
Abstract
Glioblastoma (GBM) is a highly malignant, rapidly progressive astrocytoma that is distinguished from lower grade tumors by necrosis, severe hypoxia and microvascular hyperplasia. While the development of hypoxia and necrosis are known to be ominous prognostic features, precise mechanisms that underlie their development have not been elucidated. Pathologic observations and experimental evidence now suggest that vaso-occlusion and intravascular thrombosis may initiate or propagate hypoxia and necrosis. This emerging model suggests that thrombosis arises within the vasculature of high grade gliomas secondary to the overexpression of the highly pro-thrombotic protein tissue factor. This protein is dramatically upregulated in response to EGFR activation, PTEN loss and hypoxia, which occur at the transition from grade III to grade IV astrocytoma. A pro-thrombotic enviroment also activates the family of protease-activated receptors (PARs) on tumor cells, which are G-protein coupled and enhance invasive and pro-angiogenic properties. Vaso-occlusive and pro-thrombotic mechanisms in GBM could readily explain the rapid peripheral expansion seen on neuroimaging and the dramatic shift to an acceleration in clinical progression due to hypoxia-induced angiogenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Vascular Endothelial Growth Factor
- Tissue Factor
- Tissue Factor Expression
- PTEN Loss
- Intravascular Thrombosis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Most clinicians and investigators in neuro-oncology are well aware of the features of glioblastoma (GBM), as it remains the most predictably devastating disease in the field. This tumor is the highest grade astrocytoma (WHO grade IV), characterized by widespread infiltration, necrosis, angiogenesis, rapid growth, and a dismal prognosis (CBTRUS 2002, Kleihues et al. 2000, Ohgaki and Kleihues 2005). Following the most advanced treatment, including neurosurgery, radiotherapy, and chemotherapy, mean survival of patients with GBM is only 60 weeks (Stupp et al. 2005). When patients receive only surgical resection but are not treated with adjuvant therapy, mean survival is a mere 14 weeks, underscoring the tremendous natural growth properties of these tumors (Shapiro and Young 1976). Lower grade infiltrative astrocytomas (i.e., WHO grade II and III astrocytomas) are also fatal tumors but are characterized by much slower growth rates and longer survivals (3–8 years) (Brat et al. 2002, Gupta et al. 2005). Only once these lower grade tumors have progressed to GBM do they demonstrate accelerated growth and rapid progression to death. This review explores potential mechanisms that might account for the rapid clinical progression associated with the GBM histology, emphasizing mechanisms that initiate the hypoxia and necrosis that are the hallmarks of this tumor.
2 Distinctive Features of Glioblastoma
GBMs (grade IV) are sufficiently distinct from the lower grade astrocytomas (grade II and III) in their clincial behavior and biologic potential that they can be thought of as a quantum leap in malignancy rather than a small step along a disease spectrum. The unique neuroimaging and pathologic features that emerge during the transition to GBM provide the best insight into the mechanisms that might account for these enhanced growth properties. Grade II and III astrocytomas show hyperintense T2-weighted (or FLAIR) signal abnormalities on magnetic resonance imaging (MRI), reflecting vasogenic edema generated in response to diffuse infiltration by individual tumor cells. These lower grade tumors expand the involved brain and distort its architecture to the point of producing clinical symptoms, but tissue destruction and necrosis are not observed. There is only mild or no contrast-enhancement, suggesting a relatively intact blood–brain barrier (Henson et al. 2005, Zhu et al. 2000). Radial growth rates of these tumors are modest, with annual increases in diameter of 2–4 mm/yr (Mandonnet et al. 2003, Swanson et al. 2003). Histologic sections of grade II–III tumors reflect the imaging properties: neoplastic cells are seen diffusely infiltrating between neuronal and glial processes, leading to architectural disruption and edema (Bellail et al. 2004, Gupta et al. 2005). As astrocytomas advance through the pathologic spectrum from the lower end of grade II to the upper end of grade III, the degree of nuclear anaplasia increases and the proliferative capacity edges upward, resulting in a more densely cellular tumor with greater malignant potential (Brat et al. 2002). Thus, grade II–III astrocytomas can be conceptualized as a continuum of gradually increasing tumor grade and growth, with clinical properties generally correlating with the density and malignancy of tumor cells.
Tumor imaging, histology, and dynamics change dramatically during the transition to GBM, suggesting that a fundamentally altered neoplasm has emerged. Radial growth rates can accelerate to values nearly 10 times greater than those in grade II astrocytomas (Mandonnet et al. 2003, Swanson et al. 2003). MRI typically reveals a central, contrast-enhancing component (“ring-enhancing mass”) that arises from within the infiltrative astrocytoma and rapidly expands outward, causing a much larger T2-weighted signal abnormality in the tumor’s periphery (Fig. 22.1) (Henson et al. 2005, Zhu et al. 2000). The histopathologic features that disinguish GBM from lower grade astrocytomas are found near this contrast-enhancing rim and include (1) foci of necrosis, usually with evidence of surrounding cellular pseudopalisades (“pseudopalisading necrosis”) and (2) microvascular hyperplasia, a form of angiogenesis morphologically recognized as endothelial proliferation within newly sprouted vessels (Fig. 22.2 and Color Plate 32) (Brat et al. 2002, Brat and Van Meir 2001, Burger and Green 1987, Kleihues et al. 2000). These two diagnostic findings of GBM are independent of tumor cell characteristics, yet carry an inordinant degree of prognostic power (Nelson et al. 1983, Raza et al. 2002). Given their remarkable prognostic significance, it is almost certain that these structures are mechanistically linked to the accelerated growth properties that characterize the grade III–IV transition.

Glioblastoma (WHO grade IV) has a distinct growth pattern on magnetic resonance imaging (MRI). (A) Axial post-contrast MRI of GBM shows a ring-like pattern of contrast-enhancement (arrow) surrounding a central zone of necrosis. (B) Axial T2-weighted MRI demonstrates a hyperintense region (arrow) peripheral to the central necrotic zone, which represents diffusely infiltrating tumor cells and the accompanying vasogenenic edema

Pathologic features of glioblastoma. (A) Pseudopalisades (arrowheads) are characterized by an accumulation of tumor cells around a central necrotic zone (asterisk). Microvascular hyperplasia (arrow) is a form of angiogenesis that is induced by hypoxic pseudopalisading cells and can be noted in regions adjacent to necrosis. (B) A central vessel within a GBM is occluded by an intravascular thrombus (arrow) in close proximity to an emerging region of necrosis (asterisk) with surrounding pseudopalisading cells (arrowheads). (C) H&E staining of a GBM demonstrates intravascular thrombosis occluding and distending a vessel (arrow) within the center of a pseudopalisade (arrowheads). (D) Immunohistochemistry for HIF-1α on a serial tissue section shows increased nuclear staining in pseudopalisades, indicating an adaptive response to hypoxia (arrowheads) (see Color Plate 32)
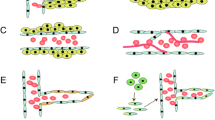
An emerging model of tumor progression may explain the development of necrosis, the relationship between necrosis and angiogenesis, and the strong association between necrosis and aggressive clinical behavior (Figs. 22.2, 22.3 and Color Plate 33) (Brat et al. 2002, Brat and Van Meir 2001, Holash et al. 1999, Zagzag et al. 2000). This model hypothesizes a sequence that begins with an infiltrating astocytoma of moderate to high cellularity (i.e., grade III astrocytoma) and continues with (see Fig. 22.3): (1) vascular occlusion within the tumor that is often associated with intravascular thrombosis; (2) hypoxia in regions surrounding vascular pathology; (3) outward migration of tumor cells away from hypoxia, creating a peripherally moving wave (pseudopalisade) and central necrosis; (4) secretion of hypoxia-inducible, pro-angiogenic factors (VEGF, IL-8) by pseudopalisading cells; (5) an exuberant angiogenic response creating microvascular proliferation in regions adjacent to central hypoxia; (6) accelerated outward expansion of tumor cells toward a new vasculature. The global growth properties of GBM within the brain reflect a coalescence of these microscopic processes and result in a peripherally expanding tumor with a large degree of central necrosis.

