
Abstract
Several experimental techniques to analyse histamine receptors are available, e.g. pharmacological characterisation of known or new compounds by different types of assays or mutagenesis studies. To obtain insights into the histamine receptors on a molecular and structural level, crystal structures have to be determined and molecular modelling studies have to be performed. It is widely accepted to generate homology models of the receptor of interest based on an appropriate crystal structure as a template and to refine the resulting models by molecular dynamic simulations. A lot of modelling techniques, e.g. docking, QSAR or interaction fingerprint methods, are used to predict binding modes of ligands and pharmacological data, e.g. affinity or even efficacy. However, within the last years, molecular dynamic simulations got more and more important: First of all, molecular dynamic simulations are very helpful to refine the binding mode of a ligand to a histamine receptor, obtained by docking studies. Furthermore, with increasing computational performance it got possible to simulate complete binding pathways of ions or ligands from the aqueous extracellular phase into the allosteric or orthosteric binding pocket of histamine receptors.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
A large number of different experimental techniques to study histamine receptors, or in general GPCRs, are available: Ligands for example represent an important tool to characterise the receptors on a macroscopic level by different assays, e.g. radioligand competition binding assay, GTPase assay, luciferase assay or GTPγS binding assay (Seifert et al. 2013; Strasser et al. 2013; Panula et al. 2015). Furthermore, the resulting experimental data allow to analyse the histamine receptors also with regard to selectivity, e.g. species or subtype selectivity (Seifert et al. 2013; Strasser et al. 2013; Panula et al. 2015). However, these methods do not provide information about distinct ligand-receptor interactions or conformational changes of the receptor during the ligand binding or receptor activation process on a molecular level. Therefore, mutagenesis studies in combination with pharmacological characterisation are one important lab experimental method of choice (Kooistra et al. 2013; Seifert et al. 2013; Strasser et al. 2013; Schneider and Seifert 2016), because those studies give information about the influence of one or more amino acids onto the pharmacological properties of the analysed receptor, which have to be interpreted on a molecular level. In contrast, the determination of crystal structures of ligand-receptor complexes gives a detailed insight into the receptor conformation and the interactions between ligand and receptor (Venkatakrishnan et al. 2013). Although more and more crystal structures of aminergic GPCRs in the inactive and in the active state are available (Venkatakrishnan et al. 2013), (http://www.rcsb.org/, access date: 16.11.2016), this method is still limited to a small number of ligand-receptor complexes because of the high experimental expense. This gap between the pharmacological data on the one hand and the structural interpretation on a molecular level on the other hand can be closed with several molecular modelling approaches, as discussed later on in more detail. However, to improve the understanding of the histamine receptors for example with regard to species, subtype or functional selectivity, all these lab experimental and in silico techniques have to be used in a combined manner, as illustrated (Fig. 1) (Strasser 2009; Munk et al. 2016). Besides molecular modelling techniques, inclusive virtual screening methods are used for lead optimisation and identifying new affine histamine receptor ligands (Heifetz et al. 2016b; Levoin et al. 2016). Meanwhile, databases represent an important tool to improve the research in the GPCR field, because they provide a large amount of data, e.g. mutagenesis data, binding data or homology models (Southan 2016).

Combination of experimental and molecular modelling techniques to obtain more detailed insight into the histamine receptors on a molecular level [modified according to Strasser (Strasser 2009)]
2 Molecular Modelling Approaches for Histamine Receptors
As illustrated in Fig. 1, a large number of different molecular modelling approaches to study GPCRs are available (Rodriguez et al. 2012; Costanzi 2013; Strasser and Wittmann 2013; Filizola 2014; Heifetz et al. 2016a). For most of these approaches, the structure of the GPCR is required. In the absence of X-ray structures, there were some attempts to model GPCRs de novo or ab initio, based on the amino acid sequence (Filizola et al. 1999; de Graaf and Rognan 2009; Xu and Zhang 2012; Zhang et al. 2015). Although there are promising results with regard to those methods, they are not established so far. Nowadays it is state of the art to generate homology models of the GPCR of interest based on an appropriate crystal structure for the in silico analysis (de Graaf and Rognan 2009; Mobarec et al. 2009; Yarnitzky et al. 2010; Costanzi 2012; Koehler Leman et al. 2015).
2.1 Crystal Structure of the Histamine H1 Receptor
Within the histamine receptors, only the crystal structure of the inactive human histamine H1 receptor co-crystallised with the H1R antagonist doxepin (pdb code: 3RZE, RCSB Protein Data Bank, http://www.rcsb.org/, access date: 16.11.2016) is available (Shiroishi and Kobayashi 2016; Shimamura et al. 2011). Thus, this crystal structure can be used, with some refinements, for in silico studies of the interactions between the human H1R and antagonists.
2.2 Homology Models of Histamine Receptors
In the very beginning, homology models of histamine receptors based on the crystal structure of bacteriorhodopsin (Henderson et al. 1990) and later on crystal structures of bovine rhodopsin (Palczewski et al. 2000) were constructed (ter Laak et al. 1995; Bakker et al. 2004; Strasser and Wittmann 2007). Although the X-ray template used for homology modelling was not an aminergic GPCR, it was possible to explain pharmacological results quite well with those models (ter Laak et al. 1995; Bakker et al. 2004; Strasser and Wittmann 2007). However, the determination of crystal structures of aminergic GPCRs in its inactive state, available at the RCSB Protein Data Bank (http://www.rcsb.org/, access date: 16.11.2016) (Table 1), enables more appropriate templates for homology modelling of histamine receptors.
Furthermore, active-state homology models of HxR-G-protein complexes, as recently described, for the hH4R-Gαβγ-complex (Geyer et al. 2016) may also be generated, using the crystal structure of the hβ2R-Gαβγ-complex (pdb code: 3SN6) as a template (Rasmussen et al. 2011b).
The percentage of identical amino acids for the TM domains of the human histamine receptors and the aminergic GPCRs with a published crystal structure is given in Table 2. The percentage of identity of the single-TM domains ranges from ~12% up to 62%, whereas the overall identity of the TM domains ranges from ~27% up to ~42%. In general, the receptor with the highest homology to the receptor of interest should be used as a template (Fiser 2010). However, a threshold of at least 30% for accurate modelling of GPCRs is recommended (Fiser 2010). Within another approach, suggested to lead to improved results, different templates for different TM domains in homology modelling are used (Fiser 2010), even considering conserved inter-residue interactions (Chaudhari et al. 2015).
However, in 2011, the crystal structure of the hH1R in the inactive state in complex with the H1R antagonist doxepin was published (Shimamura et al. 2011). A comparison of the homology model of hH1R, based on the crystal structure of hβ2R with the hH1R crystal structure, showed that the homology model was in very good accordance to the X-ray structure of the hH1R (unpublished results). Thus, carefully generated homology models represent the possibility to obtain detailed insight into histamine receptors on a molecular level, if crystal structures are not yet solved, as for H2R, H3R and H4R.
