
Abstract
Producing recombinant proteins in native conformations and functions is one of the most important aspects of structural biology. Because demand for tertiary structure information of mammalian proteins such as membrane proteins and multi-subunit complexes has been increasing in the field of biological and pharmaceutical sciences, target proteins for structural studies have shifted from prokaryote to mammalian proteins. However, since mammalian proteins are often unstable and require posttranslational modifications, it is difficult to prepare sufficient amounts of functional proteins for structural studies using bacteria expression system. Nowadays, insect and mammalian cell expression systems are widely used for overcoming such problems. In this chapter, we explain the basic concepts of these expression systems and provide examples of advanced new techniques including baculovirus-silkworm expression and HEK293 GnTI-cell expression to confer uniformed N-glycans suitable for structural studies.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Keywords
- Protein expression
- Insect cells
- Mammalian cells
- Baculovirus
- Silkworm
- Constitutive expression
- Transient expression
1 Expression of Protein in Insect Cells
1.1 Introduction
1.1.1 Overview
The production of mammalian proteins such as membrane proteins and multi-subunit complexes is often difficult in bacterial expression systems, which do not exhibit mammalian posttranslational modification. Thus, insect cell expression systems are widely used because their machineries for translation, intracellular transport, and posttranslational modification (such as glycosylation, phosphorylation, methylation, acylation, and acetylation) are similar to those in mammalian cells. In addition, insect cells can often express milligram amounts of proteins and are inexpensive compared with mammalian cells [1]. Conversely, the posttranslational modifications provided by insect cells are similar to, but partially different from, those in mammalian cells. Mammalian N-linked glycoproteins contain complex branched sugars, which are composed of mannose, galactose, N-acetylglucosamine, and neuraminic acid. In insect cells, N-linked glycosylation generally results in oligosaccharides or high-mannose-type oligosaccharides. To introduce mammalian-type glycosylation, customized cell lines known as Mimic™ Sf9 cells (Invitrogen), which express mammalian glycosyltransferases, are commercially available (see Sect. 1.1.2) [2]. Furthermore, the MultiBac system can be used for simultaneous expression of large multi-subunit complexes (see Chap. 3). Therefore, nowadays, mammalian proteins are expressed in insect cells for structural analyses [3–5].
Insect cell expression systems are mainly divided into two systems, viral and nonviral. The viral system uses baculovirus, which has strong infectivity against Lepidoptera (butterflies and moths). The polyhedrin promoter of baculovirus strongly induces expression of downstream genes; thus, the polyhedrin promoter is mostly used in baculovirus systems. At present, various insect cell expression systems are commercially available, such as Bac-to-Bac® (Invitrogen), BaculoDirect™ (Invitrogen), and flashBAC™ (OET). The details of these kits are described in Sect. 1.1.3 [6–8].
The nonviral systems are subdivided into transient and constitutive expression. For transient expression, the foreign gene is introduced only into the host cells and is not integrated into the host genome; thus, the expression does not proceed continuously. In contrast, for constitutive expression, the foreign gene is integrated into the genome so that the gene is replicated and maintained after cell division. Because the transient expression does not need the preparation of a stable clone carrying the gene of interest, it is suitable for rapid expression of the target gene. Although the constitutive expression system requires a much longer time to select a stable highly expressing clone, once the stable clone has been established, the gene of interest is continuously obtained as long as the stable clone survives. Among various constitutive expression systems, the Drosophila expression system (Invitrogen) is a well-known system. Details of this system are described in Sect. 1.1.3.4.
1.1.2 Cell Lines and Virus Types
Sf9, Mimic Sf9, Sf21, SF+, and High Five cells are basic cell lines used for recombinant protein production. Sf9, Mimic Sf9, Sf21, and SF+ cells are derived from Spodoptera frugiperda. Sf9 cells are the standard cell lines, but Sf21 cells usually show higher protein production than do Sf9 cells. SF+ cells are suitable for suspension cultures and are thus used for large-scale protein production. Mimic Sf9 cells are a derivative of Sf9 cells that stably express mammalian glycosyltransferases. Proteins produced in Mimic Sf9 cells contain terminally sialylated N-glycans different from high-mannose-type N-glycans modified in insect cells. High Five cells, derived from Trichoplusia ni, are suited for secretion of recombinant proteins. S2 cells are derived from Drosophila melanogaster and are used for both transient and constitutive protein expressions [9].
Two baculoviruses, Autographa californica multiple nuclear polyhedrosis virus (AcMNPV) and Bombyx mori nuclear polyhedrosis virus (BmNPV), are commonly used to infect the cell lines. AcMNPV can infect Sf9, Mimic Sf9, Sf21, SF+, and High Five cells, while BmNPV is used to infect Bm5 and BmN4 cells derived from silkworms. Silkworms are an attractive protein-producing factory because they have the ability to express abundant amounts of silk proteins. Because BmNPV (and not AcMNPV) directly infects the silkworm Bombyx mori, BmNPV expression in larvae and pupae of silkworms is now used for large-scale production of mammalian proteins [10–12].