Potential mechanism of pseudopalisade formation. (Upper panel) In grade III astrocytoma, tumor cells with moderate to high-density infiltrate through the CNS and receive oxygen and nutrient supplies through intact native blood vessels. (Middle panel) A vascular insult occurs as a result of tumor growth and causes endothelial injury, expression of pro-coagulant factors and intravascular thrombosis, leading to hypoxia in regions surrounding vascular pathology. Tumor cells begin to migrate away from hypoxia, creating a peripherally moving wave that is seen microscopically as pseudopalisading cells. (Lower panel) The zone of hypoxia and central necrosis expands, while the hypoxic tumor cells of pseudopalisades secrete pro-angiogenic factors (VEGF, IL-8) leading to microvascular proliferation in regions adjacent to central hypoxia causing an accelerated outward expansion of tumor cells toward a new vasculature. Illustration by Mica Duran. Reproduced with permission from Blackwell Press from Tehrani M et al. (2008) Brain Pathology 18:164–171 (see Color Plate 33)
3 The Significance of Pseudopalisades, Necrosis, and Hypoxia
It is widely agreed that pseudopalisades represent hypercellular zones that surround necrotic foci in GBM (Fig. 22.2) (Burger and Green 1987, Kleihues et al. 2000). Pseudopalisading of cells around central degeneration has been recognized for nearly a century as both a defining feature of GBM and a morphologic finding that predicts aggressive behavior (Bailey and Cushing 1926). In light of this, it is curious that there is only scattered data on their nature or their role in tumor progression. Analysis of the shapes and sizes of pseudopalisades suggests that these structures evolve and enlarge over time, giving rise to wider and wider expanses of coagulative necrosis (Brat et al. 2004). A commonly held belief has been that pseudopalisades represent a rim of residual tumor cells around a centrally degenerating clone of highly proliferative cells. The term “pseudopalisade” itself implies that cells have not truly aggregated around necrosis, but only give this impression due to the absence of a cental hypercellular zone.
Recent studies have attempted to more precisely define underlying mechanisms of necrosis and pseudopalisading in GBM (Brat et al. 2004). These began by considering that this hypercellular population around central necrosis could represent (1) a clone of rapid proliferating neoplastic cells that “outgrew its blood supply” and underwent central necrosis (clonal expansion theory); (2) a population of neoplastic cells that was resistant to apoptosis and accumulated due to increased cell survival; (3) a mixed population of neoplastic and inflammatory cells adjacent to necrosis; or (4) a population of rapidly moving tumor cells that superimposed themselves on a more stationary population, causing increased cell density. Perhaps surprisingly, these investigations determined that pseudopalisading cells were less proliferative than adjacent tumor, indicating they do not likely accumulate due to clonal expansion (Brat et al. 2004, Schiffer et al. 1995). Second, pseudopalisades are composed almost entirely of tumor cells and do not include a significant population of non-neoplastic cells (e.g., inflammatory cells). Third, pseudopalisades show increased levels of apoptosis compared to adjacent tumor, indicating they do not accumulate due to a survival advantage (Brat et al. 2004, Tachibana et al. 1996).
These investigations instead supported a model in which pseudopalisades represent a wave of actively migrating tumor cells that are moving away from an area of central hypoxia. Pseudopalisading cells are known to be hypoxic, as demonstrated by their dramatic upregulation of hypoxia-inducible factor-1 (HIF-1), a nuclear transcription factor that orchestrates the cell’s adaptive response to low oxygen (Fig. 22.2) (Brat et al. 2004, Brat and Mapstone 2003, Semenza 2001, Zagzag et al. 2000). Gene expression studies performed on microdissected pseudopalisading cells from human GBMs have also supported that pseudopalisading cells have a hypoxic gene signature (Dong et al. 2005). In one study, a total of 314 up- and 385 downregulated genes were identified in pseudopalisading cells as compared to adjacent brain tumor cells. Pathway analysis of specific gene families revealed a pattern indicating a hypoxic environment, high levels of glycolosis, and cell cycle regulation. Aldolase A, pyruvate kinase, phosphoglycerate kinase and glyceraldehyde-3-phosphate dehydrogenase,and GLUT1, a glucose transporter, were glycolytic genes that were all upregulated in pseudopalisades.
Also supporting the contention that pseudopalisades represent a highly migratory population is the demonstration that hypoxic GBM cells in culture are more motile and that HIF-1 itself can regulate critical pro-migratory mechanisms (Brat et al. 2004, Graham et al. 1999, Krishnamachary et al. 2003, Pennacchietti et al. 2003). Hypoxic upregulation of c-Met, a tyrosine kinase receptor, as well as the potentiation of hepatocyte growth factor (HGF) signaling through c-Met, could account for at least some of the pro-migratory effects of HIF-1 activation (Abounader and Laterra 2005, Pennacchietti et al. 2003). Moreover, hypoxic pseudopalisades express increased levels of extracellular matrix proteases associated with invasion, including MMP-2 and uPAR (Bellail et al. 2004, Brat et al. 2004, Graham et al. 1999, Mori et al. 2000, Pennacchietti et al. 2003, Yamamoto et al. 1996). Thus, the combined evidence suggests that the pseudopalisades in GBM are formed by a population of hypoxic, actively migrating neoplastic cells that create a hypercellular zone around an evolving area of central necrosis.