Although the transmembrane domains of GPCRs can be modelled in a good quality, it is a challenge to model loops or termini, e.g. the E2-loop or the N-terminus (Goldfeld et al. 2011; Arora et al. 2016). In a large number of the crystal structures, the E2-loop and the N-terminus are not or not completely solved. Thus, several tools have to be used to model those domains, offering a large number of different conformations. It was shown by several mutagenesis studies in combination with pharmacological analysis that amino acids of the E2-loop have influence onto affinity, potency and efficacy of ligands at the histamine receptors (Lim et al. 2008; Strasser et al. 2008b; Brunskole et al. 2011; Peeters et al. 2011; Wifling et al. 2015a). Thus, the correct modelling of the E2-loop is essential for a highly predictive homology model. However, the modelling of the loop regions, especially the E2-loop, remains quite challenging (Goldfeld et al. 2011; Arora et al. 2016). This is also reflected by comparison of the E2-loops of crystal structures of aminergic GPCRs (Fig. 2). Compared to the TM domains, the parts of the E2-loop, not being fixed by a disulphide bond, show a very high flexibility (Fig. 2a). While there are only small differences between the transmembrane domains, for the E2-loop very different conformations were found for the different receptors (Fig. 2b).

E2-loop: (a) Flexibility, (b) differences in conformation (orange: tβ1R, red: hβ2R, blue: hD3R, violet: h5-HT1BR, cyan: h5-HT2BR, green: hH1R) and (c) differences in the amino acid sequences
Another problem in modelling of E2-loops arises from the differences in length of the loops (Fig. 2c). However, the tβ1R and the hβ2R represent no appropriate template for modelling of the E2-loop of the histamine receptors, because within the E2-loop of the tβ1R and hβ2R, an additional disulphide bridge, forcing a part of the E2-loop into a helical conformation, is present (Fig. 2b, c). Due to the lack of two additional cysteines, this second disulphide bridge is missing in the E2-loops of the histamine receptors (Fig. 2c). The number of amino acids between TM4 and the highly conserved cysteine ranges from 11 to 18 in the hHxRs, but in the X-ray templates only 10 to 12 amino acids are present. Furthermore, the number of amino acids between the highly conserved cysteine of the E2-loop to TM5 ranges from 4 to 6 for the hHxRs, while it ranges from 3 to 6 for the X-ray templates (Fig. 2c). The different lengths of the E2-loops may lead to differences in the extracellular positions of TM4 and TM5, as illustrated (Fig. 2b), and have also to be considered in homology modelling. Due to these differences not only in the chemical nature of the amino acids itself, but also in the length of the parts of the loops, it is highly challenging to model an appropriate conformation of the E2-loop. Furthermore, it has to be considered that more than one conformation of the E2-loop of the receptor may exist. Instead, it has to be speculated that an E2-loop can exhibit different conformations, e.g. in dependence of the ligand bound. However, the conformation of loops can be refined by molecular dynamic simulations, taking into account the surrounding water molecules and ions, e.g. Na+ and Cl− (Arora et al. 2016).
Homology models of GPCRs can be generated manually, but meanwhile an increased number of servers and databases (Table 3) (Rodriguez et al. 2012; Koehler Leman et al. 2015) offer already prepared homology models or generate homology models. But due to the problems regarding the conformations of the loops and termini, in most cases only the TM domains are offered by servers or databases (Rodriguez et al. 2012).
2.3 Different Modelling Techniques
2.3.1 QSAR
Quantitative structure-activity relationships (QSAR) are a method to describe the relation between the ligand structure and the pharmacological property, e.g. affinity, potency or even efficacy quantitatively (Verma et al. 2010; Cherkasov et al. 2014; Damale et al. 2014). A QSAR study requires a library of ligands with high structural similarity and which bind to the same binding site of the target. Furthermore, the compounds of the library have to be separated into a training set and a test set. The training set, necessary to calculate the quantitative structure-activity relationships, should contain at least 20 or 30 compounds, with known pharmacological parameters, e.g. affinity in the range of at least two orders of magnitude. The test set, necessary to analyse the quality of the QSAR model, should contain at least ten compounds, with the same pharmacological parameter, determined experimentally under the same conditions. Of course, these requirements limit the use of QSAR methods, which can be classified as retrospective methods. However, QSAR-based methods may represent a fast tool to understand the biological effect of drugs or to predict pharmacological parameters of compounds, also in the field of histamine receptors (Strasser 2009; Istyastono et al. 2011; Sirci et al. 2012; Kooistra et al. 2014).
2.3.2 Docking
Automated docking of ligands into the binding pocket of a GPCR is a very fast method to obtain one or more suggestions for the binding mode of a ligand (Beuming and Sherman 2012; Sandal et al. 2013; Beuming et al. 2015; Yuriev et al. 2015; Irwin and Shoichet 2016). Within modern docking routines rotatable bonds of the ligand and additionally of the amino side chains of the receptor are considered, leading to improved docking results, but also to increased computational costs. Although those methods were often successfully used to describe ligand-receptor interactions or to obtain starting structures for MD simulations (Strasser 2009; Schultes et al. 2013; Darras et al. 2014; Naporra et al. 2016), one has to be aware that such methods do not consider translational or rotational movements of the backbone. Consequently, differences in receptor conformation in dependence of the bound ligand cannot be investigated. Furthermore, these methods do not provide any information about the stability of the resulting ligand-receptor complex on the time course. A large number of studies suggest that water molecules are present in the binding pocket, and even stabilise the interaction between ligand and receptor (Wagner et al. 2011; Kuhne et al. 2016). But within most docking studies, water molecules are in general not taken into account. Thus, docking results may give a first idea about a binding mode of a ligand, but should be refined by subsequent molecular dynamic studies, including approximately the physiological surrounding.
2.3.3 Molecular Interaction Fingerprint Methods
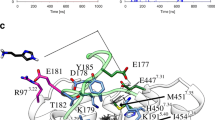
Within the computer-aided development of drugs, the data processing of a large amount of, e.g., docking results is a great challenge (Mordalski et al. 2011). To manage those data, e.g. H-bond or aromatic interactions within reasonable time, the structural or molecular interaction fingerprint approach was established (Deng et al. 2004; Mordalski et al. 2011; Vass et al. 2016), as shown exemplary for the binding mode of doxepin to the hH1R (Fig. 3) (Kooistra et al. 2016). Those fingerprint methods were shown to be helpful, e.g. in prediction of binding modes or even functional activity (Vass et al. 2016).

Fingerprint for the binding mode of the H1R antagonist doxepin in the hH1R [modified according to Kooistra et al. (2016)]
2.3.4 Molecular Dynamic Simulations
A typical simulation box for a GPCR embedded in its lipid bilayer, being surrounded by water molecules and ions in approximately physiological concentrations, contains in general more than 50,000 sites (Fig. 4). If the Gαβγ complex is included in the simulation, the simulation box contains more than 200,000 sites.

Simulation boxes for a histamine receptor in its inactive state (left) and in its active state in complex with the Gαβγ-subunit (right)
Although MD simulations (Dror et al. 2012; Ciancetta et al. 2015; Tautermann et al. 2015; McRobb et al. 2016) are a very powerful and important tool to study conformational changes of the receptor or ligand-receptor complexes, the computational time, which increases exponentially with the number of sites in the simulation box, is a crucial point. The rotation around bonds takes place in the ps scale, whereas ion transport or ligand binding is ranged in general in the ns or μs scale (Selent et al. 2010; Dror et al. 2011; Yuan et al. 2011; Wittmann and Strasser 2015; Thomas et al. 2016). Furthermore, protein folding or large conformational changes within proteins are estimated to be in the ms scale.
Nowadays it is possible to perform MD simulations of a typical GPCR simulation box (Fig. 4) up to some μs (Selent et al. 2010; Dror et al. 2011; Yuan et al. 2011; Thomas et al. 2016). Thus, it should be possible to observe conformational changes of the ligand in the binding pocket, and furthermore the binding of water or ions into the binding pocket or some conformational changes of the receptor. However, due to the limitation in computational time, it is not possible up to now to observe the whole binding process of an agonist and the subsequent activation of the receptor.
