
Abstract
Hereditary tubular transport disorders that lead to abnormal aminoaciduria and glycosuria are uncommon but are of major biologic importance. Some of these diseases can be associated with significant morbidity. In the past two decades, remarkable progress has been made in our understanding of the molecular pathogenesis of hereditary aminoaciduria and glycosuria. Molecular genetics and molecular biology studies have led to the identification of numerous mutations resulting in aminoaciduria and glycosuria, have provided important insight into the defective molecular mechanisms underlying these disorders, and have greatly increased our understanding of the physiology of renal tubular reclamation of amino acids and glucose.
This chapter summarizes the general characteristics of proximal tubular transport of amino acids and glucose, discusses the specificity and classification of amino acid and glucose transport systems, reviews the molecular pathophysiology and genetic aspects of hereditary aminoaciduria and glycosuria, describes the clinical features of these tubulopathies, and summarizes their therapy. Special emphasis is given to cystinuria, lysinuric protein intolerance, Hartnup disease, iminoglycinuria, dicarboxylic aminoaciduria, familial renal glycosuria, and Fanconi-Bickel syndrome.
Further molecular studies of inherited proximal tubular transport disorders resulting in aminoaciduria and glycosuria may shed more light on the molecular pathophysiology of these diseases and may significantly improve our understanding of the mechanisms underlying renal handling of amino acids and glucose in health and disease. The identification of the molecular defects in these inherited tubulopathies may provide a basis for future design of targeted therapeutic interventions for molecular therapy of these complex disorders.
Similar content being viewed by others
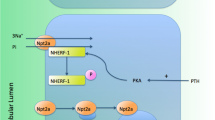
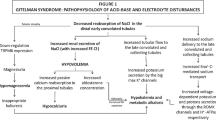
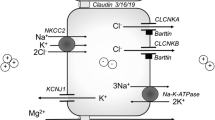
References
Zelikovic I, Chesney RW. Sodium-coupled amino acid transport in renal tubule. Kidney Int. 1989;36:351–9.
Bröer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 2008;88:249–80.
Camargo SMR, Makrides V, Kleta R, et al. Kidney transport of amino acids and oligopeptides, and aminoacidurias. In: Alpern RJ, Moe OW, Caplan M, editors. Seldin and Giebisch’s the Kidney: Physiology and pathophysiology. 5th ed: Elsevier Inc; 2013. p. 2405–23.
Roigaard –Petersen H, Jacobsen C, Iqbal Sheikh M. H+ −L-proline cotransport by vesicles from pars convoluta of rabbit proximal tubule. Am. J. Physiol (Renal Fluid Electrolyte Physiol) 1987; 253: F15–F20.
Rajendran VM, Barry JA, Kleinman JG, et al. Proton gradient –dependent transport of glycine in rabbit renal brush –border membrane vesicles. J Biol Chem. 1987;262:14974–7.
Verrey F, Ristic Z, Romeo E, et al. Novel renal amino acid transporters. Annu Rev Physiol. 2005;67:557–72.
Schafer JA, Barfuss DW. Membrane mechanisms for transepithelial amino acid absorption and secretion. Am J Phys. 1980;238:F335–46.
Zelikovic I, Chesney RW. Development of renal amino acid transport systems. Semin Nephrol. 1989;9:49–55.
Camargo SMR, Bockenhauer D, Kleta R. Aminoacidurias: clinical and molecular aspects. Kidney Int. 2008;73:918–25.
Moe OW, Wright SH, Placin M. Renal handling of organic solutes. In: Taal MW, Chertow GM, Marsden PA, editors. Brenner and Rector’s the kidney. 9th ed: Elsevier Inc; 2012. p. 252–92.
Barfuss DW, Schafer JA. Active amino acid absorption by proximal convoluted and proximal straight tubules. Am J Phys. 1979;236:F149–62.
Zelikovic I, Budreau A. Cl− and membrane potential dependence of amino acid transport across the rat renal brush border membrane. Mol Genet Metab. 1999;67:236–47.
Zelikovic I, Stejskal-Lorenz E, Lohstroh P, et al. Anion dependence of taurine transport by rat renal brush border membrane vesicles. Am J Phys. 1989;256:F646–55.
Scalera V, Corcellia A, Frassanito A, et al. Chloride dependence of the sodium-dependent glycine transport in pig kidney cortex brush-border membrane vesicles. Biochim Biophys Acta. 1987;903:1–10.
