potentially occur, or it is possible that cCMP and cUMP induce or repress mRNA expression of these proteins. So far, no proteins belonging to the Bcl-2 family could be identified using agarose-matrices as cCMP- or cUMP-binding proteins (Hammerschmidt et al. 2012). However, for cAMP-mediated apoptosis in S49 wt cells, mRNA expression of the pro-apoptotic protein Bim was reported (Zhang and Insel 2004). Increase of Bim mRNA expression is PKA-dependent, because it was not detectable in S49 kin− cells. cCMP treatment did not alter mRNA expression of Bim or Bcl-2 (Wolter et al. 2015), supporting the concept of PKA-independent mechanisms of action for cCMP. More proteins belonging to the Bcl-2 family with pro- or anti-apoptotic function need to be examined after cCMP- and cUMP-treatment and additional cell lines should be used as well.
6 Experimental Methods
6.1 Determination of Cell Number and Cell Proliferation
6.1.1 Cell Number: Cell Counting After Trypan Blue Staining
Trypan blue is one of the several stains recommended for use in dye exclusion procedures for viable cell counting and was first mentioned by Weisenthal et al. (1983). This method is based on the principle that viable cells do not take up impermeable dyes like trypan blue, whereas dead cells are permeable and take up the dye. Therefore, viable cells can be distinguished from dead cells using this method. The cell number is estimated using a hemacytometer. This method for determination of the cell number has been used for cCMP-AM-treated S49 (Wolter et al. 2015), HEL and K-562 cells (Dittmar et al. 2016).
6.1.2 Cell Proliferation: BrdU Incorporation
Four different assays are used to evaluate cell proliferation: (1) assays measuring incorporation of radioactive thymidine (3H thymidine) or its indirectly fluorescent-labeled analog (5-bromodeoxyuridine, BrdU) into newly synthesized DNA of proliferating cells, (2) assays utilizing cells labeled with a fluorescent dye (carboxyfluorescin diacetate succinimidyl ester, CFSE) that measure the loss of the fluorescent-labeled proteins during cell division, (3) assays measuring unique markers expressed in dividing cells (e.g., Ki-67) only, and (4) assays measuring biochemical activity of cells as a substitute for proliferation (MTT) (Rothaeusler and Baumgarth 2007). The advantage of BrdU labeling compared to the others is that it constitutes a non-radioactive labeling technique which avoids costly and time-consuming cell isolation procedures (Rothaeusler and Baumgarth 2007). The halogenated pyrimidine analog BrdU, which substitutes thymidine during DNA synthesis and becomes stably integrated, can thus be detected for many months either (a) cytochemically, (b) immunocytochemically, (c) by selective photolysis, or (d) by flow cytometry (Dolbeare et al. 1983).
-
(a)
Cytochemical methods of BrdU detection were previously described by Latt in 1973 (Latt 1973) and by Darzynkiewicz et al. in 1978 (Darzynkiewicz et al. 1978).
-
(b)
Immunodetection of BrdU using specific monoclonal antibodies (mAbs), developed by Gratzner in 1982 (Gratzner 1982), allows labeling of cells in the S phase of the cell cycle (Darzynkiewicz and Juan 2001). The most common technique of cell cycle analysis based on BrdU incorporation into double-stranded DNA is the immunocytochemical detection of BrdU combined with simultaneous measurement of cellular DNA content followed by bivariate data analysis (Dolbeare et al. 1983). Since incorporated BrdU in nuclear chromatin is inaccessible to the anti-BrdU antibody, certain criteria have to be considered: (1) cellular and nuclear membranes must be fixed and permeabilized to allow access of the anti-BrdU antibody to the BrdU-labeled DNA; (2) the double-stranded DNA has to be digested to generate single-stranded DNA that is accessible to binding by the detection antibody; (3) the method has to be gentle enough to avoid cell aggregation and distortion of cell morphology (Rothaeusler and Baumgarth 2007).
One major drawback of BrdU detection procedures is the necessity for fixing and permeabilizing cells (Rothaeusler and Baumgarth 2007). The partial DNA denaturation by exposure of cells to heat or strong acid often results in cell damage, aggregation (clumping), and significant cell loss (Darzynkiewicz and Juan 2001). Besides, while induction of DNA denaturation by acid may prove to be satisfactory with one cell type, it may fail with another, since some cell types require higher acid concentration (e.g., 4 M HCl vs 2 M HCl) for optimal results, which also applies for thermal DNA denaturation (Darzynkiewicz and Juan 2001). Cell adherence to tube surfaces is another critical factor (Darzynkiewicz and Juan 2001).
