
Abstract
Melanocytes make up only a tiny proportion of the skin cellular milieu but have a major impact on skin appearance as well as skin cancer risk by modulating skin pigmentation. Cutaneous melanocytes are derived from precursor cells called melanoblasts that originate from the neural crest of the developing embryo and migrate for long distances to their niches in the epidermis and hair follicles, where they differentiate into melanin pigment-producing mini-factories. Melanin is synthesized and packaged within melanosomes, which are lysosome-related organelles responsible for melanin trafficking through dendrites to interacting keratinocytes. Melanocyte development, migration, proliferation, and differentiation are regulated by a complex network of extrinsic and intrinsic signaling pathways, which are responsive to key signals, such as ultraviolet radiation that stimulates melanin production (tanning). Alterations in components of these pathways may lead to melanomagenesis. Here we present an overview of the genes and pathways that regulate different aspects of the biology of melanocytes as well as their transformation to melanoma.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Development of melanoblasts
- Regulation of specification
- Regulation of migration
- Regulation of survival and proliferation
- Differentiation of melanocytes
- Regulation of differentiation
- Regulation of survival
- Melanomagenesis
- From melanocyte to melanoma: a multistep process
- Molecular genetics: early lessons from familial and sporadic melanoma
- Melanoma: a consequence of homeostatic disruption
- Melanoma: cell of origin
- Melanoma and the environment
- Sun exposure and epidemiology
- Photobiology and melanoma
Introduction
Melanocytes are highly specialized cells that produce and distribute melanins, which are high molecular weight pigmented biopolymers responsible for pigmentation in the skin, hair, eyes, and inner ear. Although melanocytes represent a small proportion of cells in these pigmented tissues, the pigments they produce have several diverse and important functions (Hearing and Leong 2006; Nordlund 2006; Prota 1992). Melanocytes destined for the skin, hair, and choroid of the eye originate from pluripotent neural crest cells during embryonic development. In contrast, melanocytes populating the retinal pigment epithelium are derived from the optic cup (Bharti et al. 2006) and are not considered further in this chapter.
Epidermal melanocytes reside among the basal keratinocytes in an approximate ratio of 1:10 and transfer melanin via elongated dendrites that contact up to 40 overlying suprabasal keratinocytes. The melanin-carrying keratinocytes form a vital barrier against environmental assaults. Consequently, skin color is not defined solely by the melanocytes that produce the pigment but occurs in partnership with the pigment-carrying keratinocytes in the superficial skin layers. Given the intimate relationship between melanocytes and other cell types present in the surrounding microenvironment, the importance of microenvironmental cues in melanocyte development, function, and transformation to melanoma (melanomagenesis) is well established.
In many respects, the process of melanomagenesis is integrally but inversely related to the process of melanocyte development, representing a return to a proliferative, dedifferentiated, and migratory phenotype. In fact, the same essential cascades of genes that regulate development and differentiation of melanocytes are involved in the growth and eventual metastasis of melanoma cells. This chapter provides an overview of the processes critical to melanocyte development and pigmentation in mammals and describes the disruption of these processes during the transformation of melanocytes into melanoma cells.
Development of Melanoblasts
A recent curation effort by the Loftus group at the National Human Genome Research Institute (USA) has compiled a list of 650 genes that are currently known to be involved in integumentary pigmentation in humans, mice, and/or zebrafish (Baxter et al. 2018). Among these, 128 genes have been validated to have associations with human pigmentary phenotypes, while others have only been shown to exhibit phenotypes in mice and zebrafish to date. Clearly, the mechanistic network involved in the process of pigmentation is highly complex, which is exemplified by the wide spectrum of protein complexes, signaling pathways, and functions represented by the 26 different protein classes featured among the products of the pigmentary genes (Baxter et al. 2018). Disruption of the functions of these genes leads to a variety of pigmentary disorders, which can result from perturbations in melanocyte development, differentiation, and regulation. Although the process of melanocyte development is a continuous one, it is generally considered in four discrete stages: specification, migration, survival, and proliferation.
Regulation of Specification
Specification describes the process early in development when pluripotent neural crest cells, which exhibit all the key features of stem cells, begin to commit to becoming pigment cells (melanocytes) in adult tissues. Before their final specification, neural crest cells (which are specified at an earlier stage of development and originate from the dorsal edge of the neural tube) have the ability to develop into a number of distinct types of cells, including neurons, glial cells, cardiac cells, and pigment cells (Bronner and LeDouarin 2012). As one might expect, transcription factors and intracellular signaling factors play major roles in determining which pathway of development a given neural crest cell will take. Regarding those destined to become melanocytes, the precursor cells, once specified, are called melanoblasts. Early during gestation (approximately 1 month in humans), neural crest cells destined to become melanoblasts begin to express several genes encoding transcription factors (Table 1), including PAX3, LEFl, and FOXD3 (Blake and Ziman 2005; Ignatius et al. 2008; Kos et al. 2001; Silver et al. 2006; Yasumoto et al. 2002). These transcription factors regulate downstream target genes that participate in specifying further development of melanoblasts; some of those downstream genes have been identified (Blake and Ziman 2005; Hornyak et al. 2001; Ignatius et al. 2008; Kos et al. 2001). Concurrently, factors that regulate the WNT signaling pathway (WNT1 and WNT3) are also expressed and thought to trigger melanocytic differentiation through direct transcriptional targeting of the MITF gene (Dorsky et al. 2000). The canonical WNT signaling pathway is activated by extracellular WNT proteins binding to the surface receptor complex which then regulates the redistribution of β-catenin to the nucleus, where it acts to modulate transcription through activities that involve the transcription factors LEF1 and MITF (Dunn et al. 2005; Garcia-Castro et al. 2002; Hari et al. 2012; Lewis et al. 2004; Liu et al. 2014; Takeda et al. 2003). The multiple roles of MITF and WNT signaling, as well as many other intracellular signaling pathways, at virtually all phases of melanocyte development and differentiation will be a recurring theme in the discussion to follow (Kawakami and Fisher 2017; Liu et al. 2014).
At least two other transcription factors are thought to play roles in the early developmental process of specification, SOX9 (Meulemans and Bronner-Fraser 2004) and SNAI2/SLUG (LaBonne and Bronner-Fraser 1998). SOX9 has also been shown to regulate MITF expression (Passeron et al. 2007). While the transcription factor SOX10 is required for the maintenance and differentiation of neural crest cells as they migrate, SOX9 plays an essential role in neural crest generation (Guth and Wegner 2008). SNAI2 has been shown, at least in lower organisms, to regulate the expression and function of SOX10 (Jiang et al. 1998; Sanchez-Martin et al. 2002). Bone morphogenetic proteins play critical roles in determining patterning during development, and these are closely regulated by the WNT signaling pathway; in that regard, BMP2 and BMP4 have been shown to be important to melanoblast specification (Jin et al. 2001). The expression of BMP2/4 is not specific to cells destined to become melanoblasts, and other factors that are specific to melanoblasts and regulate their specification will undoubtedly be identified in the future. The majority of studies defining the genes and developmental pathways involved in melanoblast specification have been performed in lower species, including mice, chicks, zebrafish, and amphibians, although results to date suggest that these pathways and genes are highly conserved in humans (as are the defects involved in pigmentary disorders) (Baxter and Pavan 2013; Liu et al. 2014; Pavan and Raible 2012). Cells expressing these developmental markers then undergo an epithelial-mesenchymal transition and begin to migrate dorsolaterally below the ectoderm (called early migration). Expression of these same markers (and others, as described in the “Differentiation” section) is found in melanocyte stem cells in adult tissues that serve as reservoirs of cells that can be recruited to become differentiated melanocytes by appropriate environmental cues (Buac and Pavan 2007; Nishimura 2011; Osawa et al. 2005).
Regulation of Migration
As the precursor cells migrate (Fig. 1), they remain multipotent (i.e., they retain the capacity to become pigment cells, smooth muscle cells, neurons, and so on). Those destined to become melanoblasts continue to express SOX9 but now begin to express other genes that regulate their further development, including SOX10, MITF, KIT, and, a bit later, a melanogenic (pigmentation-inducing) enzyme called dopachrome tautomerase (DCT), although expression of DCT is thought to result simply from the increased function of SOX10 and MITF and is not relevant to developmental processes (Guyonneau et al. 2004). Of the newly expressed genes, SOX10 and MITF are transcription factors, whereas KIT is a surface receptor that plays a critical role in regulating intracellular signaling in melanocytes at many levels of their development and differentiation. Although these transcription factors are not specific to the pigment cell precursors, MITF is differentially expressed in different cell lineages using alternative promoters and initial exons, which are alternatively spliced to perform distinct functions and activate distinct genes, with the form of MITF specific to pigment cells designated MITF-M (Harris et al. 2010; Kawakami and Fisher 2017) (Fig. 2), although other spliced isoforms (non-tissue-specific) are typically expressed in melanocytes as well as other cell types. MITF-M (hereafter, MITF) is often referred to as the master regulator of melanocytes, because it is a central focal point through which environmental signaling modulates the behavior of melanocytes at many levels (see the “Differentiation” section). Melanoblasts move in either a ventral or a dorsolateral direction, giving rise to the peripheral nervous system and endocrine cells or to melanocytes, respectively. The full complement of signals that regulate these processes not only involves genes expressed by melanoblasts but also factors produced in the local environment in the developing embryo. For example, KIT is a tyrosine kinase receptor on the plasma membrane that is regulated by the environmentally derived stem cell factor (SCF, encoded by the KITLG gene) ligand, which works through the MAP kinase signaling pathway to regulate the expression of genes critical for melanoblast migration and survival (Wehrle-Haller 2003). Most notable among KIT-regulated genes is MITF. Yet another receptor, the endothelin receptor (EDNRB), which works through the phospholipase C (PLC) signaling cascade, is also critical for regulating melanoblast migration and proliferation, and although that receptor is expressed on the surface of melanoblasts, it is activated by endothelins (EDN) produced in the local environment (Lee et al. 2003; Saldana-Caboverde and Kos 2010). One must always bear in mind that for appropriate pigmentation patterns to occur, the signaling for melanoblast migration to begin, and later to stop, must be carefully controlled. Some melanoblasts move only short distances in the developing embryo, whereas others need to migrate long distances to reach the extremities. Defects in many of the genes involved in migration and specification are associated with developmental pigmentary diseases, often eliciting hypopigmented areas in ventral regions. For example, deficiency of the Rac-specific guanine nucleotide exchange factor (GEF) P-Rex1, encoded by the PREX1 gene, causes a white belly and feet phenotype in Prex1-knockout mice, characterized by defective melanoblast migration (Lindsay et al. 2011).


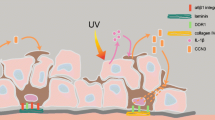
Factors involved in interactions between melanocytes and neighboring keratinocytes and fibroblasts. (Ligands and receptors are discussed in the text)
Cell-cell interactions are critically important during migration; thus, it is no surprise that the expression of various proteins regulating such interactions (e.g., cadherins and components of the extracellular matrix [ECM] , such as integrins, laminin, lectins, and fibronectin (Bonaventure et al. 2013; Haass et al. 2005; Herlyn et al. 2000; Jouneau et al. 2000; Pinon and Wehrle-Haller 2011)) is also closely involved in the regulation of melanoblast migration. Similarly, proteases associated with penetration of biologic membranes are also important to cell migration, and ADAMTS20 (a metalloproteinase) has been closely associated with melanoblast survival and migration during development, with mutations affecting this gene resulting in abnormal patterning in the skin (Rao et al. 2003; Silver et al. 2008). Studies in mice suggest that melanoblasts undergo proliferation before their initial migration and then cease proliferating while they migrate, after which they may undergo extensive proliferation once they have reached their final destination (Thomas and Erickson 2008; Wilkie et al. 2002). With respect to invasion and metastasis, migrating melanoblasts have the ability to cross the basement membrane and other biologic barriers to tissue penetration during development.
Regulation of Survival and Proliferation
Once melanoblasts reach their intended destination, they must cease migrating, then survive and proliferate, and eventually differentiate. Because survival and proliferation occur concurrently at this time during development, most genes known to be involved play roles in both processes (or at least, it is very difficult to distinguish their roles in those two processes), and they will be considered together in this section. The survival of differentiated melanocytes in the skin is discussed further on. Many of the genes mentioned above are also involved in regulating survival and/or proliferation (i.e., genes encoding MITF, SOX10, and PAX3), but additional genes act as “supporting actors,” including the transcription factor AP2, as well as additional signaling factors (basic fibroblast growth factor [bFGF, encoded by the FGF2 gene] and hepatocyte growth factor [HGF]) that join the previous list (EDN3 and KITLG). The KIT receptor and its ligand KITLG play important roles in regulating survival and proliferation, and mutations in either of these two genes are associated with pigmentary disorders of the skin, which can result in hypopigmentation (e.g., vitiligo and piebaldism) as well as in hyperpigmentation (e.g., UVR melanosis and lentigo senilis), depending on whether their function is decreased or increased, respectively (Diwakar et al. 2008; Imokawa 2004).
It is an interesting paradox that melanocytes are relatively fragile cells because of the inherent stresses they undergo, primarily as a result of their production of highly toxic intermediates during melanogenesis, yet they typically have extremely long biologic lives and seldom proliferate in adult tissues outside of the hair follicle. In fact, stimulation of their proliferation is a key to their malignant transformation, as discussed in the “Melanomagenesis” section later. Melanoblasts and melanocytes typically express AP2, BCL2, and survivin (BIRC5) (Braeuer et al. 2011; McGill et al. 2002; Nishimura et al. 2005; Raj et al. 2008), all of which are antiapoptotic, and thus prevent melanocytic cells from succumbing to the cytotoxicity of their microenvironment.
Three new sets of ligands and receptors come into play at this point. HGF and its receptor MET also work via the MAP kinase pathway (with MITF contributing as a downstream effector) to influence melanoblast survival and proliferation (Hirobe et al. 2004; Kos et al. 1999). The FGF receptor and its ligand FGF2 also work through the MAP kinase pathway to regulate proliferation and survival (Beauvais-Jouneau et al. 1999; Ito et al. 1999). Similarly, DKK1, an inhibitor of WNT signaling, also participates in regulating the survival and proliferation of melanoblasts (and later, their differentiation) (Lin and Fisher 2007; Yamaguchi et al. 2007; Yamaguchi et al. 2008).
Differentiation of Melanocytes
Regulation of Differentiation
Human melanocytes reside at the dermal-epidermal junction of the skin and are interspersed among basal keratinocytes on the basement membrane. Differentiated melanocytes produce specialized lysosome-related organelles, termed melanosomes, which contain the enzymatic and structural components required for the synthesis of melanins (Dell’Angelica 2003; Park et al. 2009; Schiaffino 2010). Melanocytes then distribute those melanosomes to the overlying suprabasal keratinocytes via their elaborate network of dendrites and cell-cell contacts (see Fig. 2). Melanocytes and keratinocytes in the basal layer of the epidermis are highly stable cell populations that proliferate extremely slowly under normal circumstances. In contrast, keratinocytes in the upper layers of the epidermis proliferate relatively rapidly and differentiate further into highly keratinized layers that eventually form the stratum corneum at the surface of the skin. Their upward proliferation carries them toward the surface of the skin along with their ingested melanin where they form a critical barrier against the environment (including ultraviolet radiation [UVR]) . In humans, keratinocytes move from the basal layer to the surface of the skin and are lost by desquamation in approximately 4–5 weeks, so it is a relatively rapid process. Because of that, transformation events in proliferating keratinocytes are not thought to pose a long-term risk for skin cancers, but rather it is the transformation of melanocytes or keratinocytes in the basal layer that have long-term consequences concerning skin cancer.
Although the various types of melanins produced by melanocytes (eumelanins and/or pheomelanins) are important for the eventual characteristic color of the skin, it is also the distribution of those melanins in the more superficial layers of the skin that has major effects. Although melanocytes in lower species often produce only one type of melanin at a given time, in humans different mixtures of eumelanins and pheomelanins are typically produced on a constitutive basis (Ito and IFPCS 2003; Micillo et al. 2016). Melanocytes in other tissues (e.g., hair follicles, eyes, and inner ears) interact with their surrounding cells in distinct manners, yet the basic processes of melanin production and melanosome biogenesis are comparable, as are some of the factors that regulate melanogenesis. Factors that influence pigmentation at sites other than the skin are covered in several reviews (Bharti et al. 2006; Hubbard et al. 2010; Lavado and Montoliu 2006; Slominski et al. 2005; Sturm and Larsson 2009; Tobin 2008) and books (Hearing and Leong 2006; Nordlund 2006).
