
Abstract
Evidenced by the three-rounds of G-protein coupled receptors (GPCR) Dock competitions, improving homology modeling methods of helical transmembrane proteins including the GPCRs, based on templates of low sequence identity, remains an eminent challenge. Current approaches addressing this challenge adopt the philosophy of “modeling first, refinement next”. In the present work, we developed an alternative modeling approach through the novel application of available multiple templates. First, conserved inter-residue interactions are derived from each additional template through conservation analysis of each template-target pairwise alignment. Then, these interactions are converted into distance restraints and incorporated in the homology modeling process. This approach was applied to modeling of the human β2 adrenergic receptor using the bovin rhodopsin and the human protease-activated receptor 1 as templates and improved model quality was demonstrated compared to the homology model generated by standard single-template and multiple-template methods. This method of “refined restraints first, modeling next”, provides a fast and complementary way to the current modeling approaches. It allows rational identification and implementation of additional conserved distance restraints extracted from multiple templates and/or experimental data, and has the potential to be applicable to modeling of all helical transmembrane proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The G-protein coupled receptors (GPCR) superfamily represents a biologically and pharmaceutically important class of membrane proteins [1]. With more than 800 members in human [2], GPCRs mediate signal transduction from outside to inside the cell through binding to a variety of cellular stimuli including light, amino acids, and peptides. Accounted for the targets of approximately 27 % of drugs available in market [3], GPCRs remain an invaluable therapeutic target for future drug discovery [4]. However, there are only about thirty X-ray structures reported for members of GPCRs and most of them belong to the Class A family except for the human glucagon receptor [5] and the human corticotropin-releasing factor receptor 1 [6] of Class B GPCR, the human mGlu1 receptor [7] of Class C GPCR, as well as the human smoothened receptor [8] of Class F GPCR. Given the paucity of available high-resolution structures, computational modeling techniques play an important role in the functional studies of the GPCR superfamily [9] and in their structure-based drug design efforts [10]. All GPCRs are believed to share a common topology of seven transmembrane (TM) helices connected by intracellular and extracellular loops, as well as amino and carboxyl terminus [11, 12]. Among these components, the TM region is involved in ligand binding and signal transduction, and is a central focus of current modeling efforts.
Various computational approaches have been applied to the modeling of the TM domains of GPCRs, which can be approximately classified into three categories: de novo methods, homology modeling methods, and hybrid methods. De novo methods such as ROSETTA [13], BiHelix [14], PconsFold [15], and MemBrain [16] construct the structural model of a GPCR from its amino acid sequence through conformational sampling and scoring; Homology modeling methods utilize the known X-ray structures of GPCRs as templates to construct the structural model of the target GPCR protein [17]. Although homology modeling remains the most accurate approach for GPCR modeling in general, the rarity of available GPCR structures limits the quality of models constructed using this approach because the accuracy of a homology model is directly related to the similarity between the template and the target protein sequence. For many GPCRs, their homology models have to be constructed using structural templates of low sequence identity. Consequently, the quality of such models is uncertain and their application is often limited.
Hybrid methods including foldGPCR [18] and LITICon [19] are proposed to improve the quality of models of GPCRs through the combined use of structural information from template proteins and/or experimental data, and limited conformational sampling [10, 20, 21, 34]. In several cases, hybrid methods have been shown to be quite successful in GPCR modeling. These methods of “modeling first, refinement next”, however, use a single best GPCR structural template of highest sequence identity. The overall success of these methods in improving the homology models of GPCRs based on the template of low sequence identity has not been assessed systematically. As evidenced by the recent GPCR Dock competitions [22, 23, 29], developing novel homology modeling approaches to improve the model quality based on the template of low sequence identity is very desirable.