2.3.5 Semi-empirical Calculations, Ab Initio Methods, QM/MM Methods
Although molecular dynamics simulations are an important tool to study histamine receptors in dependence of time, it is not possible to monitor breaking or forming of bonds. Due to the high computational costs, linked with quantum mechanical methods, e.g. semi-empirical calculations or ab initio calculations, such calculations are rarely performed in the field of histamine receptors (Kovalainen et al. 2000; Jongejan et al. 2008). An accepted alternative are combined quantum mechanical and molecular mechanical (QM/MM) methods: Here, only a small part of the receptor, e.g. the binding pocket with bound ligand, is investigated on a quantum mechanical basis, whereas the remaining larger part of the system (receptor, surrounding) is investigated on a molecular mechanical basis.
2.3.6 Comparison of Different Modelling Techniques in the Histamine Receptor Research
As described above, a lot of different molecular modelling techniques to analyse histamine receptors on a molecular level are available (Table 4). QSAR or docking methods are quite fast with low computational costs, compared to quantum mechanical calculations or simulations. On the other hand, MD simulations allow to analyse a wide area of the conformational space of histamine receptors. To solve a distinct modelling problem, a skilful combination of stationary (QSAR, docking) and dynamic (MD) methods is essential.
3 Comparison of the Orthosteric and the Allosteric Binding Site of the Four Human Histamine Receptor Subtypes Based on the Amino Acid Sequence
The analysis of the amino acid sequence alignment of the four human histamine receptor subtypes (Seifert et al. 2013; Strasser et al. 2013) shows a homology within the TM helices between ~27 and ~54%. The largest homology is found between the hH3R and hH4R, while the smallest homology is found between hH1R and hH4R as well as between hH2R and hH4R.
However, the differences in pharmacological profiles of several ligands between the four histamine receptor subtypes are in general not a consequence of the overall differences in the amino acid sequence, but rather of the differences in amino acids of the binding pocket. Based on several crystal structures of aminergic GPCRs with a bound ligand (see Sect. 2.2), the orthosteric binding pocket is known quite well. However, to obtain information if a distinct amino acid is directly or indirectly involved in ligand binding, experimental mutagenesis studies with subsequent pharmacological investigation are performed. These experimental data provide an important input for molecular modelling studies, e.g. for refinement of present models. Hundreds of mutations were analysed within the subfamily of aminergic GPCRs (http://www.gpcrdb.org, access date: 16.11.2016). However, also at histamine receptors, a large number of mutagenesis studies were performed (http://www.gpcrdb.org, access date: 16.11.2016) (Kooistra et al. 2013; Strasser et al. 2013). But not all amino acids are involved in the ligand binding. Only those amino acids close to the orthosteric (Fig. 5) binding site may have an influence on ligand binding.

Schematics of the orthosteric binding site with the main pocket I and the side pocket II. Asp3.32 and Trp6.48 are conserved within the histamine receptors; the amino acids at 2.61 and 5.46 differ between the histamine receptors and may be involved in species or subtype differences
The most important amino acids of the transmembrane domains, shown to be involved in ligand binding at the histamine receptors, are summarised in Fig. 6.

The orthosteric ligand and allosteric Na+-binding site and the most important amino acids forming both sites of the human histamine receptors (blue: negatively charged, red: positively charged, orange: polar, yellow: cysteine, green: aromatic and polar, dark grey: aromatic and lipophilic, grey: lipophilic and bulky, light grey: lipophilic and small)
A comparison of the percentage of identical amino acids of the orthosteric binding site, suggested being involved in ligand binding, shows the highest homology of 69.2% for the hH3R–hH4R. All other pairs have a clearly smaller homology in the range from 30.8 to 38.5%. This explains that a large number of ligands, e.g. thioperamide or UR-PI294 (N 1-[3-(1H-imidazol-4-yl)propyl]-N 2-propionylguanidine), have affinity to hH3R and hH4R (Seifert et al. 2013). Additionally, the extracellular domains have influence on affinity, potency and efficacy for selected ligands, as shown, e.g., for the H4R (Brunskole et al. 2011; Wifling et al. 2015b). As already mentioned, the extracellular domains, especially the E2-loop, show a very high flexibility. Thus, the prediction of amino acids of the extracellular domains being involved in ligand binding is quite a challenge (Goldfeld et al. 2011; Arora et al. 2016). Even if the influence is known by a combination of experimental mutagenesis and pharmacological studies, it is often not possible to explain the pharmacological data in a satisfactory manner, especially if extracellular domains are involved (Brunskole et al. 2011).
Besides the orthosteric ligand-binding site, the allosteric binding site near to Asp2.50 plays an important role for the binding of Na+ or other monovalent cations, as described in more detail in Sect. 4.6. It was shown by mutagenesis studies or X-ray structures for several different GPCRs that the highly conserved Asp2.50 acts as a binding site for Na+ (Katritch et al. 2014; Strasser et al. 2015). Furthermore, within the allosteric ion-binding site, the amino acids Asp2.50, Ser3.39, Asn7.45, Ser7.46 and Asn7.49, which are involved in binding of the Na+, are highly conserved within class A of the GPCRs (Katritch et al. 2014; Strasser et al. 2015). These amino acids are also present within the four human histamine receptor subtypes (Fig. 6). However, a comparison of the most important amino acids, forming the allosteric binding site and the channel, connecting the orthosteric and allosteric site, shows that about 30% of the amino acids are different within the four human histamine receptor subtypes (Fig. 6), which may explain the differences in sodium sensitivity, e.g. between the hH3R and hH4R (Schneider et al. 2009; Schnell and Seifert 2010).
4 Molecular Modelling of Histamine Receptors: Impact for Understanding the Histamine Receptors on a Molecular Level: Case Studies
4.1 Binding Mode of Histamine at the Four Human Histamine Receptor Subtypes
The binding pocket of the histamine receptors is well characterised by mutagenesis studies (Kooistra et al. 2013): It was shown by mutagenesis studies that Asp3.32, Lys5.39, Thr5.42, Asn5.46, Phe6.52 and Phe6.55 have an influence on affinity and/or potency of histamine to the H1R. The amino acids Asp3.32, Asp5.42 and Thr5.46 were shown to be involved in binding of histamine to the hH2R. Mutagenesis studies at the H3R showed that Leu5.39 has only small influence on affinity of histamine, whereas Ala5.42 and especially Glu5.46 have an influence on affinity of histamine. Furthermore, it was shown experimentally that the amino acids Asn4.57, Thr5.42, Ser5.43, Ser6.52 and especially Glu5.46 are involved in binding of histamine to the H4R.
To obtain a more detailed insight of the binding mode of histamine to the four histamine receptor subtypes on a molecular level, histamine was docked, considering experimentally determined mutagenesis and in silico data (Jongejan et al. 2005, 2008; Kooistra et al. 2013), into the orthosteric binding sites of the receptors (Fig. 7).

Comparison of the binding mode of histamine, docked into the four human histamine receptor subtypes (yellow circles: most important interactions between the respective receptor and histamine)
4.2 Binding Pathway of the Endogenous Ligand/Agonist Histamine to the Human Histamine H4 Receptor
As described above, within several studies, the binding mode of histamine at the hH4R was studied in silico by docking the histamine into the orthosteric binding site (Jongejan et al. 2008; Kiss et al. 2008). Although these studies are important, to interpret the results of mutagenesis studies on a molecular level, they give no information about the binding pathway of a ligand into its binding pocket of the receptor. However, in a recent study, the binding pathway of histamine into the orthosteric binding pocket of the hH4R was observed by unconstrained molecular dynamic simulations and could be divided into four phases (Fig. 8) (Wittmann and Strasser 2015).