4 Vascular Pathology Underlies Hypoxia, Necrosis, and Pseudopalisades
The hypoxia that leads to increased tumor cell migration to form pseudopalisades could result from limitations in vascular perfusion within the tumor (i.e., disruption of the blood supply) or from reduced oxygen diffusion within the growing neoplasm, in part due to increased metabolic demands (Brat and Van Meir 2001). A growing body of experimental and observational evidence favors the hypothesis that pseudopalisades represent tumor cells migrating away from a dysfunctional vasculature (Brat et al. 2004, Holash et al. 1999, Zagzag et al. 2000). The majority of pseudopalisades are ring-like or ovoid, but others have a long, narrow and winding (i.e., serpiginous) pattern when viewed in longitudinal tissue sections – a pattern that suggests an underlying vascular substrate (Kleihues et al. 2000). Perhaps less appreciated, abnormal vessels can often be noted within the lumina of at least a subset of pseudopalisades. A comprehensive survey of human GBM specimens found that over 50% of pseudopalisades had evidence of a central vascular lumen that was either degenerating or thrombosed (Fig. 22.2) (Brat et al. 2004). Twenty percent contained a vessel with complete luminal occlusion caused by intravascular thrombosis (Fig. 22.2). Analysis of pseudopalisade sizes and shapes led to the conclusion that tissue sampling and tangential sections result in an underestimation of the true frequency of vascular pathology and intravascular thrombosis within pseudopalisades, which has led to an under-appreciation of their relevance to necrosis in GBM. Moreover, it is worth considering that the vasculature upstream and downstream of intravascular thrombosis will not adequately perfuse the tumor in its distribution, which will ultimately lead to hypoxia and necrosis without histologic evidence of thrombus.
5 Initiators of Vascular Pathology
Experimental evidence indicates that vaso-occlusion within gliomas occurs before the development of hypoxia-induced angiogenesis. Inifiltrating tumor cells of low-grade tumors, such as those in grade II astrocytoma, gain access to oxygen and nutrients through a vascular supply by “co-opting” the host’s blood vessels (Holash et al. 1999, Zagzag et al. 2000). In response, vascular endothelial cells eventually undergo a number of changes that include hypertrophy, discohesion, and even apoptosis. One suggestion is that endothelial damage is first initiated by the effects of Angiopoietin-2 (Ang-2) on endothelial cell. Ang-2 is thought to act in an autocrine fashion on tumoral blood vessels as a Tie-2 receptor antagonist. In the absence of VEGF, Tie-2 blockage leads to vascular destabilization, endothelial cell apoptosis, and vascular regression (Holash et al. 1999, Maisonpierre et al. 1997). In human specimens, Ang-2 is expressed by endothelial cells of high-grade gliomas but not low-grade gliomas or normal brain, and its upregulation precedes endothelial apoptosis, suggesting that it could cause vascular injury (Stratmann et al. 1998, Zagzag et al. 2000, 1999). Other arguments hold that Ang-2 causes structural changes of vessels that are required for angiogenesis but does not induce apoptosis (Vajkoczy et al. 2002, Zhang et al. 2003). Factors released from glioma cells following genetic alteration (EGFR amplication or PTEN loss), such as VEGF and TNF-α, could also precipitate vascular injury and thrombosis (Shen et al. 2001). VEGF induces changes in vascular permeability, while both VEGF and TNF-α have been demonstrated to induce endothelial tissue factor expression through activation of the transcription factor Egr-1 (Mechtcheriakova et al. 2001).
6 Intravascular Thrombosis Accentuates and Propagates Tumor Hypoxia
While precise initiators of vascular pathology in GBM continue to be studied, it is becoming clear that intravascular thrombosis within these neoplasms can accentuate and propagate tumoral hypoxia and necrosis. A strong relationship between abnormal blood clotting and human malignancy is well established in gliomas and other forms of cancer (Rickles and Falanga 2001). Patients with GBM are at high risk for developing systemic disorders of coagulation, with nearly 30% suffering from deep vein thrombosis or pulmonary thrombo-embolism (Walsh and Kakkar 2001). Intravascular thrombosis within the tumoral tissue of GBM is a frequent finding at the time of operation. Even more impressively, thrombosed vessels within resected GBM specimens can almost always be identified under the microscope (Brat et al. 2004). Both the frequencies of associated systemic coagulopathy and the finding of intravascular thrombosis within neoplastic tissue are much higher in GBMs (grade IV) than AAs (grade III), indicating that critical pro-thrombotic events must occur in this transition (Brat et al. 2004, Sawaya et al. 1995, Walsh and Kakkar 2001).
In order to determine whether intravascular thrombosis was a specific finding associated with the GBM histology, Tehrani et al. investigated the histologic presence of thrombosis in a representative cross section of 297 adult CNS malignancies, including 103 GBMs (Tehrani et al. 2008). Among newly diagnosed (i.e., untreated) tumors, thrombosis was present in 92% of GBM resections, significantly greater than any of the other types of CNS neoplasm. The sensitivity of thrombosis for the diagnosis of GBM in this set of tumors was 92% and the specificity was 91%. Thus, intravascular thrombosis seemed to be a near constant feature of GBM, but not other tumor types. Interestingly, intravascular thrombosis was an uncommon finding in AAs and was only noted in a small number of stereotactic biopsies. This subset of AA patients had significantly shorter survivals than those AAs without thrombosis. This could imply that stereotactic biopsies were actually undersampled GBMs or that these AAs with thrombosis represented an aggressive subset whose progression to the GBM phenotype was imminent. The development of intravascular thrombosis within GBM and the emergence of hypoxia and necrosis are too interrelated, both spatially and temporally, to be coincidental. It is highly probable that intravascular thrombosis initiates or propagates hypoxia and necrosis in GBM (Brat and Van Meir 2004).
7 Tissue Factor, a Potent Pro-Coagulant, Is Upregulated in GBM
Multiple factors likely contribute to intravascular thrombosis in GBM, including abnormal blood flow within a distorted vasculature, increased interstitial edema and pressure resulting in vascular collapse, dysregulation of pro- and anti-coagulant factors, and access of plasma clotting factors to tumoral tissue. Normal CNS blood vessels allow only limited diffusion through their walls due to a highly restrictive blood–brain barrier, which is formed primarily by endothelial tight junctions, but also has contributions from astrocytic foot plates, extracellular matrix, and endothelial-pericytic interactions (Dinda et al. 1993). This barrier becomes breached in GBM and can be visualized radiologically by the presence of contrast-enhancement due to increased vascular permeability to contrast agents (e.g., Gadolinium) and to proteins that bind them, such as albumin (Fig. 22.1) (Henson et al. 2005, Zhu et al. 2000). Damaged vessels appear fenestrated, show detachment of pericytes, and exhibit extracellular matrix alterations (Dinda et al. 1993). All the factors that contribute to increased permeability have not been defined, but VEGF secretion by neoplastic cells is known to cause vascular leakage (Fischer et al. 1999, Senger et al. 1983). One result is the leakage of plasma coagulation factors such as Factor VII into the tissue spaces where they are activated and result in thrombosis.
