
Abstract
Cells isolated directly from tissues (primary cultures) have many applications in research and applications in the fields of toxicology, pharmacology, and virology. Where such preparations remain the only scientific option to achieve necessary results it is still possible to refine and reduce the use of animal and human sources of the tissue by cryopreservation of the primary cells for later use, this reduces the need for fresh tissues and enables improvements in standardization through the ability to provide the same cell preparation at different times and to different laboratories. The methods described provide options for adherent cultures as monolayers, harvested cell suspensions, and also lymphocytes isolated from peripheral blood.
Access provided by Autonomous University of Puebla. Download protocol PDF
Similar content being viewed by others


Key Words
1 Introduction
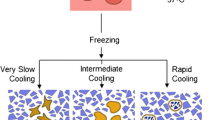
Historically the fields of toxicology, pharmacology, and virology have relied on the use of animals to provide vital research data for the development of new products and the isolation and investigation of infectious diseases. However, over a number of decades there has been a strong movement toward adopting the three Rs principle to refine, reduce, and ultimately replace the use of animals for research and testing (1). This is now supported at national and international levels (2) and much interest has been focused on the development of alternative in vitro techniques. The requirement for functional cell-based assays and virus isolation from infected material has maintained the need for tissue preparations in the form of primary cell cultures. Where such preparations remain the only scientific option to achieve necessary results it is still possible to refine and reduce the use of animal and human sources of the tissue by cryopreservation of primary cells for later use. Cryopreservation techniques provide an opportunity to reduce the need for regular fresh animal tissue and can enhance standardization of laboratory work by enabling the use of the same source of cells over a period of an experimental program and in different laboratories.
It may be tempting to reserve the use of cryopreservation methods for spare or suboptimal cultures left over from formal experiments to provide a perceived “backup” for emergencies in future experiments. However, attempts to use cryopreserved spare cultures will not enable the full benefits of the prospective establishment and qualification of cryopreserved cell stocks to be realized. It is also valuable to try to assure the quality of the primary cell obtained for preservation and the establishment of a protocol agreed with clinicians or other staff providing the cells or tissues (3). This protocol should also ensure that all legal and ethical requirements have been addressed; this is especially important for human cells and tissues where those responsible for the use of the cells should be able to demonstrate that the materials were obtained with fully informed consent (4).
Once a reliable archive of preserved cells has been established it can be quality controlled and qualified for particular purposes, and if necessary, tested for infectious agents. This provides a valuable reference stock of cells that may be considered safer for laboratory use than unqualified supplies of fresh tissue. Primary cell preparations are often very complex mixtures of cells with a very broad range of functions and morphologies. These various populations are likely to vary in their amenability to preservation. Loss of critical cell populations could therefore affect the utility of preserved cultures so it is particularly important to confirm the retention of key functional characteristics (or to identify levels of certain key cell types) in cultures recovered from a cryopreserved “bank” of cells.
Such qualified banks of stored material can provide a reliable and convenient source of fresh cells for routine experimentation. This is invaluable where primary feeder cells are required for the isolation, maintenance, and expansion of complex and potentially unstable cultures that are difficult to grow. An important example is the case of primary mouse embryonic fibroblasts used for the culture of human embryonic stem cells (5).
When carrying out the preparation of a stock of cryopreserved cells it is important to select the cultures to be used to eliminate any that do not meet certain criteria that include:
-
1.
Cultures should be subconfluent to avoid the use of cells in the plateau phase of growth that may not survive cryopreservation (e.g., ref. 6).
-
2.
Under the microscope cultures should exhibit typical, healthy morphology without cytopathic effects such as high levels of vacuolation or cell death.
-
3.
There should be no evidence of microbial contamination, which may include observation as rafts of organisms seen under the microscope (i.e., fungi and bacteria) or gross turbidity in the growth medium, positive results in sterility tests (see Note 1 ), evidence of viral contamination such as “plaques” (areas of cell death), or cell fusion or enlargement.
At the point of preparing the selected cultures for preservation it is important to eliminate any materials that might lead to loss of viability during preservation (see Note 2 ) and to ensure homogeneity of the cell bank being prepared, i.e., does vial 1 of the stock provide the same representative culture as vial 100 (see Note 3 ).
The methods described here provide options for adherent cultures as monolayers and harvested cell suspensions, and also lymphocytes isolated from peripheral blood based on the original principle of Boyum (7) as general examples of some of the more common methods used for preservation of primary cells.
2 Materials
2.1 Equipment
-
1.
Bench centrifuge with sealable biohazard-containment centrifuge buckets.
-
2.