1.1.3 Commercially Available Kits
1.1.3.1 Bac-to-Bac Expression System
The Bac-to-Bac expression system has an advantage in the production of recombinant virus [6] because it does not require recombination in insect cells. The genome of recombinant virus can be prepared using E. coli DH10Bac cells harboring both baculovirus DNA called “bacmid” and helper plasmid coding transposase. In the E. coli DH10Bac cells, a gene of interest is transferred from the original transfer vector (known as pFastBac™ series (Invitrogen)) to bacmid by transposases. The successful transposition is verified by the blue-white selection method. Inserting the mini-Tn7 sequence into the mini-attTn7 attachment site on the bacmid disrupts the expression of the LacZα peptide. Thus, the colonies containing the recombinant bacmid turn white, whereas the colonies harboring the unaltered bacmid remain blue. The recombinant bacmid and unaltered bacmid are further distinguished using the PCR method (see Note 1). Once recombinant bacmid is transfected into insect cells, recombinant viruses are generated in a week. The type of virus used in this system is not limited only to AcMNPV. E. coli cells harboring the DNA genome of BmNPV were also established and used for silkworm expression. Another unique aspect of the Bac-to-Bac system is the pFastBac™ Dual plasmid harboring a p10 promoter in addition to the conventional polyhedrin promoter, enabling co-expression. In summary, the Bac-to-Bac system establishes a fast and simple method for producing recombinant virus and avoids the need to isolate the recombinant virus from the parent, nonrecombinant virus. The simplified schematic representation of this system is illustrated in Fig. 1.

Schematic representation of the Bac-to-Bac expression system. The flowchart for producing recombinant AcMNPV is shown on the left and BmNPV is shown on the right. The pFastBac™ plasmid DNA is introduced into DH10Bac host and BmDH10Bac host. The gene of interest is colored as cyan and the transposition sequences (Tn7R and Tn7L) are colored as green. The recombinant bacmid DNA is extracted from the white colony and is transfected into Sf9 cells and B. mori larvae (pupae), to generate the recombinant virus
1.1.3.2 BaculoDirect Expression System
The BaculoDirect expression system introduces Gateway® Technology to facilitate direct transfer of the gene of interest into the baculovirus genome by site-specific recombination of lambda phage [7]. The protocol of the system is as follows: The Gateway® entry clone containing the gene of interest is mixed with BaculoDirect linearized DNA. The gene of interest is transferred from the entry clone to the virus DNA by the recombinase. The success of the recombination can be confirmed by PCR. The resulting recombinant virus DNA is transfected into insect cells, usually Sf9 cells, and then the recombinant virus containing the gene of interest is generated. The recombinant virus can be selectively isolated by ganciclovir selection, using the following method, the nonrecombinant virus expresses herpes simplex virus type 1 thymidine kinase (HSV1 tk), which phosphorylates ganciclovir. In Sf9 cells possessing nonrecombinant viruses, ganciclovir is phosphorylated and incorporated into the DNA to inhibit gene replication. Hence, recombinant virus can be selectively replicated after transfection using a BaculoDirect system. The simplified schematic representation of this system is illustrated in Fig. 2.

Schematic representation of the BaculoDirect expression system. The flowchart for producing recombinant AcMNPV is shown. The gene of interest is introduced to BaculoDirect linear DNA using Gateway® Technology. The resulting recombinant virus DNA is transfected into Sf9 cells and the recombinant virus is generated. The gene of interest is cyan and the TK gene is orange
1.1.3.3 flashBAC Expression System
The flashBAC system is a simplified method to produce recombinant baculovirus [8]. The advantage of this method is that the recombination occurs by co-transfection of insect cells with the baculovirus genome and the transfer vector containing the gene of interest. The baculovirus genome used in the flashBAC system lacks a part of an essential gene (ORF 1629) and contains a bacterial artificial chromosome (BAC) at the polyhedron gene locus, replacing the gene of interest and ORF 1629 downstream of the polyhedrin promoter of the transfer vector if the recombination is successful. Because the deletion of ORF 1629 prevents virus replication in insect cells, the parent virus does not propagate, and the recombinant virus can be obtained without plaque purification. The other advantage of this system is that viruses lacking nonessential genes for replication are commercially available. The flashBAC GOLD lacks chitinase A (chiA) and V-cathepsin (v-cath), which contributes to the productivity and secretion efficiency of the target protein. The flashBAC ULTRA lacks p10, p74, and p26 in addition to chiA and v-cath. Deletion of these nonessential genes from flashBAC improves secretion efficiency and protein yield [13, 14]. The simplified schematic representation of this system is illustrated in Fig. 3.

Schematic representation of the flashBAC expression system. The transfer vector containing the gene of interest and flashBAC DNA are co-transfected into Sf9 cells. In Sf9 cells, the gene of interest is introduced to flashBAC DNA by homologous recombination, and the recombinant virus is generated from it
1.1.3.4 Drosophila Expression System
The Drosophila expression system uses Drosophila melanogaster Schneider (S2) cells and combines the advantages of both the mammalian and baculovirus systems [15, 19]. The S2 cells grow rapidly (doubling time, 24 h) and reach high density (5.0 × 107 cells/mL) in a low-cost medium without serum and CO2. The Drosophila expression system achieves expression of mg amount of eukaryotic proteins with low cost. Although the proteins produced in S2 cells do not contain exactly the same posttranslational modifications as mammalian proteins, their activity is often maintained and sometimes higher than those in mammalian proteins. The Drosophila expression system can be applied to transient and constitutive expression. The transient expression is used for small-scale and rapid production of a target gene. In transient expression, the target gene is transfected into S2 cells and is induced by copper sulfate. Once the appropriate construction is determined, the stable expression clone should be established for large-scale expression. The establishment of the stable clone is achieved by co-transfection of the expression vector harboring the gene of interest and the selection vector harboring the antibiotic resistance gene, such as hygromycin, blasticidin, and puromycin. Optimization of the ratio of the expression and selection vectors leads to the establishment of a clone harboring high-copy number of the gene of interest.