As noted earlier, 650 distinct loci are currently known to be involved in regulating pigmentation either directly or indirectly (Baxter et al. 2018). Many of those loci/genes affect developmental processes, but others regulate either differentiation/survival of melanocytes once in situ or regulate physiological factors that affect pigmentation. The remainder of this section will provide an overview of the latter two categories. Many of the pigment regulatory genes affect the biogenesis and/or function of melanosomes, the discrete membrane-bound organelles within which melanins are synthesized. Melanosomes, which are closely related to lysosomes, are within the family of lysosome-related organelles (LROs) and require a number of specific enzymatic and structural proteins to mature and become competent to produce melanin (Chi et al. 2006; Hu et al. 2007; Yamaguchi et al. 2007). Several reviews have elaborated on processes involved in melanosome biogenesis and on the specific functions of melanosomal proteins (Barral and Seabra 2004; Schiaffino 2010). Briefly, the critical enzymes include tyrosinase (TYR), TYR-related protein 1 (TYRP1), and dopachrome tautomerase (DCT), and mutations in their encoding genes dramatically affect the quantity and quality of melanins synthesized. Important structural proteins of melanosomes include PMEL (also known as gp100) and MLANA (MART-1), both of which are required for the structural maturation of melanosomes (Berson et al. 2001; Hoashi et al. 2005). Interestingly, most (perhaps all) of those melanosomal proteins have been identified as targets of the immune system in patients with melanoma (Kawakami et al. 2000; Sakai et al. 1997). Because melanosomes are LROs, there are many proteins involved in the sorting and trafficking of proteins to melanosomes, some of which affect all LROs (such as AP3 and BLOC components), whereas others (such as OCA2 and SLC45A2) affect only melanosomes, and mutations in any of those typically lead to inherited hypopigmentary disorders (Costin and Hearing 2007; Costin et al. 2003; Nordlund 2006; Toyofuku et al. 2002). As melanosomes mature and their constituent proteins are delivered, they become cargos themselves, carried by various molecular motors (actinbased and myosin-based) from the perinuclear area to the cell periphery (Coudrier 2007; Wu and Hammer 2000), after which they are transferred to neighboring keratinocytes (Scott et al. 2003; Wu and Hammer 2014). Several critical components are involved in linking melanosomes to molecular motors responsible for their transport (e.g., RAB27A, melanophilin, and myosin Va), and mutations in those genes result in a pigmentary disease known as the Griscelli syndrome.
Constitutive skin pigmentation Human skin color ranges from extremely fair and light to extremely dark, depending on racial or ethnic background, among other factors, but the density of melanocytes in a given geographic area of the body is virtually identical in all skin types (Tadokoro et al. 2003; Yamaguchi et al. 2004). Keratinocytes in lighter skin tend to cluster less-pigmented melanosomes above the nuclei (called capping), while in darker skin, greater numbers of pigmented melanosomes are distributed individually in keratinocytes, thus maximizing their absorption of light and their photoprotective value. Constitutive melanocyte density in the skin can be affected by the environment, such as by chronic UV radiation (which can increase melanocyte density threefold or fourfold), by secreted paracrine factors in the skin (Hirobe 2011; Scott et al. 2002) (see Fig. 2), and by toxic compounds such as hydroquinone (which antagonizes the melanogenesis enzyme tyrosinase but can also permanently destroy melanocytes in the skin.) Several types of inherited pigmentary disorders also affect melanocyte density (e.g., increased numbers in freckles or decreased numbers in vitiligo (Thong et al. 2003; Whiteman et al. 1999; Yoshida et al. 2007).)
Epidermal melanocytes proliferate slowly, if at all, under normal circumstances, and they are quite resistant to apoptosis because of their expression of BCL2 and survivin (BIRC5) (McGill et al. 2002; Raj et al. 2008). Melanocyte density and differentiation are influenced by the environment, including UVR and factors secreted by neighboring keratinocytes and fibroblasts (see Fig. 2). For example, fibroblasts in the dermis of the palms/soles secrete high levels of DKK1, which suppresses melanocyte growth and function by inhibiting the WNT/β-catenin signaling pathway (Yamaguchi et al. 2004; Yamaguchi et al. 2008). DKK1 inhibition of WNT signaling in melanocytes dramatically inhibits the melanogenic pathway, ranging from effects on transcription factors (e.g., MITF, PAX3, SOX9, and SOX10) to downstream melanogenic proteins. DKK1 also affects keratinocytes in overlying epidermis, reducing their uptake of melanin and inducing a thicker less-pigmented skin phenotype (Yamaguchi et al. 2008).
One major determinant of pigment phenotype of the skin is the melanocortin 1 receptor (MC1R) , a G protein-coupled receptor which regulates virtually all functional aspects of melanocytes, including dendricity, melanogenesis, and proliferation, among other things (Herraiz et al. 2017; Wolf Horrell et al. 2016). MC1R function (and dysfunction) has critical implications for the risk for skin cancer and is considered in detail in the “Melanomagenesis” section. MC1R function is regulated by peptide agonists encoded by the POMC gene (αMSH and ACTH) and by an antagonist, agouti signaling protein (ASIP). Activation of MC1R stimulates the melanogenic cascade leading to the synthesis of eumelanin, while ASIP can reverse those effects and favor the production of pheomelanin. αMSH and ACTH also upregulate expression of the MC1R gene, thus acting in a positive feedback loop. Although MC1R is closely involved with regulating skin pigmentation, it is not the only melanocytic receptor that functions in pigmentation. In fact, virtually all of the receptor/ligand pairs discussed above that participate in developmental processes also regulate mammalian pigmentation in adult tissues, including KITLG:KIT, CSF2:CSF2RA, HGF:MET, FGF2:FGFR, and EDN:EDNRB. Those ligands can originate from neighboring cells in the epidermis (keratinocytes) and the dermis (fibroblasts) and even from more distant locations, induced by environmental factors (e.g., UVR). As seen in the schematic outline in Fig. 2, virtually all known intracellular signaling pathways are involved in responses to these factors: receptors, the common effect of many of them being to regulate the function of MITF.
In addition to regulation by a wide variety of receptor:ligands, melanin production is also modulated by multiple other factors within melanocytes. The SLC45A2 and OCA2 proteins are melanocyte-specific proteins with 12 transmembrane domains that play critical roles in the sorting/trafficking of TYR to melanosomes. If TYR is not successfully delivered to melanosomes, no melanin will be produced, even if the TYR is a functional enzyme (TYR can be abnormally degraded by proteasomes or secreted from melanocytes). SLC45A2 is an intracellular pump/transporter that regulates ion transport across intracellular membranes which regulate the sorting of TYR and thus the biosynthesis of melanin. OCA2 acts on multiple levels to influence the melanosome, the melanogenic enzymes, pH, and glutathione metabolism (Ainger et al. 2017). Mutations in any of these proteins result in the abnormal secretion of TYR from melanocytes rather than normal sorting of TYR to melanosomes. Population studies have shown that polymorphisms of the SLC45A2 or OCA2 genes (together with polymorphisms of the MC1R gene) play major roles in determining the normal range of pigmentation in the hair, skin, and eyes (Ainger et al. 2017; Graf et al. 2005; Shriver et al. 2003). A close association between a polymorphism in a sodium:calcium exchanger (SLC24A5) and melanin content of the skin has also been described (Ginger et al. 2008; Lamason et al. 2005).
Facultative skin pigmentation Facultative skin pigmentation is the term coined for increased skin color due to some type of physiologic regulation, the most obvious being UVR in what is commonly termed the tanning reaction (Eller and Gilchrest 2000; Tadokoro et al. 2005). Several studies have detailed the complex kinetics of responses of the skin to UVR, which result in tanning over the course of several weeks (Chen et al. 2014; Coelho et al. 2009). UVR is the most significant factor that influences human skin pigmentation and elicits several stages of increased skin color. Immediate pigment darkening occurs within minutes and persists for several hours, followed by persistent pigment darkening, which occurs within several hours and lasts for several days (Young 2006). These rapid increases in skin color do not result from increased melanin synthesis but rather are due to the oxidation and polymerization of existing melanin and the redistribution of existing melanosomes. A slower process, termed delayed tanning, occurs several days after UVR exposure but requires more time since it involves the activation of melanocyte function and depends on increased melanin production. As might be expected, UVR induces skin pigmentation due to its effects on melanocytes and also via indirect effects on keratinocytes. Regarding effects on melanocytes, ultimately UVR elicits increased expression of MITF and the downstream melanogenic cascade (Miyamura et al. 2007; Yamaguchi and Hearing 2006).
Keratinocytes in the skin respond to UVR by increasing their secretion of αMSH and ACTH as a result of the stimulation of POMC expression, which was shown to result from increased TP53 expression (Cui et al. 2007). Interleukin-1 (IL1) secretion by keratinocytes is also elicited by UVR and stimulates the autocrine secretion of ACTH, αMSH, EDN1, and FGF2 (Hirobe and Ootaka 2007). Each of those factors functions via receptors expressed on melanocytes, as discussed earlier. UVR can also affect fibroblasts in the dermis, and growth factors secreted from those cells in response to UVR include HGF, FGF2, and KITLG, each of which stimulates pigmentation via distinct receptors expressed by melanocytes (Imokawa 2004). The tanning response also relies on stimulation of secretion of nerve growth factor (NGF) by keratinocytes, which prevents apoptosis following UVR exposure (Botchkarev et al. 2006; Kadekaro et al. 2003). In humans, the red-hair/light-skinned phenotype is associated with an inability to tan after UVR exposure. The gene most commonly implicated in this phenotype is MC1R. The so-called “red-hair” alleles of POMC are notable for their inability to signal to adenylate-cyclase upon exposure to the ligand (αMSH). These MC1R variants thus provide genetic evidence of the rate-limiting role for MC1R signaling in the UVR-induced tanning pathway. Correspondingly, it has been demonstrated in animal models of red hair (carrying MC1R loss-of-function) that topical administration of cAMP-inducing compounds can rescue eumelanin (dark) skin pigmentation (D’Orazio et al. 2006).
In sum, skin pigmentation is determined at multiple levels, by (1) the migration of melanoblasts to various tissues during development, (2) the melanoblast survival and differentiation to melanocytes, (3) the density of melanocytes, (4) the expression/function of enzymatic and structural constituents of melanosomes, (5) the synthesis of different types of melanin (eumelanin and pheomelanin), (6) the transport of melanosomes to dendrites, (7) the transfer of melanosomes to keratinocytes, and (8) the distribution of melanin in suprabasal layers of the skin. Skin pigmentation plays a critical role in protecting the underlying skin from UVR damage that can ultimately result in various types of skin cancers, including melanoma.
Regulation of Survival
Survival is not only a critical issue for developing melanoblasts but also for melanocytes in the skin and in hair bulbs. As noted above, this is especially important to melanocytes because of their extremely low rates of proliferation, combined with the high stresses imposed by their environment and also by the potentially toxic by-products of melanin synthesis within them. In this regard, the functions of antiapoptotic proteins, such as BCL2 and survivin, are essential for melanocyte survival, as are transcription factors involved in regulating their expression (e.g., MITF and PAX3) and upstream signaling events (e.g., mediated by the actions of KITLG:KIT, FGF2:FGFR, HGF:MET, and αMSH:MC1R). Dysfunctions of many of those genes/proteins have been associated with premature hypopigmentation of the skin and hair graying, as discussed and cited previously.
Melanocyte stem cells A closely related issue is the presence and maintenance of melanocyte stem cells, i.e., cells that retain the potential to replenish melanocytes in the skin and hair as needed yet remain undifferentiated. This population has potential significance regarding the process of melanomagenesis, as discussed later in this chapter. Melanocyte stem cells typically reside in what is termed the bulge region, a hair follicle stem cell niche located at the insertion site of the arrector pili muscle (which produces a small “bulge”) along each hair shaft. During the hair growth cycle, active melanocytes are normally present exclusively in the hair bulb and function by synthesizing melanin granules and transferring them to keratinocytes within the bulb, which incorporates the melanin into the growing keratin-containing hair matrix. Hair follicles undergo cyclic patterns of growth, involution, and rest. The growth phase (anagen) has a finite lifetime (measured in years in adult humans), which is followed by the catagen stage, at which time all active keratinocytes and melanocytes within the hair bulb die. New hair bulbs require a fresh supply of functional melanocytes; else the hair will be partially or totally unpigmented. That supply of active melanocytes comes from a pool of melanocyte stem cells that remain localized in the bulge region throughout the follicle stages. Telogen is the resting phase that follows the catagen (involution) phase. During telogen, melanocyte stem cells are in a largely undifferentiated state, but they do not express significant levels of TYR and other proteins required to produce melanin; hence they are unpigmented in the hair bulge region. Although there is some argument about the exact patterns of marker expression, the melanocyte stem cells do express a number of markers that identify them. Typically they are positive for PAX3 and SOX10 and weakly positive for MITF and DCT (which is likely expressed due to PAX3/SOX10 activation); they are negative for other melanosomal proteins (enzymatic and structural) such as TYR, TYRP1, and PMEL (Buac and Pavan 2007; Lang et al. 2005; Nishikawa and Osawa 2007; Nishimura et al. 2005; Osawa et al. 2005). They retain expression of surface receptors such as KIT, MET, and EDNRB, which allows them to respond to environmental cues and start the differentiation/proliferation phase (anagen) to become functional melanocytes that incorporate into the newly formed hair bulb. Although these melanocyte stem cells are typically thought of as reservoirs required to repopulate melanocytes in hair bulbs, there is evidence to suggest that they also can be recruited to replace melanocytes at the dermal-epidermal border. The self-renewing capacity of bulge melanocyte stem cells is thought to be imperfect as depletion of these hair follicle melanocyte stem cells has been seen to correlate with age-dependent hair graying in both mice and humans (Nishimura et al. 2005).
Melanomagenesis
From Melanocyte to Melanoma: A Multistep Process
Over 20 years ago, Clark et al. (1984) devised a model for the stepwise development of melanoma from normal melanocytes based on both clinical and histopathologic features. According to this model, melanoma progression includes the following stages: (1) common acquired (benign) and congenital nevi consisting of nests of cytologically normal melanocytes, thought to exist in a senescent state; (2) dysplastic nevi characterized by structurally and architecturally atypical features and posing a risk factor for melanoma (Goldstein and Tucker 2013); (3) radial growth phase (RGP) or microinvasive malignant melanoma characterized by intraepidermal proliferation and “pagetoid spreading”; (4) vertical growth phase (VGP) or invasive malignant melanoma associated with the ability to penetrate through the basement membrane into the underlying dermis and metastasize and with a significantly poorer prognosis; and (5) metastatic melanoma, which has spread to other areas of the skin and other more distant sites. However, this sequence of progression can be histologically documented in only about a third of all melanoma cases, and malignant melanoma may also be able to arise in the absence of visually obvious pathologic progenitor lesions.
How does this aggressive, often fatal disease arise from melanocytes? What goes wrong? Based originally on extensive cytogenetic analyses of a variety of melanocytic lesions, and later confirmed by high-resolution genomic analyses (e.g., array-comparative genomic hybridization [CGH]), the degree of detectable genetic alterations increases rather dramatically as it progresses from nevus to primary melanoma to metastatic melanoma (Chin et al. 2006). There are few consistent changes associated with each of these stages, but years of such analyses have identified a number of genes and pathways that have proved critical to melanoma development and provided important hints about the transformation process (Shain et al. 2015). While numerous reports demonstrate gene expression changes in melanoma (Riker et al. 2008), we begin by examining some of the major early insights that came from extensive classic cytogenetic studies and array-CGH-based genome-wide analyses of nonrandom chromosomal alterations in melanomas (Bauer and Bastian 2006; Jonsson et al. 2007; Stark and Hayward 2007).
Molecular Genetics: Early Lessons from Familial and Sporadic Melanoma
The CDKN2A locus It has long been recognized that melanoma susceptibility can be inherited, as in familial atypical multiple mole melanoma (FAMMM) syndrome (Soura et al. 2016). Subsequent genetic analysis of large melanoma-prone families led to the identification of the melanoma susceptibility locus CDKN2A at chromosome band 9p21 (Aoude et al. 2015; Chin et al. 2006; Kamb et al. 1994). The CDKN2A locus shows loss of heterozygosity (LOH) and loss of function mutations in 25–40% of melanoma kindreds and less frequently in sporadic melanoma. Subsequent experimental studies using mice genetically engineered to carry inactivated CDKN2A have corroborated the importance of this locus in cutaneous malignant melanoma (Perez-Guijarro et al. 2017). The CDKN2A locus is quite unique, encoding two distinct proteins within overlapping DNA sequences: p16INK4A and p14ARF (p19Arf in mice). This is accomplished through the initiation of transcription from two distinct promoters upstream of two distinct first exons (lα for p16INK4A and 1β for p14ARF). Thus, the genes encoding p16INK4A and p14ARF share common second and third exons, translated in different reading frames. Although genetic deletions in this region typically inactivate both p16INK4A and p14ARF in melanoma, many cases have been documented in which expression of only one is affected, suggesting that each has a key role in melanomagenesis (Chin et al. 2006). Remarkably, both function as tumor suppressor genes, with p16INK4A functioning as a cyclindependent kinase inhibitor (CKI) to help regulate transit through the cell cycle and p14ARF stabilizing TP53 by binding and inhibiting the TP53 E3 ubiquitin ligase, HDM2 (Zhang et al. 1998).
p16INK4A binds to cyclin-dependent kinases (CDK) 4 and 6, preventing its association with D-type cyclins and inhibiting its ability to phosphorylate and inactivate RB, which in turn initiates S phase associated DNA synthesis (Fig. 3). This is a critical pathway in melanomagenesis, as evidenced by the fact that in melanoma p16INK4A expression can be suppressed by mutation, methylation, and/or polymorphic variation at both the 5′ and 3′ untranslated regions (Chin et al. 2006). Moreover, germline mutations in CDK4 have been documented in melanoma patients that prevent binding of CDK4 to p16INK4A, as have amplifications in CCND1 (encoding Cyclin D1) and occasional mutations in RB1 (Tsao et al. 2012). Notably, the various p16INK4A/CDK4/RB mutations were largely found to be mutually exclusive and can be considered redundant.