A large number of plasma clotting factors are dysregulated in GBM and favor thrombosis. For example, the pro-coagulant plasminogen activator inhibitor 1 (PAI-1) is markedly elevated in GBMs, while the expression of anti-coagulants, such as the fibrinolytic tissue type plasminogen activator (tPA), is decreased (Sawaya et al. 1991, 1995). One of the most highly upregulated pro-thrombotic factors in GBM is tissue factor (TF), a 47 kDa transmembrane glycoprotein receptor that is a critical regulator of tissue hemostasis and one of the body’s most potent stimulants of thrombosis (Versteeg et al. 2003). In normal tissue, TF is expressed almost exclusively by stromal cells and a disruption of vascular integrity is required to bring TF into contact with its activating ligand from the plasma, Factor VII/VIIa (Morrissey 2001). In turn, TF/Factor VIIa activation promotes the generation of thrombin from prothrombin, ultimately leading to platelet aggregation, fibrin deposition, and local hemostasis. The normally tight regulation of TF is lost in a variety of pathologic conditions including neoplasia and numerous cancers show increased expression by tumor cells, stroma, and endothelium (Rickles et al. 2003). A direct correlation between TF levels and tumor grade has been noted for multiple tumor types (Contrino et al. 1996, Seto et al. 2000), including gliomas (Guan et al. 2002, Hamada et al. 1996). Indeed, TF is highly expressed by over 90% of malignant astrocytomas, but only 10% of grade I and II astrocytomas (Guan et al. 2002). The pro-thrombotic effects of TF at the cell surface are largely mediated through downstream activation of coagulation proteases Factor VII (VIIa), Factor X (Xa), and thrombin.
8 PTEN, EGFR, and Hypoxia Regulate Tissue Factor Expression in GBM
Recent investigations have attempted to define the genetic and physiologic triggers that might cause increased TF expression and thrombosis in human malignancy (Boccaccio et al. 2005, Rak et al. 2006, Rong et al. 2005, Yu et al. 2005). Genetic events that arise during astrocytoma progression have been well characterized and include PTEN and TP53 mutations, p16(CDKN2A) and p14 ARF deletions, EGFR and MDM2 amplifications (Hunter et al. 2003). Among these, EGFR amplification and PTEN mutations are prime candidates to explore for regulation of pro-thrombotic factors since they occur precisely during the transition from AA to GBM, when thrombosis and pseudopalisades emerge (Rak et al. 2006, Yu et al. 2004). PTEN is a tumor suppressor located at 10q23.3 (Li et al. 1997). Inactivating mutations of PTEN occur in 30–40% of GBMs and gene inactivation through promoter methylation leads to lost expression of PTEN in over 70% (Duerr et al. 1998, Rasheed et al. 1997, Wang et al. 1997). The effects of PTEN on TF expression and pro-coagulant properties by malignant gliomas were recently studied by introducing a wild-type PTEN gene into a PTEN null glioma cell line (U87MG). The expression TF protein at the cell surface of glioma cells was dramatically suppressed by PTEN, which in turn led to prolonged plasma clotting times using in vitro measures of coagulation. Although many of PTEN’s biologic effects depend on its lipid phosphatase activity and ability to antagonize PI-3 kinase, these studies indicated that regulation of TF depended at least in part on PTEN’s protein phosphatase activity. Potential downstream signaling mechanisms relevant to the control of TF by PTEN were investigated using a series of human astrocytes that were sequentially transformed with E6/E7/hTERT, Ras, and Akt expression vectors, which have been used to recapitulate astrocytoma progression both in vitro and in vivo (Sonoda et al. 2001a, b). Cells transfected with either Akt or Ras showed upregulation of TF, while those transformed with combined Ras and Akt showed the highest TF expression, suggesting that both signaling pathways may participate as downstream regulators of PTEN (Rong et al. 2005, Yu et al. 2005). Other investigations have emphasized that activated forms of Ras are critical for the expression of TF and its tumorigenic effects (Yu et al. 2005). Thus, PTEN loss during astrocytoma progression likely leads to increased TF expression and plasma coagulation, both through Akt/Ras-dependent and protein phosphatase-dependent mechansism.
EGFR amplification is a second genetic alteration present in a subset of GBMs (40–50%) that could be responsible for TF upregulation. The most frequent EGFR mutation in GBM involves deletion of exon 2–7 resulting in the constitutively active form EGFRvIII. It has recently been shown that overexpression of either EGFR or EGFRvIII in human glioma cells in vitro consistently leads to increased basal expression of TF. When EGFR overexpressing gliomas are stimulated with EGF, there is a striking upregulation of TF that occurs within 6 hours (Fig. 22.4). In all cases, increased TF expression by GBM leads to accelerated plasma coagulation in vitro. The EGFR-mediated increases in TF expression depended most strongly on AP-1 transcriptional activity and were associated with c-Jun N-terminal kinase (JNK) and JunD activation. Interestingly, the restoration of PTEN expression in PTEN-deficient GBM cells diminished EGFR-induced TF expression by inhibiting this same JunD/AP-1 transcriptional activity. PTEN mediated this effect by antagonizing PI-3 K activity, which in turn attenuated both Akt and JNK activities. It was also suggested that these mechanisms are likely at work in vivo, since amplification of EGFR and expression of EGFR protein were associated with the expression of TF and the presence of thrombosis in human high-grade astrocytoma specimens (Fig. 22.5 and Color Plate 34). Thus, both EGFR activation and PTEN loss, which both occur in the transition to GBM, favor the upregulation of TF.