Different phases of the whole binding process of a ligand into its binding site of a receptor
After a diffusion phase of the ligand in the aqueous phase (phase I, Fig. 9), a subsequent binding onto the extracellular surface of the hH4R was observed (phase II, Fig. 9). Afterwards, the histamine bound rapidly (<1 ns) into the orthosteric binding pocket (phase IIIa, Fig. 9), followed by an orientation phase of the histamine in the orthosteric binding pocket (phase IIIb, Fig. 9) (Wittmann and Strasser 2015). During the binding process, negatively charged amino acids at the surface or within the binding channel between the extracellular surface and the orthosteric binding pocket were observed to interact with the histamine. In the orthosteric binding pocket, the positively charged amine moiety of the histamine established a stable interaction with Gln7.42 and the highly conserved Asp3.32. Furthermore, the NH of the imidazole moiety formed a stable hydrogen bond with Glu5.46. This observation is in good accordance to mutagenesis studies, because for the Glu5.46Gln mutant, the affinity of the histamine to the hH4R decreased significantly (Jongejan et al. 2008).

Binding pathway of histamine from the extracellular side into the orthosteric binding site of the hH4R by unconstrained MD simulations [modified according to Wittmann and Strasser (2015)]
One first advantage of such MD simulations is that the ligand “finds” its binding mode without any constraints. In contrast, if the ligand is docked into the binding pocket and a subsequent MD simulation is performed, the binding mode of the ligand is possibly biased by the investigator. Of course, it has to be mentioned that such calculations are in general very time consuming and are only described for the β2R (Dror et al. 2011), hH4R (Wittmann and Strasser 2015), D2R and D3R (Thomas et al. 2016) until now. A second advantage of such MD simulations is that the amino acids, being involved in the ligand binding, can be identified, which is not possible by crystal structures, because here, the ligand is already bound into the binding pocket. Although the binding pathway has to be supported by mutagenesis and pharmacological studies, MD simulation so far is the only technique, which allows to observe the dynamic behaviour of ligand and receptor on a molecular level. And thus, MD simulation is an important and powerful technique to increase the understanding of histamine receptors on a molecular level. However, MD simulations are very time consuming, and only some hundred μs can be simulated until now, which may not be enough to observe the whole agonist binding and the related receptor-activation process.
4.3 Different Orientations of Ligands in the Binding Pocket
Phenylhistamines and histaprodifens (Fig. 10), H1R partial agonists, were developed as tools to study different histamine H1 receptor species in intact cell systems and in the Sf9 expression system (Leschke et al. 1995; Malinowska et al. 1999; Elz et al. 2000; Menghin et al. 2003; Seifert et al. 2003; Strasser et al. 2008a, 2009).

Pharmacological studies showed that histaprodifen and suprahistaprodifen show higher affinity to gpH1R than to hH1R (Strasser et al. 2008a, 2009). It is important to analyse species differences on a molecular level by combined mutagenesis and molecular modelling studies: Those studies increase the understanding of histamine receptors on a molecular level, e.g. with regard to subtype or species selectivity, which is important to develop new and more efficient drugs for therapy. By mutagenesis studies, the amino acid at position 2.61 was identified to act as a selectivity switch for suprahistaprodifen between gpH1R (Ser) and hH1R (Asn), but not for histaprodifen (Bruysters et al. 2005). Molecular modelling studies and MD simulations suggest that the smaller histaprodifen is bound into the main pocket (I, Fig. 5) near to TM5 and is, in contrast to the more bulky suprahistaprodifen, not in contact with TM2 of pocket II (Fig. 5) (Bruysters et al. 2005; Strasser et al. 2008a). Furthermore, the amino acid at position 2.61 may be involved in subtype or species differences at several histamine receptors for bulky ligands, which also occupy the second part of the orthosteric binding pocket (II, Fig. 5).
From a modelling point of view, an interesting class of partial agonists at the H1R are the phenoprodifens, hybrid compounds, comprising a histaprodifen and phenylhistamine partial structure (Fig. 10) (Strasser et al. 2008a). Since histaprodifens and phenylhistamines were suggested to bind in a pocket between TM3, TM5 and TM6, phenoprodifens were assumed to be able to bind in two different orientations into the orthosteric binding site of H1R (Bruysters et al. 2004; Strasser et al. 2009; Strasser and Wittmann 2010). The MD simulations showed differences in ligand-receptor interaction energy for phenoprodifen (Strasser et al. 2009): At hH1R, orientation 1 (diphenylpropylmoiety near to TM5) is preferred compared to orientation 2 (diphenylpropylmoiety near to TM2), while at gpH1R, none of both orientations is preferred. Furthermore, it is supported by QSAR studies that the orientation of phenoprodifens and suprahistaprodifens is dependent on the ligand structure and the H1R species (Strasser and Wittmann 2010). Although it is very hard to verify two different binding orientations of a ligand by experimental studies, e.g. by X-ray crystallography, considering two different binding orientations of ligands may be an important approach in development of new ligands, especially with regard to heterobivalent ligands.
4.4 Scaffold Hopping Approach to Identify New Ligand Classes
Experimental and virtual high-throughput screening is an established, but more or less time- and cost-consuming method to identify new ligands for a distinct target (Kumari et al. 2015). By contrast, based on a scaffold hopping approach starting from the quinoxalines (Smits et al. 2008b), new quinazolines were identified as highly potent H4R inverse agonists (Smits et al. 2008a): A side pocket with hydrophobic properties within the orthosteric binding site of the H4R was proposed by a fragment-based approach (Fig. 11) (Smits et al. 2008a): Based on these findings it was suggested that the same pocket could be occupied by substituents in 2-position of the quinoxaline and 4-position of the quinazoline moiety. Furthermore, based on a structural comparison of the quinazoline and quinoxaline scaffold, it is suggested that both moieties are similar regarding their binding mode in the orthosteric binding site of the hH4R. This study is a nice example that a scaffold hopping approach may be a useful approach to identify new classes of ligands not only at the H4R, but also at the other histamine receptor subtypes.

Scaffold hopping approach to develop a new class of hH4R ligands [modified according to Smits et al. (2008a)]
4.5 Impact of Molecular Modelling Studies to Explain the Pharmacology of Phenylhistamines at the hH4R
Phenylhistamines were identified as partial agonists at the H1 receptor (Strasser et al. 2009). But recent pharmacological studies showed that N-methylated and/or CF3- or Br-substituted phenylhistamines show a higher affinity to the hH4R than to the hH1R (Wittmann et al. 2011). The exchange of R1 = H → R1 = CH3 and R2 = H → R2 = Br,CF3 leads to an increase in affinity of two orders of magnitude at the hH4R (Fig. 12) (Wittmann et al. 2011). Subsequent MD simulations of the phenylhistamines showed that the methyl group (R1) and/or the Br/CF3 (R2) bind into two small subpockets 1 (R1) and 2 (R2) of the hH4R, which are not occupied by the unsubstituted phenylhistamine. Furthermore, the predicted Gibbs energies for the transfer of the ligand from the aqueous phase into the orthosteric binding pocket are in very good correlation with the experimentally determined affinities. This is a good example to demonstrate that molecular modelling studies are able to explain pharmacological data on a molecular level. However, it has to be taken into account that the ligands investigated within this study are structurally highly related and the predictive possibilities of molecular modelling studies might decrease in case of compounds with large structural differences.