1.2 Materials
In this and the next section, the materials and methods for the production of a recombinant virus using the Bac-to-Bac system are described in detail. Expression in insect cell lines using AcMNPV and in silkworms using BmNPV is described for practical use. A simple flowchart is illustrated in Fig. 1. Green fluorescent protein (GFP) expression in B. mori larvae, pupae, and Sf9 cells is shown in Fig. 4.

GFP expression in B. mori larvae, pupae, and Sf9 cells. B. mori larvae were infected by needlepoint immersed in the BmNPV/GFP virus (a), by direct syringe injection of BmNPV bacmid/GFP DNA (b) and mock (c). B. mori pupae were infected by needlepoint immersed in the BmNPV/GFP virus (d) and by direct injection of BmNPV bacmid/GFP DNA using a pipette (e). The photographs of the larvae were taken at 96, 120, and 144 h after infection using a UV illuminator in complete darkness. (f) Sf9 cells were transfected with GFP/pFastBac1 plasmid using X-Treme GENE. The photographs of the cells were taken at 24, 48, 72, 96, 120, and 144 h after infection [10] (Reprinted from Ref. [10], with permission from Elsevier)
1.2.1 Preparation of Recombinant Bacmids
-
Escherichia coli strains: DH10Bac, BmDH10Bac (WT, CP−, CP−-Chi−).
-
Transfer vectors: pFastBac™1, pFastBac™HT, pFastBac™Dual.
-
Antibiotics: 50 g/L kanamycin, 10 g/L tetracycline, 10 g/L gentamicin, 50 g/L ampicillin.
-
Media: 2 × yeast extract tryptophan (2 × YT) media, Luria broth (LB) media.
-
Chemicals: isopropyl β-D-thiogalactopyranoside (IPTG), X-gal.
1.2.2 Expression in Insect Cells
-
Insect cell lines: Sf9, Sf21, High Five.
-
Media: Sf900 II SFM (Invitrogen), Fetal Bovine Serum (HyClone), Express Five SFM.
-
Transfection reagents: X-tremeGENE HP Transfection Reagent (Roche).
-
Flasks: Erlenmeyer flasks.
-
Safety cabinet: biological safety cabinet (Sanyo).
-
Shaker incubator: Bioshaker (Taitec).
1.2.3 Expression in Silkworm
-
Silkworms: Bombyx mori (Kinshu × Showa race) (Ehime-Sanshu).
-
Feed: synthetic diet (Ehime-Sanshu).
-
Rearing cage: plastic Tupperware.
-
Transfection reagents: DMRIE-C (Invitrogen).
-
10 × phosphate-buffered saline (PBS): 1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 18 mM KH2PO4. Adjust the pH to 7.4 with HCl.
1.3 Methods
1.3.1 Expression of Proteins in Insect Cells
1.3.1.1 Preparation of Recombinant Bacmids
-
1.
Introduce 1–50 ng recombinant plasmid DNA into DH10Bac chemically competent cells.
-
2.
Heat the cells at 42 °C for 45 s. Incubate on ice for 2 min.
-
3.
Add 1 mL room temperature LB medium.
-
4.
Shake tubes at 37 °C for 1 h.
-
5.
Add tetracycline (final concentration, 10 μg/mL) and shake tubes at 37 °C for 12–15 h.
-
6.
Add gentamicin (final concentration, 7 μg/mL) and shake tubes at 37 °C for 2 h.
-
7.
Prepare three tenfold serial dilutions of the transformed cells (10−1, 10−2, 10−3) with LB medium, and plate them on LB agar plates containing appropriate antibiotics and chemicals (50 μg/mL kanamycin, 7 μg/mL gentamicin, 200 μM IPTG, 40 μg/mL X-gal).
-
8.
Incubate the LB plates at 37 °C for 24 h.
-
9.
Pick the white colonies using sterilized chips and re-streak them on fresh LB agar plates. After re-streaking, dip the chips into a tube containing PCR reaction mixture. The reaction mixture is subjected to agarose gel electrophoresis (see Note 2).
-
10.
Isolate the bacmids from the positive clones by using a commercially available mini-prep kit (see Note 3).
1.3.1.2 Preparation of Recombinant Virus (Insect Cells)
-
1.
Inoculate Sf9 cells in six wells and culture the cells until semi-confluent.
-
2.
Prepare bacmid reagents by mixing 2 μg bacmid with 100 μL Sf900 II SFM.
-
3.
Prepare transfection reagents by mixing 8 μL X-tremeGENE with 100 μL Sf900 II SFM.
-
4.
Mix bacmid reagents with transfection reagents.
-
5.
Incubate bacmid and transfection mixture at room temperature for 30 min.
-
6.
Remove the culture medium from each well and add 800 μL Sf900 II SFM.
-
7.
