
Abstract
The important role of mitochondria in cancer biology is gaining momentum. With their regulation of cell survival, metabolism, basic cell building blocks, and immunity, among other functions, mitochondria affect not only cancer progression but also the response and resistance to current treatments. Calcium ions are constantly shuttled in and out of mitochondria; thus, playing an important role in the regulation of various cellular processes. The mitochondrial calcium uniporter (MCU) channel and its associated regulators transport calcium across the inner mitochondrial membrane to the mitochondrial matrix. Due to this central role and the capacity to affect cell behavior and fate, the MCU complex is being investigated in different cancers and cancer-related conditions. Here, we review current knowledge on the role of the MCU complex in multiple cancer types and models; we also provide a perspective for future research and clinical considerations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Mitochondrial Ca2+ uptake: functional relevance and mechanism of activation
Studies reporting that mitochondria can take up large quantities of Ca2+ were published more than 50 years ago [28]. Since then, mitochondrial Ca2+ signals have been shown to be responsible for many essential cellular functions such as proliferation, cell death, the production of cell building blocks, and energy production, e.g., ATP synthesis [4, 71]. However, most of these findings were generated in the absence of information regarding the molecular identity of the proteins responsible for Ca2+ transport across the inner mitochondrial membrane (IMM), a fact that limited the efforts to fully understand the impact of mitochondrial Ca2+ signaling in physiology and pathology. The long quest for the elusive mitochondrial Ca2+ importer was over when first a regulator and then the channel itself were identified at the beginning of this decade [5, 26, 77]. These groundbreaking studies inaugurated a new era in the field of mitochondrial Ca2+ research.
In this review article, we will briefly summarize the current knowledge on mitochondrial Ca2+ uptake, mechanisms of activation, and the molecular components belonging to the mitochondrial Ca2+ uniporter (MCU) complex. For more detailed information, we refer the interested reader to a number of comprehensive reviews on the subject [27, 34, 49, 59, 61, 64, 76]. The main goal of this review is to recapitulate the studies that link the MCU complex with cancer. Given the short period since the discovery of the MCU complex components, the number of such studies is relatively low; nevertheless, it is our belief that summarizing the current knowledge will promote and assist future efforts in understanding mitochondrial Ca2+ signaling and how it can be harnessed to curb diseases, including cancer.
In most studies that refer to the physiological and pathological roles of mitochondrial Ca2+ signaling, it was the free unbound Ca2+ within the mitochondrial matrix that was first linked to a functional outcome. The functional role of the Ca2+ dynamics within the mitochondrial intermembrane space (IMS) on the other hand was less well understood. To reach the mitochondrial matrix, Ca2+ originating from the endoplasmic reticulum (ER), cytosol, plasma membrane, or any other cellular source has to cross two membranes. The outer mitochondrial membrane (OMM), which expresses VDAC (voltage-dependent anion channels), is Ca2+-permeable; however, the IMM is not. Thus, channels and transporters are needed to carry Ca2+ between the IMS and the mitochondrial matrix. This matrix-directed Ca2+ transport is initiated when Ca2+ levels in the IMS reach concentrations in the low micromolar range or higher and largely depends on the electrochemical Ca2+ gradient that arises from the negative IMM potential (about – 180 mV) [20, 27, 51, 78, 86]. Accordingly, for mitochondria to take up Ca2+, Ca2+ hotspots are needed (i.e. microdomains in their vicinity); this is because at rest, the cytosolic free Ca2+ levels in most cells are in the lower nanomolar range. The formation of Ca2+ hotspots can be controlled by different factors, such as organellar architecture and dynamics, cell shape, signaling mechanisms, and the involvement of other Ca2+ handling proteins.
One of the major signaling mechanisms that control mitochondrial Ca2+ dynamics is the store-operated Ca2+ entry (SOCE), which is physiologically initiated by the stimulation of Gq protein-coupled receptors on the outer cell surface. Via phospholipase C and phosphatidylinositol 4,5-bisphosphate (PIP2), this signaling cascade leads to the generation of inositol 1,4,5-trisphosphate (IP3), the activation of the ER-based IP3-receptors (IP3R), the release of Ca2+ from the ER, and the subsequent opening of plasma membrane Ca2+ channels, which ultimately cause an increase in the cytosolic Ca2+ concentration [18, 35, 70]. The main molecular components that allow this final increase in cytosolic Ca2+ consist of the CRAC channels (or Ca2+ release activated Ca2+ channels), which are formed by the ER-based stromal interaction molecules (STIM1 or STIM2) and the plasma membrane-bound ORAI-channels (ORAI1, 2, or 3) [81]. During SOCE, mitochondria can take up Ca2+ originating from the ER, from the plasma membrane, or both, and how mitochondria buffer Ca2+ from the different sources can determine cellular function [69]. For example, constant low IP3R-mediated Ca2+ release from the ER and transfer to the mitochondria is crucial for the maintenance of basal levels of oxidative phosphorylation (OXPHOS), whereas mitochondrial buffering of Ca2+ ions crossing the plasma membrane via the CRAC channels is required for proper T cell activation [55, 82].
Accumulating evidence also suggests an important interaction between Ca2+-driven signals and that of reactive oxygen species (ROS), with profound implications on multiple cell activities [7, 65, 78, 90]. ROS, such as superoxide (˙O2−) and hydrogen peroxide (H2O2), are generated by mitochondria and are a group of molecules which are no longer considered simply as cytotoxic byproducts of aerobic metabolic activity [106]. On the contrary, ROS are presently well established as important second messengers and signaling molecules that link mitochondrial activity to cell biology [23]. More comprehensive information regarding the functional relevance of the Ca2+-ROS interplay for different cellular systems and organs is available elsewhere [7,8,9, 19, 30, 32, 38, 43, 45, 91].
The mitochondrial Ca2+ uniporter complex
The major Ca2+ route across the IMM is via the MCU complex, formed by the MCU protein and its regulators, found in the IMM and the IMS (see Fig. 1) [34, 49, 61, 71]. The MCU complex is a relatively bulky structure (ca. 480 kDa) composed of channel-forming subunits and regulatory elements [93]. It has been reported that the channel is organized as a pentamer, although tetrameric assembly was also proposed [66, 83]. The channel-forming subunits consist of the membrane-spanning proteins MCU and MCUb (MCU isoforms “a” and “b”) and EMRE (essential MCU regulator). The MCU isoforms have different functions; MCU enhances Ca2+ transport, while MCUb limits it; and one molecule of MCUb within the complex is sufficient to significantly decrease Ca2+ transport compared to a MCU homomer [83]. Because the expression of MCU and MCUb differs across tissues and cancer types, changes in the MCU:MCUb ratio could indicate variable Ca2+ uptake potentials for each MCU complex; however, this is a research area that still requires attention. EMRE contributes to the structural integrity of the complex; additionally, EMRE facilitates the interaction with other regulatory proteins within the MCU complex [89]. Accordingly, following EMRE depletion, mitochondrial Ca2+ uptake is inhibited, causing an effect comparable to MCU knockdown [89]. This outcome is not observed upon the depletion of other regulatory proteins, suggesting that EMRE is more than just a MCU regulator.