Inverted microscope with ×20 and ×40 objectives.
-
3.
Class II biological safety cabinet compliant with the British Standard 5726 (2005).
-
4.
1 and 10 mL plastic sterile pipets and a pipet pump.
-
5.
Trypan blue (0.4% [w/v] in PBS-A).
-
6.
Hemocytometer (Neubauer improved).
-
7.
Programmable freezer or alternative slow cooling system (see Note 4 ).
-
8.
Liquid nitrogen storage system and supply of liquid nitrogen: cryostorage containers with appropriate storage racks and an inventory system suitable for holding cryovials or other vessels described in Subheadings 3.1. – 3.3.
-
9.
Low-temperature-resistant marker pens or labeling machine.
-
10.
Biohazard waste bags and waste bag containers for contaminated plastics (all waste must be disposed of according to the local safety guidance).
2.2 Cryopreservation of Adherent Primary Cells
-
1.
Near confluent monolayer cultures of primary cells in T75-cm2 tissues culture flasks or multiwell (6 or 24 well) trays (γ irradiated and cell culture tested).
-
2.
Growth medium: 1X Eagle’s Minimal Essential Medium basal medium, 10% (v/v) fetal calf serum, 2 mM glutamine, and 1% (w/v) nonessential amino acids.
-
3.
0.5 g/L trypsin/0.2 g/L EDTA in Puck saline A (0.4 g/L potassium chloride, 8 g/L sodium chloride, 0.35 g/L sodium hydrogen carbonate, 1 g/L D-glucose, and 0.005 g/L phenol red).
-
4.
Phosphate buffered saline (PBS): 0.2 g/L potassium dihydrogen phosphate, 1.15 g/L anhydrous disodium hydrogen phosphate, 0.2 g/L potassium chloride, and 8.0 g/L sodium chloride dissolved in deionized water with a final pH of 7.4 ± 0.2 pH units).
-
5.
Humidified carbon dioxide/air (5/95% [v/v]) incubator.
-
6.
Water bath (37–40°C).
-
7.
Cryoprotectant solution: 90% (v/v) fetal calf serum and 10% (v/v) dimethyl sulfoxide (DMSO; spectroscopic grade) (see Note 5 ).
-
8.
Adhesive tape.
-
9.
Cryostorage ampoules (e.g., Cryovials from Nunc/Invitrogen).
2.3 Cryopreservation of Peripheral Blood Mononuclear Cells
-
1.
Primary cells: 10–20 mL of whole blood taken in heparin-containing tubes.
-
2.
Culture medium: RPMI-1640, 2 mM glutamine, and 10% (v/v) fetal calf serum.
-
3.
Cryoprotectant medium: culture medium with 20% fetal calf serum and 10% (v/v) DMSO without antibiotics.
-
4.
15 mL sterile “PVC” sterile conical centrifuge tubes.
-
5.
Separation medium: ACCUPSPIN™ System-HISTOPAQUE®-1077 (Sigma Aldrich-UK) (http://www.sigmaaldrich.com) tubes (one tube required for maximum volume of 15 mL blood). HISTOPAQUE-1077 consists of an aqueous solution of a high molecular weight polysaccharide and an aggregating agent sodium diatrizoate adjusted to a density of 1.077 ± 0.001.
-
6.
PBS-A: 10 g/L sodium chloride, 0.25 g/L potassium chloride, 1.44 g/L disodium hydrogen orthophosphate-anhydrous (Na2HPO4), 0.25 g/L potassium dihydrogen orthophosphate (KH2PO4) at pH 7.3–7.5.
-
7.
Benchtop centrifuge. Values for centrifugation given for use of an Eppendorf 5810 centrifuge.
-
8.
Class II biological safety cabinet compliant with the British Standard 5726 (2005).
-
9.
Plastic gown (to be worn over the laboratory coat while handling blood) and gloves.
3 Methods
3.1 Cryopreservation of Monolayer Primary Cell Cultures as Single Cell Suspensions
This method has been used by the author for preservation of a variety of primary cells including those from overnight cold trypsinization of tissues (8) and perfusion of organs (liver, kidney, and small intestine) with prewarmed collagenase or trypsin (Dowall, S., Health Protection Agency [9]).
-
1.
Inspect and select cultures that appear morphologically normal, show no signs of microbial contamination (see Note 1 ), and are subconfluent (considered to be more amenable to cryopreservation, see ref. 6.
-
2.
Aspirate the growth medium from the monolayer and wash twice in approx 10 mL PBS (i.e., approx 0.1 mL/cm2) using sterile pipets.
-
3.