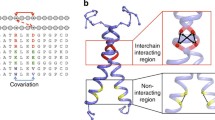
In this work, we aim to develop a complementary hybrid homology modeling method of “refined restraints first, modeling next” to help improve the inherent structural divergence in the TM region between the templates of low sequence identity and the target GPCR. Our method takes advantage of multiple crystal structures of GPCRs available in the database, and takes into account the fact that conserved inter-residue interactions exist between protein pairs of even low sequence identity [24]. Unlike traditional homology modeling approaches adopting multiple template structures [25], by which, each individual region of the target protein structure is modeled based on the aligned region of a single template structure or the average of the multiple template structures (Fig. 1a), our method aims to derive as many as possible additional inter-residue interactions existing in the target protein structure through conservation analysis of each additional template-target pairwise alignment, then incorporate them into homology modeling as additional distance restraints (Fig. 1b). This method was applied to modeling of the human β2 adrenergic receptor using the bovin rhodopsin and the human protease-activated receptor 1 as templates and should be applicable to modeling of other helical membrane proteins.

Comparison of homology modeling approach based on multiple templates. a Traditional approach by which each individual region of the target protein structure is modeled based on the aligned region of a single template structure. b Our proposed approach that incorporates only conserved inter-residue interactions from the additional templates into model construction. Top box an inter-residue interaction inherited from the primary template. Bottom box an additional inter-residue interaction derived from the additional template
Methods
The proposed method includes several steps (Fig. 2): (1) Perform database search and multiple sequence alignment; (2) Derive conserved inter-residue restraints from additional templates; and (3) Construct the homology models of the target GPCR protein while incorporating those inter-residue restraints.

A flowchart of the proposed modeling procedure
Database search and sequence alignment
First, sequences of the target and the template proteins were used as the query respectively to search the non-redundant protein sequence database at www.ncbi.nlm.nih.gov using BLAST [32]. For each query sequence, five homology sequences of 80–90 % sequence identity were identified. Next, sequences of the target and the template proteins along with their five homologous sequences were loaded in MOE (Molecular Computing Group Inc., version 2011.10) and aligned using its sequence alignment function. Finally, the above alignment was manually adjusted to remove gaps by ensuring the alignment of most conserved TM residues as identified by the Ballesteros numbering system [26]. This alignment was used for elucidation of conserved inter-residue interactions and for model construction.
Derivation of conserved inter-residue restraints
For the template structures, the loops of the soluble regions, the amino and carboxyl terminus, and the ligand were manually removed to keep only alpha helices that belong to the TM region. Similarly, for the target GPCR sequence, only those amino acids that were aligned with the alpha helices of the TM region of the selected template were preserved for homology modeling. In addition, between any two consecutive TM helices, extra five residues in the loop regions (two or three from the end of each helix) were kept when available in order to form a short loop connecting the two TM helices during model construction.
To generate conserved inter-residue restraints, the truncated structure of the second template was loaded into CMView [30] and a list of inter-residue interactions was reported. An interaction was defined between two residues if the distance between any two atoms from the two residues was ≤5 Å [31]. This list was then winnowed to preserve only those conserved interactions. A conserved interaction was defined as one for which the two residues involved in forming the interaction should be identical between the template and the target protein sequence as well as their homologous sequences. The derived list of conserved interactions was then winnowed to preserve inter-helical restraints only and converted into the Modeller [28] restraint format for the target protein.
Construction of homology models
Employing the alignment above, the homology models of the target GPCR were subsequently constructed based on the truncated template structure using Modeller (version 9.11) [28]. Modeller is a popular homology modeling software that constructs three-dimensional structures of proteins by satisfaction of spatial restraints. Using Modeller, homology models of the target protein were constructed with various functions including one template modeling, multiple template modeling, modeling with additional restraints, and multiple model generation (Table 1).
To evaluate the model quality of the target GPCR, its crystal structure and the homology models were first clipped to preserve only residues in the TM region as defined in the PDBTM database [27]. The Cα atom RMSD between each model and the crystal structure was then calculated using the Superposition function in MOE.