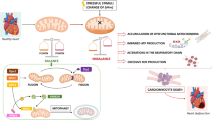
The mitochondrial Ca2+ uniporter (MCU) complex and its role in cancer. The core of the MCU channel is formed by the membrane-spanning proteins MCU and MCUb (mitochondrial Ca2+ uniporters isoforms “a” and “b”) and EMRE (essential MCU regulator). The MCU isoforms form a pentamer (or a tetramer) within the IMM that creates a Ca2+ conducting pore. The activity of this pore is further regulated by the MICU (mitochondrial Ca2+ uptake) family of proteins. The effects of the MCU complex on several cancer hallmarks and cancer-related signaling pathways are indicated with “+” for promoting effects and “–” for inhibiting effects (for details see Table 1). Chemical and endogenous inhibitors of the MCU complex are also listed
The activity of the MCU complex is also regulated by the mitochondrial Ca2+ uptake family of proteins (consisting of three isoforms: MICU1, 2, 3). The best characterized isoform, MICU1, functions as a channel gatekeeper and contains two Ca2+-binding EF hands (helix-loop-helix domain), which sense Ca2+ levels in the IMS. At low Ca2+ concentrations, the EF hands of MICU1 keep the MCU complex inactive. However, when the Ca2+ concentration in the IMS exceeds a threshold (in most cells this IMS Ca2+ concentration lies in the low micromolar range), the EF hands in MICU1 bind Ca2+ and cause a conformational change that diminishes the MCU-MICU1 interaction and opens the MCU channel [21, 58, 77, 78]. Depletion of MICU1 results in mitochondrial Ca2+ uptake even at low IMS Ca2+ concentrations, causing elevated basal Ca2+ levels in the mitochondrial matrix; it also results in dysregulated Ca2+ uptake at higher IMS Ca2+ levels, indicating a dual function for MICU1 (gatekeeper and enhancer) [21]. The absence of MICU1 and the resulting Ca2+ overload can lead to increased oxidative stress and the induction of apoptosis [58].
MICU2 was shown to inhibit the MCU complex [1, 48, 72, 80]. Overexpression of MICU2 reduces mitochondrial Ca2+ uptake, while its depletion has the opposite effect. It has to be noted that MICU1 and MICU2 form a disulfide-linked dimer that binds to the MCU complex; thus, depletion of MICU1 also results in the loss of MICU2 from the complex [72, 78]. This finding indicates that careful and well-designed experiments and interpretation of Ca2+ measurements must be undertaken since outcomes cannot be attributed to the loss of a single paralog. Additionally, MICU1 and MICU2 appear to have higher turnover rates than MCU, which hints at an additional level of MCU complex regulation and likely changes in the sensitivity of mitochondria to different Ca2+ levels. However, this also needs further investigation.
Much less is known about the function of MICU3; however, similarly to MICU1 and MICU2, MICU3 contains a cysteine in its C-terminal region, suggesting that it undergoes heterodimerization with other MICU isoforms. Indeed, a recent study examined the functional role of MICU3 in cortical neurons and showed that MICU3 forms heterodimers with MICU1, but not with MICU2. Moreover, MICU3 promoted mitochondrial Ca2+ uptake, similar to MICU1, thus regulating neuronal synaptic activity [73]. Interestingly, expression of MICU3 appears to be less pronounced compared to MICU1 and MICU2 across most tissues and cell lines, with several exceptions such as neural tissues and skeletal muscle [67, 73, 80]. Two additional IMM-based proteins, MCUR1 (MCU regulator 1) and SLC25A23, were identified as regulators of mitochondrial Ca2+ uptake [44, 57, 96, 98]. It was demonstrated that MCUR1 interacts with MCU but not with MICU1 and that silencing of MCUR1 strongly inhibits mitochondrial Ca2+ uptake. Two studies reported additional MCU-independent roles for MCUR1: as a regulator of Ca2+ sensitivity for the mitochondrial permeability transition pore and as an assembly factor for the electron transport chain (ETC) complex IV [15, 74]. Meanwhile, SLC25A23 physically interacts with MCU but also with MICU1. Interestingly, SLC25A23, similarly to the MICUs, possesses an EF-hand, which determines its role in the mitochondrial Ca2+ uptake. Further work is needed to fully understand the complex roles of MCUR1 and SLC25A23 as regulators of mitochondrial Ca2+ homeostasis. For additional comprehensive information on the MCU complex and its components, we suggest the following references (27, 34, 49, 59, 61, 64, 71).
Additional mitochondrial Ca2+ transporters
While MCU plays a major role in controlling mitochondrial Ca2+ uptake, it is not the only molecular transporter that has been proposed to do so; thus, understanding the function and contribution of additional Ca2+ regulators could better help predict cellular response to Ca2+ changes. For example, studies showed that mitochondrial Ca2+ influx and efflux rates, as well as ATP generation, are impaired in LETM1 (leucine zipper-EF-hand-containing transmembrane protein 1) knockdown or mutant cells and also in fibroblasts derived from patients with Wolf-Hirschhorn syndrome (WHS). LETM1 levels were lower in WHS-derived fibroblasts, but the MCU components MCU, MCUR1, and MICU1 were unaltered, so this suggests their independent activity (at least with regard to changes driven by LETM1) [31, 47]. Another transporter to consider is NCLX (mitochondrial Na+-Ca2+ (lithium) exchanger), which under normal conditions removes Ca2+ from the matrix and was found to be dominant to LETM1 in terms of mitochondrial Ca2+ export [25, 68]. Samanta et al. recently showed that after physiological stimulation, NCLX can also operate in reverse mode and instead transport Ca2+ into the matrix, thus generating Ca2+ oscillations [88]. The mitochondrial uncoupling proteins (UCPs) have also been proposed as regulators of the mitochondrial Ca2+ uptake in intact cells [39]. However, it has to be noted that the role of UCPs and LETM1 as Ca2+ transporters is under debate and additional work will help resolve this issue [10]. Finally, we have showed that both coenzyme Q10 (CoQ10) and its analogue CoQ1 undergo structural changes that allow them to bind and transport Ca2+ across biomimetic membranes in a redox-controlled manner [6, 40]. Additional studies are, nevertheless, needed to assess if these hydroxylated CoQ analogs regulate mitochondrial Ca2+ homeostasis in vivo.
Summarized, it is apparent that the exact function of all mitochondrial Ca2+ transporters is not yet fully understood and may in some cases change depending on the surrounding ion levels or co-regulators. Nevertheless, the crucial question is whether their aberrant expression alone or in combination with other transporters can be associated with aggressive disease or treatment resistance; additional efforts need to be invested in this direction. Novel observations in this direction could focus scientific investigations towards more clinically relevant findings in the future.
Mitochondria and cancer
Cancer cells display remarkable adaptability and robustness in order to survive, metastasize, and resist therapies. To do so, they rely on multiple molecular processes, some of which involve mitochondrial activity [87]. Mitochondria are not only sources of energy and producers of intermediates for lipid, nucleic acid, and protein synthesis but also hubs for signaling and mechanisms that control cell fate [59]. Since mitochondria play such an important yet varied role, the understanding of their function could provide useful information to treat aggressive disease. Early observations into the role of mitochondria in cancer involved unexpected metabolic switches. Almost a hundred years ago, Otto Warburg observed that cancer cells metabolize glucose differently from that of normal tissue cells and he posited that cancer cells rely on glycolysis for their ATP production even in the presence of oxygen [100]. Later studies confirmed the metabolic switch, but showed it to be reversible, and mitochondria proved to remain functional and mutation-free in many cases following cancerous transformation. We now know that mitochondria can play an important role in promoting cancer [99], and part of this mitochondrial function relies on a fine-tuned and well-localized transport and buffering system for calcium ions.
The MCU complex and cancer
According to The Human Protein Atlas (www.proteinatlas.org), a majority of cancer tissues show moderate to strong MCU immunostaining. The strongest protein expression was found in colorectal and ovarian cancers; high expression was also detected in many other cancers, such as pancreatic, stomach, and prostate cancer. For MICU1, moderate to strong immunostaining was also observed in most malignant tissues, while for MICU2, weak to moderate immunostaining was detected, with the strongest signal being observed in thyroid cancer. MICU2 was negative for several cases of malignant lymphoma, urothelial, and skin cancers (excluding melanoma). MCUb expression was weak to moderate and was found in most colorectal cancers and in breast, cervical, and ovarian cancers; most other cancers were negative. MCUR1 results are pending for human cancer tissue, but lymphoid, skin-, and breast-derived cells have the highest expression from the cell lines tested. EMRE immunostaining is highest in thyroid cancer as well as in liver, stomach, and prostate cancer. Weak to moderate expression was detected in most examined cancers with exclusion of lymphoma, testis, and skin cancer (excluding melanoma).
