
Abstract
Programmed cell death (PCD) is probably the most widely discussed subject among the topics of cancer therapy. Over the last 2 decades an astonishing boost in our perception of cell death has been seen, and its role in cancer and cancer therapy has been thoroughly investigated. A number of discoveries have clarified the molecular mechanism of PCD, thus expounding the link between PCD and therapeutic tools. Even though PCD is assumed to play a major role in anticancer therapy, the clinical relevance of its induction remains uncertain. Since PCD involves multiple death programs including programmed necrosis and autophagic cell death, it has contributed to our better understanding of cancer pathogenesis and therapeutics. In this review, we discuss a brief outline of PCD types as well as their role in cancer therapeutics. Since irregularities in the cell death process are frequently found in various cancers, key proteins governing cell death type could be used as therapeutic targets for a wide range of cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Programmed cell death is a fundamental self-destruction process in cell development and growth by which a cell can maintain homeostasis. It also has pathological implications. |
The main forms of programmed cell death are apoptosis, autophagy, and programmed necrosis, which differently contribute to the growth of cancer. |
Programmed cell death has been studied at its biomolecular key points, providing a concrete target for the development of new pharmacological therapies. |
1 Introduction
The expression programmed cell death (PCD) was introduced in 1964, and means cell death during development that is not an accidental process, but one that follows a myriad of controlled steps leading to a defined self-destruction [1].
Cancer, a complex genetic disease ensuing from mutations of oncogenes or tumor suppressor genes, resulting in the alteration of key signaling pathways, has been well known to have numerous links to PCD. Deciphering PCD in disease conditions is imperative as it not only gives new insights into the pathogenesis of such conditions, but also will help the development of new targeted anticancer therapeutic strategies. One of the main deregulated landmarks in cancer is the imbalance between cell division and cell death. Moreover, other factors that influence cell survival as well as those that control proliferation and differentiation are fundamental in cancer. Also from this perspective, any alterations in cell development or cell homeostasis can lead to dysregulation, whose fate in turn is decided by PCD, as it may hold the balance between cell death and cell survival, depending on the trophic conditions of the cell [2]. However, this is a double-edged sword; PCD can be the cause of as well as the solution to the problem [3, 4]. Hence, PCD plays an important role in both carcinogenesis and cancer treatment. Apoptosis, autophagy, and programmed necrosis/necroptosis are the three main forms of PCD that can be distinguished by their morphological and physiological differences [5, 6], and they may jointly decide the fate of the cancer cell. However, alternative types of cell death might be exploited in future to control and eliminate cancer cells, thus hinting towards a new therapeutic strategy. This article gives a comprehensive review of PCD, as well as the potential contribution of other types of cell death, and analyzes how modulation of different key cell death pathways can contribute to carcinogenesis. Finally, this review explores the cell death process as a means of targeted treatment in cancer.
2 Apoptosis
Apoptosis is probably the most investigated, highly selective and controlled cell process. The term apoptosis was coined to describe the morphological processes leading to ordered cellular self-destruction, and was first described in a publication by Kerr and collaborators [7] in the context of a thymocyte cell model. The apoptotic process has extensive biological implications, being involved in development, differentiation, normal cell turnover and, most importantly, the removal of damaged/stressed or harmful cells in a genetically determined manner [8]. Thus, dysregulation of the apoptotic program is implicated in a wide variety of pathological conditions. Apoptotic cells can be observed by stereotypical morphological changes involving cell shrinkage, pyknosis due to chromatin condensation and plasma membrane blebbing or budding. All these changes ultimately lead to cell fragmentation into membranous structures known as ‘apoptotic bodies,’ which contain cytosol, condensed chromatin, and organelles with or without nuclear fragments. The apoptotic bodies are subsequently phagocytosed by macrophages, parenchymal cells, or neoplastic cells and degraded within phagolysosomes, and are thus removed without causing inflammation. Three biochemical events are peculiar to apoptosis: (1) activation of caspases (a family of cysteine proteases), (2) DNA and protein breakdown, and (3) mitochondrial outer membrane permeabilization (MOMP) associated with the activation of catabolic hydrolases [9].
Apoptosis can occur via two core pathways, i.e., the extrinsic death receptor pathway or the intrinsic pathway (Fig. 1), which are initiated either by extracellular death receptors such as Fas cell surface death receptor (FAS), tumor necrosis factor (TNF)-α, and TNF-related apoptosis-inducing ligand (TRAIL), or by intracellular stimuli, such as hypoxia, irreparable genetic damage, nutrient deprivation, severe oxidative stress, and extremely high concentrations of cytosolic Ca2+ [10]. The signaling cascade triggering intrinsic apoptosis is highly assorted; indeed its activation can proceed in a caspase-dependent or caspase-independent manner [11]. However, data now suggests that the two pathways are allied and molecules in either pathway can influence each other [12].

Overview of Extrinsic and Intrinsic Pathways of Apoptosis. The extrinsic pathway primarily involves the binding of death ligands (e.g., TNF-α, TRAIL and FasL) to death receptors. Ligand binding to these receptors leads to receptor oligomerization and recruitment of FADD and/or TRADD, two death domain-containing adaptor proteins and subsequent recruitment of Pro-Caspase-8. Clustering of pro-caspases near a death receptor leads to formation of the death-inducing signaling complex (DISC) and the subsequent cleavage of Pro-Caspase-8 to form activated Caspase-8. The intrinsic pathway is initiated by events such as DNA damage, growth factor withdrawal, chemotherapeutic agents, irradiation leading to p53 and BH3 only proteins activation which in turn results in activation of Bax and Bak and thus modulation of mitochondrial membrane permeability. This eventually result in the release of factors from mitochondria, such as Cyto c, AIF, EndoG, Smac/DIABLO, Omi/Htra2. Cyt C forms a complex with APAF1 and procaspase 9, resulting in the cleavage and activation of procaspase 9. Caspase 8 or caspase 9 is capable of activating effector caspases such as caspase 3 or caspase 7, which then cleave apoptotic substrates leading to apoptosis. AIF, Endo G, CAD and PARP results in DNA fragmentation. Smac/DIABLO, Omi/Htra2 inhibits IAPs. A link between the extrinsic and intrinsic pathways involves the cleavage of the Bcl-2 family member Bid by caspase 8, leading to the release of cytochrome c from the mitochondria and activation of caspase 9
Extrinsic apoptosis involves transmembrane death receptor-mediated interactions, and these are members of the TNF receptor gene superfamily [13]. The best-understood ligands and corresponding death receptors can include Fas ligand (FasL)/Fas receptor (FasR), TNF-α/TNF receptor 1 (TNFR1), Apo3 ligand (Apo3L)/death receptor 3 (DR3), Apo2L/DR4, and Apo2L/DR5 [14, 15]. Extrinsic apoptosis is best characterized at the molecular level with the FasL/FasR and TNF-α/TNFR1 models. Activation of Fas, DR4, and DR5 by binding of FAS or TRAIL recruits adaptor molecules, Fas-associating protein with death domain (FADD), while it also can be stimulated by TNFR1, which can recruit TNFR1-associated death domain (TRADD). The activated FADD or TRADD lead to the formation and stimulation of death-inducing signaling complex (DISC) activating caspase-8, which in turn promotes the activation of caspase-3, a key point of apoptosis.