Chesney RW, Zelikovic I, Budreau A, et al. Chloride and membrane potential dependence of sodium ion-proline symport. J Am Soc Nephrol. 1991;2:885–93.
Scriver CR, Tenenhouse HS. Mendelian phenotypes as “probes” of renal transport systems for amino acids and phosphate. In: Windhager EE, editor. Handbook of physiology: renal physiology. New York: Oxford University Press; 1992. p. 1977–2016.
Christensen HN. Role of amino acid transport and counter transport in nutrition and metabolism. Physiol Rev. 1990;70:43–77.
Palacin M, Estevez R, Bertran J, et al. Molecular biology of mammalian plasma membrane amino acid transporters. Physiol Rev. 1998;78:969–1054.
Bröer S. Adaptation of plasma membrane amino acid transport mechanisms to physiological demands. Pflügers Arch. 2002;444:457–66.
Rabito CA. Sodium cotransport processes in renal epithelial cell lines. Miner Electrol Metab. 1986;12:32–41.
Hediger MA, Coady MJ, Ikeda TS, et al. Expression cloning and cDNA sequencing of the Na+/glucose cotransporter. Nature. 1987;330:379–81.
Hediger MA, Clemencon B, Burrier RE, et al. The ABCs of membrane transporters in health and disease (SLC series). Mol Aspects Med. 2013;34:95–107.
Makrides V, Camargo SMR, Verrey F. Transport of amino acids in the kidney. Compr Physiol. 2014;4:367–403.
Bröer S. Apical transporters for neutral amino acids: physiology and pathophysiology. Physiology (Bethesda). 2008;23:95–103.
Bröer S, Palacin M. The role of amino acid transporters in inherited and acquired diseases. Biochem J. 2011;436:193–211.
Chillarön J, Font-Llitjös M, Fort J, et al. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010;6:424–34.
Claes DJ, Jackson E. Cystinuria: mechanism and management. Ped Nephrol. 2012;27:2031–8.
Milliner DS. Cystinuria. Endocrinol Metab Clin N Am. 1990;19:889–907.
Foreman JW, Hwang SM, Segal L. Transport interactions of cystine and dibasic amino acids in isolated rat renal tubules. Metabolism. 1980;29:53–61.
Weinberger A, Sperling O, Rabinovitz M, et al. High frequency of cystinuria among Jews of Libyan origin. Hum Hered. 1974;24:568–72.
Rosenberg LE, Downing S, Durant JL, et al. Cystinuria: biochemical evidence of three genetically distinct diseases. J Clin Invest. 1966;45:365–71.
Goodyer PR, Clow C, Reade T, et al. Prospective analysis and classification of patients with cystinuria identified in a newborn screening program. J Pediatr. 1993;122:568–72.
Dello Strologo L, Pras E, Pontesilli C, et al. Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classification. J Am Soc Nephrol. 2002;13:2547–53.
Font-Llitjós M, Jiménez-Vidal M, Bisceglia L, et al. New insights into cystinuria: 40 new mutations, genotype–phenotype correlation, and digenic inheritance causing partial phenotype. J Med Genet. 2005;42:58–68.
Palacín M, Nunes V, Font-Llitjós M, et al. The genetics of heteromeric amino acid transporters. Physiology (Bethesda). 2005;20:112–24.
Palacin M, Kanai Y. The ancillary proteins of HATs: SLC3 family of amino acid transporters. Pflugers Arch. 2004;447:490–4.
Fotiadis D, Kanai Y, Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Asp Med. 2013;34:139–58.
Chillaron J, Roca R, Valencia A, et al. Heteromeric amino acid transporters: biochemistry, genetics, and physiology. Am J Phys. 2001;281:F995–F1018.
Pras E, Arber N, Aksentijevich I, et al. Localization of a gene causing cystinuria to chromosome 2p. Nat Genet. 1994;6:415–9.
Calonge MJ, Gasparini P, Chillaron J, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet. 1994;6:420–5.
Tokhmafshan F, Dickinson K, Akpa MM, et al. A no-nonsense approach to hereditary kidney disease. Pediatr Nephrol. 2020;35:2031–42.
Gaildrat P, Lebbah S, Tebani A, et al. Clinical and molecular characterization of cystinuria in a French cohort: relevance of assessing large-scale rearrangements and splicing variants. Mol Genet Genomic Med. 2017;5:373–89.