In 1992, Carayon and Bord showed that using bovine pancreatic DNase I instead of hydrochloric acid for DNA breakup and Tween-20/p-formaldehyde instead of ethanol for fixation, PE and APC fluorescent signals could be maintained and cells could be labeled with anti-BrdU antibodies (Carayon and Bord 1992). Nowadays, commercially available fixation/permeabilization solutions that fix and permeabilize in a single step are mainly used (Rothaeusler and Baumgarth 2007). Afterwards, a BrdU mAb is added to detect the incorporated BrdU and a horseradish peroxidase (HRP-) linked secondary antibody is used to recognize the bound detection antibody. Chemiluminescent reagent is then added for signal development. The magnitude of light emission, which is measured in relative light units (RLU), is proportional to the quantity of BrdU incorporated into cells and is therefore a direct indication of cell proliferation.
-
(c)
Another approach for BrdU detection is based on selective photolysis of DNA containing the incorporated BrdU. Photolytically generated DNA strand breaks are subsequently labeled with fluoresceinated nucleotides. This reaction is catalyzed by exogenous terminal transferase and also known as DNA strand break induction by photolysis (SBIP) (Li et al. 1996). Since DNA denaturation is not required in SBIP, the procedure can be combined with either immunocytochemical analysis – e.g., for cell immunophenotyping together with analysis of DNA replication – or with simultaneous detection of apoptotic cells (Li et al. 1996).
-
(d)
BrdU can be used to assess proliferating cell populations by flow cytometry or fluorescent microscopy. Therefore, DNA that has previously incorporated BrdU is detected by using a fluorochrome-conjugated anti-BrdU antibody (Leif et al. 2004). To avoid unspecific staining, it is necessary to use a dye as live/dead cell discriminator (e.g., propidium iodide, PI) (Rothaeusler and Baumgarth 2007). BrdU-immunostained nuclei can be counted to generate a labeling index analogous to thymidine autoradiography (Boulton and Hodgson 1995).
BrdU has been widely applied in vitro for cultured cells and tissues (Boulton and Hodgson 1995), but in vivo applications must be carefully controlled since toxicity issues can arise with long-term BrdU application (Reome et al. 2000). Thus, its use in human tissue is limited to ex vivo studies (Boulton and Hodgson 1995). Since BrdU is a potential carcinogen and can be absorbed through skin or by inhalation (Darzynkiewicz and Juan 2001), special precautions are required for its laboratory handling (Boulton and Hodgson 1995). Furthermore, BrdU solutions are light-sensitive and should be protected from light during storage and handling (Darzynkiewicz and Juan 2001).
6.1.3 Thymidine Incorporation
The thymidine incorporation assay uses a strategy wherein a radioactive nucleoside, 3H-thymidine or 14C-thymidine, is incorporated into the replication strands of chromosomal DNA during mitotic cell division. For measurement of the radioactivity in DNA recovered from the cells in order to define the extent of cell division, cell cultures are typically set up in microplates. The labeled DNA is usually captured with a cell harvester on glass fiber filter discs which are then placed in liquid scintillation counting vials or directly harvested into a filter plate for counting on a scintillation beta-counter. Further details are summarized by Adams (1980).
6.1.4 TUNEL Assay
The TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay is based on fluorochrome labeling of 3′-OH termini of DNA strand breaks which represent a characteristic hallmark of apoptosis and occur at later stages of the apoptotic process. This assay was first described in 1992 by Gavrieli et al. (1992). Cells undergoing apoptosis showed cleavage of their genomic DNA to approximately 180 bp fragments as a result of endonuclease activation. These strand breaks were afterwards labeled in situ, either in individually fixed, permeabilized cells or in tissue sections by in situ nick translation using the TUNEL technique. The reaction is relatively specific and detects apoptotic nuclei in tissues and at the single-cell level (Gavrieli et al. 1992). Due to difficulties in discriminating between apoptotic and necrotic cells, the assay has been improved: New methods use the incorporation of dUTPs, modified with fluorophores, haptens, biotin, or bromine, which can be detected directly, when a fluorophore is added, or indirectly with streptavidin or antibodies.