According to The Cancer Genome Atlas (TCGA; www.cancergenome.nih.gov), the highest rate of MCU genetic alterations was found in prostate and breast cancers, observed as gene amplifications. A few datasets show deletions (for example in malignant peripheral nerve sheath tumors (MPNST)), while the highest rate of MCU mutations was observed in uterine carcinosarcoma. MICU1 amplifications are also commonly identified in many cancers, with the highest rates detected in prostate and breast cancer. MICU1 deletions were also found in MPNST, while mutations were highly featured in melanoma. MICU2 was also most amplified in prostate and breast cancer and most highly mutated in breast and stomach cancer. Frequent MCUb amplifications were detected in prostate cancer, while mutations and deletions were found in pancreatic cancer (deletions were also found in MPNST). MCUR1 amplifications are also prominent and are found in breast, prostate, melanoma, ovarian, and bladder cancers, but mutations and deletions are also detected in melanoma and other cancers. Genetic modifications of EMRE mostly occur as amplifications and deep deletions in breast, pancreas, prostate, and melanoma cancers.
How these MCU complex genetic modifications affect cancer biology and why their patterns differ across cancer types is currently not understood, and this could be addressed in the future. Next, we describe the current knowledge on the role of the MCU complex in different cancers. A compact summary of the publications discussed below is presented in Table 1.
Breast cancer
Breast cancer and breast cancer models appear to be the most extensively studied so far with respect to MCU complex function. Acquired knowledge in this field not only demonstrates the involvement of MCU in cancer but also highlights its complexity and context-dependent activity. In a recent study, Tosatto et al. examined the role of MCU in cancer migration and invasion and showed that MCU expression correlated with tumor size and lymph node infiltration in triple negative breast cancer (TNBC) [97]. MCU knockdown decreased cell motility and invasion potential, as well as tumor growth in xenograft models of TNBC. Meanwhile, MCU-silencing weakened the production of mitochondrial ROS and reduced the expression of HIF1-α. Using breast cancer mRNA samples, a positive correlation of MCU levels with HIF1-α signaling was revealed. In sum, the MCU-ROS interplay was identified as an important regulator of TNBC growth and metastasis [97].
In a study featuring patient data analysis, Curry et al. investigated MCU expression in 180 human breast cancer samples using microarray analysis [22]. Results not only showed expression variability among patients but also indicated that high MCU levels can be detected in estrogen receptor (ER)-negative tumors, with the strongest enrichment found in the basal-like subtype. To further explore the contribution of MCU to breast cancer, the authors silenced MCU in the triple negative MDA-MB-231 breast carcinoma cell line and examined the effects on cell number, cell cycle, and survival. Their siRNA approach did not show significant changes in either proliferation or viability; however, MCU silencing potentiated breast cancer cell death in the presence of submaximal levels of the Ca2+ ionophore ionomycin. This cytotoxicity was not replicated with submaximal or high levels of the Bcl-2 (B cell lymphoma 2) inhibitor ABT-263, suggesting a distinct role for MCU in caspase-independent (ionomycin) vs -dependent (ABT-263) cell death. The authors also determined that MCU does not play a major role in regulating bulk cytoplasmic Ca2+ levels in MDA-MB-231 cells and suggested instead that cell outcome is influenced by either changes in mitochondrial Ca2+ uptake or alterations in localized Ca2+ signals in special domains, such as mitochondrial-associated-membranes (MAMs).
The importance of MCU in breast cancer was further confirmed by Hall et al.; their study however also warns that not all carcinomas are sensitive to mitochondrial Ca2+ uptake-based therapies [41]. In the first part of the study, the authors searched the web-based BreastMark algorithm and observed that a significantly poorer prognosis was associated with MCU overexpression and MICU1 downregulation; additionally, those with MICU1 overexpression had a better prognosis. They also used the MDA-MB-231 breast carcinoma cell line model to conduct functional studies; however, they reported that this cell line harbors mitochondrial DNA mutations and decreased oxidative metabolism, which could alter its survival mechanisms compared to other cell types. Using siRNA-mediated knockdown studies and adenoviral overexpression techniques, as well as the mitochondrial targeted genetically encoded Ca2+ sensor ratiometric-pericam, Hall et al. first showed that MCU is functional in MDA-MB-231 and that ATP-induced mitochondrial Ca2+ uptake was inhibited by MCU knockdown. Inhibition of MCU was also confirmed with the overexpression of a dominant-negative MCU mutant. Meanwhile, Ca2+ uptake was enhanced by wild-type MCU overexpression and MICU1 knockdown. On the other hand, both MCU and MICU1 silencing had minor effects on mitochondrial ROS production. Minor effects were also detected using the clonogenic survival of MDA-MB-231 cells in response to stress such as radiation, paclitaxel treatment, starvation, and ceramide treatment (which according to the authors, promotes Ca2+ leak from the ER). These findings do not fully agree with observations in HeLa cervical cancer cells, where overexpression of MCU or knockdown of MICU1 caused constitutive mitochondrial Ca2+ influx, sensitization to H2O2, and ceramide-induced cell death [58]. Hall et al. in fact proposed a spectrum of responses following MCU manipulation. This is supported by their clonogenic survival assay results using human mammary epithelial cells (HMEC), HeLa, and MDA-MB-231 cells; where HMECs were the most sensitive to MCU channel manipulation and ceramide treatment, where HeLa had an intermediate phenotype, and where MDA-MB-231 remained largely unresponsive [41]. How MDA-MB-231 cells favor survival is not understood yet, but it appears that they separate mitochondrial Ca2+ transport from other mitochondrial functions (e.g., ROS production or metabolic changes upon starvation). Another interesting observation from this study showed that knockdown of MCU reduced MICU1 protein levels but not at the mRNA level (the reverse however, was not observed for MICU1 knockdown), and the authors suggest a possible MCU-dependent post-transcriptional feedback loop. Additionally, similar to MICU1, MCUb and EMRE were shown to have favorable hazard ratios; therefore, searching patient data for co-expression patterns could provide more predictive responses than looking at MCU or MICU1 alone. The answer to understanding the spectrum of responses to MCU manipulation in different cancer samples could be partially addressed in the future by examining the expression and activity of multiple components of the MCU complex as recently reported [67].
In another breast cancer cell line model, this time featuring MDA-MB-468, Davis et al. examined whether epidermal growth factor (EGF)-induced epithelial-mesenchymal transition (EMT) involved gene expression changes for Ca2+-related channels, pumps, or exchangers. This endeavor was based on the observation that EMT is associated with altered SOCE and with changes in Ca2+ signals mediated by purinergic receptors [24]. The study concluded that MCU mRNA levels remain unchanged during EMT; however, the ER Ca2+ channels and pumps were most highly affected, notably the ryanodine receptor RYR2. We note here that the EMT process involves multiple transition states and intermediates over time, and this process is also reversible, so the role of MCU in EMT cannot be fully discounted [102]. In addition, given the heterogeneity found among breast cancer tumors, the contribution of MCU and its regulators to this process could still be further explored using additional models.
While the role of MCU in cancer cell survival appears cell line-specific, it could also be drug-specific as shown by Yoon et al. [103]. In their study, the authors showed that celastrol (an anticancer agent isolated from a Chinese vine) kills breast and colon cancer cell lines via paraptosis. Paraptosis is characterized by vacuolation, dilatation of the ER and mitochondria, and a caspase-independent cell death. Celastrol treatment caused an increase in mitochondrial Ca2+ levels and ER stress, and this was inhibited by MCU knockdown or MCU inhibition via ruthenium red pretreatment in MDA-MB-435S breast cancer cells. Similar to the study by Curry et al. [22], this study suggested that drugs that harness caspase-independent cell death mechanisms could benefit from MCU targeting.