Influence of small substituents in phenylhistamine onto affinity at the hH4R—a structural and energetical analysis [modified according to Wittmann et al. (2011)]
4.6 Influence of Monovalent Cations and Anions to the Histamine H3 and H4 Receptor
It was shown by experimental studies that the concentration of sodium ions has influence on the pharmacological data, e.g. potency or basal activity of the receptor at the hH3R or hH4R (Schneider et al. 2009; Schnell and Seifert 2010): With increasing concentration of NaCl, a decreasing basal activity of the hH3R and hH4R was observed, indicating that the inactive conformation of the receptor is stabilised. Based on experimental studies at several GPCRs (Selent et al. 2010; Katritch et al. 2014; Strasser et al. 2015), it was supported by MD simulations that Na+ is able to bind to the allosteric binding site near Asp2.50 at hH3R and hH4R (Fig. 13a–c) (Wittmann et al. 2014b). Recently, MD simulations were used to study the binding pathway of a sodium ion from the extracellular side via the orthosteric binding site into the allosteric binding site at the hH4R (Wittmann et al. 2014b; Strasser et al. 2015). The analysis of the ion entry path into the receptor showed that it is quite the same as for histamine at the hH4R (see Fig. 9) (Wittmann et al. 2014b; Strasser et al. 2015). Furthermore, the MD simulations suggest that the presence or absence of a sodium ion in the allosteric binding site may have influence on the binding mode of ligands, e.g. thioperamide at the hH3R (Wittmann et al. 2014a), which may explain differences in potencies in dependence of the NaCl concentration. In the MD simulations of a Na+ in its allosteric binding site near to Asp2.50 at hH3R and hH4R a water chain, connecting the highly conserved Asp3.32 of the orthosteric and Asp2.50 of the allosteric binding site, was observed (Wittmann et al. 2014b): While this water chain is continuous at the hH3R, it is disrupted, but bridged by Gln7.42 at hH4R. So far, it remains unclear if this water chain plays a role in receptor activation or subtype differences between hH3R and hH4R. A systematic analysis of the influence of monovalent cations (Li+, Na+, K+) and anions (Cl−, Br−, I−) on the hH3R showed that not only cations but also anions have an influence on the hH3R (Schnell and Seifert 2010), which is dependent on the chemical nature of the analysed monovalent ion. The MD simulations suggest that the depth of binding of the monovalent cation depends on its size (Fig. 13c), which may explain the different influence of cations on pharmacology of GPCRs (Schnell and Seifert 2010; Strasser et al. 2015). Furthermore, it is suggested that small positively charged ligands may be able to bind into the highly conserved Na+ pocket near to Asp2.50, e.g. the diuretic drug amiloride to the adenosine A2A receptor (Katritch et al. 2014). A similar observation was made during MD simulations of thioperamide in the binding pocket of the hH3R: In the presence of a sodium ion in the allosteric binding site, the thioperamide remains quite stable in the orthosteric binding pocket, but in the absence of a Na+ in the allosteric binding site, the positively charged imidazole moiety of the thioperamide moved between the orthosteric Asp3.32 and the allosteric Asp2.50 (Wittmann et al. 2014a). Thus, it is suggested that small molecules, able to bind in the allosteric Na+-binding site, may exhibit new functional properties or may open new opportunities in therapy (Katritch et al. 2014).

The allosteric cation-binding site near Asp2.50. (a) Interaction energy surface of Na+ with the orthosteric and allosteric binding site of the hH3R (blue: energetically preferred regions for the Na+). (b) Preferred areas for the Na+ in the allosteric binding site and water in the water channel and the orthosteric and allosteric binding site. (c) Overlay of the most preferred position of Li+, Na+, K+, Rb+ or Cs+ in the allosteric binding site of the hH3R according to MD simulations [modified according to Strasser et al. (2015)]
It was shown by MD simulations with monovalent cations and anions in the aqueous phase that the monovalent anions preferably bind between the intracellular part of the receptor, because in this region, some positively charged amino acids are located (Strasser et al. 2015). Since this is the same region for binding of the Gα-subunit, it is suggested that monovalent anions are involved in regulation of the interaction between receptor and Gα-subunit.
5 Conclusions and Future Studies
A large number of studies combining experimental (synthesis, pharmacological experiments, mutagenesis) and modelling techniques (QSAR, docking, MD simulation) addressing the histamine receptors were performed, reflecting that only a combination of several experimental and modelling techniques leads to an increased understanding of the histamine receptors on molecular level (Fig. 1) and provides synergistic input to each other. Although molecular modelling techniques are a powerful tool to obtain more detailed insights into histamine receptors (Table 5), it is necessary to proof or support the modelling results with experimental studies. However, one great advantage of modelling studies is that they allow to obtain deeper insights into the histamine receptors on a molecular level that will be complementary and even synergistic to experimental techniques.
Until now, a large number of questions in the histamine research were solved amongst others by molecular modelling studies (Table 5). However, there is a large number of remaining questions: For example, there is only little knowledge about the interactions between histamine receptors and G proteins or β-arrestin or about heterodimers on a molecular level. In future, modelling studies should focus on those questions, because they can provide important hints for mutagenesis studies to decode the interaction between a receptor and a specific G protein or for development of biased or bivalent ligands.