6.2 Detection of Apoptosis Using Flow Cytometry
6.2.1 Annexin V/Propidium Iodide Staining
Apoptotic cells change the structure by redistribution of phosphatidylserine from the internal to the external membrane surface, which can be used as an indicator for early apoptotic cells. Annexin V interacts specifically and strongly with the exposed phosphatidylserine in the presence of physiological calcium (Ca2+) concentration (Moore et al. 1998), whereas propidium iodide (PI) binds to DNA by intercalating between the bases without sequence preference. Once PI is bound to DNA, its fluorescence is enhanced up to 30-fold. Since PI is membrane-impermeable, it intercalates only in the DNA of cells with lost membrane integrity. Therefore, simultaneous staining of cells with fluorophore-labeled annexin V (e.g., annexin V-APC) and PI allow the discrimination between early apoptotic and necrotic cells by flow cytometry (Moore et al. 1998). This method was used for cCMP-AM and cUMP-AM-treated S49 (Wolter et al. 2015), HEL and K-562 cells (Dittmar et al. 2016). For other cell lines, the results regarding this method are unpublished data (Tables 1 and 2).
6.2.2 Intracellular Staining with Cytochrome c-FITC-Antibody
During mitochondrial apoptosis, cytochrome c is redistributed from mitochondria to the cytosol in intact cells (Goldstein et al. 2000). The redistribution can be determined by flow cytometry using an antibody against cytochrome c combined with a fluorophore (e.g., fluorescein isothiocyanate, FITC). Therefore, cells are fixed with formaldehyde to stabilize the cell membrane and permeabilized with a detergent like saponin or alcohol to enable antibodies against intracellular proteins to reach their antigen. A significant increase in the lower FITC-signal is a criterion for cytochrome c release from the mitochondria and indicates the involvement of the intrinsic apoptotic pathway. The results regarding cCMP-AM treated S49 cells were published in Wolter et al. (2015).
6.2.3 Cell Cycle Analysis
Cell cycle analysis by quantitation of DNA content was one of the earliest applications of flow cytometry in apoptosis. The first protocol for cell cycle analysis using PI-staining was presented in 1975 by Awtar Krishan (Krishan 1975). Besides PI, other fluorophores, e.g., 4′,6-diamidine-2′-pheylindole (DAPI), which binds also stoichiometrically to the DNA, can be used to determine the cell cycle phase of apoptotic cells (Darzynkiewicz et al. 1992). Depending on the cell cycle phase, the amount of DNA varies and correlates with the fluorescence intensity. Four distinct phases of the cell cycle can be distinguished: G1, S, G2, and M phase. During the S phase, DNA of the cells is duplicated. However, discrimination between G2 and M phase cannot be performed based on their DNA content (Nunez 2001). By means of special mathematical models, the relative amount of cells in the different phases of the cell cycle can be represented graphically (Figs. 7a, b and 8). Often, cells have to be fixed using alcohol (e.g., ethanol) or to be permeabilized with a detergent to allow the fluorophore to enter the cells. Since the dye can also bind to RNA, the cells have to be treated with RNase after fixation. This method was used for HEL cells (Dittmar et al. 2016). Unpublished data for S49 cells are shown in Fig. 8.
6.3 Determination of the Mitochondrial Membrane Potential
Mitochondria play an important role in many pathophysiological conditions and are involved in the intrinsic mitochondria-dependent apoptosis pathway (Salvioli et al. 2000). Therefore, determining the status of this organelle can be used as a tool to examine apoptosis in cells by analyzing the mitochondrial membrane potential (Δψ) which arises from the net movement of positive charge across the inner mitochondrial membrane (Mathur et al. 2000). The status of Δψ is evaluated by using the fluorescent cyanine dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimid-azolcarbocyanine iodide) (Reers et al. 1995; Smiley et al. 1991), where JC-1 stands for 1st J-aggregate-forming cationic dye found to be sensitive to Δψ. JC-1 has been generally considered as a reliable and sensitive fluorescent probe for detecting differences in Δψ due to its dual emission characteristics (Reers et al. 1991, 1995; Salvioli et al. 2000). It produces two fluorescence emission peaks congenial to the two physical forms of the dye. (1) The monomer, which is the predominant form at low concentrations of the dye or at low Δψ, has a fluorescence maximum of 520 nm after excitation at 490 nm and emits green fluorescence. (2) The so-called “J-aggregate”, which is the predominant form at high concentrations of dye or high Δψ, has a fluorescence maximum of 585 nm after excitation at 490 nm and emits orange-red fluorescence (Mathur et al. 2000; Smiley et al. 1991). Both colors can be detected using the filters commonly mounted in almost all confocal microscopes and flow cytometers, so that green emission can be analyzed in one fluorescence channel and orange-red emission in the other (Cossarizza et al. 1993).