Interestingly, the miscellaneous range of non-receptor–mediated stimuli triggers the intrinsic pathway acting directly on targets within the cell initiated by mitochondrial events. Irrespective of the stimuli, this pathway drives changes in the inner mitochondrial membrane that result in an opening of the mitochondrial permeability transition (MPT) pore and loss of the mitochondrial transmembrane potential [16]. This pathway, which finally alters/relies on the mitochondrial membrane permeability, is intricately controlled by a group of proteins belonging to the Bcl-2 family, the latter being either pro-apoptotic or anti-apoptotic. The pro-apoptotic class of proteins are, e.g., Bad, Bak, Bax, Bcl-Xs, Bid, Bik, Bim, and Hrk, whereas the anti-apoptotic ones are, e.g., Bcl-2, Bcl-W, Bcl-XL, Bfl-1, and Mcl-1 [17]. In contrast to the role of pro-apoptotic proteins facilitating the mitochondrial release of cytochrome-c, the anti-apoptotic proteins can regulate apoptosis by blocking such release. Interestingly, this fine-tuning between apoptotic and anti-apoptotic factors is regulated not by the absolute quantity, but rather the balance between the pro- and anti-apoptotic proteins that eventually determines the cell fate [17]. In support of previous reports describing the oligomerization of Bak/Bax monomer in the involvement of apoptosis, a recent study [18] revealed the molecular intricacy behind the formation of Bak/Bax clusters, accountable for releasing apoptogenic proteins from mitochondria into the cytosol. They suggested that these clusters, instead of forming proteinaceous pores, elicit mechanical perturbations in the ultrastructure of the mitochondrial membrane. These proteins respond to a wide variety of cellular conditions, and are both subjected to transcriptional and post-translational regulation [19]. Once there is formation of an MPT pore, other apoptotic factors, including apoptosis-inducing factor (AIF), second mitochondria-derived activator of caspase (Smac), direct IAP-binding protein (DIABLO), and Omi/high temperature requirement protein A2 (HtrA2), are released from the mitochondrial intermembrane space into the cytoplasm [16]. Cytoplasmic cytochrome c together with apoptotic protease activating factor 1 (Apaf-1) and pro-caspase-9 activates initiator caspase-9 via the formation of a complex known as the apoptosome by initiating a protease cascade [16]. Caspase-9 activates caspase-3, initiating a cascade in which caspase-3 cleaves different substrates, such as inhibitor of caspase-activated DNase (ICAD) and poly(ADP-ribose) polymerase (PARP), leading to nucleosomal DNA fragmentation [20, 21]. Smac/DIABLO or Omi/HtrA2 contribute to caspase activation by binding to inhibitor of apoptosis proteins (IAPs), which ultimately leads to disruption in the interaction of IAPs with caspase-3 or caspase-9 [22]. It is imperative to note that the IAPs are endogenous inhibitors of caspases that bind via their conserved domains to the active sites of caspases thus impeding their activity. This act ultimately promotes the degradation of active caspases or keeps them away from their substrates.
2.1 The Merging Pathway
The extrinsic and intrinsic pathways merge and conclude at the execution phase, which is the final pathway of apoptosis. It begins with the activation of executioner caspases, i.e., activating cytoplasmic endonuclease, which degrade nuclear material, and proteases that degrade the nuclear and cytoskeletal proteins. Caspase-3, caspase-6, and caspase-7 act as effector or executioner caspases, cleaving various substrates that include cytokeratins, PARP, cytoskeletal and nuclear proteins; together they contribute to the typical morphological changes observed in apoptotic cells [23]. Caspase-3 is the most critical of all the executioner caspases and can be activated by any of the initiator caspases (caspase-8, caspase-9, or caspase-10). Furthermore, the intrinsic and extrinsic pathways converge to caspase-3, which specifically activates the caspase-activated deoxyribonuclease (CAD). In proliferating cells, CAD is complexed with its inhibitor (ICAD). In apoptotic cells, activated caspase-3 cleaves ICAD to release CAD [24]; the latter can then degrade DNA and cause chromatin condensation. Caspase-3 also induces cytoskeletal reorganization and collapse of the cell into apoptotic bodies, which are readily engulfed by neighboring phagocytes, thereby preventing the release of cellular contents into the local tissue environment, and the consequent inflammatory response [25].
2.2 Targeting Apoptosis for Cancer Treatment
Apoptosis works as an important process both for blocking cancer growth and for inhibiting metastasis by killing altered cells. Current cancer therapy involves small molecules targeting apoptotic pathways. Each abnormality or defect in the apoptotic pathways points to an interesting target for cancer treatment; thus, restoring apoptotic signaling is one of the most researched areas in cancer therapeutics.
Preferential provocation of cell death in cancer cells as compared to normal cells via TRAIL can be considered an encouraging anticancer drug target. It is important to note that unlike TNF and FasL, TRAIL and the TRAIL receptor antibodies have been reported to be both well tolerated and safe in nonhuman primates, even at relatively high concentrations [26]. The decisive role of apoptosis regulation by Bcl-2 family anti-apoptotic proteins, such as Bcl-2, Bcl-xL, and Mcl-1, and their frequent overexpression in a variety of tumor types makes them capable targets for anticancer therapy. Many approaches target Bcl-2 family members through the inhibition or silencing of upregulated anti-apoptotic genes. In addition, use of Bcl-2-homology-3 (BH3)-only peptides or synthetic small molecule inhibitors interfering with Bcl-2–like protein function is in development [27].
Many p53-based strategies (see Sect. 4, for a discussion on p53) have been explored for cancer treatment. They can be categorized into gene therapy, drug therapy, and immunotherapy. The introduction of just the p53 gene is not sufficient to eliminate all tumor cells, so studies focused on the use of p53 gene therapy in combination with other anticancer strategies. Murine double minute 2 (MDM2), a critical negative regulator of p53 that promotes p53 ubiquitination and degradation [27, 28], is also one of the important targets for p53-based therapy. Several drugs have been investigated to target and restore the function of mutant type p53 via different mechanisms, e.g., by intercalating with DNA and altering and destabilizing the DNA–p53 core domain complex, resulting in the restoration of unstable p53 mutants, or by inhibition of the MDM2–p53 interaction and thus stabilizing p53.
Interestingly, a clinical trial has been carried out using p53 vaccine, which contained a recombinant replication-defective adenoviral vector with human wild-type p53; this was given to patients with advanced-stage cancer. When these patients were followed up at 3 months after immunization, four out of the six patients had stable disease [27].