Abbreviations
- E2-loop:
-
Extracellular loop E2
- GPCR:
-
G protein-coupled receptor
- gpH1R:
-
Guinea-pig histamine H1 receptor
- h5-HT1BR:
-
Human serotonine 5-HT1B receptor
- h5-HT2BR:
-
Human serotonine 5-HT2B receptor
- hD3R:
-
Human dopamine D3 receptor
- hH1R:
-
Human histamine H1 receptor
- hH2R:
-
Human histamine H2 receptor
- hH3R:
-
Human histamine H3 receptor
- hH4R:
-
Human histamine H4 receptor
- hM2R:
-
Human muscarinic M2 receptor
- hβ2R:
-
Human adrenergic β2 receptor
- MD:
-
Molecular dynamics
- MM:
-
Molecular mechanics
- QM:
-
Quantum mechanics
- QSAR:
-
Quantitative structure activity relationship
- tβ1R:
-
Turkey adrenergic β1 receptor
- xHxR:
-
Different species of the four histamine receptor subtypes
References
Arora B, Coudrat T, Wootten D, Christopoulos A, Noronha SB, Sexton PM (2016) Prediction of loops in G protein-coupled receptor homology models: effect of imprecise surroundings and constraints. J Chem Inf Model 56:671–686
Bakker RA, Weiner DM, ter Laak T, Beuming T, Zuiderveld OP, Edelbroek M, Hacksell U, Timmerman H, Brann MR, Leurs R (2004) 8R-lisuride is a potent stereospecific histamine H1-receptor partial agonist. Mol Pharmacol 65:538–549
Beuming T, Sherman W (2012) Current assessment of docking into GPCR crystal structures and homology models: successes, challenges, and guidelines. J Chem Inf Model 52:3263–3277
Beuming T, Lenselink B, Pala D, McRobb F, Repasky M, Sherman W (2015) Docking and virtual screening strategies for GPCR drug discovery. Methods Mol Biol 1335:251–276
Bokoch MP, Zou Y, Rasmussen SG, Liu CW, Nygaard R, Rosenbaum DM, Fung JJ, Choi HJ, Thian FS, Kobilka TS, Puglisi JD, Weis WI, Pardo L, Prosser RS, Mueller L, Kobilka BK (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463:108–112
Brunskole I, Strasser A, Seifert R, Buschauer A (2011) Role of the second and third extracellular loops of the histamine H(4) receptor in receptor activation. Naunyn Schmiedebergs Arch Pharmacol 384:301–317
Bruysters M, Pertz HH, Teunissen A, Bakker RA, Gillard M, Chatelain P, Schunack W, Timmerman H, Leurs R (2004) Mutational analysis of the histamine H1-receptor binding pocket of histaprodifens. Eur J Pharmacol 487:55–63
Bruysters M, Jongejan A, Gillard M, van de Manakker F, Bakker RA, Chatelain P, Leurs R (2005) Pharmacological differences between human and guinea pig histamine H1 receptors: Asn84 (2.61) as key residue within an additional binding pocket in the H1 receptor. Mol Pharmacol 67:1045–1052
Chaudhari R, Heim AJ, Li Z (2015) Improving homology modeling of G-protein coupled receptors through multiple-template derived conserved inter-residue interactions. J Comput Aided Mol Des 29:413–420
Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318:1258–1265
Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A (2014) QSAR modeling: where have you been? Where are you going to? J Med Chem 57:4977–5010
Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC (2010) Structure of the human dopamine D3 receptor in complex with D2/D3 selective antagonist. Science 330:1091–1095
Christopher JA, Brown J, Dore AS, Errey JC, Koglin M, Marshall FH, Myszka DG, Rich RL, Tate CG, Tehan B, Warne T, Congreve M (2013) Biophysical fragment screening of the beta1-adrenergic receptor: identification of high affinity arylpiperazine leads using structure-based drug design. J Med Chem 56:3446–3455
Ciancetta A, Sabbadin D, Federico S, Spalluto G, Moro S (2015) Advances in computational techniques to study GPCR-ligand recognition. Trends Pharmacol Sci 36:878–890
Costanzi S (2012) Homology modeling of class a G protein-coupled receptors. Methods Mol Biol 857:259–279
Costanzi S (2013) Modeling G protein-coupled receptors and their interactions with ligands. Curr Opin Struct Biol 23:185–190
Damale MG, Harke SN, Kalam Khan FA, Shinde DB, Sangshetti JN (2014) Recent advances in multidimensional QSAR (4D-6D): a critical review. Mini Rev Med Chem 14:35–55
Darras FH, Pockes S, Huang G, Wehle S, Strasser A, Wittmann HJ, Nimczick M, Sotriffer CA, Decker M (2014) Synthesis, biological evaluation, and computational studies of Tri- and tetracyclic nitrogen-bridgehead compounds as potent dual-acting AChE inhibitors and hH3 receptor antagonists. ACS Chem Nerosci 5:225–242
de Graaf C, Rognan D (2009) Customizing G Protein-coupled receptor models for structure-based virtual screening. Curr Pharm Des 15:4026–4048
Deng Z, Chuaqui C, Singh J (2004) Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J Med Chem 47:337–344
Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE (2011) Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci U S A 108:13118–13123
Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE (2012) Biomolecular simulation: a computational microscope for molecular biology. Annu Rev Biophys 41:429–452
Elz S, Kramer K, Pertz HH, Detert H, ter Laak AM, Kuhne R, Schunack W (2000) Histaprodifens: synthesis, pharmacological in vitro evaluation, and molecular modeling of a new class of highly active and selective histamine H(1)-receptor agonists. J Med Chem 43:1071–1084
Filizola ME (2014) G protein-coupled receptors—modeling and simulation. Springer, Dordrecht. ISBN 978-94-007-7423-0
Filizola M, Carteni-Farina M, Perez JJ (1999) Modeling the 3D structure of rhodopsin using a de novo approach to build G-protein-coupled receptors. J Phys Chem B 103:2520–2527
Fiser A (2010) Template-based protein structure modeling. Methods Mol Biol 673:73–94
Geyer R, Nordemann U, Strasser A, Wittmann HJ, Buschauer A (2016) Conformational restriction and enantioseparation increase potency and selectivity of cyanoguanidine-type histamine H4 receptor agonists. J Med Chem 59:3452–3470
Goldfeld DA, Zhu K, Beuming T, Friesner RA (2011) Successful prediction of the intra- and extracellular loops of four G-protein-coupled receptors. Proc Natl Acad Sci U S A 108:8275–8280
Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482:547–551
Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC (2008) A specific cholesterol binding site is established by the 2.8 Å structure of the human beta2-adrenergic receptor. Structure 16:897–905
Heifetz A, Chudyk EI, Gleave L, Aldeghi M, Cherezov V, Fedorov DG, Biggin PC, Bodkin MJ (2016a) The fragment molecular orbital method reveals new insight into the chemical nature of GPCR-ligand interactions. J Chem Inf Model 56:159–172
Heifetz A, James T, Morao I, Bodkin MJ, Biggin PC (2016b) Guiding lead optimization with GPCR structure modeling and molecular dynamics. Curr Opin Pharmacol 30:14–21
Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH (1990) Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol 213:899–929
Huang J, Chen S, Zhang JJ, Huang XY (2013) Crystal structure of oligomeric beta1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol 20:419–425
Huang CY, Olieric V, Ma P, Howe N, Vogeley L, Liu X, Warshamanage R, Weinert T, Panepucci E, Kobilka B, Diederichs K, Wang M, Caffrey M (2016) In meso in situ serial X-ray crystallography of soluble and membrane proteins at cryogenic temperatures. Acta Crystallogr D Struct Biol 72:93–112
Irwin JJ, Shoichet BK (2016) Docking screens for novel ligands conferring new biology. J Med Chem 59:4103–4120
Istyastono EP, Nijmeijer S, Lim HD, van de Stolpe A, Roumen L, Kooistra AJ, Vischer HF, de Esch IJ, Leurs R, de Graaf C (2011) Molecular determinants of ligand binding modes in the histamine H(4) receptor: linking ligand-based three-dimensional quantitative structure-activity relationship (3D-QSAR) models to in silico guided receptor mutagenesis studies. J Med Chem 54:8136–8147