Palacin M, Bertran J, Zorzano A. Heteromeric amino acid transporters explain inherited aminoacidurias. Curr Opin Nephrol Hypert. 2000;9:547–53.
Chillaron J, Estevez R, Samarzija I, et al. An intracellular trafficking defect in type I cystinuria rBAT mutants M467T and M467K. J Biol Chem. 1997;272:9543–9.
Zelikovic I. Molecular pathophysiology of tubular transport disorders. Pediatr Nephrol. 2001;16:919–35.
Palacin M, Borsani G, Sebastio G. The molecular bases of cystinuria and lysinuric protein intolerance. Curr Opin Genet Dev. 2001;11:328–35.
Wartenfeld R, Golomb E, Katz G, et al. Molecular analysis of cystinuria in Libyan Jews: exclusion of the SLC3A1 gene and mapping of a new locus on 19q. Am J Hum Genet. 1997;60:617–24.
Bisceglia L, Calonge MJ, Totaro A, et al. Localization, by linkage analysis, of the cystinuria type III gene to chromosome 19q13.1. Am J Hum Genet. 1997;60:611–6.
Feliubadalo L, Font M, Purroy J, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. Nat Genet. 1999;23:52–7.
Font M, Feliubadalo L, Estivill X, et al. Functional analysis of mutations in SLC7A9, and genotype-phenotype correlation in non-type I cystinuria. Hum Mol Genet. 2001;10:305–16.
Jaeken J, Martens K, Francois I, et al. Deletion of PREPL, a gene encoding a putative serine oligopeptidase, in patients with hypotonia-cystinuria syndrome. Am J Hum Genet. 2006;78:38–51.
Martens K, Heulens I, Meulemans S, et al. Global distribution of the most prevalent deletions causing hypotonia – cystinuria syndrome. Eur J Hum Genet. 2007;15:1029–33.
Servais A, Thomas K, Dello Strogolo L, et al. Cystinuria: clinical practice recommendation. Kidney Int. 2020;S0085–2538(20):30829–2.
Rhodes HL, Yarram-Smith L, Rice SJ, et al. Clinical and genetic analysis of patients with cystinuria in the United Kingdom. Clin J Am Soc Nephrol. 2015;10:1235–45.
Prot-Bertoye C, Lebbah S, Daudon M, et al. CKD and its risk factors among patients with cystinuria. Clin J Am Soc Nephrol. 2015;10:842–51.
Krizek V, Erben J, Lazne M, et al. Disappearance of cystinuria after kidney transplantation. Br J Urol. 1983;55:575.
Jaeger P, Portmann L, Saunders A, et al. Anticystinuric effects of glutamine and of dietary sodium restriction. N Engl J Med. 1986;315:1120–3.
Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int. 2006;69:1041–7.
Prot-Bertoye C, Lebbah S, Daudon M, et al. Adverse events associated with currently used medical treatments for cystinuria and treatment goals: results from a series of 442 patients in France. BJU Int. 2019;124:849–61.
Coulthard M, Richardson J, Fleetwood A. Captopril is not clinically useful in reducing the cystine load in cystinuria or cystinosis. Pediatr Nephrol. 1991;5:98.
Maiorino RM, Bruce DC, Aposhian HV. Determination and metabolism of dithiol chelating agents. VI. Isolation and identification of the mixed disulfides of meso-2,3-dimercaptosuccinic acid with L-cysteine in human urine. Toxicol Appl Pharmacol. 1989;97:338–49.
Simell O. Lysinuric protein intolerance and other cationic aminoacidurias. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 4933–56.
Sperandeo MP, Andria G, Sebastio G. Lysinuric protein intolerance: update and extended mutation analysis of the SLC7A7 gene. Hum Mutat. 2008;29:14–21.
de Baulny HO, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106:12–7.
Mauhin W, Habarou F, Gobin S, et al. Update on lysinuric protein intolerance, a multi- faceted disease retrospective cohort analysis from birth to adulthood. Orphanet J Rare Dis. 2017;12(1):3.
Valimahamed-Mitha S, Berteloot L, Ducoin H, et al. Lung involvement in children with lysinuric protein intolerance. J Inherit Metab Dis. 2015;38:257–63.
Desjeux JF, Rajantie J, Simell O, et al. Lysine fluxes across the jejunal epithelium in lysinuric protein intolerance. J Clin Invest. 1980;65:1382–7.
