
Abstract
Bimetallic nanoparticles have revolutionized nanomaterial-linked advances in science and technology. The key to their success, in many applications, relies largely on the superior chemical and physical properties endowed to them through synergistic effects between the two metallic constituents. Thorough studies on the formation and characterization of bimetallic nanoparticles are crucial to understand the viability of these systems in many technological applications. In this chapter, we present recent advances in the formation and characterization of bimetallic nanoparticles, especially with respect to electrochemical applications including our own efforts targeted in this direction. At first, we will outline the recent progress achieved in developing diverse structure-controllable synthesis strategies for bimetallic nanoparticles and promising characterization techniques that can provide information on atomic distribution, composition, morphology, and nanostructure. Later, emphasis will be focused on composition–activity and structure–catalytic activity relationships of bimetallic nanoparticles for specified electrochemical reactions generally involved in clean energy production. Finally, possibilities for future developments and challenges in bimetallic nanoparticle research will be discussed.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
Nanoscale metallic particles have attracted a great deal of attention as they afford myriad of opportunities in nanotechnology and nano-biotechnological applications. The rush toward the development of materials with smaller dimensions is due to the fact that nanoparticles exhibit unusual chemical and physical properties that are different to those of their bulk analogues as a result of surface and quantum size effects [1, 2]. The surface effect is related to the lower stabilization of atoms thereby forming more unsaturated bonds at the surface than the bulk region, thus making the nanoparticle more reactive. The quantum size effect is involved when the de Broglie wavelength of electron at the Fermi energy is comparable to the size of metallic particle itself and as a result particles behave electronically as zero-dimensional quantum dots relevant to quantum mechanical rules [3, 4]. Today many nanotechnological applications largely rely on the effective use of nanosized effects in fine-tuning the materials properties.
Heterogeneous catalysis is an area where development of nanomaterials is crucial. Utilization of nanomaterials as nanocatalysts and corresponding nanocatalyst-linked advances in catalysis can have a positive economic as well as environmental impact. In particular, deployment of bimetallic nanoparticles for catalyzing electrochemical reactions is gaining momentum as they play a vital role in promising technologies related to both environmental and energy-related applications such as polymer electrolyte membrane fuel cells (PEMFC) and direct methanol fuel cells (DMFC). In order to harness the potentiality of bimetallic electrocatalysts, a thorough understanding of the origin of their enhanced performance is needed. The electrocatalytic activities of nanoparticles employed in fuel cells are generally dependent on numerous factors such as particle size and particle size distribution [5–8], morphology(shape), atomic distribution (intra- and interparticle distribution) [9], composition [10, 11], and in particular surface composition, oxidation state, support, and microstructure [11–21] (Table 1).
In the synthesis of bimetallic nanoparticles, special emphasis has been given to the achievement of a high level of control over the particle’s size, morphology, atomic distribution, and dispersion over supports by means of reproducible, low cost, and scalable synthetic approaches [22, 23]. Further, rigorous characterization techniques which can provide information about the above-described nanoscale properties are required. For example, parameters such as surface structure, atomic distribution, and composition are dominant nanostructural properties that require both control and careful characterization.
Further, success in either particle design or scale-up requires a detailed knowledge of the particle’s formation mechanism and will greatly benefit the development of structure-controllable synthesis pathways for bimetallic nanoparticles. The recent advent of high-intensity tunable sources of X-rays, now available at synchrotron facilities worldwide, has made X-ray absorption spectroscopy (XAS) as a powerful tool to examine the nucleation and growth process during the formation of nanoparticles. We have recently made some significant contributions that highlight the applicability of XAS technique to study the nanoparticle formation mechanism [23–31]. The information obtained from XAS studies on local structural changes during early stages of nanoparticle formation can be utilized to control the size, shape, and surface morphology of the nanoparticles.
Certainly, understanding the formation mechanism and thorough characterization of bimetallic nanoparticles are two aspects that warrant important consideration in nanoscience and nanotechnology research. This chapter provides a comprehensive review of the recent advances in the field of bimetallic nanoparticles research pertaining to the formation and characterization of bimetallic nanoparticles and their corresponding applications toward electrocatalytic reactions. Furthermore, this chapter also examines decisive factors that are presently believed to play significant role in the electrocatalytic reactivity of bimetallic nanoparticles with a specific emphasis on understanding the relationship between the structure, chemical composition, surface composition, atomic distribution, and reactivity of bimetallic nanoparticles especially toward electrocatalytic reactions involved in PEMFCs.
Bimetallic Nanoparticles
Bimetallic nanoparticles constituted as the combination of two different metals show multiple functionalities, enhanced selectivity, catalytic activity, and stability when compared to monometallic nanoparticles [32–36]. During the past few decades, much research has been directed toward the design and synthesis of bimetallic nanoparticles due to their promising activity in catalyzing clean energy conversion reactions. Figure 1 illustrates the recent growth in the number of papers published on the formation and characterization of bimetallic nanoparticles.

Research articles published per year for bimetallic nanoparticles during the period 2000 to April 2013 (Search made through SciFinder Scholar Database)
Platinum-based bimetallic nanoparticles are widely utilized for both anodic and cathodic reactions in fuel cells. Some excellent reviews of the progress made in Pt-based fuel cell catalyst development for stationary and mobile applications have been written from both an academic and industrial perspective [37–41]. Very recently, Debe has critically reviewed the electrocatalyst approaches and challenges for automotive fuel cells [42]. The phenomenal interest in Pt-based bimetallic nanoparticles largely stems from their ability in offering the favorable electronic and geometric structures needed to catalyze small reactant molecules in fuel cells [43–47]. The use of Pt alone as an anode catalyst is detrimental to the performance of either H2 or CH3OH-fed PEMFC. For example, in the case of H2-fed PEMFC, the carbon monoxide, even when present in trace quantities, competes with H2 adsorption and poisons the Pt-active surface [48]. Similarly, in the case of the electrooxidation of CH3OH in DMFC, the CO formed as one of the intermediates covers the Pt-active surface and impedes the catalytic performance for methanol oxidation [49]. The oxygen reduction reaction (ORR) involved in PEMFC is a multi-electron reaction, and depending on the experimental conditions, it is known to take place through two different reaction pathways: the direct four-electron pathway in which O2 is reduced directly to water and the two-electron pathway in which O2 is reduced to hydrogen peroxide, followed by decomposition, or further reduction, to water [50–52]. Platinum has long been the best electrocatalyst for ORR. However, OH species formed from water decomposition on Pt electrodes inhibit oxygen reduction by blocking active surface sites which may lead to cathodic overpotentials. To overcome the difficulties associated with the use of monometallic Pt nanoparticles as electrocatalysts in PEMFC, Pt-based bimetallic nanoparticles are being extensively investigated and deployed for fuel cell applications. Several explanations have been given for the superior performance of Pt-based bimetallic nanoparticles based on structural variations caused by the presence of the second metal and will be discussed in the following sections.
Synergistic, Electronic, and Geometric Effects
Three explanations have been put forward for the higher activity offered by Pt-based bimetallic nanoparticles over Pt-alone nanoparticles: these include (i) bifunctional (or) ligand effect, (ii) electronic effect, and (iii) geometric (ensemble) effect. According to the bifunctional effect, in bimetallic nanoparticles, each metal component could promote different elementary reaction steps. For example, in the case of PtRu bimetallic nanoparticles, the intermediate CO species formed during CH3OH electrooxidation is strongly adsorbed on Pt sites. When Pt is alloyed with Ru, water dissociation occurs on Ru sites to produce Ru–OH groups at (0.2–0.3 V vs. RHE), i.e., at less positive potentials than on pure Pt surface (0.7 V vs. RHE) [53], and the Ru–OH groups react with the neighboring CO adsorbed on Pt to give carbon dioxide through the bifunctional mechanism (Eq. 1) [53–56].
Secondly, the ensemble effect (or geometric effect) largely deals with specific groupings of surface atoms that serve as active sites in the presence of secondary metal component [57]. The third explanation is based on the electronic effect. According to this effect, interactions between two metal components could improve the reactivity. In PtRu bimetallic interactions, the presence of Ru can alter the electronic state of the Pt (variations in Pt d-band vacancies) leading to the weakening of the Pt–CO bond facilitating CO oxidation. The enhancement of ORR kinetics on bimetallic nanoparticles is also explained by electronic effect [58–61]. It was reported that PtNi and Pt3Fe bimetallic systems which contain a quasi-complete Pt surface layer with more, or less, Ni or Fe in the sublayers show strong electronic modifications. The presence of an alloying component-rich second layer increases the d-electron vacancies of the thin Pt layer and improves the chemisorptive properties and corresponding electrocatalytic performances [47, 62]. In our recent X-ray absorption spectroscopic investigations, it was found that the alloying of Co10 and Fe63 with Pt induces variations in unfilled d-states (Fig. 2) which in turn significantly influences the catalyst’s electrochemical performance toward the ORR.

The variations in Pt-unfilled d-states and extent of platinum alloying in bimetallic (a) PtFe/C with various Pt-to-Fe ratios and (b) PtCo/C with various Pt-to-Co ratios (panel (a) and (b) of Fig. 2 were reproduced with permission from ref. [63], © 2010 Royal Society of Chemistry and Ref. [10] © 2009, American Chemical Society), respectively
The variations in Pt-unfilled d-states in Pt-based bimetallic nanoparticles suggest that charge transfer from one element to the other modifies the local electronic properties of pure platinum particles and makes bimetallic nanoparticles more attractive catalyst candidates. It is suggested that charge localization or transfer within a nanoparticle is strongly correlated with its structure, composition, and size. Therefore, it becomes crucial, during synthesis, to have complete size, composition, and structural control over the nanoparticles [64].
Electrochemical Aspects of Bimetallic Nanoparticles
Electrocatalytic reactions play a decisive role in emerging technologies related to environmental and energy-related applications, such as fuel cells. The efficiency and selectivity of electrocatalytic processes can be substantially improved by replacing monometallic with bimetallic nanoparticle-based catalysts. Recently, Gasteiger et al. have studied the electrooxidation of CO, formic acid, and methanol on well-characterized PtRu alloys. The authors claim that a PtRu alloy having a 50:50 surface composition presents the highest catalytic activity for CO oxidation [45, 65–67]. In another interesting work, Lin et al. [68] have made a comparative study of CO electrooxidation on different catalysts using in situ FTIR spectroscopy. From their results, it was found that, compared to Pt, the onset of CO2 formation at PtRu and Ru is shifted by about −0.1 and −0.05 V, respectively. Further, the authors noticed a faster increase of the CO2 band intensity for PtRu than for the other materials. At 0.5 V, the CO2 band intensity reaches already 97 % of its maximum value at the alloy and only 23 % and 5 % of the maximum at Ru and Pt, respectively, indicating the potentiality of bimetallic surfaces in removing the poisoning CO intermediate. From the CO band intensities versus time plots, the authors found that the CO band intensity at platinum does not change, since the onset potential for oxidation on this metal lies above 0.4 V. For the other materials, the authors observed the oxidative removal of CO: the rate being higher at PtRu than at pure Ru. It is noteworthy that the same experiments performed at 0.35 V showed CO2 formation at PtRu after 3 min, while no oxidation was observed at pure Ru even after 10 min; the authors thus concluded that the onset of oxidation is fastest on the alloy (Fig. 3).

Change of band intensity for adsorbed CO on Pt, Ru, and PtRu during CO stripping at a constant potential of 0.4 V; spectra (50 scans) were collected each 15 s. The respective surface was saturated with CO at 0.3 V after which CO was eliminated from the electrolyte by nitrogen bubbling (Reproduced with permission from Ref. [68], © 1999 American Chemical Society)
Park et al. carried out electrochemical infrared reflection absorption spectroscopy (EC-IRAS) for carbon monoxide (CO) adlayers formed by partial CO dosing on various ruthenium-decorated platinum nanoparticles [69]. The observed promotion in CO electrooxidation by the existence of a Ru-island on Pt nanoparticles, of interest to fuel cell catalysts, showed a strong relationship with Ru surface concentrations, consistent with previous studies on single-crystal or polycrystalline bimetallic surfaces. Based on the attenuation of H2-upd peak with an increase in charge in the double-layer region as the amount of Ru increases on platinum, the authors suggested that at more negative potentials, the formation of oxygen-like species takes place on bimetallic surfaces (Gasteiger et al.) [58]. The authors observed that Ru-decorated Pt nanoparticles with a composition of PtRu-48 % lower the CO stripping peak down to 0.3 V when compared to the Pt-black nanoparticles which showed a CO stripping peak at 0.45 V. However, the authors claim that if the surface Ru concentration was increased to 66 %, then the CO oxidation peak shifts positively to be centered at 0.37 V. The results show that the presence of PtRu boundaries in PtRu catalysts plays an important role in lowering the potential of irreversible CO electrooxidation.
Architectures of Bimetallic Nanoparticles
Bimetallic nanoparticles assume various types of architectures based on the conditions employed during the synthesis (Scheme 1). For example, during the simultaneous reduction process, if the standard reduction potentials of two distinct metals used are different, then the two kinds of metals thermodynamically prefer to nucleate and grow separately to generate core–shell-structured particles. By utilizing strong reducing agents which are capable of reducing simultaneously all metal precursors at optimized rates, alloyed structures can be fabricated. In another approach, by properly adjusting the redox potentials of two metals through specific adsorption or coordination by the use of selected surfactants and counterions, two distinct metal ions can be simultaneously reduced to produce alloyed nanoparticles.

Various structures of bimetallic nanoparticles
Alloy-Structured Bimetallic Nanoparticles
Bimetallic nanoparticles are formed as alloy nanoparticles when the two metals are mixed at the atomic level. Co-reduction of two different metal ions with a strong reducing agent is a widely employed method for the generation of alloy-structured bimetallic nanoparticles. Alloying Pt with suitable noble metals such Ru, Rh, Pd, Ir, Os, Ag, and Au or non-noble transition metals such as Cu, Fe, Co, or Ni has been proved to be effective in electrocatalysis of fuel cell reactions, such as the methanol oxidation reaction and the oxygen reduction reaction [35, 70–74]. It was reported that the catalytic activity and long-term stability of Pt-based alloyed structures are dependent both on composition and structure. For example, the stability of fct-FePt NPs is much better compared to fcc-FePt NPs toward ORR activity in acidic environments. Superior intermetallic stacking of Fe and Pt obtained under temperature alloying coupled with well-order structures was proposed as a reason for the enhanced catalytic activities of the fct-FePt versus fcc-FePt [75]. Among the various alloyed Pt-based catalysts studied, the PtRu alloy has been identified to be the most effective for MOR. The degree of alloying in bimetallic PtRu nanoparticles plays a crucial role in determining the CO oxidation and methanol oxidation rates. Recently, Lee et al. demonstrated that CO oxidation and methanol oxidation were enhanced as the PtRu alloying degree became higher [76].
Core–Shell Bimetallic Nanoparticles
Core–shell-structured bimetallic nanoparticles are particularly attractive for electrocatalysis in view of atom economy, which refers to the possibility to substitute the use of noble metals with other less expensive materials. A number of synthesis strategies have already been used for the production of core–shell bimetallic nanoparticles, such as redox-transmetalation [25, 77–79], ethylene glycol-assisted polyol method [80], co-reduction [81], and seed-mediated growth method [82]. In seed-mediated growth, a pair of metallic precursors is selected which differs in reduction potentials. When a reducing agent is introduced into the synthetic system containing metallic precursors, the metal cations with the highest redox potential will be reduced first by forming seeds, while the second metal will reduce and grow on the preformed metal seeds [83]. In recent years, the redox-transmetalation method has been widely used to fabricate core–shell-type nanoparticles [78]. It is an advanced process compared with the conventional reduction methodologies available for the bimetallic nanoparticles. In this method, a first core component is generated by reducing its corresponding metal ion. Later, a metal precursor solution intended for shell component will be added to the solution containing core component. When the shell-forming metal salts come into contact with the metallic core surface, they are reduced by the sacrificial oxidation of surface atoms of the metal core and deposited on the surface of the core by redox-transmetalation (Scheme 2). By properly understanding the redox chemistry, wide varieties of bimetallic nanostructures can be generated through redox-transmetalation. When compared to traditional successive reduction strategies, the redox-transmetalation process has several advantages. For example, the self-nucleation of the added second metal, which usually occurs in conventional successive reduction methods, can be avoided in redox-transmetalation, as it allows spontaneous shell layer deposition on the surface of the core nanoparticle. Various bimetallic core–shell nanoparticles, e.g., PtAu [84], PtRu [12, 85], and Pt–Co [78, 79], fabricated by redox-transmetalation are reported in the literature.

Schematic of a polyol-assisted redox-transmetalation strategy to fabricate Pt-on-Ru nanoparticles
Recently, we described the fabrication of well-defined bimetallic core–shell-structured nanoparticles using a kinetically controlled autocatalytic chemical process [23]. In this work, under kinetically controlled conditions, we carried out autocatalytic deposition of a sacrificial Cu ultrathin layer on a dimensionally stable palladium-metal core, which is later displaced to form an active ultrathin platinum-layered shell by redox-transmetalation (Fig. 4a). As can be seen from panels a and b of Fig. 4, an ultrathin Pt layer was deposited on spherical and cubic Pd nanoparticles, respectively. The striking advantages of the fabricated PdcorePtultrathin-shell nanoparticles are enhanced platinum surface-to-volume ratio and superior catalytic activities.