Critical pathways in melanomagenesis. The pathways are simplified, and arrows do not necessarily represent direct interactions (see text for more details.) Factors highlighted in red have been associated with activating mutations and/or increased copy numbers in melanoma, whereas those highlighted in green have been inactivated and/or deleted. The sun symbols represent pathways thought to be directly activated by UV radiation
p14ARF acts as a positive regulator of TP53. It targets the degradation of HDM2 (Mdm2 in mice), which destabilizes TP53. TP53 is a sequence-specific transcription factor that triggers either cell cycle arrest or programmed cell death (apoptosis) on DNA damage or other stress (Liebermann et al. 2007), depending on the cell type and the circumstances (see Fig. 3). TP53 is the most frequently mutated gene in human cancer but is a relatively infrequent target in melanoma, estimated to be mutated in fewer than 10% of melanomas (Bennett 2008; Hussein et al. 2003b; Petitjean et al. 2007). As mentioned above, p14ARF is frequently inactivated in melanoma, and HDM2 amplification has been documented as well. Genetically engineered mouse models (GEMMs) of human melanoma have corroborated the separate roles of p19ARF and p16INK4A in melanomagenesis (Ha et al. 2007; Perez-Guijarro et al. 2017). Moreover, recent evidence indicates that p19ARF has melanoma tumor suppressor functions that are independent of TP53, including a role in melanocyte senescence (Ha et al. 2007).
Cyclin-dependent kinase 4 Germline mutations at chromosome 12q14 in the CDK4 locus are oncogenic due to their effects on cell cycle control via the same pathway as p16INK4A. Two different mutations have been identified in codon 24 of exon 2 (R24C and R24H) at the CDK4 locus, both of which result in CDK4 protein acting as a dominant oncoprotein due to loss of binding to p16INK4A, which is its negative regulator (Puntervoll et al. 2013). CDK4 locus is mutated or amplified in only 5% human melanomas, albeit at a higher frequency in patients with BRAF, NRAS, and NF1 triple wild-type tumors (Cancer Genome Atlas 2015). GEMM studies have verified the oncogenic role of the CDK-R24C mutant protein in melanomagenesis (Gaffal et al. 2011; Tormo et al. 2006).
BRCA1-associated protein BAP1 is a tumor suppressor gene located on chromosome 3p21. Germline inactivating mutations at this locus were initially identified in two distinct syndromes; one was characterized by familial mesothelioma and uveal melanoma (Testa et al. 2011) and the other by cutaneous melanocytic neoplasia and uveal melanoma (Wiesner et al. 2011). Subsequently, cutaneous melanoma was included as part of the familial aggregation of cancers associated with the BAP1 syndrome (Carbone et al. 2013; Wadt et al. 2012; Wiesner et al. 2012). Approximately 5% of sporadic cutaneous melanoma have been proposed to have functional inactivation of BAP1 characterized by absence of BAP1 expression on immunohistochemistry analysis, suggesting its contribution to melanomagenesis in at least a subset of cases (Murali et al. 2013).
The PTEN-AKT pathway PTEN is localized on chromosome band 10q24, a region long associated with deletions and LOH in melanoma (Chin et al. 2006). PTEN is a dual phosphatase, containing both lipid phosphatase and protein phosphatase activities (Lee et al. 2018). Extracellular growth signals are often mediated through the intracellular second messenger lipid phosphatidylinositol-3,4,5-triphosphate (PIP3), levels of which increase upon signaling through PBK, resulting in the phosphorylation/activation of AKT (also known as protein kinase B) (see Fig. 3). The PTEN lipid phosphatase antagonizes PI3K by dephosphorylating PIP3 and therefore negatively regulates AKT activity, helping to control both cell proliferation and survival. PTEN deletions or inactivating mutations can be found in approximately 28% of melanoma cell lines, 7% of primary melanomas, and 15% of metastatic melanoma (Aguissa-Toure and Li 2012), causing sustained phosphorylation/activation of AKT; moreover, AKT3 overexpression has been reported to be associated with an increased DNA copy number in some melanomas (Robertson 2005). Despite intense study, the regulation and complex roles of PTEN are still incompletely understood. For example, PTEN has been found to be secreted into the extracellular space to be taken up by recipient cells, intriguingly suggesting a function as a cell nonautonomous tumor suppressor (Hopkins et al. 2013; Putz et al. 2012).
The WNT/β-catenin pathway β-catenin, encoded by the gene CTNNB1, is a key regulator of the WNT signaling pathway, well-documented for its involvement in the progression of many cancer types (Kaur et al. 2016; Zhan et al. 2017) and also in the physiologic differentiation of melanocytes from neural crest progenitors. In the absence of WNT signaling, β-catenin is targeted for degradation in association with a complex that includes adenomatous polyposis coli (APC) and glycogen synthase kinase 3β (GSK3β) (see Fig. 3). Upon WNT signaling, β-catenin is stabilized, accumulates, and is transported to the nucleus where it binds to LEF1 and acts as a transcriptional coactivator of LEF1 target genes. Downstream targets include the proto-oncogenes MYC and CCND1 and the gene encoding MITF (Zhan et al. 2017). Mutations have been detected in CTNNB1 that confer a resistance to β-catenin degradation in 2–23% of melanoma tissues and cell lines (Dahl and Guldberg 2007; Larue and Delmas 2006), with a higher frequency in vitro. Although infrequently mutated in melanoma, APC is more frequently characterized by decreased expression. β-catenin has also been shown to repress p16INK4A transcription (Delmas et al. 2007).
NRAS and BRAF In 2002 Davies et al. (2002) published a remarkable paper in which a genomewide cancer sequencing program was used to discover that BRAF was mutated in 66% of melanoma samples, a finding that had significant implications for the treatment of melanoma. This finding was later confirmed and extended to melanocytic nevi, where approximately 80% harbored mutant BRAF, including benign nevi (Pollock et al. 2003). Oncogenic BRAF mutations in melanoma appear to be somatic, as germline mutations are not associated with enhanced cancer risk (Niihori et al. 2006; Rodriguez-Viciana et al. 2006). The most common BRAF mutation (V600E, found in more than 90% of melanoma mutations) introduces a conformational alteration in the BRAF activation domain, causing constitutive kinase activation (Wan et al. 2004). BRAF, a member of the RAF family of genes (ARAF, BRAF, CRAF), is a serine-/threonine-specific protein kinase that activates sequentially MEK1/2 and then ERK1/2 (Garnett and Marais 2004). RAF is a downstream target of the RAS family of small guanine-nucleotide-binding proteins (NRAS, HRAS, KRAS). It was therefore not surprising to find that a RAS, almost always NRAS, is mutated in 15–25% of melanoma cell lines and tumors but rarely in melanomas harboring mutations in BRAF (Bennett 2008; Davies et al. 2002; Jonsson et al. 2007). These results strongly endorse the importance of the NRAS-BRAF-MEK-ERK (MAP kinase) pathway in melanomagenesis (see Fig. 3). It has been suggested that cancers may become overly dependent on, or “addicted” to, one or a few specific genes for maintenance of the malignant phenotype and survival (Weinstein and Joe 2008); in melanoma this may apply to oncogenic members of this pathway. In vivo mouse experiments have confirmed the key role of this pathway in melanoma, as well as the ability of members of this pathway to collaborate with loss of the CDKN2A locus in provoking melanoma development (Ackermann et al. 2005; Burd et al. 2014; Chin et al. 1997; Damsky et al. 2015; Dhomen et al. 2009; Goel et al. 2009; Kwong et al. 2012; Perez-Guijarro et al. 2017; Sharpless et al. 2003).
Receptor tyrosine kinases (RTKs) RTKs play crucial roles in regulating virtually all basic cell processes under normal physiological conditions and have been implicated in the development of most cancers. The exact phenotypic response to RTK signaling is extremely complex, depending on the RTK and its signal, the cell type, and the circumstances under which it is stimulated. In melanoma, examples of DNA copy number gains and point mutations have been documented that involve several RTKs. For example, increased copies of regions of chromosome 7, which encodes the epidermal growth factor receptor (EGFR) at 7p12, have been detected in late-stage melanomas, along with enhanced EGFR expression (Chin et al. 2006). The EGFR is an intriguing candidate because melanomas develop in Xiphophorus fish in association with aberrant expression of activated Xmrk, an EGFR-related gene. Another region of chromosome 7 (7q33-qter) that exhibits increased copy number in late-stage melanoma harbors the MET gene, encoding the HGF receptor; moreover, increased MET expression has been detected in metastatic melanoma (Natali et al. 1993). Activating point mutations in EGFR or MET have not been detected to date in melanomas. However, a recurrent L576P mutation in KIT has been reported for a small number of melanomas, with amplification and/or selective loss of the normal allele (Curtin et al. 2006; Willmore-Payne et al. 2005). Interestingly, melanomas with KIT mutations do not carry BRAF mutations and arise only from chronically sun-damaged skin (see the “Melanoma and the Environment” section). L576P is a GIST-associated, activating mutation, and as such represents a promising target for the RTK inhibitor imatinib in this subset of melanoma patients. However, clinical trials of the RTK inhibitors imatinib and sunitinib for KIT-mutated melanomas showed only modest responses (Buchbinder et al. 2015; Carvajal et al. 2011; Guo et al. 2011; Hodi et al. 2013). Some enzymes with appropriately targeted phosphatase activity counter the RTKs and may act as tumor suppressors. For example, the gene encoding protein tyrosine phosphatase receptor type D (PTPRD) was found to be homozygously deleted in 9% of melanoma cell lines (Stark and Hayward 2007). Moreover, upregulation of RTKs, e.g., PDGFR and IGF-1R, has been shown to play a key role in determining resistance of melanoma to BRAF inhibitor drugs (Nazarian et al. 2010; Villanueva et al. 2011) .
Transcription factors High-density single nucleotide polymorphism (SNP) arrays were used to detect recurrent focal amplifications of the transcription factor MITF in 10% of primary melanomas and up to 20% of metastatic melanomas (Garraway et al. 2005). A recurrent activating mutation in MITF has also been identified in the germline of kindred from multiple continents with familial melanoma (Yokoyama et al. 2011). The recurrent mutation stimulates MITF’s transcriptional activity by disrupting a SUMO-modification consensus sequence that otherwise suppresses MITF function. As discussed earlier, MITF is a melanocyte lineage-survival factor required for the commitment of immature cells to the melanocyte lineage during development and heavily involved in melanocyte survival, growth, and differentiation (Kawakami and Fisher 2017) (see Fig. 3). Melanomas resistant to BRAF inhibitors have been shown to overexpress MITF, and knocking down MITF expression reversed this resistance (Smith et al. 2013). The reliance of melanocytes on MITF makes it an intriguing candidate target for melanoma patients (Merlino 2005). However, akin to most transcription factors, the lack of a catalytic domain in MITF protein makes it a problematic therapeutic target. Consequently, a number of strategies are being employed to inhibit MITF indirectly; for example, inhibitors of HDAC, AMP-activated kinase (AMPK), and HIV1-protease have been shown to suppress MITF expression (Borgdorff et al. 2014; Smith et al. 2016; Yokoyama et al. 2008). A number of additional transcription factors have been implicated in melanoma progression (Poser and Bosserhoff 2004). The transcription factor TBX2, which can repress expression of p14ARF and p21CIP1 (product of CDKN1A gene), exhibits an increased gene copy number and overexpression in melanoma (Jacobs et al. 2000; Jonsson et al. 2007; Vance et al. 2005). Gene copy number increases up to 40% have also been reported in melanomas for the transcription factor MYC (Bennett 2008), but conflicting reports (Stark and Hayward 2007) have indicated a complex and yet unresolved role of MYC in melanoma. Recently, MYC overexpression in melanoma has been suggested to be correlated with accelerated tumor metastasis in vivo (Lin et al. 2017). The factors TP53 and β-catenin have already been discussed above.
The pro-metastasis gene NEDD9 Over the years many genes have been implicated in melanoma metastasis (Chin et al. 2006; Crowson et al. 2007; Qiu et al. 2015; Xu et al. 2008). Proceeding on the assumption that overlapping genomic changes in human and mouse melanomas arise through shared selective pressure and represent critical events in melanomagenesis, Kim et al. used genome-wide high-resolution array-CGH to identify the scaffold protein NEDD9 as a pro-metastasis factor (Kim et al. 2006). Genetically engineered mouse models of human melanoma were employed to narrow down candidates in an amplified syntenic region in the human genome to NEDD9. Loss-of-function and gain-of-function experiments were employed to confirm that NEDD9 confers enhanced invasiveness and metastatic potential to melanocytic cells. NEDD9 was found to facilitate metastasis through interaction with focal adhesion kinase (Kim et al. 2006).
Melanoma: A Consequence of Homeostatic Disruption
No cell is an island, and melanocytic cells live in dynamic and harmonic equilibrium with their microenvironment. An intricate network of interactions ensures a state of homeostasis both during development and under normal adult physiological conditions. As discussed earlier, the melanocyte microenvironment includes cellular neighbors such as keratinocytes, fibroblasts, endothelial and immune cells, as well as components of a complex ECM. In particular, melanocytes and keratinocytes (the so-called melanin unit) are intimately associated with each other in the basal layer of the epidermis of the human skin. Each melanocyte makes contact with, and sends pigment-containing melanosomes to, dozens of keratinocytes, a process that can be dramatically altered by exposure to UVR. Keratinocytes are thought to employ the pigment as protection from subsequent UVR damage. Keratinocytes regulate melanocyte growth and behavior through the activity of cell-cell adhesion molecules and paracrine growth factors (Haass et al. 2005). The initiation and progression of melanoma marks a significant disruption in this tranquil homeostasis, involving key changes within the melanocyte genome and epigenome, and in the way these melanocytes grow, survive, and interact with their microenvironment (Haass and Herlyn 2005). Moreover, it is clear that exposure to UVR, known to initiate melanomagenesis, is a powerful homeostatic disruptor.
Dysregulation of melanocyte gene expression Gene expression profiling using multiple technologies has permitted the simultaneous analysis of the entire melanoma genome. Studies have identified multiple significant changes at the level of gene expression as melanocytes are transformed into melanoma cells and further into metastatic melanomas (Haqq et al. 2005; Hoek et al. 2004; Hoek 2007; Kaufmann et al. 2008; Talantov et al. 2005). Moreover, exposure to UVR has been shown to induce significant alterations in gene expression patterns of melanocytes (Yang et al. 2006; Zaidi et al. 2011).
Several mechanisms undoubtedly contribute to the observed global changes in expression. First, the expression or activity of many important transcription factors can be altered in melanoma, including MITF, LEF1/β-catenin, TBX2, MYC, PAX3, SOX10, and SLUG (Poser and Bosserhoff 2004) (see Table 1). Another interesting transcriptional regulator, inhibitor of differentiation (ID1), has been shown to repress p16INK4A transcription and delay melanocyte senescence (Cummings et al. 2008; Polsky et al. 2001) and may play an early role in melanomagenesis. These transacting factors not only help regulate the expression of a plethora of critical downstream targets but can influence each other’s expression/activity as well, further demonstrating the complexity of this regulatory network. In addition, the modification of chromatin is another level of gene regulation that plays a major role in the development of tumors, including melanoma (Moran et al. 2018). Epigenetic changes include DNA methylation and numerous covalent modifications of histones, including methylation, acetylation, phosphorylation, sumoylation, and ubiquitination. In melanoma, promoter methylation has been documented in suppressing the expression of genes such as CDKN2A, PTEN, RASSF1A, MGMT, DAPK, RARB2, and APC.