Rajantie J, Simell O, Perheentupa J. Lysinuric protein intolerance. Basolateral transport defect in renal tubuli. J Clin Invest. 1981;67:1078–82.
Palacín M, Bertran J, Chillarón J, et al. Lysinuric protein intolerance: mechanisms of pathophysiology. Mol Gen Metab. 2004;81:S27–37.
Sabastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet. 2011;157:54–62.
Simell O. Diamino acid transport into granulocytes and liver slices of patients with lysinuric protein intolerance. Pediatr Res. 1975;9:504–8.
Smith DW, Scriver CR, Tenenhouse HS, et al. Lysinuric protein intolerance mutation is expressed in the plasma membrane of cultured skin fibroblasts. Proc Natl Acad Sci U S A. 1987;84:7711–5.
Tanner LM, Näntö-Salonen K, Niinikoski H, et al. Nephropathy advancing to end-stage renal disease: a novel complication of lysinuric protein intolerance. J Pediatr. 2007;150:631–4.
Bröer S. Lysinuric protein intolerance: one gene, many problems. Am J Physiol Cell Physiol. 2007;293:C540–1.
Lauteala T, Sistonen P, Savontaus ML, et al. Lysinuric protein intolerance (LPI) gene maps to the long arm of chromosome 14. Am J Hum Genet. 1997;60:1479–86.
Lauteala T, Mykkanen J, Sperandeo MP, et al. Genetic homogeneity of lysinuric protein intolerance. Eur J Hum Genet. 1998;6:612–5.
Torrents D, Estevez R, Pineda M, et al. Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J Biol Chem. 1998;273:32437–45.
Estevez R, Camps M, Rojas AM, et al. The amino acid transport system y+L/4F2hc is a heteromultimeric complex. FASEB J. 1998;12:1319–29.
Borsani G, Bassi MT, Sperandeo MP, et al. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat Genet. 1999;21:297–301.
Torrents D, Mykkanen J, Pineda M, et al. Identification of SLC7A7, encoding y+LAT−1, as the lysinuric protein intolerance gene. Nat Genet. 1999;21:293–6.
Font- Llitjö M, Rodriguez-Santiago B, Espino M, et al. Novel SLC7A7 large rearrangements in lysinuric protein intolerance patients involving the same AluY repeat. Eur J Hum Genet. 2009;17:71–9.
Mykkanen J, Torrents D, Pineda M, et al. Functional analysis of novel mutations in y+LAT-1 amino acid transporter gene causing lysinuric protein intolerance (LPI). Hum Mol Genet. 2000;9:431–8.
Feral CC, Nishiya N, Fenczik CA, et al. CD98hc (SLC3A2) mediates integrin signaling. Proc Natl Acad Sci U S A. 2005;102:355–60.
Tsumura H, Suzuki N, Saito H, et al. The targeted disruption of the CD98 gene results in embryonic lethality. Biochem Biophys Res Commun. 2003;308:847–51.
Whelan DT, Scriver CR. Hyperdibasic aminoaciduria: an inherited disorder of amino acid transport. Pediatr Res. 1968;2:525–34.
Kihara H, Valente M, Porter MT, et al. Hyperdibasic aminoaciduria in a mentally retarded homozygote with a peculiar response to phenothiazines. Pediatrics. 1978;51:223–9.
Omura K, Yamanaka N, Higami S, et al. Lysine malabsorption syndrome: a new type of transport defect. Pediatrics. 1976;57:102–5.
Dirckx JH. Julius Caesar and the Julian emperors: a family cluster with Hartnup disease? Am J Dermatopathol. 1986;8:351–7.
Levy HL. Hartnup disorder. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 4957–69.
Bröer S. The role of the neutral amino acid transporter B0AT1 (SLC6A19) in Hartnup disorder and protein nutrition. IUBMB Life. 2009;61:591–9.
Milne MD, Crawford MA, Girao CB, et al. The metabolic disorder in Hartnup disease. Q J Med. 1960;29:407–21.
Asatoor AM, Cheng B, Edwards KDG, et al. Intestinal absorption of two dipeptides in Hartnup disease. Gut. 1970;11:380–7.
Bröer A, Klingel K, Kowalczuk S, et al. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J Biol Chem. 2004;279:24467–76.
Kleta R, Romeo E, Ristic Z, et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet. 2004;36:999–1002.
Seow HF, Bröer S, Bröer A, et al. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet. 2004;36:1003–7.
