
Abstract
Many fundamentally important biological processes rely on the mechanical responses of membrane proteins and their assemblies in the membrane environment, which are multiscale in nature and represent a significant challenge in modeling and simulation. For example, in mechanotransduction, mechanical stimuli can be introduced through macroscopic-scale contacts, which are transduced to mesoscopic-scale (micron) distances and can eventually lead to microscopic-scale (nanometer) conformational changes in membrane-bound protein or protein complexes. This is a fascinating process that spans a large range of length scales and time scales. The involvement of membrane environment and critical issues such as cooperativity calls for the need for an efficient multi-scale computational approach. The goal of the present research is to develop a hierarchical approach to study the mechanical behaviors of membrane proteins with a special emphasis on the gating mechanisms of mechanosensitive (MS) channels. This requires the formulation of modeling and numerical methods that can effectively bridge the disparate length and time scales. A top-down approach is proposed to achieve this by effectively treating biomolecules and their assemblies as integrated structures, in which the most important components of the biomolecule (e.g., MS channel) are modeled as continuum objects, yet their mechanical/physical properties, as well as their interactions, are derived from atomistic simulations. Molecular dynamics (MD) simulations at the nanoscale are used to obtain information on the physical properties and interactions among protein, lipid membrane, and solvent molecules, as well as relevant energetic and temporal characteristics. Effective continuum models are developed to incorporate these atomistic features, and the conformational response of macromolecule(s) to external mechanical perturbations is simulated using finite element (FEM) analyses with in situ mechanochemical coupling. Results from the continuum mechanics analysis provide further insights into the gating transition of MS channels at structural and physical levels, and specific predictions are proposed for further experimental investigations. It is anticipated that the hierarchical framework is uniquely suited for the analysis of many biomolecules and their assemblies under external mechanical stimuli.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
- Mechanotransduction
- Multi-scale simulation
- Mechanosensitive channels
- Gating mechanism
- Continuum mechanics
- Continuum solvation
Introduction
Occurring over large time and length scales, various biological signal transduction processes rely on the mechanical response of biomolecules and their assemblies to external stimuli. Muscle contraction or stretch, as a prominent example, involves the cooperative mechanical response of a large number of myosin molecules (Geeves and Holmes 1999, 2005), and structural changes from molecular scale to organ scales of muscle contribute greatly to its various and remarkable adaptations under different mechanical stimuli (Wisdom et al. 2014). Another example is mechanosensation (Hamill and Martinac 2001), during which the mechanosensitive (MS) channels play important roles in living cells of diverse phylogenetic origin (Martinac 2004) and have been identified in more than 30 cell types (Sackin 1995). By converting mechanical forces exerted on the cell membrane into biochemical or electrical signals, MS channels are involved in a wide range of cellular processes including cell growth and differentiation (Wang and Thampatty 2006) and blood pressure and cell volume regulation (Hamill and Martinac 2001; Martinac and Kloda 2012; Sun et al. 2009) and are essential to sensations such as touching, balance, and hearing (Hamill and Martinac 2001; Ingber 2006; Martinac 2004). A direct link between the lipid membrane and the structure/function of some MS channels has been revealed (Phillips et al. 2009). And in eukaryotic cells, the cytoskeleton was shown to play a similar role in the activation of MS channels (Hayakawa et al. 2008). With an increasing number of MS channels being identified, their atomic structures, gating characteristics, and functional mechanisms have been studied extensively in the past decades. Among the families of MS channels, a much-studied system is the mechanosensitive channel of small/large conductance (MscS/MscL) in Escherichia coli (E. coli), which serves as a paradigm for understanding the gating behaviors of the MS family of ion channels. Functioning as the “safety valve” of bacteria that regulates turgor pressure, MscS/MscL is sensitive to tension in the membrane, and the opening of MscS/MscL allows exchange of ions (nonselective between anions and cations) and small molecules (including water) between the cytoplasm and the environment (Berrier et al. 1996; Blount et al. 1997; Martinac et al. 2014; Saimi et al. 1992).
A Brief Review of MscS Studies
The first crystal structure for E. coli-MscS was solved at 3.95 Å resolution (Bass et al. 2002) and subsequently refined to a higher resolution of 3.7 Å (Steinbacher et al. 2007). The structure has a pore of less than 5 Å in diameter, and because of its hydrophobic constriction, the pore is thus considered as nonconducting or closed (Steinbacher et al. 2007; Vora et al. 2006). The open form of the A106V mutant of MscS was subsequently crystallized at 3.45 Å resolution (Wang et al. 2008). The crystal structures of E. coli-MscS have been constantly challenged, mainly due to the large voids between the TM3 helix (the third transmembrane helix) and the closely packed TM1 and TM2 helices (also referred as TM pockets). Using extrapolated motion dynamics (EMD) (Akitake et al. 2007) and continuous wave electron paramagnetic resonance (cwEPR) (Vasquez et al. 2008a, b), alternative MscS structures of both closed and open forms were independently generated, in which the apparent voids in the crystal structures were absent. The three approaches lead to three sets of mutually incompatible models of the closed and open structures and thus models for the gating transition. A pulsed electron-electron double resonance (PELDOR) approach reevaluated these competing structural models both in detergent (Pliotas et al. 2012) and in bilayer mimics (Ward et al. 2014); the results supported the arrangement of helices seen in the crystal structures. Another study reported the crystal structure of β-dodecylmaltoside-solubilized wild-type E. coli-MscS at 4.4 Å resolution and further supported that the A106V structure resembles the open state (Lai et al. 2013). Finally, a higher-resolution structure of the E. coli-MscS identified alkyl chains inside the pockets/voids formed by the transmembrane helices (Pliotas et al. 2015), strongly support that the voids in E. coli-MscS crystal structures are realistic (Pliotas and Naismith 2016). Based on the above evidence, the present study starts with the assumption that the crystal structure at 3.7 Å resolution (Steinbacher et al. 2007) and the A106V mutant at 3.45 Å resolution (Wang et al. 2008) represent the closed (resting) and open states of MscS, respectively, but noting that the specific functional states of these structures of MscS may be still in debate. Nevertheless, despite the available closed and open structures, little is known about the partially expanded intermediate structures during MscS gating transition.
Numerous studies have explored residues and interactions that are important to the gating characteristics of MscS. Some of the established cases include the Asp62-Arg131/Arg128 salt bridges (Nomura et al. 2008) and the Phe68-Leu111/Leu115 (Belyy et al. 2010) apolar interaction, which affect channel gating and inactivation; Leu105 and Leu109 form a hydrophobic lock at the channel pore (Anishkin and Sukharev 2004; Vora et al. 2006); the interaction between the lower part of TM3 and the cytoplasmic β domain and Gly113 is crucial to inactivation (Edwards et al. 2008; Koprowski et al. 2011; Petrov et al. 2013); and a number of residues were shown to influence force transmission at the protein-lipid interface (Malcolm et al. 2011; Nomura et al. 2006). The physical origins for the importance of these interactions are not always well understood. Another much studied mechanistic issue concerns inactivation of MscS: under prolonged exposure to subthreshold membrane tension, the channel desensitizes into an inactivated and nonconducting state from which it must relax back to the closed state in lower membrane tension before reactivation can be induced (Akitake et al. 2005; Edwards et al. 2008; Koprowski and Kubalski 1998; Levina et al. 1999). Up to now, while multiple residues and interactions are known to be important to inactivation (Vasquez 2013), the structural mechanism underlying inactivation remains elusive.
Furthermore, it has been reported that the open probability of MscS can be significantly increased by membrane depolarization (Cui et al. 1995; Martinac et al. 1987). A later study, however, showed that the activation of E. coli-MscS by membrane tension is essentially independent of the transmembrane voltage (Akitake et al. 2005), though depolarizing membrane voltage strongly promotes inactivation (Akitake et al. 2005). In addition, a recent study reported that the arginine residues at positions of 46, 54, and 74 in TM1 and TM2 helices are not responsible for the voltage dependence of inactivation (Nomura et al. 2016). The structural response and inactivation mechanism under membrane potential thus remain to be better clarified.
Atomistic simulations have made valuable contributions to the understanding of ion channels, including MscS, in recent years (Akitake et al. 2007; Anishkin and Sukharev 2004; Deplazes et al. 2012; Masetti et al. 2016; Pliotas et al. 2015; Sotomayor and Schulten 2004; Sotomayor et al. 2006). Nevertheless, such simulations remain computationally intensive, making it difficult to study gating transitions that occur on the millisecond time scale and explore contributions of specific structural motifs and interactions.
A Brief Review of MscL Studies
A large body of experimental, theoretical, and simulation work has focused on elucidating the molecular mechanism of MscL gating (Booth et al. 2007; Haswell et al. 2011). Experiments directly probing the gating transition of MscL were primarily patch-clamp measurements, which simultaneously monitor the membrane tension and ionic currents through the channel (thus opening probability) (Sukharev et al. 1997, 1999). An important clue from these studies on MscL reconstituted into purified lipid bilayers is that the mechanical property of the membrane plays a principal role in determining the gating behaviors of MscL. A model with five subconducting states was established (Sukharev et al. 1999), in which the tension-dependent conformational transition was primarily attributed to the pore area variation that occurs between the closed state and a low subconductance state. In addition, other experimental studies have been used to probe MscL’s conformational transition, and these include electron paramagnetic resonance spectroscopy (EPR) with site-directed spin labeling (SDSL) (Perozo et al. 2002a, b) for monitoring the structural rearrangements, cysteine scanning for identifying residue contacts in the transmembrane helices (Levin and Blount 2004), and numerous mutation studies for probing the importance of residues in different structural motifs (Anishkin et al. 2005; Blount et al. 1997; Levin and Blount 2004; Tsai et al. 2005). Besides, a single-molecule fluorescence resonance energy transfer (FRET) method (Wang et al. 2014) or the combination of data from FRET spectroscopy and simulations (Corry et al. 2010) has enabled a more detailed description of the open form of MscL in the natural lipid environment. Based on geometrical constraints provided by various measurements and the crystal structure of MscL in Mycobacterium tuberculosis (Tb) (Chang et al. 1998), structural models for the closed-open transition of E. coli-MscL were constructed (Sukharev et al. 2001a, b); revised models were proposed subsequently where the conformational changes of the S3 helices are much smaller in scale (Sukharev and Anishkin 2004; Sukharev and Corey 2004). Although highly valuable, these structural models need to be evaluated for validity in a systematic and physical manner (Chen et al. 2008; Tang et al. 2006, 2008).
Meanwhile, analytical models for the gating transition in MscL have been developed by several groups. Markin and Sachs presented a general thermodynamic model for mechanotransduction that relates the probability of channel opening to membrane properties such as thickness, curvature, and stiffness (Markin and Sachs 2004). An analytical continuum model was developed by Wiggins and Phillips to characterize the free energy of the protein-bilayer system (Wiggins and Phillips 2004); the model highlighted that the competition of hydrophobic mismatch could be a physical gating mechanism. As an alternative to dilatational gating, a gating-by-tilt model was proposed (Turner and Sens 2004) in which the gate opening is due to the swinging of the lipids near the channel with respect to a pivot. Although these analytical models are valuable for highlighting the potential contribution of specific physical factors (e.g., hydrophobic mismatch), they lack sufficient structural details to make specific connection with experimental studies.
Numerical simulation is a powerful approach for exploring the fundamental principles of mechanotransduction. To properly assign structural features to important intermediate states along the closed-open transition, it is important to simulate the structural response of the channel to mechanical perturbation consistent with the experimental protocol. Due to the large length scale and time scale involved, however, this is usually beyond the capability of atomistic simulations despite the rapid progresses being made (Deplazes et al. 2012; Dror et al. 2012; Gullingsrud and Schulten 2003; Karplus and Kuriyan 2005; Klepeis et al. 2009; Monticelli et al. 2008; Sawada et al. 2012; Snow et al. 2005; Yefimov et al. 2008). Hence, developing coarse-grained models to access longer time scales has become an important topic in the simulation community (Ingolfsson et al. 2014; Marrink et al. 2007; Marrink and Tieleman 2013; Monticelli et al. 2008; Praprotnik et al. 2008; Saunders and Voth 2012; Shi et al. 2006; Shinoda et al. 2012; Yefimov et al. 2008). Most of these efforts have focused on developing particle-based models in which one bead represents a group of atoms. Specifically for MscL, building upon their success in developing an effective coarse-grained model for lipids, Marrink et al. developed a coarse-grained model (Yefimov et al. 2008) for MscL based on the transfer free energy of amino acids between water and lipids. The gating transition was successfully observed in the simulation although the pore radius in the final state is somewhat smaller than that estimated in the literature (Sukharev et al. 2001b).
Molecular Dynamics-Decorated Finite Element Method (MDeFEM)
In light of the limitations of previous experimental/theoretical/numerical efforts concerning the structural rearrangements of MS channels during gating, it is worthwhile pursuing the alternative approach of continuum mechanical modeling, which has been used in a broad set of mechanics problems (Scarpa et al. 2010; Tang et al. 2006; Tserpes and Papanikos 2009; Zeng et al. 2012). Along this line, the establishment of molecular dynamics-decorated finite element method (MDeFEM) (Chen et al. 2008; Tang et al. 2006, 2008) represents an attractive alternative to atomistic and particle-based coarse-grained simulations, allowing the analysis of bimolecular systems at long time scales while maintaining sufficient molecular details to capture the most essential characteristics of the system under study. In~MDeFEM,~biomolecules are modeled as integrated continuum motifs and the finite element simulation framework allows efficient treatment of deformations at large length scales and complex deformation modes inaccessible to conventional all-atom simulations.
In this chapter, MDeFEM (Chen et al. 2008; Tang et al. 2006, 2008) is firstly adapted to study the gating mechanism of MscS. The high computational efficiency of MDeFEM allows us to analyze the contributions of various structural motifs and interactions to the gating process as well as the effect of voltage. The observation of different gating characteristics upon perturbation of material properties of the helical kink region in TM3 at Gly113 also leads to the proposal of a mechanism for inactivation. Overall, the current simulations not only provide new insights into the gating transition of MscS with structural details but also lead to specific predictions that can be tested by future experimental studies.
Secondly, a number of major limitations of previous MDeFEM models of MscL (Chen et al. 2008; Tang et al. 2006, 2008) (or the MDeFEM models of MscS) need to be alleviated for more quantitative analysis. These limitations include (1) not sufficient structural/energetic details of MscL. The helices were represented by rounded sticks, and the inter-component interactions were computed based on surface-to-surface interactions; the nonbonded interactions among loops and those between the loops and the helices were not considered. (2) The lipid bilayer was treated as an elastic solid slab, whereas the realistic membrane should be fluidic and does not sustain large shear stress. (3) No solvation contribution was considered. It has been proposed that solvation plays a major role in stabilizing the open conformation of MscL due to the exposure of hydrophilic residues (Anishkin et al. 2005; Anishkin and Kung 2005). These limitations may have led to the exceedingly high membrane strain required for the full-gating transition of MscL in the previous studies (Chen et al. 2008; Tang et al. 2008). The MDeFEM approach is here improved significantly to address these issues through a coupled mechanical-chemical approach. More realistic models for the MscL molecule and the membrane are developed. To include solvation effects, a force-based protocol is established to integrate a continuum mechanics model for the mechanical properties of the macromolecule with a continuum treatment of solvation. A similar approach has been applied to study the salt concentration dependence of DNA bendability (Ma et al. 2009). It is envisioned that the high computational efficiency and flexibility will make this hierarchical multi-scale framework uniquely applicable to the study of mechanical behaviors of various biological systems, interpreting existing and stimulating new experimental investigations.
Gating and Inactivation of E. coli-MscS
Models and Methods
In MDeFEM, the protein structure is described by continuum FEM models, and nonbonded interactions between different components are represented by nonlinear distance-dependent pressures or point-to-point connectors. In the following, the main continuum construction procedures of E. coli-MscS are introduced briefly with the commercial package ABAQUS. More details can be found in our previous works (Chen et al. 2008; Zhu et al. 2016). In the subsequent discussion, unless otherwise specified, MscS refers to E. coli-MscS.
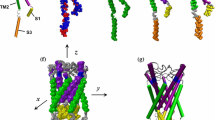
A MscS channel (Bass et al. 2002) is formed by seven chains assembled as a heptamer with a large cytoplasmic region that exhibits a mixed α/β structure with several strands and α-helices intertwined together to form a balloon-like osmolyte filter cage (Gamini et al. 2011). In the transmembrane domain (Fig. 1c, e), each monomer (Fig. 1a) of MscS consists of three helices, referred to as TM1, TM2, and TM3, the last of which has a pronounced kink at the Gly113 region and is thus split into TM3a and TM3b. Each TM1 helix and its nearest neighbor, a TM2 helix, are assembled into a pair through a periplasmic loop, forming the outer boundary that interacts extensively with the lipid membrane; the TM3a helices form the inner boundary for the pore, and TM3b helices splay out to be nearly parallel to the membrane plane.