Finally, a relatively newly recognized category of powerful gene regulators is the microRNAs (miRNAs), which has been shown to participate in tumorigenesis, including metastasis (Di Leva et al. 2014). miRNAs are short molecules of RNA of about 22 nucleotides in length and are the most expressed class of noncoding RNAs in eukaryotic cells. Their main gene regulatory function is through silencing or degrading mRNAs, and a single miRNA can potentially regulate several target mRNAs simultaneously. Consequently, miRNAs can have oncogenic or tumor suppressor effects depending on the types of genes they regulate. miRNAs have been extensively studied in melanoma cells and clinical samples, and dysregulation of a relatively large number of melanoma-specific miRNAs has been identified, with regulatory effects on gene expression affecting a variety of pathways (Fattore et al. 2017). Next-generation sequencing studies have identified another category of regulatory RNAs called long noncoding RNAs (lncRNAs) , which are defined as transcripts >200 nucleotides in length. Several lncRNAs have been characterized to play key roles in cellular proliferation, survival, migration, and genome stability, and their aberrant expression is associated with many cancer types, including melanoma (Huarte 2015; Leucci et al. 2016). Together, genetic and epigenetic alterations can cooperate to disrupt normal homeostasis as melanocytes move along the path to malignancy.
Dysregulation of melanocyte proliferation/differentiation Adult melanocytes are built to manufacture pigment, to respond to environmental challenges through enhanced differentiation but controlled growth, and to survive. In contrast, melanoma cells proliferate autonomously and resist differentiation and growth inhibition signals. How does this happen? It is important to appreciate that these critical melanocytic functions are regulated more through highly interactive webs or networks and not simple linear pathways (Palmieri et al. 2015) (see Fig. 3). What sits at the center of this web is the melanocyte master regulator MITF (Kawakami and Fisher 2017; Palmieri et al. 2015). MITF can regulate virtually all known melanocyte-specific genes, including TYR, DCT, and TYRP1. MITF is absolutely required for melanocyte survival, partly by regulating expression of survival factors such as BCL2, and a number of MITF-mutant mouse strains have been described that are devoid of melanocytes. Both a positive and a negative influence, MITF is also required for melanocyte proliferation but depending on its level of expression or posttranslational modification can also be growth inhibitory (Kawakami and Fisher 2017). For example, MITF can transactivate stimulators of growth such as CDK2 and TBX2 but can also directly transactivate growth inhibitors such as p16INK4A and p21CIP1 and indirectly activate the CDK inhibitor p27KIP1 (product of CDKN1B gene). The expression or activity of MITF can in turn be regulated by numerous key melanocytic factors, including the transcription factors PAX3, SOX9, SOX10, CREB, and LEF1; the cofactors CBP, RB1, p300, SWI/SNF, and β-catenin; and the MAPK pathway through EDN and EDNRB. The network centering on MITF radiates out to myriad pathways regulating virtually every aspect of melanocyte function. Notably, MITF amplification or mutation can occur within melanomas, suggesting that under the appropriate circumstances, MITF can function as a lineage-survival oncogene (Garraway and Sellers 2006). The ability of melanocytes to produce pigment is also heavily regulated through MC1R and its ligands, POMC-derived αMSH, and ACTH (Rouzaud et al. 2005). Polymorphic variations and mutations in MC1R in the general population determine the type of pigment that is produced (eumelanin or pheomelanin) and therefore the color of the skin (dark skinned or fair skinned). Notably, fair-skinned individuals have a greater risk for melanoma, linking MC1R to melanomagenesis. A significant association was detected between germline MCIR variants, BRAF mutations, and a subset of melanomas (Landi et al. 2006).
As in other types of cells, the key to controlling proliferation is through regulation of transit through the cell cycle. Entry into the cell cycle is normally promoted by growth factors, which in melanocytes include FGF2, HGF, KITLG, EDN3, IGF-1, and the WNTs; these factors can of course regulate other melanocyte behaviors as well, such as survival, differentiation, and migration (see Table 1). By triggering cascading signaling pathways leading to the activation of downstream cell cycle-associated transcription factors (e.g., MYC, FOS, JUN, and ATF2), these growth factors help overcome the G1-S cell cycle checkpoint. These factors stimulate CDK4/6-cyclin D phosphorylation/inhibition of RB and thereby the release of E2F to transactivate critical cell cycle control genes, such as those encoding the E and A cyclins, MYC, and CDK1. CKIs normally serve as key regulators of G1 cell cycle progression and include p16INK4A, p15INK4B, p21CIP1, and p27KIP1. An important part of the melanoma story is written in the deregulation of these pathways. We have already noted above that mutations or altered expression characterizes virtually every key cell cycle regulator. Moreover, melanoma cells often become growth factor-independent, constitutively initiating these important pathways through, for example, activating mutations in RTKs such as KIT. Another mechanism of deregulated growth frequently described in melanoma is through the creation of autocrine signaling loops, wherein melanoma cells express a growth factor or cytokine (e.g., HGF, FGF2, TGFα, VEGF, MGSA/GRO, IL-8/CXCL8), as well as its cognate receptor (e.g., MET, FGFR1, EGFR, VEGFR1/2, CXCR1/2).
A critical intermediary between the initial phosphorylation event triggered by the binding of a growth factor to its receptor and the activation of downstream cell cycle-associated transcription factors is the MAPK pathway. RAS gene products are 21 kD GTPase proteins that serve as molecular switches converting the kinase activation events at the cell membrane to nuclear events and ultimately to altered melanocyte behavior (Simanshu et al. 2017). There are three different RAS genes, NRAS, HRAS, and KRAS. Despite their structural similarity, these three RAS genes have distinct functions. In fact, genetically engineered mouse models have confirmed that NRAS is significantly more effective than HRAS at transforming melanocytes in vivo (Perez-Guijarro et al. 2017). Based on the strong preference for NRAS mutations in melanomas, it has been shown that NRAS is the key regulatory G-protein in melanocytes (Burd et al. 2014). RAS activating point mutations cause impaired GTPase activity resulting in a constitutively activated G-protein and deregulated growth control. Similarly, the most common BRAF mutation in melanomas, V600E, is also forced into a state of perpetual activation. The obligatory nature of the deregulated MAPK pathway in melanomagenesis is evident from the high percentage of melanomas with either NRAS or BRAF mutations, which together range from 80% to 90%. Because mutant BRAF and NRAS can be detected in nevi (Damsky and Bosenberg 2017), it is assumed that these mutations are early events in melanomagenesis and are important in clonal expansion. Although the MAPK pathway is most central to melanocyte growth control, a second key RAS effector, PBK, also helps regulate proliferation. Although not as frequently mutated in melanomas, the PTEN-PBK-AKT pathway also plays a critical role in other melanocyte behaviors, including regulation of migration and apoptosis. A mouse model employing grafted human skin tissue containing melanocytes genetically engineered to express specific mutations has confirmed the importance of activated PBK and NRAS, but notably not that of BRAF in melanomagenesis (Chudnovsky et al. 2005).
In an extreme example of growth control, cells can enter an irreversible state of growth arrest called senescence. This phenomenon was described in cultured cells many years ago and was initially argued to be an in vitro artifact; however, a number of impressive examples have been reported demonstrating the relevance of in vivo senescence in various types of cancers, including melanoma (Liu et al. 2018; Mooi and Peeper 2006). Early benign pigmented nevi are now thought to represent collections of mostly senescent melanocytes, and senescence is considered a significant barrier that melanoma cells must overcome to become proliferative melanomas (Bennett 2008). Cellular senescence can be induced through two distinct mechanisms. Replicative senescence occurs after extensive proliferation and is associated with telomere exhaustion. The shortening of telomeres is a natural by-product of replication in normal cells with limited telomerase, a heterodimer consisting of the protein TERT and the RNA TERC. After enough replication cycles, chromosome ends become so short that their protective cap structure is compromised, resulting in the activation of TP53-mediated DNA damage signaling, chromosome fusion, and mitotic disruption, termed crisis (Shay 2016). Cancer cells cannot therefore grow indefinitely without employing a mechanism to overcome telomere shortening, as they would become senescent or die. A mechanism that is often exploited by cancer cells, including melanoma cells, to gain the ability to grow indefinitely, or become immortalized, is reactivation of telomerase expression.
Senescence can also be induced by oncogene activation or an equally dramatic cellular stress (Ben-Porath and Weinberg 2005; Lowe et al. 2004; Mooi and Peeper 2006). Ironically, despite the fact that mutant BRAF and NRAS appear to be nearly obligatory for the development of human melanomas, these same activated oncogenes can induce a senescence response in melanocytes (Michaloglou et al. 2005). A number of factors have been implicated in overcoming early oncogene-induced senescence, most notably the p16INK4A-CDK4/6-RB and ARF-HDM2-TP53 pathways, and the CDKN2A locus in particular. p16INK4A has long been considered a key melanocyte senescence gene; BRAFV600E induces p16INK4A expression, and p16Ink4a deficiency in genetically engineered mice confers immortality to melanocytes and induces melanomagenesis (Liu and Sharpless 2012; Perez-Guijarro et al. 2017). Evidence in mice indicates that the other member encoded by the CDKN2A locus, p19ARF, is also very important in melanocyte senescence, highlighting the importance of the simultaneous loss of p16INK4A and p19ARF function when CDKN2A experiences deletions. Notably, p19ARF was found to induce senescence independently of TP53 in melanocytes (Ha et al. 2007). However, the view that human nevi undergo oncogene-induced senescence has been challenged in the light of the observations that the biomarkers widely used to define senescence are not exclusive to the senescence program (e.g., p16INK4A and SA-β-gal). p16INK4A was found to be abundantly expressed in both nevi and primary melanomas, and SA-β-gal, the most widely accepted senescence marker, was detected in both nevi and metastatic melanoma (Tran and Rizos 2013). These observations cast doubt on human nevi being defined as truly senescent (Tran and Rizos 2013).
Dysregulation of melanocyte survival The ability of cancer cells to overcome built-in programming designed to trigger cell death (apoptosis) in response to DNA damage, depletion of survival factors, or disruption of interactions with the microenvironment is a hallmark of successful progression (Hanahan and Weinberg 2011). Apoptosis is activated when a sensor, usually TP53, detects a deathinducing signal, which in turn transmits an execution order to the cell death machinery, often associated with mitochondria. Pro-apoptotic signals, including TP53, NOXA, PUMA, BAX, BAD, tumor necrosis factor (TNF), TNF-related apoptosis-inducing ligand (TRAIL /TNFSF10), and the Fas ligand (FASLG), induce the release of cytochrome c, which in concert with apoptotic protease-activating factor 1 (APAF1) activates caspase-9 and the resulting effector protease cascade (Lowe and Lin 2000). To achieve balance, these pathways are countered by pro-survival factors, such as BCL2, BCL2L1, nuclear factor-κB (NFKB), survivin (BIRC5), and livin (BIRC7) (Hussein et al. 2003a). Melanocytes have evolved to survive the adverse environment created from the production of melanin and exposure to UVR. Therefore, it is not surprising that melanoma cells utilize several effective means to avoid apoptotic destruction (see Fig. 3) (Hussein et al. 2003a). Although TP53 is infrequently mutated in melanomas, loss of its positive regulator, p14ARF, indirectly hampers the TP53-mediated DNA damage response. Melanoma cells characteristically produce elevated levels of BCL2, limiting cytochrome c-mediated caspase-9 activation. Caspase-9 activity is also inhibited through the influence of the kinase AKT, often highly active in melanomas. Advanced melanomas have been found to overexpress survivin, which binds to and inhibits effector caspase-3 and caspase-7, acting downstream of caspase-9 (Grossman et al. 1999). Similarly, livin is overexpressed in melanomas and can inactivate caspase-3, caspase-7, and caspase-9 (Kasof and Gomes 2001). Loss of APAF1 expression has been associated with melanoma cell survival and chemoresistance (Soengas et al. 2001).
Caspases are also triggered by activation of the death receptors, through binding of TNF, FASLG, and TRAIL (TNFSF10) (Ivanov et al. 2003). For example, TRAIL can induce apoptosis through interactions with the death receptors TRAIL-R1 (TNFRSF10A) and TRAIL-R2 (TNFRSF10B), which are expressed in most normal tissues as well as in melanomas; normal tissues survive by expressing the inhibitory decoy receptors TRAIL-R3/DcR1 and TRAIL-R4/DcR2 (Lowe and Lin 2000). However, these survival pathways are complex, and death receptor activation also triggers NF-κB, which is frequently upregulated in melanoma and is an essential survival-promoting factor in melanoma. Countering the ability of melanoma cells to evade apoptotic destruction is an attractive therapeutic approach and is currently being actively pursued (Fulda 2015).
Dysregulation of cell-cell and cell-ECM interactions Melanocytic cells, particularly melanoblasts, are highly migratory by nature. It has therefore been suggested that melanoma cells become metastatic by coopting and enhancing this innate migratory tendency (Gupta et al. 2005). In the initial phase of this cellular relocation, melanocytes must first sever their molecular ties with keratinocytes, ties consisting of cell surface molecules that promote interactions with keratinocytes (Haass et al. 2005). Alterations in expression of cell adhesion molecules such as cadherins, integrins, and immunoglobulin superfamily members can significantly disrupt the homeostatic balance between melanocytes and keratinocytes, facilitating melanoma progression. Cadherins are multifunctional transmembrane proteins that act as both receptor and ligand to maintain appropriate cell-cell contacts. The switch in expression from E-cadherin (encoded by CDH1) to N-cadherin (CDH2), first observed in RGP melanoma, redirects gap junction formation from E-cadherin-expressing keratinocytes, which inhibit melanocyte proliferation and maintain a differentiated morphology, to N-cadherin-expressing fibroblasts and vascular endothelial cells, encouraging migration from the epidermis to the dermis (McGary et al. 2002). N-cadherin also enhances melanoma survival by stimulating β-catenin signaling, LEF1 activity, and subsequent activation of MITF and CCND1. E-cadherin is downregulated in part by the transcription factor Snail (SNAI1) (Tsutsumida et al. 2004), whereas N-cadherin expression is upregulated by the Notch signaling pathway (Liu et al. 2006), already implicated in melanomagenesis (Nickoloff et al. 2005). Loss of E-cadherin is often accompanied by changes in other cell adhesion molecules, including integrins.
Integrins are heterodimeric cell surface receptors, consisting of 1 of 20 different α chains and 1 of 9 different β chains. The anchorage of melanocytes to the dermal-epidermal basement membrane can be weakened through changes in melanocytic integrins. Melanomas express specific integrins (e.g., αvβ3 and α4β1) that have been implicated in adhesion to ECM, promoting motility, invasion, and metastasis, which correlates closely with poor clinical outcome in melanoma patients (Kuphal et al. 2005). For example, integrin αvβ3, the vitronectin receptor, can facilitate melanoma cell growth, survival, and matrix invasion and marks the progression of RGP to VGP melanoma. Expression of integrin αvβ3 has also been shown to correlate with the activity of the matrix metalloproteinases (MMPs) MMP-1 and MMP-2 (Hofmann et al. 2000). Melanomas also overexpress immunoglobulin superfamily members, including MCAM (MUC-18/CD146), ICAM-1 (CD54), and ALCAM (CD166) (Crowson et al. 2007; McGary et al. 2002). MCAM is overexpressed in more than 80% of melanomas and mediates interactions between melanoma cells and both the matrix and other cell types such as endothelial cells. In a three-dimensional epidermal skin model, melanoma cells expressing MCAM exhibited enhanced separation from the epidermis and invasion through the basement membrane and into the dermis (Satyamoorthy et al. 2001).
Melanoma cell invasiveness can be accounted for in part by their ability to induce basement membrane breakdown and other forms of remodeling through secretion of proteolytic enzymes such as hyaluronidase, heparanase, and MMPs (Edward and MacKie 1993). Melanoma cells can also recruit surrounding fibroblasts to aid and abet invasion by encouraging secretion of their proteolytic enzymes. The degraded ECM in turn releases additional factors that can induce melanoma cell and endothelial cell growth, stimulating angiogenesis. The MMPs are broadly acting, zinc-dependent enzymes capable of functioning as collagenases, gelatinases, or stromelysins and are classified accordingly. MMP activity is carefully regulated, partly through their tissue inhibitors (TIMPs). Increased expression of a number of MMPs has been documented in invasive melanomas, including MMP-1, MMP-2, and MMP-9 and MT1-MMP (Crowson et al. 2007). One focal point in melanoma research has been MMP-2, which localizes to the melanomastromal interface in primary and metastatic lesions and correlates with progression. Of interest, MMP-2 is activated in association with a complex that includes MT1-MMP, TIMP-2, and integrin αvβ3 (Hofmann et al. 2000).