Bröer A, Cavanaugh JA, Rasko JE, et al. The molecular basis of neutral aminoacidurias. Pflugers Arch. 2006;451:511–7.
Azmanov DN, Kowalczuk S, Rodgers H, et al. Further evidence for allelic heterogeneity in Hartnup disorder. Hum Mutat. 2008;29:1217–21.
Scriver CR, Mahon B, Levy H, et al. The Hartnup phenotype: Mendelian transport disorder, multifactorial disease. Am J Hum Genet. 1987;40:401–12.
Camargo SMR, Singer D, Makrides V, et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with Hartnup mutations. Gastroenterology. 2009;136:872–82.
Kowalczuk S, Bröer A, Tietze N, et al. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22:2880–7.
Singer D, Camargo SMR. Collectrin and ACE2 in renal and intestinal amino acid transport. Channels. 2011;5:410–23.
Jonas AJ, Butler IJ. Circumvention of defective neutral amino acid transport in Hartnup disease using tryptophan ethyl ester. J Clin Invest. 1989;84:200–4.
Smith AJ, Strang LB. An inborn error of metabolism with the urinary excretion of α-hydroxybutyric acid and phenylpyruvic acid. Arch Dis Child. 1958;33:109–13.
Hooft C, Timmermans J, Snoeck J, et al. Methionine malabsorption syndrome. Ann Pediatr. 1965;205:73–84.
Sabater J, Ferre C, Puliol M, et al. Histidinuria: a renal and intestinal histidine transport deficiency found in two mentally retarded children. Clin Genet. 1976;9:117–24.
Holmgren G, Hambraeus L, De Chateau P. Histidinemia and "normohistidinemic histidinuria": report of three cases and the effect of different protein intakes on urinary excretion of histidine. Acta Paediatr Scand. 1974;63:220–4.
Kamoun PP, Parvy P, Chathelineau L, et al. Renal histidinuria. J Inherited Metab Dis. 1981;4:217–9.
Chesney RW. Iminoglycinuria. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 4971–81.
Turner B, Brown DA. Amino acid excretion in infancy and early childhood. A survey of 200,000 infants. Med J Aust. 1972;1:62–5.
Lasley L, Scriver CR. Ontogeny of amino acid reabsorption in human kidney. Evidence for the homozygous infant with familial renal iminoglycinuria for multiple proline and glycine systems. Pediatr Res. 1979;13:65–70.
Scriver CR. Renal tubular transport of proline, hydroxyproline and glycine. III. Genetic basis for more than one mode of transport in human kidney. J Clin Invest. 1968;47:823–35.
Greene ML, Lietman PS, Rosenberg LE, et al. Familial hyperglycinuria: new defect in renal tubular transport of glycine and imino acids. Am J Med. 1973;54:265–71.
Bröer S, Bailey CG, Kowalczuk S, et al. Iminoglycinuria and hyperglycinuria are discrete human phenotypes resulting from complex mutations in proline and glycine transporters. J Clin Invest. 2008;118:3881–92.
deVries A, Kochwa S, Lazebnik J, et al. Glycinuria, a hereditary disorder associated with nephrolithiasis. Am J Med. 1957;23:408–15.
Kaser H, Cottier P, Antener I. Glucoglycinuria, a new familial syndrome. J Pediatr. 1962;61:386–94.
Tiejema HL, Van Gelderen HH, Giesberts MAH, et al. Dicarboxylic aminoaciduria: an inborn error of glutamate and aspartate transport with metabolic implications in combination with hyperprolinemia. Metabolism. 1974;23:115–23.
Melancon SB, Dallaire L, Lemieux B, et al. Dicarboxylic aminoaciduria: an inborn error of amino acid conservation. J Pediatr. 1977;91:422–7.
Auray-Blais C, Cyr D, Drouin R. Quebec neonatal mass urinary screening programme: from micromolecules to macromolecules. J Inherit Metab Dis. 2007;30:515–21.
Bailey CG, Ryan RM, Thoeng AD, et al. Loss-of-function mutations in the glutamate transporter SLCA1 cause human dicarboxylic aminoaciduria. J Clin Invest. 2011;121:446–53.
Melancon SB, Grenier B, Dallaire L, et al. Dicarboxylic amino acid uptake in normal, Friedreich’s ataxia, and dicarboxylic aminoaciduria fibroblasts. J Can Sci Neurol. 1979;6:262–73.