Test case
To demonstrate the effectiveness of our proposed approach, we purposely chose two dissimilar GPCR proteins of known X-ray structures, the bovin rhodopsin (PDB ID: 1U19) and the human β2 adrenegic GPCR (PDB ID: 2RH1). The sequence identity between the TM regions of the two proteins is ~22 %. For this exercise, the structure of the bovin rhodopsin was adopted as the template, and the goal was to construct a model of the human β2 adrenegic GPCR based on it. To study the effect of multiple templates using our proposed approach, the human protease-activated receptor 1 (PDB ID: 3VW7) was chosen as the second template due to again its sequence and structure dissimilarity with the β2 adrenegic receptor. The sequence identity between the TM regions of the two proteins is ~23 %.
Results
Conserved distance restraints
From the first template structure of the bovin rhodopsin receptor (PDB ID: 1U19), and by applying the strict rule, 17 inter-helical interactions were identified as conserved between this template and the target GPCR (Supplementary Table I). Similarly from the second template structure of the human protease-activated receptor 1 (PDB ID: 3VW7), 23 unique inter-helical interactions were identified as conserved (Supplementary Table II). All these inter-helical restraints were adopted for model construction whenever indicated (Table 1).
Comparison of multiple-template and single-template modeling approaches
Using the structures of the bovin rhodopsin receptor and the human protease-activated receptor 1 as templates and employing the standard multiple-template modeling function in Modeller, the homology model of the human β2 adrenegic GPCR showed the Cα RMSD value of 2.92 Å. This value was even worse than the homology model generated using the single template of the bovin rhodopsin receptor structure (Fig. 3).

Cα RMSD value of the homology model of the human β2 adrenegic GPCR using Modeller compared to the crystal structure. Models I–V correspond to conditions listed in Table 1
In contrast, using the structure of the bovin rhodopsin receptor as the template and by incorporating the 23 additional restraints derived from the second template structure of the human protease-activated receptor 1, the homology model of the human β2 adrenegic GPCR had the Cα RMSD value of 2.26 Å. This result was significantly better than the model generated using the standard multiple-template approach and was also better than the models generated using either single template (3.18 and 2.31 Å). These results clearly demonstrated the effectiveness of the proposed multiple-template modeling approach.
Insight from the generation of multiple models
To gain further insight into the effect of the various modeling approaches, 500 homology models were generated in Modeller using the single-template approach with the bovin rhodopsin template which had the better Cα RMSD value, as well as the two multiple-template approaches. For the standard single-template modeling approach, the Cα RMSD values of the 500 models varies from 2.22 to 2.45 Å (Fig. 4a), In addition, all of the models had the RMSD value <2.50 Å. In contrast, for the standard multiple-template modeling approach, the Cα RMSD values of the 500 models varies significantly from 2.24 to 3.19 Å (Fig. 4b), with most of the models having the RMSD value >2.50 Å.

Cα RMSD value of the 500 generated models of the human β2 adrenegic GPCR using Modeller compared to the crystal structure. a Modeling using a single template as VI in Table 1; b Modeling using the traditional multiple-template approach as VII in Table 1; c Modeling using the proposed multiple-template approach as VIII in Table 1
For our proposed approach, the variation of the Cα RMSD values of the 500 models was much smaller from 2.19 to 2.50 Å, a range similar to the single-template approach (Fig. 4c). Furthermore, the best model from our approach (2.19 Å) is again better than either the single-template or the standard multiple-template approach (Fig. 5). Considering the fact that discriminating structural models with current scoring functions remains a challenge [33, 38], the smaller model variation resulted from our approach certainly helps with obtaining a good homology model.

Structural superimposition between the best conformation of the human β2 adrenegic GPCR by Cα RMSD value (in gray) and the crystal structure (in black)
Discussion
Homology modeling remains an important tool in the structure–function studies of GPCRs as well as in their structure-based drug-discovery efforts [9, 10]. With few high-resolution crystal structures available for members of this biologically and therapeutically important class, it is very desirable to develop novel approaches to improve the model quality of GPCRs constructed based on the template structures of low sequence identity. Universally conserved amino acid residues are observed for every TM helix of the GPCR superfamily [26], they can serve as the anchor residue to obtain the optimal alignment between the template and the target GPCR sequence. Therefore, the major remaining challenge is to develop effective computational approaches to improve the inherent structural divergence in the TM region between the template of low sequence identity and the target GPCR. Hybrid methods that construct the homology model of GPCRs using standard methods followed by limited conformational sampling represents a promising approach [10, 18–21]. In this work, we present a complementary hybrid method that utilizes structural information from multiple GPCR crystal structures in the database in a novel way.