In viable cells whose Δψ is polarized, JC-1 is rapidly taken up by mitochondria, leading to an increase of JC-1 concentration and the formation of the so-called J-aggregates, whereas in mitochondria with depolarized Δψ (which is associated with apoptosis) JC-1 does not accumulate but remains in the cytoplasm as monomers. Therefore, apoptotic cells emit mostly green fluorescence, whereas viable cells emit relatively high levels of both green and orange-red fluorescence (Facompré et al. 2000; Mathur et al. 2000; Petit et al. 1995).
The poor water solubility of JC-1 makes it hard to use it for some applications. Therefore, JC-10 has been developed as an alternative when high dye concentration is desired. Compared to JC-1, JC-10 has a much better water solubility. In some cell lines, JC-10 has even superior performance to JC-1. This method was used for cCMP-AM- and cUMP-AM-treated HEL cells (Dittmar et al. 2016).
6.4 Detection of Caspases by Western Blotting and Fluorometric Assays
Caspases are the most important enzymes in apoptosis (Elmore 2007). They recognize a 4 or 5 amino sequence of the target substrate which includes an aspartic acid residue and cleave the substrate protein. This occurs at the carbonyl end of the aspartic acid residue. Activation of specific initiator caspases indicates the three different apoptotic pathways. Identification of caspases can be performed by immunoprecipitation or western blotting techniques. With specific antibodies against cleaved caspase products or against the full-length proteins, the different caspases can be identified. This requires very specific antibodies which can discriminate between the different caspases; sources of antibodies are documented in Dittmar et al. (2016) and Wolter et al. (2015). Western blotting was used for S49 and HEL cells treated with cCMP-AM and cUMP-AM (Dittmar et al. 2016; Wolter et al. 2015).
Additional methods for determination of the involvement of specific caspases are calorimetric or fluorometric assays. For calorimetric assays, a substrate with a specific recognition sequence (e.g., a tetrapeptide sequence DEVD) of the respective caspase linked to a chromophore (e.g., p-nitroaniline) is used (Gurtu et al. 1997). After cleavage, light emission of the chromophore increases and can be quantified using a spectrophotometer. For fluorometric assays, fluorochrome substrates are used which become fluorescent after cleavage of the substrate. For example, a caspase 8 fluorometric assay is based on the detection of AFC (7-amino-4-trifluoromethyl coumarin) after cleavage of the labeled substrate IETD-AFC. For detection of caspase 3 activation, the substrate DEVD-AFC can be used. Measurement of caspase 3 and caspase 8 activity was used for HEL and S49 cells, respectively (Dittmar et al. 2016; Wolter et al. 2015).
7 Conclusion and Further Perspectives
Recently, Wilderman et al. published new insights regarding cAMP-mediated apoptosis (Wilderman et al. 2015). They used quantitative proteomic analysis of mitochondria-enriched fractions and found some upregulated genes for enzymes involved in BCAA (branched-chain amino acid) and fatty acid oxidation in S49 wt cells after stimulation with a cAMP analog. cAMP acts via PKA and causes mitochondrial perturbation which leads to apoptosis in S49 wt cells (Wilderman et al. 2015).
Based on this new level of knowledge, further experiments concerning the cCMP mechanism could be performed, although the required high concentrations of cCMP-AM would be a limitation, when similarly adapted to the cAMP concentrations used by Wilderman et al. (2015). Therefore, it is necessary to develop additional cCMP analogs that can be used in lower concentrations than cCMP-AM. Interestingly, Wilderman et al. performed a proteomic approach, but could not find a single gene whose expression is altered in S49 kin− cells. Such an approach using S49 kin− cells and cCMP-AM should be performed, because in contrast to cAMP, cCMP induced apoptosis in both S49 cell lines and hitherto known cCMP targets are excluded in S49 kin− cells. The identification of unique cCMP target molecules would support the establishment of cCMP as an independent second messenger.