Schematic of architecture of PdCore–PtUltrathin–shell/C nanoparticles (a), TEM images of different morphologies (b and c), and corresponding Pd@Pt nanocrystals (insets) (Reproduced with permission from ref. [23], © 2011 American Chemical Society)
Adzic et al. [86] and Zhang et al. [87] synthesized Pt monolayers on various core structures in an effort to stabilize Pt and Pt group metals. In their experiments, either carbon-supported Pt or a noble single-crystal metal was used as the core component [88, 89]. Later, a monolayer of another noble metal was deposited on top of the core component to generate core–shell-structured nanocatalysts. These catalytic platinum monolayers on various core structures were tested for ORR activity and stability. By properly tuning the compositions of the core and monolayer components, the authors achieved remarkable ORR activity on monolayer catalysts. For example, in rotating ring-disk (RRDE) experiments in 0.1 M HClO4 supporting electrolyte solutions, the observed Pt mass-specific activities for the ORR of PtML/Pd/C catalyst, with a typical loading of 3.4 μgPt/cm2, were 5–8 times higher than that of Pt/C. Oxygen reduction followed a four-electron reduction pathway O2 to H2O, with the first-charge transfer as the rate-determining step.
Hollow-Structured Bimetallic Nanoparticles
In recent years, there has been a growing interest in the fabrication of hollow-structured bimetallic nanoparticles for catalytic applications because of their unique void structure, increased surface areas, and reduced densities [90–92]. The interior voids that are usually present in hollow-structured bimetallic NPs play a dual role by serving both as a tiny container for encapsulating multifunctional active materials and as a nanoreactor. Many synthesis strategies including hard-templating, soft-templating, and sacrificial templating approaches have been developed for the synthesis of hollow-structured bimetallic nanoparticles. Silica colloids [93–95], selenium colloids [96], polystyrene colloids [97], and Ag nanoparticles [98] are some of the commonly utilized templates for the fabrication of hollow structures. Au–Pt and Au–Pd bimetallic hollow structures have been conveniently prepared by utilizing silica colloidal templates [96]. Zhang et al. emphasized a facial colloidal templating method to monodisperse hollow Ag and Ag/Au submicrometer spheres by using silica colloidal templates [99]. Xia and coworkers have proposed a galvanic replacement reaction for the fabrication of various noble metal-based NPs [90–92, 100]. In this process, a Ag-based sacrificial template reacts with a precursor compound of the desired metal such as Au, Pd, and Pt. It has been demonstrated that by tuning the synthesis conditions and structures of starting Ag nanoparticle templates, different end products can be made [100, 101]. The Kirkendall effect which deals with the creation of “Kirkendall voids” close to the interface of a bimetallic couple has been used as a new fabrication route to designed new hollow nano-objects [102]. Puntes and coworkers [36] demonstrated fabrication of bimetallic Au–Ag hollow nanoparticles by the simultaneous or sequential action of galvanic replacement and the Kirkendall effect using a silver template and HAuCl4 · 4H2O as an oxidizing agent. Li and coworkers demonstrated a one-pot synthesis for bimetallic Pt/Cu [103] and Pd/Sn [104] hollow nanostructures with large surface areas that potentially open up new opportunities for catalytic applications.
Formation of Bimetallic Nanoparticles
Structure-Controllable Synthesis Methodologies
The performance of the fuel cell is dependent on how robust the electrocatalyst is for each electrode. The electrocatalyst must exhibit high activity and durability while being cost-effective. Regardless of being an anode or cathode, the electrocatalyst’s activity is surface atom sensitive only, which means that a high surface area-to-volume ratio must be achieved. Therefore, the ability to manipulate the size and shape of the nanocatalyst is crucial to achieving high activity without losing durability. However, the challenge still lies in fabricating highly active and durable catalysts while also having homogeneity in size and good particle dispersion.
Various methods have been employed for the preparation of bimetallic nanocatalysts, based on colloidal chemistry [105, 106], reverse micelles [107, 108], microwave irradiation, [109] and alcohol reduction [74, 110]: a brief summary of each is given below. No single method is universally superior; thus depending on the situation, the reader is advised to consider the intrinsic advantages and disadvantages of each approach when deciding which method to employ.
Colloidal Chemistry Methods
The colloidal methods, which offer a good capability for structural control, are widely used for the preparation of metal nanoparticles. The starting material, or precursor, is a metallic salt solution mixed with protective agents, which are usually surfactant molecules. The metal precursors are chemically reduced by the addition of reducing agents to form metal nanoparticles. A narrow size distribution can be achieved by the colloidal metal nanoparticles stabilized by stereo-hindrance effects or electrostatic charge.
The major drawback is the presence of protecting ligands (stereo-hindrance) such as NR4 +, PPh3, PVP, PNI, PAAm, and PVA, which may also block the catalytic active sites of the nanoparticles. Therefore, the removal of the protecting shells is necessary and important for realistic applications. The protecting shells can be removed by washing in an appropriate solvent or by decomposition at high temperature in an inert atmosphere. However, these processes result in unpredictable phenomena such as the surface segregation or cluster growth due to sintering effects during decomposition.
It is preferable to use an alternative route to prepare colloidal metal nanoparticles without protecting agents. The synthesis of bimetallic nanoparticles by this approach was shown to be effective; see Christina et al. [111]: they proved that size-selected PtRu bimetallic nanoparticles can be synthesized by reducing Pt4+ and Ru3+ ions in ethylene glycol (EG) without a need for protection agents. The size of nanoparticles can be easily manipulated by varying the pH value of EG solvent. Shape-controlled synthesis of nanocrystals can also be achieved by the colloidal route.
Reverse Micelles Methods
Metal nanoclusters can be prepared by reverse micelle methods with oil, water, and surfactant. The inverse micelle solution is used to solubilize the metal salt and provide a microreactor for the nucleation, growth, and stabilization of the nanometer-sized clusters. For example, the oil component (n-heptane) and surfactant ((2-ethylhexyl) sulfosuccinate (AOT)) are first well-mixed, and an aqueous solution containing metal ions is subsequently added to form well-defined microemulsion phase with the metallic complex in the “water pool.”
An important parameter in characterizing the microemulsion is the water-to-surfactant molar ratio, W0 = [water]/[surfactant], which plays a crucial role in controlling the equilibrium state of phase in a microemulsion system. The microemulsion of the same composition of oil, water, and the surfactant that contained the reducing agent was also prepared. An appropriate amount of reducing agents containing microemulsion was then gradually added to the microemulsion containing metal ions, reducing them to metal nanoparticles. For a binary system, the final composition of the bimetallic nanoparticles can be easily controlled by the molar ratio of the metal precursors [25, 29–31]. Therefore, the materials synthesized from micellar solutions of surfactants, which combine the advantages of both high specific surface area and uniform particle size distribution, are of great interest for applications in catalysis and fuel cells. However, the disadvantage of this method is its cost since the surfactant and oil used are expensive. Additionally, both surfactant and oil have negative environmental impacts, and the removal of the surfactants is also necessary for further catalytic applications.
Microwave Irradiation Methods
Microwaves are electromagnetic waves. Dielectric materials in a microwave field will be heated by the amount tanδ, the dielectric loss tangent, to define the ability of a material to convert electromagnetic energy into heat energy at a given frequency. The major advantage of microwave irradiation is that it can heat a substance uniformly through a glass or plastic reaction container, leading to a more homogeneous nucleation and shorter crystallization time compared to those obtained from conventional heating.
Conductive heating is often used, but microwave dielectric loss heating may be a better synthesis approach in view of its energy efficiency, speed, uniformity, and implementation simplicity; hence, the microwave-assisted method is beneficial to the formation of metal colloids. Liu et al. [112] have prepared the PtRu nanoparticles supported on Vulcan XC-72 carbon and carbon nanotubes by a microwave-assisted polyol process. An ethylene glycol solution containing Pt and Ru salts was heated in a household microwave oven for several seconds. The resulting suspension was filtered, and the residue was washed with acetone and dried at 100 °C overnight in a vacuum oven. It was found that the PtRu nanoparticles (2–6 nm in diameter) were uniformly dispersed on carbon. It was also found that both PtRu/C catalysts exhibited higher electrocatalytic activities for methanol oxidation than a comparative Pt/C catalyst.
Alcohol-Reduction Methods
The alcohol-reduction method has been developed for the preparation of metal colloids for both homogeneous and heterogeneous catalysis. In both cases, well monodispersed metal colloids can be formed and stabilized in aqueous solution with the existence of polymers. The successful application of the alcohol-reduction method for the preparation of supported Pt and PtRu catalysts for fuel cell applications has been reported by Hsing et al. [110].
The prepared metal colloids were stabilized with a surfactant (SB12) without influencing the deposition of the colloids on the carbon support during the reduction process. Hwang et al. [74] have synthesized a nanosized PtRu/C catalyst using a modified alcohol-reduction method by incorporating small amount of Nafion in the preparation step. The addition of Nafion eliminates the use of stabilizers, which are commonly used to prevent cluster agglomeration. Even though the addition of stabilizers prevents agglomeration and coalescence of the metal particles on the supports, their removal prior to the electrochemical measurements requires complex procedures. However, the addition of Nafion in the present investigation serves a dual role. It disperses the catalyst particles on the carbon support and controls the size of the PtRu particles. Additionally, it is found that the Nafion addition into the catalytic layer can enhance the activity of PtRu catalyst for the electrooxidation of methanol by acting as a dispersing agent while increasing the ionic conductivity.
Electrochemical Deposition
Electrochemical deposition can be utilized to deposit a variety of metal nanoparticles, such as Pt and Cu, onto various substrates. The deposition system consists of an electric conductive working electrode, counter electrode, and a reference electrode. A metal salt was dissolved in the electrolyte, by applying suitable potential on the substrate, upon which metal salts can be deposited to form metal nanoparticles.
The 2-D or 3-D growth of metal can be controlled by the selection of applied potentials. 2-D growth can be fabricated by using underpotential deposition (UPD), in which the potential used for depositing the metal is higher than its reversible thermodynamic potential. For 3-D growth, overpotential deposition (OPD) can be used for bulk-like metal film. Adzic et al. [87, 113, 114] has utilized the UPD method to fabrication a variety of core–shell nanocatalysts. For example, they deposited Cu onto the substrate of Pd nanoparticles using an UPD method, in which a thin layer of Cu was formed on the surface the Pd substrate. By using redox-transmetalation, Pt metal can be deposited onto the surface of Pd nanoparticles to fabricate the Pdcore–Ptshell nanocatalyst. Fabrication of nanocatalysts by using electrochemical method may not be as attractive as chemical methods in which the large-scale synthesis of catalyst can be achieved. Fabrication of nanocatalysts using electrochemical methods may not be attractive as chemical methods in which the large-scale synthesis of catalyst can be achieved.
Formation Mechanism Studies
Understanding structural variations during the nucleation and growth process, especially in the early stages of nanoparticle formation, are extremely important to achieve structural as well as size control of the nanoparticles. For metal nanoparticles, X-ray absorption spectroscopy (XAS) has proved to be one of the most suitable methods for investigating structural evolution, and in many cases structural properties of metal particles can be probed in situ during the different preparation steps [20, 115–118]. Other techniques such as X-ray diffraction (XRD) and transmission electron microscopy (TEM) [119] are difficult to employ in situ because the nanoparticle’s structure changes during preparation of the sample or due to the lack of long-range ordering [120]. UV–Vis spectroscopy has also been utilized for probing the nanoparticle formation mechanism [121–125]. By following the shift in the absorbance band and accompanying changes in the intensity in absorbance, information about the formation of nanoparticles can be obtained.
X-ray Absorption Spectroscopy
It has been shown in the literature that the study at the X-ray absorption near-edge spectroscopy (XANES) region (conventionally from below the edge up to ~ 30 – 50 eV) provides information about the oxidation state, fractional d-electron density, and electronic environment of the absorbing atom. Spectra obtained from the region extending from the XANES region to as high as 2 keV above the edge are known as the extended X-ray absorption fine structure (EXAFS) and are primarily due to the scattering of the photoelectron by near-neighbor atoms. The amplitude of the EXAFS function χ(k), where k is the wave vector, is proportional to the number of nearest neighbors, and the change of phase with the wavelength of the photoelectron depends on the distance between the emitter and the backscattering atom. The backscattering strength also depends on the type or atomic number of atoms evolved in the backscattering process. Thus, an analysis of EXAFS data yields structural details about the absorbing atom and its local environment. In recent years, XAS studies have been explored on bimetallic nanoparticles. In our group, we explored XAS methodologies to understand the formation of bimetallic nanoparticles.
Formation mechanism of PtRu/C bimetallic nanoparticles synthesized by a modified Watanabe’s method: We reported the formation mechanism of carbon-supported bimetallic PtRu nanoparticles by performing XAS measurements at each stage of synthesis [27]. The synthesis process involves adjustment of the pH of equimolar aqueous H2PtCl6 and RuCl3 followed by reduction with NaHSO3 to their corresponding intermediate compounds. Later, to each compound, hydrogen peroxide was added, and again the pH was adjusted to 5 using 1 M NaOH. These two solutions were then mixed, and the pH was maintained at 5. Later, an appropriate amount of Vulcan XC-72R carbon was mixed in, and the mixture was heated at 100 °C for 8 h. The resulting colloidal product was then washed with ultrapure water and dried. Hydrogen reduction was performed on the colloidal product at 300 °C for 2 h to achieve carbon-supported PtRu bimetallic NPs.
Based on the XAS parameters, formation mechanism of bimetallic PtRu/C NPs was proposed. From Pt LIII-edge XAS, we observed that, for the beginning compound H2PtCl6, the coordination number of Pt, i.e., N Pt–Cl, is 6.0 showing that Pt4+ ion is surrounded by six chloride ions. Upon addition of NaHSO3, we observed the change in Pt neighbors with sulfur contribution around Pt (N Pt–S, 3.9). From the EXAFS results, we confirmed that the species formed at this stage is in the form of [Pt(SO3)4]6− in which Pt2+ ion is surrounded by four \( {\mathrm{SO}}_3^{2-} \) ionic groups. The addition of H2O2 to these species increases N Pt–O coordination to 5.9 with a bond length of 2.00 Å which is comparable with that of the Pt–OH bond length indicating that Pt2+ ions are oxidized to Pt4+ and surrounded by six hydroxide ions. Ru K-edge XAS analysis reveals that N Ru–Cl coordination is 6.0 for RuCl3, and after the addition of NaHSO3, the contribution from Ru–O coordination with N Ru–O is 3.9 similar to the [Ru(OH)4]2− species, while upon addition of H2O2, the Ru–O coordination with N Ru–O is 6.0 similar to the RuO2 species. Later, when we mixed the RuO2 species with the H2Pt(OH)6 and heated this mixture at 100 °C for about 8 h, it produced Ru and Pt coordination values of around Ru as 1.6 and 1.2, respectively. Similarly, Pt and Ru coordination around Pt is found to be 0.8 and 1.3, respectively. The oxygen contribution around Pt and Ru is found to be 2.8 and 4.1, respectively. After hydrogen reduction, the N Ru–Pt is increased to 1.9, and N PtRu is increased to 1.9 revealing the formation of PtRu bimetallic NPs. From the compilation of XAS data, a schematic of PtRu formation process was proposed as shown in Scheme 3.

Schematic of formation of carbon-supported bimetallic PtRu nanoparticles synthesized by a modified Watanabe’s method (Reproduced with permission from ref. [27], © 2006 American Chemical Society)
XAS results of this study revealed that the mixing of Pt4+ ions with a ligand environment of OH− groups and Ru4+ surrounded by oxygen groups at a reaction temperature of 100 °C for 8 h prior to H2 reduction initiated the reduction reaction as evidenced by the presence of Pt and Ru bimetallic and ionic contributions that enhance the atomic distribution and dispersion in PtRu bimetallic nanoparticles.
Formation of PtRu/C bimetallic nanoparticles in ethylene glycol solutions: Very recently, by using XAS, we deduced the formation process of PtRu nanoparticles in ethylene glycol solutions [26]. The reduction of Pt4+ and Ru3+ metallic ions was performed in ethylene glycol solutions at 160 °C for four time periods, i.e., 0.5, 1, 2, and 4 h. After reflux at each time period, the reaction mixture was cooled to room temperature and was taken for the XAS measurements.
By comparing the FT-EXAFS spectra and fitting results of both Pt LIII-edge and Ru K-edge, the formation mechanism of PtRu nanoparticles in EG solutions was understood (Scheme 4). From the Pt LIII-edge XAS and Ru K-edge XAS, we observed that upon mixing H2PtCl6 with RuCl3 in EG solution, the coordination numbers N Pt–Cl and N Ru–Cl were found to be 5.6 and 6.0, respectively. However, after controlling the pH at 11, the N Pt–Cl are found to be 3.8 with a Pt–Cl distance of 2.311 Å related to anionic \( {\mathrm{PtCl}}_4^{2-} \) ions, respectively. However, at the same step, around Ru, a contribution from OH− appeared (N Ru–O = 4.9) and the contribution from Cl − is largely decreased (N Ru–Cl = 0.5).

Formation of PtRu bimetallic NPs in EG solutions (Reproduced with permission from ref. [26], © 2007 American Chemical Society)
Hence, higher OH− coordination around Ru indicated that the species produced at this stage are in the form of [Ru(OH)6]3−. Later refluxing the mixture containing \( {\mathrm{PtCl}}_4^{2-} \) and [Ru(OH)6]3−ions at 160 °C for 0.5 h produces Pt and Ru coordination around Pt of 6.5 and 2.7, respectively. Similarly, the Ru and Pt coordinations around Ru are 2.8 and 2.7, respectively. No change in coordination numbers and interatomic distances were found after increasing the reflux time, i.e., to 1, 2, and 4 h, indicating good stability of PtRu NPs formed in EG solutions.
Formation of Pd–Au/C bimetallic nanoparticles in AOT reverse micelles: We have recently demonstrated the unique application of XAS as a fundamental characterization tool to help in designing and controlling the architecture of Pd–Au bimetallic nanoparticles within a water-in-oil microemulsion system of water/sodium bis(2-ethylhexyl)sulfosuccinate (AOT)/n-heptane [25]. By properly performing hydrazine reduction and redox-transmetalation reactions sequentially within water-in-oil microemulsions, we have generated Pd–Au bimetallic clusters with various Pd–Au atomic stackings. At first by performing a redox-transmetalation reaction between Pdnuclei NPs and the Au3+ ions, Pdnuclei–Austack-1 NPs were generated. By following hydrazine reduction reaction, Pd2+ ions were reduced on the preformed Pdnuclei–Austack-1 NPs in order to fabricate (Pdnuclei–Austack-1)–Pdsurf NPs (Scheme 5). Both the redox-transmetalation and hydrazine reduction reactions were repeated to manipulate the Pd–Au stacking until we reach (Pdnuclei–Austack-3)–Pdsurf NPs.

Schematic of architecture of Pd–Au bimetallic NPs as investigated by X-ray absorption spectroscopy (Modified and reproduced with the permission from the original figure of ref. [29], © 2007 American Chemical Society)
From the XAS structural parameters, the degree of alloying and extent of atomic distribution were evaluated. In the case of (Pdnuclei–Austack-1)–Pdsurf-stacked Pd–Au bimetallic NPs formed during the second hydrazine reduction reaction, the N Au–Au and N Au–Pd were determined as 8.17 and 1.37, respectively, giving the total coordination number of Au and Pd around Au \( \left(\varSigma {N}_{\mathrm{Au}{-} \mathrm{i}}={N}_{\mathrm{Au}{-} \mathrm{A}\mathrm{u}}+{N}_{\mathrm{Au}{-} \mathrm{P}\mathrm{d}}\right) \) as 9.54. Similarly, the N Pd–Pd and N Pd–Au are determined as 6.37 and 0.87, respectively, providing the total coordination number of Pd and Au around Pd \( \left(\varSigma {N}_{\mathrm{Pd}{-} \mathrm{i}}={N}_{\mathrm{Pd}{-} \mathrm{P}\mathrm{d}}+{N}_{\mathrm{Pd}{-} \mathrm{A}\mathrm{u}}\right) \) as 7.24. From these values, the structural parameters \( {P}_{\mathrm{observed}}\left(={N}_{\mathrm{Au}{-} \mathrm{P}\mathrm{d}}/\varSigma {N}_{\mathrm{Au}{-} \mathrm{i}}\right) \) and \( {R}_{\mathrm{observed}}\left(={N}_{\mathrm{Pd}{-} \mathrm{A}\mathrm{u}}/\varSigma {N}_{\mathrm{Pd}{-} \mathrm{i}}\right) \) are calculated as 0.14 and 0.12, respectively. From the calculated degree of alloying of Au and Pd, i. e., J Au (22.9 %) and J Pd (30.8 %), by our developed methodology, low J Au and J Pd indicated that both Pd and Au atoms are segregated in the cluster. Thus, XAS can be conveniently utilized to characterize the segregation in nanomaterials once we properly extract the coordination number parameters.
Nashner et al. have studied the formation process of carbon-supported PtRu5 bimetallic nanoparticles synthesized from molecular cluster precursors, i.e., PtRu5C(CO)16 [117, 126]. By following in situ EXAFS studies, scanning transmission electron microscopy, the authors have understood the atomic distribution and surface segregation phenomena. Authors have shown that during the nucleation and growth process, while the metallic character increases, the bond between the CO ligands and metals should gradually break. Further, it was demonstrated that Pt exhibits a marked preference for segregation to the particle’s surfaces under an ambient H2 atm. However, in the presence of O2, the authors observed the formation of a metal oxide surface over a metal core which can revert back to the initial metal core structure on exposure to H2.
The examples discussed above indicate that XAS can be conveniently employed to understand the early stage formation of bimetallic nanoparticles. The information obtained from XAS can be utilized to develop structure-controllable synthesis strategies for a wide variety of bimetallic nanoparticles.
UV–Vis Absorbance Spectroscopy
The characteristic surface plasmon absorption of metallic species is a convenient tool for following the formation of nanoparticles and their reaction kinetics. In addition, when following the simultaneous reduction of two metal systems, UV–Vis spectroscopy offers information whether the resultant system is a bimetal, an alloy, or a physical mixture [127, 128]. Torigoe et al. studied the formation of Ag–Pt alloy colloidal nanoparticles with optical absorption spectra [123]. The Ag–Pt alloy colloidal nanoparticles were prepared by step-by-step reduction of Ag2[Pt(C2O4)2] by dropwise addition of NaBH4 in ethylene glycol solution. The authors recorded optical spectra of Ag2[Pt(C2O4)2] at various stages of the reduction (Fig. 5). The authors pointed out that the reduction reaction is composed of at least three stages. In the first stage (a–c), colloidal Ag particles were formed as evidenced by the growth of the absorption band at 407 nm with increasing concentrations of the reductant and lower extent of shift in the λmax (less than 3 nm). In the second stage (d–f), the authors observed a red shift of the plasmon band and an accompanying decrease in the absorbance. The authors opinioned that this is probably due to the hole injection and the accompanying decrease in the density of conduction electron. In the final stage (g), the plasmon band abruptly reestablishes at further shorter wavelengths than the initial position, i.e., at 340 nm, with a long tail toward longer wavelengths. At this stage, the plasmon band has only one peak, with a bandwidth broader than that of the silver colloid observed in the first stage. This observation was taken as evidence for the formation of Ag–Pt alloy colloid.

Optical spectra of Ag2[Pt(C2O4)2] at various reduction stages with NaBH4. (Reproduced with permission from ref. [123], © 1993 American Chemical Society)
In another interesting study, M. A. El-Sayed and coworkers followed the formation of bimetallic gold–silver alloy nanoparticles with UV–Vis spectroscopy [121]. The authors prepared Au–Ag bimetallic nanoparticles by the co-reduction of chloroauric acid (HAuCl4) and silver nitrate (AgNO3) with sodium citrate. From the UV–Vis absorption spectra (Fig. 6), the authors noticed that the plasmon maximum blue-shifts linearly with increasing silver content. Further, as the optical absorption spectra of their solutions shows only one plasmon absorption, the authors concluded that co-reduction of chloroauric acid and silver nitrate with sodium citrate leads to a homogeneous formation of alloy nanoparticles.

UV–Vis absorption spectra of gold and gold–silver alloy nanoparticles with varying gold mole fractions xAu. The inset shows how the absorption maximum of the plasmon band depends on the composition (Reprinted with permission from Ref. [121], © 1999 American Chemical Society)
Zhang et al. followed the evolution of Ag@AgAu metal core–alloy shell bimetallic nanoparticles by UV–Vis spectroscopy [129]. The authors prepared Ag@AgAu metal core–alloy shell bimetallic nanoparticles by a replacement reaction between Ag nanoparticles and HAuCl4. The authors recorded absorption spectra at various stages of nanoparticle formation (Fig. 7). The surface plasmon resonance peak appeared at 409 nm for the initial Ag nanoparticles was slightly red shifted with a decrease in peak intensity. At the same time the authors noticed the appearance of a new peak at 565 nm which began to red shift with reaction time (panel A of Fig. 6). The authors pointed out that the position of the peak at 565 nm was a significant red shift from the SPR of non-alloyed Ag@Au core–shell nanoparticles which normally exhibit a SPR peak at 530 nm. From this observation, the authors surmise that Au atom deposition and Ag atom dissolution were not uniform across the nanoparticle’s surface. By comparing the locations of two peaks with the theoretical calculations derived from plasmon hybridization theory, the intermediate bimetallic nanoparticle was a hybrid of a Ag nanosphere and a hexapod-like branched particle. The authors also noticed that the position of the second peak did not change 4 to 6 min after the HAuCl4 addition, while the first peak underwent a red shift (spectrum 360 s in Fig. 6a). This observation was related to the deposition of Au atoms and alloying with the underlying Ag atoms. The authors observed a blue shift of the second peak 9 min after the addition of HAuCl4 (spectrum 540 s, Fig. 6b) and pointed out that this happened due to the atomic rearrangement by Ostwald ripening. Finally, the red shifting of the first peak and the blue shifting of second peak finally collapsed into a common peak (spectrum 1200s and 6000 s in Fig. 6b), signaling the complete formation of Ag@AgAu bimetallic nanoparticles.