Traditional multiple-template homology modeling approaches have been reported which model the individual region of the target protein structure based on the aligned region of a single template structure or the average of the multiple template structures [35]. The use of multiple templates is regarded to naturally improve the accuracy of homology models. However, a later study showed that the demonstrated improvement is mainly due to the extension of the models based on the multiple templates [25]. As demonstrated here (Fig. 3), constructing the same region of the target protein by averaging the multiple template structures does not automatically improve the model accuracy. To some extent, this is understandable.
A folded protein structure contains many inter-residue interactions that play a crucial role in driving protein folding. It has been shown that the use of native-like inter-residue interaction based distance restraints is able to reproduce structural model essentially identical to the crystal structure of the target protein [36]. From the perspective of improving model quality, the crucial step is to obtain a complete and accurate set of inter-residue interactions within the structure of the target protein. When the sequence identity between the template and the target protein is low, there could be inherent structural divergence between them. Consequently, the intersection of their inter-residue interaction sets is small and simply averaging the interactions sets obtained from multiple templates does not automatically improve the quality of the interaction set for the target protein. Therefore, constructing the homology model by such a traditional approach as implemented in Modeller won’t necessarily result in the improvement in the accuracy of the target model.
For a protein structure, its inter-residue interactions can be classified into three categories: (1) Global interactions existing in all the protein structures within the same family. For homology modeling, these interactions are naturally carried over by any single template; (2) Pair-specific interactions shared by two or more homologous structures, but not all structures within the same family (Fig. 6); and (3) Individual-specific interactions existing only within one structure. For our approach, we chose to identify as many as possible pair-specific inter-residue interactions existing in both the target protein structure and each additional template structure. Hence, unlike the multiple-template modeling approach in Modeller, which derives all three types of spatial restraints from both templates and treats those restraints equally in term of modeling; our approach utilizes only a small subset of the spatial restraints from the second template by applying the strict criterion to filter out the third type of inter-residue interactions. As shown in the Result section, this approach showed improvement in the final model accuracy even though the template of the bovin rhodopsin receptor is quite similar to the target protein structurally (Fig. 3).

Illustration of pair-specific inter-residue interactions
Another advantage of our method is the flexibility of incorporating additional data from more templates or experimental data into the modeling process. However, it is necessary to emphasize that care needs to be practiced to ensure that the type of the experimental data to be incorporated can be reliably converted into distance or torsional angle restraints. In this regard, photo affinity labeling or FRET data are probably straightforward to implement. Mutagenesis data are less so because the impact of a mutagenesis could be due to the spatial closeness or the long-range effect. Hence, its implementation is less reliable. Nevertheless, attempt can be made and the final model can be assessed to determine the usefulness of such data.
Several hybrid methods have been reported to improve the model accuracy of GPCRs [10, 18–21]. These methods use a single best GPCR structural template of highest sequence identity and their application has only been demonstrated in individual GPCR modeling cases. Hence, it is difficult to compare the performance of our approach with them systematically, in particular, for modeling of GPCRs based on the template of low sequence identity. However, methodologically, there are clear differences. For instance, those reported hybrid approaches adopt the “modeling first, refinement next” philosophy and focus primarily on the development of new sampling algorithms. In contrast, our method focuses on extracting more reliable structural information of the target protein from multiple temple structures in a novel way, adopting the “refined restraints first, modeling next” philosophy. Given the complementarity of these methods, it will be of interest to integrating them together in the future. For instance, a model generated using our approach can be still subjected to MD simulations for further sampling and refinement.