Melanoma: Cell of Origin
Pluripotent stem cells are defined by their abilities to indefinitely self-renew and to give rise to an organ-specific differentiated repertoire of cell types. These stem cells tend to be relatively quiescent and to express drug resistance and anti-apoptotic genes. Based mostly on data from hematologic malignancies and a few solid tumors, it has long been suggested that tumors harbor relatively rare cells with similar properties, called cancer stem cells or tumor-initiating cells (Batlle and Clevers 2017). Several critical questions followed upon acceptance of this hypothesis. One involved the cancer cell of origin: Do cancers arise from stem cells that have become malignant, or do cancers arise from more differentiated cell types, which then acquire the ability to self-renew? A second question concerned the ability of tumors to recur after drug treatment: Would cancer stem cells be intrinsically resistant to conventional therapeutic treatment? Adult melanocyte stem cells have been identified and reside within a specialized niche within the hair follicle called the bulge region (Nishimura 2011; Nishimura et al. 2005; Nishimura et al. 2002). During cyclic hair renewal, dormant bulge melanocyte stem cells are activated by specific signaling molecules and then give rise to transiently amplifying progeny, which proliferate, differentiate into pigment-producing melanocytes, and migrate to the base of the hair follicle. Transiently amplifying melanocytes can also migrate from the bulge region to the epidermis where they function as differentiated, pigmented melanocytes. Interestingly, a number of factors implicated in regulating melanocyte stem cell function – MITF, PAX3, NOTCH, and SLUG (SNAI2) – were also linked to melanomagenesis (Garraway and Sellers 2006; Gupta et al. 2005; Hoek et al. 2004; Nickoloff et al. 2005; Plummer et al. 2008), supporting the idea that a cancer stem cell was the melanoma cell of origin. Fang et al. (2005) cultured nonadherent spheroids from human melanoma cells or from metastases in an ES-like medium and identified a CD20-rich subpopulation that could be induced to differentiate into multiple phenotypes, consistent with stem cell properties. Frank and colleagues (Frank et al. 2005; Schatton et al. 2008) identified a subpopulation of human melanoma cells expressing the chemoresistance mediator ABCB5, which was capable of self-renewal and differentiation as well as serial and limiting-dilution transplantation. It was postulated that the ABCB5-expressing cancer stem cells would represent a population highly resistant to conventional melanoma therapy. Recently, however, it has been reported that differentiated pigment-producing melanocytes located in the interfollicular epidermis can give rise to melanoma (Kohler et al. 2017). Using a BrafV600E-driven mouse melanoma model, Kohler et al. showed that the melanocyte stem cells residing in the hair follicle bulge region are refractory to melanomagenesis, and in fact, melanoma originates from the pigment-producing, but not amelanotic, melanocytes located in the interfollicular regions. These pigment-producing mature melanocytes exhibited loss of differentiation markers before dermal invasion, suggesting that a transcriptomic reprogramming had caused a reversion to a dedifferentiated tumor-initiating state (Kohler et al. 2017). It is unclear what microenvironmental cues play a role in dictating this reprogramming.
Melanoma and the Environment
Sun Exposure and Epidemiology
Melanoma rates continue to rise in most populations at a time when the incidence of many other cancers is on the decline (Cronin et al. 2018; Nikolaou and Stratigos 2014). Extensive epidemiologic studies have provided strong support for the notion that UVR is a significant environmental carcinogen for melanoma (Garibyan and Fisher 2010; Moan et al. 2008a; Tran et al. 2008). The relentless rise in skin cancer incidence despite broad knowledge of the carcinogenic activity of UVR is likely a manifestation of recent evidence demonstrating that release of beta-endorphin and consequent opiate-like responses occur as by-products of the UVR-tanning response (Fell et al. 2014). Supporting experimental evidence for UVR’s carcinogenic activity has come from studies of the effects of UVR on a variety of animal models, including the opossum, the fish Xiphophorus , the Sinclair swine, and several lines of genetically engineered mice (Jhappan et al. 2003; Zaidi et al. 2008). However, significant questions remain as to how UVR causes melanoma and under what circumstances (Maddodi and Setaluri 2008). For example, unlike other skin cancers, melanoma risk does not appear to be associated with cumulative lifetime exposure to UVR; instead, intermittent burning doses of UVR represent a more significant risk factor. Other epidemiologic data draw a critical association between melanoma and childhood UVR exposure (Green et al. 2011; Whiteman et al. 2001). Immunologic differences between children and adults, or differences in their proportion of melanocytic progenitors, have been suggested as possible explanations for this observation (Zaidi et al. 2012). So, while the association between melanoma and sunlight exposure has long been appreciated, the relationship between exposure and risk is complex, and the underlying mechanisms are largely unknown.
In contrast, great strides have been made in classifying melanomas based on epidemiologic and molecular associations (Bastian 2014; Cancer Genome Atlas 2015). In perhaps the best example, Curtin et al. (2005) demonstrated that melanomas arising from skin sites not subjected to chronic sun damage (e.g., those receiving intermittent sun exposure) were highly likely to carry characteristic BRAF mutations, whereas in melanomas arising from skin with chronic sun damage, or regions that virtually never see sunlight (e.g., acral and mucosal melanomas), such mutations were very rare. In support of this relationship, congenital nevi arising without UVR exposure also lacked BRAF mutations (Bauer et al. 2007). Interestingly, as if to compensate for the lack of BRAF mutations, melanomas from chronically sundamaged skin frequently harbor activating oncogenic mutations in KIT, a receptor tyrosine kinase upstream of BRAF (Curtin et al. 2006).
Despite the general agreement on the causal role of UVR in melanoma, the efficacy of sunscreens in the prevention of melanoma remained controversial; in fact, some earlier epidemiologic data suggested that people who used sunscreens were more likely to develop skin cancer, not less (Bastuji-Garin and Diepgen 2002). Part of this controversy can be traced to the fact that the early sunscreens were designed to block UVB, and not UVA, because erythema (the reddening accompanying burning) is caused by UVB. If in fact UVA played a significant role in melanomagenesis, early epidemiologic data would reflect the failure of early sunscreens to effectively filter out this waveband. However, the role of UVA in melanoma was itself controversial. Another interpretation of these data was that they reflected improper use of sunscreens in terms of frequency and thickness of sunscreen application by those spending significant time out in the sun. Recent studies have started to shed light on these controversies. Klug et al. (2010) performed a matched logistic regression analysis of sunscreen use adjusted for average UVB intensity, sun exposure, skin type, number of sunburns, age group, study site, and gender to show that sunscreen use was not associated with melanomagenesis. Moreover, utilizing the HGF transgenic mouse model of UVR-induced melanomagenesis, they showed that SPF-15 sunscreen-treated mice developed significantly fewer melanomas than the vehicle-treated control mice; and this effect was associated with significant reduction in UVB-induced DNA damage, indicating a distinct protective effect of sunscreen (Klug et al. 2010). Using a Braf-mutant mouse model, Viros et al. showed evidence that sunscreen at least partially protected against UVR-induced melanomagenesis (Viros et al. 2014). In another in vivo study using the UVR-susceptible NRas61R mutant mouse model of melanoma, Hennessey et al. recently showed that aerosol sunscreens effectively blocked UVR-induced DNA damage and delayed melanoma formation (Hennessey et al. 2017).
Yet another controversy surrounded the opposing values of restricting sunlight to prevent the development of melanoma and other skin cancers and of receiving enough sunlight to ensure optimal production of vitamin D, which in adequate amounts prevents a variety of diseases (Moan et al. 2008b). For example, in a post-hoc analysis of a randomized controlled trial, vitamin D supplementation was not associated with reduced melanoma risk (Tang et al. 2011). On the other hand, vitamin D deficiency has been shown to be associated with a worse prognosis in metastatic melanoma patients (Timerman et al. 2017). Extensive studies in the last two decades have provided strong evidence that vitamin D deficiency is associated with reduced survival of melanoma patients and single-nucleotide polymorphisms (SNPs) in the vitamin D receptor (VDR) gene affect melanomagenesis and/or disease outcome (Slominski et al. 2017). Active forms of vitamin D3 protect against oxidative DNA damage by upregulating genes such as TP53 and GADD45 (Fleet et al. 2012), and its newly discovered derivatives exhibit VDR-mediated antiproliferative effects on melanoma cell lines (Janjetovic et al. 2011; Slominski et al. 2012). Based on these and other extensive studies, multiple randomized clinical trials are currently underway to evaluate whether vitamin D supplementation improves survival outcome in melanoma patients. Importantly, however, these questions do not lead to the conclusion that exposure to UVR would be an optimal means of maintaining healthy vitamin D levels, as oral vitamin supplements are effective, inexpensive, and unlike UVR, not associated with any known carcinogenic activity.
Photobiology and Melanoma
UVB versus UVA To begin to understand how UVR affects biological systems (photobiology), one needs to appreciate the complexity of UVR itself. UVR from sunlight is subdivided into shorter wavelength UVC (200–280 nm), middle wavelength UVB (280–320 nm), and longer wavelength UVA (320–400 nm). UVC is biologically irrelevant, because it is absorbed by the atmospheric ozone layer. UVB and UVA are very different in their properties and biologic consequences. UVB penetrates poorly into the skin but is efficiently absorbed directly by DNA and proteins (Bruls et al. 1984). The two major types of DNA damage induced by UVB are cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts, which are repaired via the nucleotide excision repair pathway. The UVB-induced CPDs are characterized by signature C to T or CC to TT transitions. However, in melanomas such CPD signature mutations are relatively rare in the oncogenes or tumor suppressors that have been implicated in melanomagenesis. For example, 90% of BRAF mutations contain a T to A transversion (T1799A) (Wan et al. 2004). Only UVB can cause skin erythema. In contrast to shortwave UVB, UVA penetrates much more deeply into the skin but is poorly absorbed by DNA. UVA does not directly damage DNA, instead exerting its effects through photosensitizers. Thus, UVA induces oxidative DNA damage, such as the formation of 8-oxo-7,8-dihydroguanine through the generation of species such as reactive melanin radicals (Hill and Hill 2000).
UVR and ROS In eukaryotic cells, mitochondrial-mediated oxidative phosphorylation produces energy and, because electron transfer is not completely efficient, also generates potentially harmful side products called reactive oxygen species (ROS) (Wittgen and van Kempen 2007). Some ROS are required as secondary messengers of signaling cascades and for other normal cell functions, so cells have developed an antioxidant network that allows excess ROS to be scavenged, resulting in a state of homeostasis. However, melanocytes face a daunting ROS burden because of their ability to produce melanin and because they are routinely exposed to UVR, a situation that can have melanomagenic consequences. UVR stimulates the synthesis of melanin that, although designed for protection from further UVR damage, can become a prooxidant under oxidative stress conditions. Specifically, UVR exposure can induce the formation of oxidized melanin, forming a variety of radicals and disrupting ROS homeostasis. UVR, and UVA in particular, can damage DNA through the production of these melaninderived radical species. Using the HGF transgenic mouse model, Noonan et al. (2012) showed that UVA was indeed melanomagenic but required the presence of melanin pigment and was associated with oxidative DNA damage, whereas UVB-induced melanomagenesis was independent of melanin.
Interestingly, ROS activity is related to the hypoxic response, mediated by the transcription factor hypoxia-inducible factor-1 (HIF-1α/β) (Wittgen and van Kempen 2007). The epidermis of the normal skin exists in a mildly hypoxic environment, an environment that can promote melanomagenesis. Bedogni et al. (2005) demonstrated that AKT-mediated transformation of melanocytes only occurs under HIF-lα-inducing hypoxic conditions similar to those found in the normal skin. In addition, AKT hyperactivation can bring about ROS generation by inducing NOX4 and by inhibiting apoptosis, thereby damaging mitochondria (Govindarajan et al. 2007).
UVR and the microenvironment UVR is known to induce an increase in the number of melanocytes and to enhance melanin synthesis, and ultimately UVR stimulates an increased transfer of melanin to those keratinocytes that it contacts (Miyamura et al. 2007; Zaidi et al. 2011). UVR can alter the melanocyte microenvironment, which subsequently influences the melanocytic response. D’Orazio et al. (2006) reported that UVR induces enhanced synthesis and secretion of αMSH by keratinocytes, which in turn triggers an appropriate melanocyte response through MC1R and the cAMP pathway, activating melaninsynthesizing enzymes and melanin production. In fact, UVR affects most of the components of the skin including melanocytes, keratinocytes, fibroblasts, and elements of the vascular and immune systems (Slominski and Pawelek 1998). UVR induces secretion of multiple factors by multiple cellular components, including neuropeptides (POMCderived αMSH, ACTH, and β-endorphin), growth factors (FGF2, IGF-1, TGFα), and numerous cytokines (TNF-α, CSF2, IL-1, IL-6, IL-8, IL-10) (Slominski and Pawelek 1998).
UVR triggers inflammatory reactions and has an overall immunoinhibitory effect on the skin (Hart and Norval 2017; Kripke 1990). Moreover, the effect of UVR on the immune system in mouse models may be most influential at the neonatal stage, perhaps explaining the elevated risk associated with childhood UVR exposure (Zaidi et al. 2011, 2012). More specifically, UVR suppresses the ability of antigen-presenting Langerhans cells in the skin to induce immunity by decreasing their number and their expression of MHC. UVR also stimulates an influx of macrophages and a decrease in the viability of T-lymphocytes. It has also been shown that inflammation can have positive influences on tumorigenesis. Together, it is evident that the abilities of UVR to induce mutagenesis, inflammation, and long-term immunosuppression can all profoundly influence melanomagenesis. Importantly, however, there is evidence to suggest some UVR-independent melanoma risk in certain circumstances. In individuals with the red-hair/light-skin phenotype, melanoma risk is elevated in both sun-exposed and non-sun-exposed skin (in contrast to their risk of non-melanoma skin cancers whose incidence is elevated only in sun-exposed skin). Mitra et al. (2012) utilized mouse models of red-hair/Mc1r-deficiency to show that red pigment (pheomelanin) is intrinsically carcinogenic – elevating the spontaneous incidence of BRAFV600E-associated melanomas in the complete absence of UVR exposure. When red-haired mice were crossed to albinos (carrying Tyr mutations, thus unable to make any melanins), the melanoma risk was fully blocked. Significant levels of pheomelanin are dominantly present in the skin of not only redheads but also other Caucasians, suggesting that it may contribute chemically to melanomagenesis. However, it is also very likely that UVR amplifies the ROS-inducing capacity of pheomelanin, suggesting that UVR significantly exacerbates skin carcinogenicity in humans with fair skin.
Conclusion
It has been more than 200 years since the Parisian physician René Laennec first reported “melanoma” in Europe, while back in 1857 William Norris (1857) recognized that melanoma had a familial component. It is unlikely that either could have foreseen that by the twenty-first century, cutaneous malignant melanoma would become one of the fastest increasing cancers. In the United States alone, melanoma incidence has more than quadrupled over the past four decades, and it is estimated that more than 90,000 new cases will be diagnosed in 2018 (Cronin et al. 2018). Melanoma remains one of only a few major malignancies demonstrating significant positive rates of increase in the United States. We have come a long way with respect to our understanding of the origins of melanoma and the mechanisms by which it develops. As documented in this chapter, the first decade of the twenty-first century featured tremendous progress in the molecular technologic weaponry with which to address the most outstanding questions about the melanoma disease initiation, progression, and metastasis. Utilizing these impressive tools, researchers generated a treasure trove of data, gathered from both broadly global and exquisitely detailed analyses of melanocytic lesions from patients and from genetically engineered mouse models. In the second decade of this century, these data have advanced our understanding of the fundamental mechanisms of the disease, which has fueled an explosive expansion of therapeutic strategies that have revolutionized the clinical management of melanoma. There is excited anticipation within the melanoma research community that the holy grail of eliminating melanoma-related death is within reach.