References
Adams RLP (1980) Use of radioactive isotopes in cell culture. In: Adams RLP (ed) Cell culture for biochemists, vol 11(24). Elsevier/North-Holland Biomedical Press, Amsterdam, pp 181–203
Beckert U, Grundmann M, Wolter S, Schwede F, Rehmann H, Kaever V, Kostenis E, Seifert R (2014) cNMP-AMs mimic and dissect bacterial nucleotidyl cyclase toxin effects. Biochem Biophys Res Commun 451(4):497–502
Bloch A (1974) Cytidine 3ˈ,5ˈ-monophosphate (cyclic CMP). I. Isolation from extracts of leukemia L-1210 cells. Biochem Biophys Res Commun 58(3):652–659
Bloch A, Dutschman G, Maue R (1974) Cytidine 3ˈ,5ˈ-monophosphate (cyclic CMP). II. Initiation of leukemia L-1210 cell growth in vitro. Biochem Biophys Res Commun 59(3):955–959
Boulton RA, Hodgson HJ (1995) Assessing cell proliferation: a methodological review. Clin Sci 88(2):119–130
Carayon P, Bord A (1992) Identification of DNA-replicating lymphocyte subsets using a new method to label the bromo-deoxyuridine incorporated into the DNA. J Immunol Methods 147(2):225–230
Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C (1993) A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cationic 5,5′,6,6′-tetrachloro-1,1′,3,3′- tetraethylbenzimidazolcarbocyanine iodide (JC-1). Biochem Biophys Res Commun 197(1):40–45
Darzynkiewicz Z, Juan G. (2001) Analysis of DNA content and BrdU incorporation. Curr Protoc Cytom (Chapter 7:Unit 7.7). doi:10.1002/0471142956.cy0707s02
Darzynkiewicz Z, Andreeff M, Traganos F, Sharpless T, Melamed MR (1978) Discrimination of cycling and non-cycling lymphocytes by BUdR-suppressed acridine orange fluorescence in a flow cytometric system. Exp Cell Res 115(1):31–35
Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca, Hotz MA, Lassota P, Traganos F (1992) Features of apoptotic cells measured by flow cytometry. Cytometry 13:795–808
Darzynkiewicz Z, Galkowski D, Zhao H (2008) Analysis of apoptosis by cytometry using TUNEL assay. Methods 44(3):250–254
DeBerg HA, Brzovic PS, Flynn GE, Zagotta WN, Stoll S (2016) Structure and energetics of allosteric regulation of HCN2 ion channels by cyclic nucleotides. J Biol Chem 291(1):371–381
Desch M, Schinner E, Kees F, Hofmann F, Seifert R, Schlossmann J (2010) Cyclic cytidine 3′,5′-monophosphate (cCMP) signals via cGMP kinase I. FEBS Lett 584(18):3979–3984
Dittmar F, Wolter S, Seifert R (2016) Regulation of apoptosis by cyclic nucleotides in human erythroleukemia (HEL) cells and human myelogenous leukemia (K-562) cells. Biochem Pharmacol 112:13–23
Dlamini Z, Tshidino SC, Hull R (2015) Abnormalities in alternative splicing of apoptotic genes and cardiovascular diseases. Int J Mol Sci 16(11):27171–27190
Dolbeare F, Gratzner H, Pallavicini MG, Gray JW (1983) Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc Natl Acad Sci U S A 80(18):5573–5577
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516
Facompré M, Wattez N, Kluza J, Lansiaux A, Bailly C (2000) Relationship between cell cycle changes and variations of the mitochondrial membrane potential induced by etoposide. Mol Cell Biol Res Commun 4(1):37–42
Fallahian F, Karami-Tehrani F, Salami S, Aghaei M (2011) Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J 278(18):3360–3369
Feller N, Broxterman HJ, Währer DC, Pinedo HM (1995) ATP-dependent efflux of calcein by the multidrug resistance protein (MRP): no inhibition by intracellular glutathione depletion. FEBS Lett 368(2):385–388
Francis SH, Corbin JD (1999) Cyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP action. Crit Rev Clin Lab Sci 36(4):275–328
Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119(3):493–501
Goldstein C, Waterhouse NJ, Juin P, Evan GI, Green DR (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2(3):156–162
Gratzner HG (1982) Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science 218(4571):474–475
Gurtu V, Kain SR, Zhang G (1997) Fluorometric and calorimetric detection of caspases activity associated with apoptosis. Anal Biochem 255(1):98–102