Continuum mechanics modeling of MscS. (a) The backbone structure of one MscS monomer with two key interaction pairs (Asp62-Arg131 and Phe68-Leu111) highlighted by arrows. (b) The corresponding FEM model of one monomer. The Young’s modulus, E (GPa), and Poisson’s ratio, v, of each component in one monomer are indicated aside. (c–d) Side views of the crystal structure and FEM model. (e–f) Top views of the crystal structure and FEM model. Examples of several lowest eigenmodes and frequencies of helices and loops are displayed in (g) and (h), respectively. Results of molecular mechanics are compared to those of the finite element simulations
Based on the closed backbone structure of MscS (Bass et al. 2002; Steinbacher et al. 2007), within the continuum mechanics framework, each helix is modeled as a three-dimensional elastic cylinder of 5 Å diameter and the loops as quasi-one-dimensional winding beams with cross-sectional diameter of 2.5 Å (Fig. 1b) (Tang et al. 2006; Zhu et al. 2016). For simplicity, only the transmembrane domain of MscS is considered (Anishkin et al. 2008b). Although there is evidence supporting that the cytoplasmic domain swells up (Machiyama et al. 2009) during gating, it is also suggested to be nonessential but only responsible for increased stability and activity (Schumann et al. 2004); the cytoplasmic domain is not expected to undergo large changes during the gating transition (Pliotas et al. 2012; Wang et al. 2008). The endpoints where the cytoplasmic domain is truncated are softly restrained in the continuum model (1 kcal/mol/Å2) (Anishkin et al. 2008a, b; Spronk et al. 2006). Furthermore, the first 26 residues (in the N-termini) of the 286 amino acids of each MscS monomer were not resolved in the crystal structure (Bass et al. 2002) and therefore are also excluded in the continuum model; an experimental study showed that MscS can tolerate small deletions at the N-terminus (Miller et al. 2003).
Material properties of each component of the continuum model are calibrated by matching results of normal mode analysis (NMA) at the atomistic and continuum levels (Fig. 1g–h). The key mechanical properties for the helices and loops are shown in Fig. 1b, and these are much larger than the estimated range of the Young’s modulus of MscL α-helices (0.2 to 12.5 GPa) in Martinac et al.’s recent work (Bavi et al. 2017) with constant-force steered molecular dynamics (SMD). This is because, in the present study, the helices, for instance, are modeled as much thinner elastic cylinders (with a diameter of 5 Å) by considering only the main chain. The bending stiffness of the helices in the present continuum model and that of MscS helices by SMD are expected to be consistent. As a qualitative comparison, for example, the bending stiffness EI (the product of Young’s modulus and moment of inertia) of MscS helices in vacuum is in the range of 70∼200 (10−10 N Å2) in this work, while based on Martinac et al.’s study (Bavi et al. 2017), the bending stiffness of MscL helices in water can be calculated to be in the range of 1∼80 (10−10 NÅ2). For simplicity, the softening effect of the helices due to hydration (Anishkin and Sukharev 2017; Bavi et al. 2017) is not considered here, which may be one of the reasons for the difference in the above comparison of the bending stiffness. In both MscL (Bavi et al. 2017) and MscS, the second helix, TM2, is the stiffest one among the α-helices in each channel.
The MscS continuum model is embedded into an elastic membrane modeled as a sandwich panel that consists of three layers to mimic the lipid head and tail regions. A flat square membrane with a size of 400 × 400 Å is employed. To embed the channel (Fig. 1) into the continuum membrane, a cavity with the shape of a multi-petal flower (Fig. 2) is created in the middle of the membrane with the size and shape of the cavity conforming to those of MscS transmembrane helices in the closed state with an equilibrium distance of ∼5.5 Å (Chen et al. 2008) from the surface of the cavity to the surfaces of the helices. The lipids buried in the voids within the transmembrane helices (Pliotas et al. 2015) are neglected; their specific role in gating is not quite clear. The membrane model is parameterized based on MD calculations of the density map of water and lipids and the lateral pressure profiles of the POPE lipid membrane (Chen et al. 2008; Gullingsrud and Schulten 2004). Mechanical properties of the lipid membrane are shown in Fig. 2, and the in-plane shear modulus of the continuum membrane model is reduced (Zhu et al. 2016) to take into account the fluidity of the lipid membrane, i.e., its incapability of sustaining a large shear stress. It would be of interest to investigate the effect of the buried lipids (Pliotas et al. 2015) or more complex lipid properties (such as viscoelasticity (Deseri et al. 2016)) on gating, but we leave it to future studies since more sophisticated continuum models are required.

Schematic of the continuum protein-lipid membrane model. A zoomed-in view is shown to illustrate the multi-petal lipid hole encompassing the MscS protein
The assembled protein-membrane continuum model is shown in Fig. 2. Helices of MscS are meshed by four-node tetrahedron finite elements and loops by one-dimensional beam elements (Fig. 1). Without over-resolving the system, appropriate mesh density is ensured through convergence studies, and a typical element size of 1.5 Å is chosen. Four-node tetrahedron finite elements are also used to mesh the lipid membrane model with the mesh gradually more refined toward the boundary of the inner hole where it interacts with the protein directly (Fig. 2). To be consistent with the simple description of the protein-lipid continuum model, solvent molecules are not included, though the hydration effect can be further studied with a more sophisticated continuum mechanics-solvation coupled approach (Zhu et al. 2016) which is to be introduced in section “Coupled Continuum Mechanical-Continuum Solvation Approach with Application to Gating Mechanism of MscL.”
In this work, we explicitly consider two key interaction pairs known to be essential to MscS gating, i.e., the Asp62-Arg131 salt bridge and the Phe68-Leu111 apolar interaction (Fig. 1a) mentioned in the section “Introduction.” The nonbonded interaction energy (electrostatic and van der Waals) between each pair of residues is calculated using the CHARMM 36 force field (Best et al. 2012; Brooks et al. 1983) as a function of distance along the center-of-mass separation vector (Fig. 3b). An electrostatic screening factor of 6 is used when calculating the electrostatic interaction for the Asp62-Arg131 salt bridge, considering that the gap between Asp62-Arg131 residues in the same monomer or across monomers is spanned mainly by protein atoms or lipid molecules rather than water (Pliotas et al. 2015); varying this factor from 3 to 10 generally has little impact on the computed gating behavior (data not show). The nonlinear distance-dependent interaction force between two residues in each pair is derived accordingly from the energy profile and applied to the closest finite element nodes by invisible nonlinear connector elements (arrows in Fig. 1b) in ABAQUS (Zhu et al. 2016). The connector element representing a pair of key interaction is defined both within the same monomer and across neighbor monomers.

Examples of the fitting of nonbonded interactions between helices of E. coli-MscS (a) and the interaction energy for two key interaction pairs (b). In (a), the distance between two components is normalized by their equilibrium distances. Data points are obtained through molecular mechanics (MM)
Apart from the above two specific residue pairs, interactions between different helices and between the helix and the lipid membrane are described by a pair-wise effective pressure-distance relationship where the atom-to-atom interaction in the atomic structure are averaged to the surfaces of different components (Tang et al. 2006) (see Fig. 3a). The nonbonded (electrostatic and van der Waals) interactions are calculated without any cutoff. Incorporating an implicit membrane environment through the GBSW model (Im et al. 2003) in CHARMM in the inter-helical calculations has a negligible impact on the energy profiles. Taking the first derivative of the interaction energy with respect to distance, the pressure-distance relationship between two surfaces (adopting the sign convention that repulsive pressure is positive) is obtained and takes the following form:
where p and d0 are the interaction pressure and equilibrium distance between two surfaces, respectively, C is the energy well-depth, and αi is the instantaneous distance between two deformed surfaces for the i-th element. This nonbonded pressure model implicitly includes both electrostatic and van der Waals (VDW) interactions and has been successfully applied to study the deformation and buckling of carbon nanotubes (Cao and Chen 2011; Chen et al. 2006; Pantano et al. 2003), as well as gating transition of the E. coli and Mycobacterium tuberculosis MscL (Bavi et al. 2016b; Chen et al. 2008; Tang et al. 2006). Figure 3a shows examples of the interaction energy between different protein components in closed and open crystal structures and the fitted curves used in FEM. The consistency between the interaction energy curves of the closed and open states suggests that the fitted FEM parameters are fairly transferable. Table 1 summarizes the fitted values of C, d0, and the exponents (m, n) for the closed crystal structure. When calculating the interaction between TM2 and TM3a, the Phe68-Leu111 pair is excluded since it is already considered separately as discussed above. The constitutive interaction relationship is called in each increment through the analysis based on the relative position between two surfaces that are continuously updated. Similar to the connector elements, the pressure-based nonlinear interaction is defined both within the same monomer and across neighbor monomers as long as the distance between two components is smaller than 16 Å.
The different treatment of the key interaction pairs (described by node-to-node connectors) and other “less important” interactions (described by the averaged surface-to-surface pressure) allows a simple description of the continuum model while providing the opportunity to explore how these key interactions affect gating. A typical tension simulation of the gating transition of MscS takes only about 1 h, on a Thinkpad laptop with four 2.5 GHz CPUs and 8 GB of RAM.
Results and Discussion
In this section, gating transition of MscS in response to membrane stretch (tension) is firstly obtained. The open FEM structure is compared to the crystal open structure, and the intermediate structure is identified in FEM. Similar analysis is then conducted with some key interaction pairs (Asp62-Arg131 or Phe68-Leu111) or loops excluded to explore their role in the gating transition. Next, the kink between TM3a and TM3b is considered as helical (thus having larger cross section and Young’s modulus) rather a loop, leading to the discussion of a plausible inactivation mechanism. Finally, the effects of transmembrane voltage are analyzed.
MscS Gating Pathway
Shown in Fig. 4 are several snapshots of MscS during the membrane tension-driven gating process in comparison to the crystal structures for the closed (Bass et al. 2002) and open states (Wang et al. 2008). The results indicate that, during gating, all helices shift radially away from the center of the pore in a manner reminiscent of a mechanical camera iris, similar to the gating transition described for MscL (Betanzos et al. 2002; Tang et al. 2008). The TM1 and TM2 helices in the same monomer move concurrently almost as a rigid body due to the strong inter-helical interactions. The TM3a helices rotate around and move outward from the central axis and finally become parallel to each other and normal to the membrane plane. Notably, the loop that connects TM1 and TM2 in the same monomer transitions from interacting with the end of TM3b in the neighboring chain (in the counterclockwise direction) to interact with the end of TM3b within the same monomer. Besides, significant rotation of TM3b helices around the central pore axis is observed (Fig. 4), though the average distance from TM3b ends to the pore axis is essentially unchanged. The conformational transitions discussed above are reversible in the FEM simulations once the membrane strain is removed.

Comparison between MscS gating pathways under equi-biaxial membrane tension between FEM results and the crystal models in both top (the top panel) and side views (the lower panel)
On a qualitative level, the current FEM results are in agreement with the crystal structure models, regarding both the orientation and displacement of the transmembrane helices and connecting loops (Fig. 4). Small differences lie in the titling of TM1 helices; from the top view (fist panel in Fig. 4), the TM1 helices form a more compact bundle in the open crystal structure than in the FEM model. In addition to the open structure, intermediate structures during the gating process are obtained in the FEM simulation, and an example is given in Fig. 4.
In the FEM model of MscS in the closed form, the radius of the area lined by TM3a has a radius R′ ≈ 7.0Å (inset in Fig. 5), while the actual pore radius is R ≈ 2.4Å (Wang et al. 2008) considering the pore’s irregular inner surfaces. In the following, the pore radius (R) is estimated by R = R′ − 4.6Å based on the FEM results of R′, and the pore diameter is thus 2R. Figure 5 depicts the evolution of pore diameter during the gating process. As the membrane strain (εm) increases, the pore size of MscS increases slowly until εm reaches 5%, after which the pore diameter (2R) experiences a rapid increase leading to pore opening (2R = 13 Å) (Wang et al. 2008). Since the hydrophobic pore remains nonconducting until approximately 2R > 9 Å (the threshold diameter for hydration) (Beckstein et al. 2001), this observation is in qualitative agreement with the patch-clamp study of MscS (Martinac and Kloda 2003), in which the gating membrane tension for full opening of MscS was measured to be about half of that of MscL (Martinac and Kloda 2003). The gating membrane strain for McsL was estimated to be ∼10% (Zhu et al. 2016), thus for MscS, the gating strain is expected to be 5%; the lipid membrane strain required for full opening of MscS in the present FEM simulation is 6.6% (Fig. 5), which is slightly larger than the 5% estimation.