Focusing on the role of MCU in metastatic breast cancer, Tang et al. examined the expression of MCU mRNA in the Oncomine database and observed a correlation between MCU, metastasis, and invasive breast cancer [95]. Similar to previous studies, mechanistic follow-up on these observations was conducted with the MDA-MB-231 cell line. In this cell model, MCU inhibition halted serum-induced migration and serum- or thapsigargin-induced SOCE. Interestingly, SOCE inhibitors were also able to inhibit serum-induced MDA-MB-231 cell migration. Thus, it appears that MCU’s role in MDA-MB-231 cells includes SOCE activity with consequences on cell migration.
Given the accumulated evidence for the presence of high levels of MCU in aggressive disease, Yu et al. investigated the effects of MCU knockdown and overexpression on cell migration, invasion, and glucose metabolism using the human breast carcinoma lines ZR-75-30, MDA-MB-231, MCF7, and BT-474. Highlights of the study indicate that MCU silencing in MDA-MB-231 cells decreased migration and invasion in vitro and reduced lung metastasis in vivo; conversely, overexpression in the less aggressive MCF7 potentiated these effects in vitro and in vivo. These findings increasingly support the role of MCU in invasive and metastatic processes. Additionally, Yu et al. showed enhanced glycolysis following MCU overexpression in MCF7 and they proposed a novel mechanism whereby this MCU-driven process is negatively regulated by microRNA-340. Finally, MCU expression was evaluated in 60 human breast cancer samples using immunohistochemistry techniques, and data showed that MCU protein levels are significantly increased in metastatic samples [104].
Using breast and prostate cancer cell lines, as well as transformed primary human fibroblasts, Cardenas et al. demonstrated that Ca2+ communication between the ER and mitochondria is essential for tumor cell survival, but this is not the case for normal cells, i.e., some cancer cells are addicted to mitochondrial Ca2+ [13, 55]. Briefly, the authors’ findings showed that inhibition of mitochondrial Ca2+ uptake by knockdown of MCU or its regulator MCUR1 caused a decrease in ATP levels, caused AMPK activation, and engaged autophagy in normal cells, permitting cell survival. Additionally, genetic or drug inhibition of MCU decreased TCA cycle activity, ATP levels, and metabolic products, also leading to autophagy. However, only normal cells survived this process, while tumor cells died by necrosis. Interestingly, all the effects induced by interference with the ER-mitochondria Ca2+ transfer (in both normal and cancer cells) were reversed by the addition of pyruvate or alpha-ketoglutarate, indicating that a lack of metabolic intermediates was the main cause of cell death.
Together, the previously described studies suggest that the role of the MCU complex in breast cancer is important yet complex and so far, cell-type dependent; they also advocate that certain cells can acquire MCU-independent survival properties. However, the MCU complex works as a multi-component channel and it is not yet fully understood how mitochondria adapt and compensate for the knockdown of its single components. Additionally Ca2+ buffering systems, aside from the MCU complex, can also contribute a survival advantage. For example, MCU functionally depends on VDAC channels, and according to Liao et al., it also mediates VDAC overexpression-induced cell death in cerebellar granule neurons (CGNs). Thus, understanding the MCU complex and VDAC status could better predict mitochondrial Ca2+ homeostasis and possibly stress-induced apoptosis [53]. Finally, the MCU complex is not likely to be targeted alone therapeutically; thus, this highlights the need to study this Ca2+ regulator in the context of a broader signaling landscape. We note here that the studies listed above do not fully recapitulate the heterogeneity of breast cancer or the response in a more in vivo environment; however, they provide a useful base on which future studies can build and can help narrow the focus as we move forward.
Hepatocellular carcinoma
Similar to breast cancer, expression studies of the MCU complex in hepatocellular carcinoma (HCC) tissues (by microarray and immunohistochemical analyses) indicated that MCU is frequently upregulated, MICU1 is downregulated, while no significant differences were observed for MICU2, MCUb, or EMRE [84]. In this study, high MCU or low MICU1 expression were also associated with poor overall survival, recurrence-free survival, as well as with metastatic tissue. Using HCC cell lines, the authors demonstrated that mitochondrial Ca2+ uptake was enhanced in a MCU-dependent manner, also that MICU2 expression was not affected by MICU1 knockdown or overexpression in this cancer type and that both MICU isoforms played non-redundant roles in Ca2+ regulation. Alterations in MCU expression did not appear to have significant effects on the expression levels of sarcoendoplasmic reticulum Ca2+ transport ATPase pumps (SERCA) or IP3R (both ER-related Ca2+ regulators). On the other hand, MCU changes affected mitochondrial ROS production via a nicotinamide adenine dinucleotide (NAD+), sirtuin 3 (SIRT3), and superoxide dismutase 2 (SOD2)-driven pathway; this enhanced metastasis (as also observed in breast cancer models). More precisely, high MCU levels induced matrix metalloproteinase 2 (MMP2) production and cell motility, confirmed by increased intrahepatic and distal lung metastases in vivo.
In a second publication from the same group, the authors showed that aside from MCU, the regulator MCUR1 was also often upregulated in HCC cells and caused increased mitochondrial Ca2+ uptake, tumor cell survival, and proliferation [85]. In their model, the authors suggested that following increased Ca2+ uptake, higher mitochondrial ROS were produced, which caused an increase in AKT/MDM2-mediated p53 degradation, and subsequent changes in the expression of apoptosis and cell-cycle related proteins. In vivo, overexpressed MCUR1 decreased cell death and increased proliferation; conversely, MCUR1 reduction impaired tumor growth in nude mice. The role of both p53 and MCU in driving cancer biology was also explored by Giorgi et al. [36, 37]. In these studies featuring colon, breast, and non-small cell lung cancer cell lines, the authors showed that upon adriamycin or H2O2 exposure, wild type p53 localized to the ER and to the mitochondrial associated membranes. Herein, p53 was shown to directly bind SERCA pumps, thereby altering the redox state and thus causing increases in Ca2+ load, mitochondria Ca2+ overload, and cell death. The authors also showed that this does not occur in cell lines harboring p53 mutations. Together, these studies on multiple cancer cell types suggest the need to monitor p53 status when manipulating Ca2+ flux and MCU for therapeutic purposes.
Another study flagged MCU as being relevant to HCC and this followed microarray analyses of sublethally heat-treated HCC cells. This sublethal heat treatment model is suggested to reflect a transition zone found in radiofrequency ablation (RFA) tumor treatment; treatment conditions in this transition zone are insufficient to kill tumor cells and are expected to cause local recurrence. While the primary goal of this study was to measure long non-coding RNAs involved in treatment resistance, differentially expressed mRNAs were also investigated and flagged MCU as being important in the SMMC-7721 cell line [29]. While more work is required to validate the role of MCU in this model, it highlights the potential contribution of MCU to acquired treatment resistance and initial response to overcoming stress. Studies exploring the role of the MCU complex in dynamic processes, such as drug resistance, would be interesting to explore in the future.