Our approaches can be further improved from several perspectives. Just like traditional homology modeling techniques, the choice of the original template makes difference in the quality of the final model. Using the structure of the bovin rhobopsin as the template, the model had the Cα RMSD value of 2.26 Å; while using the structure of the human protease-activated receptor 1 as the template, the model had the Cα RMSD value of 3.09 Å. This is understandable since the additional restraints derived from the second template were limited to inter-helical interactions. The quality of the individual TM helices themselves certainly matters to the final model quality. In practice, attempts should be made to construct the homology models using each template structure as the primary template while incorporating additional restraints from other templates. These resultant models can be ranked to identify the best one. Also, more template structures could be used to derive more distance restraints.
In addition, further improvement to the quality of the individual helix model of the target protein can be implemented. First, instead of modeling the individual helix from a single template structure, the modeling can be done based on the individual TM helix in the PDB database that has the highest sequence identity to the TM helix in the target protein. Secondly, several computational tools for the prediction of kinks in TM helices have been reported [37]. Potential kinks in the target protein can be predicted using these tools and the results can be used to adjust the model accordingly.
Furthermore, in the current implementation, the definition of residue conservation is simply based on identity. Over the years, many different quantitative ways have been proposed to score residue conservation [39]. By comparing the existing inter-residue interactions in homologous membrane protein structures, the best definition of conserved residues can be derived that will result in a list of conserved inter-helical restraints between each template-target pair with best overall specificity and sensitivity.
Conclusions
In summary, we proposed an innovative, easy to implement homology modeling strategy that helps improve the model accuracy of GPCRs constructed based on the template structures of low sequence identity. This strategy derives structural information from multiple templates and/or experimental data and incorporates them into model construction. It complementarities current hybrid homology modeling approaches, and can be easily integrated into those approaches. This approach can be potentially applied to the modeling of other membrane proteins.
References
Lin HH (2013) G-protein-coupled receptors and their (Bio) chemical significance win 2012 Nobel Prize in Chemistry. Biomed J 36:118–124
Foord SM, Bonner TI, Neubig RR, Rosser EM, Pin JP, Davenport AP, Spedding M, Harmar AJ (2005) International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol Rev 57:279–288
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996
Lagerstrom MC, Schioth HB (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov 7:339–357
Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, Zhou C, Xu Q, Wacker D, Joseph JS, Liu W, Lau J, Cherezov V, Katritch V, Wang MW, Stevens RC (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499:444–449
Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, Cooke RM, Weir M, Marshall FH (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499:438–443
Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V, Conn PJ, Stevens RC (2014) Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344:58–64
Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC (2013) Structure of the human smoothened receptor bound to an antitumour agent. Nature 497:338–343
Levit A, Barak D, Behrens M, Meyerhof W, Niv MY (2012) Homology model-assisted elucidation of binding sites in GPCRs. Methods Mol Biol 914:179–205
Katritch V, Rueda M, Lam PC, Yeager M, Abagyan R (2010) GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins 78:197–211
Baldwin JM, Schertler GF, Unger VM (1997) An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J Mol Biol 272:144–164
Ballesteros JA, Shi L, Javitch JA (2001) Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol 60:1–19
Subbotina J, Yarov-Yarovoy V, Lees-Miller J, Durdagi S, Guo J, Duff HJ, Noskov SY (2010) Structural refinement of the hERG1 pore and voltage-sensing domains with ROSETTA-membrane and molecular dynamics simulations. Proteins 78:2922–2934