Evolution of the absorption spectra of bimetallic nanoparticles (Reproduced with permission from ref. [129], © 2008 Wiley-VCH)
Characterization of Bimetallic Nanoparticles
Electrochemical energy conversion for technical applications relies largely on the high catalytic reactivity of electrocatalysts. The optimum catalytic activity is strongly dependent on various factors such as atomic distribution of catalytic metal sites in the catalyst matrix [20, 111, 130, 131], as well as the surface structure, composition, and particle size distribution of the nanocatalysts. In order to select the proper electrocatalyst materials for energy applications, characterization methods play an important role in energy-related research. Ideally, the characteristics of the electrocatalyst materials should be used as selection criteria, and they should allow researchers to forecast the corresponding PEMFCs performance. The physical characterization and electrochemical characterization will be introduced and discussed in this section.
Physical Characterization
Several experimental techniques, including microscopy, diffraction, and numerous spectroscopies, have been successfully applied to deduce the structural aspects of bimetallic nanoparticles. Size distribution, morphology, segregation, alloying extent, atomic distribution, degree of alloying, and surface composition are some of the key properties given considerable attention in the characterization of bimetallic alloy or core–shell-structured electrocatalysts.
X-Ray Diffraction (XRD)
XRD has been widely employed to study supported and unsupported electrocatalyst nanoparticles to gather information on structure, crystallinity, lattice spacing, and grain size. In practice, the average particle size of PtRu-based electrocatalysts is calculated from the broadening of the (220) peak using the Scherrer equation [132], as shown in Eq. 2.
where d is the average particle diameter, λkα1 is the wavelength of X-ray radiation, θB is the angle of the (220) peak, and B(2θ) is the width in radians of the diffraction peak at half-height. The determination of average particle diameter using (220) peak broadening in 2θ ~ 67° is particularly reliable for carbon-supported PtRu electrocatalysts, since in this region there are no reflection signals associated with the carbon support. By careful XRD measurements, several authors have followed the changes in the lattice constant caused by alloying in electrocatalysts in order to obtain information about the alloying degree [133–136]. Antolini and coworkers have proposed an equation for determining the alloying degree of a PtRu catalyst which is defined as the Ru atomic fraction (x Ru) through Eq. 3 [137, 138]:
where a 0 is the lattice constant of pure Pt. For unsupported pure Pt, a 0 has the value of 0.39231 nm, whereas for supported pure Pt, a 0 = 0.39155 nm reported for Pt/C catalyst of E-TEK [137]. Radmilović and coworkers also proposed a similar type of relationship for single-phase PtRu bulk alloys: a = 0.39262 – 0.124 x Ru (or a = 0.38013 + 0.1249 x Pt) [132]. Antolini and Cardellini utilized the peak height ratio of the Pt[111] crystal face and the C[0015] reflection of the carbon in order to evaluate the thermal crystallization considered as a crystallinity degree index of PtRu/C nanoparticles [138]. In some cases, XRD has been also used to calculate the surface area of the catalyst if the shape of the catalyst particles is spherical by using Eq. 4:
where S is the surface area (m2 g−1), d is the average particle size (nm), and ρ is the Pt density (21.4 g cm−3). Information on chemical composition can be obtained first by constructing a calibration graph of lattice parameter versus atomic fraction and later by quantifying a composition based on the measured lattice parameter. Gasteiger et al. carried out detailed studies on the correlation of the lattice parameter with the alloy composition in the binary PtRu system [58] and found a linear dependence, according to the Vegard’s law. These studies allowed the authors to establish the relationship between the electrode composition and electrocatalytic activity. However, XRD is a bulk method and reveals information on the bulk structure of the catalyst, and its support and its application to the interpretation of nanosized particles are rather difficult. By using simulation calculations, Debye function analysis (DFA) offers a convenient approach to determine the size distribution and structure of small clusters [139]. In contrast to the analysis restricted to the limited regions such as the integral intensities of single Bragg peaks, the DFA provides detailed structural information in a range of dispersion (approx. > 40 %). In a typical DFA analysis, the measured diffraction curves are fitted by a set of Debye functions for clusters with the “magic” numbers N = 13, 55, 147, … (for cuboctahedra and icosahedra) and N = 54, 181 (for decahedra) and N = 13, 57, 154 (for hexagonal close-packed clusters) [140]. A histogram plotting the mass fraction of specific structural units present within the sample against their average size can then be constructed through the evaluation of the full pattern. By carrying out the systematic numerical simulations using Debye functions, Vogel et al. obtained the intrinsic structure including the average lattice constant and the size distribution of surfactant-stabilized PtRu catalysts and silica-supported PtRu colloids in the as-synthesized state and after several heat treatments under various atmospheres [106].
Transmission Electron Microscopy (TEM)
TEM in which the electrons pass through the sample generally requires the electrocatalyst particles to be dispersed onto an electron-transparent substrate such as a thin carbon film-coated copper microgrid. TEM is particularly useful because of the high contrast between the metal atoms (especially heavy metals) and gives information about the size, size distribution, dispersion, and even the morphology of various shapes of particles. In general, for fuel cell catalysts, microscopic investigation is combined with other spectroscopic and diffraction techniques to obtain a comprehensive understanding on real structure. Radmilovic et al. [132] reported a detailed study of carbon-supported nanoparticles by TEM and XRD. The focus of their work was put on the characterization of a commercially available carbon-supported PtRu (1:1) catalyst in terms of both particle size and completeness of alloy formation. In the authors’ opinion, many of the difficulties of XRD can be addressed by TEM. Especially, the lattice structure can be studied by high-resolution electron microscopy (HRTEM), including the presence of defects such as dislocations, twins, etc. HRTEM presents an interesting tool in catalyst characterization, as it can be used to determine the geometric shape of faceting planes, the presence of surface steps, the surface roughness, as well as the size and distribution of electrocatalyst nanoparticles. HRTEM offers resolution down to the Ångstrom level and enables information to be obtained on the structure (atomic packing) rather than just morphology of the nanoparticles. Zhang and Chan presented TEM images along with the selected area electron diffraction patterns of PtRu nanoparticles synthesized by a two-microemulsion route in which the metal precursors and reducing agent formed two individual microemulsion systems [141]. Based on the presence of only diffractions from the face-centered cubic (fcc) in the electron diffraction pattern of PtRu nanoparticles, authors have concluded the formation of binary PtRu alloy with the fcc structure. Further evidence of the formation of fcc-structured PtRu binary alloy was given through the established linear relationship of the root of the sum of squares of the lattice coordinates versus the radius of the concentric rings with the lattices (111), (200), (220), (311), and (222). The authors have indicated that the calculated lattice cell constant through such a relationship is 3.862 Å which is in between those of platinum and ruthenium and is in agreement with that of a 1:1 PtRu alloy. Once the particle size distributions were obtained through TEM images, the mean particle size d m can be calculated with the following formula (5) [142]:
where n i is the number of particles with diameter d i. It is also possible to estimate the dispersion (ratio of surface atoms to total number of atoms) of the spherically shaped PtRu clusters through the information of cluster composition and particle size distribution.
Scanning Electron Microscopy (SEM)
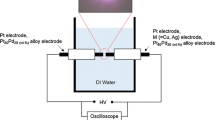
In SEM, the surface of the sample is scanned in a raster pattern with a beam of energetic electrons. The SEM image is produced due to secondary electrons emitted by the sample surface following excitation by the primary electron beam [143]. Bi and Lu utilized SEM to follow the growth process and morphological control of platinum nanostructure, nanofiber, and nanotube junction structures [144]. These Pt nanostructures with various anisotropies were obtained by the galvanic replacement reaction between Ag nanowires and platinum salt solution in the presence of CTAB solution. From SEM observations, the authors have found that the platinum nanostructure growth follows three steps; at first, platinum nanoparticles will grow on the surface of Ag nanowire, and then Ag–Pt composite nanowires will be formed, and finally the Pt nanofibers and nanotubes will grow. From the field-emission SEM images, the authors were able to determine the length and the inner and outer diameters of the platinum nanotubes. Additionally, information related to Pt nanotube uniformity was conveniently obtained from the SEM images. Kawaguchi et al. studied the process of particle growth for Pt, Ru, and binary PtRu supported on carbon as a function of pyrolysis time [145]. The catalyst nanoparticles were prepared by an impregnation-reductive pyrolysis method at various temperatures. The authors have discussed the particle growth behavior from high-resolution SEM images. Although SEM images have lower resolution than TEM, SEM offers better three-dimensional images of the electrocatalysts [146, 147].
Atomic Force Microscopy (AFM)
AFM is a nondestructive method for investigating the microscopic surface topography of nanostructures. In this method, a probe scans the surface of a material with a sharp tip in order to clearly image the features of a sample and senses the small (approximately 1 nN) repulsive force between the probe tip and the surface. Rodríguez-Nieto et al. utilized AFM in order to obtain morphological and microscopic surface characterization of PtRu electrodeposits produced on activated highly ordered pyrolytic graphite (HOPG) substrates [148]. The authors were able to deduce the surface roughness of PtRu electrodeposits in nanometer scale from AFM images. Schmidt et al. employed UHV-AFM to determine the particle size distribution and corresponding dispersion of PtRu nanoparticles from height measurements of the imaged PtRu nanoparticles [149].
X-ray Absorption Spectroscopy (XAS)
In order to understand the structure of either alloy or core–shell-structured bimetallic electrocatalysts, especially in the sub-nm 2–3 nm range, combinations of characterization techniques are required. In general, the X-ray absorption spectrum of a sample can be divided into two regions: the near-edge region (XANES, 0–50 eV above the absorption edge) and the oscillatory part of the spectrum (EXAFS, > 50 eV above the absorption edge). The capability of tuning the X-ray energy to the absorption edge of each participating metal in bimetallic systems makes EXAFS as an attractive technique to elucidate the local structure and provide information on the environment about a particular atom. By analyzing the EXAFS spectrum of each metal in bimetallic nanocatalysts concurrently, valuable structural and chemical information (e.g., interatomic distance, coordination number, oxidation state of chemical species) about the nanostructure can be conveniently obtained, and this information can supplement the microscopy data. From XANES measurements, information about oxidation states, valence states, valence bond vacancies, and adsorption geometries of molecules at the surface can be obtained. However, as the evaluation of the spectra is quite complex, due to multiple scattering processes, EXAFS analysis is generally preferred. Several researchers have successfully used EXAFS to study the bonding, geometry, and surface structure of many electrocatalysts nanoparticles (e.g., PtRu, Pt–Co, and PtMo.), from which the shape, size, and short-range order in atomic distributions occurring within the particles can be reliably obtained [126, 150–154]. Recently, Russell and Rose thoroughly reviewed the capabilities of XAS with respect to the analysis of structural aspects of low-temperature fuel cell catalysts [118].
By collecting the XAS data at the absorption edges corresponding to each element in the bimetallic nanocatalysts under investigation, the extent of intermixing (alloying extent) and homogeneity (atomic distribution) of bimetallic nanocatalysts may be assessed [20, 155]. In general one can assess the alloyed or core–shell structure of nanomaterials simply from the coordination numbers of participating elements in core–shell materials. For a homogeneous bimetallic system of Acore–Bshell cluster in which the core of the cluster is composed of N atoms of A (N A) and the surface is made of N atoms of B (N B), the total coordination number (N AA + N AB) for the A atom will be greater than the total coordination number (N BB + N BA) for the B atom [156, 157]. If bimetallic nanocatalysts possess a random alloyed structure, the ratios of coordination number of A and B coordination, N AA/N AB and N BA/N BB, should be consistent with the ratio of atomic fraction x A/x B.
The alloy or core–shell-type structure of metallic nanostructures from XAS measurements can be better understood if we can obtain knowledge about the atomic distribution and alloying extent of the participating elements. This is particularly important since, among the various structural aspects, it is most important to control the homogeneity, dispersion, and alloying extent as they have profound influence on the surface properties which affect the activity and stability of the bimetallic NPs. Hence, methods to gain more insights into structural aspects are highly needed. Even though alloying is a well-known phenomenon, detailed studies on the quantitative assessment of alloying extent in bimetallic NPs have been lacking so far. In our research group, by deriving the structural parameters from X-ray absorption spectroscopy (XAS) analysis, a general methodology to estimate the alloying extent or atomic distribution in bimetallic NPs has been developed.
By estimating the ratio of the coordination number (CN) of A around B and also the CN of B around A to the total CNs, one can conveniently estimate the alloying extent of A (J A ) and B (J B ) in A–B bimetallic NPs. The parameters that are needed to derive the extent of alloying are represented as P observed, R observed, P random, and R random. The parameter P observed can be defined as a ratio of the scattering atoms “B” CN around absorbing “A” atoms (N A–B ) to the total CN of absorbing atoms \( \left({\displaystyle \sum }{N}_{A{-} i}\right) \), \( \left({P}_{\mathrm{observed}}={N}_{A{-} B}/{\displaystyle \sum }{N}_{A{-} i}\right) \). Similarly, R observed can be defined as a ratio of the scattering atoms “A” CN around absorbing “B” atoms (N B–A ) to the total CNs of absorbing atoms \( \left({\displaystyle \sum }{N}_{B{-} i}\right) \), \( \left({R}_{\mathrm{observed}}={N}_{B{-} A}/{\displaystyle \sum }{N}_{B{-} i}\right) \), whereas P random and R random can be taken as 0.5 for perfect alloyed bimetallic NPs if the atomic ratio of “A” and “B” is 1:1. The J A and J B for 1:1 A–B bimetallic NPs can then be estimated by using Eqs. 6 and 7, respectively.
Based on \( {\displaystyle \sum }{N}_{A{-} i} \), \( {\displaystyle \sum }{N}_{B{-} i} \), J A , and J B , it is possible to develop structural models of NPs. For example, if \( {\displaystyle \sum }{N}_{A{-} i}>{\displaystyle \sum }{N}_{B{-} i} \), the core is rich in “A” atoms and shell is rich in “B” atoms. In this case if both J A < J B , then the bimetallic NPs structure is almost pure Acore–Bshell (case 1, Fig. 8). However, if J B > J A with a coordination parameter relationship \( {\displaystyle \sum }{N}_{A{-} i}>{\displaystyle \sum }{N}_{B{-} i} \), then the bimetallic NPs possess an “A” rich in core–“B” rich in shell structure (case 2, Fig. 8). If \( {\displaystyle \sum }{N}_{A{-} i}={\displaystyle \sum }{N}_{B{-} i} \) and J A and J B ≈ 100 %, then bimetallic NPs adopt an alloy structure (case 3, Fig. 8).

Structural models of bimetallic nanoparticles with core–shell and alloy structures deduced from XAS structural parameters (key: blue, A; pink, B)
It is possible to construct structural models emphasizing the atomic distribution in the bimetallic NPs with knowledge of the \( {\displaystyle \sum }{N_A}_{{-} i} \), \( {\displaystyle \sum }{N}_{B{-} i} \), J A , and J B values derived from XAS. With the help of alloying extent values and structural parameters extracted from EXAFS, it is possible to generate structural models for PtRu/C catalysts. We have calculated the alloying extent of Pt (JPt) and Ru (JRu) for commercial 30 wt% PtRu/C catalysts.
In the case of JM 30 catalyst, the coordination numbers of Pt and Ru atoms around the Pt atom are found to be 5.6 ± 0.3 and 1.4 ± 0.1, respectively, and the total coordination number \( {\displaystyle \sum }{\mathrm{N}}_{\mathrm{Pt}{-} \mathrm{i}} \) is 7.0. The coordination numbers of Ru and Pt atoms around the Ru atom were determined as 3.4 ± 0.2 and 2.2 ± 0.3, respectively, and the total coordination number \( {\displaystyle \sum }{\mathrm{N}}_{\mathrm{Ru}{-} \mathrm{i}} \) is calculated as 5.6. From these values, Pobserved and Robserved are determined as 0.20 and 0.39, respectively, and JPt and JRu values are calculated as 40 and 78 %, respectively. For E-TEK 30 catalyst, we have calculated the coordination numbers of Pt and Ru atoms around the Pt atom as 6.2 ± 0.3 and 0.9 ± 0.1, respectively, and ∑NPt–i as 7.1; the coordination numbers of Ru and Pt atoms around the Ru atom are determined as 3.7 ± 0.2 and 1.2 ± 0.2, respectively, and the \( {\displaystyle \sum }{\mathrm{N}}_{\mathrm{Ru}{-} \mathrm{i}} \) as 4.9. The other two structural parameters Pobserved and Robserved in the case of E-TEK 30 are calculated as 0.13 and 0.24, respectively, and the JPt and JRu values are calculated as 26 and 48 %, respectively. It is clear from the structural coordination parameter values of both the catalysts that \( {\displaystyle \sum }{\mathrm{N}}_{\mathrm{Pt}{-} \mathrm{i}}>{\displaystyle \sum }{\mathrm{N}}_{\mathrm{Ru}{-} \mathrm{i}} \) and JRu > JPt and indicates that the catalysts adopt a Pt rich in core and Ru rich in shell structure.
From the quantitative extent of alloying values, we can see that in both the catalysts, a considerable amount of Ru is segregated on the shell layer, but the extent of segregation of Ru is higher in E-TEK 30 when compared to the JM 30. The increased value of JRu in JM 30 catalyst indicated that most of the Ru is involved in alloying and hence less segregation of Ru in the shell, whereas in the case of E-TEK 30 catalyst, lesser extent of Ru is involved in the alloying and considerable extent of segregation of Ru can be expected in the shell region. The segregation of Ru in the case of E-TEK 30 may in part be responsible for its lower methanol oxidation activity compared to JM 30. Recent infrared measurements on the PtRu alloy particle electrodes indicate that two modes of adsorbed CO vibrations related to both Pt and Ru domains present on the surface support the surface segregation of Ru in commercial catalysts [69]. The XAS results support the Pt-rich core and Ru-rich shell structure for commercial carbon-supported PtRu catalysts. Increase in JPt and JRu values in JM 30 compared to E-TEK 30 indicates that the atomic distribution of Pt and Ru atoms are much facilitated, while an increase in atomic distribution can be taken as a measure for enhanced homogeneity.
Lin et al. investigated the commercial and in-house prepared PtRu catalysts by ex situ EXAFS [158]. From the observed EXAFS parameters, the authors have proposed that the two catalysts differed in the degree of PtRu alloying. The EXAFS data of the in-house prepared catalyst indicated the signatures of PtRu bonds, whereas no significant contributions were found in the commercial catalyst. These observations lead the authors to conclude that the catalyst synthesized in-house was at least partially alloyed, while the commercial catalyst system seemed to contain mixed phases of Pt and RuO x . Greegor and Lytle demonstrated the feasibility of EXAFS technique for measuring the size and shape of small metal particles [159]. This methodology relies on developing a two-region model for various geometrical shapes like spheres, cubes, and disks and calculating the EXAFS average coordination number for first, second, and third coordination spheres as a function of cluster size. Nuzzo and coworkers have also elaborated the modeling nanoparticle size and shape with EXAFS [116]. They considered two model particles characterized by a common average first-shell coordination number (a value of ~ 8 for a 92 atom hemispherical and a 55 atom spherical cubooctahedral cluster). The authors have emphasized that the geometry of these two clusters is significantly different with different sizes, shapes, or lattice symmetries, so each cluster can generate a unique sequence of average coordination numbers in the first few nearest-neighbor shells. Once such a sequence is obtained experimentally, then the corresponding cluster size, shape, and symmetry may be conveniently determined. Several authors have studied the effect of particle size on the XANES region of the XAS spectra for Pt/C catalysts [116, 160, 161]. In their potential-dependent XANES studies on Pt/C catalyst particles with a 3.7 and ≤ 1.0 nm in diameter, Yoshitake et al. have observed that the white line intensity was increased for both particle sizes as the potential increased. In general the white line at the Pt LIII-edge is an absorption threshold resonance, attributed to electronic transitions from 2p3/2 to unoccupied states above the Fermi level and is sensitive to changes in electron occupancy in the valence orbitals of the absorber [162]. Hence, changes in the white line intensity have been directly related to the density of unoccupied d-states and indicate the changes in the oxidation state of the Pt absorber. In general, if the white line intensity decreases, lower the density of unoccupied d-states and lower the oxidation state of Pt. The lower white line intensity observed at negative potentials thus corresponds to a more metallic state. Mansour and coworkers [163] have proposed that by comparing the white line intensities of Pt L3 and Pt L2 edges of a sample with those of a reference metal foil, one can determine the fractional d-electron occupancy (f d) of the absorber atoms in the sample by the following formula (8):
where A3, r and A2, r represent the areas under the white line at the LIII-edge and LII-edge, respectively, of the reference foil spectrum.
with x = 2 or 3 and Ax, s the area under the white line at the Lx edge of the sample spectrum.
f d can then be used to calculate the total number of unoccupied d-states per Pt atom in the samples:
where (h J)t, r t = total for Pt has been shown to be 0.3 [164]. A large (h J)t, s value thus indicates a smaller d-electron density and an increased d-band vacancy as compared to those for bulk Pt.
Further, Mukerjee et al. [165] and Min et al. [166] studied detailed particle size effects in several binary anode and cathode electrocatalysts. Mukerjee et al. calculated the values for Pt/C particles with four different diameters at potentials corresponding to the hydrogen adsorption (0.0 V vs. RHE), the double layer (0.54 vs. RHE), and the oxide formation (0.84 V vs. RHE) regions. With the decreasing particle size, authors have observed an increased widening of the white line. The authors observed an increase in Pt LIII white line intensity at 0.84 V vs. RHE due to the adsorption of OH species at higher potentials, whereas the broadening of the white line at 0 V vs. RHE is related to adsorbed hydrogen. It has been shown that with increasing particle size, the d-band vacancy decreases, indicating that the electronic effects due to adsorption of H and OH are more pronounced for smaller particles. The authors have proposed that as the adsorption strength of H, OH, and CO is increased with decreasing particle size, however, below a certain size, there is a reduction methanol oxidation activity. It has been proposed that the intrinsic activity of Pt-based electrocatalysts for ORR in acidic solutions depends on both the shape, size of the particles, and the adsorption strength of oxygen intermediates [167]. Min and coworkers carried out detailed investigations on the particle size and alloying effects in Pt-based Pt–Co, Pt–Ni, and Pt–Cr catalysts [166]. From the XANES region of the spectra, the authors observed a decrease of the d-band vacancy with increasing particle size which is in agreement with Mukerjee et al. This observation suggests lowered adsorption strength of adsorbed oxygen species, thus facilitating the ORR reaction at larger particles.
Nashner and coworkers reported X-ray absorption spectroscopy characterization of carbon-supported PtRu nanoparticles with exceptionally narrow size and compositional distributions synthesized from the molecular cluster precursor PtRu5C(CO)16 [126]. The authors have deduced structural variations in the PtRu nanoparticles exposed to different gaseous atmospheres such as hydrogen and oxygen on the basis of ex situ EXAFS measurements in combination with transmission electron microscopy. In case of PtRu nanoparticles exposed to H2 atm, the authors found that the ratio of PtRu bonds to Ru–M (N Ru–Pt/N Ru–M) as well as PtRu bonds to Pt–M (N PtRu/N Pt–M) obtained from experimental EXAFS data is always lower than the statistically predicted ratios indicating stronger weighting of the homometallic coordination in nanoparticles. The authors proposed that Pt shows a pronounced preference for segregation to the particle surfaces based on the fact that N PtRu/N Pt–M > N Ru–Pt/N Ru–M. Upon chemisorption of oxygen, the authors found an increase in the disorder in the first-shell metal bond lengths accompanied by the average bonding of two oxygens to both Pt and Ru with bond distances similar to those found in structures with binding oxygen atoms. In another interesting study, Nuzzo and coworkers utilized XAS to follow core–shell inversion in PtRu nanoparticles during hydrogen treatment at various temperatures [117]. Based on XAS structural parameters, the authors found that the incipient PtRu nanoparticles initially formed a disordered structure at 473 K in which Pt is found preferentially at the core of condensing particle. After exposure to high-temperature treatment to 673 K, the nanoparticle undergoes a core–shell inversion leading to the migration of Pt to the equilibrated bimetallic nanoparticle.
Very recently, by utilizing the XAS, we examined heat-induced changes in the surface population of Pt and Ru in PtRu/C catalyst NPs and correlated them with the electrocatalytic activity [168]. In this study, the thermal treatment procedure was designed in such a way that the particle size of initial NPs was not altered upon thermal treatment but can change only the surface population of Pt and Ru allowing us to deduce the structural information independent of particle size effect. We utilized X-ray absorption spectroscopy (XAS) to deduce the structural parameters that can provide information on atomic distribution (or) alloying extent as well as surface population of Pt and Ru in PtRu/C NPs. The PtRu/C catalyst sample obtained from Johnson Matthey was subjected to heat treatment in two environments. At first the as-received catalyst was reduced in 2 % H2 and 98 % Ar gas mixture at 300 °C for 4 h (PtRu/C as-reduced). Later this sample was subjected to either oxygen (PtRu/C-O2-300) or hydrogen thermal treatment (PtRu/C-H2-350). XAS results reveal that when the as-reduced PtRu/C catalyst was exposed to the O2 thermal treatment strategy, considerable amount of Ru was moved to the catalyst surface. In contrast, H2 thermal treatment strategy led to the higher population of Pt on the PtRu/C surface. Characterization of the heat-treated PtRu/C samples by XRD and TEM reveals that there are no significant changes in the particle size of thermal-treated samples when compared to the as-received PtRu/C sample. Both XAS and electrochemical COads stripping voltammetry results suggested that the PtRu/C-H2-350 sample exhibits significant enhancement in reactivity toward CO oxidation as a result of the increased surface population of the Pt when compared to the PtRu/C-O2-300 and PtRu/C as-reduced samples.
X-ray Photoelectron Spectroscopy (XPS)
XPS analyses are commonly used in the characterization of fuel cell electrocatalysts. XPS works based on the photoelectric effect where the energy of X-ray beam is sufficient to overcome the binding energy of electron of the analyte atom, molecule, or solid/surface, allowing the electrons to be ejected. In general, both valence and core electrons can be ejected by X-ray radiation. The composition of materials can be determined using the peal areas under the binding energy curves of the core electron which is characteristic of each element present in the sample [34]. Further, information on chemical bonding can also be conveniently obtained through XPS since the peak shape and binding energy are sensitive to the oxidation and chemical state of the emitting atom [169, 170]. XPS is also particularly useful in determining the particle size effects in fuel cell catalysts. In their studies, Kao et al. observed a 0.3 eV increase of the Pt 4f binding energy when compared to bulk systems [171]. Eberhard et al. also found a continuous increase of the Pt 4f binding energy with decreasing particle size [172]. Zhang and Chan presented XPS analyses of PtRu nanoparticles prepared in water-in-oil reverse microemulsion [173]. The Pt 4f7/2 and Pt 4f5/2 lines that appeared at 71.30 eV and 74.57 eV, respectively, were attributed to metallic Pt0. The peaks that appeared at 72.49 eV and 75.88 eV were assigned to PtII in PtO and Pt(OH)2, respectively. Based on the relative height of the peaks, the authors suggested that metallic Pt0 is the predominant species in the nanoparticles. The authors observed three components with binding energies of 461.32, 463.41, and 465.72 eV in the corresponding Ru 3p3/2 spectrum corresponding to the Ru0 metal, RuIV (e.g., RuO2), and RuVI (in RuO3), respectively. From these results, the authors concluded that the surface of nanoparticles contains metal and Ru oxides species.
Although XPS is suitable for obtaining the chemical state and bonding in electrocatalysts, its application toward the determination of surface composition is limited. For particle size < 3 nm, not less than half of the atoms in the cluster belong to the surface. Hence, the surface-specific XPS with the escaping depth of an electron of about 3 nm becomes a bulk method for small particles [174]. Recently, the work done by Tao et al. demonstrated that using a synchrotron-based X-ray light source with tunable incident X-ray energy allows for investigation of the composition of bimetallic nanoparticles [34, 175].
Auger Electron Spectroscopy (AES)
AES is a powerful tool for determining the composition of the top few layers of a surface. In AES, the sample of interest is irradiated with a high-energy (2 – 10 keV) primary electron beam. This bombardment results in the emission of backscattered, secondary, and Auger electrons that can be detected and analyzed. The backscattered and the secondary electrons are used for imaging purposes similar to that in scanning electron microscopy (SEM). The Auger electrons are emitted at discrete energies that are characteristic of the elements present on the sample surface. When analyzed as a function of energy, the peak positions are used to identify the elements and the chemical states present. AES is widely employed on electrocatalysts to realize the surface structure. Stamenkovic et al. investigated the surface structure of PtM (M = Co, Ni, Fe) polycrystalline alloys with the combination of AES, low energy ion scattering (LEIS) and ultraviolet photoemission spectroscopy (UPS) [176]. By careful modeling of emission from several subsurface layers with dynamic scattering of the outgoing Auger electron, the authors have observed that the Co775/Pt237 AES peak ratio for Pt3Co sample is different on sputtered and annealed surface indicating that the concentration profile of Pt and Co atoms in the surface region may depend on the respective UHV treatment of the alloy sample. From the combination of spectroscopic results, the authors found that in the case of annealed Pt3Co sample at 1,000 K, a completely Pt-skin surface was formed, and due to complete segregation of Pt atoms, the surface composition of Pt is calculated as 100 at.%. In the case of ion-sputtered Pt3Co sample, the surface composition corresponds to the ratio of alloying elements in the bulk, i.e., 75 at.% Pt and 25 % Co. In another interesting study, Tremiliosi-Filho et al. utilized AES as a primary characterization technique to investigate the ruthenium coverage on Pt (111) surface [177]. The authors have calculated the amount of Ru monolayers formed on Pt(111) surface from the intensity of AES peak observed at 274 eV as a result of Pt(111) exposure to RuCl3 solution. With the increasing concentration of RuCl3 solution, the intensity of AES peak was found to be increased, and the corresponding amount of Ru monolayer coverage on Pt(111) was calculated to be higher.
Electrochemical Nuclear Magnetic Resonance (EC-NMR) Spectroscopy
Electrochemical nuclear magnetic resonance (EC-NMR) spectroscopy which combines both solid state NMR and electrochemistry has emerged as a powerful technique to elucidate the electronic properties of metal surfaces [178]. In particular, EC-NMR provides an electronic level description based on the Fermi level local density of states (Ef-LDOS) [179]. The Wieckowski group carried out detailed EC-NMR studies in order to explore the structure of electrocatalyst nanoparticles, to estimate various Ef-LDOS that are involved in construction of the metal-adsorbate bonds, and to examine the diffusional behavior of CO on PtRu bimetallic catalysts as well as interesting relationship between electrochemical current generation and the Ef-LDOS of CO on Pt [180, 181]. 13C and 195Pt are particularly useful nuclei for investigating electrochemical interfaces. Quantitative information about the Ef-LDOS of both 5σ and 2π* orbitals of the chemisorbed CO on Pt nanoparticles can be conveniently achieved by the 13C EC-NMR. This analysis is particularly based on metal and ligand Knight shifts and spin–lattice relaxation rates, and it is important since the variation of these Ef-LDOS reflects the changes in Pt–CO chemisorption bonds. Similarly, from the 195Pt EC-NMR, the 6 s and 5d Ef-LDOS of Pt surfaces can be obtained. The electronic alterations of the metal surfaces can be understood through variations in Ef-LDOS. In elegant work, Wieckowski and coworkers carried out thorough 195Pt EC-NMR measurements on commercial PtRu alloy nanoparticles and 13C EC-NMR for CO chemisorbed on these catalysts [180]. The authors showed 195Pt EC-NMR spectra of a Pt-black sample (with an average particle diameter of 2.8 nm) and PtRu nanoparticles (with an average particle diameter of 2–3 nm). The authors found that for the Pt-black sample, the Pt NMR spectrum extends from 1.095 to 1.14 G/kHz, whereas for PtRu nanoparticles, a much narrower NMR signal extending only from 1.095 to 1.115 G/kHz was found. Based on the observation that the whole spectrum is shifted toward lower Knight shifts, the authors arrived to a conclusion that there are no Pt atoms whose electronic properties resemble those of bulk Pt. The authors have suggested that if the nanoparticle retains a homogeneous composition, then the corresponding NMR spectra of bimetallic catalysts can be expected to show broad, layer-like structures. For example, the 195Pt NMR spectrum of a 2.5 nm-sized PtPd bimetallic catalyst extended from 1.09 to 1.13 G/kHz [182]. In contrast, the presence of relatively narrow peak can be found if there is a surface segregation of one component in bimetallic catalysts. Based on the fact that the 195Pt NMR spectrum of PtRu nanoparticles exhibited a relatively narrow peak centered at about 1.104 G/kHz, the authors have suggested that there is a major surface enrichment of Pt atoms in the PtRu alloy nanoparticles. From the spin–lattice relaxation measurements, the authors have found significant reduction in Ef-LDOS at Pt sites and also on the C-sites of adsorbed CO due to Ru addition, indicating a decrease in the total DOS at EF for the Pt atoms. Thus EC-NMR is useful to evaluate the electronic effects in bimetallic electrocatalysts and for investigating electrochemical interfaces.
Temperature-Programmed Reduction (TPR)
Temperature-programmed reduction (TPR) has been successfully explored to evaluate the surface composition of bimetallic PtRu/C catalysts [183–185]. In their work, the authors suggested that upon calcination, surface platinum (Pts) on reduced Pt crystallites is oxidized to PtsO and PtsO2 as shown in Eqs. 11 and 12:
After calcinations, the state of PtsO x can easily be characterized with the TPR technique by reducing the calcined catalysts by flowing H2 as shown in Eq. 13:
Similarly, the state of Ru also can be characterized by calcinations followed by the reduction. The authors found that oxygen chemisorbed on Ru exhibited a higher reduction temperature (Tr = 300 K) than that chemisorbed on Pt (Tr = 250 K). In case of bimetallic PtRu alloy nanoparticles, the experimental results suggested that Tr varies with PtRu surface composition [183]. For example, a Pt-rich surface displays a lower Tr (~300 K) when compared to Ru-rich surface (Tr = 320 K). Based on these observations, the authors evaluated the surface enrichment in bimetallic PtRu catalysts.
Electrochemical Characterization
Characterizing the electrocatalytic activity of bimetallic nanocatalysts is an important step en route to performance optimization that requires several electrochemical methods. For instance, cyclic voltammetry (potential cycling), linear scan voltammetry (LSV), rotating disk electrode (RDE), rotating ring-disk electrode (RRDE), and CO stripping voltammetry are widely employed to get structural as well as electrocatalytic activity information about electrocatalysts. An electrochemical reaction generally involves sequence of steps: it usually starts with the transport and adsorption of the reactants on the surface of the electrode/catalyst, followed by charge transfer related to either oxidation or reduction on the surface of the electrode/catalyst, and finishing with the transport of product(s) from the surface of the electrode/catalyst.
In this section, we attempt to cover several electrochemical techniques commonly employed in evaluating the electrochemical properties of bimetallic nanoparticles, with emphasis on applied aspects. We have also considered the following aspects: the electrochemical cell and its instrumentation in order to obtain valid results of the characterization and evaluation of the electrocatalyst activity will be presented where appropriate but not described in detail.
Cyclic Voltammetry (CV)
CV is a type of potentiodynamic electrochemical measurement and generally used to study the basic characteristics of the studied system regarding mainly the mechanism of electrode reactions and their kinetic parameters. It offers a rapid determination of redox potentials of electroactive species. CV is characterized by the linear sweep of a working electrode potential from one starting potential to high (or low) limit and back to low (or high) limit. In that case, the current at the working electrode is plotted versus the applied potential to give the cyclic voltammogram trace [186].
In general, the trace or feature of cyclic voltammogram is contributed from the sequence of electrochemical process steps, which can be described as follows. First, when the potential goes higher (or lower) enough to cause the oxidation (or reduction) of an electrochemically active species, we may observe an appearance of anodic (or cathodic) current. An increase in anodic (or cathodic) current as the potential goes higher (or lower) is because of the reaction kinetics becoming faster. Second, the concentrations of the oxidized and the reduced forms of electrochemically active species become equal on the surface of the electrode when the potential reaches the standard reduction potential. The highest anodic (or cathodic) current is obtained when the potential reaches a value at which all the reduced (or oxidized) form of the electrochemically active species at the electrode surface is consumed. At this particular moment, the highest anodic (or cathodic) current is obtained because the mass transport rate of the electrochemically active species reaches a maximum rate, which is driven by the largest concentration gradient between the bulk and surface concentration of the electrochemically active species. Third, when the potential goes higher (or lower) beyond this point, the current starts to fall off because the diffusion thickness increases, resulting in a less steep concentration gradient of the electrochemically active species. Therefore, an anodic (or a cathodic) peak develops. Finally, when the potential reaches the set high (or low) limit, it reverses direction and scans toward the set low limit. During this reverse potential scan, the oxidized (or reduced) form of the electrochemically active species reacts and develops a cathodic (or anodic) peak. The cathodic (anodic) peak is located at a slightly lower (higher) potential than the anodic (cathodic) peak. If the redox couple is reversible, the cathodic and anodic peaks are of equal height (or equal area), and their peak positions do not change with the potential scan rate. In order to characterize electrocatalysts through CV, three-electrode electrochemical cells are widely used. The schematic diagram of a typical three-electrode electrochemical cell is presented in Fig. 9.