Colon cancer
Marchi et al. identified in silico a cancer-related MCU-targeting microRNA-25 and showed that its overexpression in HeLa cells reduced MCU levels and mitochondrial Ca2+ uptake, thus increasing cell survival following apoptotic challenges (such as H2O2 and C2-ceramide) [60, 62]. Additional members of the microRNA-25 family, such as microRNA-92a and microRNA-363, were proposed to have similar effects on MCU and Ca2+ signaling. The authors also demonstrated that the effect of microRNA-25 is focused on mitochondrial Ca2+ uptake alone and that no changes occurred in mitochondrial membrane potential, mitochondrial volume, number, or ER contact sites. However, aside from its effect on mitochondrial Ca2+, it is not excluded that additional microRNA-25 targets, such as PTEN (phosphatase and tensin homolog), the pro-apoptotic protein Bim, and TRAIL (tumor necrosis factor related apoptosis inducing ligand), also contributed to the antiapoptotic effects. The authors then showed that multiple colon and prostate cancer cell lines displayed high microRNA-25 levels and low MCU expression compared to primary normal cells. This observation was confirmed in human samples of colonic adenocarcinoma using immunohistochemistry and microarray analyses. Finally, the authors used the PC3 prostate cancer cell line to show that increased MCU activity reduced soft agar colony formation, while overexpressed anti-microRNA-25 in HCT116 and PC3 cells led to increased sensitivity to H2O2 and C2-ceramide. To summarize, the authors suggested that microRNA-25, through MCU downregulation, reduced the sensitivity of cancer cells to apoptosis-inducing agents. This study sets the stage for further studies on the role of microRNAs in Ca2+ homeostasis and opens the possibility of targeting microRNAs instead of MCU directly in order to regulate its activity.
Pancreatic cancer
Previous studies have shown that the histidine triad nucleotide-binding protein (HINT2) sensitizes HepG2 HCC cells to mitochondrial apoptosis following cytotoxic drug treatment [63]. In pancreatic cancer, HINT2 also promoted cell death and this was proposed to involve MCU regulation and Ca2+ influx [16]. In this study, HINT2 expression was shown to be reduced in pancreatic cancer tissue compared to adjacent normal tissue (assessed via immunohistochemistry and microarray analyses). This downregulation was not surprising given that HINT2 was shown to inhibit tumor growth and invasion in pancreatic carcinoma models. Functional studies revealed that HINT2-mediated apoptosis could be blocked using the MCU inhibitor ruthenium red. Meanwhile, HINT2 overexpression using an adenoviral vector increased mitochondrial Ca2+ and changed the expression profile of MCU regulators such as MICU1, MICU2 (downregulation), and EMRE (upregulation), which could contribute to this Ca2+ overload. The role of MCU cannot be discounted in the anticancer effects of HINT2; however, the study by Chen et al. also highlighted 1240 differentially expressed genes following HINT2 overexpression, indicating MCU changes alone are not responsible for the anticancer effects. Of interest in the future is to potentially use the expression levels of MCU and its regulators to predict patient survival and combine them with additional cancer biomarkers.
Head and neck squamous cell carcinoma (HNSCC)
Unlike HINT2, the enhancer of zeste homolog 2 (EZH2) is overexpressed or activated in many human cancers including head and neck squamous cell carcinoma (HNSCC), where it was shown to be associated with high tumor grade and poor prognosis (TCGA data). Zhou et al. confirmed such observations in a Chinese HNSCC cohort; in addition to showing that the EZH2 inhibitor DZNep and siRNA against EZH2 decreased MICU1 expression in human oral cancer cell lines. Additionally, siRNA against MICU1 decreased cell viability in the SCC25 and Cal27 cell lines, while western blot analyses showed that Bcl-2 was decreased and BAX and cleaved caspase-3 were increased. Finally, a HNSCC xenograft model using the Cal27 cell line was used to test the in vivo effects of the EZH2 inhibitor DZNep. Immunohisochemistry analysis showed decreased MICU1 and Bcl-2 expression, increased proapoptotic BAX expression and cleaved caspase-3, and showed decreased tumor volume [105]. The authors mentioned that EZH2 inhibition triggers cytoplasmic Ca2+ accumulation, loss of membrane potential, and changes in mitochondrial proteins involved in cell death; however, more focused mitochondrial studies could shed light on the role of MICU1 in HNSCC and whether it should be targeted in combinatorial treatment strategies.
Multiple myeloma
The role of mitochondria in drug-induced cytotoxicity or resistance was explored in multiple myeloma (MM) cell lines displaying varied sensitivity to the proteasome inhibitor bortezomib by Song et al. [92]. Findings demonstrated that all cells increased mitochondrial Ca2+ levels in response to bortezomib, but what differentiated sensitive from resistant cells was the ratio between the basal and induced mitochondrial Ca2+ (the higher the fold increase in mitochondrial Ca2+, the more cytotoxicity was observed). Differences were also observed in membrane potential, mitochondrial ROS levels (which were suggested to contribute to cytotoxicity), oxygen consumption, and mitochondrial ATP; all suggested to contribute to the drug response in MM cells. The authors next explored regulators of mitochondrial activity and showed that MCU expression was highly upregulated following bortezomib treatment, but only in the sensitive cells. Additional genes, such as cyclophilin D (CYPD) and SOD2, were highlighted as important regulators of cell death, thus suggesting the involvement of mitochondrial ROS in the process. Concluding remarks suggested the combined use of mitochondria-targeting agents with bortezomib to obtain maximal MM cell apoptosis. These findings support those of Curry et al. in breast cancer and that of Giorgi et al. in multiple cancer cell types, in that MCU status alone as a biomarker of drug response may not be sufficient to predict cell outcome; however, in a wider signaling or genetic context, it could be of use clinically [22, 37].
Supporting the role of MCU in drug-induced cytotoxicity, Madreiter-Sokolowski et al. showed that siRNA against MCU and LETM1 could prevent resveratrol/piceatannol-induced cancer cell death [56]. This study featured HeLa cells, human umbilical vein endothelial cells (HUVEC), and EA.hy926 cells (established by fusing HUVEC with a thioguanine-resistant clone of the A549 human lung carcinoma line); however, additional human cancer models could be used to confirm observations. Moreover, the authors proposed that enhanced mitochondrial Ca2+ sequestration within ER-mitochondrial contact sites in cancer cells made them susceptible to resveratrol/piceatannol SERCA pump inhibition, resulting in enhanced mitochondrial Ca2+ uptake, overload, and ultimately cell death. This would suggest that ER-mitochondrial contact sites might control cancer cell drug sensitivity. Thus, understanding the role of contact sites, or proteins involved in organellar tethering, could provide additional indicators of response if drugs affect Ca2+ homeostasis. For additional reading on the role of mitochondria-ER contact sites in cancer, please refer to [2, 11, 12, 33, 50, 75, 79].
Prostate cancer, osteosarcoma, and melanoma
In a study predominantly featuring prostate cancer cell lines, Loubiere et al. proposed that the antidiabetic drugs and metabolic disruptors metformin and phenformin regulate intracellular Ca2+ flux. In brief, these drugs were shown to induce ER stress, ER Ca2+ release, mitochondrial Ca2+ uptake, organelle swelling, and apoptosis; however, this was reversed when MCU was inhibited. Supporting the mitochondrial Ca2+ uptake increase, MCU mRNA was also increased following metformin treatment (among other Ca2+ handling proteins). In vivo, using different mouse models, metformin efficiently reduced tumor growth and increased mitochondrial areas in tumor cells [54]. Our own work in melanoma showed that phenformin blocked the emergence of a subpopulation of tumor-maintaining drug-resistant cells during anti-melanoma treatment and these cells displayed upregulated mitochondrial ETC proteins, consumed more oxygen, and generated more ATP [87]. Moreover, we found that these aggressive cells had significantly upregulated mitochondrial Ca2+ levels (unpublished observations). Whether a phenformin-induced increase in mitochondrial Ca2+ was sufficient to push this subpopulation of cells towards apoptosis was not examined. In a different study, we also demonstrated that increased mitochondrial activity and enhanced ROS production could be used as an Achilles’ heel for these drug-resistant tumor-maintaining melanoma cells [17]. Although it is evident that mitochondrial Ca2+ can tune the effects of ETC inhibitors such as phenformin, it must be considered that anti-metabolic drugs have activities beyond that of regulating Ca2+ homeostasis; thus, careful dissection of multiple biological processes, including metabolic changes, needs consideration.