EGFR upregulates TF in human GBM cells and accelerates plasma coagulation, while PTEN suppresses TF expression (A) Western blot of cell lysates from U87MG, U87MG-EGFRvIII, and U87MG-wt-EGFR cells demonstrates higher basal TF expression by U87MG-EGFRvIII cells (lane 2) and U87MG-EGFRwt (lane 3). U87MG-EGFRvIII has a high level of constitutively active tyrosine kinase activity as demonstrated by the phosphorylated form of EGFR. (B) Western blot of cell lysates from U87MG-wt-EGFR cells following Lentiviral infection of PTEN-HA or GFP shows reduced TF expression under both basal and EGF-stimulated (50 ng/ml, 24 h) conditions. Western blot of U87MG-EGFRvIII cell lysates following Lentiviral infection of PTEN-HA or GFP shows reduced expression of TF in PTEN-HA infected cells. (C) The addition of 106 U87MG-EGFRwtglioma cells to human plasma causes plasma clotting in 30 sec (control, neoplastin causes plasma clotting in 12 sec). Stimulation of these cells with EGF (50 ng/ml) causes a significant shortening of the clotting time to 23 sec (#p < 0.001). An inhibitory antibody directed at TF (TFiab) causes a dramatic prolongation of plasma clotting time in both the unstimulated and the EGF-stimulated conditions (*, ** p < 0.001), while nonspecific IgG has no effect on plasma clotting

Fluorescence in situ hybridization (FISH) analysis of EGFR gene status and IHC analysis of EGFR and TF protein in human GBM and AA specimens. (A, B) Representative fluorescence images showing the EGFR (orange) and chromosome 7 centromeric (green) signals in interphase GBM cells. (A) A GBM specimen that is non-amplified for EGFR shows roughly equal numbers of EGFR and centromeric signals per nuclei (typically 2 EGFR signals per nucleus). (B) GBM with EGFR gene amplification shows greatly increased signals for EGFR compared to centromeric signal (>10 EGFR signals per nucleus). (C, D) IHC for TF protein expression in AA and GBM specimens using DAB (brown) for detection. (C) Representative image of AA showing weak TF expression (1+, brown color) in the cytoplasm on scattered tumor cells. (D) GBM specimen showing strong TF staining (3+ brown color) within the cytoplasm of nearly all tumor cells (see Color Plate 34)
In addition to genetic regulation, TF is also strongly upregulated by hypoxia in GBM (Rong et al. 2005). Hypoxic GBM cells placed directly into human plasma cause a marked acceleration of plasma clotting times compared to normoxic cells. This effect can be prevented by both the pre-incubation of cells with inhibitory antibodies to TF (Fig. 22.4C) and by using plasma that lacks Factor VII (not shown), strongly implicating TF-dependent mechanisms. Similar hypoxic conditions also cause a rapid increase in TF mRNA and protein expression by GBM cells in vitro, which are modestly suppressed by PTEN expression (Rong et al. 2005). Within human GBM specimens, the severely hypoxic pseudopalisading cells around necrosis show the highest level of TF expression, corroborating in vitro studies (Rong et al. 2005). Mechanisms responsible for the hypoxic upregulation of TF are challenging to investigate since the TF promoter contains binding sites for a variety of transcriptional regulators that can be induced by hypoxia, including Egr-1, Sp1, NF-κB, and AP-1 (Mackman 1997). The accumulated evidence in animal models and in vitro indicates that Egr-1 is the transcription factor that is most important to the hypoxic upregulation of TF and that these mechanisms do not depend on the increased expression of HIF-1 (Rong et al. 2006, Yan et al. 1999a, b). Thus, both PTEN loss and hypoxia upregulate TF expression and promote plasma clotting by GBM cells in vitro, which might suggest that these mechanisms promote intravascular thrombosis and pseudopalisading necrosis in the transition from AA to GBM.
9 TF Intitiates Signaling Through Its Cytoplasmic Tail and Through PARs
As a transmembrane receptor, TF is activated by binding to Factor VII/VIIa and transduces independent intracellular signals through its cytoplasmic tail and through interactions with the G-protein-coupled protease-activated receptors (PARs) that promote tumorigenesis. Activation of pro-coagulant function of TF is accompanied by the formation of stabilizing disulfide bonds between two cystein residues within its extracellular domain (Rehemtulla et al. 1991). A cell surface protein-disulfide isomerase (PDI) targets the disulfide bond and inactivates pro-coagulant activity but maintains intracellular signaling throughout the cytoplasmic tail (Ahamed et al. 2006). Activation of TF following Factor VIIa binding leads to the downstream activation of the mitogen-activated protein (MAP) kinase members p38 MAP kinase, p42/p44 MAP kinase, and c-Jun N-terminal kinase (JNK) (Versteeg and Ruf 2006). Other downstream pathways activated include Src-like kinases, PI3-kinase, and c-Akt/PKB Rac and Cdc42. Many of these signaling cascades cause interactions with cytoskeletal elements including actin to induce a pro-migratory phenotype (Ott et al. 1998, 2005, Versteeg et al. 2000). Intracellular signaling mechanisms induced by TF strongly promote tumorigenesis through pro-angiogenic and pro-metastatic mechanisms (Belting et al. 2004, Versteeg et al. 2003, Versteeg and Ruf 2006, Versteeg et al. 2008, 2004).
The activation of TF by Factor VIIa also has biologic significance beyond specific pro-coagulant function and signaling through its cytoplasmic tail. For example, thrombin, Factor VIIa, and Factor Xa are proteases that act as potent physiologic activators of protease-activated receptors (PARs), a family of G-protein-coupled, transmembrane receptors (Versteeg and Ruf 2006). PAR1, the family’s prototype, is activated most strongly by thrombin, which cleaves the amino-terminal extracellular domain of PAR1 and unmasks a new N-terminus, which then serves as the receptor’s ligand (Coughlin 2000). Both PAR1 and PAR2 can also be activated by Factors VIIa and Factor Xa. Activated PARs tranduce intracellular signals by coupling through G-proteins, predominantly Gαi, Gαq, and Gα12/13. Secondary signals are generated through Rho, phopholipase C (IP3 and diacylglycerol), and inhibition of adenyl cyclase. Although PARs are expressed at low levels in most normal epithelia, they are aberrantly overexpressed by a variety of carcinomas including those of breast, colon, lung, and stomach (Darmoul et al. 2003, Even-Ram et al. 1998, 2001). PAR1 activation can transform cells and is able to enhance tumorigenicity, in large part by signaling through Gαq and Gα13 (Marinissen et al. 2003, Wang et al. 2002). It is also clear that PAR1 and PAR2 activation by coagulation factors promotes invasive and metastatic properties of malignant cells (Even-Ram et al. 1998, Hjortoe et al. 2004, Morris et al. 2006, Nguyen et al. 2005, Versteeg et al. 2008). Mechanisms of increased invasion include its ability to direct cytoskeletal actin rearrangements, phosphorylation of focal adhesion kinases, and recruitment of αvβ5 integrin to contact sites (Even-Ram et al. 2001).
PAR1 protein is present in both the human and the mouse central nervous system, mostly in astrocytes, where it can be activated by thrombin (Junge et al. 2004, 2003). Investigations of human GBM cell lines and short-term cultures of resected human GBM specimens have demonstrated that PAR1 is present on the surface of tumor cells where it can be activated by both thrombin and PAR1 agonists. Such activation leads to increased phospho-inositol (PI) hydrolysis and calcium mobilization, presumably coupling through Gαq (Junge et al. 2004). Although more evidence is required to determine the biologic relevance of TF activation of thrombin and consequent PAR1 signaling in gliomas, it is highly probable that the activation of PAR1 by pro-coagulant proteases directs the migration of tumor cells in a manner similar to other malignancies. In the context of human GBM, activation would be expected to direct migration away from vaso-occlusion and hypoxia to form pseudopalisades.
10 Angiogenesis Supports Peripheral Tumor Growth
The emerging models of GBM progression indicate that vascular pathology may underlie the development of hypoxia and necrosis in GBM. While necrosis has long been recognized as a marker of aggressive behavior in diffuse gliomas, it does not explain rapid tumor progression per se (Burger and Green 1987, Raza et al. 2002). Instead, pseudopalisades that surround necrosis in GBM are intimately related to microvascular hyperplasia, a defining morphologic feature of GBM that is most often noted in regions directly adjacent to pseudopalisades (Figs. 22.2, 22.3) (Brat et al. 2002, Brat and Mapstone 2003, Brat and Van Meir 2001). This angiogenic response attempts to lay down a new vasculature for rapid neoplastic expansion, yet the proper function of these distorted vessels has not been established.