Add 200 μL transfection reagent as droplets.
-
8.
Incubate at 27 °C for 5 h.
-
9.
Add 1 L Sf900 II SFM supplemented with 10 % fetal bovine serum (FBS).
-
10.
Incubate the plate at 27 °C for 5 days.
-
11.
Collect the supernatant in sterilized tubes.
-
12.
Centrifuge the tubes for 5 min at 1000 × g to remove extra cells.
-
13.
Collect the supernatant and store at 4 °C until use (P1 virus).
1.3.1.3 Amplification of Recombinant Virus
-
1.
Inoculate Sf9 cells in six wells and culture the cells until semi-confluent.
-
2.
Remove the culture medium and add the P1 virus stock.
-
3.
Incubate the plate at 27 °C for 5–7 days.
-
4.
Collect the supernatant from the wells in sterilized tubes.
-
5.
Centrifuge the tubes for 5 min at 1000 × g to remove extra cells.
-
6.
Collect the supernatant and store at 4 °C until use (P2 virus).
1.3.1.4 Expression Check
-
1.
Inoculate Sf9/Sf21/High Five cells in six wells and culture the cells until semi-confluent.
-
2.
Remove the culture medium and add the P2 virus stock.
-
3.
Incubate the plate at 27 °C for 3–5 days.
-
4.
Collect both the supernatant and the cells from the wells in the same sterilized tubes.
-
5.
Centrifuge the tubes for 5 min at 1000 × g and collect the supernatant and cells separately.
-
6.
Check for protein expression in the medium.
-
(a)
Mix the supernatant with SDS-PAGE sample buffer.
-
(b)
If the expression of the protein is low, the supernatant should be concentrated using trichloroacetic acid (TCA).
-
(a)
-
7.
Check for protein expression in the cell lysate.
-
(a)
Add 100 μl appropriate buffer to the collected cell (e.g., 50 mM HEPES-NaOH pH 7.5, 250 mM NaCl, 10 % glycerol, 1 mM β-ME, 1 % NP40, 1 × protease inhibitor cocktail).
-
(b)
Vortex briefly and incubate at 4 °C for 10 min.
-
(c)
Repeat step (b) for two times.
-
(d)
Centrifuge the cells for 30 min at 20,000 × g at 4 °C.
-
(e)
Collect both the supernatant (soluble fraction) and the precipitant (insoluble fraction), and mix them with SDS-PAGE sample buffer.
-
(a)
1.3.2 Expression in Silkworm
1.3.2.1 Preparation of Recombinant Bacmid Using BmDH10Bac Cells
Same as the method described above (see Sect. 1.3.1.1).
1.3.2.2 Injection of Recombinant Bacmid into Silkworm Bombyx mori
-
1.
Mix 50 μL recombinant bacmid (20 ng/μL) with 3 μL DMRIE-C (Invitrogen).
-
2.
Incubate the bacmid mixture at room temperature for 45 min.
-
3.
Inject 50 μl bacmid mixture on the first day of the fifth instar larvae using a syringe with a 26-gauge needle and on the fourth day of pupae using a pipette.
-
4.
Cultivate the infected silkworm larvae and pupae at 25 °C for 5–7 days (see Note 4).
1.3.2.3 Collection of the Hemolymph and Fat Body from Infected Silkworm Larvae and Pupae
1.3.2.3.1 Isolation of the Hemolymph Fraction from Silkworms
-
1.
Transfer the infected silkworm onto ice for 2 min to stop their activities.
-
2.
Make a small hole at the center of the larvae using a syringe with a 26-gauge needle.
-
3.
Isolate the hemolymph to tubes containing 50 μl 5 % (w/v) sodium thiosulfate per one larva (see Note 5).
-
4.
Centrifuge the tubes for 10 min at 20,000 × g at 4 °C.
-
5.
Transfer the supernatant to new tubes and freeze them using liquid nitrogen and store them at −80 °C until use.
1.3.2.3.2 Isolation of the Fat Body Fraction from Silkworm
-
1.
Collect the hemolymph fraction using the same method as above (see section “Isolation of the hemolymph fraction from silkworms”).
-
2.
Secure the larvae onto the rubber plate using pins to prepare them for dissection.
-
3.
Slice the larvae down its back using scissors and pin the skin of larvae onto the rubber plate.
-
4.
Discard the internal organs and wash the skin using 1 × PBS with 0.5 % (w/v) sodium thiosulfate.
-
5.
Scrape off the fat bodies from the skin and collect them into tubes containing 0.5 % (w/v) sodium thiosulfate.
-
6.
Centrifuge the tubes for 10 min at 10,000 × g at 4 °C.
-
7.
Discard the supernatant and freeze the fat bodies using liquid nitrogen and store them at −80 °C until use.
2 Expression in Mammalian Cells
2.1 Introduction
We have described the recombinant protein expression in bacteria and in insect cells (Chaps. 1 and 2). At present, both of the expression systems are generally more productive and less costly than mammalian cell systems. However, using these systems, we sometimes face difficulties in producing recombinant proteins such as secreted and/or membrane proteins, which require posttranslational modifications and usually result in accumulation of inclusion bodies. In these cases, mammalian cells would be another option to consider, because mammalian cells can produce high molecular weight proteins, membrane proteins, and posttranslationally modified proteins derived from eukaryotes. Here we provide guidance for overexpression of recombinant proteins in mammalian cells.