A recent study by Takata et al. examined the role of mitochondrial Ca2+ dynamics in osteosarcoma and melanoma and showed that Ca2+ protects cancer cells from TRAIL cytotoxicity. The authors also showed that acute TRAIL treatment increased cytosolic and mitochondrial Ca2+ concentrations. Calcium chelators, the MCU inhibitor ruthenium 360, the mitochondrial permeability transition pore opener atractyloside, capsazepine, and AMG9810, all decreased mitochondrial Ca2+ and sensitized tumor cells to TRAIL treatment in an apoptotic and non-apoptotic way. The study points to the important role of mitochondrial Ca2+, and thus MCU activity, in overcoming cancer cell resistance to TRAIL cytotoxicity [94].
Alternative, cancer-related, and cancer-like pathologies
To be of use clinically, targeting MCU does not have to be exclusively focused on cancer cells per se. For example, a cancer-like disease where excessive pulmonary artery smooth muscle cells proliferate, migrate, and resist apoptosis also relies on MCU activity; this vasculopathy leads to pulmonary arterial hypertension (PAH) and was shown to display impaired MCU complex function, downregulation of MCU and upregulation of MICU1 protein [46]. MCU dysfunction not only increased cytosolic Ca2+ that then stimulated proliferation and migration, but also decreased mitochondrial Ca2+, thus inhibiting pyruvate dehydrogenase and glucose oxidation. Encouragingly, microRNA-mediated MCU complex regulation could be targeted. It would be interesting to examine if other pathologies with cancer-like properties, or early stages of disease, rely on MCU activity for progression. An indication of such potential comes from the study of Wiel et al. who identified MCU as a regulator of oncogene-induced senescence (OIS) through a loss-of-function genetic screen in human endothelial cells. In this study, loss of MCU or the Ca2+ channel ITPR2 (a member of the IP3 receptor family) allowed cells to escape OIS by reducing mitochondrial Ca2+ accumulation, or Ca2+ release from the ER, respectively. The authors also suggested that this mechanism could be involved in replicative senescence [101].
Another cancer-related challenge arises in the form of cardiotoxicity in cancer patients treated with chemotherapeutics, such as anthracyclines, taxanes, or fluoropyrimidines. Chemotherapeutics can cause the upregulation of an atypical G protein Gβ5 in the myocardium. If targeted, Gβ5 loss can maintain membrane potential, basal MCU expression, and mitochondrial Ca2+ levels; it can also reduce drug-induced proinflammatory cytokines, hypertrophic, and profibrotic factors [14]. Thus, while treatment strategies revolving around targeting MCU and its regulators directly hold merit, understanding the upstream and downstream signals can also reveal novel treatment strategies, some of which may not eradicate the cancer fully, but can certainly improve patients’ responses and quality of life.
The advantage of dealing with less aggressive or advanced disease when targeting the MCU complex is that the genetic landscape of these cells may be easier to navigate therapeutically. On the other hand, little is known about long-term MCU targeting, especially as it appears to have different functions in different tissues; thus, long-term adaption of cells to MCU complex manipulation is a research area still in need of attention.
Pharmacology of MCU
From a clinical point of view, an essential prerequisite to pharmacologically target a specific molecule or a signaling cascade in order to impair cancer growth and invasion is having a specific drug with minimal side effects. Based on the data summarized in this review, inhibiting rather than activating MCU should be beneficial for, at least some, cancer patients. However, all known inhibitors of MCU are currently non-specific, do not cross cellular membranes, and have a plethora of side effects. Ruthenium red (RuR) and its derivative ruthenium 360 (Ru360) were, until recently, more or less the only choices to directly manipulate MCU activity (for details see [27]). In addition, compounds such as the NCX inhibitor KB-R7943 or the antibiotic minocycline have been reported to also block MCU. Similar to RuR however, these drugs were shown to be unspecific and displayed a number of disadvantages. Accordingly, the clinical use of any of these drugs as inhibitors of MCU does not seem feasible.
A recent study by Arduino et al. characterized Mitoxantrone, an anthracenedione-derived cytostatic agent used in hematological malignancies, as a specific inhibitor of MCU [3]. However, Mitoxantrone also acts cytostatically by DNA intercalation, which may limit its use in in vivo disease models even though the antineoplastic and anti-MCU properties are supposed to be independent. Chemical modifications of Mitoxantrone that will separate the MCU-blocking properties from the antineoplastic ones might provide a new generation of specific MCU inhibitors with distinct anticancer properties [3]. An additional recent study utilized a screen of 120,000 small molecule compounds to identify DS16570511 as a specific membrane-permeant MCU inhibitor [52]. Besides inhibiting agonist-induced mitochondrial Ca2+ uptake at a cellular level, DS16570511 enhanced cardiac contractility in perfused rat hearts. A potential utility of DS16570511 or one of its derivatives for cancer treatment needs to be explored in the future. A list of chemical and endogenous MCU inhibitors is displayed in Fig. 1.
Conclusion
As we have learned from studies on targeted therapies and immunotherapies so far, single biomarkers and drug targets can be useful clinically; however, they must be used in the right context and with the right strategy in order to be effective and prevent adverse reactions [42]. The importance of MCU and its regulators in cancer is increasingly supported by multiple scientific studies and models; nevertheless, its function and role in pathobiological conditions are yet to be fully understood and reconciled across experimental conditions. To date, not all studies comprehensively examined MCU effects on proliferation, migration, invasion, adhesion, starvation, or treatment with various drugs, or examined Ca2+ and redox-dependent processes, so work is still needed to predict if targeting the MCU complex will be advantageous or not (Fig. 1). Additionally, many studies still feature only a handful of cell lines that do not fully recapitulate disease heterogeneity or function in vivo. This does not mean that the only path forward is to dissect the role of the MCU complex and each regulator in each cancer type, stage, environment, and mutational background before there are clinical insights and applications. Instead, there is a possibility to build on the knowledge gained from the MCU complex so far and focus future studies on research areas that show a more universal response. For example, as we have seen for breast cancer, aberrant expression of MCU and its key regulators is an indicator of poor prognosis; if combined with HIF1α- or p53-related information, results could be even more robust and predictive. Targeting MCU could also potentiate current therapeutic strategies to prevent resistance and this is a research area that still needs attention. The recent discovery of MCU’s structural makeup, combined with further chemical engineering of known, but also of new specific compounds targeting the MCU complex, should help in such future endeavors and will hopefully pave the way for exploiting the mitochondrial Ca2+ signaling machinery in treating cancer.