One of the most critical pro-angiogenic factors produced by hypoxic pseudopalisades that is responsible for directing nearby angiogenesis in GBM is vascular endothelial growth factor (VEGF). The VEGF gene contains an hypoxia responsive element (HRE) within its promoter that binds HIF-1, thereby activating transcription (Plate 1999, Plate et al. 1992, Semenza 2001, Shweiki et al. 1992). VEGF concentrations in the cystic fluid of human GBMs can reach levels that are 200- to 300-fold higher than in serum (Takano et al. 1996). Inhibition of the HIF/VEGF pathway suppresses tumor growth experimentally (Kung et al. 2000). Once expressed and secreted, extracellular VEGF binds to its high-affinity receptors, VEGFR-1 and VEGFR-2, which are upregulated on endothelial cells of high-grade gliomas but not present in normal brain (Plate 1999). Receptor activation then leads to angiogenesis in regions adjacent to pseudopalisades, eventually leading to a vascular density in GBMs that is among the highest of all human neoplasms (Figs. 22.2, 22.3).
A second pro-angiogenic factor that is highly upregulated in GBMs is interleukin-8 (IL-8, CXCL8) (Brat et al. 2005). Much like VEGF, hypoxia/anoxia strongly stimulates IL-8 expression and its expression is also found at highest levels within the pseudopalisades of GBM (Desbaillets et al. 1999, 1997). Unlike VEGF, IL-8 has a more punctate distribution within pseudopalisades and it remains unclear if tumor cells or scattered infiltrating macrophages are responsible for the majority of its expression. Hypoxic upregulation of the IL-8 gene is not directly due to HIF activation, since there is no HRE within its promoter. Rather, the IL-8 promoter contains binding sites for other transcription factors, including NF-κB, AP-1, and C-EBP/NF-IL-6. AP-1 appears to mediate much of IL-8’s upregulation by hypoxia/anoxia (Desbaillets et al. 1999, 1997, Garkavtsev et al. 2004). IL-8 is also strongly upregulated by tumor cells in response to activation of Factor VIIa by TF. Such overexpression of IL-8 by neoplastic cells may have autocrine effects on the malignant behavior of tumor cells (i.e., invasion or metastasis) in addition to inducing angiogenesis (Hjortoe et al. 2004). The IL-8 receptors that could potentially contribute to IL-8-mediated tumorigenic and angiogenic responses in GBM include CXCR1 and CXCR2, both of which are G-protein coupled.
The precise type of angiogenesis that is most evident in GBM, microvascular hyperplasia, is characterized by numerous enlarged, rapidly dividing endothelial cells, pericytes, and smooth muscle cells that form tufted micro-aggregates at the leading edge of sprouting vessels (Fig. 22.2A) (Brat and Van Meir 2001). In its most florid form, angiogenesis takes the shape of “glomeruloid bodies” – a feature that is most characteristic of GBM but is also an independent marker of poor prognosis in other forms of cancer (Straume et al. 2002). Since necrosis and hypoxia are located in the GBM’s core and near the contrast-enhancing rim, hypoxia-induced angiogenesis occurs further peripherally, favoring neoplastic growth outward. The permissive nature of the CNS parenchymal matrix to diffuse infiltration by individual glioma cells allows for this burst of peripheral expansion (Bellail et al. 2004).
11 Conclusion
The development of hypoxia and necrosis within a diffuse glioma has critical biologic implications and results in a highly aggressvie tumor, GBM. The pseudopalisades around necrosis and the ensuing microvascular hyperplasia may suggest an underlying mechanism responsible for this accelerated growth. We have proposed that a sequence of events that includes vaso-occlusion and intravascular thrombosis leads to the development of the biologically aggressive hypoxic growth phase in GBM. These mechanisms could readily explain the dramatic change in behavior as tumors transition to the GBM histology. Since both necrosis and vascular proliferation are also markers of poor prognosis in other types of cancer, the identification of their underlying mechanisms may have more general implications for tumor angiogenesis and malignant progression. Once identified, the pathophysiologic triggers underlying vaso-occlusion will become attractive, novel targets for anti-tumor therapy.
References
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 7:436–451
Ahamed J, Versteeg HH, Kerver M, et al. (2006) Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA 103:13932–13937
Bailey P, Cushing H (1926) Tumors of the glioma group. Lippincott Philadelphia
Bellail AC, Hunter SB, Brat DJ, et al. (2004) Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol 36:1046–1069
Belting M, Dorrell MI, Sandgren S, et al. (2004) Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med 10:502–509
Boccaccio C, Sabatino G, Medico E, et al. (2005) The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature 434:396–400
Brat DJ, Bellail AC, Van Meir EG (2005) The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol 7:122–133
Brat DJ, Castellano-Sanchez A, Kaur B, et al. (2002) Genetic and biologic progression in astrocytomas and their relation to angiogenic dysregulation. Adv Anat Pathol 9:24–36
Brat DJ, Castellano-Sanchez AA, Hunter SB, et al. (2004) Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 64:920–927
Brat DJ, Mapstone TB (2003) Malignant glioma physiology: cellular response to hypoxia and its role in tumor progression. Ann Intern Med 138:659–668
Brat DJ, Van Meir EG (2001) Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: a new world of angiogenesis research. Am J Pathol 158:789–796
Brat DJ, Van Meir EG (2004) Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab Invest 84:397–405
Burger PC, Green SB (1987) Patient age, histologic features, and length of survival in patients with glioblastoma multiforme. Cancer 59:1617–1625
Central Brain Tumor Registry of the United States (CBTRUS) (2002) Statistical report: primary brain tumors in the United States 1995–1999. Central Brain Tumor Registry of the United States (CBTRUS), Chicago, IL
Contrino J, Hair G, Kreutzer DL, et al. (1996) In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med 2:209–215
Coughlin SR (2000) Thrombin signalling and protease-activated receptors. Nature 407:258–264
Darmoul D, Gratio V, Devaud H, et al. (2003) Aberrant expression and activation of the thrombin receptor protease-activated receptor-1 induces cell proliferation and motility in human colon cancer cells. Am J Pathol 162:1503–1513
Desbaillets I, Diserens AC, de Tribolet N, et al. (1999) Regulation of interleukin-8 expression by reduced oxygen pressure in human glioblastoma. Oncogene 18:1447–1456