Use a 5-mL pipet to add 2–4 mL (i.e., 0.5 mL/25cm2) prewarmed (approx 37°C) trypsin solution to cover the cell monolayer, recap the flask, and incubate at approx 37°C for 5 min.
-
4.
Examine the monolayer and tap the flask to dislodge cells. If the large majority cells are not dislodged, incubate for up to a further 10 min.
-
5.
Recover cells in trypsin in a sterile pipet and add to approx 10 mL PBS in a conical-based centrifuge tube and take a 1-mL sample to determine the total number of viable cells by Trypan blue dye exclusion ([10,11]; and the Cell Biology and Imaging Laboratory Manual of Basic Techniques [http://www.nibsc.ac.uk/aboutus/cell-techniques.html]) or an appropriate alternative method (12).
-
6.
Meanwhile centrifuge the remaining suspension at approx 80–100g for 5 min.
-
7.
Aspirate the supernatant and resuspend the cell pellet in 1 mL of culture medium.
-
8.
Add cryoprotectant medium to the cells to give a final cell concentration of approx 5 × 106–107 cells/mL.
-
9.
Mix the cell suspension gently to provide a homogenous suspension and carefully aliquot the cell/cryoprotectant mixture into 1- to 2-mL aliquots in sterile cryovials, sealing each tube as it is filled.
-
10.
Transfer all sealed vials to a programmable cooler. One vial should be taken to determine postcryoprotection viability (see Notes 3 and 6 ).
-
11.
Prepare a reference vial containing the cryoprotectant solution used for the cells and a thermocouple, and set the machine to cool as follows:
-
a.
Step 1: hold for 4°C for 10 min.
-
b.
Step 2: −3°C/min to −10°C.
-
c.
Step 3: −15°C/min to −30°C.
-
d.
Step 4: −3°C/min to −60°C.
-
e.
Step 5: −10°C/min to −140°C and hold at −140°C.
-
a.
-
12.
Using an appropriate cryoprotective mask, gloves, and apron, the cooling machine is opened, according to the manufacturer’s instructions, and vials are transferred to a temporary storage rack in the vapor phase of liquid nitrogen prior to archiving frozen vials in the vapor phase of liquid nitrogen.
-
13.
Vials of cells for quality control should be recovered for by rapid thawing at 37°C (e.g., in a water bath at 37–40°C) followed by gradual dilution of thawed cells by dropwise addition of prewarmed growth medium.
-
14.
A sample ampoule should be removed from the liquid nitrogen storage after overnight equilibration and a viability assay again performed and compared with pre- and postcryoprotection treatment results to determine the overall success of the cryopreservation process (see Note 3 ). The remainder of this sample should also be recovered in antibiotic-free medium to demonstrate the appropriate growth and morphology of the cells, as well as the absence of contamination (see Note 1 ).
3.2 Cryopreservation of Adherent Monolayers of Primary Cells
This method has been used for hepatocyte and kidney epithelial cell cultures.
-
1.
Seed fresh primary cells into culture vessels (e.g., 2 mL of growth medium at 2 × 104 cells/mL in each well of a 24-well plate) and incubate the cultures in a humidified atmosphere of 95% air/5% carbon dioxide for several days until the culture has almost formed a confluent monolayer.
-
2.
Inspect and select primary cultures that appear morphologically typical, show no signs of microbial contamination, and are subconfluent.
-
3.
Hold the cultures at room temperature (18–24°C) for 15 min.
-
4.
Aspirate the growth medium supernatant, replace with cryoprotectant medium, and incubate for 15 min (see Note 7 ).
-
5.
Aspirate almost all of the cryoprotectant medium, leaving the cell monolayer with sufficient medium such that its meniscus against the side walls of the wells is still visible.
-
6.
Secure the lid of each 24-well plate with adhesive tape to provide a seal that will protect against ingress of nitrogen vapor during storage.
-
7.
Check that each culture tray is labeled with the date, growth medium, cell type, and any other key information.
-
8.
Transfer the plates to a programmable freezer set to cool its chamber as follows (see Note 8 ):
-
a.
Step 1: initial hold at 4°C for 15 min to allow chamber and vessels to equilibrate.
-
b.
Step 2: cool to −5°C at −1°C/min.
-
c.
Step 3: cool to −12°C at −3°C/min.
-
d.
Step 4: cool to between −10 and −14°C at −5°C/min (see Note 9 ).
-
e.
Step 5: cool to −20°C at −7.5°C/min.
-
f.
Step 6: cool to −25°C at −6.5°C/min and hold at −25°C for 2 min.
-
g.