Another potential target linked to apoptosis includes IAPs, targeted by antisense strategies and short interfering RNA (siRNA) so as to disable them to inhibit apoptosis. Moreover, these strategies have been reported to result in improved in vivo tumor control by radiotherapy by increasing radiation sensitivity. Interestingly, this approach when used together with anticancer drugs has been demonstrated to exhibit enhanced chemotherapeutic activity [27].
Furthermore, upregulation of caspases appears to be a triggering mechanism for tumors displaying therapeutic resistance to radiation and chemotherapy. Several caspase-based drug and gene therapies have been reported in different studies, in combination with other treatments. For example, human caspase-3 gene therapy was used in an AH130 liver tumor model in combination with etoposide administration, and was found to reduce tumor volume by extensive apoptosis [27], except for Bcl-2 overexpressing tumors. Therefore, caspase-3 gene transduction accompanied by an additional death stimulus may be a useful method in anticancer gene therapy. Moreover, another target, caspase-8, has prompted significant clinical interest. It is possible to upregulate the transcription of caspase-8 in some cellular contexts via treatment with therapeutic agents, cytokines, and agents of differentiation, thereby promoting the cells towards apoptosis [29].
3 Autophagy
Autophagy, a term derived from the Greek words “auto” (self) and “phagy” (to eat), refers to an indispensable, regulated, and conserved catabolic process, which principally mediates the recycling and turnover of various cytoplasmic eukaryotic cell constituents. It shares many elements of the pathophysiological processes that occur in malignant cells [30], and is strictly regulated by autophagy-related genes (ATGs) [31]. Three forms of autophagy have been identified based on the delivery method into the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [32].
Macroautophagy, the most functional and best characterized form of autophagy, involves the formation of double membrane autophagosomes, which clear the damaged organelles or unwanted cellular components by delivering them to lysosomes for degradation and recycling. The role of macroautophagy in cancer is still perplexing, as numerous reports present macroautophagy and macroautophagic cell death as anti-tumoral responses, whereas macroautophagy is also related to enhanced cancer-cell metabolism in hypoxic and nutrient-deficient environments and chemotherapeutics resistance [33].
Microautophagy, the non-selective lysosomal degradative process, is mediated by direct lysosomal (mammals) or vacuolar (plants and fungi) engulfment of the cytoplasmic cargo at a boundary membrane by autophagic tubes, which mediate both invagination and vesicle scission into the lumen. This pathway is especially important for survival of cells under starvation.
Interestingly, CMA signifies the chaperone-dependent selection of soluble cytosolic proteins to be targeted to lysosomes, which are then translocated across the lysosome membrane for degradation. The unique features of this type of autophagy are the selectivity of the proteins that are degraded and the direct shuttling of these proteins without the requirement for the formation of additional vesicles. Upregulation of CMA has been linked to the survival and proliferation of cancer cells [34].
It is imperative to note that, although the dependence of cancer cells on CMA suggests a pro-oncogenic function for CMA, its effect in normal cells is almost the opposite, where it protects cells from the damage caused by extracellular and intracellular injuries. Recently, it has been shown that CMA-mediated degradation takes on an anti-oncogenic role in non-proliferating tumor cells by reducing the cellular levels of mutant p53 [35]. A recent study has shown that cross-talk between CMA and ATG5-dependent macroautophagy could regulate breast cancer cell metastasis [36].
It has been shown that autophagy activates in response to either extra- or intracellular stress, i.e., nutrient starvation, differentiation, metabolic stress, and developmental triggers. It plays a dual role in the regulation of cell death signaling pathways: a pro-survival function under certain circumstances and a pro-death process in a variety of diseases, including cancer [37]. Although the regulatory mechanisms of autophagy are only partially known, still many controversies remain around whether it protects cell survival or contributes to cell death. An emerging hypothesis suggests that its role depends on tumor development stage. For instance, autophagy can limit tumor formation in cancer early stages, but favors tumor cell survival, invasion, and metastasis after tumor growth [37, 38]. Moreover, the unambiguity of autophagic cell death in molecular and functional processes was clarified by Shimizu et al. [39], who reported that the term autophagic cell death should be used in circumstances where inhibition of autophagy (via chemical inhibitors or genetic extirpation) leads to cell death suppression. On the contrary, if such inhibition of autophagy does not prevent cell death, the progression should not be referred to as autophagic cell death.
ATGs are implicated in the formation of autophagosomes and are closely linked to cancer initiation and progression. A silencing approach to crucial ATGs, such as ATG3, ATG4, Beclin1/ATG6, ATG10, and ATG12, can induce tumor transformation, especially when cells are forced into stressful conditions [40]. Recently, it has been reported that ATG5-dependent cell death contributes to the embryonic development of Bax/Bak double-knockout (DKO) mice, which is resistant to apoptosis, which would suggest that autophagy compensates for the deficiency in apoptosis [41]. To determine the role of ATG5, ATG5/Bax/Bak triple-knockout (TKO) mice were developed. In these mice autophagy is greatly suppressed, when compared with in DKO mice. Embryonic fibroblasts and thymocytes endured autophagy much less frequently, and when exposed to cellular stressors their viability was higher than DKO cells. This provides genetic evidence that DKO cells undergo Atg5-dependent death [41].
Beside ATGs functions, there are a number of protein kinases capable of regulating the induction of protective autophagy in cancer cells in response to cytotoxic agents, these include glycogen synthase kinase 3 beta (GSK3B), AMP-activated protein kinase (AMPK), extracellular signal-regulated kinases 1 and 2 (ERK1/2), and eukaryotic elongation factor-2 kinase (eEF2 K) [42,43,44,45].
In mammals, two homologues of ATG1 (ULK1 and ULK2), mammalian autophagy-related protein 13 (mATG13) and scaffold protein FIP200 (an ortholog of yeast ATG17), are able to form a complex (Fig. 2), although FIP200 can localize with ULK into pre-autophagosomal structures for recruitment of other ATG proteins [46, 47]. Under nutrient starvation conditions, mammalian target of rapamycin (mTOR), an evolutionarily conserved serine/threonine kinase, disrupts the binding of ATG13 to ULK and destabilizes it, thereby inhibiting the ULK-dependent phosphorylation of FIP200 and autophagy induction by phosphorylation of ULK and ATG13 [48].