The schematic diagram of the structure of a three-electrode cell
Materials with good electronic conductivity can be used as the working electrode, such as glassy carbon, gold, or platinum. The surfaces of these materials are exposed to the electrolyte, and the other parts are usually covered by an inert material, such as Teflon. The reference electrode is an electrode which has a stable and well-known electrode potential, is typically placed in the Luggin capillary, and is constructed in a way that its fine tip can reach the surface of working electrode in order to minimize the influence of the uncompensated electrolyte resistance, without hindering the electrochemical reaction on the surface of working electrode. The most common reference electrodes are Pt/H2/H+ (standard/normal/dynamic hydrogen electrode), Ag/AgCl/Cl− (silver/silver chloride electrode), and Hg/Hg2Cl2/Cl− (calomel electrode). A counter electrode in three-electrode system often has a surface area much larger than that of the working electrode. For example, Pt was chosen as the counter electrode in the DMFCs field when the Pt-based catalysts were under characterization [186–189].
A catalyst is usually made in the form of a slurry before it is applied on the surface of the electrode. A catalyst is first well-mixed with other components: solvent and additives, through ultrasonication. A solvent could be typically water and short chain alcohols such as ethanol. Naturally, the catalyst particles adhere to the surface of working electrode. However, in order to increase the adhesion, a typical additive, e.g., DuPont’s perfluorinated ionomers (Nafion), may be used. Nafion can also be used as a binding material between each catalyst particles, which may lead to higher catalyst utilization. The dispersion of the catalyst particles is also crucial regarding with the optimum catalyst utilization; therefore, it is important to find the optimum loading of the metal: we found that an optimum Pt loading for the working electrode preparation was 0.22 mg-Pt/cm2 [190]. The working electrode was made of unsupported or supported Pt-based catalysts immobilized on glassy carbon (GC) electrode surface (0.1964 cm2). The procedure for electrode fabrication involved three steps: first, the preparation of a clear suspension by sonication of a known amount of catalyst powder dispersed in 0.5 % Nafion; second, placing an aliquot of the suspension (7 μL of 6.2 μg-Pt mL−1 of the catalyst) on the GCE disk; and third, air-drying about 5 min at room temperature and then at 80 °C to yield a uniform thin film of the catalyst [24, 168, 188, 191].
Typically, dilute aqueous acid solutions, such as sulfuric acid (H2SO4) and perchloric acid (HClO4), are usually applied as an electrolyte in the study of DMFCs. However, it is also important to carefully choose a proper electrolyte in the fuel cell system. Although, sulfuric acid has been commonly used in the experiments, their sulfate anions (SO4 2−) can be absorbed on the surface of Pt catalyst. That situation is completely different from perchloric anion (ClO4 −), where this anion will not be absorbed onto the surface of Pt catalyst and will not influence the reaction kinetics.
Linear Sweep Voltammetry (LSV)
As mentioned in the previous section, it is appropriate to continue our discussion on linear scan voltammetry (LSV) experiment performed on a RDE to study the intrinsic kinetics of the catalyst [105, 192–194]. The knowledge of the velocities in radial and vertical direction, which can be obtained via the Navier–Stokes equations, allows the calculations of the mass transport to the disk surface through a diffusion layer with the thickness of δ according to Eq. 14:
where v represents the kinematic viscosity of the electrolyte. As mentioned earlier, according to Eq. 14, the thickness of the diffusion layer of a chosen system strongly depends on the rotation rate. The potential of the working electrode in the LSV experiment is scanned from a potential in which no reaction occurs to a potential that leads to the occurrence of a reaction. Furthermore, when the overpotential is high enough, the reaction rate will be determined by the diffusion of the reactant at a given electrode rotation rate. In this condition, a diffusion-limiting current is achieved and can be described as a function of the diffusion layer thickness as shown in Eq. 15, by assuming that Fick’s law can be applied.
n, A, and F are the number of electrons involved, geometric electrode area, and Faraday constant, respectively. The combination of Eqs. 14 and 15 results in Levich’s equation for the diffusion-limited current on a RDE. The diffusion-limited current is for a given system only determined by the rotation rate with B being the Levich constant: [187, 189]
It is clearly seen in Eq. 16 that a linear relationship exists in the plotting of i d versus \( {\omega}^{{1}/ {2}} \) and goes through the (0, 0) origin. Indeed, we also found a same linear relationship in part of our recent result as can be observed in Fig. 10b. It is of interest to point out that at the onset potential, the current is controlled mainly by reaction kinetics rather than mass transport rate and is expressed in Eq. 17 [186, 189].

Evaluation of intrinsic kinetics activity toward oxygen reduction reaction for commercial carbon-supported Pt3-Cr1 catalyst. (a) LSV recorder at 1 mV/s under various rotation rates in oxygen-saturated 0.5 M sulfuric acid at 25 °C. (b) Plot of i d versus ω½. (c) Koutecky–Levich plots at various potentials
where k η is the rate constant and is a function of overpotential (η). Furthermore, i k is the current that would flow under the kinetic limitation if the mass transfer was efficient enough to keep the concentration at the electrode’s surface equal to the bulk value, regardless of the electrode reaction.
In an entire potential scan range, the overall current (i) in Eq. 18 is described by the Koutecky–Levich equation, as the partition of the overall current in a kinetically determined and a diffusion determined part as shown below[186, 189, 195]:
It is clearly seen in Eq. 18 that \( {1}/ {{\omega}^{{1}/ {2}}C} \) is a constant only when i k is very large. Otherwise, a plot of i versus \( {\omega}^{{1}/ {2}} \) will be curved and tend toward the limit \( i={i}_k \) as \( {\omega}^{{1}/ {2}}\to \infty \) (Fig. 10a). The plot of \( {i}^{-1} \) versus \( {\omega}^{-{1}/ {2}} \) as observed in Figs. 10c and 11b will yield a straight line, where its slope can be used to determine Levich constant of B, from which the number of electrons involved in the reaction can be calculated using known values of solubility and the diffusion of particular reactant in the medium under investigation. The intercept of the plot on the ordinate axis at \( {\omega}^{{1}/ {2}}=0 \) gives the values of \( {i}_k^{-1} \), which can be used for further determination of the kinetic parameter k η according to Eq. 18.

(a) Variation of i with \( {\omega}^{{1}/ {2}} \) in the RDE experiment under a constant E D for the slow reaction on the electrode. (b) Koutecky–Levich plots at potential E 1 , where the rate of electron transfer is sufficiently slow to act as limiting factor, and at E 2 , where electron transfer is rapid
The combination of LSV and RDE methods can be utilized to obtain several intrinsic catalyst parameters, such as kinetic parameters of the Tafel slope, mass activity, and specific activity which together define the catalyst’s activity. Figure 12 shows a sequence of steps for evaluating the activity of carbon-supported Pt and Pt–Co catalysts toward oxygen reduction reaction (ORR) [80].

Evaluation of electroactivity toward oxygen reduction reaction for various carbon-supported Pt and Pt–Co catalysts. (a) LSV recorder at 1 mV/s under rotation rate of 2,500 rpm in oxygen-saturated 0.5 M sulfuric acid at 25 °C. (b) Tafel plots. Currents are per gram of Pt used in the electrode preparation. (c) Mass activity measured at 0.95 VRHE (Reprinted from ref. [191], © 2007 American Association for the Advancement Science)
It is possible to take an advantage of these observed phenomena: one can measure the value of diffusion-limited current by linearizing the observed current of the LSV region 0–0.7 V. Between 0 and 0.7 V, the observed current commonly shows almost a constant value, thus one can easily make a linearization of that observed current. The result of the linearization is known as the diffusion-limited current and is denoted as i d . We believe that the normalized current in Fig. 12a is more appropriate for qualitative comparisons of the catalysts ORR activity. The normalized current is obtained by simply dividing the measured currents with the obtained i d . In the Tafel region (higher than 0.85 V) and the mixed potential region, the ORR activities show a significant difference in their magnitudes. It can be visualized from the inset of Fig. 12a that E-tek Pt–Co/C and sample-2 show a drastically enhanced activity in raising a portion of the curves compared to that for E-tek Pt/C. On the other hand, sample-1 displays comparatively poor activity toward ORR.
Adsorptive CO Stripping Voltammetry (COads-SV)
The second electrochemical method for the estimation of ECSA is CO stripping voltammetry [105, 196–201]. Figure 13a shows two CVs obtained on the bimetallic homemade PtRu catalyst with a CO-adsorbed adlayer. CO can be strongly adsorbed onto the surface of Pt to form a monolayer; however, this irreversible monolayer of CO will be removed quickly and completely by electrochemical oxidation at a high enough potential in the first cycle of CV.