References
Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F (2005) Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res 65:4005–4011. https://doi.org/10.1158/0008-5472.CAN-04-2970
Aguissa-Toure AH, Li G (2012) Genetic alterations of PTEN in human melanoma. Cell Mol Life Sci 69:1475–1491. https://doi.org/10.1007/s00018-011-0878-0
Ainger SA, Jagirdar K, Lee KJ, Soyer HP, Sturm RA (2017) Skin pigmentation genetics for the clinic. Dermatology 233:1–15. https://doi.org/10.1159/000468538
Aoude LG, Wadt KA, Pritchard AL, Hayward NK (2015) Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res 28:148–160. https://doi.org/10.1111/pcmr.12333
Barral DC, Seabra MC (2004) The melanosome as a model to study organelle motility in mammals. Pigment Cell Res 17:111–118. https://doi.org/10.1111/j.1600-0749.2004.00138.x
Bastian BC (2014) The molecular pathology of melanoma: an integrated taxonomy of melanocytic neoplasia. Annu Rev Pathol 9:239–271. https://doi.org/10.1146/annurev-pathol-012513-104658
Bastuji-Garin S, Diepgen TL (2002) Cutaneous malignant melanoma, sun exposure, and sunscreen use: epidemiological evidence. Br J Dermatol 146(Suppl 61):24–30
Batlle E, Clevers H (2017) Cancer stem cells revisited. Nat Med 23:1124–1134. https://doi.org/10.1038/nm.4409
Bauer J, Bastian BC (2006) Distinguishing melanocytic nevi from melanoma by DNA copy number changes: comparative genomic hybridization as a research and diagnostic tool. Dermatol Ther 19:40–49. https://doi.org/10.1111/j.1529-8019.2005.00055.x
Bauer J, Curtin JA, Pinkel D, Bastian BC (2007) Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol 127:179–182. https://doi.org/10.1038/sj.jid.5700490
Baxter LL, Pavan WJ (2013) The etiology and molecular genetics of human pigmentation disorders. Wiley Interdiscip Rev Dev Biol 2:379–392. https://doi.org/10.1002/wdev.72
Baxter LL, Watkins-Chow DE, Pavan WJ, Loftus SK (2018) A curated gene list for expanding the horizons of pigmentation biology Pigment Cell & Melanoma Research 32(3):348–358. https://doi.org/10.1111/pcmr.12743
Beauvais-Jouneau A et al (1999) A novel model to study the dorsolateral migration of melanoblasts. Mech Dev 89:3–14
Bedogni B, Welford SM, Cassarino DS, Nickoloff BJ, Giaccia AJ, Powell MB (2005) The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell 8:443–454. https://doi.org/10.1016/j.ccr.2005.11.005
Bennett DC (2008) How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res 21:27–38. https://doi.org/10.1111/j.1755-148X.2007.00433.x
Ben-Porath I, Weinberg RA (2005) The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37:961–976. https://doi.org/10.1016/j.biocel.2004.10.013
Berson JF, Harper DC, Tenza D, Raposo G, Marks MS (2001) Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell 12:3451–3464. https://doi.org/10.1091/mbc.12.11.3451
Bharti K, Nguyen MT, Skuntz S, Bertuzzi S, Arnheiter H (2006) The other pigment cell: specification and development of the pigmented epithelium of the vertebrate eye. Pigment Cell Res 19:380–394. https://doi.org/10.1111/j.1600-0749.2006.00318.x
Blake JA, Ziman MR (2005) Pax3 transcripts in melanoblast development. Develop Growth Differ 47:627–635. https://doi.org/10.1111/j.1440-169X.2005.00835.x
Bonaventure J, Domingues MJ, Larue L (2013) Cellular and molecular mechanisms controlling the migration of melanocytes and melanoma cells. Pigment Cell Melanoma Res 26:316–325. https://doi.org/10.1111/pcmr.12080
Borgdorff V et al (2014) A chemical biology approach identifies AMPK as a modulator of melanoma oncogene MITF. Oncogene 33:2531–2539. https://doi.org/10.1038/onc.2013.185
Botchkarev VA et al (2006) Neurotrophins in skin biology and pathology. J Invest Dermatol 126:1719–1727. https://doi.org/10.1038/sj.jid.5700270
Braeuer RR, Zigler M, Villares GJ, Dobroff AS, Bar-Eli M (2011) Transcriptional control of melanoma metastasis: the importance of the tumor microenvironment. Semin Cancer Biol 21:83–88. https://doi.org/10.1016/j.semcancer.2010.12.007
Brenner M, Hearing VJ (2008) Modifying skin pigmentation-approaches through intrinsic biochemistry and exogenous agents. Drug Discov Today 5:e189–e199
Bronner ME, LeDouarin NM (2012) Development and evolution of the neural crest: an overview. Dev Biol 366:2–9. https://doi.org/10.1016/j.ydbio.2011.12.042
Bruls WA, van Weelden H, van der Leun JC (1984) Transmission of UV-radiation through human epidermal layers as a factor influencing the minimal erythema dose. Photochem Photobiol 39:63–67
Buac K, Pavan WJ (2007) Stem cells of the melanocyte lineage. Cancer Biomark 3:203–209
Buchbinder EI et al (2015) Phase 2 study of sunitinib in patients with metastatic mucosal or acral melanoma. Cancer 121:4007–4015. https://doi.org/10.1002/cncr.29622
Burd CE et al (2014) Mutation-specific RAS oncogenicity explains NRAS codon 61 selection in melanoma. Cancer Discov 4:1418–1429. https://doi.org/10.1158/2159-8290.CD-14-0729
Cancer Genome Atlas N (2015) Genomic classification of cutaneous melanoma. Cell 161:1681–1696. https://doi.org/10.1016/j.cell.2015.05.044
Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G (2013) BAP1 and cancer. Nat Rev Cancer 13:153–159. https://doi.org/10.1038/nrc3459
Carvajal RD et al (2011) KIT as a therapeutic target in metastatic melanoma. JAMA 305:2327–2334. https://doi.org/10.1001/jama.2011.746
Chen H, Weng QY, Fisher DE (2014) UV signaling pathways within the skin. J Invest Dermatol 134:2080–2085. https://doi.org/10.1038/jid.2014.161
Chi A et al (2006) Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. J Proteome Res 5:3135–3144. https://doi.org/10.1021/pr060363j
Chin L et al (1997) Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev 11:2822–2834
Chin L, Garraway LA, Fisher DE (2006) Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev 20:2149–2182. https://doi.org/10.1101/gad.1437206
Chudnovsky Y, Adams AE, Robbins PB, Lin Q, Khavari PA (2005) Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet 37:745–749. https://doi.org/10.1038/ng1586
Clark WH Jr, Elder DE, Guerry D, Epstein MN, Greene MH, Van Horn M (1984) A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol 15:1147–1165
Coelho SG et al (2009) Short- and long-term effects of UV radiation on the pigmentation of human skin. J Investig Dermatol Symp Proc 14:32–35. https://doi.org/10.1038/jidsymp.2009.10
Costin GE, Hearing VJ (2007) Human skin pigmentation: melanocytes modulate skin color in response to stress. FASEB J 21:976–994. https://doi.org/10.1096/fj.06-6649rev
Costin GE, Valencia JC, Vieira WD, Lamoreux ML, Hearing VJ (2003) Tyrosinase processing and intracellular trafficking is disrupted in mouse primary melanocytes carrying the underwhite (uw) mutation. A model for oculocutaneous albinism (OCA) type 4. J Cell Sci 116:3203–3212. https://doi.org/10.1242/jcs.00598
Coudrier E (2007) Myosins in melanocytes: to move or not to move? Pigment Cell Res 20:153–160. https://doi.org/10.1111/j.1600-0749.2007.00376.x
Cronin KA et al (2018) Annual report to the nation on the status of Cancer, part I: national cancer statistics. Cancer 124:2785–2800. https://doi.org/10.1002/cncr.31551
Crowson AN, Magro C, Miller A, Mihm MC Jr (2007) The molecular basis of melanomagenesis and the metastatic phenotype. Semin Oncol 34:476–490. https://doi.org/10.1053/j.seminoncol.2007.09.007
Cui R et al (2007) Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell 128:853–864. https://doi.org/10.1016/j.cell.2006.12.045
Cummings SD, Ryu B, Samuels MA, Yu X, Meeker AK, Healey MA, Alani RM (2008) Id1 delays senescence of primary human melanocytes. Mol Carcinog 47:653–659. https://doi.org/10.1002/mc.20422
Curtin JA et al (2005) Distinct sets of genetic alterations in melanoma. N Engl J Med 353:2135–2147. https://doi.org/10.1056/NEJMoa050092
Curtin JA, Busam K, Pinkel D, Bastian BC (2006) Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol 24:4340–4346. https://doi.org/10.1200/JCO.2006.06.2984
D’Orazio JA et al (2006) Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 443:340–344. https://doi.org/10.1038/nature05098
Dahl C, Guldberg P (2007) The genome and epigenome of malignant melanoma. APMIS 115:1161–1176. https://doi.org/10.1111/j.1600-0463.2007.apm_855.xml.x
Damsky WE, Bosenberg M (2017) Melanocytic nevi and melanoma: unraveling a complex relationship. Oncogene 36:5771–5792. https://doi.org/10.1038/onc.2017.189
Damsky W et al (2015) mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 27:41–56. https://doi.org/10.1016/j.ccell.2014.11.014
Davies H et al (2002) Mutations of the BRAF gene in human cancer. Nature 417:949–954. https://doi.org/10.1038/nature00766
Dell’Angelica EC (2003) Melanosome biogenesis: shedding light on the origin of an obscure organelle. Trends Cell Biol 13:503–506
Delmas V et al (2007) Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev 21:2923–2935. https://doi.org/10.1101/gad.450107
Dhomen N et al (2009) Oncogenic Braf induces melanocyte senescence and melanoma in mice Cancer. Cell 15:294–303. https://doi.org/10.1016/j.ccr.2009.02.022
Di Leva G, Garofalo M, Croce CM (2014) MicroRNAs in cancer. Annu Rev Pathol 9:287–314. https://doi.org/10.1146/annurev-pathol-012513-104715
Diwakar G, Zhang D, Jiang S, Hornyak TJ (2008) Neurofibromin as a regulator of melanocyte development and differentiation. J Cell Sci 121:167–177. https://doi.org/10.1242/jcs.013912
Dorsky RI, Raible DW, Moon RT (2000) Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev 14:158–162
Dunn KJ, Brady M, Ochsenbauer-Jambor C, Snyder S, Incao A, Pavan WJ (2005) WNT1 and WNT3a promote expansion of melanocytes through distinct modes of action. Pigment Cell Res 18:167–180. https://doi.org/10.1111/j.1600-0749.2005.00226.x
Edward M, MacKie RM (1993) Cell-cell and cell-extracellular matrix interactions during melanoma cell invasion and metastasis. Melanoma Res 3:227–234
Eller MS, Gilchrest BA (2000) Tanning as part of the eukaryotic SOS response. Pigment Cell Res 13(Suppl 8):94–97
Fang D et al (2005) A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res 65:9328–9337. https://doi.org/10.1158/0008-5472.CAN-05-1343
Fattore L et al (2017) MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 8:22262–22278. https://doi.org/10.18632/oncotarget.14763
Fell GL, Robinson KC, Mao J, Woolf CJ, Fisher DE (2014) Skin beta-endorphin mediates addiction to UV light. Cell 157:1527–1534. https://doi.org/10.1016/j.cell.2014.04.032
Fleet JC, DeSmet M, Johnson R, Li Y (2012) Vitamin D and cancer: a review of molecular mechanisms. Biochem J 441:61–76. https://doi.org/10.1042/BJ20110744
Frank NY et al (2005) ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 65:4320–4333. https://doi.org/10.1158/0008-5472.CAN-04-3327
Fulda S (2015) Targeting apoptosis for anticancer therapy. Semin Cancer Biol 31:84–88. https://doi.org/10.1016/j.semcancer.2014.05.002
Gaffal E, Landsberg J, Bald T, Sporleder A, Kohlmeyer J, Tuting T (2011) Neonatal UVB exposure accelerates melanoma growth and enhances distant metastases in Hgf-Cdk4(R24C) C57BL/6 mice. Int J Cancer 129:285–294. https://doi.org/10.1002/ijc.25913
Garcia-Castro MI, Marcelle C, Bronner-Fraser M (2002) Ectodermal Wnt function as a neural crest inducer. Science 297:848–851
Garibyan L, Fisher DE (2010) How sunlight causes melanoma. Curr Oncol Rep 12:319–326. https://doi.org/10.1007/s11912-010-0119-y
Garnett MJ, Marais R (2004) Guilty as charged: B-RAF is a human oncogene. Cancer Cell 6:313–319. https://doi.org/10.1016/j.ccr.2004.09.022
Garraway LA, Sellers WR (2006) Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer 6:593–602. https://doi.org/10.1038/nrc1947
Garraway LA et al (2005) Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436:117–122. https://doi.org/10.1038/nature03664
Ginger RS et al (2008) SLC24A5 encodes a trans-Golgi network protein with potassium-dependent sodium-calcium exchange activity that regulates human epidermal melanogenesis. J Biol Chem 283:5486–5495. https://doi.org/10.1074/jbc.M707521200
Goel VK et al (2009) Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 28:2289–2298. https://doi.org/10.1038/onc.2009.95
Goldstein AM, Tucker MA (2013) Dysplastic nevi and melanoma. Cancer Epidemiol Biomark Prev 22:528–532. https://doi.org/10.1158/1055-9965.EPI-12-1346
Govindarajan B et al (2007) Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest 117:719–729. https://doi.org/10.1172/JCI30102
Graf J, Hodgson R, van Daal A (2005) Single nucleotide polymorphisms in the MATP gene are associated with normal human pigmentation variation. Hum Mutat 25:278–284. https://doi.org/10.1002/humu.20143
Green AC, Wallingford SC, McBride P (2011) Childhood exposure to ultraviolet radiation and harmful skin effects: epidemiological evidence. Prog Biophys Mol Biol 107:349–355. https://doi.org/10.1016/j.pbiomolbio.2011.08.010
Grossman D, McNiff JM, Li F, Altieri DC (1999) Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J Invest Dermatol 113:1076–1081. https://doi.org/10.1046/j.1523-1747.1999.00776.x
Guo J et al (2011) Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J Clin Oncol 29:2904–2909. https://doi.org/10.1200/JCO.2010.33.9275
Gupta PB et al (2005) The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet 37:1047–1054. https://doi.org/10.1038/ng1634
Guth SI, Wegner M (2008) Having it both ways: sox protein function between conservation and innovation. Cell Mol Life Sci 65:3000–3018. https://doi.org/10.1007/s00018-008-8138-7
Guyonneau L, Murisier F, Rossier A, Moulin A, Beermann F (2004) Melanocytes and pigmentation are affected in dopachrome tautomerase knockout mice. Mol Cell Biol 24:3396–3403
Ha L et al (2007) ARF functions as a melanoma tumor suppressor by inducing p53-independent senescence. Proc Natl Acad Sci U S A 104:10968–10973. https://doi.org/10.1073/pnas.0611638104
Haass NK, Herlyn M (2005) Normal human melanocyte homeostasis as a paradigm for understanding melanoma. J Investig Dermatol Symp Proc 10:153–163. https://doi.org/10.1111/j.1087-0024.2005.200407.x
Haass NK, Smalley KS, Li L, Herlyn M (2005) Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res 18:150–159. https://doi.org/10.1111/j.1600-0749.2005.00235.x
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. https://doi.org/10.1016/j.cell.2011.02.013
Haqq C et al (2005) The gene expression signatures of melanoma progression. Proc Natl Acad Sci USA 102: 6092–6097. https://doi.org/10.1073/pnas.0501564102
Hari L et al (2012) Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development 139:2107–2117. https://doi.org/10.1242/dev.073064
Harris ML, Baxter LL, Loftus SK, Pavan WJ (2010) Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res 23:496–513. https://doi.org/10.1111/j.1755-148X.2010.00711.x
Hart PH, Norval M (2017) Ultraviolet radiation-induced immunosuppression and its relevance for skin carcinogenesis. Photochem Photobiol Sci. https://doi.org/10.1039/c7pp00312a
Hearing VJ, Leong SPL (2006) From melanocytes to melanoma: the progression to malignancy. Humana Press, Totowa
Hennessey RC et al (2017) Ultraviolet radiation accelerates NRas-mutant melanomagenesis: a cooperative effect blocked by sunscreen. Pigment Cell Melanoma Res 30:477–487. https://doi.org/10.1111/pcmr.12601
Herlyn M, Berking C, Li G, Satyamoorthy K (2000) Lessons from melanocyte development for understanding the biological events in naevus and melanoma formation. Melanoma Res 10:303–312
Herraiz C, Garcia-Borron JC, Jimenez-Cervantes C, Olivares C (2017) MC1R signaling. Intracellular partners and pathophysiological implications. Biochim Biophys Acta 1863:2448–2461. https://doi.org/10.1016/j.bbadis.2017.02.027
Hill HZ, Hill GJ (2000) UVA, pheomelanin and the carcinogenesis of melanoma. Pigment Cell Res 13(Suppl 8):140–144
Hirobe T (2011) How are proliferation and differentiation of melanocytes regulated? Pigment Cell Melanoma Res 24:462–478. https://doi.org/10.1111/j.1755-148X.2011.00845.x