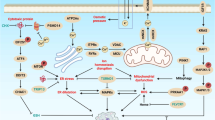
Schematic representation of Autophagy. Autophagy is stimulated by nutrient deprivation, hypoxia, cytokines, hormones, and DNA damage. mTORC1 is downstream of PI3K and is activated in response to mitogenic stimuli or nutrient availability. mTORC1 inhibits autophagy by phosphorylating ULK1-ATG13-FIP200 complex. The inhibition of mTORC1 by rapamycin (an mTORC1 inhibitor) strongly induces autophagy. The kinase activity of TOR is inhibited by TSC1 and TSC2, which form a complex, with GAP activity, against the small GTPase Rheb, a direct activator of TOR. The TSC1/TSC2 complex, in turn, is regulated by several upstream protein kinases, including Akt and AMPK. Reactive oxygen species (ROS) are generated in response to starvation and are required for activation of the autophagy-specific protease Atg4. Starvation and the phosphorylation of Bcl-2 by JNK also inhibits the association of Beclin 1 with Bcl2, leading to autophagy. p53 incites autophagy in a transcription-dependent manner by modifying the expression of a number of regulators that inhibit the mTOR pathway like AMPK. Nutrient deprivation and PARP activation in response to DNA damage leads to ATP depletion that leads to elevated AMP/ATP ratios ultimately triggering the energy-sensing serine/threonine kinase 1(LKB1)-AMPK signaling axis. Raised AMP/ATP ratio activates the LKB1 which subsequently phosphorylates and activates AMPK. This activation of AMPK facilitates the phosphorylation of TSC, which results in the inactivation of mTORC1 and induction of autophagy
In addition, mTOR acts as the main negative regulator of autophagy in cancer cells by controlling the balance between cell growth and autophagy in response to specific signals, i.e., nutritional status, growth factor and stress. mTOR consists of two distinct complexes named mTOR complex 1 (mTORC1) and 2 (mTORC2) in mammalian cells. Many components of the phosphoinositide 3-kinase (PI3K) signaling pathway, ahead of both mTORC1 and mTORC2, are reported to be mutated in human cancers. Moreover, the loss of p53, a very common event in cancer, promotes mTORC1 activation [49]. It should be noted that mTORC1-inhibited autophagy is frequently observed in malignant cells, because the signaling pathways that promote mTORC1 activity are induced by oncoproteins and/or loss of tumor suppressors. Moreover, PI3K antagonizes autophagy by activating Akt. In turn Akt can phosphorylate tuberous sclerosis complex 2 (TSC2) protein, destabilizing it and disrupting its interaction with TSC1 to abolish the negative regulatory effect of the TSC2/TSC1 complex on mTORC1 [45]. Also, the phosphorylation of TSC2 by AMPK can increase GTP-ase–activating protein (GAP) (an active portion of TSC2) activity, which stabilizes the TSC2/TSC1 complex and results in the inactivation of the mTORC1 interacting protein Ras homolog enriched in brain (Rheb), which inactivates mTORC1 and leads to the initiation of autophagy [50]. Moreover, nutrient deprivation and PARP activation in response to DNA damage lead to ATP depletion that causes an elevated AMP/ATP ratio that triggers the energy-sensing serine/threonine kinase 1 (LKB1)–AMPK signaling axis. Raised AMP/ATP ratio activates the LKB1, which consequently phosphorylates and activates AMPK. Activated AMPK mediates the phosphorylation of TSC, resulting in the inactivation of mTORC1 and induction of autophagy. Interestingly, activation of autophagy is indispensable, but not adequate for autophagic cell death, as it requires additional death signals like c-Jun N-terminal kinase (JNK) [51]. JNK is activated in response to various stress signals, and its elevated level has been reported to contribute in autophagy. JNK signaling regulates the expression of multiple ATGs [52]. Recently, insights into the interaction between apoptosis and autophagy have been reported that hint at the important role of autophagy in resistance to apoptosis when MG63 cells are incubated with curcumin [53]. In this report, inhibition of apoptosis enhanced curcumin-induced autophagy by the upregulation of the JNK signaling pathway. Moreover, JNK exhibits dual tumor-promoting and tumor-suppressive roles subjected to the genetic perspective of the tumor cells [54]. Additionally, the extent of JNK activation varies between normal and cancer cells exposed to apoptosis, being comparatively lower in the latter [39]. Thus, insufficient activation of JNK in such cancer cells and subsequent failure of autophagic cell death may possibly contribute to the unrestrained growth of cells that ultimately attain the malignancy.
Beclin 1, the mammalian homolog of yeast ATG6, is a haploinsufficient tumor suppressor gene, while Beclin 1 protein is an important point of convergence between apoptosis and autophagy. It has also been shown to interact with anti-apoptotic Bcl-2-like proteins, and recently, it has been reported to be a BH3-only protein. The phosphorylation of Bcl-2 by JNK results in the subsequent dissociation of Beclin 1 from Bcl-2, and the induction of autophagy. Furthermore, mTORC1 regulates autophagy by mediating protein translation and cell growth through phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and ribosomal protein S6 kinase beta-1 (S6K1). When mTOR activity is low, 4E-BP1 is hypophosphorylated, which allows efficient binding to eIF4E and blocks translation initiation, whereas when mTOR activity is high, 4E-BP1 is phosphorylated, resulting in the release of eIF4E, thus allowing cap-dependent translation to begin.
Interestingly, the role of mTORC2 in cancer has been reported in gliomas that overexpress the mTORC2 subunit rictor, promoting mTORC2 assembly and activity, thus enhancing the increased proliferative and invasion potential of cancer cells [55].
Epithelial cell adhesion molecule (EpCAM), is a transmembrane glycoprotein mediating Ca2+-independent cell–cell adhesion. It is known to be highly expressed in a variety of epithelial carcinomas, and it is involved in cell adhesion and proliferation. Recently, it has been reported that its overexpression regulates epithelial–mesenchymal transition, stemness, and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway [56]. EpCAM can regulate the AMPK signaling pathway and consequently inner cellular energy availability. Indeed, the connection between EpCAM and AMPK is under the control of a complex formed by EpCAM and CD147, CD98 heavy chain, and several amino acid transporters such as LAT1 and ASCT2 at the plasma membrane. This complex can control cellular energy homeostasis and indirectly affects cell energy sensor AMPK, at least in prostate cancer cell models [57].
To sum up, there is context-dependent effects and ‘doubled-edged swords’ when it comes to autophagy and cancer because both autophagy-enhancing and autophagy-inhibiting agents may elicit beneficial effects in the treatment of cancer. Broadly speaking, autophagy may protect against cancer development, but it may also be important for cancer progression and treatment [55].
3.1 Targeting Autophagy for Cancer Treatment
Of the potential targets in autophagy, the Akt–mTOR pathway is the most well studied. Since the last decade, new promising druggable autophagy molecular targets for cancer therapy include Beclin-1, ULK1, ATG4, ATG7, and Vps34 [58]. Clinical trials involving hydroxychloroquine (HCQ) were the first deliberate attempt to modulate autophagy therapeutically in cancer patients [58]. DEP domain-containing mTOR-interacting protein (Deptor), also known as DEP-domain containing protein 6 (DEPDC6), an inhibitor of mTORC1 and mTORC2, inhibits mTORC1 and mTORC2 by directly binding to them both. Deptor has been reported to be highly expressed in some cancer types where it can act either as an oncogene or oncosuppressor, depending on the cell or tissue contexts [59]. Recently, it has been reported that overexpression of Deptor is necessary for the proliferation, migration, invasion, and survival of osteosarcoma cells and may be a prospective target for the treatment of osteosarcoma [60]. As mentioned in Sect. 2.2 regarding apoptosis, Bcl-2 family members also have an important regulatory role in autophagy. As a result, the modulation of Bcl-2 family proteins leads to not only apoptotic, but also autophagic cell death, thus serving as a possible target.