Rozen R, Scriver CR, Mohyuddin F. Hypertaurinuria in the C57BL/6J mouse: altered transport at the renal basolateral membrane. Am J Phys. 1983;244:F150–5.
Mandla S, Scriver CR, Tenenhouse HS. Decreased transport in renal basolateral membrane vesicles from hypertaurinuric mice. Am J Phys. 1988;255:F88–95.
Hummel CS, Wright EM. Glucose reabsorption in the kidney. In: Alpern RJ, Moe OW, Caplan M, editors. Seldin and Giebisch’s the kidney: physiology and pathophysiology. 5th ed: Elsevier Inc; 2013. p. 2393–3404.
Sacktor B. Sodium-coupled hexose transport. Kidney Int. 1989;36:342–50.
Elsas LJ, Longo N. Glucose transporters. Annu Rev Med. 1992;43:377–93.
Elsas LJ, Rosenberg LE. Familial renal glycosuria: a genetic reappraisal of hexose transport by kidney and intestine. J Clin Invest. 1969;48:1845–54.
Brodehl J, Franken A, Gellissen K. Maximal tubular reabsorption of glucose in infants and children. Acta Paediatr Scand. 1972;61:413–20.
Barfuss DW, Schafer JA. Differences in active and passive glucose transport along the proximal nephron. Am J Phys. 1981;241:F322–32.
Turner RJ, Moran A. Stoichiometric studies of the renal cortical brush border membrane D-glucose transporter. J Membr Biol. 1982;67:73–80.
Turner RJ, Moran A. Further studies of proximal tubular brush-border membrane. D-glucose transport heterogeneity. J Membr Biol. 1982;70:37–45.
Wright EM, Turk E. The sodium/glucose cotransport family SLC5. Pflugers Arch. 2004;447:510–8.
Wright EM, Loo DD, Hirayama BA, et al. Surprising versatility of Na+-glucose cotransporters: SLC5. Physiology (Bethesda). 2004;19:370–6.
Wright EM, Loo DDF, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011;91:733–94.
Hediger MA, Turk E, Wright EM. Homology of the human intestinal Na+/glucose and Escherichia coli Na+/proline cotransporters. Proc Natl Acad Sci U S A. 1989;86:5748–52.
Hediger MA, Budard ML, Emanual BS, et al. Assignment of the human intestinal Na+/glucose gene (SGLT 1) to the q11.2lqter regions of chromosome 22. Genomics. 1989;4:297–300.
Pajor AM, Hirayama BA, Wright EM. Molecular evidence for two renal Na+/glucose cotransporters. Biochim Biophys Acta. 1992;1106:216–20.
Lee WS, Kanai Y, Wells RG, et al. The high affinity Na+/glucose cotransporter. Re-evaluation of function and distribution of expression. J Biol Chem. 1994;269:12032–9.
Kanai Y, Lee WS, You G, et al. The human kidney low affinity Na+/glucose cotransporter SGLT2, delineation of the major renal reabsorptive mechanism for D-glucose. J Clin Invest. 1994;93:397–404.
Wells RG, Mohandas TK, Hediger MA. Localization of the Na+/glucose cotransporter gene SGLT2 to human chromosome 16 close to the centromere. Genomics. 1993;17:787–9.
Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Asp Med. 2013;34:121–38.
Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298:E141–5.
Thorens B, Cheng ZQ, Brown D, et al. Liver glucose transporter: a basolateral protein in hepatocytes and intestine and kidney epithelial cells. Am J Phys. 1990;259:C279–85.
Thorens B, Lodish HF, Brown D. Differential localization of two glucose transporter isoforms in kidney nephron. Am J Phys. 1990;259:C286–95.
Wright E, Martin MG, Turk E. Familial glucose-galactose malabsorption and hereditary glycosuria. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 4891–908.
Elsas LJ, Busse D, Rosenberg LE. Autosomal recessive inheritance of renal glycosuria. Metabolism. 1971;20:968–75.
Woolf LI, Goodwin BL, Phelps CE. Tm-limited renal tubular reabsorption and the genetics of renal glucosuria. J Theor Biol. 1966;11:10–21.
Oemar BS, Byrd DJ, Brodehl J. Complete absence of tubular glucose reabsorption: a new type of renal glucosuria (type 0). Clin Nephrol. 1987;27:156–60.
van den Heuvel LP, Assink K, Willemsen M, et al. Autosomal recessive renal glucosuria attributable to a mutation in the sodium glucose cotransporter (SGLT2). Hum Genet. 2002;111:544–7.