Pore diameter evolution for different FEM models
Effect of the Key Interaction Pairs
Mutation studies have identified two key interaction pairs that greatly affect the gating behavior of MscS, and these involve the Asp62-Arg131 salt-bridge (Nomura et al. 2008) and the apolar interaction between Phe68-Leu111 (Belyy et al. 2010). For example, when the negatively charged Asp62 was replaced with either a neutral (Cys or Asn) or basic (Arg) amino acid, the gating threshold increased significantly (Nomura et al. 2008). Both F68S and L111S substitutions also led to severe loss-of-function phenotypes (Belyy et al. 2010). To provide a structural understanding of how these interactions influence the gating transition of MscS, we conduct FEM simulations with either pair of interactions excluded.
Firstly, the electrostatic interaction between Asp62-Arg131 in the continuum model (Fig. 1) is removed, while the van der Waals interaction between them is preserved (Fig. 6a) so as to mimic the replacement of Asp62 with a charge neutral amino acid. The structural transition upon membrane stretch up to 6.6% is shown in Fig. 6b–c. Without the strong electrostatic interaction, the TM1-TM2 helices tend to detach from the end of TM3b. Under equi-biaxial membrane tension, all helices are lifted up and the kink in TM3 is straightened. As a result, the size of the channel pore surrounded by TM3a helices remains essentially unchanged despite the outward displacements of TM1 and TM2. Therefore, opening of the pore in this “mutant” requires an exceedingly large membrane strain (data not shown).

FEM results of the structural transition of MscS under equi-biaxial membrane tension without electrostatic interaction between Asp62 and Arg131 (a–c) or without the Phe68-Leu111 traction (d–f)
Similarly, a simulation without the apolar interaction between Phe68-Leu111 is conducted (Fig. 6d–f). During lipid membrane stretching, the TM1 and TM2 helices move outward, but the TM3a helices do not follow and the kink angle in TM3 remains unchanged. With a membrane strain of 6.6%, the pore size actually becomes smaller since the lower ends of TM3a helices are more closely packed because of the loss of TM2-to-TM3a attraction (F68-L111).
In short, the FEM simulations recapitulate the expected behaviors of the relevant mutants in which the key interactions are perturbed (Belyy et al. 2010; Nomura et al. 2008). The observed structural evolution in the FEM simulations also provides a physical understanding of how these interactions contribute to the gating transition. These results support the use of the FEM model to explore other contributions, to which we turn to next.
Effect of Structural Motifs (Loops) in Force Transmission
In this section, we explore the roles that periplasmic loops play in MscS gating by repeating FEM simulations with specific loops excluded from the model. When the loops connecting TM1 and TM2 helices are removed (with the Asp62-Arg131 interaction reserved), the structural response (data not shown) closely resembles that of the full channel model; likewise, the pore diameter evolution is not significantly perturbed (see Fig. 5). When the loops connecting TM2 and TM3 helices are removed, while the rearrangements of TM1 and TM2 helices are similar to those of the full channel model (Fig. 4), the TM3a helices collapse at the upper ends, leading to an essentially closed pore (Fig. 7). This suggests that the loops linking TM2 and TM3 helices are essential to the force transmission from lipid membrane to the pore-lining helices during MscS gating; this is quite different from the situation of MscL (Tang et al. 2008), in which loops that connect the transmembrane helices generally constrain channel opening (Ajouz et al. 2000; Tang et al. 2008). To the best of our knowledge, the importance of the loop between TM2 and TM3 helices to MscS gating has not been pointed in the literature, and this prediction can be further tested by studying loop deletion mutants in patch-clamp experiments as done for MscL (Ajouz et al. 2000; Bavi et al. 2016b).

Structural transition of MscS under equi-biaxial membrane tension with the TM2-TM3 loop removed
Effect of the Helical Propensity of the TM3a-TM3b Kink: Inactivation
The Gly113 introduces a kink in TM3, and the region exhibits a weak helical propensity; thus, the kink segment of G1y113 is modeled as a loop (with an arc length of ∼8 Å corresponding to the length of the backbone of Gly113) with a small cross-section and a lower Young’s modulus (Fig. 1) in models described so far. To explore the significance of the helical kink, FEM simulations are performed in which the Gly113 region in TM3 is treated as helical but remained (enforced) as the bent shape and modeled as a curved solid stick (Fig. 8a) whose material properties take that of the TM3b helix. The structural response of this modified channel model under membrane tension is depicted in Fig. 8a and the pore diameter evolution process in Fig. 5 (square curve). When the membrane strain reaches 6.6%, the pore expands from 4.8 Å to 8 Å in diameter (Fig. 8a). At this point, the expanded pore still remains nonconducting since, for a hydrophobic pore, the threshold diameter for hydration is ∼9 Å (Beckstein et al. 2001). To fully open the channel (2R=13 Å), a high membrane strain of 11.4% is required (Fig. 8a); this value is comparable to the gating strain of MscL (Zhu et al. 2016). Combining Fig. 4 and Fig. 8a, it seems that a flexible end of TM3a at the kink allows a more effective pulling by the Phe68-Leu111 interaction, thus leading to a lower gating strain. The interaction between TM3b and the cytoplasmic β domain where the TM3b-β interface is quite hydrophobic (Koprowski et al. 2011) promotes the kink around Gly113 (Koprowski et al. 2011; Petrov et al. 2013), which, in the closed state, may prevent backbone hydrogen bonding in the kink area, thus weakening the constraint at the lower ends of TM3a. The TM3b-β interface is indicated in Fig. 8a (ellipse), though noting that the cytoplasmic part is not explicitly included in the present study. Introducing higher polarity (charged residues) into the TM3b-β interface (e.g., the N117 K or G168D mutant) results in a weaker interaction between TM3b and the cytoplasmic β domain (Koprowski et al. 2011), which could be in favor of the backbone hydrogen bonding in the kink area. It has been observed that neither N117 K nor G168D mutant shows visible inactivation behavior, while both of them require a much larger membrane pressure for gating, which is consistent with the present results (Fig. 8a).

Structural transition of MscS under equi-biaxial membrane tension with the TM3a-TM3b kink considered as helical (a) and the proposed mechanism for gating, desensitizing, and inactivation of MscS. The purple ellipse in (a) indicates the TM3b-β interface. The increased helical propensity of the kink could be induced by weakening the interaction between TM3b and the cytoplasmic β domain (e.g., the G168D mutant), thus, to an extent, relaxing the kink
With these observations, it is tempting to speculate an inactivation mechanism for MscS: in the resting state, the kink of TM3 behaves like a loop, and upon rapid membrane tension, the channel is easily opened (Fig. 4 and the circle curve in Fig. 5). Under prolonged exposure to subthreshold membrane tension, however, the channel desensitizes (dashed arrow in Fig. 5) into a nonconductive state (the circle curve); this occurs because of the local transition of the kink region into a helical conformation that hinders further pore opening. The helical transition of the kink could be induced by the weakening of the TM3b-β interface because of the increased hydration around the interface domain caused by the rotation and outward movement of TM3 helices (Wang et al. 2008) or the reduction of lipids in the TM pockets (Pliotas et al. 2015).
To further test this hypothesis, a FEM simulation is conducted in which, at a subthreshold membrane strain, the Young’s modulus of the TM3a-b loop is increased to the extent that its bending stiffness is equal to that of the TM3b helix. In Fig. 8b, after stretching the initial model (model in Fig. 5) by 5% (εm=5%), the TM3a-b loop is stiffened, which leads to reduced pore size; further opening the “inactivated structure” requires a very large membrane strain (9%) that is close to the gating strain of MscL. Increasing the stiffness of the TM3a-b loop at other reduced values (<5%) of membrane strain leads to similar results. By contrast, if the stiffening of the TM3a-b loop occurs at a much larger membrane strain (e.g., εm=6.6%), the impact on the subsequent pore evolution is minimal. These results are in qualitative agreement with the experimental observation of Sukharev et al. (Kamaraju et al. 2011) that MscS inactivates primarily from the nonconducting state and channel opening prevents inactivation. It should be noted that the present quasi-static approach does not show the dependence of inactivation on (long) time (Kamaraju et al. 2011), which could be a result of desensitization against hydration inside the TM3a pore.
The mechanism for MscS inactivation has remained a mystery for decades. Though some key interactions and residues have been proposed to be involved, how these interactions and residues induce inactivation is poorly understood. The current study suggests that this behavior of MscS could be related to the conformational state of the helical kink at Gly113; a loop conformation allows other interactions to effectively pull TM3a helices outward thus opening the channel, while a more ordered helical conformation hinders the pulling, leading to channel inactivation. Previous experimental studies highlighted the importance of the Gly113 region as well. For example, another MscS-like channel from Silicibacter pomeroyi (MscSP) has a conserved glycine residue at the position equivalent to Gly113 in MscS (Petrov et al. 2013). However, the N117 residue in MscS is replaced by a charged residue Glu in MscSP based on the alignments of MscS and MscSP (Petrov et al. 2013), which leads to a weaker interaction between TM3b and the cytoplasmic β domain in MscSP (Koprowski et al. 2011; Petrov et al. 2013) and may facilitate the helical transition of the kink region, resulting in a higher gating threshold (square curve in Fig. 5); indeed, it was reported that the threshold gating strain of MscSP is ∼1.5 times of that of MscS (Petrov et al. 2013). In another two MscS-type channels, MscMJ of M. jannaschii (Kloda and Martinac 2001) and MscCG of C. glutamicum (Borngen et al. 2010), inactivation was not observed (Petrov et al. 2013); this might be explained by the observation that Gly113 in MscS is replaced in the equivalent position by Asp and Ser in MscMJ and MscCG, respectively (Petrov et al. 2013).
In addition to the connection to these previous studies, the inactivation mechanism proposed here (Fig. 8b) can be tested by replacing Gly113 with amino acids of higher helical propensity or extending the kink area (e.g., multiple Gly insertion) to further increase its structural flexibility; the latter was done by Martinac et al. for MscL (Bavi et al. 2016b). Both mutations are expected to result in the absence of inactivation in MscS.
Interestingly, an earlier work (Akitake et al. 2007) has conducted a series of mutation studies both at the Gly113 kink region and the TM3b-β interface area (Gly121), though that study was aimed to show that the Gly113 kink is a unique feature of the inactivated state and the closed structure favors buckling at Gly121, which was not supported by later studies (Pliotas et al. 2012, 2015; Ward et al. 2014). Thus we here attempt to interpret the results of mutation studies in Akitake et al. (2007) from the perspective of our proposed inactivation mechanism. First, the G113A mutant in Akitake et al. (2007) had increased helical propensity in the kink region and indeed showed no inactivation behavior; this agrees with our inactivation mechanism. The gating tension of G113A mutant was, however, observed to be comparable to that of the wild-type MscS in Akitake et al. (2007); this observation is not consistent with the predicted gating pathway in Fig. 5 (square curve), probably because the Ala substitution interacts with phospholipids inside the TM pockets (Pliotas et al. 2015) (not considered in the present model), thus strengthens the apolar interaction between the lower end of TM2 and that of TM3a (the main traction that pulls TM3a bundle open (Fig. 6)); alternatively, the Ala substitution may lead to structural rearrangements not considered in the present model (e.g., a certain extent of kink straightening (Akitake et al. 2007)) that enhance the interaction between the ends of TM2 and TM3a. To test these possibilities, a simulation is conducted where the kink region is treated as helical and modeled as a curved solid stick (Fig. 8a), while at the same time the TM2-TM3a traction force (green arrow in Fig. 8a) is increased by 50%. The gating strain is found to be 6.0% in this case, close to the gating strain (6.6%) of the model in Fig. 4 where the kink is modeled as a loop.
Second, the Q112G mutation (Akitake et al. 2007) increased the flexibility of the kink area, but instead of resulting in the absence of inactivation as suggested in our proposed inactivation mechanism, the Q112G mutant exhibited faster inactivation (Edwards et al. 2008). We conjecture that the Q112G mutation may weaken the apolar interaction between the lower end of TM2 and that of TM3a and may make the kink area “too flexible” that upon channel opening or even in the initial state, water molecules enter the TM pocket between TM1-TM2 and TM3a thus further causing the detachment between TM2 and TM3a. To test this assumption, a simulation is conducted for a modified Q112G mutant model, where the kink region is treated as a loop and modeled as a flexible quasi-one-dimensional winding beam (Fig. 1b), while at the same time the TM2-TM3a traction force (green arrow in Fig. 1b) is decreased by 10%. The simulation results in a structure close to that in Fig. 6 where the Phe68-Leu111 traction is missing and the channel cannot be opened, which may explain the faster inactivation of Q112G. As replacing one residue could change a set of interactions nearby (Wang et al. 2008), especially for the subtle situation at the Gly113 area which also affects the TM2-TM3a apolar interaction or possibly the hydrophobic lock of the channel pore (Anishkin and Sukharev 2004; Vora et al. 2006), mutation studies in this region require careful analysis and interpretation. The two simulations mentioned above serve as exploratory models by controlling the state of the kink (helix vs. loop) and the traction between the ends of TM2 and TM3a. More detailed experimental and simulation works are required in future to determine the structural and interaction changes around the kink area due to mutations.
At the TM3b-β interface, two mutations were conducted in Akitake et al. (2007), G121A and A120G. The G121A substitution, as discussed above, can reinforce the hydrophobic TM3b-β interface, promoting the kink around Gly113 (Koprowski et al. 2011; Petrov et al. 2013) and resulting in a more flexible kink region at Gly113. As expected from our mechanism, the G121A mutant was observed to open easily and showed no inactivation (Akitake et al. 2007). The A120G mutation, on the other hand, weakened TM3b-β interface, thus promoting the helical propensity at the kink area. Again as expected from our mechanism, the A120G mutant exhibited a high degree of inactivation (Akitake et al. 2007). Therefore, the results for both G121A and A120G mutants are supportive of the inactivation mechanism proposed here.
Effect of Transmembrane Voltage
Transmembrane voltage is applied to the continuum model to investigate its effects on MscS gating (Akitake et al. 2005; Nomura et al. 2016). For simplicity, we only consider the charged residues in the atomic model which are mapped to the finite element nodes in the continuum model (inset of Fig. 9). There are only a few charged residues in the transmembrane domain, which are Arg46, Arg54, Arg59, Lys60, Asp62, Asp67, Arg74, Arg88, and Arg128. Transmembrane voltages from −100 mV to +100 mV (Akitake et al. 2005) are applied with the exterior of the cell being positive with respect to its interior. No evident dependence of the activation pathways (Fig. 9) on transmembrane voltages is observed in the FEM simulations. This finding is consistent with recent experimental results (Akitake et al. 2005; Nomura et al. 2016). The rectifying behavior that the conductance is larger at positive voltages observed in some studies (Martinac et al. 1987; Petrov et al. 2013) could be a result of the inside-out nature in patch-clamp experiments and the slight anion preference of MscS (Martinac et al. 1987; Sukharev 2002).