One of the biggest advantages of using mammalian expression systems is the appropriate posttranslational modifications including glycosylation, disulfide formation, phosphorylation, and acetylation, which provide appropriate biological activities to the proteins of interest—one of the main concerns for biological research and medical use. Another advantage is that the proteins can be secreted in the culture supernatant by adding a mammalian signal sequence to the gene of interest. This approach ensures the quality of the proteins because each export step checks whether they are properly folded and modified. It is also helpful for reducing the number of steps required for protein purification, because the culture supernatants, which have relatively low contaminants, can be directly applied to tag-based affinity chromatography. Finally, mammalian expression systems are adopted to produce high molecular weight proteins, which are often expressed in insoluble forms in different systems such as E. coli.
Similar to the other expression systems, the selection of vectors and host mammalian cell lines is important for producing recombinant proteins. At present, several mammalian promoters are available; CMV (human cytomegalovirus), CAG (chicken β-actin promoter coupled with CMV), SV40 (simian virus 40), human EF-1α (elongation factor-1 α), and human UbC (human ubiquitin C) are utilized for the constitutive expression, and tTA/Tet (a tetracycline-controlled transactivator protein (tTA); the expression of the transactivator can be regulated with the different concentrations of tetracycline (Tc)), GLVP/TAXI (GAL4-PR-LBD) (GLVP is a regulator controlled by the GAL4 DNA binding, consisting of a human progesterone receptor ligand-binding domain (PR-LBD) fused to the yeast GAL4 DNA-binding domain), and GAL4-E1b (consisting of the binding sites for the yeast GAL4 DNA-binding domain followed by the adenovirus E1b promoter TATA box) are utilized for inducible expression. Constitutive promoters are usually chosen for overproduction of recombinant proteins unless strict control of its expression is necessary because of the toxicity to cell growth. There are several cell lines commonly used for protein expression, including COS7 (derived from monkey kidney tissue), CHO (derived from Chinese hamster ovary), and HEK293 (derived from human embryonic kidney). The heterogeneity of the posttranslational modification that occurred in mammalian cells, especially glycosylation, sometimes affects the quality of the protein crystals. To overcome this problem, the glycosylation modified cell lines are also available, such as CHO Lec1− and HEK293S GnTI−, both of which are unable to synthesize complex N-glycans due to the deficiency in N-acetylglucosaminyltransferase I (GnTI) [16, 17]. Recently, further modified cell lines, such as Expi293F™ cell line (Thermo Fisher), are available to achieve greater protein yield. Usually, the expression level in mammalian cells is not high, and the addition of affinity tags is necessary to resolve some issues in protein preparation by increasing the yield, enhancing folding, and reducing the number of purification steps. The affinity tag technology is described in a later chapter (see Chap. 4).
Transfection of mammalian cells with plasmid vectors is quite different from transformation of E. coli. In the case of E. coli, a single-plasmid DNA enters into a single cell, and the number of copies of the DNA increases as a result of the host cell enzymes. In principle, each E. coli clone transformed with the same plasmid vector is identical. In contrast, transfection with popular transfection reagents typically introduces multiple-plasmid DNAs into a cell. Unlike typical plasmids for E. coli, plasmids for mammalian cells do not replicate in host cells (unless they have a viral replication origin). Among transfectants, a small percentage of cells, usually less than 10 % or even lower, possess the gene of interest randomly integrated into the host cell genome. The number of the integrated genes and the locus (loci) of the integrated gene(s) differs between individual cells. Therefore, each clone is not identical after selection with drugs and varies significantly in protein productivity.
Protein expression in mammalian cells can be performed using two procedures, transient and constitutive expression, as described earlier for insect cell expression. For transient expression, the transfection efficiency directly alters the yield. We usually harvest cells expressing recombinant protein from a few days up to a week after the transfection because the yield decreases after longer periods. To overcome this problem, constitutive expression with stable cell lines is required. To establish stable cell lines, transfection is carried out in the same way for transient expression, and the cells are grown without a selection drug for a few days. As a next step, cells were cultured and selected in the presence of appropriate drugs. For this purpose, the plasmid for transfection must contain a drug-resistant gene. Within 1–7 days, most of the cells die in the presence of the drug and floating cells may be visible. This is either because the cells are not transfected or the drug-resistant gene is not integrated into the host genomes. For drug selection, the medium containing the drug should be changed to a fresh medium every 3–5 days until certain cells survive and form a colony(ies). This drug selection process usually takes a month or more to obtain a single colony. These selected clones have potential for high productivity. Many factors that determine the productivity, such as the genome location and the number of genes integrated, are unpredictable. Thus, it is recommended to select several clones. However, this would lead to consistent overproduction of the target protein once a stable cell line is established.
Another choice of gene delivery into mammalian cells is viruses: lentiviruses, adenoviruses, and insect viruses (baculoviruses). The lentiviral vectors are derived from the human immunodeficiency virus (HIV-1). The advantage of the lentiviral system is the capability to mediate transduction and constitutive expression of the gene of interest into dividing and nondividing cells. Thus, it is useful to establish the stable cell lines. However, generation and transduction of the lentiviruses require biosafety level 3 (BSL3). The adenoviruses are also useful for the recombinant protein production since it can be prepared at high titer. Moreover, the viruses are not usually integrated into the host genome so that it is relatively safe for the researchers.