(a) CO stripping voltammograms for the homemade Pt3–Ru1/C catalysts in N2-saturated 0.5 M sulfuric acid electrolyte at a scan rate of 10 mV/s and 25 ± 1 °C. (b) Cu deposition in N2-saturated 0.5 M sulfuric acid and in the presence of 32 mM CuSO4 on commercial carbon-supported Pd (20 wt%)/GC electrode under various deposition potentials for 120 s at 30 ± 1 °C. Scan rate for Pd/C in the absence of CuSO4 solution was 20 mV/s, while the Cu deposition was scanned at 10 mV/s. Note: Edep-1 < Edep-2 < Edep-3
During the first forward step, the peak characteristics of hydrogen/desorption (H ads/des ) were suppressed due to the presence of adsorbed CO. Moreover, the observed peaks at 0.45–0.50 V correspond to oxidative stripping of the adsorbed CO layer. On the second sweep after electrooxidation of CO, the voltammograms return to those observed in the absence of adsorbed CO. The calculated charge under the CO oxidation peak (Q CO) is related to the following oxidation process converts CO to CO2:
CO oxidation to CO2 involves 2 electrons as shown in Eq. 19. Therefore, if one CO molecule bonded with one Pt atom in a linear adsorption configuration (Pt–COad), then the charge required to oxidize a monolayer of CO adsorbed on Pt is equal to 420μC/cm2. ECSA can then be calculated by using Eq. 20:
However, a bridge adsorption configuration (2Pt–COad) may happen if 1 CO molecule occupies 2 Pt atoms. In that case, the charge required to oxidize a monolayer CO adsorbed on Pt is equal to 210μC/cm2. Those two CO adsorption configurations are strongly affected by the applied potential. A linear adsorption may dominate if the CO adsorption occurs at a potential close to 0 V, as shown in a recent study [202].
Pozio et al. [203] have suggested that ECSA calculated by means of CO adsorption seems not to be dependent on the platinum loading. This may due to the stripping mechanism being dominated by the electronic conduction of the oxidation reaction involving the hydroxyl group from water (the electronic conduction is not to be influenced by the Nafion layer resistance in the catalyst). In this study of bimetallic PtRu, the CO stripping method basically gives a separate determination of platinum and second metal (Ru) in Pt-based alloy catalysts to bring a true ECSA, as desorption of CO from “Pt” and “Ru” usually takes place at different potentials [105, 200, 201].
Underpotential Deposition (UPD)
Up to now, we have described two typical electrochemical methods which are used for the estimation of ECSA. However, other studies have reported an alternative electrochemical method based on foreign-applied metal ad-atoms, such as copper (Cu) [196, 198, 204–207], silver (Ag) [208, 209], lead (Pb) [210], and antimony (Sb) [211]. This technique, the so-called underpotential deposition (UPD) of metal is extremely useful for carbon-supported catalysts as the ad-atoms can only be formed on metallic portion in the complex surface. In that case, an advantage of this method over H ads/des methods is that there is no spillover effect. Indeed, in a very recent study, Green and Kucernak [206, 207] showed the viability of underpotential deposition of copper (Cu-UPD) to characterize supported and unsupported Pt, Ru, and PtRu catalysts. In their particular study of Pt, Ru, and PtRu catalysts, the choice of Cu ad-atoms as a probe was motivated by the close atomic radii of Cu (0.128 nm), Ru (0.134 nm), and Pt (0.138 nm) and a suitable potential region of Cu desorption at relatively low potential. This can introduce a correction for double-layer charging and oxygen adsorption. Furthermore, they also showed that it is also possible to determine the surface composition of PtRu catalyst using a Cu-UPD method due to the difference in adsorption energies for Cu on either Pt or Ru. The UPD studies have been performed using a variety of metals that include Cu, Pb, Sn, and Fe deposited on particular Pt catalyst [212].
The phenomenon of UPD itself refers to the deposition of metals on foreign metal substrates at the potentials more positive than that predicted by the Nernst equation for bulk deposition. This implies that the depositing ad-atoms are more strongly bonded to the foreign metal electrode [213–215]. In the case of Cu-UPD, the metal deposition processes at the electrode surface can be represented by Eq. 21:
The Nernst equation was applied in order to predict the equilibrium potential (E eq ) at which the deposition and dissolution of the bulk-metal phase happens:
where E 0 is the standard potential and a is the activity. R, T, and F are the molar gas constant, temperature, and Faraday constant, respectively. The formation of the first monolayer is inferred from the pronounced current peaks at the potential \( E>{E}_{eq}, \) while the bulk deposition occurs at \( E<{E}_{eq}. \) Taking an example of Cu on Au system, UPD Cu on Au occurs at ~0.46 V, while bulk deposition occurs at ~0.25 V.
The cyclic voltammogram of platinum in solution composed of H2SO4 and CuSO4 at wide scan range of 0–1.5 V commonly exhibited four main peaks related to different mechanisms. The CV is dominated by the deposition and stripping of both bulk and underpotential-deposited copper. In a forward anodic scan, one may find a distinct sharpened peak that represents the bulk copper stripping, while in the backward cathodic scan, the bulk copper deposition occurs at almost similar potential with bulk copper deposition. From detailed observations, bulk copper deposition occurs at around 0.25 V, which is a slightly lower potential than its bulk copper stripping. At the platinum double-layer region, we may able to observe several peaks associated with the UPD processes of copper on platinum. It is indicating that the Cu stripped from different sites, which has different adsorption energies, on the surface of platinum. In a forward anodic scan, UPD of copper stripping occurs at much positive potentials compared to that of bulk copper stripping. Furthermore, UPD of copper deposition is also shifted to much positive potentials compared to the deposition of bulk copper. In addition, the hydrogen adsorption region is suppressed due to the presence of copper, and the oxide reduction is distorted due to the onset of copper UPD [206].
It is worth mentioning that Green and Kucernak used the plot of the ratio of copper stripping charge to hydrogen charge \( \left({Q}_{Cu-UPD}/{Q}_{H-des}\right) \) as a function of deposition potential on platinum electrode in order to obtain an optimum condition for the formation of well-ordered monolayer of UPD copper without the possibility of a three-dimensional growth of bulk copper [206]. The charge of 420 μC/cm2 in the copper metal deposition reaction is used in the calculation of \( {Q}_{Cu-UPD}, \) while \( {Q}_{Cu-UPD} \) itself is obtained after subtracting the total measured UPD copper stripping charges with the platinum background. The ratio of \( {Q}_{Cu-UPD}/{Q}_{H-des} \) is to be expected 2, where a copper atoms adsorbs on platinum surface at the same sites with hydrogen to form a completely UPD copper layer. Their results showed that \( {Q}_{Cu-UPD}/{Q}_{H-des}\approx 2 \) was achieved when the deposition potential is in the range of 0.25–0.3 V. When E dep is lower than 0.25 V, the deposition of bulk copper occurs, and when higher than 0.3 V, the UPD layer does not form completely. This technique can be also widely applied in finding an optimum deposition time of UPD copper.
In our recent results which are shown in Fig. 13b, we also found a similar trend, where the UPD of Cu occurs at the potentials more positive than that of bulk Cu deposition. The bulk copper deposition on commercial Pd/C occurs at the deposition potential of Edep.-1. In a detailed observation, an increase in deposition potentials at Edep.-2 and Edep.-3 is followed by a decrease in current density corresponding to bulk copper stripping while the UPD copper starts to grow, as is shown with black arrow.
In any study of Pt-based catalysts, the method used for the determination of ECSA from Cu-UPD should consider: (i) applied deposition potential and time for the formation of UPD copper, (ii) applied potential scan range for both Pt background and UPD copper stripping, and (iii) a correction for double-layer charging, oxygen adsorption, and further possibility of adsorbed anion. In our earlier discussion, we mentioned that an optimum condition for the formation of well-ordered monolayers of UPD copper can be achieved when the ratio of \( {Q}_{Cu-UPD}/{Q}_{H-des} \) is nearly equal to 2, as suggested by Green and Kucernak [206]. Thus, for the first important aspect, the plot between \( {Q}_{Cu-UD}/{Q}_{H-des} \) versus potential and (or) time deposition should be made in order to find out which potential and (or) time deposition region shows a value of \( {Q}_{Cu-UPD}/{Q}_{H-des}\approx 2. \) In the second aspect, the CV for bare Pt (as background) should be scanned from 0.05 to 0.85 V. The end potential in forward scan was chosen to be 0.85 V, at the point where oxide growth commonly starts on platinum, and thus only a very small oxide reduction peak was expected to occur at the backward scan. The linear potential scan for the Cu-UPD stripping should be also scanned until the end potential of 0.85 V. In case of PtRu catalyst, the linear potential scan for Cu-UPD stripping could be started from 0.3 V to 0.85 V. An applied potential at 0.3 V was chosen as the starting point of the scan due to a completion of monolayer of UPD copper while 0.85 V as the end potential which was due to a completion of oxidative removal of UPD copper layer and also a suppression of oxide adsorption/desorption. Once the oxide adsorption/desorption mechanism was suppressed, we may consider it as an advantage for the calculation of \( {Q}_{Cu-UPD} \) as a very small correction would be expected and will be discussed in the following third aspect.
In the third aspect, the charge of \( {Q}_{Cu-UPD} \) should result after subtracting the total measured charge from experiment (Q exp) with the charge due to the charging of double-layer capacitance (Q DL ), the charge due to the growth of any oxide and (or) oxygenated species (Q ox ), and the charge due to adsorption/desorption of any adsorbed anion (Q anion) as shown in Eq. 23 [207]:
Therefore, it is important to include the CV for bare platinum as a background for the matter of correction to \( {Q}_{Cu-UPD}. \) ECSA can be calculated by assuming that a charge required to oxidize a monolayer of Cu adsorbed on each metal surface is equal to 420 μC/cm2 as shown in the following equation [207]:
Once the ECSA was obtained either from H-UPD, CO stripping, and Cu-UPD methods, it was possible to determine the average particle size (d) by assuming that the shape of the catalyst particle is spherical, as shown below:
where ρ is the average particle density, and it was 21.4 g/cm3 for Pt. However for the bimetallic system, the average particle density could be obtained by using the following relationship:
where x bulk Pt and x bulk M are the bulk compositions of Pt and second metal “M,” respectively, while ρ M is the average particle density of second metal “M.”
Surface composition of Pt-based catalysts through the Cu-UPD method. It is of interest to describe the feature for the stripping of Cu-UPD layer formed on particularly dispersed bimetallic PtRu catalyst. Commonly, a peak at low potential around 0.42 V is accompanied by a shoulder that continues to a much higher potential. The first peak at around 0.42 V was due to the oxidative removal of adsorbed Cu-UPD layer, while the shoulder was the removal from the Pt sites. This information strongly supports the differing adsorption energies for Cu on either Pt or Ru that could be used to quantify the Ru metal surface content of PtRu catalyst. Further deconvolution of linear anodic stripping of Cu-UPD should be made to separate a charges contributed by Ru and Pt sites by assuming that the feature of CU-UPD has a Gaussian line shape. The Ru coverage on the surface of bimetallic PtRu catalyst (x s Ru ) can be quantified from the ratio of charge in first peak contributed by Ru (\( {Q}_{Ru}^{Cu-UPD} \)) to \( {Q}_{Cu-UPD} \) as shown below [207]:
Rotating Disk Electrode (RDE) Method
It is well known that the steady-state current generated by cyclic voltammetry is mainly determined by the diffusion of substrate around the electrode’s surface when immersed in a stagnant/unstirred electrolyte solution. A minor natural convection from the impact of environmental fluctuation in some cases may appear, but its effect can be neglected. It is possible to increase the mass transport by introducing a forced convection in which the analyte solution flows relative to a working electrode. A popular method for creating such a relative movement is by rotating the working electrode. Such a method is called a rotating disk electrode (RDE). Therefore, RDE could also be called as a hydrodynamic working electrode, where the steady-state current is determined by solution flow rather than diffusion [186, 188].
In common three-electrode cells, when a working electrode spins, the reactant in the solution is dragged to the surface of the working electrode, and the resulting centrifugal flings the product away from it. However, one should notice that there is still a stagnant reaction layer covering that surface of the working electrode and the reactants transport through this layer by diffusion. There is a strong dependency of thickness of diffusion layer (δ) with applied rotation rate (ω) of working electrode as shown in Eq. 14. An increase in rotation rate will lead to a thinner diffusion layer. A rotation rate between 5 and 10,000 rpm can control the flow of the reactant through the surface of working electrode that follows a laminar pattern. Figure 14 shows a schematic diagram of RDE setup. The working electrode was connected to the electrode rotator that has very fine control of the electrode’s rotation rate.

The RDE setup of Pine instrument and its schematic diagram. 1 Rotator body; 2 bearing, 2; 3 rubber housing, 2; 4 washer; 5 spring, 4; 6 spindle for electrode; 7 pulley; 8 top lock nut; 9 bottom lock nut; 10 bushing/brush contact; and 11 retainer
The flow pattern under the rotating disk electrode was obtained by numerically solving the Navier–Stokes equation and continuity equation under the following conditions:
-
(i)
The radius of the disk on the working electrode is large enough compared to that of boundary layer thickness, and thus the small distortion of flow pattern at the center and edge can be neglected.
-
(ii)
The roughness of the disk surface of the working electrode is small enough compared to that of the boundary layer thickness.
-
(iii)
The radius of the electrochemical cell is large enough compared to that of the disk on the working electrode, and thus the reflection of the flow at the vessel of electrochemical cell’s wall does not affect the flow pattern under the disk.
-
(iv)
The rotation speed is sufficiently greater than the lower critical value in which the effect of natural convection is negligible. Furthermore, the rotating speed is also smaller than that of the higher critical value in which the flow gets to be turbulent.
The solution of the Navier–Stokes equation and continuity equation under those mentioned conditions may bring two possibilities of the net mass transport rate of a reactant to the surface of the working electrode. The first net mass transport is the convection which can control the thickness of the diffusion layer, while the second one is the diffusion which can control the reactant through the diffusion layer. The flux normal to the electrode surface due to diffusion is given by \( D\left({{\partial}^2C}/ {\partial {x}^2}\right) \) and that due to convection is given by \( {v}_x\left({\partial C}/ {\partial x}\right) \), where D is the diffusion coefficient of the species, C is the bulk concentration of the species, and v x is the solution velocity in the x direction, which is normal to the electrode surface [186, 189].
The rotating disk electrode is becoming one of the most powerful methods for studying both the diffusion in electrolytic solutions and the kinetics of moderately fast electrode reaction because the hydrodynamics and the mass-transfer characteristics are well understood and the current density on the disk electrode is supposed to be uniform. V. G. Levich [195] solved the family of equations and provided an empirical relationship between diffusion-limiting current (i d ) and rotation rate (ω) as shown in Eq. 15. In particular application in fuel cells, the empirical relationship which is given by Levich is also applicable in linear scan voltammetry (LSV) experiment performed on a RDE to study the intrinsic kinetics of the catalyst [105, 192].
Electrocatalytic Applications: Electrochemical Studies of Fuel Cell Reactions on Bimetallic Nanoparticles
Success in fuel cell technology largely relies on electrocatalysts. Electrocatalysts employed for fuel cell reactions are required to possess well-controlled structures, dispersions, and compositional homogeneities. Further, in order to realize fuel cells as viable future power-generating technology, electrocatalysts must meet performance, durability, and cost targets. Recently, Mark K. Debe critically reviewed electrocatalytic approaches and challenges for automotive fuel cells [42]. The author pointed out that a clear understanding of electrocatalyst surface area and activity loss mechanisms and insights into durability issues associated with externally and internally generated impurities should significantly assist the development of fuel cell technology. Critical factors that are presently believed to play significant role in the electrocatalysts need to be thoroughly understood in order to develop newer catalyst system that can meet the targeted performance and durability.
Decisive Factors Influencing Electrocatalytic Activity
Structural Effects
With regard to electrocatalysts, structure-dependent catalytic activity toward methanol oxidation reaction (MOR) [12, 216–218], formic acid oxidation reaction (FAOR) [17, 219], and oxygen reduction reaction (ORR) [220–222] have been observed. Maillard et al. studied the influence of the catalyst’s structure on the reactivity of electrooxidation of methanol [218]. The authors noticed that the electrocatalytic activity of a PtRu catalyst with a Ru-decorated Pt surface was two orders of magnitude higher when compared to the commercial Johnson Matthey PtRu black (Pt:Ru = 1:1)-alloyed structured catalyst. In our recent investigations, we found that Pt-decorated Ru catalyst exhibited higher mass activity for MOR than commercial Johnson Matthey PtRu black (Pt:Ru = 1:1)-alloyed structured catalyst [12]. From the X-ray absorption spectroscopy measurements, we observed more Pt d-band vacancies in the case of Pt-on-Ru nanoparticles causing weaker CO adsorption on the synthesized catalyst surface. Zeng et al. showed that a core–shell-structured Ag@Pt/C bimetallic nanoparticle exhibits enhanced specific activity in methanol electrooxidation when compared to similarly prepared bimetallic Ag–Pt catalyst. The authors attributed the observed activity enhancement to the presence of Au underneath a very thin Pt shell where electron exchange between Au and Pt had promoted the formation of active oxygen species on Pt, which facilitated the removal of inhibiting CO-like reaction intermediates [84]. The core–shell-structured bimetallic nanoparticles available for MOR reveal that the core material influences the outer shell layer and optimizes its surface electronic and structural properties thereby exhibiting improved catalytic activities. Zhang et al. studied the formic acid electrooxidation on Pt-around-Au nanocomposite structures [223]. From the electrochemical measurements in supporting electrolyte containing formic acid solutions, the authors found that formic acid oxidation on Pt-around-Au/C nanocomposites was about 3.0 times than on Pt/C catalyst. Based on cyclic voltammetric analyses, the authors concluded that the possible reason for the unexpected high activity for HCOOH oxidation on the Pt-around-Au nanocomposite is due to the efficient spillover of HCOO from Au to the surrounding Pt nanoparticles where HCOO is further oxidized to CO2. Based on voltammetric studies, the authors found that the specific activity of Pt atoms increased with the decrease of Pt surface coverage. Authors found a 3 % Pt-covered-Au/C surface that showed 17.0- and 23.9-folds more activity toward formic acid oxidation compared to 75 % and 100 % Pt-covered surfaces, respectively. In the case of oxygen reduction reaction, Pt-based bimetallic nanoparticles with various structures have been studied. Several efforts have been made to improve the dispersion and stability of Pt-based ORR catalysts, e.g., by creating a near-surface region with a Pt-skin structure in Pt-alloys via acid treatment [176, 224] and by forming core–shell structures [87]. In a recent study, Stamenkovic et al. showed that the Pt3Ni(111) surface is about 90 times more active than the state-of-the-art Pt/C catalysts for ORR [21]. In another interesting study, Chen et al. [225] demonstrated that by creating a percolated structure with Pt-rich and Pt-poor regions within individual Pt3Co nanoparticles through acid treatment, their ORR activity could be increased by up to two orders of magnitude when compared to Pt nanoparticles. These results clearly indicate that significant improvements in electrocatalytic activity can be achieved by properly fine-tuning the structure of bimetallic nanoparticles.
Composition/Surface Composition Effects
The composition of bimetallic nanoparticles in either bulk or surface need to be properly controlled during the fabrication process as the bulk/surface composition strongly influences the electrocatalytic activity. Further, variations in composition are very important in modifying the surface structure of bimetallic nanoparticles. Ball et al. studied the effect of atomic ratio of Pt–M/C (M = Co, Cr, Fe, Ni, and Mn) catalysts on the fuel cell activity and stability [226]. The authors found that bimetallic Pt–M/C catalysts with Pt-to-M ratio 3:1 showed higher electrocatalytic activities. Very recently, in our group Pt x Ru100−x /C with various alloy compositions was prepared by a microwave-assisted ethylene glycol method [9]. Among the Pt x Ru100−x /C with various Pt:Ru atomic ratios (x = 25, 50, and 75), the Pt75Ru25/C NPs were shown to be superior in MOR activity on account of their favorable alloying extent, Pt d-band vacancy, and Pt electrochemical active surface area (ECASA). In another work, we also studied ORR on Pt x -Fe1−x nanoparticles with various Pt:Fe atomic compositions (3:1, 1:1, and 1:3) and found that Pt1Fe1 nanocatalyst showed a greater enhancement in ORR than Pt/C. The higher alloying extent of platinum coupled with promising electronic structure offered by the lower Pt d-band vacancies was given as the reason for the enhanced ORR activity [63]. Stamenkovic et al. observed that the arrangement of surface atoms in the near-surface region of a Pt3Ni (111) surface drastically improves its catalytic activity toward ORR [21]. From the crystal truncation rod (CTR) experiments, they showed that the first layer entirely consists of Pt, whereas the second layer was Ni-rich with 52 % Ni and the third layer is Pt-enriched with a Pt content of 87 %. With this surface configuration, the Pt3Ni(111) surface exhibited 10 times higher mass activity toward ORR when compared to Pt(111) surfaces and 90 times higher ORR activity than the current state-of-the-art Pt/C catalysts in 0.1 M HClO4 solutions. Further the authors showed that different low-index Pt3Ni surfaces exhibited different ORR activities with the order, Pt3Ni(100)-skin < Pt3Ni(110)-skin < Pt3Ni(111)-skin [21]. The authors pointed out that the observed differences in the ORR activities of three low-indexed planes of Pt3Ni were attributable to the variations in d-band center and surface atomic arrangement of platinum and decreased coverage of nonreactive oxygenated species (OHads), due to the incorporation of Ni atoms. These results suggested that surface engineering strategies offer a big help in fine-tuning the electrocatalytic activity of bimetallic nanoparticles.
Morphological Effects
In recent years, morphology-controlled synthesis of various Pt-based catalysts with shapes such as nanodendrites, polyhedral, hollow structures, and core–shell has been explored for fuel cell reactions. It was reported that different crystallographic facets stabilized on particles with different shapes exhibit different reactivities and selectivities [227, 228]. Commonly found low-index planes or facets on single-crystal surfaces of bulk Pt appear to possess lower reactivity than the high-index planes. Armed with high density of atomic steps, ledges, and kinks, which usually serve as active sites for breaking chemical bonds, the high-index planes exhibit much higher catalytic activity. Sun and coworkers developed a novel electrochemical approach to prepare tetrahexahedral (THH) Pt nanocrystals (NCs) with high-index facets on glass carbon substrates [19]. The THH Pt nanocrystals developed in this study exhibited much enhanced catalytic activity per unit surface compared to the other Pt morphologies like Pt-nanospheres and commercial Pt/C catalysts for the oxidation of formic acid and ethanol which are considered to be promising alternative fuels for direct fuel cells. The enhanced catalytic activity was attributed to the higher extent of stepped atoms on the surface of THH Pt NCs. In a recent study, Kim et al. reported the synthesis protocol for heterogeneous bimetallic nanocrystals consisting of Pt multibranches on Au nanocrystal cores with well-defined morphologies (cubes, rods, and octahedral) [229]. The authors showed the TEM and HRTEM images of the formation of Pt-islands on various Au morphologies (Fig. 15). The ORR activity of Au@Pt heteronanostructures with various morphologies recorded in O2-saturated 0.1 M HClO4 solutions showed that at 0.8 V versus RHE, the mass activities follows the order: Aucube@Pt < Aurod@Pt < Auoctahedron@Pt. The higher electrocatalytic activity observed in the case of Auoctahedron@Pt was attributed to its (111)-orientation-rich Pt surface. The above examples illustrate that specific facets of metallic component along with its shape play a crucial role in determining the electrocatalytic activity.