Pore diameter evolutions of MscS under equi-biaxial membrane tension and different transmembrane voltages. Residue charges and electric field are incorporated into the continuum model based on the charge distribution in the crystal structure (inset). Red particles represent positive residues and blue particles negative residues
Another observation of depolarizing voltage-dependent inactivation (Akitake et al. 2005), however, remains unsettled. How negative voltages promote inactivation rate must await further studies with more sophisticated models combined with experimental analyses. It is possible that, though the electric field causes only small displacements on the charges in MscS during activation (Akitake et al. 2005), its presence may disrupt some key interactions like the TM3b-β interface (e.g., Asn117). Besides, voltage-dependent inactivation may be related to the conformational changes of the cytoplasmic domain (not included in the present study) as indicated in Rowe et al.’s work (Rowe et al. 2014) where the G168D mutant is insensitive to applied voltages.
Coupled Continuum Mechanical-Continuum Solvation Approach with Application to Gating Mechanism of MscL
Models and Methods
In this section, the construction process of the continuum mechanics model for E.~coli-MscL is presented in detail. Relevant material and interaction parameterizations are either calibrated by matching results at the atomistic and continuum level or obtained from a previous study (Chen et al. 2008). The computational framework includes essentially two components: continuum mechanics (CM) simulations based on finite element method (FEM), which solve for the deformed structure under specific external loads, and the continuum solvation model (CS), which computes solvation forces based on structural information from the CM model. The commercial software ABAQUS (2011) (for CM calculations) and open-source software APBS (Baker et al. 2001) (for CS model) are used in this fully integrated (CM/CS) simulation framework. Detailed ABAQUS-APBS co-simulation protocols are presented.
Continuum Modeling of E. coli-MscL and Interactions Within the Protein
Although E. coli-MscL is one of the most studied MS channels, the only available X-ray crystal structure in the literature is for the MscL from bacteria Mycobacterium tuberculosis (Tb), which was captured in its closed state by Rees Lab (Chang et al. 1998). By retaining the main features of the crystal structure of Tb-MscL, the atomic structure of E. coli-MscL was developed based on homology modeling along with other experimental constraints (Sukharev and Anishkin 2004; Sukharev et al. 2001a; Sukharev and Corey 2004); the closed state model is shown in Fig. 10g. The crystal structure of Tb-MscL was further refined by Stefan Steinbacher et al. (2007), where its N-terminal (termed S1) was an amphipathic helix positioned approximately parallel to the cytoplasmic surface of the membrane. This refined structure for Tb-MscL was supported by later studies (Iscla et al. 2008). Based on the new structure for Tb-MscL and MD simulations, S1 domain was suggested to interact closely with the lipid membrane, which may facilitate the opening of the channel (Iscla and Blount 2012; Vanegas and Arroyo 2014). While developing the new atomic E. coli-MscL structure based on the revived Tb-MscL crystal structure (Steinbacher et al. 2007) is beyond the scope of the present study, it should be noted that the effect of S1 domain as a “sliding anchor” to the lipid membrane thus helping channel opening (Iscla and Blount 2012; Vanegas and Arroyo 2014) is not available for studying in this work since we are using Sukharev S. et al.’s model (Sukharev and Anishkin 2004) whose S1 domain assembles as a helix bundle. The S1 domain’s interaction to the lipid and its hydration process may be to some extent different from what we present in the following context but is not expected to bias the principal outcomes of the current model.

Illustration of various steps in constructing the FEM Model of E. coli-MscL . (a, b) the atomic structure of one chain of E. coli-MscL in cartoon and van der Waals (VDW) representation where blue indicates hydrophobic and red hydrophilic; (c) the triangularized molecular surface; (d) the simplified triangularized molecular surface and the volume enclosed by this surface are discretized into tetrahedral elements; (e) the coarse-grained model of one chain of E. coli-MscL; (f) the FEM model of E. coli-MscL; (g) structural model of E. coli-MscL based on homology modeling (Sukharev and Anishkin 2004). Zhu et al. (2016), reprinted with permission of Springer
An E. coli-MscL molecule is formed by five chains (from chain-1 to chain-5) assembled as a fivefold structure around its symmetry axis, and each single chain (Fig. 10a) of MscL consists of four helices (referred as TM1, TM2, S1, and S3) and several loops. Within the transmembrane helix bundles, the five TM2 helices form the outer boundary that interact extensively with the lipid membrane, while the longer TM1 helices form the inner boundary for the pore and have limited contact with the lipid. The TM1 and TM2 helix bundles share the same fivefold symmetry axis, denoted as the z-axis here, which is also the direction of the membrane normal. Each TM1 helix and its nearest neighbor, a TM2 helix, are assembled into a pair through a periplasmic loop. A closer view of the TM1 shows that there is a break near the top of the helix due to Pro-43; thus, the segment above Pro-43 is also referred to as the S2 helix. The cytoplasmic region contains two different types of helix bundles, referred to as the S1 helices and S3 helices; each bundle contains five subunits, and each subunit of the S1 or S3 bundles is connected, respectively, to a TM1 or TM2 helix through a loop. The TM1, TM2, S1, and S3 helices correspond to residues Asn-15–Gly-50, Val-77–Glu-107, Ile-3–Met-12, and Lys-117–Arg-135, respectively (Sukharev and Anishkin 2004). In the subsequent discussion, unless otherwise specified, MscL refers to the E. coli-MscL.
In the continuum model, we start from the backbone structure with side chains removed because when side chains are presented, some subcomponents are unable to be separated, such as the TM1 and TM2 helices in the same chain which are very close to each other. More dedicated calculation of the surfaces of the protein could be considered in future work to get a more precise model. The molecular surface (Fig. 10c) of each single chain of MscL molecule is calculated and triangularized using the MSMS program (Sanner et al. 1996) with appropriate probe radius to avoid overlapping of subcomponents (such as TM1 and TM2 helices). Among the multiple subcomponents of MscL, helices are modeled to have larger cross sections because they are strengthened by the hydrogen bonding between residues, while the loops are modeled much thinner (Fig. 10c). The triangularized surface is then simplified by the QSLIM program (Heckbert and Garland 1999) to reduce the number of the surface triangles to 2000 (Fig. 10d). The volume enclosed by this simplified surface is subsequentially discretized into a 3-D mesh consisting of tetrahedral elements. The final FEM model consists of 2,685 nodes and 9,699 finite elements for each chain of MscL.
Assuming that the mechanical properties of each component vary little with respect to sequence, the properties of each component are assumed to be homogeneous, isotropic, and constant during the gating process. The only exception is for the break between S2 and TM1 (Pro43, illustrated by the blue segment in Fig. 10a), whose properties are determined separately and less stiff than the rest of the helix. The material properties of each component are calibrated by matching results of normal mode analysis (NMA) at the continuum and atomistic levels. The NMA for individual components of the channel is conducted in vacuum using the Gromacs MD simulation package with the Gromos96 vacuum parameter set (Van Der Spoel et al. 2005) so as to be consistent with the continuum calculations which are also conducted in vacuum. A possible way to get more realistic elastic properties is to conduct NMA considering the water and lipid environment in both continuum and atomistic calculations though this kind of NMA at the continuum level is highly complex. The Young’s modulus is then varied at the continuum calculations such that the eigenvalues and eigenvectors for the three lowest-frequency modes best fit the results from the atomistic normal mode calculations. The lowest eigenmode of the TM1 or TM2 helices, for example, is essentially flexural bending as shown with both continuum and atomistic configurations in Fig. 11. The lowest frequencies are 136.2 and 96.1 GHz for TM1 and TM2 helices, respectively, which lead to the fitting of their effective Young’s moduli as 69 GPa (TM1) and 54 Gpa (TM2). Summarized in Table 2 are the key mechanical properties for the helices and loops. These are smaller than estimated in the previous work (Chen et al. 2008) where the helices, for instance, were modeled as thinner (with a diameter of 5 Å) elastic cylinders by considering only the main chain and larger than the estimated range of the Young’s modulus of MscL α-helices (0.2 to 12.5 GPa) in Martinac et al.’s recent work (Bavi et al. 2017) with constant-force steered molecular dynamics (SMD).

Examples of several lowest eigenmodes and frequencies of helices and loops. Results of molecular mechanics are compared to those of the finite element simulations. Here the TM1 helix only includes to the segment below Pro43. Zhu et al. (2016), reprinted with permission of Springer
The nonbonded interactions within MscL are treated by adopting a sufficiently detailed yet computationally efficient protocol to replace the oversimplified surface-to-surface contact model used previously (Chen et al. 2008; Tang et al. 2006). To reduce computational cost, the new approach first simplifies the full-atomic structure into a coarse-grained (CG) model consisting of a much smaller number of particles based on the Martini force field v2.1 (Monticelli et al. 2008). This CG process reduces the number of particles from 2166 to 283 for each chain of E.~coli-MscL with side chains included (Fig. 10e). The coarse-grained particles are then mapped to the nodes of the continuum MscL model so that it consists of two types of nodes: ordinary finite element (FE) nodes and the “chemical nodes.” The “chemical nodes” in the continuum MscL model refer to the FE nodes that have the same coordinates to that of the CG particles (Fig. 10d, e). Interactions between the Martini particles are then applied to the corresponding FE nodes in the continuum mechanics simulation. These interactions include the particle-to-particle interactions between different components within the same chain and between components in different chains. For example, within chain-1, one particle on the TM1 may interact with another particle on S3, TM2, S1, or loops of chain-1, and as for different chains, one particle on the TM1 of chain-1 may interact with any particle on chain-2 (from S3 to S1). In the MDeFEM framework, the interactions among atoms within each continuum component (e.g., particles on TM1 of chain-1) are not computed explicitly because the corresponding energy is already included in the elastic representation of the continuum components; this is one reason that the computational cost associated with the continuum framework is substantially lower than all-atom simulations. The nonbonded interactions between CG particles in different continuum components are calculated using pair-wise terms following the standard cutoff schemes commonly used in atomistic simulations. Specifically, we adopt a group of “connector elements” in the mechanical space, each of which characterizes the nonbonded interaction between a pair of interacting CG particles. These connector elements are not actually FEM elements but a special kind of invisible connectors in ABAQUS used to define force-based nonlinear interactions, just like the nonbonded interactions in MD. The connector element behavior is nonlinear elastic including two possible forms:
where ε and σ are the energy and distance parameters, respectively, to characterize the Lennard-Jones interactions. ε0 is the vacuum dielectric constant, εr = 15 is the relative dielectric constant for electrostatic screening in the Martini force field v2.1 (Monticelli et al. 2008), and qi is the partial charge of the i-th particle. l denotes the distance between a pair of interacting particles. We note that with the polarizable MARTINI water model (Yesylevskyy et al. 2010), the relative dielectric constant is substantially smaller (2.5); test calculations are also done with this value, and the results are generally very similar (data not shown) since the behavior of MscL appears to be largely dictated by the nonpolar solvation (see discussions below). For the interactions among the MscL components, all the parameters involved in the Lennard-Jones interaction are obtained from the Martini force field v2.1; all distances are calculated based on the CG particle coordinates that are continuously updated and mapped from the continuum model. Also continuously updated are the connector elements to include newly emerged interactions when two remote particles come closer during the simulation process. The Lennard-Jones interaction cutoff length is set to be 1.0 nm and the electrostatic cutoff length 1.2 nm.
The major advantage of the irregular molecular surface FEM model over the highly simplified cylindrical stick model (Chen et al. 2008; Tang et al. 2006) is that they take into account the molecular nature of MscL, such as its irregular shape. In addition, the mapping between CG particles and corresponding FEM nodes makes it straightforward to define “chemical nodes,” which encodes the key chemical characteristics of the molecule (i.e., charge distribution and solute/solvent interface) that are required in the solvation calculations.
Continuum Modeling of Lipid Bilayers and Interactions Between MscL and Lipid
Motivated by the natural difference in the chemical and physical properties of these regions, the lipid membrane bilayer is modeled as a sandwich panel that consists of three layers (Fig. 12): a soft layer in the middle with a thickness of 2.5 nm and two hard layers in two sides with a thickness of 0.5 nm each (Chen et al. 2008). These values for thickness estimation are based on density map of water and lipids from a MD simulation of the POPE lipid system (Gullingsrud and Schulten 2004), and both the derived head group thickness and the tail group thickness are consistent with general thickness estimations for the POPE lipid membrane. Meanwhile, the work of Andrew M. Powl et al. identified the hydrophobic thickness of E. coli-MscL associated with the lipid membrane as 25 Å (Powl et al. 2005b). Thus, there is a good match between the hydrophobic thickness of helix and bilayer; and hydrophobic mismatch is not considered in the present model.