Recently, oncology drug re-positioning came to the fore, with reports that conventional drugs used to treat non-malignant diseases can exhibit anticancer effects by activating/suppressing autophagy [61, 62]. For instance, chloroquine, which is used to treat malaria and autoimmune disorders, blocks autolysosome formation by impairing the fusion between mature autophagosomes and lysosomes, eventually inducing apoptosis primarily via excessive ER stress. Temozolomide (TMZ) is an alkylating agent that induces formation of O6-methylguanine in DNA, and thus inducing mismatch pair with thymine during DNA replication. Chloroquine and TMZ therefore show a synergistic curative effect for cancer cells [63].
4 Role of p53 in Apoptosis and Autophagy: Twin Regulatory Function
One of the most frequently mutated genes in human cancer, p53, was originally discovered as a transformation-linked protein [64,65,66,67] and was later found to be involved in the regulation of a wide range of cellular processes, including cell cycle control, differentiation, senescence, DNA repair, genome stability, apoptosis, and autophagy [68].
It contributes to apoptosis induction predominantly by its transcription-dependent effects. MDM2 oncogenic protein serves as the chief cellular antagonist of the p53 tumor suppresser gene. The activity of p53 needs to be regulated to provoke proper responses to differential cellular stress conditions. MDM2 negatively regulates p53 activity through the induction of p53 protein degradation by serving as an E3 ubiquitin ligase. It catalyzes polyubiquitination and subsequently induces proteasome degradation to downregulate p53 protein level. Stress signals such as DNA damage interrupt Mdm2-mediated inhibition of p53, leading to accumulation of p53 both in the nucleus and in the cytoplasm. Interestingly, p53 has a very short half-life in normal cells, whereas its half-life is dramatically prolonged in human tumor cells [69].
Multiple mechanisms have been proposed to explain how p53 triggers MOMP. It is reported that in apoptotic cells, p53 co-interacts with Bcl2, Bcl-XL, and Bak. Alternatively, cytoplasmic p53 can also induce cell death via direct activation of cytosolic Bax and subsequent mitochondria permeabilization and apoptosis. Oncogene expression, DNA damage, or other forms of stress lead to stabilization of p53 protein by phosphorylation or other modifications [69]. p53 modifications, including phosphorylation, ubiquitination, acetylation, methylation, sumoylation, and neddylation, contribute a very complex epigenetic code that complicatedly modulates p53 functions. Stabilized p53 accumulates in the nucleus and binds to specific DNA sequences, leading to transactivation of a number of proapoptotic genes like Bax, Noxa and Puma. A number of other proapoptotic genes, such as Bid, Apaf1, and p53-induced protein with a death domain (PIDD), are also defined as transcriptional targets of p53.
It has been reported that genotoxic and metabolic stress (nutrient deprivation) also activates p53-dependent autophagy [70]. Two mechanisms have been proposed. In the first, in a p53-dependent manner, genotoxic stress induces the activation of AMPK (a kinase that serves as a fuel sensor in the cell by assessing the ratio of AMP to ATP), which inhibits mTOR. Additionally, p53 transcriptionally activates negative regulators of mTOR in a tissue-specific manner. Moreover, recently, it has been reported that the products of two p53 target genes, Sestrin1 and Sestrin2, can bind and activate AMPK, causing it to phosphorylate TSC2 and thereby inhibit mTOR [71]. Nutrient starvation significantly increases the expression of Sestrin2, but its loss will markedly reduce the level of p53-mediated autophagy [71]. The other mechanism whereby p53 induces autophagy was suggested when it was identified that p53 directly transactivates the gene encoding damage-regulated autophagy modulator (DRAM), which encodes a highly evolutionarily conserved protein that co-localizes with cathepsin D in the lysosome [72]. The gene is transactivated when p53 binds directly to its consensus binding site in the DRAM promoter following genotoxic stress [72]. It has also been shown that p73, another member of the p53 family, can induce autophagy by transactivating DRAM, but this is not essential for p73 to induce autophagy [73]. Moreover, the roles of p53 in autophagy appear contradictory, as stress-activated p53 induces autophagy, while unstressed p53 represses basal levels of autophagy. The ability to inhibit autophagy and sensitize tumors to metabolic stress present a promising new approach for cancer therapy.
5 Programmed Necrosis/Necroptosis
Regulated/programmed necrosis or necroptosis is activated under particular conditions, for example, when a portion of apoptotic machinery is imperfect, when cells suffer from severe stress, or when they are treated with chemotherapy or inflammatory factors and cannot follow apoptotic process [74]. As in apoptosis, in necroptosis, cells are committed to die in an ordered and orchestrated manner [75]. It differs in necrosis that is caused by physical trauma [76] and can be discriminated from other death types by undetectable caspase activation and lysosome-independent characteristics that involve swelling of subcellular organelles including endosomes, Golgi bodies and mitochondria at an early stage and eventual functional loss of cell membrane [77]. Regulated necrosis can be further divided into different types characterized by (but not limited to) necroptosis, MPT-dependent regulated necrosis, phosphoribosyl pyrophosphate (PPRP)-1-mediated necrotic death, pyroptosis and ferroptosis [77].
Necroptosis is activated in an organized manner that requires the activation of the serine/threonine kinase receptor-interacting protein 1 (RIP1) via a cascade of signaling pathways involving activation of the TNF receptor superfamily [78], T cell receptors [79, 80], interferon receptors [81], Toll-like receptors (TLRs) [82], cellular metabolic and genotoxic stresses, or various anticancer agents. Interestingly, necroptosis has been reported to be pharmacologically inhibited by chemical compounds such as necrostatin-1 (Nec-1) [83].
In TNFR1 signaling (Fig. 3), TNF-α activates TNFR1 and leads to the recruitment of RIP1 kinase, TRAF2, TRADD, and cIAP1/2, forming a transitory complex referred to as complex I [84, 85]. In this complex, RIP1 gets modified by polyubiquitination mediated by E3 ligases, cIAP1 and cIAP2. It can consequently be deubiquitinated by the enzyme cylindromatosis (CYLD) [86] and thus form complex II, which encompasses RIP1, FADD, TRADD, and caspase-8 [87] as key components. The choice as to whether complex II initiates cell death via apoptosis or necroptosis is determined at this step. Inhibition of cIAP1 leads to formation of complex IIa to stimulate the caspase cascade and to induce apoptosis [88]. However, when caspase-8 activation is repressed (by genetic or pharmacological stimuli), RIP1 together with RIP3 forms a cytoplasmic necroptotic protein complex (complex IIb/necrosome) that leads to the necroptotic signal transduction pathway [89]. The mixed lineage kinase domain-like protein (MLKL), a downstream effector of necroptosis, is also detected in complex IIb [90].