Left and right panels features low- and high-magnification TEM images, respectively, of (a, b) Aucube@Pt, (c, d) Aurod@Pt, and (e, f) Auoctahedron@Pt nanoparticles (Reproduced with permission from ref. [229], © 2010 Wiley – VCH Verlag GmbH & Co. KGaA)
Size Effects
Substantial efforts have been directed to the investigation of the influence of particle size on the catalytic activity after the breakthrough experiments of Haruta et al. on Au nanoparticles [230]. Park et al. [231] demonstrated the electrooxidation of formic acid, formaldehyde, and methanol in acidic environment on Pt/C nanoparticles with the varying particle diameters in the range of 2–9 nm. Authors carried out electrooxidation of formic acid, formaldehyde, and methanol in acidic electrolyte on Pt/C nanoparticles with varying particle diameters in the range of 2–9 nm. From the voltammetry and infrared spectroscopy measurements, the authors found size-dependent chemisorption of CO formed from three reactants. The observed rates of methanol electrooxidation decrease for Pt nanoparticle diameters below 4 nm. In contrast, the rate of formic acid electrooxidation increases for Pt nanoparticle diameters above 4 nm. Different roles played by Pt ensemble effect were given as the reason for the observed size-dependent CO chemisorptions and corresponding CO2 production. The rate of formaldehyde electrooxidation found little sensitivity to the Pt size. Yamamoto and coworkers investigated size-specific catalytic activity of Pt clusters synthesized in G-3 dendrimers toward oxygen reduction reaction [5]. Three kinds of Ptn@3 (n = 12, 28, 60) clusters, viz., Pt12@3 (particle size = 0.9 ± 0.1 nm), Pt28@3 (particle size = 1.0 ± 0.1 nm), and Pt60@3 (particle size = 1.2 ± 0.1 nm) were synthesized. Authors found smallest cluster Pt12@3 exhibited 13 times higher ORR activity than that of commercial Pt/C catalyst. Sun’s group studied the particle size and catalytic activity relationship of Co60Pd40 nanoparticles toward formic acid oxidation [232]. The authors observed that the catalytic activity of Co60Pd40 nanoparticles for formic acid oxidation increases with the decrease in particle size.
Atomic Distribution/Alloying Extent
In bimetallic nanoparticles, the atomic distribution between two different metals and the degree of alloying play significant role in catalyst activity [10, 20, 24, 63, 233]. In our group, X-ray absorption spectroscopy methods were developed to determine the atomic distribution in bimetallic nanoparticles [20]. By collecting the XAS data at the Pt and Ru absorption edges of two commercial PtRu catalysts, the extent of intermixing of Pt and Ru (alloying extent) and Pt and Ru atom distribution was determined. The extent of both Pt and Ru alloying was found to be higher in commercial Johnson Matthey PtRu catalyst than with commercial E-TEK PtRu catalyst, which was reflected in higher methanol electrooxidation activity. In another study, we observed enhanced ORR activities in bimetallic PtFe catalysts with higher extent of platinum and iron alloying [63].
Hydrogen Fuel Cells
Proton exchange membrane fuel cells (PEMFCs) powered by hydrogen from secure and renewable sources are considered to be a promising energy source for various applications [234–237]. In a typical H2-fed PEMFC, hydrogen is oxidized on an anode leaving protons and electrons. The protons generated at anode travel through the proton exchange membrane to the cathode where they react with oxygen to form water. The overall electrochemical reaction between hydrogen and oxygen to form water gives a theoretical potential of 1.23 V versus SHE at room temperature. The principal reactions are shown in Eqs. 28, 29, and 30:
Pt is the best-known material for the hydrogen oxidation. However, CO present even in trace quantities will impede hydrogen adsorption on Pt surfaces and decreases its rate of oxidation popularly known as CO-poisoning effect. Experiments illustrate that CO oxidation on bimetallic Pt–M-based electrodes, where M is a promoter metal (e.g., Ru), occurs at potentials lower than that of a pure Pt electrode [238]. The enhanced CO oxidation on such Pt–M formulations may be achieved by two metals working synergistically as explained by the so-called bifunctional (or) reactant pair mechanism. Higher PEMFC performance was reported on PtRu bimetallic nanoparticles for fuel streams containing CO [239, 240]. The development of bimetallic electrocatalysts helps in the reduction of Pt loadings as well as CO tolerance of the anode. In the case of bimetallic catalysts, strong interaction of participating components is required. It was reported that the catalytic performance of PtRu nanoparticles is strongly dependent on the distribution of Pt and Ru sites at the atomic level and believed to be sensitive to the particle’s shape and size and to the nature of the support. In the past few years, our research group has actively worked on developing suitable electrocatalysts for both CO oxidation and oxygen reduction reactions for fuel cell applications by focusing on size, structure, and atomic distribution manipulation strategies [24, 26, 27, 130, 168, 241, 242]. In a recent study by studying the stepwise formation mechanism employing X-ray absorption spectroscopy, we established a structure-controllable synthesis strategy based on a modified Watanabe’s process for bimetallic PtRu/C nanoparticles [27]. With this methodology, PtRu/C NPs with a particle size of about 2.9 nm were conveniently achieved.
Many other bimetallic electrocatalysts such as PtMo [154, 243–248], PtSn [249, 250] PtPd [251–253], and Pt–Co [254] have been successfully explored for CO-tolerant HOR. In most cases, electrocatalyst particles were dispersed on conductive supports of high surface area. The support for the catalyst nanoparticles turns out to be a major factor in influencing their dispersion and stability. Characteristics such as electrical conductivity, surface area, hydrophobicity, morphology, porosity, and corrosion resistance are most considered in the choice of a good catalyst support. Based on these considerations, carbon is the best catalyst support material for PEMFC electrocatalysts. Carbon black and activated carbons have been extensively studied as catalyst supports, with Vulcan 72 being the most representative. In the last decade, a number of new synthetic carbons with various mesostructures and nanostructures have been reported. These include carbon nanotubes [254], aerogel carbon [255, 256], and meso-carbon [257] with or without a high degree of order. With the developments in attaining high performance bimetallic electrocatalysts and high surface area carbon supports, bottlenecks such as higher catalyst loadings and costs associated with fuel cell technology were nearly resolved.
Methanol Fuel Cells
Direct methanol fuel cells (DMFCs) are advantageous over H2-fed PEMFCs due to easy fuel transportation, energy storage, and high-energy conversion efficiency and are found as promising power sources for electric vehicles and portable devices [258, 259]. In a typical DMFC, methanol and water molecules are simultaneously electrooxidized at the anode to produce CO2, electrons, and protons called as methanol oxidation reaction (MOR). Protons generated at the anode pass through the proton exchange membrane to cathode where they combine with electrons and the oxidant air or oxygen simultaneously reducing to water called as oxygen reduction reaction (ORR). The principal reactions involved in DMFCs are show in Eqs. 31, 32, and 33:
Methanol oxidation reaction:
Oxygen reduction reaction:
Overall cell reaction:
Pt is the best material for the adsorption and dehydrogenation of methanol. However, the formation of intermediate species such as CO, formic acid, and formaldehyde poisons the platinum anode and critically lowers the performance. During methanol oxidation, the efficient catalyst should allow a complete oxidation to CO2. Presently, carbon-supported PtRu catalysts have been shown to be the best candidates for electrochemical oxidation of methanol [74, 260]. May studies confirm that Ru could produce OHads species at lower potential to react with adsorbed intermediates on neighboring Pt sites [260–263]. Furthermore, the presence of Ru can induce variations in the electronic structure of Pt. This electronic or ligand effect enhances methanol dehydrogenation on Pt and facilitates weakening of Pt–CO bond, and thus an easy removal of poisoned CO on Pt is expected.
Many other bimetallic Pt-based nanocatalysts have been explored as low cost alternatives to PtRu catalysts for MOR. For example, alloys of Pt with Fe [264], Ni [265], Co [266], Mo [154, 247], Sn [267, 268], Pb [269], V [270], or Ir [271] show promise in generating OHads at lower potentials than Pt with improved activity toward MOR. In most cases, catalytic nanoparticles were dispersed on high surface carbon structures. The advantage of using a supported metal catalyst lies in the possibility to reduce the metal loadings drastically. The development in highly CO-resistant Pt-based bimetallic nanoparticles as electrocatalysts for MOR is a big help in making DMFC a viable power-generating technology. However, significant improvements are still required in increasing the durability of bimetallic Pt-based catalysts for the widespread implementation of DMFCs.
Electrochemical Oxidation of Formic Acid
Formic acid-based direct formic acid fuel cells (DFAFCs) are another attractive power source for portable electronic applications due to their promising energy densities, power system simplicities, and the convenient storage and transport of liquid formic acid [272, 273]. Further, DFAFCs claim lower fuel crossover than DMFCs due to the repulsive nature of formate ions with sulfuric acid groups of the Nafion membrane so that higher concentrations of formic acid can be used to get the higher energy density of the cell [274]. On conventional platinum surfaces, formic acid is electrooxidized directly to produce CO2 (dehydrogenation path), or it may leave adsorbed CO species (dehydration pathway) as shown below:
Although pure Pt can be easily poisoned by adsorbed CO species, one tactic to improve its activity is to form bimetallic Pt-based catalysts. Many efforts have been made to enhance the oxidation rates of formic acid by developing PtAu [223, 274–277], PtPd [278, 279], PtFe [280], PtBi [273, 281, 282], PtAg [283], Pt–Co [284], and PtSb [285] bimetallic catalysts. Pt-based bimetallic catalysts display improved catalytic activity toward formic acid as a result of the ensemble effect. According to this, at least two ensembling binding Pt sites are required for the dissociative adsorption of formic acid to CO. However, the direct oxidation of formic acid via dehydration pathway does not require the same Pt atom ensemble [231, 286].
In recent years, much interest has been focused on the development of PtAu bimetallic nanoparticles as electrocatalysts for formic acid electrooxidation. The addition of Au to Pt promises the enhancement of formic acid oxidation kinetics by operating a combined ensemble and electronic effect which helps in diminishing the COads formation and increasing the intrinsic activity, respectively [219]. Further, Au is believed to promote the first electron transfer during the direct dehydrogenation process of formic acid oxidation which generates HCOOads species on Au [287–289]. If Pt is around Au, efficient spillover of HCOOads from Au to the neighboring Pt occurs, which is further oxidized to CO2 [223]. Various carbon-based supports have been utilized for the purpose of depositing Pt-based bimetallic nanoparticles, including carbon [290], carbon nanotubes [291], carbon nanofibers [276], and graphene [275], which have led to improved kinetics during formic acid electrooxidation. It was widely demonstrated that the use of formic acid as a fuel is more advantageous than hydrogen and methanol due to its low-temperature operation, storage, and convenient transport.
Conclusions and Future Perspective
In conclusion, the content reviewed here underscores the importance of understanding the formation of bimetallic nanoparticles in particle design and in establishing structure-controllable synthesis methodologies and scaling-up processes. X-ray absorption spectroscopy has a striking advantage in revealing the underlying chemistry involved in the nucleation and growth process of bimetallic nanoparticles. Further, XAS can be utilized to follow the formation of alloy and core–shell structures which are believed to influence bimetallic nanoparticles electrocatalytic activities toward fuel cell reactions. In addition, the capabilities of XAS can be used to provide atomic level information on alloying extent and atomic distribution. So far, significant developments have been made in the bimetallic nanoparticle synthesis with achievements such as synthesizing more complicated structures with an easy control over composition, size, and morphology. The relationship between the catalytic activity and the selectivity of bimetallic nanoparticles with their structural characteristics in fuel cell reactions such as the methanol oxidation reaction, oxygen reduction reaction, and formic acid oxidation reactions has been discussed. Key influencing factors including surface structure, composition, size, and morphology have been investigated by many researchers. These studies strengthen our fundamental understanding of bimetallic catalysis and the rational selection of novel materials for power-generating devices such as fuel cells. Although great progress has been made in bimetallic nanoparticle research, the common challenges of achieving long-lasting, highly active, selective, and environmentally benign bimetallic nanoparticles are still remain. A promising hope is that by systematically minimizing the experimental trial-and-error strategies by integrating theoretical and practical approaches, great strides in the development bimetallic nanoparticle-linked catalysis can be expected.
References
Roduner E (2006) Size matters: why nanomaterials are different. Chem Soc Rev 35(7):583–592
Jortner J (1992) Cluster size effects. Z Phys D: At Mol Clusters 24(3):247–275
Valden M, Lai X, Goodman DW (1998) Onset of catalytic activity of gold clusters on Titania with the appearance of nonmetallic properties. Science 281(5383):1647–1650
Valden M, Pak S, Lai X, Goodman DW (1998) Structure sensitivity of CO oxidation over model Au/TiO2 2 catalysts. Catal Lett 56(1):7–10
Yamamoto K, Imaoka T, Chun W-J, Enoki O, Katoh H, Takenaga M, Sonoi A (2009) Size-specific catalytic activity of platinum clusters enhances oxygen reduction reactions. Nat Chem 1(5):397–402
Saida T, Ogiwara N, Takasu Y, Sugimoto W (2010) Titanium oxide nanosheet modified PtRu/C electrocatalyst for direct methanol fuel cell anodes. J Phys Chem C 114(31):13390–13396
Cherstiouk OV, Simonov PA, Savinova ER (2003) Model approach to evaluate particle size effects in electrocatalysis: preparation and properties of Pt nanoparticles supported on GC and HOPG. Electrochim Acta 48(25–26):3851–3860
Mukerjee S, McBreen J (1998) Effect of particle size on the electrocatalysis by carbon-supported Pt electrocatalysts: an in situ XAS investigation1. J Electroanal Chem 448(2):163–171
Taufany F, Pan C-J, Rick J, Chou H-L, Tsai M-C, Hwang B-J, Liu D-G, Lee J-F, Tang M-T, Lee Y-C, Chen C-I (2011) Kinetically controlled autocatalytic chemical process for bulk production of bimetallic core–shell structured nanoparticles. ACS Nano 5(12):9370–9381
Lai F-J, Sarma LS, Chou H-L, Liu D-G, Hsieh C-A, Lee J-F, Hwang B-J (2009) Architecture of bimetallic PtxCo1−x electrocatalysts for oxygen reduction reaction as investigated by X-ray absorption spectroscopy. J Phys Chem C 113(29):12674–12681
Koffi RC, Coutanceau C, Garnier E, Léger JM, Lamy C (2005) Synthesis, characterization and electrocatalytic behaviour of non-alloyed PtCr methanol tolerant nanoelectrocatalysts for the oxygen reduction reaction (ORR). Electrochim Acta 50(20):4117–4127
Chen C-H, Sarma LS, Wang D-Y, Lai F-J, AlAndra C-C, Chang S-H, Liu D-G, Chen C-C, Lee J-F, Hwang B-J (2010) Platinum-decorated ruthenium nanoparticles for enhanced methanol electrooxidation. ChemCatChem 2(2):159–166
Xu CW, Wang H, Shen PK, Jiang SP (2007) Highly ordered Pd nanowire arrays as effective electrocatalysts for ethanol oxidation in direct alcohol fuel cells. Adv Mater 19(23):4256–4259
Wang C, Daimon H, Onodera T, Koda T, Sun S (2008) A general approach to the size-and shape-controlled synthesis of platinum nanoparticles and their catalytic reduction of oxygen. Angew Chem Int Ed 47(19):3588–3591
Mayrhofer KJJ, Juhart V, Hartl K, Hanzlik M, Arenz M (2009) Adsorbate-induced surface segregation for core–shell nanocatalysts. Angew Chem Int Ed 48(19):3529–3531
Cheng F, Wang H, Sun Z, Ning M, Cai Z, Zhang M (2008) Electrodeposited fabrication of highly ordered Pd nanowire arrays for alcohol electrooxidation. Electrochem Commun 10(5):798–801
Zhang S, Shao Y, Liao H-G, Liu J, Aksay IA, Yin G, Lin Y (2011) Graphene decorated with PtAu alloy nanoparticles: facile synthesis and promising application for formic acid oxidation. Chem Mater 23(5):1079–1081
Sun S, Zhang G, Geng D, Chen Y, Li R, Cai M, Sun X (2011) A highly durable platinum nanocatalyst for proton exchange membrane fuel cells: multiarmed starlike nanowire single crystal. Angew Chem Int Ed 50(2):422–426
Tian N, Zhou Z-Y, Sun S-G, Ding Y, Wang ZL (2007) Synthesis of tetrahexahedral platinum nanocrystals with high-index facets and high electro-oxidation activity. Science 316(5825):732–735
Hwang B-J, Sarma LS, Chen J-M, Chen C-H, Shih S-C, Wang G-R, Liu D-G, Lee J-F, Tang M-T (2005) Structural models and atomic distribution of bimetallic nanoparticles as investigated by X-ray absorption spectroscopy. J Am Chem Soc 127(31):11140–11145
Stamenkovic VR, Fowler B, Mun BS, Wang G, Ross PN, Lucas CA, Marković NM (2007) Improved oxygen reduction activity on Pt3Ni(111) via increased surface site availability. Science 315(5811):493–497
Cuenya BR (2010) Synthesis and catalytic properties of metal nanoparticles: size, shape, support, composition, and oxidation state effects. Thin Solid Films 518(12):3127–3150
Taufany F, Pan C-J, Rick J, Chou H-L, Tsai M-C, Hwang B-J, Liu D-G, Lee J-F, Tang M-T, Lee Y-C, Chen C-I (2011) Kinetically controlled autocatalytic chemical process for bulk production of bimetallic core–shell structured nanoparticles. ACS Nano 5(12):9370–9381
Hwang BJ, Senthil Kumar SM, Chen C-H, Chang R-W, Liu D-G, Lee J-F (2008) Size and alloying extent dependent physiochemical properties of Pt−Ag/C nanoparticles synthesized by the ethylene glycol method. J Phys Chem C 112(7):2370–2377
Chen C-H, Sarma LS, Chen J-M, Shih S-C, Wang G-R, Liu D-G, Tang M-T, Lee J-F, Hwang B-J (2007) Architecture of Pd–Au bimetallic nanoparticles in sodium bis(2-ethylhexyl)sulfosuccinate reverse micelles as investigated by X-ray absorption spectroscopy. ACS Nano 1(2):114–125
Sarma LS, Chen C-H, Kumar SMS, Wang G-R, Yen S-C, Liu D-G, Sheu H-S, Yu K-L, Tang M-T, Lee J-F, Bock C, Chen K-H, Hwang B-J (2007) Formation of Pt−Ru nanoparticles in ethylene glycol solution: an in situ X-ray absorption spectroscopy study. Langmuir 23(10):5802–5809
Hwang BJ, Chen C-H, Sarma LS, Chen J-M, Wang G-R, Tang M-T, Liu D-G, Lee J-F (2006) Probing the formation mechanism and chemical states of carbon-supported Pt−Ru nanoparticles by in situ X-ray absorption spectroscopy. J Phys Chem B 110(13):6475–6482
Chen C-H, Sarma LS, Wang G-R, Chen J-M, Shih S-C, Tang M-T, Liu D-G, Lee J-F, Chen J-M, Hwang B-J (2006) Formation of bimetallic Ag−Pd nanoclusters via the reaction between Ag nanoclusters and Pd2+ ions. J Phys Chem B 110(21):10287–10295
Chen C-H, Hwang B-J, Wang G-R, Sarma LS, Tang M-T, Liu D-G, Lee J-F (2005) Nucleation and growth mechanism of Pd/Pt bimetallic clusters in sodium bis(2-ethylhexyl)sulfosuccinate (AOT) reverse micelles as studied by in situ X-ray absorption spectroscopy. J Phys Chem B 109(46):21566–21575
Tsai YW, Tseng YL, Sarma LS, Liu DG, Lee JF, Hwang BJ (2004) Genesis of Pt clusters in reverse micelles investigated by in situ X-ray absorption spectroscopy. J Phys Chem B 108(24):8148–8152
Hwang B-J, Tsai Y-W, Sarma LS, Tseng Y-L, Liu D-G, Lee J-F (2004) Genesis of bimetallic Pt−Cu clusters in reverse micelles investigated by in situ X-ray absorption spectroscopy. J Phys Chem B 108(52):20427–20434
Yu W, Porosoff MD, Chen JG (2012) Review of Pt-based bimetallic catalysis: from model surfaces to supported catalysts. Chem Rev 112(11):5780–5817
Liu X, Li X, Wang D, Yu R, Cui Y, Peng Q, Li Y (2012) Palladium/tin bimetallic single-crystalline hollow nanospheres. Chem Commun 48(11):1683–1685
Tao F, Grass ME, Zhang Y, Butcher DR, Renzas JR, Liu Z, Chung JY, Mun BS, Salmeron M, Somorjai GA (2008) Reaction-driven restructuring of Rh-Pd and Pt-Pd core-shell nanoparticles. Science 322(5903):932–934
Lim B, Jiang M, Camargo PHC, Cho EC, Tao J, Lu X, Zhu Y, Xia Y (2009) Pd-Pt bimetallic nanodendrites with high activity for oxygen reduction. Science 324(5932):1302–1305
González E, Arbiol J, Puntes VF (2011) Carving at the nanoscale: sequential galvanic exchange and Kirkendall growth at room temperature. Science 334(6061):1377–1380
Gasteiger HA, Kocha SS, Sompalli B, Wagner FT (2005) Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal B Environ 56(1–2):9–35
Norskov JK, Bligaard T, Rossmeisl J, Christensen CH (2009) Towards the computational design of solid catalysts. Nat Chem 1(1):37–46
Wagner FT, Lakshmanan B, Mathias MF (2010) Electrochemistry and the future of the automobile. J Phys Chem Lett 1(14):2204–2219
Greeley J, Stephens IEL, Bondarenko AS, Johansson TP, Hansen HA, Jaramillo TF, Rossmeisl J, Chorkendorff I, Nørskov JK (2009) Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat Chem 1(7):552–556
Chen A, Holt-Hindle P (2010) Platinum-based nanostructured materials: synthesis, properties, and applications. Chem Rev 110(6):3767–3804
Debe MK (2012) Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486(7401):43–51
Lamy C, Lima A, LeRhun V, Delime F, Coutanceau C, Léger J-M (2002) Recent advances in the development of direct alcohol fuel cells (DAFC). J Power Sources 105(2):283–296
Marković NM, Schmidt TJ, Stamenković V, Ross PN (2001) Oxygen reduction reaction on Pt and Pt bimetallic surfaces: a selective review. Fuel Cells 1(2):105–116
Gasteiger HA, Markovic N, Ross PN, Cairns E (1994) J Electrochim Acta 39:1825
Marković NM, Ross PN Jr (2002) Surface science studies of model fuel cell electrocatalysts. Surf Sci Rep 45(4–6):117–229
Toda T, Igarashi H, Uchida H, Watanabe M (1999) Enhancement of the electroreduction of oxygen on Pt alloys with Fe, Ni, and Co. J Electrochem Soc 146(10):3750–3756