Desbaillets I, Diserens AC, Tribolet N, et al. (1997) Upregulation of interleukin 8 by oxygen-deprived cells in glioblastoma suggests a role in leukocyte activation, chemotaxis, and angiogenesis. J Exp Med 186:1201–1212
Dinda AK, Sarkar C, Roy S, et al. (1993) A transmission and scanning electron microscopic study of tumoral and peritumoral microblood vessels in human gliomas. J Neurooncol 16:149–158
Dong S, Nutt CL, Betensky RA, et al. (2005) Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol 64:948–955
Duerr EM, Rollbrocker B, Hayashi Y, et al. (1998) PTEN mutations in gliomas and glioneuronal tumors. Oncogene 16:2259–2264
Even-Ram S, Uziely B, Cohen P, et al. (1998) Thrombin receptor overexpression in malignant and physiological invasion processes. Nat Med 4:909–914
Even-Ram SC, Maoz M, Pokroy E, et al. (2001) Tumor cell invasion is promoted by activation of protease activated receptor-1 in cooperation with the alpha vbeta 5 integrin. J Biol Chem 276:10952–10962
Fischer S, Clauss M, Wiesnet M, et al. (1999) Hypoxia induces permeability in brain microvessel endothelial cells via VEGF and NO. Am J Physiol 276:C812–820
Garkavtsev I, Kozin SV, Chernova O, et al. (2004) The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 428:328–332
Graham CH, Forsdike J, Fitzgerald CJ, et al. (1999) Hypoxia-mediated stimulation of carcinoma cell invasiveness via upregulation of urokinase receptor expression. Int J Cancer 80:617–623
Guan M, Jin J, Su B, et al. (2002) Tissue factor expression and angiogenesis in human glioma. Clin Biochem 35:321–325
Gupta M, Djalilvand A, Brat DJ (2005) Clarifying the diffuse gliomas: an update on the morphologic features and markers that discriminate oligodendroglioma from astrocytoma. Am J Clin Pathol 124:755–768
Hamada K, Kuratsu J, Saitoh Y, et al. (1996) Expression of tissue factor correlates with grade of malignancy in human glioma. Cancer 77:1877–1883
Henson JW, Gaviani P, Gonzalez RG (2005) MRI in treatment of adult gliomas. Lancet Oncol 6:167–175
Hjortoe GM, Petersen LC, Albrektsen T, et al. (2004) Tissue factor-factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood 103:3029–3037
Holash J, Maisonpierre PC, Compton D, et al. (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284:1994–1998
Hunter SB, Brat DJ, Olson JJ, et al. (2003) Alterations in molecular pathways of diffusely infiltrating glial neoplasms: application to tumor classification and anti-tumor therapy (Review). Int J Oncol 23:857–869
Junge CE, Lee CJ, Hubbard KB, et al. (2004) Protease-activated receptor-1 in human brain: localization and functional expression in astrocytes. Exp Neurol 188:94–103
Junge CE, Sugawara T, Mannaioni G, et al. (2003) The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc Natl Acad Sci USA 100:13019–13024
Kleihues P, Burger PC, Collins VP, et al. (2000) Pathology and genetics of tumours of the nervous systems. International Agency for Research on Cancer Lyon
Krishnamachary B, Berg-Dixon S, Kelly B, et al. (2003) Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res 63:1138–1143
Kung AL, Wang S, Klco JM, et al. (2000) Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med 6:1335–1340
Li J, Yen C, Liaw D, et al. (1997) PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275:1943–1947
Mackman N (1997) Regulation of the tissue factor gene. Thromb Haemost 78:747–754
Maisonpierre PC, Suri C, Jones PF, et al. (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55–60
Mandonnet E, Delattre JY, Tanguy ML, et al. (2003) Continuous growth of mean tumor diameter in a subset of grade II gliomas. Ann Neurol 53:524–528
Marinissen MJ, Servitja JM, Offermanns S, et al. (2003) Thrombin protease-activated receptor-1 signals through Gq- and G13-initiated MAPK cascades regulating c-Jun expression to induce cell transformation. J Biol Chem 278:46814–46825
Mechtcheriakova D, Schabbauer G, Lucerna M, et al. (2001) Specificity, diversity, and convergence in VEGF and TNF-alpha signaling events leading to tissue factor up-regulation via EGR-1 in endothelial cells. Faseb J 15:230–242
Mori T, Abe T, Wakabayashi Y, et al. (2000) Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-alpha or basic fibroblast growth factor. J Neurooncol 46:115–123
Morris DR, Ding Y, Ricks TK, et al. (2006) Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res 66:307–314
Morrissey JH (2001) Tissue factor: an enzyme cofactor and a true receptor. Thromb Haemost 86:66–74
Nelson JS, Tsukada Y, Schoenfeld D, et al. (1983) Necrosis as a prognostic criterion in malignant supratentorial, astrocytic gliomas. Cancer 52:550–554
Nguyen QD, De Wever O, Bruyneel E, et al. (2005) Commutators of PAR-1 signaling in cancer cell invasion reveal an essential role of the Rho-Rho kinase axis and tumor microenvironment. Oncogene 24:8240–8251
Ohgaki H, Kleihues P (2005) Epidemiology and etiology of gliomas. Acta Neuropathol (Berl) 109:93–108
Ott I, Fischer EG, Miyagi Y, et al. (1998) A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. J Cell Biol 140:1241–1253
Ott I, Weigand B, Michl R, et al. (2005) Tissue factor cytoplasmic domain stimulates migration by activation of the GTPase Rac1 and the mitogen-activated protein kinase p38. Circulation 111:349–355
Pennacchietti S, Michieli P, Galluzzo M, et al. (2003) Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 3:347–361
Plate KH (1999) Mechanisms of angiogenesis in the brain. J Neuropathol Exp Neurol 58:313–320
Plate KH, Breier G, Weich HA, et al. (1992) Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 359:845–848
Rak J, Milsom C, May L, et al. (2006) Tissue factor in cancer and angiogenesis: the molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin Thromb Hemost 32:54–70
Rasheed BK, Stenzel TT, McLendon RE, et al. (1997) PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res 57:4187–4190
Raza SM, Lang FF, Aggarwal BB, et al. (2002) Necrosis and glioblastoma: a friend or a foe? A review and a hypothesis. Neurosurgery 51:2–12; discussion 12–13
Rehemtulla A, Ruf W, Edgington TS (1991) The integrity of the cysteine 186-cysteine 209 bond of the second disulfide loop of tissue factor is required for binding of factor VII. J Biol Chem 266:10294–10299
Rickles FR, Falanga A (2001) Molecular basis for the relationship between thrombosis and cancer. Thromb Res 102:V215–224
Rickles FR, Patierno S, Fernandez PM (2003) Tissue factor, thrombin, and cancer. Chest 124:58S–68S
Rong Y, Hu F, Huang R, et al. (2006) Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res 66:7067–7074
Rong Y, Post DE, Pieper RO, et al. (2005) PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res 65:1406–1413