Schematic Overview of Necroptosis: Various stimuli including DNA damage, engagement of receptors, such as TCR, TLR, IFNR or TNFR, lead to RIPK3 activation and formations of divergent signaling complexes, eventually leading to the activation of NF-κB, apoptosis and necroptosis. Binding of ligands to receptors leads to the formation of complex I. cIAP ligase mediated Polyubiquitination of RIP1 (in complex I) results in cell survival through the activation of NF-κB and MAPKs. Deubiquitination of RIP1 by CYLD or inhibition of cIAP proteins leads to the conversion of complex I to complex IIa, activating the caspase cascade for further apoptosis induction. In case when caspase 8 activity is inhibited or RIP3 is highly expressed, RIP1 interacts with RIP3 to form complex IIb (necrosome), which facilitates necroptosis. The formation of complex IIb necessitates the kinase activity of RIP1. Activation of RIPK3 leads to its oligomerization and downstream phosphorylation of MLKL. This also results in mitochondria-dependent ROS production. Upon phosphorylation, MLKL oligomerises and translocates to the plasma membrane and causes its lysis. RIPK3- and MLKL-containing necrosome has been found to translocate to mitochondrially associated endoplasmic reticulum (ER) membranes (MAMs). Two mitochondrial proteins, PGAM5 (a mitochondrial phosphatase) and Drp-1 (a protein required for mitochondrial fission) acts as downstream components of necrosome signaling. Following RIPK3-dependent phosphorylation, the PGAM5 activates Drp-1 by its dephosphorylation, leading to extensive mitochondrial fission, ROS production and necroptosis
RIP3-mediated phosphorylation of the MLKL is a critical event for necroptosis as made evident by the fact that obstructing MLKL activity leads to necroptosis inhibition. Furthermore, phosphorylation of MLKL by RIP3 activates the mitochondrial phosphatase phosphoglycerate mutase 5 (PGAM5), a crucial downstream effector of the necrosomal complex. PGAM5 in turn begins the dephosphorylation of GTPase dynamin-related protein 1 (DRP1), a mitochondrial fission regulator, which leads to mitochondrial fission and further fragmentation.
Another necroptosis-inducing complex, ripoptosome, has also been reported [91]. The core components of this complex, FADD, RIP1, and caspase-8, are ubiquitinated and then degraded by IAPs, which will suppress ripoptosome formation. Nevertheless, when exposed to Smac mimetics or genotoxic stress, IAPs are downregulated, which results in the spontaneous formation of the ripoptosome; this then triggers caspase-8–mediated apoptosis or caspase-independent necroptosis [92].
Necroptosis has been reported to play a key role in the development of a variety of human diseases, including cancer [93]. Several studies have reported genetic or epigenetic alterations in crucial necroptosis regulators during tumor progression, thus contributing to tumor cell survival. In most cases of acute myeloid leukemia, chronic lymphocytic leukemia, breast cancer, and colon cancer, in tumor tissue, RIP 1 and RIP3 are significantly reduced, resulting in decreased apoptosis/necroptosis [77]. Interestingly, downregulation of the key executor of necroptosis, MLKL, in pancreatic adenocarcinomas and ovarian cancers has also been reported [77].
Studies have linked necroptosis to metastasis [94]; indeed, the induction of a high level of reactive oxygen species (ROS) via necroptosis might represent one factor that restricts cancer cell metastasis [95]. In particular, RIP3 has been observed to be critical for regulating ROS production during necroptosis [96]. RIP1 and RIP3 may promote anti-metastatic effects by regulating oxidative stress and thus eliminating tumor cells. For a successful metastatic process, tumor cells must overcome necroptosis key points [81].
A new form of programmed necrosis known as ‘ferroptosis’ has been reported, which requires the accumulation of cellular ROS in an iron-dependent manner [97]. Although it is caused by the loss of cellular redox homeostasis, lipid ROS/peroxides, not cytosolic ROS, play more crucial roles in ferroptosis. Furthermore, inactivation of glutathione peroxidase 4 (GPX4), an enzyme mandatory for the clearance of lipid ROS, can induce ferroptosis even when cellular levels of cysteine and GSH are normal [98].
Although its physiological function is not well explored, ferroptosis has been reported to be involved in cancer [97] and contributes to the tumor suppressive function of p53 [99]. Recently, ferroptosis has been reported as a form of autophagic cell death that is mechanically facilitated via a form of cargo-specific autophagy known as ferrotinophagy [100]. Upon cystine deprivation, autophagy is activated to degrade the cellular iron storage/stock protein ferritin (increasing cellular labile iron) via cargo receptor NCOA4, thus promoting the accumulation of cellular ROS and resulting ferroptotic cell death.
5.1 Targeting Necroptosis for Cancer Treatment
Along with apoptosis, necroptosis may serve as a promising secondary cell death process for sensitizing tumor cells to anticancer drugs that are particularly apoptosis resistant. Interestingly, it has been reported that necroptosis is impaired during tumorigenesis [101]. Defects in necroptosis regulators, including RIP3 and CYLD, and RIP3 polymorphisms in non-Hodgkin lymphoma have been demonstrated to be correlated with tumor progression [101]. Potential targets for necroptosis include RIP1 and RIP3, necrosome complex, mitotic kinase polo-like kinase 1, MLKL, and PARP-1 [77, 101].
To sum up the involvement of all these types of cell death on cancer cell fate, Fig. 4 describes the representative cross-talk between apoptosis, autophagy, and programmed necrosis.

Cross-talk between apoptosis, autophagy and programmed necrosis
6 Interlinking: Apoptotic, Programmed Necrosis and Autophagic Pathways
In Sect. 4, we focused on interconnections between apoptosis and autophagic pathways. The interconnection between apoptosis, necroptosis, and autophagy are outlined in Fig. 5. The proteins of the extrinsic death receptor pathways can also influence autophagy when apoptosis is blocked, i.e., by caspase inhibition, suggesting that autophagy and apoptosis are induced concurrently by the FADD death domain. But given that apoptotic cell death progresses faster, this is dominantly observed, whereas the full implementation of autophagy (or necrosis) emerges only when caspase inhibitors are present. The caspase inhibitor zVAD-fmk, mostly used for blocking apoptotic cell death, may be implicated in all three main cell death pathways. It may not only regulate the balance in favor of autophagy, but in some cell types, it also shifts apoptosis towards necrosis [102].