Häcki J, Egger L, Monney L, Conus S, Rossé T, Fellay I, Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19(19):2286–2295
Hammerschmidt A, Chatterji B, Zeiser J, Schröder A, Genieser HG, Pich A, Kaever V, Schwede F, Wolter S, Seifert R (2012) Binding of regulatory subunits of cyclic AMP-dependent protein kinase to cyclic CMP agarose. PLoS One 7(7):e39848
Hartwig C, Bähre H, Wolter S, Beckert U, Kaever V, Seifert R (2014) cAMP, cGMP, cCMP and cUMP concentrations across the tree of life: high cCMP and cUMP levels in astrocytes. Neurosci Lett 579:183–187
Hasan A, Danker KY, Wolter S, Bähre H, Kaever V, Seifert R (2014) Soluble adenylyl cyclase accounts for high basal cCMP and cUMP concentrations in HEK293 and B103 cells. Biochem Biophys Res Commun 448(2):236–240
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407(6805):770–776
Hofmann F (2005) The biology of cyclic GMP-dependent protein kinases. J Biol Chem 280(1):1–4
Insel PA, Zhang L, Murray F, Yokouchi H, Zambon AC (2012) Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol 204(2):277–287
Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13(10):1211–1233
Krishan A (1975) Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol 66(1):188–193
Latt SA (1973) Microfluorometric detection of deoxyribonucleic acid replication in human metaphase chromosomes. Proc Natl Acad Sci U S A 70(12):3395–3399
Laue S, Winterhoff M, Kaever V, van der Heuvel JJ, Russel FG, Seifert R (2014) cCMP is a substrate for MRP5. Naunyn Schmiedeberg’s Arch Pharmacol 387(9):893–895
Leif RC, Stein JH, Zucker RM (2004) A short history of the initial application of anti-5-BrdU to the detection and measurement of S phase. Cytometry A 58(1):45–52
Li X, Melamed MR, Darzynkiewicz Z (1996) Detection of apoptosis and DNA replication by differential labeling of DNA strand breaks with fluorochromes of different color. Exp Cell Res 222(1):28–37
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408
Lozzio CB, Lozzio BB (1975) Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45(3):321–334
Martin P, Papayannopoulou T (1982) HEL cells: a new human erythroleukemia cell line with spontaneous and induced globin expression. Science 216(4551):1233–1235
Martinou JC, Youle RJ (2011) Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell 21(1):92–101
Mathur A, Hong Y, Kemp BK, Barrientos AA, Erusalimsky JD (2000) Evaluation of fluorescent dyes for the detection of mitochondrial membrane potential changes in cultured cardiomyocytes. Cardiovasc Res 46(1):126–138
Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2(8):599–609
Monzel M, Kuhn M, Bähre H, Seifert R, Schneider E (2014) PDE7A1 hydrolyzes cCMP. FEBS Lett 588(18):3469–3474
Moore A, Donahue CJ, Bauer KD, Mather JP (1998) Simultaneous measurement of cell cycle and apoptotic cell death. Methods Cell Biol 57:265–278
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403(6765):98–103
Nunez R (2001) DNA measurement and cell cycle analysis by flow cytometry. Curr Issues Mol Biol 3(3):67–70
Orellana SA, McKnight GS (1990) The S49 Kin− cell line transcribes and translates a functional mRNA coding for the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem 265(6):3048–3053
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK (2012) Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif 45(6):487–498
Petit PX, Lecoeur H, Zorn E, Dauguet C, Mignotte B, Gougeon ML (1995) Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J Cell Biol 130(1):157–167
Reers M, Smith TW, Chen LB (1991) J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30(18):4480–4486
Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol 260:406–417
Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryot Cell 1(1):11–21
Reinecke D, Burhenne H, Sandner P, Kaever V, Seifert R (2011) Human cyclic nucleotide phosphodiesterases possess a much broader substrate-specificity than previously appreciated. FEBS Lett 585(20):3259–3262
Reome JB, Johnston DS, Helmich BK, Morgan TM, Dutton-Swain N, Dutton RW (2000) The effects of prolonged administration of 5-bromodeoxyuridine on cells of the immune system. J Immunol 165(8):4226–4230