Finally, we describe baculoviruses modified to be used in mammalian cells. The baculoviruses modified by engineering of a mammalian expression cassette for transgene expression in mammalian cells are commonly referred to as BacMam viruses. There is concern that mammalian viruses could be harmful to not only mammalian cells but also the researchers themselves. Conversely, insect viruses are much safer because of their inability to replicate in mammalian cells. Various mammalian cell lines have been reported to be transducible with baculovirus [18]. The procedure for virus generation and amplification is the same as those for the baculovirus insect cell system (see Sect. 1).
Here we provide an example of transient protein expression in mammalian cells using human signaling lymphocytic activation molecule (hSLAM), a cellular receptor of measles virus that mediates important regulatory signals in immune cells [19]. Overproduction and purification procedures were slightly modified from the reported ones [19].
2.2 Materials
2.2.1 Transfection and Cell Harvesting
Materials
PBS (phosphate-buffered saline), incomplete Dulbecco’s Modified Eagle’s Medium (DMEM) “serum-free medium,” “complete DMEM,” DMEM supplemented with 10 % fetal bovine serum (FBS).
Cells
HEK293T cells, grown in complete DMEM. HEK293T cells contain the SV40 large T antigen, which allows episomal replication of transfected plasmids containing the SV40 origin of replication. This allows amplification of transfected plasmids and extended temporal expression of the desired gene products.
Plasmids
The pCA7-hSLAM plasmid, for the overproduction of hSLAM protein extracellular domain [19]. The plasmid was constructed by ligating PCR-amplified fragments of the hSLAM extracellular domain into the expression vector pCA7. This vector contains the signal sequence derived from the pHLsec vector [20], which is located upstream of the protein-coding sequence. Therefore, the hSLAM is expected to be secreted to the culture medium because of the signal sequence.
Reagents
Polyethylenimine (PEI) max (Polysciences) is used for transfection.
Equipment
Basic tissue culture facilities, e.g., tissue culture hood, 37 °C, 5 % CO2 tissue culture incubator, cell culture dish and/or flask, centrifuge for harvesting cells, aspirator, 0.45-μm pore size filter unit.
Buffers
10 × wash buffer, 500 mM Na2HPO4, 1.5 M NaCl, 100 mM imidazole.
2.2.2 His-Tag Purification
Colum
HisTrap™ FF 5 mL (GE Healthcare)
Buffers
Wash buffer (1 × wash buffer), 50 mM Na2HPO4, 150 mM NaCl, 10 mM imidazole. Elution buffer, 50 mM NaH2PO4, 150 mM NaCl, 500 mM imidazole.
Make sure that all buffers for column purification are degassed before use.
Equipment
Peristaltic pump, stand to hold a column, SDS-PAGE, Western blot apparatus, and power supplies.
2.3 Methods
2.3.1 Transfection
Transfection is carried out using PEI-mediated gene delivery strategy (Fig. 5). PEI is a synthetic polycation that can bind tightly to DNA. Thus, PEI forms a stable complex with the plasmid DNA, which enters the nucleus via endocytosis without significant cellular obstacles. The typical protocol for PEI-mediated transfection is described below and should be optimized depending on the purpose (see Notes 6, 7, 8, and 9).

PEI-mediated transfection
-
1.
Seed the HEK293T cells in DMEM supplemented with 10 % FBS, L-glutamine, and nonessential amino acids 1 day before transfection. Grow the cells in a 15-cm dish until they were approximately 80 % confluent.
-
2.
Add 1 mL each of DMEM without serum into two tubes. Add 50 μg plasmid encoding the hSLAM extracellular domain (Fig. 6a) to one tube containing medium and PEI Max with twice the amount of the plasmid (100 μg in this case) to another tube. The amount of medium and DNA described above is for one 15-cm dish. Mix thoroughly by tapping tubes.
Fig. 6 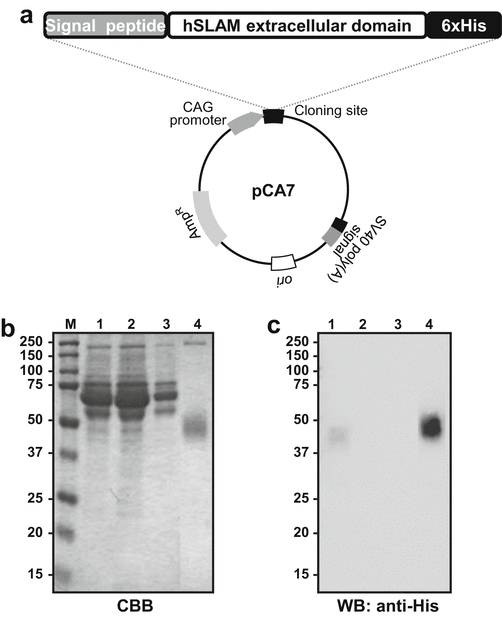
Expression of the hSLAM in mammalian cells. (a) The map of the hSLAM expression plasmid, pCA7-hSLAM. (b) SDS-PAGE of the protein of interest. M Protein size marker, 1 culture supernatant, 2 flow-through fraction, 3 wash fraction, 4 elution fraction. (c) Western blot probed with anti-His antibody. The loaded samples are identical to (b)
-
3.