Interlinking and regulation of apoptosis and programmed necrosis and autophagy with metastasis. Ligation of death receptor with ligands leads to caspase-8 activation. This results in the caspase-8- dependent cleavage of effector caspases and activation of apoptosis with proteolytic inhibition of RIPK1 and RIPK3 that inhibits autophagy and Programmed necrosis and favors apoptosis. The full commencement of autophagy (or necrosis) emerges only when caspase inhibitors (zVAD-fmk) are applied. Metastatic cells face numerous unfavorable conditions i.e., increased cellular ROS, DNA damage and insufficient energy status. It leads to apoptosis or necroptosis depending upon the intensity of death signals, resulting in the hindrance in metastasis. Autophagy plays the dual role as on one hand, autophagy mends the aptness of metastatic cells under stressful conditions by counteracting apoptosis and necroptosis, but on the other hand, autophagy reduces metastasis by inducing the death of metastasizing cells
Another molecule, receptor-interacting protein kinase-1 (RIPK1), may connect all three major death pathways. RIPK1 has an important role to play in the instigation of caspase- independent death. Autophagic cell death starts as a survival challenge by blocking necrosis and a clear-out of oxidative damaged mitochondria [103], whereas necrotic cell death may exhibit a rapid onset, involving ROS production, cytoplasmic ATP reduction, and other cellular events. Both the necrotic and autophagic cell death pathways are connected by a signaling cascade, involving RIPK1, which is negatively regulated by caspase-8. It is imperative to note that the caspase-dependent apoptosis elicited by death receptor ligation involves the induction of apoptosis through caspase-8-dependent activation of effector caspases and subsequent activation of the mitochondrial death pathway. Activity of RIPK1 and RIPK3, which modulates necrosis and autophagy, in turn is controlled by their cleavage regulated by caspase-8. A stable complex between RIPK1 and RIPK3, formed only in the absence of caspase-8 activity, promotes programmed necrosis and autophagy, instead of apoptosis [104].
Another pivotal factor in cell death fate is the energy/ATP level status. ATP exhaustion activates autophagy. However, if autophagy fails to maintain the energy levels, necroptosis occurs [93].
During metastatic processes, malignant cells must overwhelm a sequence of unfavorable disorders, including hypoxia, invasion by immune cells, and detachment from the ECM, which can lead to increased ROS production, DNA damage, and insufficient energy status. Although autophagy can control the aptness of cancer cells under traumatic conditions (and thus attenuating apoptosis and necroptosis), on the other hand autophagy can antagonize metastasis by inducing metastasizing cell death and restricting tumor necrosis. Since imperfections in the machinery of one type of cell death may not be influenced by each other, pharmacological targeting of a single type of PCD may not be sufficient for an efficient treatment of cancer. An ideal approach using combined inducers acting on different cell death pathways may help to overcome drug resistance or to enhance metastatic cell killing.
7 Conclusion
PCD is crucial to innumerable biological processes and involves not just the traditional mode of death, apoptosis, but also numerous other death pathways. Since PCD is involved in numerous human diseases [105,106,107,108,109], many death regulatory genes are common to more than one pathway; therefore, PCD should be regarded as a network of interconnected modules whose targeting may have therapeutic benefit. Interestingly, current research is now investigating the mechanisms that regulate other under-explored forms of cell death and how these pathways could be mapped and integrated with each other. This review has explored pivotal studies regarding apoptosis, necroptosis, and other forms of cellular death and can provide more insights into how PCD pathways play decisive roles as possible drug targets in cancer therapy.
References
Lockshin RA, Williams CM. Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol. 1964;10:643–9.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Jensen M, Engert A, Weissinger F, Knauf W, Kimby E, Poynton C, et al. Phase I study of a novel pro-apoptotic drug R-etodolac in patients with B-cell chronic lymphocytic leukemia. Invest New Drugs. 2008;26:139–49.
Baritaki S, Militello L, Malaponte G, Spandidos DA, Salcedo M, Bonavida B. The anti-CD20 mAb LFB-R603 interrupts the dysregulated NF-κB/Snail/RKIP/PTEN resistance loop in B-NHL cells: role in sensitization to TRAIL apoptosis. Int J Oncol. 2011;38:1683–94.
Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–19.
Tan ML, Ooi JP, Ismail N, Moad AIH, Muhammad TST. Programmed cell death pathways and current antitumor targets. Pharm Res. 2009;26:1547–60.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta. 2010;1805:53–71.
Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94.
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516.
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, et al. Anti- and pro-tumor functions of autophagy. Nature. 1999;397:441–6.
Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–88.
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501.
Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8.
Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem. 1997;272:32401–10.
Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163.
Reed JC. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Semin Hematol. 1997;34:9–19.
Nasu Y, Benke A, Arakawa S, Yoshida GJ, Kawamura G, Manley S, Shimizu S, Ozawa T. In situ characterization of Bak clusters responsible for cell death using single molecule localization microscopy. Sci Rep. 2016;6:27505.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310.
Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50.
Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, et al. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–9.
Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64:7183–90.
Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55:178–94.
Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–9.
Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006;11:1709–26.
Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev. 2008;19:325–31.
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87.
Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–55.
Stupack DG. Caspase-8 as a therapeutic target in cancer. Cancer Lett. 2013;332:133–40.
Wang S, Yu Q, Zhang R, Liu B. Core signaling pathways of survival/death in autophagy-related cancer networks. Int J Biochem Cell Biol. 2011;43:1263–6.
Pyo JO, Nah J, Jung YK. Molecules and their functions in autophagy. Exp Mol Med. 2012;44:73–80.
Liu B, Bao J-K, Yang J-M, Cheng Y. Targeting autophagic pathways for cancer drug discovery. Chin J Cancer. 2013;32:113–20.
Grasso D, Vaccaro MI. Macroautophagy and the oncogene-induced senescence. Front Endocrinol (Lausanne). 2014;5:157.
Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104.
Vakifahmetoglu-Norberg H, Kim M, Xia H-G, Iwanicki MP, Ofengeim D, Coloff JL, et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013;27:1718–30.
Han Q, Deng Y, Chen S, Chen R, Yang M, Zhang Z, et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep. 2017;7:4759.
Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75.
Morselli E, Galluzzi L, Kepp O, Vicencio J-M, Criollo A, Maiuri MC, et al. Anti- and pro-tumor functions of autophagy. BBA-Mol Cell Res. 2009;1793:1524–32.
Shimizu S, Yoshida T, Tsujioka M, Arakawa S. Autophagic cell death and cancer. Int J Mol Sci. 2014;15:3145–53.
Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906.
Arakawa S, Tsujioka M, Yoshida T, Sakurai HT, Nishida Y, Matsuoka Y, et al. Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 2017;24:1598–608.
Corcelle EA, Puustinen P, Jäättelä M. Apoptosis and autophagy: targeting autophagy signalling in cancer cells -’trick or treats’? FEBS J. 2009;276:6084–96.
Bhutia SK, Kegelman TP, Das SK, Azab B, Su Z-Z, Lee S-G, et al. Astrocyte elevated gene-1 induces protective autophagy. Proc Natl Acad Sci USA. 2010;107:22243–8.
Yang J, Takahashi Y, Cheng E, Liu J, Terranova PF, Zhao B, et al. GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death. J Cell Sci. 2010;123:861–70.
Wu WKK, Cho CH, Lee CW, Wu YC, Yu L, Li ZJ, et al. Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy. 2010;6:228–38.
Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150:1507–13.
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan J-L, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510.
Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12:209–18.
Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA. 2005;102:8204–9.
Inoki K, Li Y, Zhu T, Wu J, Guan K-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57.
Shimizu S, Konishi A, Nishida Y, Mizuta T, Nishina H, Yamamoto A, Tsujimoto Y. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29:2070–82.