Rothaeusler K, Baumgarth N (2007) Assessment of cell proliferation by 5-bromodeoxyuridine (BrdU) labeling for multicolor flow cytometry. Curr Protoc Cytom 40:7.31.1–7.31.13
Salvioli S, Dobrucki J, Moretti L, Troiano L, Fernandez MG, Pinti M, Pedrazzi J, Franceschi C, Cossarizza A (2000) Mitochondrial heterogeneity during staurosporine-induced apoptosis in HL60 cells: analysis at the single cell and single organelle level. Cytometry 40(3):189–197
Schultz C (2003) Prodrugs of biologically active phosphate esters. Biorg Med Chem 11(6):885–898
Schwede F, Maronde E, Genieser H, Jastorff B (2000) Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol Ther 87(2–3):199–226
Seifert R (2015) cCMP and cUMP: emerging second messengers. Trends Biochem Sci 40(1):8–15
Siddiqui WA, Ahad A, Ahsan H (2015) The mystery of BCL2 family: Bcl-2 proteins and apoptosis: an update. Arch Toxicol 89(3):289–317
Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD Jr, Chen LB (1991) Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A 88(9):3671–3675
Szegezdi E, Fitzgerald U, Samali A (2003) Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci 1010:186–194
Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G (2004) PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta 1697(1–2):259–269
Taylor SS, Kim C, Cheng CY, Brown SH, Wu J, Kannan N (2008) Signaling through cAMP and cAMP-dependent protein kinase: diverse strategies for drug design. Biochim Biophys Acta 1784(1):16–26
van Daalen Wetters T, Coffino P (1987) Cultered S49 mouse T lymphoma cells. Methods Enzymol 151:9–19
Weisenthal LM, Marsden JA, Dill PL, Macaluso CK (1983) A novel dye exclusion method for testing in vitro chemosensitivity of human tumors. Cancer Res 43(2):749–757
Wilderman A, Guo Y, Divakaruni AS, Perkins G, Zhang L, Murphy AN, Taylor SS, Insel PA (2015) Proteomic and metabolic analyses of S49 lymphoma cells reveal novel regulation of mitochondria by cAMP and protein kinase A. J Biol Chem 290(36):22274–22286
Wolfertstetter S, Reinders J, Schwede F, Ruth P, Schinner E, Schlossmann J (2015) Interaction of cCMP with the cGK, cAK and MAPK kinases in murine tissues. PLoS One 10(5):e0126057
Wolter S, Golombek M, Seifert R (2011) Differential activation of cAMP- and cGMP-dependent protein kinases by cyclic purine and pyrimidine nucleotides. Biochem Biophys Res Commun 415(4):563–566
Wolter S, Kloth C, Golombek M, Dittmar F, Försterling L, Seifert R (2015) cCMP causes caspase-dependent apoptosis in mouse lymphoma cell lines. Biochem Pharmacol 98(1):119–131
Yan L, Herrmann V, Hofer JK, Insel PA (2000) Beta-adrenergic receptor/cAMP-mediated signaling and apoptosis of S49 lymphoma cells. Am J Physiol Cell Physiol 279(5):C1665–C1674
Zhang L, Insel PA (2004) The pro-apoptotic protein Bim is a convergence point for cAMP/protein kinase A- and glucocorticoid-promoted apoptosis of lymphoid cells. J Biol Chem 279(20):20858–20865
Zhang L, Zambon AC, Vranizan K, Pothula K, Conklin BR, Insel PA (2008) Gene expression signatures of cAMP/protein kinase A (PKA)-promoted, mitochondrial-dependent apoptosis. Comparative analysis of wild-type and cAMP-deathless S49 lymphoma cells. J Biol Chem 283(7):4304–4313
Zong X, Krause S, Chen CC, Krüger J, Gruner C, Cao-Ehlker X, Fenske S, Wahl-Schott C, Biel M (2012) Regulation of hyperpolarization-activated cyclic nucleotide-gated (HCN) channel activity by cCMP. J Biol Chem 287(32):26506–26512
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG, Cham
About this chapter
Cite this chapter
Wolter, S., Dittmar, F., Seifert, R. (2017). cCMP and cUMP in Apoptosis: Concepts and Methods. In: Seifert, R. (eds) Non-canonical Cyclic Nucleotides. Handbook of Experimental Pharmacology, vol 238. Springer, Cham. https://doi.org/10.1007/164_2016_5007
Download citation
DOI: https://doi.org/10.1007/164_2016_5007
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-52671-3
Online ISBN: 978-3-319-52673-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)