Jongejan A, Bruysters M, Ballesteros JA, Haaksma E, Bakker RA, Pardo L, Leurs R (2005) Linking agonist binding to histamine H1 receptor activation. Nat Chem Biol 1:98–103
Jongejan A, Lim HD, Smits RA, de Esch IJ, Haaksma E, Leurs R (2008) Delineation of agonist binding to the human histamine H4 receptor using mutational analysis, homology modeling, and ab initio calculations. J Chem Inf Model 48:1455–1463
Katritch V, Fenalti G, Abola EE, Roth BL, Cherezov V, Stevens RC (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem Sci 39:233–244
Kiss R, Noszal B, Racz A, Falus A, Eros D, Keseru GM (2008) Binding mode analysis and enrichment studies on homology models of the human histamine H4 receptor. Eur J Med Chem 43:1059–1070
Koehler Leman J, Ulmschneider MB, Gray JJ (2015) Computational modeling of membrane proteins. Proteins 83:1–24
Kooistra AJ, Kuhne S, De Esch IJ, Leurs R, De Graaf C (2013) A structural chemogenomics analysis of aminergic GPCRs: lessons for histamine receptor ligand design. Br J Pharmacol 170:101–126
Kooistra AJ, de Graaf C, Timmerman H (2014) The receptor concept in 3D: from hypothesis and metaphor to GPCR-ligand structures. Neurochem Res 39:1850–1861
Kooistra AJ, Vischer HF, McNaught-Flores D, Leurs R, de Esch IJ, de Graaf C (2016) Function-specific virtual screening for GPCR ligands using a combined scoring method. Sci Rep 6:28288
Kovalainen JT, Christiaans JAM, Ropponen R, Poso A, Perakyla M, Vepsalainen J, Laatikainen R, Gynther J (2000) A proton relay process as the mechanism of activation of the histamine H3-receptor determined by 1H NMR and ab initio quantum mechanical calculations. J Am Chem Soc 122:6989–6996
Kuhne S, Kooistra AJ, Bosma R, Bortolato A, Wijtmans M, Vischer HF, Mason JS, de Graaf C, de Esch IJ, Leurs R (2016) Identification of ligand binding hot spots of the histamine H1 receptor following structure-based fragment optimization. J Med Chem 59:9047–9061
Kumari P, Ghosh E, Shukla AK (2015) Emerging approaches to GPCR ligand screening for drug discovery. Trends Mol Med 21:687–701
Leschke C, Elz S, Garbarg M, Schunack W (1995) Synthesis and histamine H1 receptor agonist activity of a series of 2-phenylhistamines, 2-heteroarylhistamines, and analogues. J Med Chem 38:1287–1294
Leslie AG, Warne T, Tate CG (2015) Ligand occupancy in crystal structure of beta1-adrenergic G protein-coupled receptor. Nat Struct Mol Biol 22:941–942
Levoin N, Labeeuw O, Billot X, Calmels T, Danvy D, Krief S, Berrebi-Bertrand I, Lecomte JM, Schwartz JC, Capet M (2016) Discovery of nanomolar ligands with novel scaffolds for the histamine H4 receptor by virtual screening. Eur J Med Chem 125:565–572
Lim HD, Jongejan A, Bakker RA, Haaksma E, de Esch IJ, Leurs R (2008) Phenylalanine 169 in the second extracellular loop of the human histamine H4 receptor is responsible for the difference in agonist binding between human and mouse H4 receptors. J Pharmacol Exp Ther 327:88–96
Malinowska B, Piszcz J, Schlicker E, Kramer K, Elz S, Schunack W (1999) Histaprodifen, methylhistaprodifen, and dimethylhistaprodifen are potent H1-receptor agonists in the pithed and in the anaesthetized rat. Naunyn Schmiedebergs Arch Pharmacol 359:11–16
McRobb FM, Negri A, Beuming T, Sherman W (2016) Molecular dynamics techniques for modeling G protein-coupled receptors. Curr Opin Pharmacol 30:69–75
Menghin S, Pertz HH, Kramer K, Seifert R, Schunack W, Elz S (2003) N(alpha)-imidazolylalkyl and pyridylalkyl derivatives of histaprodifen: synthesis and in vitro evaluation of highly potent histamine H(1)-receptor agonists. J Med Chem 46:5458–5470
Miller-Gallacher JL, Nehme R, Warne T, Edwards PC, Schertler GF, Leslie AG, Tate CG (2014) The 2.1 A resolution structure of cyanopindolol-bound beta1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One 9:e92727
Mobarec JC, Sanchez R, Filizola M (2009) Modern homology modeling of G-protein coupled receptors: which structural template to use? J Med Chem 52:5207–5216
Mordalski S, Kosciolek T, Kristiansen K, Sylte I, Bojarski AJ (2011) Protein binding site analysis by means of structural interaction fingerprint patterns. Bioorg Med Chem Lett 21:6816–6819
Moukhametzianov R, Warne T, Edwards PC, Serrano-Vega MJ, Leslie AG, Tate CG, Schertler GF (2011) Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc Natl Acad Sci U S A 108:8228–8232
Munk C, Harpsoe K, Hauser AS, Isberg V, Gloriam DE (2016) Integrating structural and mutagenesis data to elucidate GPCR ligand binding. Curr Opin Pharmacol 30:51–58
Naporra F, Gobleder S, Wittmann HJ, Spindler J, Bodensteiner M, Bernhardt G, Hubner H, Gmeiner P, Elz S, Strasser A (2016) Dibenzo[b,f][1,4]oxazepines and dibenzo[b,e]oxepines: Influence of the chlorine substitution pattern on the pharmacology at the H1R, H4R, 5-HT2AR and other selected GPCRs. Pharmacol Res 113:610–625
Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289:739–745
Panula P, Chazot PL, Cowart M, Gutzmer R, Leurs R, Liu WL, Stark H, Thurmond RL, Haas HL (2015) International union of basic and clinical pharmacology. XCVIII. Histamine receptors. Pharmacol Rev 67:601–655
Peeters MC, van Westen GJ, Li Q, IJzerman AP (2011) Importance of the extracellular loops in G protein-coupled receptors for ligand recognition and receptor activation. Trends Pharmacol Sci 32:35–42
Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VRP, Sansihvili R, Fischetti RF, Schertler GFX, Weis WI, Kobilka BK (2007) Crystal structure of the human beta 2 adrenergic G protein-coupled receptor. Nature 450:383–387
Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK (2011a) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469:175–180
Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK (2011b) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477:549–555
Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK (2013) Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502:575–579
Rodriguez D, Bello X, Gutierrez-de-Teran H (2012) Molecular modelling of G protein-coupled receptors through the web. Mol Inform 31:334–341
Rosenbaum DM, Zhang D, Lyons J, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi H-J, Devree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK (2011) Structure and function of an irreversible agonist-beta2 adrenoceptor complex. Nature 469:236–240
Sandal M, Duy TP, Cona M, Zung H, Carloni P, Musiani F, Giorgetti A (2013) GOMoDo: a GPCRs online modeling and docking webserver. PLoS One 8:e74092
Schneider EH, Seifert R (2016) Pharmacological characterization of human histamine receptors and histamine receptor mutants in the Sf9 cell expression system. Handb Exp Pharmacol. doi:10.1007/164_2016_124
Schneider EH, Schnell D, Papa D, Seifert R (2009) High constitutive activity and a G-protein-independent high-affinity state of the human histamine H(4)-receptor. Biochemistry 48:1424–1438
Schnell D, Seifert R (2010) Modulation of histamine H(3) receptor function by monovalent ions. Neurosci Lett 472:114–118
Schultes S, Nijmeijer S, Engelhardt H, Kooistra AJ, Vischer HF, de Esch IJP, Haaksma EEJ, Leurs R, de Graaf C (2013) Mapping histamine H4 receptor-ligand binding modes. Med Chem Commun 4:193–204
Seifert R, Wenzel-Seifert K, Bürckstümmer T, Pertz HH, Schunack W, Dove S, Buschauer A, Elz S (2003) Multiple differences in agonist and antagonist pharmacology between human and guinea-pig histamine H1-receptor. J Pharmacol Exp Ther 305:1104–1115
Seifert R, Strasser A, Schneider EH, Neumann D, Dove S, Buschauer A (2013) Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol Sci 34:33–58