Hirobe T, Ootaka H (2007) Interleukin-1alpha stimulates the differentiation of melanocytes but inhibits the proliferation of melanoblasts from neonatal mouse epidermis. Zool Sci 24:959–970. https://doi.org/10.2108/zsj.24.959
Hirobe T, Osawa M, Nishikawa S (2004) Hepatocyte growth factor controls the proliferation of cultured epidermal melanoblasts and melanocytes from newborn mice. Pigment Cell Res 17:51–61
Hoashi T, Watabe H, Muller J, Yamaguchi Y, Vieira WD, Hearing VJ (2005) MART-1 is required for the function of the melanosomal matrix protein PMEL17/GP100 and the maturation of melanosomes. J Biol Chem 280:14006–14016. https://doi.org/10.1074/jbc.M413692200
Hodi FS et al (2013) Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J Clin Oncol 31:3182–3190. https://doi.org/10.1200/JCO.2012.47.7836
Hoek KS (2007) DNA microarray analyses of melanoma gene expression: a decade in the mines. Pigment Cell Res 20:466–484. https://doi.org/10.1111/j.1600-0749.2007.00412.x
Hoek K et al (2004) Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res 64:5270–5282. https://doi.org/10.1158/0008-5472.CAN-04-0731
Hofmann UB, Westphal JR, Van Kraats AA, Ruiter DJ, Van Muijen GN (2000) Expression of integrin alpha(v)beta(3) correlates with activation of membrane-type matrix metalloproteinase-1 (MT1-MMP) and matrix metalloproteinase-2 (MMP-2) in human melanoma cells in vitro and in vivo. Int J Cancer 87:12–19
Hopkins BD et al (2013) A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341:399–402. https://doi.org/10.1126/science.1234907
Hornyak TJ, Hayes DJ, Chiu LY, Ziff EB (2001) Transcription factors in melanocyte development: distinct roles for Pax-3 and Mitf. Mech Dev 101:47–59
Hu ZZ et al (2007) Comparative bioinformatics analyses and profiling of lysosome-related organelle proteomes. Int J Mass Spectrom 259:147–160. https://doi.org/10.1016/j.ijms.2006.09.024
Huarte M (2015) The emerging role of lncRNAs in cancer. Nat Med 21:1253–1261. https://doi.org/10.1038/nm.3981
Hubbard JK, Uy JA, Hauber ME, Hoekstra HE, Safran RJ (2010) Vertebrate pigmentation: from underlying genes to adaptive function. Trends Genet 26:231–239. https://doi.org/10.1016/j.tig.2010.02.002
Hussein MR, Haemel AK, Wood GS (2003a) Apoptosis and melanoma: molecular mechanisms. J Pathol 199:275–288. https://doi.org/10.1002/path.1300
Hussein MR, Haemel AK, Wood GS (2003b) p53-related pathways and the molecular pathogenesis of melanoma. Eur J Cancer Prev 12:93–100. https://doi.org/10.1097/01.cej.0000062797.86004.35
Ignatius MS, Moose HE, El-Hodiri HM, Henion PD (2008) colgate/hdac1 repression of foxd3 expression is required to permit mitfa-dependent melanogenesis. Dev Biol 313:568–583. https://doi.org/10.1016/j.ydbio.2007.10.045
Imokawa G (2004) Autocrine and paracrine regulation of melanocytes in human skin and in pigmentary disorders. Pigment Cell Res 17:96–110. https://doi.org/10.1111/j.1600-0749.2003.00126.x
Ito S, IFPCS (2003) The IFPCS presidential lecture: a chemist’s view of melanogenesis. Pigment Cell Res 16:230–236
Ito M et al (1999) Removal of stem cell factor or addition of monoclonal anti-c-KIT antibody induces apoptosis in murine melanocyte precursors. J Invest Dermatol 112:796–801. https://doi.org/10.1046/j.1523-1747.1999.00552.x
Ivanov VN, Bhoumik A, Ronai Z (2003) Death receptors and melanoma resistance to apoptosis. Oncogene 22:3152–3161. https://doi.org/10.1038/sj.onc.1206456
Jacobs JJ et al (2000) Senescence bypass screen identifies TBX2, which represses Cdkn2a (p19(ARF)) and is amplified in a subset of human breast cancers. Nat Genet 26:291–299. https://doi.org/10.1038/81583
Janjetovic Z et al (2011) High basal NF-kappaB activity in nonpigmented melanoma cells is associated with an enhanced sensitivity to vitamin D3 derivatives. Br J Cancer 105:1874–1884. https://doi.org/10.1038/bjc.2011.458
Jhappan C, Noonan FP, Merlino G (2003) Ultraviolet radiation and cutaneous malignant melanoma. Oncogene 22:3099–3112. https://doi.org/10.1038/sj.onc.1206450
Jiang R, Norton CR, Copeland NG, Gilbert DJ, Jenkins NA, Gridley T (1998) Genomic organization, expression and chromosomal localization of the mouse Slug (Slugh) gene. Biochim Biophys Acta 1443:251–254
Jin EJ, Erickson CA, Takada S, Burrus LW (2001) Wnt and BMP signaling govern lineage segregation of melanocytes in the avian embryo. Dev Biol 233:22–37. https://doi.org/10.1006/dbio.2001.0222
Jonsson G et al (2007) Genomic profiling of malignant melanoma using tiling-resolution arrayCGH. Oncogene 26:4738–4748. https://doi.org/10.1038/sj.onc.1210252
Jouneau A, Yu YQ, Pasdar M, Larue L (2000) Plasticity of cadherin-catenin expression in the melanocyte lineage. Pigment Cell Res 13:260–272
Kadekaro AL, Kavanagh RJ, Wakamatsu K, Ito S, Pipitone MA, Abdel-Malek ZA (2003) Cutaneous photobiology. The melanocyte vs. the sun: who will win the final round? Pigment Cell Res 16:434–447
Kamb A et al (1994) Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet 8:23–26. https://doi.org/10.1038/ng0994-22
Kasof GM, Gomes BC (2001) Livin, a novel inhibitor of apoptosis protein family member. J Biol Chem 276:3238–3246. https://doi.org/10.1074/jbc.M003670200
Kaufmann WK et al (2008) Defective cell cycle checkpoint functions in melanoma are associated with altered patterns of gene expression. J Invest Dermatol 128:175–187. https://doi.org/10.1038/sj.jid.5700935
Kaur A, Webster MR, Weeraratna AT (2016) In the Wnt-er of life: Wnt signalling in melanoma and ageing. Br J Cancer 115:1273–1279. https://doi.org/10.1038/bjc.2016.332
Kawakami A, Fisher DE (2017) The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Investig 97:649–656. https://doi.org/10.1038/labinvest.2017.9
Kawakami Y et al (2000) T cell immune responses against melanoma and melanocytes in cancer and autoimmunity. Pigment Cell Res 13(Suppl 8):163–169
Kim M et al (2006) Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell 125:1269–1281. https://doi.org/10.1016/j.cell.2006.06.008
Klug HL et al (2010) Sunscreen prevention of melanoma in man and mouse. Pigment Cell Melanoma Res 23:835–837. https://doi.org/10.1111/j.1755-148X.2010.00756.x
Kohler C et al (2017) Mouse cutaneous melanoma induced by mutant BRaf arises from expansion and dedifferentiation of mature pigmented melanocytes. Cell Stem Cell 21(679-693):e676. https://doi.org/10.1016/j.stem.2017.08.003
Kos L, Aronzon A, Takayama H, Maina F, Ponzetto C, Merlino G, Pavan W (1999) Hepatocyte growth factor/scatter factor-MET signaling in neural crest-derived melanocyte development. Pigment Cell Res 12:13–21
Kos R, Reedy MV, Johnson RL, Erickson CA (2001) The winged-helix transcription factor FoxD3 is important for establishing the neural crest lineage and repressing melanogenesis in avian embryos. Development 128:1467–1479
Kripke ML (1990) Effects of UV radiation on tumor immunity. J Natl Cancer Inst 82:1392–1396
Kuphal S, Bauer R, Bosserhoff AK (2005) Integrin signaling in malignant melanoma. Cancer Metastasis Rev 24:195–222. https://doi.org/10.1007/s10555-005-1572-1
Kwong LN et al (2012) Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 18:1503–1510. https://doi.org/10.1038/nm.2941
LaBonne C, Bronner-Fraser M (1998) Neural crest induction in Xenopus: evidence for a two-signal model. Development 125:2403–2414
Lamason RL et al (2005) SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310:1782–1786. https://doi.org/10.1126/science.1116238
Landi MT et al (2006) MC1R germline variants confer risk for BRAF-mutant melanoma. Science 313:521–522. https://doi.org/10.1126/science.1127515
Lang D et al (2005) Pax3 functions at a nodal point in melanocyte stem cell differentiation. Nature 433:884–887. https://doi.org/10.1038/nature03292
Larue L, Delmas V (2006) The WNT/Beta-catenin pathway in melanoma. Front Biosci 11:733–742
Lavado A, Montoliu L (2006) New animal models to study the role of tyrosinase in normal retinal development. Front Biosci 11:743–752
Lee HO, Levorse JM, Shin MK (2003) The endothelin receptor-B is required for the migration of neural crest-derived melanocyte and enteric neuron precursors. Dev Biol 259:162–175
Lee YR, Chen M, Pandolfi PP (2018) The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. https://doi.org/10.1038/s41580-018-0015-0
Leucci E, Coe EA, Marine JC, Vance KW (2016) The emerging role of long non-coding RNAs in cutaneous melanoma. Pigment Cell Melanoma Res 29:619–626. https://doi.org/10.1111/pcmr.12537
Lewis JL, Bonner J, Modrell M, Ragland JW, Moon RT, Dorsky RI, Raible DW (2004) Reiterated Wnt signaling during zebrafish neural crest development. Development 131:1299–1308. https://doi.org/10.1242/dev.01007
Liebermann DA, Hoffman B, Vesely D (2007) p53 induced growth arrest versus apoptosis and its modulation by survival cytokines. Cell Cycle 6:166–170. https://doi.org/10.4161/cc.6.2.3789
Lin JY, Fisher DE (2007) Melanocyte biology and skin pigmentation. Nature 445:843–850. https://doi.org/10.1038/nature05660
Lin X et al (2017) C-myc overexpression drives melanoma metastasis by promoting vasculogenic mimicry via c-myc/snail/Bax signaling. J Mol Med (Berl) 95:53–67. https://doi.org/10.1007/s00109-016-1452-x
Lindsay CR et al (2011) P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat Commun 2:555. https://doi.org/10.1038/ncomms1560
Liu W, Sharpless NE (2012) Senescence-escape in melanoma. Pigment Cell Melanoma Res 25:408–409
Liu ZJ et al (2006) Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res 66:4182–4190. https://doi.org/10.1158/0008-5472.CAN-05-3589
Liu J, Fukunaga-Kalabis M, Li L, Herlyn M (2014) Developmental pathways activated in melanocytes and melanoma. Arch Biochem Biophys 563:13–21. https://doi.org/10.1016/j.abb.2014.07.023
Liu XL, Ding J, Meng LH (2018) Oncogene-induced senescence: a double edged sword in cancer. Acta Pharmacol Sin. https://doi.org/10.1038/aps.2017.198
Lowe SW, Lin AW (2000) Apoptosis in cancer. Carcinogenesis 21:485–495
Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432:307–315. https://doi.org/10.1038/nature03098
Maddodi N, Setaluri V (2008) Role of UV in cutaneous melanoma. Photochem Photobiol 84:528–536. https://doi.org/10.1111/j.1751-1097.2007.00283.x
McGary EC, Lev DC, Bar-Eli M (2002) Cellular adhesion pathways and metastatic potential of human melanoma. Cancer Biol Ther 1:459–465
McGill GG et al (2002) Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109:707–718
Merlino G (2005) Cancer biology: the weakest link? Nature 436:33–35. https://doi.org/10.1038/436033a
Meulemans D, Bronner-Fraser M (2004) Gene-regulatory interactions in neural crest evolution and development. Dev Cell 7:291–299. https://doi.org/10.1016/j.devcel.2004.08.007
Michaloglou C et al (2005) BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436:720–724. https://doi.org/10.1038/nature03890
Micillo R, Panzella L, Koike K, Monfrecola G, Napolitano A, d’Ischia M (2016) “Fifty shades” of black and red or how carboxyl groups fine tune Eumelanin and Pheomelanin properties. Int J Mol Sci 17. https://doi.org/10.3390/ijms17050746
Mitra D et al (2012) An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 491:449–453. https://doi.org/10.1038/nature11624
Miyamura Y et al (2007) Regulation of human skin pigmentation and responses to ultraviolet radiation. Pigment Cell Res 20:2–13. https://doi.org/10.1111/j.1600-0749.2006.00358.x
Moan J, Porojnicu AC, Dahlback A (2008a) Ultraviolet radiation and malignant melanoma. Adv Exp Med Biol 624:104–116. https://doi.org/10.1007/978-0-387-77574-6_9
Moan J, Porojnicu AC, Dahlback A, Setlow RB (2008b) Addressing the health benefits and risks, involving vitamin D or skin cancer, of increased sun exposure. Proc Natl Acad Sci USA 105:668–673. https://doi.org/10.1073/pnas.0710615105
Mooi WJ, Peeper DS (2006) Oncogene-induced cell senescence – halting on the road to cancer. N Engl J Med 355:1037–1046. https://doi.org/10.1056/NEJMra062285
Moran B, Silva R, Perry AS, Gallagher WM (2018) Epigenetics of malignant melanoma. Semin Cancer Biol 51:80–88. https://doi.org/10.1016/j.semcancer.2017.10.006
Murali R et al (2013) BAP1 expression in cutaneous melanoma: a pilot study. Pathology 45:606–609. https://doi.org/10.1097/PAT.0b013e3283653818
Natali PG, Nicotra MR, Di Renzo MF, Prat M, Bigotti A, Cavaliere R, Comoglio PM (1993) Expression of the c-Met/HGF receptor in human melanocytic neoplasms: demonstration of the relationship to malignant melanoma tumour progression. Br J Cancer 68:746–750
Nazarian R et al (2010) Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468:973–977. https://doi.org/10.1038/nature09626
Nickoloff BJ, Hendrix MJ, Pollock PM, Trent JM, Miele L, Qin JZ (2005) Notch and NOXA-related pathways in melanoma cells. J Investig Dermatol Symp Proc 10:95–104. https://doi.org/10.1111/j.1087-0024.2005.200404.x
Niihori T et al (2006) Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 38:294–296. https://doi.org/10.1038/ng1749
Nikolaou V, Stratigos AJ (2014) Emerging trends in the epidemiology of melanoma. Br J Dermatol 170:11–19. https://doi.org/10.1111/bjd.12492
Nishikawa S, Osawa M (2007) Generating quiescent stem cells. Pigment Cell Res 20:263–270. https://doi.org/10.1111/j.1600-0749.2007.00388.x
Nishimura EK (2011) Melanocyte stem cells: a melanocyte reservoir in hair follicles for hair and skin pigmentation. Pigment Cell Melanoma Res 24:401–410. https://doi.org/10.1111/j.1755-148X.2011.00855.x
Nishimura EK et al (2002) Dominant role of the niche in melanocyte stem-cell fate determination. Nature 416:854–860. https://doi.org/10.1038/416854a
Nishimura EK, Granter SR, Fisher DE (2005) Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science 307:720–724. https://doi.org/10.1126/science.1099593
Noonan FP et al (2012) Melanoma induction by ultraviolet a but not ultraviolet B radiation requires melanin pigment. Nat Commun 3:884. https://doi.org/10.1038/ncomms1893
Nordlund JJ (2006) The pigmentary system: physiology and pathophysiology, 2nd edn. Blackwell Publication, Malden
Norris W (1857) Eight cases of melanosis with pathological and therapeutical remarks on that disease. Longman, London
Osawa M et al (2005) Molecular characterization of melanocyte stem cells in their niche. Development 132:5589–5599. https://doi.org/10.1242/dev.02161
Palmieri G et al (2015) Multiple molecular pathways in Melanomagenesis: characterization of therapeutic targets. Front Oncol 5:183. https://doi.org/10.3389/fonc.2015.00183
Park HY, Kosmadaki M, Yaar M, Gilchrest BA (2009) Cellular mechanisms regulating human melanogenesis. Cell Mol Life Sci 66:1493–1506. https://doi.org/10.1007/s00018-009-8703-8
Passeron T et al (2007) SOX9 is a key player in ultraviolet B-induced melanocyte differentiation and pigmentation. Proc Natl Acad Sci USA 104:13984–13989. https://doi.org/10.1073/pnas.0705117104
Pavan WJ, Raible DW (2012) Specification of neural crest into sensory neuron and melanocyte lineages. Dev Biol 366:55–63. https://doi.org/10.1016/j.ydbio.2012.02.038
Perez-Guijarro E, Day CP, Merlino G, Zaidi MR (2017) Genetically engineered mouse models of melanoma. Cancer 123:2089–2103. https://doi.org/10.1002/cncr.30684
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, Olivier M (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28:622–629. https://doi.org/10.1002/humu.20495
Pinon P, Wehrle-Haller B (2011) Integrins: versatile receptors controlling melanocyte adhesion, migration and proliferation. Pigment Cell Melanoma Res 24:282–294. https://doi.org/10.1111/j.1755-148X.2010.00806.x
Plummer RS, Shea CR, Nelson M, Powell SK, Freeman DM, Dan CP, Lang D (2008) PAX3 expression in primary melanomas and nevi. Mod Pathol 21:525–530. https://doi.org/10.1038/modpathol.3801019