Abrol R, Bray JK, Goddard WA 3rd (2011) Bihelix: towards de novo structure prediction of an ensemble of G-protein coupled receptor conformations. Proteins 80:505–518
Michel M, Hayat S, Skwark MJ, Sander C, Marks DS, Elofsson A (2014) PconsFold: improved contact predictions improve protein models. Bioinformatics 30:i482–i488
Yang J, Jang R, Zhang Y, Shen HB (2013) High-accuracy prediction of transmembrane inter-helix contacts and application to GPCR 3D structure modeling. Bioinformatics 29:2579–2587
Kelm S, Shi J, Deane CM (2010) MEDELLER: homology-based coordinate generation for membrane proteins. Bioinformatics 26:2833–2840
Michino M, Chen J, Stevens RC, Brooks CL 3rd (2010) FoldGPCR: structure prediction protocol for the transmembrane domain of G protein-coupled receptors from class A. Proteins 78:2189–2201
Bhattacharya S, Lam AR, Li H, Balaraman G, Niesen MJ, Vaidehi N (2013) Critical analysis of the successes and failures of homology models of G protein-coupled receptors. Proteins 81:729–739
Obiol-Pardo C, Lopez L, Pastor M, Selent J (2011) Progress in the structural prediction of G protein-coupled receptors: D3 receptor in complex with eticlopride. Proteins 79:1695–1703
Malo M, Persson R, Svensson P, Luthman K, Brive L (2013) Development of 7TM receptor-ligand complex models using ligand-biased, semi-empirical helix-bundle repacking in torsion space: application to the agonist interaction of the human dopamine D2 receptor. J Comput Aided Mol Des 27:277–291
Michino M, Abola E, GPCR Dock 2008 Participants, Brooks CL 3rd, Dixon JS, Moult J, Stevens RC (2009) Community-wide assessment of GPCR structure modelling and ligand docking: GPCR Dock 2008. Nat Rev Drug Discov 8:455–463
Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R, GPCR Dock 2010 Participants (2010) Status of GPCR modeling and docking as reflected by community-wide GPCR Dock 2010 assessment. Structure 19:1108–1126
Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194
Larsson P, Wallner B, Lindahl E, Elofsson A (2008) Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci 17:990–1002
Ballesteros JA, Weinstein H (1995) Integrated methods for the construction of three dimensional models and computational probing of structure function relations in G protein-coupled receptors. In: Sealfon SC, Conn PM (eds) Methods in neurosciences. Academic Press, San Diego, p 428
Kozma D, Simon I, Tusnady GE (2013) PDBTM: protein data bank of transmembrane proteins after 8 years. Nucleic Acids Res 41:D524–D529
Webb B, Sali A (2014) Protein structure modeling with MODELLER. Methods Mol Biol 1137:1–15
Kufareva I, Katritch V, Participants of GPCR Dock, Stevens RC, Abagyan R (2013) Advances in GPCR modeling evaluated by the GPCR Dock 2013 assessment: meeting new challenges. Structure 22:1120–1139
Vehlow C, Stehr H, Winkelmann M, Duarte JM, Petzold L, Dinse J, Lappe M (2011) CMview: interactive contact map visualization and analysis. Bioinformatics 27:1573–1574
del Sol A, Fujihashi H, Amoros D, Nussinov R (2006) Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci 15:2120–2128
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res 25(17):3389–3402
Heim AJ, Li Z (2012) Developing a high-quality scoring function for membrane protein structures based on specific inter-residue interactions. J Comput Aided Mol Des 26:301–309
Chen K-YM, Sun J, Salvo JS, Baker D, Barth P (2014) High-resolution modeling of transmembrane helical protein structures from distant homologues. PLoS Comput Biol 10(5):e1003636
Venclovas C (2003) Comparative modeling in CASP5: progress is evident, but alignment errors remain a significant hindrance. Proteins 53(Suppl 6):380–388
Kukic P, Mirabello C, Tradigo G, Walsh I, Veltri P, Pollastri G (2014) Toward an accurate prediction of inter-residue distances in proteins using 2D recursive neural networks. BMC Bioinformatics 15:6
Mai TL, Chen CM (2014) Computational prediction of kink properties of helices in membrane proteins. J Comput Aided Mol Des 28:99–109
Ray A, Lindahl E, Wallner B (2010) Model quality assessment for membrane proteins. Bioinformatics 26:3067–3074
Valdar WS (2002) Scoring residue conservation. Proteins 48:227–241
Acknowledgments
We thank our anonymous reviewers for constructive comments. This work was supported by the NIH Grant R15-GM084404 and NSF 1229564 award.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chaudhari, R., Heim, A.J. & Li, Z. Improving homology modeling of G-protein coupled receptors through multiple-template derived conserved inter-residue interactions. J Comput Aided Mol Des 29, 413–420 (2015). https://doi.org/10.1007/s10822-014-9823-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-014-9823-2