Schematic of equi-biaxial tension of the lipid membrane model. A zoomed-in view is shown to illustrate the 10-petal lipid hole that encompasses the MscL protein in the CM model. Zhu et al. (2016), reprinted with permission of Springer
For the case of in-plane membrane stretching, which is the major driving force for MscL gating (Tang et al. 2006), a flat square membrane (within the x-y plane) with a size of 400 × 400 Å is employed. The equi-biaxial membrane tension is most likely induced by osmotic pressure: assuming the liposome is spherical with a typical diameter of 1.0 μm, a patch of membrane with the size of 400 × 400 Å corresponds to a center angle of ∼0.08, which suggests that the curvature of the patch is negligible. To embed the channel into the continuum membrane, a cavity (hole) with the shape of a 10-petal flower (Fig. 12) is created in the middle of the membrane with the size and shape of the cavity conform to those of MscL transmembrane helices in the closed state with an equilibrium distance of ∼5.5 Å, as was measured from the trajectories in previous all-atom simulation (Gullingsrud and Schulten 2003).
The three-layer phenomenological model (with a thickness of ∼35 Å (Gullingsrud and Schulten 2003)) for the palmitoyloleoylphosphatidylethanolamine (POPE) lipid bilayer is first assumed to be homogeneous and isotropic, and its mechanical properties are obtained from a previous study (Chen et al. 2008) where the effective elastic properties of the head group layers and the tail group layer were derived from MD simulations (Gullingsrud and Schulten 2004). And these parameters are overall consistent with general simulation or experimental estimations (Binder and Gawrisch 2001; Chacon et al. 2015; Venable et al. 2015), though they may be further optimized through recent developments about the new force decomposition methods (Torres-Sánchez et al. 2015; Vanegas et al. 2014). For isotropic linear elasticity, the constitutive relation (stress-strain relationship) is given by
where σij and εij are the stress and strain tensor components. For isotropic linear elastic materials, the shear moduli \( {G}_{12}={G}_{23}={G}_{13}=\frac{E}{2\left(1+\nu \right)} \); thus, the elastic properties for each layer of the continuum lipid bilayer model are governed by Young’s modulus, E, and Poisson’s ratio, v, listed in Table 2. To take into account the prominent characteristics of the fluidic lipid membrane, i.e., its incapability of sustaining a large shear stress, we model each layer of the lipid membrane as orthotropic and reduce the in-plane shear modulus of the continuum lipid model, G12 (Eq. 3), to a small value (without losing generality, \( \frac{E}{2\left(1+\nu \right)}/1000 \) is adopted), while all other parameters are inherited from the previous isotropic continuum lipid model. Such a change does not affect the area compressibility under equi-biaxial tension, which is used to estimate the lipid membrane’s Young’s modulus and Poisson’s ratio (Chen et al. 2008) because of the decoupling between tension and shear. It is expected that the in-plane fluidity of the lipid bilayer allows a closer interaction between the surface of the 10-petal lipid hole and the MscL molecule, thus facilitating the gating process since the in-plane lipid tension has been regarded as the major driving force that pulls the channel open (Moe and Blount 2005; Tang et al. 2006). Upon applied tension, the behavior of a real membrane may not be strictly elastic or orthotropic as described, and it was found in a molecular dynamics study that MscL inclusion may increase the rigidity of the membrane (Jeon and Voth 2008). For simplicity, these details are ignored in the present work and are not expected to have a significant influence on the structure or behavior of MscL based on our previous studies (Chen et al. 2008; Tang et al. 2008).
The nonbonded interactions between the lipid hole and the MscL molecule are treated as in Chen et al. (2008) where the interactions between helix and lipid are represented by a pair-wise effective pressure-distance relationship in the following form:
where p and d0 are the interaction pressure and equilibrium distance between two surfaces, respectively, and αi is the instantaneous distance between two deformed surfaces for the i-th element. This nonbonded interaction model implicitly includes both electrostatic and van der Waals interactions and has been successfully applied to study the radial elastic properties of multiwalled carbon nanotubes (Chen et al. 2006) and the deformation and buckling of double-walled carbon nanotubes (Pantano et al. 2003) as well as nanoindentation of nanotubes (Cao and Chen 2006). To estimate the helix-lipid interactions, the insertion energy profile of a single helix (TM1, TM2, or S1) is calculated with an implicit membrane model; i.e., the helix is gradually transferred from the implicit membrane to the implicit bulk solution. Determined from molecular mechanics calculations (Chen et al. 2008), shown in Table 3 are the well-depth D and the exponents (m, n) which are fairly transferable to the current model since we are using the same atomic structure of E. coli-MscL.
It is noted the irregular surface of the lipid membrane is not included in this work, and the particle-to-particle lipid-channel interaction force is averaged to the surfaces of them; thus, the heterogeneous binding of the lipid to the channel is lost. Further refinements of the continuum mechanics lipid model and its heterogeneous interaction to the channel could be considered in future work with different levels of sophistication.
Continuum Solvation Modeling
As described above, the new continuum MscL model is meshed in a way so that it consists of two types of nodes: ordinary finite element (FE) nodes and the “chemical nodes” which are also FE nodes but correspond to the CG particles. While all FE nodes contribute to the mechanical deformation of MscL, the “chemical nodes” are subjected to additional force contributions from solvation and inter-component interactions. Since the new continuum representation adopted here retains the information about the irregular shape of the protein and spatial distribution of the amino acid residues, the solvation contribution (electrostatic plus apolar solvation forces) can be readily calculated using a popular continuum solvation model. Shown in Fig. 10e are the CG particles of E. coli-MscL that bear solvation forces in the continuum solvation calculations. Following the mapping of these CG particles to the nodes of the continuum MscL model, the solvation forces are also transferred from the CG particles to the corresponding “chemical nodes” and included in the subsequent continuum mechanics simulations.
The total solvation free energy is usually decomposed into apolar and electrostatic contributions. For the electrostatic component, the nonlinear Poisson-Boltzmann (NLPB) (Davis and Mccammon 1990; Honig and Nicholls 1995) model is used. The van der Waals radius (21/6σ/2) of each CG particle is used to approximately determine the dielectric boundary between MscL and the solvent, and ions in solution have a finite radius of 2.0 Å. The spline-based (Im et al. 1998) molecular surface, which permits stable solvation force calculations, is used with a 0.3 Å spline window. The channel is surrounded by the nonpolar membrane environment, which is represented crudely by a low dielectric slab following the procedure in APBS (Baker et al. 2001). Accordingly, the MscL protein-lipid system has three dielectric regions: the high dielectric solvent exterior (80.0), the intermediate dielectric protein interior (10.0), and the low dielectric interior of the membrane (2.0). The membrane environment is applied except for the hole in which the MscL is located. Test calculations indicate that for MscL, the results are not sensitive to the value of the protein dielectric used.
As tests of the electrostatic solvation protocol, we compare the computed polar solvation free energy of two systems to values obtained based on the standard solvation protocol in APBS using atomistic structures. One system is a short peptide (with 27 residues and a length of ∼4 nm) from APBS, for which the computed polar transferring free energy from bulk water into the membrane is 127.1 and 120.9 KJ/mol for the atomistic protocol and current CG-based protocol, respectively. The second system is the closed state of E. coli-MscL, for which the computed polar transferring free energy with the atomistic and CG models are 61.8 and 70.9 KJ/mol, respectively. These two examples suggest that the Martini/NLPB combination appears to provide a reasonable estimate for the polar solvation effects.
The apolar solvation force calculations follow the very generic framework described in Wagoner and Baker (2006) where the solvent-accessible surface area contribution is supplemented with volume and dispersion integral terms. A 0.3 spline window is used for the spline-based molecular surface, and the coefficients for surface tension and the volume term of the apolar calculations are 0.0042 KJ/mol/Å2 and 0.23 KJ/mol/Å3, respectively. It is noted that the lipid membrane is not present in apolar solvation force calculations, while in fact most of the residues of TM2 and some residues on TM1 are buried in the membrane. Therefore, the apolar solvation forces on the outer particles of the transmembrane helices are not included in the continuum mechanics calculation (the interactions between these particles and lipids are included explicitly as discussed above). Care is taken to identify these particles based on the coordinates and van der Waals radius of each CG particle in the transmembrane helices.
Besides the outer particles of the transmembrane helices buried in lipids, the inner pore constriction at the closed state for the wild-type (WT) MscL is actually not hydrated at the first stage of gating until the pore gets large enough (> 0.45 nm (Beckstein et al. 2001)). In the above calculations, the channel pore is assumed to be already wetted at the beginning, which is probably more close to the gain-of-function (GOF) mutant MscL (e.g., the V23 T GOF mutant (Anishkin et al. 2005)). The hydration of the channel pore depends on the effective pore radius, and since the pore has different radii along the symmetry axis, at the beginning, only part of the pore surface is hydrated and the rest is not. In the closed state of MscL, about half of the TM1 bundle’s inner surface is exposed to solvent and the lower half is not. When considering the hydration process depending on the effective pore radius as a more realistic model for the WT MscL, similar to those outer residues buried in lipids, the apolar solvation forces on the inner hydrophobic constriction region are not included in the continuum mechanics calculations. This is likely a reasonable approximation in the context of continuum modeling, although the hydration of MscL is a complex case that requires thorough MD studies (Beckstein et al. 2001; Beckstein and Sansom 2004).
In the following context, when considering hydrophobic interactions between water and the channel, the effect of gradual hydration is not included first; and the pore radius depended hydration process is considered in the section of “Different Pathways for the GOF (Gain-of-Function) mutant and WT (Wild-Type) MscL.”
During the continuum calculations, the positions of the CG model are constantly updated based on the coordinates of the “chemical nodes” of continuum mechanics MscL model, while the particle type, radius, charge, and interaction parameters are retained. And solvation forces of the constantly updated CG model calculated through the above protocols in turn participate in the FEM calculations. Solvation effects on the lipid membrane are neglected.
A Force-Based CM/CS Co-simulation Protocol
Shown in Fig. 13 is the assembled continuum model of E. coli-MscL under equi-biaxial tension with zoomed-in views near the protein (the 10-petal lipid hole surface and the E. coli-MscL). Four-node tetrahedron finite elements are used to mesh both the helices and the lipid membrane with the commercial package ABAQUS (2011). Each chain of the continuum MscL model consists of 2,685 nodes and 9,699 finite elements. The lipid bilayer incorporates 2,412 nodes and 10,680 elements, with the mesh gradually more refined toward the boundary of the inner hole where it interacts with the protein extensively. As a reference calculation, MscL gating is modeled without including the solvation effects. Through ABAQUS simulation, the structural deformation of the lipid-protein system is explicitly calculated in response to an external load, where the lipid membrane is stretched (Fig. 13) by applying equi-biaxial displacement on its outer boundary (relevant to osmotic pressure).

Force-based ABAQUS-APBS co-simulation protocol: schematic (with a zoomed-in view near the protein) and flowchart representation. Zhu et al. (2016), reprinted with permission of Springer
To explicitly explore the solvation effect on MscL gating pattern, continuum solvation forces (electrostatic or apolar) need to be integrated into the continuum model that solves for lipid-protein system deformation under external force. This requires an iterative procedure that alternates between CM simulation and CS calculations (illustrated in Fig. 13):
-
1.
Set up nonbonded interactions within MscL and those between MscL and lipid membrane.
-
2.
A small equi-biaxial displacement on the lipid’s outer boundary is applied, and a deformed conformation of the system is obtained by finite element analysis using ABAQUS.
-
3.
The positions of all “chemical nodes” of MscL molecule are extracted from the deformed mesh, and then continuum solvation forces are calculated by APBS.
-
4.
At the same time, the connector elements (interactions within MscL) are updated: new connector elements are added for pairs of two “chemical nodes” that are originally remote but come close during the gating process. The forces exerted by the connector element are directly applied to the corresponding nodes in the continuum model without interrupting the integrity of the element community. Since the gating mechanism of MscL is a gradually expanding process, there are only a few of newly applied connector elements (forces).
-
5.
The solvation forces on the chemical nodes and forces from the newly emerged connector elements are included in the FEM simulation, and a new deformed structure is obtained in the next time step, leading to a new iteration.
-
6.
Dozens of iterations (steps) are carried out until the channel is fully opened, i.e., when the channel pore radius of the continuum model reaches the experimentally estimated value (∼19 Å) for the fully opened state (Sukharev et al. 2001b). Note that the solvation contribution includes both electrostatic and apolar components, and their effects are explored separately below. With different salt concentrations, the procedure above can be repeated to validate Sukharev et al.’s experimental observation (Sukharev et al. 1999) that there is no significant change in the gating pattern for salt concentrations between 0.05 and 1.0 M.
For a typical tension simulation of the gating of E. coli-MscL, the computational time is ∼8 h on a Dell workstation with 3.2 GHz CPU and 4 Gb RAM, highlighting the efficiency of the MDeFEM framework compared to atomistic MD simulations, which take from days to months for large membrane protein systems.
Results and Discussion
In this section, we compare the gating pathways of three systems: (1) a pure CM model with isotropic membrane properties (i.e., in absence of membrane fluidity and solvation effects), (2) a CM model with membrane fluidity but lacking solvation effects, and (3) a complete model that incorporates the electrostatic/apolar solvation effect through the ABAQUS-APBS co-simulation protocol developed above. Simulation results are compared to structural models, previous all-atom simulations, as well as available experimental results.
Gating Pathway of MscL Without Membrane Fluidity and Solvation Effects
As a reference model, same as our previous MDeFEM study (Chen et al. 2008; Tang et al. 2008), we first treat each layer of the lipid as an isotropic slab without any explicit solvation effects; this is referred to as the preliminary model below. Shown in Fig. 14 are several snapshots of E. coli-MscL during the tension-driven gating process in comparison to the structural model of Sukharev and Anishkin (2004) at closed, half-, and fully opened states. Not surprisingly, the transmembrane region (TM1 and TM2 helices) exhibits the most striking conformational changes, in which both TM1 and TM2 helices shift radially away from the fivefold symmetry axis. Gating is primarily realized through the nonbonded interactions between transmembrane helices and the lipid membrane, in which the pore enclosed by TM1s is pulled open. The S1 bundle expands in the radial direction following the path of TM1 though it does not open as large as the TM1 bundle. At the current maximum lipid strain (εm=21%), no disassembly of S3 helices is observed; although the top region of the S3 bundle is expanded slightly, the lower ends remain assembled. This observation is in agreement with the revised version of the structural model (Sukharev and Anishkin 2004), as opposed to the earlier one (Sukharev et al. 2001b). Beside radial expansion of the TM helices, visible shrinking in the height of MscL is also observed, which is correlated with the significant titling of the helices and uplifting of the S3 bundle. Similar to previous studies (Tang et al. 2006, 2008), the loops and transmembrane helices are considerably stretched and bent during the gating process to maintain mechanical equilibrium; these features may be verified from experimental studies with sufficient resolution. On a qualitative level, the current MDeFEM results are in good agreement with the structural models (Sukharev and Anishkin 2004), regarding both the orientation and displacement of the helices and loops (Fig. 14). The only exception regards the periplasmic loops that link TM1 and TM2 helices; they remain well packed in our simulations but expand radially in the structural model of Sukharev and Anishkin (2004) along with the tips of the transmembrane helices.

Gating pathway of E. coli-MscL at closed (a), half- (b), and fully opened (c) states. Configurations are illustrated by continuum model in mechanical space and atomic model (VDW) in chemical space with comparison to the structural models (Sukharev and Anishkin 2004). The dashed lines indicate the approximate location of membrane/water interface. Zhu et al. (2016), reprinted with permission of Springer
The conductance of MscL is directly correlated with the size of the gate, and it is commonly believed that the TM1 helices in the core of the transmembrane bundle constitute the most important gate of the channel (Sukharev and Anishkin 2004; Tang et al. 2006), which is pentagon-shaped (insert of Fig. 15) when projected onto the x-y (membrane) plane. To characterize the pore size, we define an effective pore radius (denoted as r) as the radius of a circle that has the same area as the pentagon-shaped TM1 pore (see insert of Fig. 15). To be consistent with previous studies (Chen et al. 2008; Tang et al. 2008), the area of the pentagon-shaped TM1 pore is calculated as that surrounded by the five TM1 helical axes. From the closed state to the fully opened state, the effective radius increases from ∼6.5 Å to ∼19 Å, echoing the structural model (Sukharev and Anishkin 2004). Considering the pore’s irregular inner surface, the actual pore’s radius ranges from <1 Å to ∼14 Å which is consistent with the estimation from fluorescence resonance energy transfer (FRET) experiments (Corry et al. 2010; Wang et al. 2014). For the preliminary model, the percentage of increment of the effective pore radius and the actual pore radius is depicted in Fig. 15 as a function of membrane strain (the inverted triangle curve). Due to the more detailed representation of MscL and the nonbonded interactions, the relationship between the membrane strain and the lipid cavity expansion is much less linear as found in the previous study (the triangle curve (Tang et al. 2008)). As mentioned in the Introduction, the surface-based nonbonded interaction model used in the previous study is oversimplified, and most importantly, it neglects all the nonbonded interactions involving the loops that connect helices. The current work demonstrates that as the membrane strain increases, the pore size of MscL is firstly stabilized at a small value and then experiences a rapid increase leading to pore opening when the membrane strain becomes sufficiently large. This observation is in qualitative agreement with the patch-clamp experiment of Sukharev et al. (1999), in which the tension-activation data was fitted to a Boltzmann form.