References
Ahuja M, Muallem S (2014) The gatekeepers of mitochondrial calcium influx: MICU1 and MICU2. EMBO Rep 15:205–206
Akl H, Bultynck G (2013) Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta 1835:180–193
Arduino DM, Wettmarshausen J, Vais H, Navas-Navarro P, Cheng YM, Leimpek A, Ma ZM, Delrio-Lorenzo A, Giordano A, Garcia-Perez C, Medard G, Kuster B, Garcia-Sancho J, Mokranjac D, Foskett JK, Alonso MT, Perocchi F (2017) Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol Cell 67:711–723.e7
Balaban RS (2009) The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta 1787:1334–1341
Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–U111
Bogeski I, Gulaboski R, Kappl R, Mirceski V, Stefova M, Petreska J, Hoth M (2011) Calcium binding and transport by coenzyme Q. J Am Chem Soc 133:9293–9303
Bogeski I, Kappl R, Kummerow C, Gulaboski R, Hoth M, Niemeyer BA (2011) Redox regulation of calcium ion channels: chemical and physiological aspects. Cell Calcium 50:407–423
Booth DM, Enyedi B, Geiszt M, Varnai P, Hajnoczky G (2016) Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol Cell 63:240–248
Brookes PS, Yoon YS, Robotham JL, Anders MW, Sheu SS (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Phys Cell Phys 287:C817–C833
Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP, Gunter TE, Nicholls DG, Bernardi P, Lemasters JJ, Brand MD (2008) UCPs—unlikely calcium porters. Nat Cell Biol 10:1235–1237; author reply 1237–1240
Bustos G, Cruz P, Lovy A, Cárdenas C (2017) Endoplasmic reticulum–mitochondria calcium communication and the regulation of mitochondrial metabolism in cancer: a novel potential target. Front Oncol 7:199
Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, Ridky TW, Foskett JK (2016) Selective vulnerability of Cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep 14:2313–2324
Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, Ridky TW, Foskett JK (2016) Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep 15:219–220
Chakraborti S, Pramanick A, Saha S, Roy SS, Chaudhuri AR, Das M, Ghosh S, Stewart A, Maity B (2018) Atypical G protein beta5 promotes cardiac oxidative stress, apoptosis, and fibrotic remodeling in response to multiple cancer chemotherapeutics. Cancer Res 78:528–541
Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE (2016) Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A 113:E1872–E1880
Chen L, Sun Q, Zhou D, Song W, Yang Q, Ju B, Zhang L, Xie H, Zhou L, Hu Z, Yao H, Zheng S, Wang W (2017) HINT2 triggers mitochondrial Ca(2+) influx by regulating the mitochondrial Ca(2+) uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett 411:106–116
Cierlitza M, Chauvistre H, Bogeski I, Zhang X, Hauschild A, Herlyn M, Schadendorf D, Vogt T, Roesch A (2015) Mitochondrial oxidative stress as a novel therapeutic target to overcome intrinsic drug resistance in melanoma cell subpopulations. Exp Dermatol 24:155–157
Clapham DE (2007) Calcium signaling. Cell 131:1047–1058
Csordas G, Hajnoczky G (2009) SR/ER-mitochondrial local communication: calcium and ROS. BBA-Bioenergetics 1787:1352–1362
Csordas G, Thomas AP, Hajnoczky G (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J 18:96–108
Csordas G, Golenar T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, Moffat C, Weaver D, de la Fuente Perez S, Bogorad R, Koteliansky V, Adijanto J, Mootha VK, Hajnoczky G (2013) MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab 17:976–987
Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR (2013) Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun 434:695–700
D'Autreaux B, Toledano MB (2007) ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8:813–824
Davis FM, Parsonage MT, Cabot PJ, Parat MO, Thompson EW, Roberts-Thomson SJ, Monteith GR (2013) Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int 13:76
De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N (2014) NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem 289:20377–20385
De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–U104
De Stefani D, Rizzuto R, Pozzan T (2016) Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85, edited by Kornberg RD2016:161–192
Deluca HF, Engstrom GW (1961) Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A 47:1744–1750
Deng Q, Chen S, Fu C, Jiang J, Zou M, Tan Y, Wang X, Xia F, Feng K, Ma K, Bie P (2017) Long noncoding RNA expression profiles in sub-lethal heat-treated hepatoma carcinoma cells. World J Surg Oncol 15(136):136
Di A, Mehta D, Malik AB (2016) ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 60:163–171
Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, Rajan S, Vallem S, Chen X, Foskett JK, Cheung JY, Houser SR, Madesh M (2014) LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J 28:4936–4949
Esterberg R, Linbo T, Pickett SB, Wu P, Ou HC, Rubel EW, Raible DW (2016) Mitochondrial calcium uptake underlies ROS generation during aminoglycoside-induced hair cell death. J Clin Investig 126:3556–3566
Filadi R, Theurey P, Pizzo P (2017) The endoplasmic reticulum-mitochondria coupling in health and disease: molecules, functions and significance. Cell Calcium 62:1–15
Foskett JK, Philipson B (2015) The mitochondrial Ca2+ uniporter complex. J Mol Cell Cardiol 78:3–8
Foskett JK, White C, Cheung KH, Mak DOD (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 87:593–658
Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, Morganti C, Pandolfi PP, Mammano F, Pinton P (2015) Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget 6:1435–1445
Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di Virgilio F, Zito E, Pandolfi PP, Wieckowski MR, Mammano F, Del Sal G, Pinton P (2015) p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A 112:1779–1784
Gorlach A, Bertram K, Hudecova S, Krizanova O (2015) Calcium and ROS: a mutual interplay. Redox Biol 6:260–271
Graier WF, Trenker M, Malli R (2008) Mitochondrial Ca2+, the secret behind the function of uncoupling proteins 2 and 3? Cell Calcium 44:36–50
Gulaboski R, Bogeski I, Mirceski V, Saul S, Pasieka B, Haeri HH, Stefova M, Stanoeva JP, Mitrev S, Hoth M, Kappl R (2013) Hydroxylated derivatives of dimethoxy-1,4-benzoquinone as redox switchable earth-alkaline metal ligands and radical scavengers. Sci Rep 3:1865
Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME (2014) Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One 9:e96866
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS, Malek S (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464:431–435
Hempel N, Trebak M (2017) Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 63:70–96
Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, Malliankaraman K, Guo S, Rajan S, Elrod JW, Koch WJ, Cheung JY, Madesh M (2014) SLC25A23 augments mitochondrial Ca(2)(+) uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell 25:936–947
Holzmann C, Kilch T, Kappel S, Dorr K, Jung V, Stockle M, Bogeski I, Peinelt C (2015) Differential redox regulation of Ca(2)(+) signaling and viability in normal and malignant prostate cells. Biophys J 109:1410–1419
Hong Z, Chen KH, DasGupta A, Potus F, Dunham-Snary K, Bonnet S, Tian L, Fu J, Breuils-Bonnet S, Provencher S, Wu D, Mewburn J, Ormiston ML, Archer SL (2017) MicroRNA-138 and MicroRNA-25 down-regulate mitochondrial calcium uniporter, causing the pulmonary arterial hypertension Cancer phenotype. Am J Respir Crit Care Med 195:515–529
Jiang D, Zhao L, Clish CB, Clapham DE (2013) Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A 110:E2249–E2254
Kamer KJ, Mootha VK (2014) MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep 15:299–307
Kamer KJ, Sancak Y, Mootha VK (2014) The uniporter: from newly identified parts to function. Biochem Biophys Res Commun 449:370–372
Kerkhofs M, Giorgi C, Marchi S, Seitaj B, Parys JB, Pinton P, Bultynck G, Bittremieux M (2017) Alterations in Ca(2+) signalling via ER-mitochondria contact site remodelling in cancer. Adv Exp Med Biol 997:225–254
Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427:360–364
Kon N, Murakoshi M, Isobe A, Kagechika K, Miyoshi N, Nagayama T (2017) DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov 3:17045
Liao Y, Hao Y, Chen H, He Q, Yuan Z, Cheng J (2015) Mitochondrial calcium uniporter protein MCU is involved in oxidative stress-induced cell death. Protein Cell 6:434–442
Loubiere C, Clavel S, Gilleron J, Harisseh R, Fauconnier J, Ben-Sahra I, Kaminski L, Laurent K, Herkenne S, Lacas-Gervais S, Ambrosetti D, Alcor D, Rocchi S, Cormont M, Michiels JF, Mari B, Mazure NM, Scorrano L, Lacampagne A, Gharib A, Tanti JF, Bost F (2017) The energy disruptor metformin targets mitochondrial integrity via modification of calcium flux in cancer cells. Sci Rep 7(5040):5040