Oetjen HF, Schmidt VM, Stimming U, Trila F (1996) Performance data of a proton exchange membrane fuel cell using H2/CO as fuel gas. J Electrochem Soc 143(12):3838–3842
Beden BLC, Leger J-M (1992) Modern aspects of electrochemistry. Plenum Press, New York, 97
Adžić RR, Wang JX (1998) Configuration and site of O2 adsorption on the Pt(111) electrode surface. J Phys Chem B 102(45):8988–8993
Shukla AK, Raman RK (2003) Methanol-resistant oxygen-reduction catalysts for direct methanol fuel cells. Annu Rev Mater Res 33(1):155–168
Damjanovic A, Brusic V (1967) Electrode kinetics of oxygen reduction on oxide-free platinum electrodes. Electrochim Acta 12(6):615–628
Ticanelli E, Beery JG, Paffett MT, Gottesfeld S (1989) An electrochemical, ellipsometric, and surface science investigation of the PtRu bulk alloy surface. J Electroanal Chem Interfacial Electrochem 258(1):61–77
Frelink T, Visscher W, van Veen JAR (1995) On the role of Ru and Sn as promotors of methanol electro-oxidation over Pt. Surf Sci 335:353–360
Watanabe M, Motoo S (1975) Electrocatalysis by ad-atoms: part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J Electroanal Chem Interfacial Electrochem 60(3):267–273
Davies JC, Hayden BE, Pegg DJ (1998) The electrooxidation of carbon monoxide on ruthenium modified Pt(110). Electrochim Acta 44(6–7):1181–1190
Maroun F, Ozanam F, Magnussen OM, Behm RJ (2001) The role of atomic ensembles in the reactivity of bimetallic electrocatalysts. Science 293(5536):1811–1814
Gasteiger HA, Markovic N, Ross PN, Cairns EJ (1993) Methanol electrooxidation on well-characterized platinum-ruthenium bulk alloys. J Phys Chem 97(46):12020–12029
Jiang X, Parmeter JE, Estrada CA, Goodman DW (1991) The adsorption and decomposition of methanol on copper overlayers on the Rh(100) surface. Surf Sci 249(1–3):44–60
Goodenough JB, Manoharan R, Shukla AK, Ramesh KV (1989) Intraalloy electron transfer and catalyst performance: a spectroscopic and electrochemical study. Chem Mater 1(4):391–398
Goodenough JB, Hamnett A, Kennedy BJ, Manoharan R, Weeks SA (1988) Methanol oxidation on unsupported and carbon supported Pt + Ru anodes. J Electroanal Chem Interfacial Electrochem 240(1–2):133–145
Bertolini JC (1996) Local order at the surface of binary alloys in relation to their chemical reactivity. Surf Rev Lett 03(05n06):1857–1868
Lai F-J, Chou H-L, Sarma LS, Wang D-Y, Lin Y-C, Lee J-F, Hwang B-J, Chen C-C (2010) Tunable properties of PtxFe1-x electrocatalysts and their catalytic activity towards the oxygen reduction reaction. Nanoscale 2(4):573–581
Lisiecki I (2005) Size, shape, and structural control of metallic nanocrystals. J Phys Chem B 109(25):12231–12244
Gasteiger HA, Markovic N, Ross PN, Cairns EJ (1994) Carbon monoxide electrooxidation on well-characterized platinum-ruthenium alloys. J Phys Chem 98(2):617–625
Markovic NM, Gasteiger HA, Ross PN (1995) Oxygen reduction on platinum low-index single-crystal surfaces in sulfuric acid Solution: rotating ring-Pt(hkl) disk studies. J Phys Chem 99(11):3411–3415
Alexandridis P, Holzwarth JF, Hatton TA (1995) Thermodynamics of droplet clustering in percolating AOT water-in-oil microemulsions. J Phys Chem 99(20):8222–8232
Lin WF, Iwasita T, Vielstich W (1999) Catalysis of CO electrooxidation at Pt, Ru, and PtRu alloy. An in situ FTIR study. J Phys Chem B 103(16):3250–3257
Park S, Wieckowski A, Weaver MJ (2003) Electrochemical infrared characterization of CO domains on ruthenium-decorated platinum nanoparticles. J Am Chem Soc 125(8):2282–2290
Paulus UA, Wokaun A, Scherer GG, Schmidt TJ, Stamenkovic V, Radmilovic V, Markovic NM, Ross PN (2002) Oxygen reduction on carbon-supported Pt−Ni and Pt−Co alloy catalysts. J Phys Chem B 106(16):4181–4191
Stamenković V, Schmidt TJ, Ross PN, Marković NM (2003) Surface segregation effects in electrocatalysis: kinetics of oxygen reduction reaction on polycrystalline Pt3Ni alloy surfaces. J Electroanal Chem 554–555:191–199
Gupta G, Slanac DA, Kumar P, Wiggins-Camacho JD, Wang X, Swinnea S, More KL, Dai S, Stevenson KJ, Johnston KP (2009) Highly stable and active Pt−Cu oxygen reduction electrocatalysts based on mesoporous graphitic carbon supports. Chem Mater 21(19):4515–4526
Peng Z, Yang H (2009) Synthesis and oxygen reduction electrocatalytic property of Pt-on-Pd bimetallic heteronanostructures. J Am Chem Soc 131(22):7542–7543
Sarma LS, Lin TD, Tsai Y-W, Chen JM, Hwang BJ (2005) Carbon-supported Pt–Ru catalysts prepared by the Nafion stabilized alcohol-reduction method for application in direct methanol fuel cells. J Power Sources 139(1–2):44–54
Kim J, Lee Y, Sun S (2010) Structurally ordered FePt nanoparticles and their enhanced catalysis for oxygen reduction reaction. J Am Chem Soc 132(14):4996–4997
Lee K-S, Jeon T-Y, Yoo SJ, Park I-S, Cho Y-H, Kang SH, Choi KH, Sung Y-E (2011) Effect of PtRu alloying degree on electrocatalytic activities and stabilities. Appl Catal B Environ 102(1–2):334–342
Lee W-R, Kim MG, Choi J-R, Park J-I, Ko SJ, Oh SJ, Cheon J (2005) Redox−transmetalation process as a generalized synthetic strategy for core−shell magnetic nanoparticles. J Am Chem Soc 127(46):16090–16097
Park J-I, Kim MG, Jun Y-W, Lee JS, Lee W-R, Cheon J (2004) Characterization of superparamagnetic “core−shell” nanoparticles and monitoring their anisotropic phase transition to ferromagnetic “solid solution” nanoalloys. J Am Chem Soc 126(29):9072–9078
Park J-I, Cheon J (2001) Synthesis of “solid solution” and “core-shell” type cobalt−platinum magnetic nanoparticles via transmetalation reactions. J Am Chem Soc 123(24):5743–5746
Harpeness R, Gedanken A (2004) Microwave synthesis of core−shell gold/palladium bimetallic nanoparticles. Langmuir 20(8):3431–3434
Nath S, Praharaj S, Panigrahi S, Ghosh SK, Kundu S, Basu S, Pal T (2005) Synthesis and characterization of N,N-dimethyldodecylamine-capped Aucore-Pdshell nanoparticles in toluene. Langmuir 21(23):10405–10408
Srnová-Šloufová I, Vlčková B, Bastl Z, Hasslett TL (2004) Bimetallic (Ag)Au nanoparticles prepared by the seed growth method: two-dimensional assembling, characterization by energy dispersive X-ray analysis, X-ray photoelectron spectroscopy, and surface enhanced raman spectroscopy, and proposed mechanism of growth. Langmuir 20(8):3407–3415
Huang X, Tang S, Liu B, Ren B, Zheng N (2011) Enhancing the photothermal stability of plasmonic metal nanoplates by a core-shell architecture. Adv Mater 23(30):3420–3425
Zeng J, Yang J, Lee JY, Zhou W (2006) Preparation of carbon-supported core−shell Au−Pt nanoparticles for methanol oxidation reaction: the promotional effect of the Au core. J Phys Chem B 110(48):24606–24611
Hwang B-J, Sarma LS, Chen C-H, Bock C, Lai F-J, Chang S-H, Yen S-C, Liu D-G, Sheu H-S, Lee J-F (2008) Controlled synthesis and characterization of Rucore−Ptshell bimetallic nanoparticles. J Phys Chem C 112(50):19922–19929
Adzic RR, Zhang J, Sasaki K, Vukmirovic MB, Shao M, Wang JX, Nilekar AU, Mavrikakis M, Valerio JA, Uribe F (2007) Top Catal 46:249
Zhang J, Lima FHB, Shao MH, Sasaki K, Wang JX, Hanson J, Adzic RR (2005) Platinum monolayer on nonnoble metal−noble metal core−shell nanoparticle electrocatalysts for O2 reduction. J Phys Chem B 109(48):22701–22704
Zhang J, Vukmirovic MB, Xu Y, Mavrikakis M, Adzic RR (2005) Controlling the catalytic activity of platinum-monolayer electrocatalysts for oxygen reduction with different substrates. Angew Chem Int Ed 44(14):2132–2135
Zhang J, Mo Y, Vukmirovic MB, Klie R, Sasaki K, Adzic RR (2004) Platinum monolayer electrocatalysts for O2 reduction: Pt monolayer on Pd(111) and on carbon-supported Pd nanoparticles. J Phys Chem B 108(30):10955–10964
Sun Y, Xia Y (2004) Mechanistic study on the replacement reaction between silver nanostructures and chloroauric acid in aqueous medium. J Am Chem Soc 126(12):3892–3901
Chen J, Wiley B, McLellan J, Xiong Y, Li Z-Y, Xia Y (2005) Optical properties of Pd−Ag and Pt−Ag nanoboxes synthesized via galvanic replacement reactions. Nano Lett 5(10):2058–2062
Chen J, McLellan JM, Siekkinen A, Xiong Y, Li Z-Y, Xia Y (2006) Facile synthesis of gold−silver nanocages with controllable pores on the surface. J Am Chem Soc 128(46):14776–14777
Graf C, van Blaaderen A (2001) Metallodielectric colloidal core−shell particles for photonic applications. Langmuir 18(2):524–534
Lu L, Sun G, Xi S, Wang H, Zhang H, Wang T, Zhou X (2003) A colloidal templating method to hollow bimetallic nanostructures. Langmuir 19(7):3074–3077
Kim S-W, Kim M, Lee WY, Hyeon T (2002) Fabrication of hollow palladium spheres and their successful application to the recyclable heterogeneous catalyst for Suzuki coupling reactions. J Am Chem Soc 124(26):7642–7643
Mayers B, Jiang X, Sunderland D, Cattle B, Xia Y (2003) Hollow nanostructures of platinum with controllable dimensions can be synthesized by templating against selenium nanowires and colloids. J Am Chem Soc 125(44):13364–13365
Chen Z, Zhan P, Wang ZL, Zhang JH, Zhang WY, Ming NB, Chan CT, Sheng P (2004) Two-and three-dimensional ordered structures of hollow silver spheres prepared by colloidal crystal templating. Adv Mater 16(5):417–422
Sun Y, Mayers BT, Xia Y (2002) Template-engaged replacement reaction: a one-step approach to the large-scale synthesis of metal nanostructures with hollow interiors. Nano Lett 2(5):481–485
Zhang J, Zhan P, Liu H, Wang Z, Ming N (2006) A facile colloidal templating method to monodisperse hollow Ag and Ag/Au submicrometer spheres. Mater Lett 60(2):280–283
Lu X, Tuan H-Y, Chen J, Li Z-Y, Korgel BA, Xia Y (2007) Mechanistic studies on the galvanic replacement reaction between multiply twinned particles of Ag and HAuCl4 in an organic medium. J Am Chem Soc 129(6):1733–1742
Qingbo Z, Jim Yang L, Jun Y, Chris B, Jixuan Z (2007) Size and composition tunable Ag–Au alloy nanoparticles by replacement reactions. Nanotechnology 18(24):245605
Fan HJ, Gösele U, Zacharias M (2007) Formation of nanotubes and hollow nanoparticles based on Kirkendall and diffusion processes: a review. Small 3(10):1660–1671
Yu X, Wang D, Peng Q, Li Y (2011) High performance electrocatalyst: Pt-Cu hollow nanocrystals. Chem Commun 47(28):8094–8096
Liu X, Wang D, Li Y (2012) Synthesis and catalytic properties of bimetallic nanomaterials with various architectures. Nano Today 7(5):448–466
Schmidt TJ, Noeske M, Gasteiger HA, Behm RJ, Britz P, Brijoux W, Bönnemann H (1997) Electrocatalytic activity of PtRu alloy colloids for CO and CO/H2 electrooxidation: stripping voltammetry and rotating disk measurements. Langmuir 13(10):2591–2595
Vogel W, Britz P, Bönnemann H, Rothe J, Hormes J (1997) Structure and chemical composition of surfactant-stabilized PtRu alloy colloids. J Phys Chem B 101(51):11029–11036
Zhang X, Chan K-Y (2002) Water-in-oil microemulsion synthesis of platinum−ruthenium nanoparticles, their characterization and electrocatalytic properties. Chem Mater 15(2):451–459
Liu Y, Qiu X, Chen Z, Zhu W (2002) A new supported catalyst for methanol oxidation prepared by a reverse micelles method. Electrochem Commun 4(7):550–553
Boxall DL, Deluga GA, Kenik EA, King WD, Lukehart CM (2001) Rapid synthesis of a Pt1Ru1/carbon nanocomposite using microwave irradiation: a DMFC anode catalyst of high relative performance. Chem Mater 13(3):891–900
Wang X, Hsing IM (2002) Surfactant stabilized Pt and Pt alloy electrocatalyst for polymer electrolyte fuel cells. Electrochim Acta 47(18):2981–2987
Bock C, Paquet C, Couillard M, Botton GA, MacDougall BR (2004) Size-selected synthesis of PtRu nano-catalysts: reaction and size control mechanism. J Am Chem Soc 126(25):8028–8037
Liu Z, Lee JY, Chen W, Han M, Gan LM (2003) Physical and electrochemical characterizations of microwave-assisted polyol preparation of carbon-supported PtRu nanoparticles. Langmuir 20(1):181–187
Knupp S, Vukmirovic M, Haldar P, Herron J, Mavrikakis M, Adzic R (2010) Platinum monolayer electrocatalysts for O2 reduction: Pt monolayer on carbon-supported PdIr nanoparticles. Electrocatalysis 1(4):213–223
Koenigsmann C, Santulli AC, Gong K, Vukmirovic MB, Zhou W-P, Sutter E, Wong SS, Adzic RR (2011) Enhanced electrocatalytic performance of processed, ultrathin, supported Pd–Pt core–shell nanowire catalysts for the oxygen reduction reaction. J Am Chem Soc 133(25):9783–9795
Frenkel AI, Yevick A, Cooper C, Vasic R (2011) Modeling the structure and composition of nanoparticles by extended X-ray absorption fine-structure spectroscopy. Annu Rev Anal Chem 4(1):23–39
Frenkel AI, Hills CW, Nuzzo RG (2001) A view from the inside: complexity in the atomic scale ordering of supported metal nanoparticles. J Phys Chem B 105(51):12689–12703
Nashner MS, Frenkel AI, Somerville D, Hills CW, Shapley JR, Nuzzo RG (1998) Core shell inversion during nucleation and growth of bimetallic Pt/Ru nanoparticles. J Am Chem Soc 120(32):8093–8101
Russell AE, Rose A (2004) X-ray absorption spectroscopy of low temperature fuel cell catalysts. Chem Rev 104(10):4613–4636
Ashcroft AT, Cheetham AK, Harris PJF, Jones RH, Natarajan S, Sankar G, Stedman NJ, Thomas JM (1994) Particle size studies of supported metal catalysts: a comparative study by X-ray diffraction, EXAFS and electron microscopy. Catal Lett 24(1–2):47–57
Cao D, Bergens SH (2004) Pt–Ruadatom nanoparticles as anode catalysts for direct methanol fuel cells. J Power Sources 134(2):170–180
Link S, Wang ZL, El-Sayed MA (1999) Alloy formation of gold−silver nanoparticles and the dependence of the plasmon absorption on their composition. J Phys Chem B 103(18):3529–3533
Torigoe K, Esumi K (1993) Preparation of bimetallic silver-palladium colloids from silver(I) bis(oxalato)palladate(II). Langmuir 9(7):1664–1667
Torigoe K, Nakajima Y, Esumi K (1993) Preparation and characterization of colloidal silver-platinum alloys. J Phys Chem 97(31):8304–8309
Zhao D, Xu B-Q (2006) Enhancement of Pt utilization in electrocatalysts by using gold nanoparticles. Angew Chem Int Ed 45(30):4955–4959
Henglein A (2000) Preparation and optical aborption spectra of AucorePtshell and PtcoreAushell colloidal nanoparticles in aqueous solution. J Phys Chem B 104(10):2201–2203
Nashner MS, Frenkel AI, Adler DL, Shapley JR, Nuzzo RG (1997) Structural characterization of carbon-supported platinum−ruthenium nanoparticles from the molecular cluster precursor PtRu5C(CO)16. J Am Chem Soc 119(33):7760–7771
Mulvaney P (1996) Surface plasmon spectroscopy of nanosized metal particles. Langmuir 12(3):788–800
D’Souza L, Bera P, Sampath S (2002) Silver-palladium nanodispersions in silicate matrices: highly uniform, stable, bimetallic structures. J Colloid Interface Sci 246(1):92–99
Zhang Q, Xie J, Lee JY, Zhang J, Boothroyd C (2008) Synthesis of Ag@AgAu metal core/alloy shell bimetallic nanoparticles with tunable shell compositions by a galvanic replacement reaction. Small 4(8):1067–1071
Chen J-M, Sarma LS, Chen C-H, Cheng M-Y, Shih S-C, Wang G-R, Liu D-G, Lee J-F, Tang M-T, Hwang B-J (2006) Multi-scale dispersion in fuel cell anode catalysts: role of TiO2 towards achieving nanostructured materials. J Power Sources 159(1):29–33
Iwasita T, Hoster H, John-Anacker A, Lin WF, Vielstich W (1999) Methanol oxidation on PtRu electrodes. influence of surface structure and Pt−Ru atom distribution. Langmuir 16(2):522–529
Radmilovic V, Gasteiger HA, Ross PN (1995) Structure and chemical composition of a supported Pt-Ru electrocatalyst for methanol oxidation. J Catal 154(1):98–106
Wang D, Zhuang L, Lu J (2007) An alloying-degree-controlling step in the impregnation synthesis of PtRu/C catalysts. J Phys Chem C 111(44):16416–16422
Aricò AS, Cretı P, Modica E, Monforte G, Baglio V, Antonucci V (2000) Investigation of direct methanol fuel cells based on unsupported Pt–Ru anode catalysts with different chemical properties. Electrochim Acta 45(25–26):4319–4328
Rolison DR, Hagans PL, Swider KE, Long JW (1999) Role of hydrous ruthenium oxide in Pt−Ru direct methanol fuel cell anode electrocatalysts: the importance of mixed electron/proton conductivity. Langmuir 15(3):774–779
Gurau B, Viswanathan R, Liu R, Lafrenz TJ, Ley KL, Smotkin ES, Reddington E, Sapienza A, Chan BC, Mallouk TE, Sarangapani S (1998) Structural and electrochemical characterization of binary, ternary, and quaternary platinum alloy catalysts for methanol electro-oxidation 1. J Phys Chem B 102(49):9997–10003
Antolini E, Cardellini F, Giorgi L, Passalacqua E (2000) Effect of Me (Pt+Ru) content in Me/C catalysts on PtRu alloy formation: an XRD analysis. J Mater Sci Lett 19(23):2099–2103
Antolini E, Cardellini F (2001) Formation of carbon supported PtRu alloys: an XRD analysis. J Alloys Compd 315(1–2):118–122
Gnutzmann V, Vogel W (1990) Structural sensitivityof the standard platinum/silica catalyst EuroPt-1 to hydrogen and oxygen exposure by in situ x-ray diffraction. J Phys Chem 94(12):4991–4997
Vogel W, Rosner B, Tesche B (1993) Structural investigations of gold (Au55) organometallic complexes by x-ray powder diffraction and transmission electron microscopy. J Phys Chem 97(45):11611–11616
Bommarius AS, Holzwarth JF, Wang DIC, Hatton TA (1990) Coalescence and solubilizate exchange in a cationic four-component reversed micellar system. J Phys Chem 94(18):7232–7239
Dobrosz I, Jiratova K, Pitchon V, Rynkowski JM (2005) Effect of the preparation of supported gold particles on the catalytic activity in CO oxidation reaction. J Mol Catal A Chem 234(1–2):187–197
Ferrando R, Jellinek J, Johnston RL (2008) Nanoalloys: from theory to applications of alloy clusters and nanoparticles. Chem Rev 108(3):845–910
Bi Y, Lu G (2008) Facile synthesis of platinum nanofiber/nanotube junction structures at room temperature. Chem Mater 20(4):1224–1226
Kawaguchi T, Sugimoto W, Murakami Y, Takasu Y (2005) Particle growth behavior of carbon-supported Pt, Ru, PtRu catalysts prepared by an impregnation reductive-pyrolysis method for direct methanol fuel cell anodes. J Catal 229(1):176–184
Bauer A, Gyenge EL, Oloman CW (2006) Electrodeposition of Pt–Ru nanoparticles on fibrous carbon substrates in the presence of nonionic surfactant: application for methanol oxidation. Electrochim Acta 51(25):5356–5364
Park K-W, Sung Y-E, Han S, Yun Y, Hyeon T (2004) Origin of the enhanced catalytic activity of carbon nanocoil-supported PtRu alloy electrocatalysts. J Phys Chem B 108(3):939–944
Rodrıguez-Nieto FJ, Morante-Catacora TY, Cabrera CR (2004) Sequential and simultaneous electrodeposition of Pt–Ru electrocatalysts on a HOPG substrate and the electro-oxidation of methanol in aqueous sulfuric acid. J Electroanal Chem 571(1):15–26
Schmidt TJ, Noeske M, Gasteiger HA, Behm RJ, Britz P, Bönnemann H (1998) PtRu alloy colloids as precursors for fuel cell catalysts: a combined XPS, AFM, HRTEM, and RDE study. J Electrochem Soc 145(3):925–931
Liu D-G, Lee J-F, Tang M-T (2005) Characterization of Pt-Ru/C catalysts by X-ray absorption spectroscopy and temperature-programmed surface reaction. J Mol Catal A Chem 240(1–2):197–206
Russell AE, Maniguet S, Mathew RJ, Yao J, Roberts MA, Thompsett D (2001) In situ X-ray absorption spectroscopy and X-ray diffraction of fuel cell electrocatalysts. J Power Sources 96(1):226–232
O’Grady WE, Hagans PL, Pandya KI, Maricle DL (2001) Structure of Pt/Ru catalysts using X-ray absorption near edge structure studies. Langmuir 17(10):3047–3050
Harada M, Toshima N, Yoshida K, Isoda S (2005) Aggregated structure analysis of polymer-protected platinum/ruthenium colloidal dispersions using EXAFS, HRTEM, and electron diffraction measurements. J Colloid Interface Sci 283(1):64–78
Mukerjee S, Urian RC (2002) Bifunctionality in Pt alloy nanocluster electrocatalysts for enhanced methanol oxidation and CO tolerance in PEM fuel cells: electrochemical and in situ synchrotron spectroscopy. Electrochim Acta 47(19):3219–3231
Frenkel AI (2012) Applications of extended X-ray absorption fine-structure spectroscopy to studies of bimetallic nanoparticle catalysts. Chem Soc Rev 41(24):8163–8178
Bazin DC, Sayers DA, Rehr JJ (1997) Comparison between X-ray absorption spectroscopy, anomalous wide angle X-ray scattering, anomalous small angle X-ray scattering, and diffraction anomalous fine structure Techniques applied to nanometer-scale metallic clusters. J Phys Chem B 101(51):11040–11050
Via GH, Sinfelt JH (1996) X-ray absorption fine structure for catalysts and surfaces (ed: Iwasawa Y). World Scientific, London
Lin SD, Hsiao T-C, Chang J-R, Lin AS (1999) Morphology of carbon supported Pt−Ru electrocatalyst and the CO tolerance of anodes for PEM fuel cells. J Phys Chem B 103(1):97–103
Greegor RB, Lytle FW (1980) Morphology of supported metal clusters: determination by EXAFS and chemisorption. J Catal 63(2):476–486
Yoshitake H, Yamazaki O, Ota K (1994) In situ X‐ray absorption fine structure study on structure transformation and electronic state of various Pt particles on carbon electrode. J Electrochem Soc 141(9):2516–2521
Siepen K, Bönnemann H, Brijoux W, Rothe J, Hormes J (2000) EXAFS/XANES, chemisorption and IR investigations of colloidal Pt/Rh bimetallic catalysts. Appl Organomet Chem 14(10):549–556
Horsley JA (1982) Relationshipbetween the area of L2,3X-ray absorption edge resonances and the d orbital occupancy in compounds of platinum and iridium. J Chem Phys 76(3):1451–1458
Mansour AN, Sayers DE, Cook JW, Short DR, Shannon RD, Katzer JR (1984) X-ray absorption studies of some platinum oxides. J Phys Chem 88(9):1778–1781
Brown M, Peierls RE, Stern EA (1977) White lines in x-ray absorption. Phys Rev B 15(2):738–744
Mukerjee S, Srinivasan S, Soriaga MP, McBreen J (1995) Role of structural and electronic properties of Pt and Pt alloys on electrocatalysis of oxygen reduction: an in situ XANES and EXAFS investigation. J Electrochem Soc 142(5):1409–1422
Min M-K, Cho J, Cho K, Kim H (2000) Particle size and alloying effects of Pt-based alloy catalysts for fuel cell applications. Electrochim Acta 45(25–26):4211–4217
Markovic N, Gasteiger H, Ross PN (1997) Kinetics of oxygen reduction on Pt(hkl) electrodes: implications for the crystallite size effect with supported Pt electrocatalysts. J Electrochem Soc 144(5):1591–1597
Hwang B-J, Sarma LS, Wang G-R, Chen C-H, Liu D-G, Sheu H-S, Lee J-F (2007) Heat-induced alterations in the surface population of metal sites in bimetallic nanoparticles. Chem Eur J 13(21):6255–6264
Reetz MT, Lopez M, Grünert W, Vogel W, Mahlendorf F (2003) Preparation of colloidal nanoparticles of mixed metal oxides containing platinum, ruthenium, osmium, and iridium and their use as electrocatalysts. J Phys Chem B 107(30):7414–7419
Park K-W, Choi J-H, Sung Y-E (2003) Structural, chemical, and electronic properties of Pt/Ni thin film electrodes for methanol electrooxidation. J Phys Chem B 107(24):5851–5856