Predicted behavior of MscL upon equi-biaxial tension (a) and a representative illustration of the apolar solvation forces (directions only) during MscL gating (b). Evolution of the effective pore radius and approximate actual radius of MscL is shown as a function of membrane strain. The results obtained currently are compared with those of the previous MDeFEM model (Tang et al. 2008). The refined model includes the effect of the lipid’s fluidity, and the full model further includes the apolar solvation effects. Zhu et al. (2016), reprinted with permission of Springer
Gating Pathway of MscL with Fluidic Membrane and Apolar Solvation Effects
As emphasized above, two major limitations of our previous work (Chen et al. 2008; Tang et al. 2008) and the preliminary model concern the lack of membrane fluidity and solvation effects. In the following, we first include the fluidic properties of the membrane, leading to the “refined model.” Next, we further include solvation effects, leading to the “full model.” Since we expect that the interactions between the apolar surface of the MscL pore and the solvent make a dominant contribution to the gating process, in this section we only include effects of apolar solvation; the relative importance of electrostatic and apolar solvation forces will be discussed in the next section.
With the fluidic lipid model and continuum apolar solvation model employed successively, new effective pore radius evolution curves are obtained and shown in Fig. 15 (square curve for the refined model and circle curve for the full model with apolar solvation). The overall trend of the gating pathway remains the same as that of the preliminary model (inverted triangle curve) in that the pore size is firstly stabilized at a small value and then experiences a rapid increase as the lipid strain further increases. Compared to the preliminary model, the most notable difference lies in the lipid membrane strain required for fully opening MscL. With the more realistic (orthotropic) model of the lipid, the membrane strain for full opening of MscL is reduced slightly (∼10%); with the apolar solvation included, the gating strain is further reduced by as much as ∼50%. Figure 15b shows the solvation forces (directions only) on the “hydrophobic chemical nodes (residues)” on TM1s and TM2s in one step of the simulation of MscL gating. Since most of the residues on the transmembrane helices are hydrophobic rather than hydrophilic (Fig. 10b), the interactions between water- and solvent-exposed residues are expected to push the channel outward, providing another driving force (in addition to the membrane tension) for the gating transition of MscL.
Take the hydrophobic residues on the constriction area as an example. In the closed state, these residues are very close, and hydrophobic confinement is thermodynamically favorable since exposure of these hydrophobic residues is energetically unfavorable. But when the pore constriction is opened wide enough (mainly by membrane stretch), water molecules will be “driven” to fill in the constriction space despite the fact that exposure of hydrophobic residues on pore constriction area is still energetically unfavorable. This “driving” factor that compel the water to fill into the opened space is probably the system’s internal pressure, change of which is usually not considered in MD simulations. Thus the “unfavorable” energy required to expose the hydrophobic residues on the pore constriction is compensated by drop of the internal pressure which may be very small on the scope of the whole system but critical for MscL gating. After that, the repulsion interaction between water and these hydrophobic residues is helping channel gating.
It is known that the gating of MscL can be triggered solely by membrane stretching, although the importance of apolar solvation to gating has only been indirectly probed. The release of content through Tb-MscL has been studied by a pioneering coarse-grained MD simulation work (Louhivuori et al. 2010) which, however, did not explicitly elucidate the importance of the hydrophobic effect in MscL gating. A molecular dynamics study on Tb-MscL (Jeon and Voth 2008) observed that the water chain formation across the channel pore took place at the same time as the channel pore radius increased to a certain value, and they suggested that the two processes may provide positive feedback to each other. The results we present here provide direct evidence for this point. Though the importance of hydrophobic interactions in channel gating has been demonstrated previously by hydrophilic mutations or molecular dynamics studies (Anishkin et al. 2005; Jeon and Voth 2008), this work, to the best of our knowledge, represents the first explicit evaluation of the hydrophobic contribution.
Another work that concerned hydration process of MscL gating is conducted by Anishkin et al. (2010) who focused on the hydrophilic interaction between water and buried hydrophilic residues instead of hydrophobic interaction as we present in this work. Based on the analysis of the hydration energy of the pore constriction and the fact that hydrophilic mutations in the pore constriction area make the channel opening more easy, a conclusion is drawn that the process of the glycine (hydrophilic residue) exposure with pore expansion and their favorable hydration create disjoining pressure that assists opening (Anishkin et al. 2010).
Based on Anishkin et al.’s hydration energy analysis and the analysis of the results in our present work, detailed effects of the hydrophilic or hydrophobic residues on channel gating are elucidated as follows. In the process of the exposure of initially buried hydrophilic residues in the pore constriction area or buried in other area, due to their favorable hydration, disjoining pressure will be generated that assists conducting as suggested by Anishkin et al. (2010). But once these hydrophilic residues are exposed, their interaction with water molecules will resist opening of the channel based on our analysis primarily due to the hydrogen bonding between the polar residue and water molecules. On the other hand, the effects of the hydrophobic residues also act in a similar but contrary way. Before exposure of the hydrophobic residues in the hydrophobic constriction area, these hydrophobic residues are closely packed and the interaction within helps the channel keep closed. But once these hydrophobic residues are exposed, their interaction with water molecules will facilitate opening of the channel primarily due to the repulsion interaction between water and nonpolar residues. Collectively, that is, the effects of the hydrophobic or hydrophilic residues are reversed before and after exposure to water. These mechanisms may be further verified by future experimental and simulation studies.
Shown in Fig. 16 are the configurations at half-/fully opened states of the refined and full models in comparison to the structural model of Sukharev and Anishkin (2004). As with the preliminary model, the conformational transitions observed with the refined or full model are also in qualitative agreement with the structural model. Closer inspection shows that opening of the S1 bundle with the full model is much more significant and the pore size surrounded (not enclosed) by S1 helices are also comparable with that of the TM1 pore (Fig. 16). Note that the position of the S1 domain in the opened state may be different from the real case for biological MscL molecule since the S1 domain of the initial MscL structure (Sukharev and Anishkin 2004) we use may not be appropriately positioned (Iscla et al. 2008). The S3 bundle also separates more in the full model especially for the upper ends though its lower ends remain assembled together. Similar to the preliminary model, the loops linking TM1-TM2 remain essentially unseparated, and this result is consistent with that of the SMD study (Gullingsrud and Schulten 2003) in which the periplasmic loop region in all simulations remained stable. Experimentally, it was found that the channel remains mechanosensitive even with the external loops cleaved (Ajouz et al. 2000); thus, the precise functions of these loops remain unclear. The stable behavior of the periplasmic loops during the simulated gating process suggests that they may function as a filter screen to prevent large molecules from entering and occluding the channel while at the same time prolonging channel opening.

MscL gating pathway of the refined model (a) and the full model (b). Results at half- and fully opened states are shown with comparison to the structural model (c) (Sukharev and Anishkin 2004). The dashed lines indicate the approximate location of membrane/water interface. Here, the refined model takes into account the fluidity of the lipid membrane, and the full model further includes the apolar solvation effects. Zhu et al. (2016), reprinted with permission of Springer
Respective Effects of Electrostatic and Apolar Solvation Forces
Previous patch-clamp experiments (Sukharev et al. 1999) showed that there was no significant change in the MscL gating pattern in the range of salt concentration between 0.05 and 1 M. This observation suggests that electrostatic interactions are unlikely to dominate in MscL gating. To further explore this hypothesis quantitatively, we simulate MscL gating at several ion concentrations (0.05 M and 2.0 M) in the framework of continuum solvation.
As shown in Fig. 17, there are minor differences between the effective pore radius evolution curves of the refined model, the full model with electrostatic solvation contributions with ion concentration of 0.05 M or 2.0 M, respectively. Configurations from the full model with electrostatic solvation effects (not shown) are also very close to those from the refined model (ion concentration, 0.00 M) shown in Fig. 16. Compared to the refined model where no solvation contributions are included, the effective pore radius evolution does not change much when only the electrostatic solvation forces are considered. The apolar solvation forces, on the other hand, contribute significantly to the gating process (circle curve in Fig. 17 or Fig. 15) as described in the above section.

Evolution of the effective pore radius of MscL versus membrane strain for different models. The different effects of the apolar and electrostatic solvation forces on the gating of E. coli MscL are obvious. The full model includes the effects of the electrostatic solvation (ES) (with ion concentration C) or apolar solvation (AS) forces. Zhu et al. (2016), reprinted with permission of Springer
Different Pathways for the GOF (Gain-of-Function) mutant and WT (Wild-Type) MscL
The hydrophobic core of the TM1 bundle of WT MscL appears to be dehydrated according to the EPR data and molecular dynamics results (Gullingsrud and Schulten 2003; Perozo et al. 2002a). The energy landscape for WT MscL indicates that the major energy cost for MscL opening is between the closed state and the substate S0.13 (Anishkin et al. 2005) (0.13 means the relative conductance to the fully opened state) and this substate is believed to be already well hydrated (Anishkin et al. 2005). Experiments showed that hydrophilic substitutions in the hydrophobic restriction of TM1s led to a reduction of the gating tension, while a more hydrophobic substitution resulted in a channel that requires a greater tension to open (Anishkin et al. 2005). Based on these data, one could associate the passage of the main energy barrier with the hydration of the largely hydrophobic pore (Anishkin et al. 2005), which is a prerequisite for conduction. The hydrophilic substitution in the hydrophobic restriction of TM1s may disrupt this initial hydrophobic restriction, leading to facile hydration of the pore and reduction of the first free energy barrier.
In the present simulation, the hydration of the channel pore depends on the effective pore radius as described in the “Models and methods” section. The threshold radius for hydration is taken to be 0.45 nm (Beckstein et al. 2001) as indicated in the right bottom inset of Fig. 18. In the closed state of MscL, about half of the TM1’s inner surface is exposed to solvent and the lower half is not. When considering the hydration process here, accordingly, the apolar solvation forces on the inner hydrophobic constriction region are not included in the continuum mechanics calculations.

Effective radius evolution of simulations for the GOF mutant and WT MscL. The GOF mutant MscL has a hydrophilic substitution in its hydrophobic constriction causing full hydration in the initial state, while the initial state of the WT MscL is only half hydrated. The WT channel won’t get fully hydrated until the membrane strain reaches ∼2/3 of the gating strain (indicated by the dashed line) which agrees well with Sukharev et al.’s experimental study (Sukharev et al. 1999). Zhu et al. (2016), reprinted with permission of Springer
Figure 18 shows the effective radius evolution curves from full model simulations (including apolar solvation effects) for the WT and the GOF mutant MscL (hypothetical hydrophilic substitution in the hydrophobic constriction of TM1s). A rightward shift of the effective radius evolution curve is observed for the WT MscL as compared to the GOF mutant. This result is in agreement with experimental observation that an initially better hydrated state (GOF mutant MscL caused by hydrophilic substitution) is easier to open (Anishkin et al. 2005). We note that the TM1 helices are not fully hydrated during the simulation until the membrane strain gets to as large as ∼2/3 of the strain required for full gating (indicated by the dashed line in Fig. 18), suggesting that MscL is nonconductive until two thirds of its gating tension is reached. This result is in agreement with Sukharev et al.’s experimental observation (Sukharev et al. 1999) that the open probability remains zero until the membrane tension reaches ∼10 dyne/m and the gating tension is about 15 dyne/m. Thus, when the membrane strain reaches half of the gating strain for MscL, the mechanosensitive channel of small conductance (MscS) reaches its fully conducting and opened state (Martinac and Kloda 2003), while MscL is not yet conductive, emphasizing MscL’s role of a “safety valve” in prokaryotes as a last-ditch effort for survival (Berrier et al. 1996).
The effective radius evolution of these two full models is compared to that in the steered molecular dynamics (SMD) study (Gullingsrud and Schulten 2003) (rhombus curve in Fig. 18). The maximum membrane strain in SMD is estimated to be ∼5.4%, and we assume that its increment is proportional to the time step used in SMD. Although the channel is far from fully open in the nanosecond SMD simulation, as shown in Fig. 18, the effective pore radius evolution in SMD is in good agreement with that of the continuum full model at small strain.
The final lipid strain (εm) of the full model for the complete opening of MscL is ∼11.76% (gating strain). The strain in the direction normal to the membrane is (Tang et al. 2006)
which leads to a reduction in the membrane thickness of
where \( {\overline{v}}_t=0.5 \) is the equivalent Poisson’s ratio of the orthotropic lipid membrane model and h = 35Å is the initial thickness of the membrane. With the membrane strain of 11.76%, the thickness is reduced from 35 Å to 28.77 Å, which is a 23.5% change. This value is in close agreement with the 20% reduction in the thickness of membrane spanning part of MscL measured by experiment (Perozo et al. 2002a) since the flatness of MscL is directly caused by thinning of the lipid membrane during gating.
Another prominent feature of the full model’s gating pathway is the stepwise nature of the pore radius evolution curve, as shown in Fig. 18 the circle curve or the square curve. The initial survey of MscL in liposome patches (Sukharev et al. 1999) has recorded three short-lived subconducting states where the pore size stabilizes even in the presence of tension, and a later study of Chiang et al. (2004) identified nine subconducting states. In the present study, when the membrane strain is below ∼2/3 of the gating strain, the channel is not fully hydrated and remains nonconducting. Afterward, the channel becomes conductive, and the effective pore radius increases very rapidly with the membrane strain. A closer inspection of the data shows some plateaus (marked with arrows in Fig. 18) of the curve where the pore size stabilizes. Although the current continuum model may not be sufficient enough to capture some delicate molecular phenomena, such as the subconducting states of MscL gating, nevertheless, the results here may imply that the subconducting states may be in part due to the intricate “overall” mechanical interactions of the multiple components of the system, which complement their biophysical functions.
The actual pore radius is about 4.75 Å for the first substate in Fig. 18, and the radius of the inner pore surface in the open state is ∼14 Å, which is consistent with the estimation from fluorescence resonance energy transfer (FRET) experiments (Corry et al. 2010; Wang et al. 2014). Assuming that the conductance of the MscL pore is proportional to its size (area enclosed by the inner surfaces of TM1s) (Steinbacher et al. 2007), the relative conductance in the present study can be estimated as the ratio of the pore area to that of the full open state. For example, the relative conductance for the first plateaus in present work would be (4.75/14)2 = 0.115, which is in good agreement of the experimental estimate of 0.13 for the first subconducting state (Anishkin et al. 2005). Their proposal that the main energy costing substate is well hydrated is also consistent with results shown in Fig. 18.
Analytical Effort
An important goal of this section is to establish a closed-form and simple analytical model (Chen et al. 2008), as an alternative approach that can capture the most essential features of MscL gating that might be broadly applicable to MS channels, such as the different contributions of solvation and membrane stretch to gating. An analytical model that couples MscL to both lipid membrane stretch (outer boundary) and apolar solvation effects (inner boundary) is developed as follows.
For the square lipid membrane (Fig. 19a) with a length of 2 l (400 Å) and a central cavity radius of c, its outer boundary pressure is -plipid, and the interface pressure between the lipid and MscL is -plipid-MscL. The whole lipid membrane is treated as an isotropic plate. Since the in-plane fluidity of the lipid membrane contributes only slightly (less than 10%) to the gating process, this treatment is considered a reasonable approximation. During gating, the deformation of the membrane cavity is mainly transferred to the closest TM2 helices via nonbonded interactions in the radial direction. The nonlinear interaction pressure-distance relationship is analogous to a nonlinear elastic medium between the lipid cavity and the TM2 bundle. After the TM2 helices are pulled open, the TM1/TM2 nonbonded interactions (another effective nonlinear medium) may perturb the MS channel radius, which is enclosed by the five TM1 helices. Therefore, a simple analytical model can be established in which the details of protein structures are ignored and the nonbonded interactions are described by effective elastic media. A schematic of such plane stress effective continuum medium model (ECMM) is given in Fig. 19b with E. coli-MscL as an example. The inner effective annular medium I accounts for the TM1-TM1 interactions in hoop direction and TM1-TM2 interactions in radial direction, and the outer continuum medium II incorporates TM2-TM2 interactions in hoop direction and TM2-lipid interactions in radial direction. The inner radius, interface radius, and outer radius of the ECMM are denoted by a, b, and c, respectively. Here, a is the effective radius of the closed MscL (consistent with the definition in previous sections), which corresponds to the smallest “through” capacity of the TM1 bundle; b is defined similarly for the TM2 bundle; and c is the interface radius between MscL/lipid cavity. From the closed homology structure of E. coli-MscL, a, b, and c are equal to 6.5 Å, 17 Å, and 22 Å, respectively.