Lovy A, Foskett JK, Cardenas C (2016) InsP3R, the calcium whisperer: maintaining mitochondrial function in cancer. Mol Cell Oncol 3:e1185563
Madreiter-Sokolowski CT, Gottschalk B, Parichatikanond W, Eroglu E, Klec C, Waldeck-Weiermair M, Malli R, Graier WF (2016) Resveratrol specifically kills cancer cells by a devastating increase in the Ca2+ coupling between the greatly tethered endoplasmic reticulum and mitochondria. Cell Physiol Biochem 39:1404–1420
Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Mueller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14:1336–1343
Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M (2012) MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151:630–644
Mammucari C, Gherardi G, Rizzuto R (2017) Structure, activity regulation, and role of the mitochondrial calcium uniporter in health and disease. Front Oncol 7:139
Marchi S, Pinton P (2013) Mitochondrial calcium uniporter, MiRNA and cancer: live and let die. Commun Integr Biol 6:e23818
Marchi S, Pinton P (2014) The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol Lond 592:829–839
Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P (2013) Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol 23:58–63
Martin J, Maurhofer O, Bellance N, Benard G, Graber F, Hahn D, Galinier A, Hora C, Gupta A, Ferrand G, Hoppeler H, Rossignol R, Dufour JF, St-Pierre MV (2013) Disruption of the histidine triad nucleotide-binding hint2 gene in mice affects glycemic control and mitochondrial function. Hepatology 57:2037–2048
Mishra J, Jhun BS, Hurst S, OU J, Csordas G, Sheu SS (2017) The mitochondrial Ca(2+) uniporter: structure, function, and pharmacology. Handb Exp Pharmacol 240:129–156
OU J, Ryu SY, Jhun BS, Hurst S, Sheu SS (2014) Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid Redox Signal 21:987–1006
Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK, Chou JJ (2016) Architecture of the mitochondrial calcium uniporter. Nature 533:269–273
Paillard M, Csordas G, Szanda G, Golenar T, Debattisti V, Bartok A, Wang N, Moffat C, Seifert EL, Spat A, Hajnoczky G (2017) Tissue-specific mitochondrial decoding of cytoplasmic Ca(2+) signals is controlled by the stoichiometry of MICU1/2 and MCU. Cell Rep 18:2291–2300
Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A 107:436–441
Parekh AB (2008) Mitochondrial regulation of store-operated CRAC channels. Cell Calcium 44:6–13
Parekh AB, Putney JW Jr (2005) Store-operated calcium channels. Physiol Rev 85:757–810
Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R (2013) The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. J Biol Chem 288:10750–10758
Patron M, Checchetto V, Raffaello A, Teardo E, Reane DV, Mantoan M, Granatiero V, Szabo I, De Stefani D, Rizzuto R (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53:726–737
Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D (2018) MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. https://doi.org/10.1038/s41418-018-0113-8
Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA (2015) CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab 21:109–116
Pedriali G, Rimessi A, Sbano L, Giorgi C, Wieckowski MR, Previati M, Pinton P (2017) Regulation of endoplasmic reticulum–mitochondria Ca2+ transfer and its importance for anti-cancer therapies. Front Oncol 7:180
Pendin D, Greotti E, Pozzan T (2014) The elusive importance of being a mitochondrial Ca2+ uniporter. Cell Calcium 55:139–145
Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467:291–U267
Petrungaro C, Zimmermann KM, Kuttner V, Fischer M, Dengjel J, Bogeski I, Riemer J (2015) The Ca(2+)-dependent release of the Mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metab 22:721–733
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27:6407–6418
Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8:e55785
Prakriya M, Lewis RS (2015) Store-operated calcium channels. Physiol Rev 95:1383–1436
Quintana A, Schwindling C, Wenning AS, Becherer U, Rettig J, Schwarz EC, Hoth M (2007) T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci U S A 104:14418–14423
Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabo I, Rizzuto R (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32:2362–2376
Ren T, Zhang H, Wang J, Zhu J, Jin M, Wu Y, Guo X, Ji L, Huang Q, Zhang H, Yang H, Xing J (2017) MCU-dependent mitochondrial Ca(2+) inhibits NAD(+)/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 36:5897–5909
Ren T, Wang J, Zhang H, Yuan P, Zhu J, Wu Y, Huang Q, Guo X, Zhang J, Ji L, Li J, Zhang H, Yang H, Xing J (2018) MCUR1-mediated mitochondrial calcium signaling facilitates cell survival of hepatocellular carcinoma via reactive oxygen species-dependent P53 Degradation. Antioxid Redox Signal 28(12):1120–1136
Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP(3)-sensitive channels that are sensed by neighboring mitochondria. Science 262:744–747
Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, Korbel C, Laschke MW, Gimotty PA, Philipp SE, Krause E, Patzold S, Villanueva J, Krepler C, Fukunaga-Kalabis M, Hoth M, Bastian BC, Vogt T, Herlyn M (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23:811–825
Samanta K, Mirams GR, Parekh AB (2018) Sequential forward and reverse transport of the Na(+) Ca(2+) exchanger generates Ca(2+) oscillations within mitochondria. Nat Commun 9(156):156
Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342:1379–1382
Saul S, Gibhardt CS, Schmidt B, Lis A, Pasieka B, Conrad D, Jung P, Gaupp R, Wonnenberg B, Diler E, Stanisz H, Vogt T, Schwarz EC, Bischoff M, Herrmann M, Tschernig T, Kappl R, Rieger H, Niemeyer BA, Bogeski I (2016) A calcium-redox feedback loop controls human monocyte immune responses: the role of ORAI Ca2+ channels. Sci Signal 9:ra26
Sena LA, Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48:158–167
Song IS, Kim HK, Lee SR, Jeong SH, Kim N, Ko KS, Rhee BD, Han J (2013) Mitochondrial modulation decreases the bortezomib-resistance in multiple myeloma cells. Int J Cancer 133:1357–1367
De Stefani D, Patron M, Rizzuto R (2015) Structure and function of the mitochondrial calcium uniporter complex. Biochim Biophys Acta 1853:2006–2011
Takata N, Ohshima Y, Suzuki-Karasaki M, Yoshida Y, Tokuhashi Y, Suzuki-Karasaki Y (2017) Mitochondrial Ca2+ removal amplifies TRAIL cytotoxicity toward apoptosis-resistant tumor cells via promotion of multiple cell death modalities. Int J Oncol 51:193–203
Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F (2015) Mitochondrial Ca(2)(+) uniporter is critical for store-operated Ca(2)(+) entry-dependent breast cancer cell migration. Biochem Biophys Res Commun 458:186–193
Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M (2016) MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep 15:1673–1685
Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C (2016) The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol Med 8:569–585
Vais H, Tanis JE, Muller M, Payne R, Mallilankaraman K, Foskett JK (2015) MCUR1, CCDC90A, is a regulator of the mitochondrial calcium uniporter. Cell Metab 22:533–535
Vyas S, Zaganjor E, Haigis MC (2016) Mitochondria and cancer. Cell 166:555–566
Warburg O (1956) On the origin of cancer cells. Science 123(3191):309–314
Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D, Bernard D (2014) Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 5:3792
Wu Y, Sarkissyan M, Vadgama JV (2016) Epithelial-mesenchymal transition and breast cancer. J Clin Med 5(2). https://doi.org/10.3390/jcm5020013
Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, Choi KS (2014) Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget 5:6816–6831
Yu C, Wang Y, Peng J, Shen Q, Chen M, Tang W, Li X, Cai C, Wang B, Cai S, Meng X, Zou F (2017) Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 8:83831–83844
Zhou X, Ren Y, Kong L, Cai G, Sun S, Song W, Wang Y, Jin R, Qi L, Mei M, Wang X, Kang C, Li M, Zhang L (2015) Targeting EZH2 regulates tumor growth and apoptosis through modulating mitochondria dependent cell-death pathway in HNSCC. Oncotarget 6:33720–33732
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950
Funding
This work was supported by the German Research Foundation (DFG) projects: SFB1190 project 17, SFB1027 Project C4, IRTG1816, and BO3643/3-2.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the special issue on Mitochondrial Signalling in Pflügers Archiv—European Journal of Physiology
Rights and permissions
About this article

Cite this article
Vultur, A., Gibhardt, C.S., Stanisz, H. et al. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch - Eur J Physiol 470, 1149–1163 (2018). https://doi.org/10.1007/s00424-018-2162-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-018-2162-8