Combine DNA mixture and PEI Max mixture in an appropriate tube, and mix well.
-
4.
Incubate for 15 min at room temperature. Discard the medium and replace it with fresh DMEM medium containing 2 % (v/v) FBS. DMEM containing 2 % FBS is prepared by mixing complete DMEM containing 10 % FBS and serum-free DMEM.
-
5.
Add the plasmid DNA-PEI Max mixture to the cells. After adding the reagent mixture, mix well by swirling gently.
-
6.
Incubate overnight in 5 % CO2 incubator, and change the medium to complete DMEM the next day.
-
7.
Incubate in 5 % CO2 incubator for a further 3–5 days. The protein of interest is secreted to the medium via signal sequencing.

2.3.2 Harvesting of Secreted Protein Containing the Culture Supernatant
-
1.
Harvest the culture supernatant.
-
2.
Add 10× wash buffer, about 1/10 volume of the supernatant, and incubate overnight at 4 °C.
-
3.
Centrifuge at 5000 × g for 10 min.
-
4.
Filter the medium using a 0.45-μm pore size filter. The filtration is performed by aspirator if the total volume is more than 100 mL or using a syringe with an attached filter for smaller volumes.
2.3.3 His-Tag Purification
Purify the protein of interest from the culture supernatant using His-tag affinity chromatography. The details of affinity chromatography are described in Chap. 4.
-
1.
Centrifuge the cultured medium at 5000 × g for 10 min to remove cell debris.
-
2.
Filter the harvested supernatant with a 0.45-μm membrane filter.
-
3.
Attach a HisTrap™ FF column to a peristaltic pump and equilibrate it with wash buffer (more than three times the column volume).
-
4.
Inject the culture supernatant into the column, and collect the liquid fraction.
-
5.
After injection, equilibrate the column with wash buffer (more than five times the column volume). Collect the wash fraction.
-
6.
Elute the protein using the elution buffer (one to five column volume). Pool the fractions containing the protein.
-
7.
Analyze the protein samples using SDS-PAGE with Coomassie Brilliant Blue (CBB) staining and Western blotting (Fig. 6b, c) (see Note 10). Further purification steps should be considered if good quality is required for structural studies including X-ray crystallography.
Notes
-
1.
Recombinant bacmid DNA is extracted from the white colonies and is further analyzed by PCR using M13 Forward (−40) and M13 Reverse primers. As these primers are complementary to either end of the transposition site, the success of the transposition is verified by PCR analysis.
-
2.
PCR is performed using GoTaq® Master Mix (Promega). The length of the amplified DNA from a positive clone becomes 2300 base pairs (bp) plus the length of the target gene. The length of amplified DNA from a negative clone becomes 300 bp.
-
3.
Bacmid DNA is sometimes damaged by freezing and thawing. Bacmid DNA should be stored at 4 °C.
-
4.
The silkworm is cultivated in the incubator at 25 °C. A beaker containing water should be placed on the top shelf of the incubator to maintain an appropriate humidity in the incubator. Feed the synthetic diet twice a day.
-
5.
The collected hemolymph immediately turns black without sodium thiosulfate. 1-phenyl-2-thiourea could be used as a substitute for sodium thiosulfate.
-
6.
We would suggest that the optimization of the transfection condition at a small scale using a 24- or 6-well plate is better initially. Several factors can be considered: the amount of plasmid DNA, the ratio of DNA-PEI, the incubation time of the DNA-PEI mixture, the medium (serum percentage) and host cells, and the cultivation time after transfection (see Fig. 7).
Fig. 7 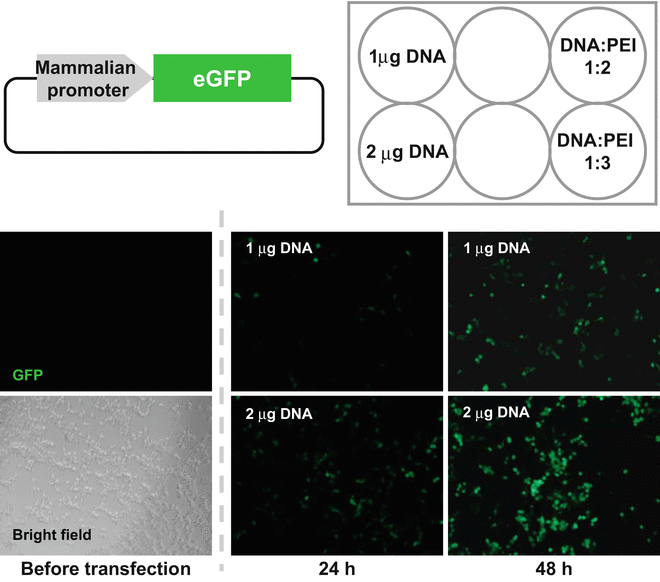
Example of the optimization of transfection condition. HEK293T cells were transfected with the plasmid encoding mammalian promoter and eGFP as a reporter. Images used to examine the amount of DNA and DNA-PEI ratio are shown
-
7.
Other transfection reagents, such as FuGENE HD (Promega), can be used instead of polyethylenimine (PEI) when the transfection efficiency is low.
-
8.