Calado J, Sznajer Y, Metzger D, et al. Twenty-one additional cases of familial renal glucosuria: absence of genetic heterogeneity, high prevalence of private mutations and further evidence of volume depletion. Nephrol Dial Transplant. 2008;23:3874–9.
Cannizzaro M, Jarosova J, De Paepe B. Relevance of solute carrier family 5 transporter defects to inherited and acquired human disease. J Appl Genet. 2019;60:305–17.
Magen D, Sprecher E, Zelikovic I, et al. A novel missense mutation in SLC5A2 encoding SGLT2 underlies autosomal-recessive renal glucosuria and aminoaciduria. Kidney Int. 2005;67:34–41.
Ellard S. Hepatocyte nuclear factor 1 alpha (HNF-1 α) mutations in maturity-onset diabetes of the young. Hum Mutat. 2000;16:377–85.
Pontoglio M, Barra J, Hadchouel M, et al. Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell. 1996;84:575–85.
Fukui K, Yang Q, Cao Y, et al. The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab. 2005;2:373–84.
Elsas LJ, Hillman RE, Patterson JH, et al. Renal and intestinal hexose transport in familial glucose-galactose malabsorption. J Clin Invest. 1970;49:576–85.
Turk E, Zabel B, Mundlos S, et al. Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature. 1991;350:354–6.
Santer R, Calado J. Familial renal glucosuria and SGLT2: from a Mendelian trait to a therapeutic target. Clin J Am Soc Nephrol. 2010;5:133–41.
Zelikovic I. Hereditary tubulopathies. In: Oh W, Baum M, editors. Nephrology and fluid/electrolyte physiology: neonatology questions and controversies. 3rd ed. Philadelphia: Elsevier; 2019. p. 315–44.
Vallon V, Platt K, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. Am J Soc Nephrol. 2011;22:104–12.
Ly J, Onay T, Sison K, et al. The sweet pee model for SGLT2 mutation. Am J Soc Nephrol. 2011;22:113–23.
Manz F, Bickel H, Brodehl J, et al. Fanconi-Bickel syndrome. Pediatr Nephrol. 1987;1:509–18.
Santer R, Schneppenheim R, Dombrowski A, et al. Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat Genet. 1997;17:324–6.
Santer R, Steinmann B, Schaub J. Fanconi-Bickel syndrome- a congenital defect of facilitative glucose transport. Curr Mol Med. 2002;2:213–27.
Sharari S, Abou-Alloul M, Hussain K, et al. Fanconi-Bickel syndrome: a review of the mechanisms that lead to the dysglycaemia. Int J Mol Sci. 2020;21:6286–307.
Santer R, Groth S, Kinner M, et al. The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi-Bickel syndrome. Hum Genet. 2002;110:21–9.
Mannstadt M, Magen D, Segawa H, et al. Fanconi-Bickel syndrome and autosomal recessive proximal tubulopathy with hypercalciuria (ARPTH) are allelic variants caused by GLUT2 mutations. J Clin Endocrinol Metab. 2012;97:E1978–86.
Enogieru OJ, Ung PMU, Yee SW, et al. Functional and structural analysis of rare SLC2A2 variants associated with Fanconi-Bickel syndrome and metabolic traits. Hum Mutat. 2019;40:983–95.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer-Verlag GmbH Germany, part of Springer Nature
About this entry

Cite this entry
Zelikovic, I., Servais, A. (2021). Aminoaciduria and Glycosuria in Children. In: Emma, F., Goldstein, S., Bagga, A., Bates, C.M., Shroff, R. (eds) Pediatric Nephrology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-27843-3_33-2
Download citation
DOI: https://doi.org/10.1007/978-3-642-27843-3_33-2
Received:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-27843-3
Online ISBN: 978-3-642-27843-3
eBook Packages: Springer Reference MedicineReference Module Medicine
Publish with us
Chapter history
-
Latest
Aminoaciduria and Glycosuria in Children- Published:
- 18 March 2022
DOI: https://doi.org/10.1007/978-3-642-27843-3_33-2
-
Original
Aminoaciduria and Glycosuria in Children- Published:
- 10 February 2015
DOI: https://doi.org/10.1007/978-3-642-27843-3_33-1