Kao CC, Tsai SC, Bahl MK, Chung YW, Lo WJ (1980) Electronic properties, structure and temperature-dependent composition of nickel deposited on rutile titanium dioxide (110) surfaces. Surf Sci 95(1):1–14
Eberhardt W, Fayet P, Cox DM, Fu Z, Kaldor A, Sherwood R, Sondericker D (1990) Photoemission from mass-selected monodispersed Pt clusters. Phys Rev Lett 64(7):780–783
Zhang X, Chan K-Y (2003) Water-in-oil microemulsion synthesis of platinum−ruthenium nanoparticles, their characterization and electrocatalytic properties. Chem Mater 15(2):451–459
Phung X, Groza J, Stach EA, Williams LN, Ritchey SB (2003) Surface characterization of metal nanoparticles. Mater Sci Eng A 359(1–2):261–268
Tao F, Grass ME, Zhang Y, Butcher DR, Aksoy F, Aloni S, Altoe V, Alayoglu S, Renzas JR, Tsung C-K, Zhu Z, Liu Z, Salmeron M, Somorjai GA (2010) Evolution of structure and chemistry of bimetallic nanoparticle catalysts under reaction conditions. J Am Chem Soc 132(25):8697–8703
Stamenkovic VR, Mun BS, Mayrhofer KJJ, Ross PN, Markovic NM (2006) Effect of surface composition on electronic structure, stability, and electrocatalytic properties of Pt-transition metal alloys: Pt-skin versus Pt-skeleton surfaces. J Am Chem Soc 128(27):8813–8819
Tremiliosi-Filho G, Kim H, Chrzanowski W, Wieckowski A, Grzybowska B, Kulesza P (1999) Reactivity and activation parameters in methanol oxidation on platinum single crystal electrodes ‘decorated’ by ruthenium adlayers. J Electroanal Chem 467(1–2):143–156
Tong Y, Oldfield E, Wieckowski A (1998) Peer reviewed: exploring electrochemical interfaces with solid-state NMR. Anal Chem 70(15):518A–527A
Babu PK, Tong YY, Kim HS, Wieckowski A (2002) Nanostructured electrode surfaces studied by electrochemical NMR. J Electroanal Chem 524–525:157–167
Babu PK, Kim HS, Oldfield E, Wieckowski A (2003) Electronic alterations caused by ruthenium in Pt−Ru alloy nanoparticles as revealed by electrochemical NMR. J Phys Chem B 107(31):7595–7600
Lu C, Rice C, Masel RI, Babu PK, Waszczuk P, Kim HS, Oldfield E, Wieckowski A (2002) UHV, electrochemical NMR, and electrochemical studies of platinum/ruthenium fuel cell catalysts. J Phys Chem B 106(37):9581–9589
Tong YY, Yonezawa T, Toshima N, van der Klink JJ (1996) 195Pt NMR of polymer-protected Pt/Pd bimetallic catalysts. J Phys Chem 100(2):730–733
Huang S-Y, Chang S-M, Yeh C-T (2005) Characterization of surface composition of platinum and ruthenium nanoalloys dispersed on active carbon. J Phys Chem B 110(1):234–239
Huang S-Y, Chang S-M, Lin C-L, Chen C-H, Yeh C-T (2006) Promotion of the electrochemical activity of a bimetallic platinum−ruthenium catalyst by oxidation-induced segregation. J Phys Chem B 110(46):23300–23305
Wang K-W, Huang S-Y, Yeh C-T (2007) Promotion of carbon-supported platinum−ruthenium catalyst for electrodecomposition of methanol. J Phys Chem C 111(13):5096–5100
Bard AJ, Faulkner LR (2000) Electrochemical methods: fundamentals and applications, 2nd edn. Wiley, New York
Zoski CG (2006) Handbook of electrochemistry, 1st edn. Elsevier
Sawyer DT, Roberts JL (1974) Experimental electrochemistry for chemist. Wiley, New York
Rieger PH (1994) Electrochemistry, 2nd edn. Prentice-Hall
Pan C-J, Su W-N, Kumar SMS, Al Andra CC, Yang S-J, Chen H-Y, Hwang BJ (2012) An electrochemical approach for estimation of intrinsic active area and activation of Pt/C nano-catalysts. J Chin Chem Soc 59(10):1303–1312
Hwang BJ, Kumar SMS, Chen C-H, Monalisa XX, Cheng M-Y, Liu D-G, Lee J-F (2007) An Investigation of structure−catalytic activity relationship for Pt−Co/C bimetallic nanoparticles toward the oxygen reduction reaction. J Phys Chem C 111(42):15267–15276
Schmidt TJ, Gasteiger HA, Stäb GD, Urban PM, Kolb DM, Behm RJ (1998) Characterization of high‐surface‐area electrocatalysts using a rotating disk electrode configuration. J Electrochem Soc 145(7):2354–2358
Mayrhofer KJJ, Strmcnik D, Blizanac BB, Stamenkovic V, Arenz M, Markovic NM (2008) Measurement of oxygen reduction activities via the rotating disc electrode method: from Pt model surfaces to carbon-supported high surface area catalysts. Electrochim Acta 53(7):3181–3188
Wang JX, Markovic NM, Adzic RR (2004) Kinetic analysis of oxygen reduction on Pt(111) in acid solutions: intrinsic kinetic parameters and anion adsorption effects. J Phys Chem B 108(13):4127–4133
Levich VG (1962) Physical hydrodynamics. Prentice-Hall, Englewood Cliffs
Watt-Smith MJ, Friedrich JM, Rigby SP, Ralph TR, Walsh FC (2008) Determination of the electrochemically active surface area of Pt/C PEM fuel cell electrodes using different adsorbates. J Phys D Appl Phys 41(17):174004
Vidaković T, Christov M, Sundmacher K (2007) The use of CO stripping for in situ fuel cell catalyst characterization. Electrochim Acta 52(18):5606–5613
Nagel T, Bogolowski N, Baltruschat H (2006) Towards a determination of the active surface area of polycrystalline and nanoparticle electrodes by Cu upd and CO oxidation. J Appl Electrochem 36(11):1297–1306
Jusys Z, Kaiser J, Behm RJ (2002) Composition and activity of high surface area PtRu catalysts towards adsorbed CO and methanol electrooxidation: a DEMS study. Electrochim Acta 47(22–23):3693–3706
Dinh HN, Ren X, Garzon FH, Piotr Z, Gottesfeld S (2000) Electrocatalysis in direct methanol fuel cells: in-situ probing of PtRu anode catalyst surfaces. J Electroanal Chem 491(1–2):222–233
Paseka I (2007) Adsorption of CO on Ru and Pt blacks and catalysts and the possibility of its utilization for the determination of the ruthenium-free surface. J Solid State Electrochem 11(1):52–58
Rush BM, Reimer JA, Cairns EJ (2001) Nuclear magnetic resonance and voltammetry studies of carbon monoxide adsorption and oxidation on a carbon-supported platinum fuel cell electrocatalyst. J Electrochem Soc 148(2):A137–A148
Pozio A, De Francesco M, Cemmi A, Cardellini F, Giorgi L (2002) Comparison of high surface Pt/C catalysts by cyclic voltammetry. J Power Sources 105(1):13–19
Thiel KO, Hintze M, Vollmer A, Donner C (2008) A new approach on the Cu UPD on Ag surfaces. J Electroanal Chem 621(1):7–12
Hernandez F, Baltruschat H (2007) Hydrogen evolution and Cu UPD at stepped gold single crystals modified with Pd. J Solid State Electrochem 11(7):877–885
Green CL, Kucernak A (2002) Determination of the platinum and ruthenium surface areas in platinum−ruthenium alloy electrocatalysts by underpotential deposition of copper. I. Unsupported catalysts. J Phys Chem B 106(5):1036–1047
Green CL, Kucernak A (2002) Determination of the platinum and ruthenium surface areas in platinum−ruthenium electrocatalysts by underpotential deposition of copper. 2. Effect of surface composition on activity. J Phys Chem B 106(44):11446–11456
Mascaro LH, Santos MC, Machado SAS, Avaca LA (1997) Underpotential deposition of silver on polycrystalline platinum studied by cyclic voltammetry and rotating ring-disc techniques. J Chem Soc Faraday Trans 93(22):3999–4003
Takakusagi S, Kitamura K, Uosaki K (2008) In situ real-time monitoring of electrochemical Ag deposition on a reconstructed Au(111) surface studied by scanning tunneling microscopy. J Phys Chem C 112(8):3073–3077
Uhm S, Chung ST, Lee J (2007) Activity of Pt anode catalyst modified by underpotential deposited Pb in a direct formic acid fuel cell. Electrochem Commun 9(8):2027–2031
Lee JK, Jeon H, Uhm S, Lee J (2008) Influence of underpotentially deposited Sb onto Pt anode surface on the performance of direct formic acid fuel cells. Electrochim Acta 53(21):6089–6092
Lamy-Pitara E, Barbier J (1997) Platinum modified by electrochemical deposition of adatoms. Appl Catal A Gen 149(1):49–87
Kolb DM, Przasnyski M, Gerischer H (1974) Underpotential deposition of metals and work function differences. J Electroanal Chem Interfacial Electrochem 54(1):25–38
Kolb DM (1978) Advances in electrochemistry and electrochemical engineering, vol 11. John Wiley Interscience, New York, p 125
Aramata A (1997) Underpotential deposition on single-crystal metals, modern aspects of electrochemistry, vol 31. Plenum Press, New York, p 181
Kaplan D, Alon M, Burstein L, Rosenberg Y, Peled E (2011) Study of core–shell platinum-based catalyst for methanol and ethylene glycol oxidation. J Power Sources 196(3):1078–1083
Jiang X, Gür TM, Prinz FB, Bent SF (2010) Atomic layer deposition (ALD) co-deposited Pt−Ru binary and pt skin catalysts for concentrated methanol oxidation. Chem Mater 22(10):3024–3032
Maillard F, Lu GQ, Wieckowski A, Stimming U (2005) Ru-decorated Pt surfaces as model fuel cell electrocatalysts for CO electrooxidation. J Phys Chem B 109(34):16230–16243
Kristian N, Yu Y, Gunawan P, Xu R, Deng W, Liu X, Wang X (2009) Controlled synthesis of Pt-decorated Au nanostructure and its promoted activity toward formic acid electro-oxidation. Electrochim Acta 54(21):4916–4924
Ramirez-Caballero GE, Balbuena PB (2008) Surface segregation of core atoms in core–shell structures. Chem Phys Lett 456(1–3):64–67
Zhang J, Vukmirovic MB, Sasaki K, Nilekar AU, Mavrikakis M, Adzic RR (2005) Mixed-metal Pt monolayer electrocatalysts for enhanced oxygen reduction kinetics. J Am Chem Soc 127(36):12480–12481
Zhang J, Sasaki K, Sutter E, Adzic RR (2007) Stabilization of platinum oxygen-reduction electrocatalysts using gold clusters. Science 315(5809):220–222
Zhang S, Shao Y, Yin G, Lin Y (2010) Electrostatic self-assembly of a pt-around-au nanocomposite with high activity towards formic acid oxidation. Angew Chem Int Ed 49(12):2211–2214
Stamenkovic VR, Mun BS, Arenz M, Mayrhofer KJJ, Lucas CA, Wang G, Ross PN, Markovic NM (2007) Trends in electrocatalysis on extended and nanoscale Pt-bimetallic alloy surfaces. Nat Mater 6(3):241–247
Chen S, Sheng W, Yabuuchi N, Ferreira PJ, Allard LF, Shao-Horn Y (2008) Origin of oxygen reduction reaction activity on “Pt3Co” nanoparticles: atomically resolved chemical compositions and structures. J Phys Chem C 113(3):1109–1125
Ball SC, Hudson SL, Leung JH, Russell AE, Thompsett D, Theobald BR (2007) Mechanisms of activity loss in PtCo alloy systems. ECS Trans 11(1):1247–1257
Narayanan R, El-Sayed MA (2004) Effect of nanocatalysis in colloidal solution on the tetrahedral and cubic nanoparticle SHAPE: electron-transfer reaction catalyzed by platinum nanoparticles. J Phys Chem B 108(18):5726–5733
Narayanan R, El-Sayed MA (2004) Shape-dependent catalytic activity of platinum nanoparticles in colloidal solution. Nano Lett 4(7):1343–1348
Kim Y, Hong JW, Lee YW, Kim M, Kim D, Yun WS, Han SW (2010) Synthesis of AuPt heteronanostructures with enhanced electrocatalytic activity toward oxygen reduction. Angew Chem Int Ed 49(52):10197–10201
Masatake H (1997) Size-and support-dependency in the catalysis of gold. Catal Today 36(1):153–166
Park S, Xie Y, Weaver MJ (2002) Electrocatalytic pathways on carbon-supported platinum nanoparticles: comparison of particle-size-dependent rates of methanol, formic acid, and formaldehyde electrooxidation. Langmuir 18(15):5792–5798
Mazumder V, Chi M, Mankin MN, Liu Y, Metin Ö, Sun D, More KL, Sun S (2012) A facile synthesis of MPd (M = Co, Cu) nanoparticles and their catalysis for formic acid oxidation. Nano Lett 12(2):1102–1106
Chang S-H, Su W-N, Yeh M-H, Pan C-J, Yu K-L, Liu D-G, Lee J-F, Hwang B-J (2010) Structural and electronic effects of carbon-supported PtxPd1−x nanoparticles on the electrocatalytic activity of the oxygen-reduction reaction and on methanol tolerance. Chem Eur J 16(36):11064–11071
Steele BCH, Heinzel A (2001) Materials for fuel-cell technologies. Nature 414(6861):345–352
Faur Ghenciu A (2002) Review of fuel processing catalysts for hydrogen production in PEM fuel cell systems. Curr Opinion Solid State Mater Sci 6(5):389–399
Costamagna P, Srinivasan S (2001) Quantum jumps in the PEMFC science and technology from the 1960s to the year 2000: part II. Engineering, technology development and application aspects. J Power Sources 102(1–2):253–269
Costamagna P, Srinivasan S (2001) Quantum jumps in the PEMFC science and technology from the 1960s to the year 2000: part I. Fundamental scientific aspects. J Power Sources 102(1–2):242–252
Ianniello R, Schmidt VM, Stimming U, Stumper J, Wallau A (1994) CO adsorption and oxidation on Pt and Pt-Ru alloys: dependence on substrate composition. Electrochim Acta 39(11–12):1863–1869
Castro Luna AM, Camara GA, Paganin VA, Ticianelli EA, Gonzalez ER (2000) Effect of thermal treatment on the performance of CO-tolerant anodes for polymer electrolyte fuel cells. Electrochem Commun 2(4):222–225
Divisek J, Oetjen HF, Peinecke V, Schmidt VM, Stimming U (1998) Components for PEM fuel cell systems using hydrogen and CO containing fuels. Electrochim Acta 43(24):3811–3815
Hwang BJ, Sarma LS, Chen CH, Tang MT, Liu DG, Lee JF (2007) Depth profile of alloying extent and composition in bimetallic nanoparticles investigated by in situ x-ray absorption spectroscopy. Appl Phys Lett 91(2):023108
Sarma LS, Chen C-H, Wang G-R, Hsueh K-L, Huang C-P, Sheu H-S, Liu D-G, Lee J-F, Hwang B-J (2007) Investigations of direct methanol fuel cell (DMFC) fading mechanisms. J Power Sources 167(2):358–365
Janssen GJM, de Heer MP, Papageorgopoulos DC (2004) Bilayer anodes for improved reformate tolerance of PEM fuel cells. Fuel Cells 4(3):169–174
Grgur BN, Markovic NM, Ross PN (1999) The electro‐oxidation of H2 and H2/CO mixtures on carbon‐supported Pt x Mo y alloy catalysts. J Electrochem Soc 146(5):1613–1619
Lebedeva NP, Janssen GJM (2005) On the preparation and stability of bimetallic PtMo/C anodes for proton-exchange membrane fuel cells. Electrochim Acta 51(1):29–40
Ball S, Hodgkinson A, Hoogers G, Maniguet S, Thompsett D, Wong B (2002) The proton exchange membrane fuel cell performance of a carbon supported PtMo catalyst operating on reformate. Electrochem Solid-State Lett 5(2):A31–A34
Götz M, Wendt H (1998) Binary and ternary anode catalyst formulations including the elements W, Sn and Mo for PEMFCs operated on methanol or reformate gas. Electrochim Acta 43(24):3637–3644
Santiago EI, Camara GA, Ticianelli EA (2003) CO tolerance on PtMo/C electrocatalysts prepared by the formic acid method. Electrochim Acta 48(23):3527–3534
Lee SJ, Mukerjee S, Ticianelli EA, McBreen J (1999) Electrocatalysis of CO tolerance in hydrogen oxidation reaction in PEM fuel cells. Electrochim Acta 44(19):3283–3293
Gasteiger HA, Markovic NM, Ross PN (1995) Electrooxidation of CO and H2/CO mixtures on a well-characterized Pt3Sn electrode surface. J Phys Chem 99(22):8945–8949
Wee J-H, Lee K-Y (2006) Overview of the development of CO-tolerant anode electrocatalysts for proton-exchange membrane fuel cells. J Power Sources 157(1):128–135
Garcia AC, Paganin VA, Ticianelli EA (2008) CO tolerance of PdPt/C and PdPtRu/C anodes for PEMFC. Electrochim Acta 53(12):4309–4315
Stonehart P (1984) Electrocatalyst advances for hydrogen oxidation in phosphoric acid fuel cells. Int J Hydrog Energy 9(11):921–928
Uchida H, Izumi K, Aoki K, Watanabe M (1771–1779) Temperature-dependence of hydrogen oxidation reaction rates and CO-tolerance at carbon-supported Pt, Pt-Co, and Pt-Ru catalysts. Phys Chem Chem Phys 2009:11(11)
Guilminot E, Fischer F, Chatenet M, Rigacci A, Berthon-Fabry S, Achard P, Chainet E (2007) Use of cellulose-based carbon aerogels as catalyst support for PEM fuel cell electrodes: electrochemical characterization. J Power Sources 166(1):104–111
Marie J, Berthon-Fabry S, Achard P, Chatenet M, Pradourat A, Chainet E (2004) Highly dispersed platinum on carbon aerogels as supported catalysts for PEM fuel cell-electrodes: comparison of two different synthesis paths. J Non-Cryst Solids 350:88–96
Zhou J-H, He J-P, Ji Y-J, Dang W-J, Liu X-L, Zhao G-W, Zhang C-X, Zhao J-S, Fu Q-B, Hu H-P (2007) CTAB assisted microwave synthesis of ordered mesoporous carbon supported Pt nanoparticles for hydrogen electro-oxidation. Electrochim Acta 52(14):4691–4695
Hwan Jung D, Hyeong Lee C, Soo Kim C, Ryul Shin D (1998) Performance of a direct methanol polymer electrolyte fuel cell. J Power Sources 71(1–2):169–173
McNicol BD, Rand DAJ, Williams KR (1999) Direct methanol–air fuel cells for road transportation. J Power Sources 83(1–2):15–31
Herrero E, Franaszczuk K, Wiecskowski A (1993) A voltammetric identification of the surface redox couple effective in methanol oxidation on a ruthenium-covered platinum (110) electrode. J Electroanal Chem 361(1–2):269–273
Ma L, Liu C, Liao J, Lu T, Xing W, Zhang J (2009) High activity PtRu/C catalysts synthesized by a modified impregnation method for methanol electro-oxidation. Electrochim Acta 54(28):7274–7279
Watanabe M, Motoo S (1975) Electrocatalysis by ad-atoms: part III. Enhancement of the oxidation of carbon monoxide on platinum by ruthenium ad-atoms. J Electroanal Chem Interfacial Electrochem 60(3):275–283
Watanabe M, Motoo S (1975) Electrocatalysis by ad-atoms: part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J Electroanal Chem Interfacial Electrochem 60(3):267–273
Hsieh C-T, Lin J-Y (2009) Fabrication of bimetallic Pt–M (M = Fe, Co, and Ni) nanoparticle/carbon nanotube electrocatalysts for direct methanol fuel cells. J Power Sources 188(2):347–352
Jiang Q-Z, Wu X, Shen M, Ma Z-F, Zhu X-Y (2008) Low-Pt content carbon-supported Pt–Ni–TiO2 nanotube electrocatalystfor direct methanol fuel cells. Catal Lett 124(3–4):434–438
Page T, Johnson R, Hormes J, Noding S, Rambabu B (2000) A study of methanol electro-oxidation reactions in carbon membrane electrodes and structural properties of Pt alloy electro-catalysts by EXAFS. J Electroanal Chem 485(1):34–41
Shubina TE, Koper MTM (2002) Quantum-chemical calculations of CO and OH interacting with bimetallic surfaces. Electrochim Acta 47(22–23):3621–3628
Dupont C, Jugnet Y, Loffreda D (2006) Theoretical evidence of PtSn alloy efficiency for CO oxidation. J Am Chem Soc 128(28):9129–9136
Ghosh T, Matsumoto F, McInnis J, Weiss M, Abruña H, DiSalvo F (2009) PtPb nanoparticle electrocatalysts: control of activity through synthetic methods. J Nanoparticle Res 11(4):965–980
Justin P, Rao GR (2009) Enhanced activity of methanol electro-oxidation on Pt–V2O5/C catalysts. Catal Today 141(1–2):138–143
Wang J, Holt-Hindle P, MacDonald D, Thomas DF, Chen A (2008) Synthesis and electrochemical study of Pt-based nanoporous materials. Electrochim Acta 53(23):6944–6952
Yu X, Pickup PG (2008) Recent advances in direct formic acid fuel cells (DFAFC). J Power Sources 182(1):124–132
Uhm S, Lee HJ, Kwon Y, Lee J (2008) A stable and cost-effective anode catalyst structure for formic acid fuel cells. Angew Chem Int Ed 47(52):10163–10166
Zhang S, Shao Y, Yin G, Lin Y (2010) Facile synthesis of PtAu alloy nanoparticles with high activity for formic acid oxidation. J Power Sources 195(4):1103–1106
Wang S, Wang X, Jiang SP (2011) Self-assembly of mixed Pt and Au nanoparticles on PDDA-functionalized graphene as effective electrocatalysts for formic acid oxidation of fuel cells. Phys Chem Chem Phys 13(15):6883–6891
Huang J, Hou H, You T (2009) Highly efficient electrocatalytic oxidation of formic acid by electrospun carbon nanofiber-supported PtxAu100−x bimetallic electrocatalyst. Electrochem Commun 11(6):1281–1284
Yu Y, Hu Y, Liu X, Deng W, Wang X (2009) The study of Pt@Au electrocatalyst based on Cu underpotential deposition and Pt redox replacement. Electrochim Acta 54(11):3092–3097
Liu B, Li HY, Die L, Zhang XH, Fan Z, Chen JH (2009) Carbon nanotubes supported PtPd hollow nanospheres for formic acid electrooxidation. J Power Sources 186(1):62–66
Lee H, Habas SE, Somorjai GA, Yang P (2008) Localized Pd overgrowth on cubic Pt nanocrystals for enhanced electrocatalytic oxidation of formic acid. J Am Chem Soc 130(16):5406–5407
Hu C, Zhao Y, Cheng H, Hu Y, Shi G, Dai L, Qu L (2012) Ternary Pd2/PtFe networks supported by 3D graphene for efficient and durable electrooxidation of formic acid. Chem Commun 48(97):11865–11867
Casado-Rivera E, Volpe DJ, Alden L, Lind C, Downie C, Vázquez-Alvarez T, Angelo ACD, DiSalvo FJ, Abruña HD (2004) Electrocatalytic activity of ordered intermetallic phases for fuel cell applications. J Am Chem Soc 126(12):4043–4049
Tripković AV, Popović KD, Stevanović RM, Socha R, Kowal A (2006) Activity of a PtBi alloy in the electrochemical oxidation of formic acid. Electrochem Commun 8(9):1492–1498
Xu JB, Zhao TS, Liang ZX (2008) Synthesis of active platinum−silver alloy electrocatalyst toward the formic acid oxidation reaction. J Phys Chem C 112(44):17362–17367
Chi N, Chan K-Y, Phillips D (2001) Electrocatalytic oxidation of formic acid by Pt/Co nanoparticles. Catal Lett 71(1–2):21–26
Yang Y-Y, Sun S-G, Gu Y-J, Zhou Z-Y, Zhen C-H (2001) Surface modification and electrocatalytic properties of Pt(100), Pt(110), Pt(320) and Pt(331) electrodes with Sb towards HCOOH oxidation. Electrochim Acta 46(28):4339–4348
Kristian N, Yan Y, Wang X (2008) Highly efficient submonolayer Pt-decorated Au nano-catalysts for formic acid oxidation. Chem Commun 3:353–355
Zhao D, Wang Y-H, Xu B-Q (2009) Pt flecks on colloidal Au (Pt∧Au) as nanostructured anode catalysts for electrooxidation of formic acid. J Phys Chem C 113(49):20903–20911
Ojeda M, Iglesia E (2009) Formic acid dehydrogenation on Au-based catalysts at near-ambient temperatures. Angew Chem Int Ed 48(26):4800–4803
Xiang J, Wu B-L, Chen S-L (2001) Investigation of the mechanism of the electrochemical oxidation of formic acid at a gold electrode in sulfuric acid solution. J Electroanal Chem 517(1–2):95–100
Wang J, Yin G, Wang G, Wang Z, Gao Y (2008) A novel Pt/Au/C cathode catalyst for direct methanol fuel cells with simultaneous methanol tolerance and oxygen promotion. Electrochem Commun 10(6):831–834
Wang J, Yin G, Liu H, Li R, Flemming RL, Sun X (2009) Carbon nanotubes supported Pt–Au catalysts for methanol-tolerant oxygen reduction reaction: a comparison between Pt/Au and PtAu nanoparticles. J Power Sources 194(2):668–673
Acknowledgments
Financial support from the National Science Council (NSC) (101-3113-E-011-002, 101-2923-E-011-001-MY3, 100-2221-E-011-105-MY3), the Ministry of Economic Affairs (MOEA) (101-EC-17-A-08-S1-183), and the Top University Projects of Ministry of Education (MOE) (100H451401) and the facilities’ supports from the National Taiwan University of Science and Technology (NTUST) and the National Synchrotron Radiation Research Center (NSRRC) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this entry
Cite this entry
Pan, CJ., Sarma, L.S., Hwang, BJ. (2016). Formation and Characterization of Bimetallic Nanoparticles in Electrochemistry. In: Aliofkhazraei, M., Makhlouf, A. (eds) Handbook of Nanoelectrochemistry. Springer, Cham. https://doi.org/10.1007/978-3-319-15266-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-15266-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-15265-3
Online ISBN: 978-3-319-15266-0
eBook Packages: Chemistry and Materials ScienceReference Module Physical and Materials ScienceReference Module Chemistry, Materials and Physics