Pollock PM et al (2003) High frequency of BRAF mutations in nevi. Nat Genet 33:19–20. https://doi.org/10.1038/ng1054
Polsky D, Young AZ, Busam KJ, Alani RM (2001) The transcriptional repressor of p16/Ink4a, Id1, is up-regulated in early melanomas. Cancer Res 61:6008–6011
Poser I, Bosserhoff AK (2004) Transcription factors involved in development and progression of malignant melanoma. Histol Histopathol 19:173–188. https://doi.org/10.14670/HH-19.173
Prota G (1992) Melanins and melanogenesis. Academic, San Diego
Puntervoll HE et al (2013) Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. J Med Genet 50:264–270. https://doi.org/10.1136/jmedgenet-2012-101455
Putz U, Howitt J, Doan A, Goh CP, Low LH, Silke J, Tan SS (2012) The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal 5:ra70. https://doi.org/10.1126/scisignal.2003084
Qiu T, Wang H, Wang Y, Zhang Y, Hui Q, Tao K (2015) Identification of genes associated with melanoma metastasis. Kaohsiung J Med Sci 31:553–561. https://doi.org/10.1016/j.kjms.2015.10.002
Raj D, Liu T, Samadashwily G, Li F, Grossman D (2008) Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 29:194–201. https://doi.org/10.1093/carcin/bgm219
Rao C et al (2003) A defect in a novel ADAMTS family member is the cause of the belted white-spotting mutation. Development 130:4665–4672. https://doi.org/10.1242/dev.00668
Riker AI et al (2008) The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genet 1:13. https://doi.org/10.1186/1755-8794-1-13
Robertson GP (2005) Functional and therapeutic significance of Akt deregulation in malignant melanoma. Cancer Metastasis Rev 24:273–285. https://doi.org/10.1007/s10555-005-1577-9
Rodriguez-Viciana P et al (2006) Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311:1287–1290. https://doi.org/10.1126/science.1124642
Rouzaud F, Kadekaro AL, Abdel-Malek ZA, Hearing VJ (2005) MC1R and the response of melanocytes to ultraviolet radiation. Mutat Res 571:133–152. https://doi.org/10.1016/j.mrfmmm.2004.09.014
Sakai C, Kawakami Y, Law LW, Furumura M, Hearing VJJ (1997) Melanosomal proteins as melanoma-specific immune targets. Melanoma Res 7:83–95
Saldana-Caboverde A, Kos L (2010) Roles of endothelin signaling in melanocyte development and melanoma. Pigment Cell Melanoma Res 23:160–170. https://doi.org/10.1111/j.1755-148X.2010.00678.x
Sanchez-Martin M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I (2002) SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet 11:3231–3236
Satyamoorthy K, Muyrers J, Meier F, Patel D, Herlyn M (2001) Mel-CAM-specific genetic suppressor elements inhibit melanoma growth and invasion through loss of gap junctional communication. Oncogene 20:4676–4684. https://doi.org/10.1038/sj.onc.1204616
Schatton T et al (2008) Identification of cells initiating human melanomas. Nature 451:345–349. https://doi.org/10.1038/nature06489
Schiaffino MV (2010) Signaling pathways in melanosome biogenesis and pathology. Int J Biochem Cell Biol 42:1094–1104. https://doi.org/10.1016/j.biocel.2010.03.023
Scott MC, Suzuki I, Abdel-Malek ZA (2002) Regulation of the human melanocortin 1 receptor expression in epidermal melanocytes by paracrine and endocrine factors and by ultraviolet radiation. Pigment Cell Res 15:433–439
Scott G, Leopardi S, Parker L, Babiarz L, Seiberg M, Han R (2003) The proteinase-activated receptor-2 mediates phagocytosis in a Rho-dependent manner in human keratinocytes. J Invest Dermatol 121:529–541. https://doi.org/10.1046/j.1523-1747.2003.12427.x
Shain AH et al (2015) The genetic evolution of melanoma from precursor lesions. N Engl J Med 373:1926–1936. https://doi.org/10.1056/NEJMoa1502583
Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L (2003) Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene 22:5055–5059. https://doi.org/10.1038/sj.onc.1206809
Shay JW (2016) Role of telomeres and telomerase in aging and Cancer. Cancer Discov 6:584–593. https://doi.org/10.1158/2159-8290.CD-16-0062
Shriver MD et al (2003) Skin pigmentation, biogeographical ancestry and admixture mapping. Hum Genet 112:387–399. https://doi.org/10.1007/s00439-002-0896-y
Silver DL, Hou L, Pavan WJ (2006) The genetic regulation of pigment cell development. Adv Exp Med Biol 589:155–169. https://doi.org/10.1007/978-0-387-46954-6_9
Silver DL, Hou L, Somerville R, Young ME, Apte SS, Pavan WJ (2008) The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet 4:e1000003. https://doi.org/10.1371/journal.pgen.1000003
Simanshu DK, Nissley DV, McCormick F (2017) RAS proteins and their regulators in human disease. Cell 170:17–33. https://doi.org/10.1016/j.cell.2017.06.009
Slominski A, Pawelek J (1998) Animals under the sun: effects of ultraviolet radiation on mammalian skin. Clin Dermatol 16:503–515
Slominski A, Wortsman J, Plonka PM, Schallreuter KU, Paus R, Tobin DJ (2005) Hair follicle pigmentation. J Invest Dermatol 124:13–21. https://doi.org/10.1111/j.0022-202X.2004.23528.x
Slominski AT et al (2012) Novel vitamin D hydroxyderivatives inhibit melanoma growth and show differential effects on normal melanocytes. Anticancer Res 32:3733–3742
Slominski AT et al (2017) Vitamin D signaling and melanoma: role of vitamin D and its receptors in melanoma progression and management. Lab Investig 97: 706–724. https://doi.org/10.1038/labinvest.2017.3
Smith MP et al (2013) Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J Natl Cancer Inst 105:33–46. https://doi.org/10.1093/jnci/djs471
Smith MP et al (2016) Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 29:270–284. https://doi.org/10.1016/j.ccell.2016.02.003
Soengas MS et al (2001) Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409:207–211. https://doi.org/10.1038/35051606
Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H (2016) Hereditary melanoma: update on syndromes and management: genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol 74:395–407; quiz 408-310. https://doi.org/10.1016/j.jaad.2015.08.038
Stark M, Hayward N (2007) Genome-wide loss of heterozygosity and copy number analysis in melanoma using high-density single-nucleotide polymorphism arrays. Cancer Res 67:2632–2642. https://doi.org/10.1158/0008-5472.CAN-06-4152
Sturm RA, Larsson M (2009) Genetics of human iris colour and patterns. Pigment Cell Melanoma Res 22:544–562. https://doi.org/10.1111/j.1755-148X.2009.00606.x
Tadokoro T et al (2003) UV-induced DNA damage and melanin content in human skin differing in racial/ethnic origin. FASEB J 17:1177–1179. https://doi.org/10.1096/fj.02-0865fje
Tadokoro T et al (2005) Mechanisms of skin tanning in different racial/ethnic groups in response to ultraviolet radiation. J Invest Dermatol 124:1326–1332. https://doi.org/10.1111/j.0022-202X.2005.23760.x
Takeda K, Yokoyama S, Yasumoto K, Saito H, Udono T, Takahashi K, Shibahara S (2003) OTX2 regulates expression of DOPAchrome tautomerase in human retinal pigment epithelium. Biochem Biophys Res Commun 300:908–914
Talantov D et al (2005) Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res 11:7234–7242. https://doi.org/10.1158/1078-0432.CCR-05-0683
Tang JY et al (2011) Calcium plus vitamin D supplementation and the risk of nonmelanoma and melanoma skin cancer: post hoc analyses of the women’s health initiative randomized controlled trial. J Clin Oncol 29:3078–3084. https://doi.org/10.1200/JCO.2011.34.5967
Testa JR et al (2011) Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet 43:1022–1025. https://doi.org/10.1038/ng.912
Thomas AJ, Erickson CA (2008) The making of a melanocyte: the specification of melanoblasts from the neural crest. Pigment Cell Melanoma Res 21:598–610. https://doi.org/10.1111/j.1755-148X.2008.00506.x
Thong HY, Jee SH, Sun CC, Boissy RE (2003) The patterns of melanosome distribution in keratinocytes of human skin as one determining factor of skin colour. Br J Dermatol 149:498–505
Timerman D et al (2017) Vitamin D deficiency is associated with a worse prognosis in metastatic melanoma. Oncotarget 8:6873–6882. https://doi.org/10.18632/oncotarget.14316
Tobin DJ (2008) Human hair pigmentation – biological aspects. Int J Cosmet Sci 30:233–257. https://doi.org/10.1111/j.1468-2494.2008.00456.x
Tormo D et al (2006) Rapid growth of invasive metastatic melanoma in carcinogen-treated hepatocyte growth factor/scatter factor-transgenic mice carrying an oncogenic CDK4 mutation. Am J Pathol 169:665–672. https://doi.org/10.2353/ajpath.2006.060017
Toyofuku K et al (2002) The etiology of oculocutaneous albinism (OCA) type II: the pink protein modulates the processing and transport of tyrosinase. Pigment Cell Res 15:217–224
Tran S, Rizos H (2013) Human nevi lack distinguishing senescence traits. Aging (Albany NY) 5:98–99. https://doi.org/10.18632/aging.100537
Tran TT, Schulman J, Fisher DE (2008) UV and pigmentation: molecular mechanisms and social controversies. Pigment Cell Melanoma Res 21:509–516. https://doi.org/10.1111/j.1755-148X.2008.00498.x
Tsao H, Chin L, Garraway LA, Fisher DE (2012) Melanoma: from mutations to medicine. Genes Dev 26:1131–1155. https://doi.org/10.1101/gad.191999.112
Tsutsumida A et al (2004) Epigenetic silencing of E- and P-cadherin gene expression in human melanoma cell lines. Int J Oncol 25:1415–1421
Vance KW, Carreira S, Brosch G, Goding CR (2005) Tbx2 is overexpressed and plays an important role in maintaining proliferation and suppression of senescence in melanomas. Cancer Res 65:2260–2268. https://doi.org/10.1158/0008-5472.CAN-04-3045
Villanueva J, Vultur A, Herlyn M (2011) Resistance to BRAF inhibitors: unraveling mechanisms and future treatment options. Cancer Res 71:7137–7140. https://doi.org/10.1158/0008-5472.CAN-11-1243
Viros A et al (2014) Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature 511:478–482. https://doi.org/10.1038/nature13298
Wadt K et al (2012) A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res 25:815–818. https://doi.org/10.1111/pcmr.12006
Wan PT et al (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867
Wehrle-Haller B (2003) The role of Kit-ligand in melanocyte development and epidermal homeostasis. Pigment Cell Res 16:287–296
Weinstein IB, Joe A (2008) Oncogene addiction. Cancer Res 68:3077–3080; discussion 3080. https://doi.org/10.1158/0008-5472.CAN-07-3293
Whiteman DC, Parsons PG, Green AC (1999) Determinants of melanocyte density in adult human skin. Arch Dermatol Res 291:511–516
Whiteman DC, Whiteman CA, Green AC (2001) Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies. Cancer Causes Control 12:69–82
Wiesner T et al (2011) Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet 43: 1018–1021. https://doi.org/10.1038/ng.910
Wiesner T et al (2012) Toward an improved definition of the tumor spectrum associated with BAP1 germline mutations. J Clin Oncol 30:e337–e340. https://doi.org/10.1200/JCO.2011.41.2965
Wilkie AL, Jordan SA, Jackson IJ (2002) Neural crest progenitors of the melanocyte lineage: coat colour patterns revisited. Development 129:3349–3357
Willmore-Payne C, Holden JA, Tripp S, Layfield LJ (2005) Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum Pathol 36:486–493. https://doi.org/10.1016/j.humpath.2005.03.015
Wittgen HG, van Kempen LC (2007) Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res 17:400–409. https://doi.org/10.1097/CMR.0b013e3282f1d312
Wolf Horrell EM, Boulanger MC, D’Orazio JA (2016) Melanocortin 1 receptor: structure, function, and regulation. Front Genet 7:95. https://doi.org/10.3389/fgene.2016.00095
Wu X, Hammer JA (2014) Melanosome transfer: it is best to give and receive. Curr Opin Cell Biol 29:1–7. https://doi.org/10.1016/j.ceb.2014.02.003
Wu X, Hammer JA 3rd (2000) Making sense of melanosome dynamics in mouse melanocytes. Pigment Cell Res 13:241–247
Xu L et al (2008) Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res 6:760–769. https://doi.org/10.1158/1541-7786.MCR-07-0344
Yamaguchi Y, Hearing VJ (2006) Melanocyte distribution and function in human skin. In: Hearing VJ, Leong SPL (eds) From melanocytes to melanoma. Humana Press, Totowa
Yamaguchi Y et al (2004) Mesenchymal-epithelial interactions in the skin: increased expression of dickkopf1 by palmoplantar fibroblasts inhibits melanocyte growth and differentiation. J Cell Biol 165:275–285. https://doi.org/10.1083/jcb.200311122
Yamaguchi Y, Brenner M, Hearing VJ (2007) The regulation of skin pigmentation. J Biol Chem 282:27557–27561. https://doi.org/10.1074/jbc.R700026200
Yamaguchi Y et al (2008) Dickkopf 1 (DKK1) regulates skin pigmentation and thickness by affecting Wnt/beta-catenin signaling in keratinocytes. FASEB J 22:1009–1020. https://doi.org/10.1096/fj.07-9475com
Yang G, Zhang G, Pittelkow MR, Ramoni M, Tsao H (2006) Expression profiling of UVB response in melanocytes identifies a set of p53-target genes. J Invest Dermatol 126:2490–2506. https://doi.org/10.1038/sj.jid.5700470
Yasumoto K, Takeda K, Saito H, Watanabe K, Takahashi K, Shibahara S (2002) Microphthalmia-associated transcription factor interacts with LEF-1, a mediator of Wnt signaling. EMBO J 21:2703–2714. https://doi.org/10.1093/emboj/21.11.2703
Yokoyama S et al (2008) Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment Cell Melanoma Res 21:457–463. https://doi.org/10.1111/j.1755-148X.2008.00480.x
Yokoyama S et al (2011) A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 480:99–103. https://doi.org/10.1038/nature10630
Yoshida Y et al (2007) Functional analysis of keratinocytes in skin color using a human skin substitute model composed of cells derived from different skin pigmentation types. FASEB J 21:2829–2839. https://doi.org/10.1096/fj.06-6845com
Young AR (2006) Acute effects of UVR on human eyes and skin. Prog Biophys Mol Biol 92:80–85. https://doi.org/10.1016/j.pbiomolbio.2006.02.005
Zaidi MR, Day CP, Merlino G (2008) From UVs to metastases: modeling melanoma initiation and progression in the mouse. J Invest Dermatol 128:2381–2391. https://doi.org/10.1038/jid.2008.177
Zaidi MR et al (2011) Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature 469: 548–553. https://doi.org/10.1038/nature09666
Zaidi MR, De Fabo EC, Noonan FP, Merlino G (2012) Shedding light on melanocyte pathobiology in vivo. Cancer Res 72:1591–1595. https://doi.org/10.1158/0008-5472.CAN-11-2586
Zhan T, Rindtorff N, Boutros M (2017) Wnt signaling in cancer. Oncogene 36:1461–1473. https://doi.org/10.1038/onc.2016.304
Zhang Y, Xiong Y, Yarbrough WG (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 92:725–734
Acknowledgments
This chapter has been updated from the previous edition, which was written by Drs. Glenn Merlino and Vince Hearing. Because of space limitations, we have relied heavily on the citation of many review articles and therefore apologize to the authors of numerous significant primary papers we were unable to reference. M.R.Z. acknowledges research support from the National Institutes of Health (K22CA163799 and R01CA193711), US Department of Defense (CA150492), Melanoma Research Foundation, and W.W. Smith Charitable Trust. D.E.F. acknowledges research support from the National Institutes of Health (5P01 CA163222, 1R01CA222871, R01AR072304, and 5R01 AR043369-22), the Melanoma Research Alliance, and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. D.E.F. has a financial interest in Soltego, Inc., a company developing SIK inhibitors for topical skin-darkening treatments.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Crown
About this entry

Cite this entry
Zaidi, M.R., Fisher, D.E., Rizos, H. (2020). Biology of Melanocytes and Primary Melanoma. In: Balch, C., et al. Cutaneous Melanoma. Springer, Cham. https://doi.org/10.1007/978-3-030-05070-2_42
Download citation
DOI: https://doi.org/10.1007/978-3-030-05070-2_42
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-05068-9
Online ISBN: 978-3-030-05070-2
eBook Packages: MedicineReference Module Medicine