Sawaya R, Ramo OJ, Shi ML, et al. (1991) Biological significance of tissue plasminogen activator content in brain tumors. J Neurosurg 74:480–486
Sawaya R, Yamamoto M, Ramo OJ, et al. (1995) Plasminogen activator inhibitor-1 in brain tumors: relation to malignancy and necrosis. Neurosurgery 36:375–380; discussion 380–371
Schiffer D, Cavalla P, Migheli A, et al. (1995) Apoptosis and cell proliferation in human neuroepithelial tumors. Neurosci Lett 195:81–84
Semenza GL (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7:345–350
Senger DR, Galli SJ, Dvorak AM, et al. (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219:983–985
Seto S, Onodera H, Kaido T, et al. (2000) Tissue factor expression in human colorectal carcinoma: correlation with hepatic metastasis and impact on prognosis. Cancer 88:295–301
Shapiro WR, Young DF (1976) Treatment of malignant glioma. A controlled study of chemotherapy and irradiation. Arch Neurol 33:494–450
Shen BQ, Lee DY, Cortopassi KM, et al. (2001) Vascular endothelial growth factor KDR receptor signaling potentiates tumor necrosis factor-induced tissue factor expression in endothelial cells. J Biol Chem 276:5281–5286
Shweiki D, Itin A, Soffer D, et al. (1992) Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359:843–845
Sonoda Y, Ozawa T, Aldape KD, et al. (2001a) Akt pathway activation converts anaplastic astrocytoma to glioblastoma multiforme in a human astrocyte model of glioma. Cancer Res 61:6674–6678
Sonoda Y, Ozawa T, Hirose Y, et al. (2001b) Formation of intracranial tumors by genetically modified human astrocytes defines four pathways critical in the development of human anaplastic astrocytoma. Cancer Res 61:4956–4960
Stratmann A, Risau W, Plate KH (1998) Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol 153:1459–1466
Straume O, Chappuis PO, Salvesen HB, et al. (2002) Prognostic importance of glomeruloid microvascular proliferation indicates an aggressive angiogenic phenotype in human cancers. Cancer Res 62:6808–6811
Stupp R, Mason WP, van den Bent MJ, et al. (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Swanson KR, Bridge C, Murray JD, et al. (2003) Virtual and real brain tumors: using mathematical modeling to quantify glioma growth and invasion. J Neurol Sci 216:1–10
Tachibana O, Lampe J, Kleihues P, et al. (1996) Preferential expression of Fas/APO1 (CD95) and apoptotic cell death in perinecrotic cells of glioblastoma multiforme. Acta Neuropathol (Berl) 92:431–434
Takano S, Yoshii Y, Kondo S, et al. (1996) Concentration of vascular endothelial growth factor in the serum and tumor tissue of brain tumor patients. Cancer Res 56:2185–2190
Tehrani M, Friedman, T, Olson, JJ, Brat, DJ. (2008) Intravascular thrombosis in central nervous system malignancies: a potential role in astrocytoma progression. Brain Pathol 18:164–171
Vajkoczy P, Farhadi M, Gaumann A, et al. (2002) Microtumor growth initiates angiogenic sprouting with simultaneous expression of VEGF, VEGF receptor-2, and angiopoietin-2. J Clin Invest 109:777–785
Versteeg HH, Hoedemaeker I, Diks SH, et al. (2000) Factor VIIa/tissue factor-induced signaling via activation of Src-like kinases, phosphatidylinositol 3-kinase, and Rac. J Biol Chem 275:28750–28756
Versteeg HH, Peppelenbosch MP, Spek CA (2003) Tissue factor signal transduction in angiogenesis. Carcinogenesis 24:1009–1013
Versteeg HH, Ruf W (2006) Emerging insights in tissue factor-dependent signaling events. Semin Thromb Hemost 32:24–32
Versteeg HH, Schaffner F, Kerver M, et al. (2008) Inhibition of tissue factor signaling suppresses tumor growth. Blood 111:190–199
Versteeg HH, Spek CA, Peppelenbosch MP, et al. (2004) Tissue factor and cancer metastasis: the role of intracellular and extracellular signaling pathways. Mol Med 10:6–11
Walsh DC, Kakkar AK (2001) Thromboembolism in brain tumors. Curr Opin Pulm Med 7:326–331
Wang H, Ubl JJ, Stricker R, et al. (2002) Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am J Physiol Cell Physiol 283:C1351–1364
Wang SI, Puc J, Li J, et al. (1997) Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res 57:4183–4186
Yamamoto M, Mohanam S, Sawaya R, et al. (1996) Differential expression of membrane-type matrix metalloproteinase and its correlation with gelatinase A activation in human malignant brain tumors in vivo and in vitro. Cancer Res 56:384–392
Yan SF, Lu J, Zou YS, et al. (1999a) Hypoxia-associated induction of early growth response-1 gene expression. J Biol Chem 274:15030–15040
Yan SF, Mackman N, Kisiel W, et al. (1999b) Hypoxia/Hypoxemia-Induced activation of the procoagulant pathways and the pathogenesis of ischemia-associated thrombosis. Arterioscler Thromb Vasc Biol 19:2029–2035
Yu JL, May L, Klement P, et al. (2004) Oncogenes as regulators of tissue factor expression in cancer: implications for tumor angiogenesis and anti-cancer therapy. Semin Thromb Hemost 30:21–30
Yu JL, May L, Lhotak V, et al. (2005) Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 105:1734–1741
Zagzag D, Amirnovin R, Greco MA, et al. (2000) Vascular apoptosis and involution in gliomas precede neovascularization: a novel concept for glioma growth and angiogenesis. Lab Invest 80:837–849
Zagzag D, Hooper A, Friedlander DR, et al. (1999) In situ expression of angiopoietins in astrocytomas identifies angiopoietin-2 as an early marker of tumor angiogenesis. Exp Neurol 159:391–400
Zagzag D, Zhong H, Scalzitti JM, et al. (2000) Expression of hypoxia-inducible factor 1alpha in brain tumors: association with angiogenesis, invasion, and progression. Cancer 88:2606–2618
Zhang L, Yang N, Park JW, et al. (2003) Tumor-derived vascular endothelial growth factor up-regulates angiopoietin-2 in host endothelium and destabilizes host vasculature, supporting angiogenesis in ovarian cancer. Cancer Res 63:3403–3412
Zhu XP, Li KL, Kamaly-Asl ID, et al. (2000) Quantification of endothelial permeability, leakage space, and blood volume in brain tumors using combined T1 and T2* contrast-enhanced dynamic MR imaging. J Magn Reson Imaging 11:575–585
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Humana Press, a part of Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Rong, Y., Brat, D.J. (2009). Vaso-occlusive Mechanisms that Intiate Hypoxia and Necrosis in Glioblastoma: The Role of Thrombosis and Tissue Factor. In: Meir, E. (eds) CNS Cancer. Cancer Drug Discovery and Development. Humana Press. https://doi.org/10.1007/978-1-60327-553-8_22
Download citation
DOI: https://doi.org/10.1007/978-1-60327-553-8_22
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-60327-552-1
Online ISBN: 978-1-60327-553-8
eBook Packages: MedicineMedicine (R0)