Selent J, Sanz F, Pastor M, De Fabritiis G (2010) Induced effects of sodium ions on dopaminergic G-protein coupled receptors. PLoS Comput Biol 6(8) pii: e1000884
Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S (2011) Structure of the human histamine H-1 receptor complex with doxepin. Nature 475:65–U82
Shiroishi M, Kobayashi T (2016) Structural analysis of the histamine H1 receptor. Handb Exp Pharmacol. doi:10.1007/164_2016_10
Sirci F, Istyastono EP, Vischer HF, Kooistra AJ, Nijmeijer S, Kuijer M, Wijtmans M, Mannhold R, Leurs R, de Esch IJ, de Graaf C (2012) Virtual fragment screening: discovery of histamine H3 receptor ligands using ligand-based and protein-based molecular fingerprints. J Chem Inf Model 52:3308–3324
Smits RA, de Esch IJ, Zuiderveld OP, Broeker J, Sansuk K, Guaita E, Coruzzi G, Adami M, Haaksma E, Leurs R (2008a) Discovery of quinazolines as histamine H4 receptor inverse agonists using a scaffold hopping approach. J Med Chem 51:7855–7865
Smits RA, Lim HD, Hanzer A, Zuiderveld OP, Guaita E, Adami M, Coruzzi G, Leurs R, de Esch IJ (2008b) Fragment based design of new H4 receptor-ligands with anti-inflammatory properties in vivo. J Med Chem 51:2457–2467
Southan C (2016) Retrieving GPCR data from public databases. Curr Opin Pharmacol 30:38–43
Strasser A (2009) Molecular modeling and QSAR-based design of histamine receptor ligands. Expert Opin Drug Discovery 4:1061–1075
Strasser A, Wittmann HJ (2007) LigPath: a module for predictive calculation of a ligand’s pathway into a receptor-application to the gpH1-receptor. J Mol Model 13:209–218
Strasser A, Wittmann HJ (2010) 3D-QSAR CoMFA study to predict orientation of suprahistaprodifens and phenoprodifens in the binding-pocket of four histamine H1-receptor species. Mol Inform 29:333–341
Strasser A, Wittmann H-J (2013) Modelling of GPCRs—a practical handbook. Springer, Dordrecht
Strasser A, Striegl B, Wittmann HJ, Seifert R (2008a) Pharmacological profile of histaprodifens at four recombinant H1-receptor species isoforms. J Pharmacol Exp Ther 324:60–71
Strasser A, Wittmann HJ, Seifert R (2008b) Ligand-specific contribution of the N terminus and E2-loop to pharmacological properties of the histamine H1-receptor. J Pharmacol Exp Ther 326:783–791
Strasser A, Wittmann HJ, Kunze M, Elz S, Seifert R (2009) Molecular basis for the selective interaction of synthetic agonists with the human histamine H1-receptor compared with the guinea pig H1-receptor. Mol Pharmacol 75:454–465
Strasser A, Wittmann H-J, Buschauer A, Schneider EH, Seifert R (2013) Species-dependent activities of GPCR ligands: lessons from histamine receptor orthologs. Trends Pharmacol Sci 34:13–32
Strasser A, Wittmann HJ, Schneider EH, Seifert R (2015) Modulation of GPCRs by monovalent cations and anions. Naunyn Schmiedebergs Arch Pharmacol 388:363–380
Tautermann CS, Seeliger D, Kriegel JM (2015) What can we learn from molecular dynamic simulations for GPCR drug design? Comput Struct Biotechnol J 13:111–121
ter Laak AM, Timmerman H, Leurs R, Nederkoorn PH, Smit MJ, Donne-Op den Kelder GM (1995) Modelling and mutation studies on the histamine H1-receptor agonist binding site reveal different binding modes for H1-agonists: Asp116 (TM3) has a constitutive role in receptor stimulation. J Comput Aided Mol Des 9:319–330
Thomas T, Fang Y, Yuriev E, Chalmers DK (2016) Ligand binding pathways of clozapine and haloperidol in the dopamine D2 and D3 receptors. J Chem Inf Model 56:308–321
Vass M, Kooistra AJ, Ritschel T, Leurs R, de Esch IJ, de Graaf C (2016) Molecular interaction fingerprint approaches for GPCR drug discovery. Curr Opin Pharmacol 30:59–68
Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GFX, Babu MM (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194
Verma J, Khedkar VM, Coutinho EC (2010) 3D-QSAR in drug design—a review. Curr Top Med Chem 10:95–115
Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC (2010) Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc 132:11443–11445
Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC (2013) Structural features for functional selectivity at serotonin receptors. Science 340:615–619
Wagner E, Wittmann HJ, Elz S, Strasser A (2011) Mepyramine-JNJ7777120-hybrid compounds show high affinity to hH(1)R, but low affinity to hH(4)R. Bioorg Med Chem Lett 21:6274–6280
Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE (2013) Structural basis for molecular recognition at serotonin receptors. Science 340:610–614
Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454:486–491
Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG (2011) The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469:241–244
Warne T, Edwards PC, Leslie AG, Tate CG (2012) Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure 20:841–849
Weichert D, Kruse AC, Manglik A, Hiller C, Zhang C, Hubner H, Kobilka BK, Gmeiner P (2014) Covalent agonists for studying G protein-coupled receptor activation. Proc Natl Acad Sci U S A 111:10744–10748
Wifling D, Löffel K, Nordemann U, Strasser A, Bernhardt G, Dove S, Seifert R and Buschauer A (2015a) Molecular determinants for the high constitutive activity of the human histamine H4 receptor: functional studies on orthologues and mutants. Br J Pharmacol 172:785–798
Wifling D, Bernhardt G, Dove S, Buschauer A (2015b) The extracellular loop 2 (ECL2) of the human histamine H4 receptor substantially contributes to ligand binding and constitutive activity. PLoS One 10:e0117185
Wittmann HJ, Strasser A (2015) Binding pathway of histamine to the hH4R, observed by unconstrained molecular dynamics. Bioorg Med Chem Lett 25:1259–1268
Wittmann HJ, Elz S, Seifert R, Strasser A (2011) N (alpha)-methylated phenylhistamines exhibit affinity to the hH(4)R-a pharmacological and molecular modelling study. Naunyn Schmiedebergs Arch Pharmacol 384:287–299
Wittmann HJ, Seifert R, Strasser A (2014a) Mathematical analysis of the sodium sensitivity of the human histamine H3 receptor. In Silico Pharmacol 2:1
Wittmann HJ, Seifert R, Strasser A (2014b) Sodium binding to hH3R and hH 4R—a molecular modeling study. J Mol Model 20:2394
Xu D, Zhang Y (2012) Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80:1715–1735
Yarnitzky T, Levit A, Niv MY (2010) Homology modeling of G-protein-coupled receptors with X-ray structures on the rise. Curr Opin Drug Discov Devel 13:317–325
Yuan S, Vogel H, Filipek S (2011) The role of water and sodium ions in the activation of the μ-opioid receptor. Angew Chem Int Ed 52:10112–10115
Yuriev E, Holien J, Ramsland PA (2015) Improvements, trends, and new ideas in molecular docking: 2012-2013 in review. J Mol Recognit 28:581–604
Zhang J, Yang J, Jang R, Zhang Y (2015) GPCR-I-TASSER: a hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure 23:1538–1549
Zou Y, Weis WI, Kobilka BK (2012) N-terminal T4 lysozyme fusion facilitates crystallization of a G protein coupled receptor. PLoS One 7:e46039
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Strasser, A., Wittmann, HJ. (2017). Molecular Modelling Approaches for the Analysis of Histamine Receptors and Their Interaction with Ligands. In: Hattori, Y., Seifert, R. (eds) Histamine and Histamine Receptors in Health and Disease. Handbook of Experimental Pharmacology, vol 241. Springer, Cham. https://doi.org/10.1007/164_2016_113
Download citation
DOI: https://doi.org/10.1007/164_2016_113
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58192-7
Online ISBN: 978-3-319-58194-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)