Schematic of the linear lipid membrane model (a) and the effective continuum medium model (ECMM) of MscL (b)
The outer boundary of MscL ECMM (Fig. 19b) is coupled to the inner cavity of the lipid model (Fig. 19a), i.e., \( {\sigma}_r^{\mathrm{II}}(c)=-{p}_{\mathrm{lipid}-\mathrm{MscL}},\ \ {u}_r^{\mathrm{II}}(c)={u}_r(c) \). Continuity of radial stress and displacement at the interface between medium I and II (r = b) requires \( {\sigma}_r^{\mathrm{I}}(b)={\sigma}_r^{\mathrm{I}\mathrm{I}}(b),\ \ {u}_r^{\mathrm{I}}(b)={u}_r^{\mathrm{I}\mathrm{I}}(b) \). The inner boundary pressure of medium I is, \( {\sigma}_r^{\mathrm{I}}(a)=-{p}_{\mathrm{water}} \), from apolar solvation effects. Following the theoretical analysis in Chen et al. (2008) but considering the coupling between MscL ECMM and the lipid model and the coupling between MscL ECMM and the solvation contribution, a closed-form solution of the MscL pore radius increment can be obtained as
where the units for pwater and Δa are MPa and Å, respectively. On the right of Eq. (7), the first term is the contribution of apolar solvation effects to MscL gating and the second the contribution of membrane stretch. The two constants before pwater and εm depend on the elastic properties of medium I and II (Chen et al. 2008). The value of pwater depends on the exposed residues of the TM1 bundle and the state of the hydrophobic constriction. At the initial state, pwater = 0. Averagely, based on the continuum solvation forces calculated in the above sections and the exposed pore surface area, pwater ≈ 68 MPa. Accordingly, the final membrane strain to fully open the channel is 11.16%, which is in close agreement with that (11.76%) calculated by the coupled CM/CS MDeFEM approach.
Limitations of the Current Implementation and Future Directions
Despite our tremendous endeavor and that the current model has been able to help us gain some useful perspectives for channel gating, there are considerable limitations for the present coupled continuum mechanical-continuum solvation approach, and the room for future improvement is still large.
One of the major limitations concerns the atomic structure of E. coli-MscL whose S1 domain may need revision (Iscla et al. 2008) based on the revised crystal structure of Tb-MscL (Steinbacher et al. 2007). And the crystal structure of Tb MscL could also be subjected to the above developed continuum-based approach to study the effects of the S1 domain on the gating behavior. Through these improvements, the proposed facilitation effect of the S1 domain on gating due to its close interaction to the membrane (Iscla and Blount 2012; Vanegas and Arroyo 2014) could be verified and more specifically quantified.
Another limitation of the current work is the treatment of the lipid membrane and its surface-based interaction to the channel. The particle-to-particle lipid-channel interaction force is averaged to the surfaces of them; thus, the heterogeneous binding of the lipid to the channel is lost. Interfacial lipid-channel interactions are exceedingly complicated due to hydrophobic mismatch, electrostatic effects, dynamic nature of the lipid environment, hydrogen bonding, variation during conformational transition, asymmetry, localization, etc. (Argudo et al. 2016; Powl et al. 2003, 2005a, 2008; Vanegas and Arroyo 2014). While emerging evidence has shown the importance of the interaction of the membrane with membrane channels for many biological processes, it is still difficult to elucidate these interactions in a detailed and comprehensive manner both experimentally and computationally. These characteristics of lipid-channel interactions impose great challenge for continuum modeling of the lipid membrane and its interaction to the channel. For future work, one may consider developing particle-to-particle-based interactions and geometry refinement of the lipid membrane (especially for the lipid closely around the channel), which could be included in a similar way as how we treat the channel in the current work. And solvation forces can then be applied to the lipid membrane model as well to mimic a hydration environment for the lipid model. Thereafter, this well-established lipid model can be exploited to explore the contribution or mechanism of different lipid-channel interactions for channel gating in future work. For example, how and how much does the “anchor” effect between S1 domain and the lipid membrane affects gating (Iscla and Blount 2012)? How and how much does the electrostatic interaction between the lipid head group and the charged transmembrane helix residues affect channel gating? Is the distortion of the lipid membrane around the channel helping channel gating, or is it just a spontaneous adaptation behavior for the system to reach a low energy state? Will the distortion of the membrane around the channel disappear or diminish during gating? Besides, this new lipid membrane is also much more reasonable for a study of the effect of negative or positive hydrophobic mismatch (Perozo et al. 2002b), although the current lipid model can also catch some of the basic principle that, as an example, for the wild-type MscL full model with apolar solvation effects, increasing the hydrophobic (tail group) layer of the current lipid membrane model by 10% will lead to a 2.8% increase of gating tension for channel opening.
Moreover, some other subtle but potentially important molecular details such as inclusion of side chains in the FEM model or initial turgor pressure in living E. coli cells (Deng et al. 2011) may also play a part in the gating process and need further refinement in future continuum modeling. We hopefully expect that these refinements will elaborately improve the accuracy of the continuum-based model and greatly expand the application scope and universality of this coupled continuum mechanics-continuum solvation approach.
Concluding Remarks
The analysis of MS channel gating provides us with an effective window to explore how mechanical stimuli induce adaptive cellular behaviors through protein structural transitions across different time and length scales. As shown by the results of the present work, their gating patterns highlight the roles of large-scale helical movements, pore hydration, and protein-lipid interactions during ion channel gating transitions. These processes occur on time and length scales that are too large to be studied directly by regular atomistic simulations. In this chapter, we have modified a molecular dynamics-decorated finite element (MDeFEM) method to incorporate key interaction pairs (e.g., Asp62-Arg131 and Phe68-Leu111) into a continuum mechanics model; this allows us to explore the gating pathway of MscS and how specific interactions and structural motifs impact the gating transition. Besides, a novel simulation protocol is developed that effectively integrates continuum solvation contributions (CS) into continuum mechanics (CM) calculations to study the gating pathway of MscL.
A complete gating transition trajectory of MscS from the closed to the open state along with partially open intermediates is obtained, and the open structure is close to the available structural model from crystallographic studies. It is observed that removing either the Asp62-Arg131 salt bridge or the Phe68-Leu111 nonpolar interaction leads to essentially nonconducting structures. The loop connecting TM2 (the second transmembrane helix) and TM3 is found to be essential for force transmission during gating, while the loop connecting TM1 and TM2 does not make any major contribution. Based on the different structural evolutions observed when the TM3 kink is treated as a loop or a helical segment, we propose that the helical propensity of the kink plays a central role in inactivation. Gating transition of MscS under different transmembrane voltages is also explored.
A novel computational framework is further developed by using MscL as a template. The continuum mechanics is closely coupled with the continuous solvent model. The influence of the solvent and the chemical coupling force is obtained by real-time iteration between the mechanical and chemical spaces. Compared to previous continuum mechanics studies, the present model is capable of capturing the most essential features of the gating process in a much more realistic fashion: due mainly to the apolar solvation contribution, the membrane tension for full opening of MscL is reduced substantially to the experimental measured range. A significant fraction (∼2/3) of the gating membrane strain is required to reach the first subconducting state of our model, which is featured with a relative conductance of 0.115 to the fully opened state. These trends agree well with experimental observations.
We expect that many of the mechanical principles discussed here are also likely to play a role in various membrane-mediated biomechanical processes. The successful application of the MDeFEM approach to MscS/MscL suggests similar studies of the growing families of sensory channels (Chen et al. 2015; Clapham 2003; Dhaka et al. 2006; Krishtal 2003; Pruitt et al. 2014) and their modulations by lipids, lipid-soluble factors, temperature, cell volume, and membrane tension. The approach is particularly powerful in cases that involve large length and long time scales, which are usually not easily accessible to standard particle-based simulations.
References
ABAQUS 6.11 User’s Manual. ABAQUS Inc., Providence, RI, (2011)
B. Ajouz, C. Berrier, M. Besnard, B. Martinac, A. Ghazi, J. Biol. Chem. 275, 1015 (2000)
B. Akitake, A. Anishkin, S. Sukharev, J. Gen. Physiol. 125, 143 (2005)
B. Akitake, A. Anishkin, N. Liu, S. Sukharev, Nat. Struct. Mol. Biol. 14, 1141 (2007)
A. Anishkin, C. Kung, Curr. Opin. Neurobiol. 15, 397 (2005)
A. Anishkin, S. Sukharev, Biophys. J. 86, 2883 (2004)
A. Anishkin, S. Sukharev, Channels (Austin) 11, 173 (2017)
A. Anishkin, C.S. Chiang, S. Sukharev, J. Gen. Physiol. 125, 155 (2005)
A. Anishkin, B. Akitake, S. Sukharev, Biophys. J. 94, 1252 (2008a)
A. Anishkin, K. Kamaraju, S. Sukharev, J. Gen. Physiol. 132, 67 (2008b)
A. Anishkin, B. Akitake, K. Kamaraju, C. Chiang, S. Sukharev, J. Phys. Condens. Matter 22, 454120 (2010)
D. Argudo, N.P. Bethel, F.V. Marcoline, M. Grabe, Biochim. Biophys. Acta Biomembr. 1858, 1619 (2016)
N.A. Baker, D. Sept, S. Joseph, M.J. Holst, J.A. McCammon, Proc. Natl. Acad. Sci. U. S. A. 98, 10037 (2001)
R.B. Bass, P. Strop, M. Barclay, D.C. Rees, Science 298, 1582 (2002)
N. Bavi, O. Bavi, M. Vossoughi, R. Naghdabadi, A.P. Hill, B. Martinac, Y. Jamali, Channels (Austin) 11, 209, (2017)
N. Bavi et al., Nat. Commun. 7, 11984 (2016b)
O. Beckstein, M.S. Sansom, Phys. Biol. 1, 42 (2004)
O. Beckstein, P.C. Biggin, M.S.P. Sansom, J. Phys. Chem. B 105, 12902 (2001)
V. Belyy, A. Anishkin, K. Kamaraju, N. Liu, S. Sukharev, Nat. Struct. Mol. Biol. 17, 451 (2010)