It is advisable to select the plasmid containing a drug-resistant gene if drug selection and/or establishment of stable cell lines is important. G418, also known under the trade name Geneticin, is often used for this purpose. The drug is inactivated by the neomycin phosphotransferase encoded in NeoR or KanR, originally isolated from a bacterial transposon, Tn5. To be resistant to G418, the gene must be expressed by the appropriate promoter and have the appropriate expression for the host cells.
-
9.
If the secretion of the protein is not sufficient, the signal peptide sequence for secretion can be replaced with another signal peptide. While we provided an example using the pHLsec signal sequence (MGILPSPGMPALLSLVSLLSVLLMGCVAE), other signal sequences are commercially available vectors, such as pDISPLAY (Invitrogen) or pSecTag2 (Invitrogen).
-
10.
It is strongly recommended, especially for a preliminary experiment, to keep aliquots utilized at various stages of expression and purification. If there is a problem in the yield or/and purity of the target protein, the aliquoted samples should be analyzed, for example, the cell pellets and cell debris in step 1, Sect. 2.3.2, and the flow-through fraction in step 4, Sect. 2.3.3.

References
Kollewe C, Vilcinskas A (2013) Production of recombinant proteins in insect cells. Am J Biochem Biotechnol 9:255–271. doi:10.3844/ajbbsp.2013.255.271
Growth and maintenance of mimic™ Insect cells. Invitrogen, Thermo Fisher Scientific
Massotte D (2003) G protein-coupled receptor overexpression with the baculovirus–insect cell system: a tool for structural and functional studies. Biochim Biophys Acta Biomembr 1610:77–89. doi:10.1016/S0005-2736(02)00720-4
Bieniossek C, Imasaki T, Takagi Y, Berger I (2012) MultiBac: expanding the research toolbox for multiprotein complexes. Trends Biochem Sci 37:49–57. doi:10.1016/j.tibs.2011.10.005
Berger I, Fitzgerald DJ, Richmond TJ (2004) Baculovirus expression system for heterologous multiprotein complexes. Nat Biotechnol 22:1583–1587. doi:10.1038/nbt1036
Bac-to-Bac Baculovirus Expression System. Invitrogen, Thermo Fisher Scientific
BaculoDirect™ baculovirus expression system. Invitrogen, Thermo Fisher Scientific
flashBAC one-step baculovirus protein expression. Oxford Expression Technologies (2008)
Moraes AM, Jorge SAC, Astray RM et al (2012) Drosophila melanogaster S2 cells for expression of heterologous genes: from gene cloning to bioprocess development. Biotechnol Adv 30:613–628. doi:10.1016/j.biotechadv.2011.10.009
Motohashi T, Shimojima T, Fukagawa T et al (2005) Efficient large-scale protein production of larvae and pupae of silkworm by Bombyx mori nuclear polyhedrosis virus bacmid system. Biochem Biophys Res Commun 326:564–569. doi:10.1016/j.bbrc.2004.11.060
Kato T, Kajikawa M, Maenaka K, Park EY (2010) Silkworm expression system as a platform technology in life science. Appl Microbiol Biotechnol 85:459–470. doi:10.1007/s00253-009-2267-2
Kajikawa M, Sasaki K, Wakimoto Y et al (2009) Efficient silkworm expression of human GPCR (nociceptin receptor) by a Bombyx mori bacmid DNA system. Biochem Biophys Res Commun 385:375–379. doi:10.1016/j.bbrc.2009.05.063
Kaba SA, Salcedo AM, Wafula PO et al (2004) Development of a chitinase and v-cathepsin negative bacmid for improved integrity of secreted recombinant proteins. J Virol Methods 122:113–118. doi:10.1016/j.jviromet.2004.07.006
Hitchman RB, Possee RD, Siaterli E et al (2010) Improved expression of secreted and membrane-targeted proteins in insect cells. Biotechnol Appl Biochem 56:85–93. doi:10.1042/BA20090130
Drosophila expression system. Invitrogen, Thermo Fisher Scientific
Stanley P, Chaney W (1985) Control of carbohydrate processing: the lec1A CHO mutation results in partial loss of N-acetylglucosaminyltransferase I activity. Mol Cell Biol 5:1204–1211. doi:10.1128/MCB.5.6.1204
Reeves PJ, Callewaert N, Contreras R, Khorana HG (2002) Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc Natl Acad Sci U S A 99:13419–13424. doi:10.1073/pnas.212519299
Kost TA, Condreay JP, Jarvis DL (2005) Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat Biotechnol 23:567–575. doi:10.1038/nbt1095
Hashiguchi T, Ose T, Kubota M et al (2011) Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol 18:135–141. doi:10.1038/nsmb.1969
Aricescu AR, Assenberg R, Bill RM et al (2006) Eukaryotic expression: developments for structural proteomics. Acta Crystallogr D Biol Crystallogr 62:1114–1124
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this protocol
Cite this protocol
Kita, S., Maenaka, K., Tadokoro, T. (2016). Expression of Proteins in Insect and Mammalian Cells. In: Senda, T., Maenaka, K. (eds) Advanced Methods in Structural Biology. Springer Protocols Handbooks. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56030-2_2
Download citation
DOI: https://doi.org/10.1007/978-4-431-56030-2_2
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56028-9
Online ISBN: 978-4-431-56030-2
eBook Packages: Springer Protocols