Zhou Y-Y, Li Y, Jiang W-Q, Zhou L-F. MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep. 2015;35(3):e00199.
Zhang Y, Chen P, Hong H, Wang L, Zhou Y, Lang Y. JNK pathway mediates curcumin-induced apoptosis and autophagy in osteosarcoma MG63 cells. Exp Ther Med. 2017;14:593–9.
Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49.
Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, et al. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007;67:11712–20.
Wang MH, Sun R, Zhou XM, Zhang MY, Lu JB, Yang Y, et al. Epithelial cell adhesion molecule overexpression regulates epithelial-mesenchymal transition, stemness and metastasis of nasopharyngeal carcinoma cells via the PTEN/AKT/mTOR pathway. Cell Death Dis. 2018;9:2.
Ni J, Cozzi P, Hao J, Beretov J, Chang L, Duan W, et al. Epithelial cell adhesion molecule (EpCAM) is associated with prostate cancer metastasis and chemo/radioresistance via the PI3K/Akt/mTOR signaling pathway. Int J Biochem Cell Biol. 2013;45(12):2736–48.
Rebecca VW, Amaravadi RK. Emerging strategies to effectively target autophagy in cancer. Oncogene. 2016;35:1–11.
Catena V, Fanciulli M. Deptor: not only a mTOR inhibitor. J Exp Clin Cancer Res. 2017;36:12.
Hu B, Lv X, Gao F, Chen S, Wang S, Qing X, et al. Downregulation of DEPTOR inhibits the proliferation, migration, and survival of osteosarcoma through PI3K/Akt/mTOR pathway. Onco Targets Ther. 2017;10:4379.
Langedijk J, Mantel-Teeuwisse AK, Slijkerman DS, Schutjens MH. Drug repositioning and repurposing: terminology and definitions in literature. Drug Discov Today. 2015;20(8):1027–34.
Tommasino C, Gambardella L, Buoncervello M, Griffin RJ, Golding BT, Alberton M, et al. New derivatives of the antimalarial drug Pyrimethamine in the control of melanoma tumor growth: an in vitro and in vivo study. J Exp Clin. Cancer Res. 2016;35(1):137.
Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. J Hematol Oncol. 2017;10:67.
Chang C, Simmons DT, Martin MA, Mora PT. Identification and partial characterization of new antigens from simian virus 40-transformed mouse cells. J Virol. 1979;31:463–71.
DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci USA. 1979;76:2420–4.
Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31:472–83.
Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3.
Chaabane W, User SD, El-Gazzah M, Jaksik R, Sajjadi E, Rzeszowska-Wolny J, et al. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp (Warsz.). 2013;61:43–58.
Rotter V. p53, a transformation-related cellular-encoded protein, can be used as a biochemical marker for the detection of primary mouse tumor cells. Proc Natl Acad Sci USA. 1983;80:2613–7.
Balaburski GM, Hontz RD, Murphy ME. p53 and ARF: unexpected players in autophagy. Trends Cell Biol. 2010;20:363–9.
Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–60.
Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34.
Crighton D, O’Prey J, Bell HS, Ryan KM. p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ. 2007;14:1071–9.
Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem. 2016;85:743–63.
Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48.
Cho YS, Park SY. Harnessing of programmed necrosis for fighting against cancers. Biomol Ther (Seoul). 2014;22:167–75.
Wang T, Jin Y, Yang W, Zhang L, Jin X, Liu X, et al. Necroptosis in cancer: an angel or a demon? Tumour Biol. 2017;39:1–11.
Chan FK-M. Programmed necrosis/necroptosis: an inflammatory form of cell death. In: Wu H, editor. Cell death: mechanism and disease. New York: Springer; 2014. p. 211–28.
Ch’en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–41.
Lu JV, Chen HC, Walsh CM. Necroptotic signaling in adaptive and innate immunity. Semin Cell Dev Biol. 2014;35:33–9.
Dempsey LA. Interferon-induced necroptosis. Nat Immunol. 2013;14:892.
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108:20054–9.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:4.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14.
Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–16.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90.
Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–26.
Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–8.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–48.
Xu YZ, Kanagaratham C, Youssef M, Radzioch D. New frontiers in cancer chemotherapy—targeting cell death pathways. In: Najman S, editor. Cell biology—new insights. Rijeka: InTech; 2016. p. 93–140.
Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer. 2015;14:48.
Fu Z, Deng B, Liao Y, Shan L, Yin F, Wang Z, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13:580.
Buchheit CL, Rayavarapu RR, Schafer ZT. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol. 2012;23:402–11.
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–32.
Cho YS, Park HL. Exploitation of necroptosis for treatment of caspase-compromised cancers. Oncol Lett. 2017;14:1207–14.
Los M, Mozoluk M, Ferrari D, Stepczynska A, Stroh C, Renz A, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13:978–88.
Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;6:pe44.
Lu JV, Weist BM, van Raam BJ, Marro BS, Nguyen LV, Srinivas P, et al. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci USA. 2011;108:15312–7.
Nigam M, Ranjan V, Srivastava S, Sharma R, Balapure AK. Centchroman induces G0/G1 arrest and caspase-dependent apoptosis involving mitochondrial membrane depolarization in MCF-7 and MDA MB-231 human breast cancer cells. Life Sci. 2008;82:577–90.
Nigam M, Singh N, Ranjan V, Zaidi D, Sharma R, Nigam D, et al. Centchroman mediated apoptosis involves cross-talk between extrinsic/intrinsic pathways and oxidative regulation. Life Sci. 2010;87:750–8.
Singh N, Nigam M, Ranjan V, Sharma R, Balapure AK, Rath SK. Caspase mediated enhanced apoptotic action of cyclophosphamide- and resveratrol-treated MCF-7 cells. J Pharmacol Sci. 2009;109:473–85.
Sharifi-Rad J, Sureda A, Tenore GC, Daglia M, Sharifi-Rad M, Valussi M, Tundis R, Sharifi-Rad M, Loizzo MR, Ademiluyi AO, Sharifi-Rad R, Ayatollahi SA, Iriti M. Biological activities of essential oils: from plant chemoecology to traditional healing systems. Molecules. 2017;22:70.
Singh N, Nigam M, Ranjan V, Zaidi D, Garg VK, Sharma S, et al. Resveratrol as an adjunct therapy in cyclophosphamide-treated MCF-7 cells and breast tumor explants. Cancer Sci. 2011;102:1059–67.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Abhay P. Mishra, Bahare Salehi, Mehdi Sharifi-Rad, Raffaele Pezzani, Farzad Kobarfard, Javad Sharifi-Rad and Manisha Nigam declare no conflict of interest.
Funding
The authors have no funding to declare.
Rights and permissions
About this article

Cite this article
Mishra, A.P., Salehi, B., Sharifi-Rad, M. et al. Programmed Cell Death, from a Cancer Perspective: An Overview. Mol Diagn Ther 22, 281–295 (2018). https://doi.org/10.1007/s40291-018-0329-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-018-0329-9