C. Berrier, M. Besnard, B. Ajouz, A. Coulombe, A. Ghazi, J Membr. Biol. 151, 175 (1996)
R.B. Best, X. Zhu, J. Shim, P.E. Lopes, J. Mittal, M. Feig, A.D. Mackerell Jr., J. Chem. Theory Comput. 8, 3257 (2012)
M. Betanzos, C.S. Chiang, H.R. Guy, S. Sukharev, Nat. Struct. Biol. 9, 704 (2002)
H. Binder, K. Gawrisch, J. Phys. Chem. B 105, 12378 (2001)
P. Blount, M.J. Schroeder, C. Kung, J. Biol. Chem. 272, 32150 (1997)
I.R. Booth, M.D. Edwards, S. Black, U. Schumann, S. Miller, Nat. Rev. Microbiol. 5, 431 (2007)
K. Borngen, A.R. Battle, N. Moker, S. Morbach, K. Marin, B. Martinac, R. Kramer, Biochim. Biophys. Acta 1798, 2141 (2010)
B.R. Brooks, R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, M. Karplus, J. Comput. Chem. 4, 187 (1983)
G.X. Cao, X. Chen, J. Mater. Res. 21, 1048 (2006)
G. Cao, X. Chen, J. Mater. Res. 21, 1048 (2011)
E. Chacon, P. Tarazona, F. Bresme, J. Chem. Phys. 143, 034706 (2015)
G. Chang, R.H. Spencer, A.T. Lee, M.T. Barclay, D.C. Rees, Science 282, 2220 (1998)
X. Chen, Y. Tang, G. Cao, Proc. Inst. Mech. Eng. Part N 219, 73 (2006)
X. Chen, Q. Cui, Y. Tang, J. Yoo, A. Yethiraj, Biophys. J. 95, 563 (2008)
B. Chen, B. Ji, H. Gao, Annu. Rev. Biophys. 44, 1 (2015)
C.S. Chiang, A. Anishkin, S. Sukharev, Biophys. J. 86, 2846 (2004)
D.E. Clapham, Nature 426, 517 (2003)
B. Corry, A.C. Hurst, P. Pal, T. Nomura, P. Rigby, B. Martinac, J. Gen. Physiol. 136, 483 (2010)
C. Cui, D.O. Smith, J. Adler, J. Membr. Biol. 144, 31 (1995)
M.E. Davis, J.A. Mccammon, Chem. Rev. 90, 509 (1990)
Y. Deng, M. Sun, J.W. Shaevitz, Phys. Rev. Lett. 107, 158101 (2011)
E. Deplazes, M. Louhivuori, D. Jayatilaka, S.J. Marrink, B. Corry, PLoS Comput. Biol. 8, e1002683 (2012)
L. Deseri, P. Pollaci, M. Zingales, K. Dayal, J. Mech. Behav. Biomed. Mater. 58, 11 (2016)
A. Dhaka, V. Viswanath, A. Patapoutian, Annu. Rev. Neurosci. 29, 135 (2006)
R.O. Dror, R.M. Dirks, J.P. Grossman, H. Xu, D.E. Shaw, Annu. Rev. Biophys. 41, 429 (2012)
M.D. Edwards, W. Bartlett, I.R. Booth, Biophys. J. 94, 3003 (2008)
R. Gamini, M. Sotomayor, C. Chipot, K. Schulten, Biophys. J. 101, 80 (2011)
M.A. Geeves, K.C. Holmes, Annu. Rev. Biochem. 68, 687 (1999)
M.A. Geeves, K.C. Holmes, Adv. Protein Chem. 71, 161 (2005)
J. Gullingsrud, K. Schulten, Biophys. J. 85, 2087 (2003)
J. Gullingsrud, K. Schulten, Biophys. J. 86, 3496 (2004)
O.P. Hamill, B. Martinac, Physiol. Rev. 81, 685 (2001)
E.S. Haswell, R. Phillips, D.C. Rees, Structure 19, 1356 (2011)
K. Hayakawa, H. Tatsumi, M. Sokabe, J. Cell Sci. 121, 496 (2008)
P.S. Heckbert, M. Garland, Comp Geom. Theor. Appl. 14, 49 (1999)
B. Honig, A. Nicholls, Science 268, 1144 (1995)
W. Im, D. Beglov, B. Roux, Comput. Phys. Commun. 111, 59 (1998)
W. Im, M. Feig, C.L. Brooks, Biophys. J. 85, 2900 (2003)
D.E. Ingber, FASEB J. 20, 811 (2006)
H.I. Ingolfsson, C.A. Lopez, J.J. Uusitalo, D.H. de Jong, S.M. Gopal, X. Periole, S.J. Marrink, Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 225 (2014)
I. Iscla, P. Blount, Biophys. J. 103, 169 (2012)
I. Iscla, R. Wray, P. Blount, Biophys. J. 95, 2283 (2008)
J. Jeon, G.A. Voth, Biophys. J. 94, 3497 (2008)
K. Kamaraju, V. Belyy, I. Rowe, A. Anishkin, S. Sukharev, J. Gen. Physiol. 138, 49 (2011)
M. Karplus, J. Kuriyan, Proc. Natl. Acad. Sci. U. S. A. 102, 6679 (2005)
J.L. Klepeis, K. Lindorff-Larsen, R.O. Dror, D.E. Shaw, Curr. Opin. Struct. Biol. 19, 120 (2009)
A. Kloda, B. Martinac, EMBO J. 20, 1888 (2001)
P. Koprowski, A. Kubalski, J. Membr. Biol. 164, 253 (1998)
P. Koprowski, W. Grajkowski, E.Y. Isacoff, A. Kubalski, J. Biol. Chem. 286, 877 (2011)
O. Krishtal, Trends Neurosci. 26, 477 (2003)
J.Y. Lai, Y.S. Poon, J.T. Kaiser, D.C. Rees, Protein science: A publication of the protein. Society 22, 502 (2013)
G. Levin, P. Blount, Biophys. J. 86, 2862 (2004)
N. Levina, S. Totemeyer, N.R. Stokes, P. Louis, M.A. Jones, I.R. Booth, EMBO J. 18, 1730 (1999)
M. Louhivuori, H.J. Risselada, E. van der Giessen, S.J. Marrink, Proc. Natl. Acad. Sci. U. S. A. 107, 19856 (2010)
L. Ma, A. Yethiraj, X. Chen, Q. Cui, Biophys. J. 96, 3543 (2009)
H. Machiyama, H. Tatsumi, M. Sokabe, Biophys. J. 97, 1048 (2009)
H.R. Malcolm, Y.Y. Heo, D.E. Elmore, J.A. Maurer, Biophys. J. 101, 345 (2011)
V.S. Markin, F. Sachs, Phys. Biol. 1, 110 (2004)
S.J. Marrink, D.P. Tieleman, Chem. Soc. Rev. 42, 6801 (2013)
S.J. Marrink, H.J. Risselada, S. Yefimov, D.P. Tieleman, A.H. de Vries, J. Phys. Chem. B 111, 7812 (2007)
B. Martinac, J. Cell Sci. 117, 2449 (2004)
B. Martinac, A. Kloda, Prog. Biophys. Mol. Biol. 82, 11 (2003)
B. Martinac, A. Kloda, Comprehensive Biophysics (Elsevier, Amsterdam, 2012), p. 108
B. Martinac, M. Buechner, A.H. Delcour, J. Adler, C. Kung, Proc. Natl. Acad. Sci. U. S. A. 84, 2297 (1987)
B. Martinac et al., Antioxid. Redox Signal. 20, 952 (2014)
M. Masetti, C. Berti, R. Ocello, G.P. Di Martino, M. Recanatini, C. Fiegna, A. Cavalli, J. Chem. Theory Comput. 12, 5681 (2016)
S. Miller, W. Bartlett, S. Chandrasekaran, S. Simpson, M. Edwards, I.R. Booth, EMBO J. 22, 36 (2003)
P. Moe, P. Blount, Biochemistry 44, 12239 (2005)
L. Monticelli, S.K. Kandasamy, X. Periole, R.G. Larson, D.P. Tieleman, S.J. Marrink, J. Chem. Theory Comput. 4, 819 (2008)
T. Nomura, M. Sokabe, K. Yoshimura, Biophys. J. 91, 2874 (2006)
T. Nomura, M. Sokabe, K. Yoshimura, Biophys. J. 94, 1638 (2008)
T. Nomura, M. Sokabe, K. Yoshimura, BioMed Res. Int. 2016, 2401657 (2016)
A. Pantano, M.C. Boyce, D.M. Parks, Phys. Rev. Lett. 91, 145504 (2003)
E. Perozo, D.M. Cortes, P. Sompornpisut, A. Kloda, B. Martinac, Nature 418, 942 (2002a)
E. Perozo, A. Kloda, D.M. Cortes, B. Martinac, Nat. Struct. Biol. 9, 696 (2002b)
E. Petrov, D. Palanivelu, M. Constantine, P.R. Rohde, C.D. Cox, T. Nomura, D.L. Minor Jr., B. Martinac, Biophys. J. 104, 1426 (2013)
R. Phillips, T. Ursell, P. Wiggins, P. Sens, Nature 459, 379 (2009)
C. Pliotas, J.H. Naismith, Curr. Opin. Struct. Biol. 45, 59 (2016)
C. Pliotas et al., Proc. Natl. Acad. Sci. U. S. A. 109, E2675 (2012)
C. Pliotas et al., Nat. Struct. Mol. Biol. 22, 991 (2015)
A.M. Powl, J.M. East, A.G. Lee, Biochemistry 42, 14306 (2003)
A.M. Powl, J.M. East, A.G. Lee, Biochemistry 44, 5873 (2005a)
A.M. Powl, J.N. Wright, J.M. East, A.G. Lee, Biochemistry 44, 5713 (2005b)
A.M. Powl, J.M. East, A.G. Lee, Biochemistry 47, 12175 (2008)
M. Praprotnik, L.D. Site, K. Kremer, Annu. Rev. Phys. Chem. 59, 545 (2008)
B.L. Pruitt, A.R. Dunn, W.I. Weis, W.J. Nelson, PLoS Biol. 12, e1001996 (2014)
I. Rowe, A. Anishkin, K. Kamaraju, K. Yoshimura, S. Sukharev, J. Gen. Physiol. 143, 543 (2014)
H. Sackin, Annu. Rev. Physiol. 57, 333 (1995)
Y. Saimi, B. Martinac, A.H. Delcour, P.V. Minorsky, M.C. Gustin, M.R. Culbertson, J. Adler, C. Kung, Methods Enzymol. 207, 681 (1992)
M.F. Sanner, A.J. Olson, J.C. Spehner, Biopolymers 38, 305 (1996)
M.G. Saunders, G.A. Voth, Curr. Opin. Struct. Biol. 22, 144 (2012)
Y. Sawada, M. Murase, M. Sokabe, Channels (Austin) 6, 317 (2012)
F. Scarpa, S. Adhikari, A.J. Gil, C. Remillat, Nanotechnology 21, 125702 (2010)
U. Schumann, M.D. Edwards, C. Li, I.R. Booth, FEBS Lett. 572, 233 (2004)
Q. Shi, S. Izvekov, G.A. Voth, J. Phys. Chem. B 110, 15045 (2006)
W. Shinoda, R. DeVane, M.L. Klein, Curr. Opin. Struct. Biol. 22, 175 (2012)
C.D. Snow, E.J. Sorin, Y.M. Rhee, V.S. Pande, Annu. Rev. Biophys. Biomol. Struct. 34, 43 (2005)
M. Sotomayor, K. Schulten, Biophys. J. 87, 3050 (2004)
M. Sotomayor, T.A.v.d. Straaten, U. Ravaioli, K. Schulten, Biophys. J. 90, 3496 (2006)
S.A. Spronk, D.E. Elmore, D.A. Dougherty, Biophys. J. 90, 3555 (2006)
S. Steinbacher, R. Bass, P. Strop, D.C. Rees, Mechanosens. Ion Channels Part A 58, 1 (2007)
S. Sukharev, Biophys. J. 83, 290 (2002)
S. Sukharev, A. Anishkin, Trends Neurosci. 27, 345 (2004)
S. Sukharev, D.P. Corey, Sci. Signal. 2004, re4 (2004)
S.I. Sukharev, P. Blount, B. Martinac, C. Kung, Annu. Rev. Physiol. 59, 633 (1997)
S.I. Sukharev, W.J. Sigurdson, C. Kung, F. Sachs, J. Gen. Physiol. 113, 525 (1999)
S. Sukharev, M. Betanzos, C.S. Chiang, H.R. Guy, Nature 409, 720 (2001a)
S. Sukharev, S.R. Durell, H.R. Guy, Biophys. J. 81, 917 (2001b)
H. Sun, D.P. Li, S.R. Chen, W.N. Hittelman, H.L. Pan, J. Pharmacol. Exp. Ther. 331, 851 (2009)
Y. Tang, G. Cao, X. Chen, J. Yoo, A. Yethiraj, Q. Cui, Biophys. J. 91, 1248 (2006)
Y. Tang, J. Yoo, A. Yethiraj, Q. Cui, X. Chen, Biophys. J. 95, 581 (2008)
A. Torres-Sánchez, J.M. Vanegas, M. Arroyo, Phys. Rev. Lett. 114, 258102 (2015)
I.J. Tsai, Z.W. Liu, J. Rayment, C. Norman, A. McKinley, B. Martinac, Eur. Biophys. J. 34, 403 (2005)
K.I. Tserpes, P. Papanikos, Compos. Struct. 91, 131 (2009)
M.S. Turner, P. Sens, Phys. Rev. Lett. 93, 118103 (2004)
D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A.E. Mark, H.J. Berendsen, J. Comput. Chem. 26, 1701 (2005)
J.M. Vanegas, M. Arroyo, PLoS One 9, e113947 (2014)
J.M. Vanegas, A. Torres-Sánchez, M. Arroyo, J. Chem. Theory Comput. 10, 691 (2014)
V. Vasquez, Biophys. J. 104, 1391 (2013)
V. Vasquez, M. Sotomayor, J. Cordero-Morales, K. Schulten, E. Perozo, Science 321, 1210 (2008a)
V. Vasquez, M. Sotomayor, D.M. Cortes, B. Roux, K. Schulten, E. Perozo, J. Mol. Biol. 378, 55 (2008b)
R.M. Venable, F.L. Brown, R.W. Pastor, Chem. Phys. Lipids 192, 60 (2015)
T. Vora, B. Corry, S.H. Chung, Biochim. Biophys. Acta 1758, 730 (2006)
J.A. Wagoner, N.A. Baker, Proc. Natl. Acad. Sci. 103, 8331 (2006)
H.C. Wang, B.P. Thampatty, Biomech. Model. Mechanobiol. 5, 1 (2006)
W. Wang, S.S. Black, M.D. Edwards, S. Miller, E.L. Morrison, W. Bartlett, C. Dong, J.H. Naismith, I.R. Booth, Science 321, 1179 (2008)
Y. Wang, Y. Liu, H.A. Deberg, T. Nomura, M.T. Hoffman, P.R. Rohde, K. Schulten, B. Martinac, P.R. Selvin, elife 3, e01834 (2014)
R. Ward et al., Biophys. J. 106, 834 (2014)
P. Wiggins, R. Phillips, Proc. Natl. Acad. Sci. U. S. A. 101, 4071 (2004)
K.M. Wisdom, S.L. Delp, E. Kuhl, Biomech. Model. Mechanobiol. 14, 195 (2014)
S. Yefimov, E. van der Giessen, P.R. Onck, S.J. Marrink, Biophys. J. 94, 2994 (2008)
S.O. Yesylevskyy, L.V. Schafer, D. Sengupta, S.J. Marrink, PLoS Comput. Biol. 6, e1000810 (2010)
Y. Zeng, A.K. Yip, S.K. Teo, K.H. Chiam, Biomech. Model. Mechanobiol. 11, 49 (2012)
L. Zhu, J. Wu, L. Liu, Y. Liu, Y. Yan, Q. Cui, X. Chen, Biomech. Model. Mechanobiol. 15, 1557 (2016)
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this entry

Cite this entry
Zhu, L., Cui, Q., Liu, Y., Yan, Y., Xiao, H., Chen, X. (2019). Molecular Dynamics-Decorated Finite Element Method (MDeFEM): Application to the Gating Mechanism of Mechanosensitive Channels. In: Voyiadjis, G. (eds) Handbook of Nonlocal Continuum Mechanics for Materials and Structures. Springer, Cham. https://doi.org/10.1007/978-3-319-58729-5_46
Download citation
DOI: https://doi.org/10.1007/978-3-319-58729-5_46
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-58727-1
Online ISBN: 978-3-319-58729-5
eBook Packages: EngineeringReference Module Computer Science and Engineering