Step 7: raise temperature to −20°C at 3°C/min (see Note 9 ).
-
h.
Step 8: cool to −50°C at −1°C/min.
-
i.
Step 9: cool to −130°C at −10°C/min and hold at this temperature.
-
a.
-
9.
Transfer the cryopreserved cells to appropriate and secure storage containers in the vapor phase of liquid nitrogen being careful to avoid cell rewarmth.
3.3 Cryopreservation of Peripheral Mononuclear Cells
-
1.
Equilibrate blood sample(s) and the required number of mononuclear cell separation tubes to room temperature. If necessary, protect tubes from excessive light (in exceptional cases of exposure to strong sunlight, wrap in aluminium foil while on the open bench).
-
2.
Centrifuge tubes at 1000 rcf (∼2230 rpm Eppendorf 5810 centrifuge) for 30 s at room temperature to insure all HISTOPAQUE-1077 is above the “frit” prior to use (see Fig.1 ; Note 10 ).
-
3.
Working in a class II safety cabinet, pour 5–15 mL of whole blood (previously mixed with an anticoagulant such as heparin) into the upper chamber of each prefilled ACCUPSPIN tube.
-
4.
Dilute blood directly in the upper chamber of the ACCUPSPIN System-HISTOPAQUE-1077 tube with an equal volume of PBS-A (maximum final volume of 30 mL per tube).
-
5.
Centrifuge at 800 rcf (∼2000 rpm Eppendorf 5810 centrifuge) for 15 min. Set centrifuge to low-brake deceleration (setting 0 on the Eppendorf 5810) (see Note 11 ).
-
6.
After centrifugation, carefully aspirate and discard the plasma layer with a Pasteur pipet to within 0.5 cm of the opaque interface containing the mononuclear cells (see Fig. 1 ).
-
7.
Carefully transfer the mononuclear cell band using a sterile Pasteur pipet into a sterile, 15-mL centrifuge tube.
-
8.
Add PBS-A to a total volume of 15 mL and gently mix by inversion.
-
9.
Centrifuge at 290 rcf (1200 rpm; Eppendorf Centrifuge 5810) for 10 min.
-
10.
Check to see that a cell pellet has formed before discarding the supernatant (see Note 11 ).
-
11.
Wash the mononuclear cells by adding 10 mL PBS-A and resuspend cells by gentle aspiration with a sterile Pasteur pipet.
-
12.
Centrifuge at 1200 rpm (290 rcf; Eppendorf Centrifuge 5810) for up to 5 min and discard the PBS-A supernatant.
-
13.
Resuspend the pellet in 5 mL PBS-A, take a sample (0.2–0.5 mL) to perform a total and viable cell count ([10,11]; or go to http://www.nibsc.ac.uk/aboutus/research.html and click on Cell Biology and Imaging Laboratory Manual of Basic Techniques), and repeat step 9 .
-
14.
Resuspend the mononuclear cell pellet in freeze medium at approx 1 × 106 cells/mL (approx 5–10 × 106 lymphocytes can be expected from a fresh 10-mL blood sample).
-
15.
Mix and aliquot the cell/cryoprotectant mix to prelabeled cryovials.
-
16.
Transfer to a programmable freezer and set to cool according to the program given in Subheading 3.1. , step 11 or alternatively by a passive preservation method as follows ( steps 17 – 19 ).
-
17.
Wrap the vials of cells in insulated paper towels inside a polystyrene box (e.g., protective boxes used by manufacturers to ship individual 500-mL bottles of cell culture medium) and tape the box lid in place.
-
18.
Place the box in a freezer (at or below −70°C) overnight (minimum of 16 h).
-
19.
Recover the box, place it in the vapor phase of liquid nitrogen, and transfer all vials (taking care to avoid rewarming) to a storage location in the vapor phase of liquid nitrogen (see Note 12 ). An atypical cooling profile obtained by this method is given in ref. 13.

Separation of mononuclear cells from whole blood using the ACCUSPINTM system. (A) Diluted blood prior to centrifugation. (B) Appearance of cell layers after centrifugation. MNC, mononuclear cells; red cells, erythrocyte pellet. (Figure kindly provided by Byrne, E., National Institute for Biological Standards and Control [NIBSC].)
4 Notes
-
1.
Sterility tests for fungi and bacterial contaminants can prove highly useful to give confidence to workers in the quality of their aseptic technique, and are an important element in the quality control of stocks of primary cells. There are various methods for carrying out sterility tests, including standard Pharmacopeial tests. A typical method is given in Stacey and Stacey (14) and there are also industrial standards published in Pharmacopeia (15). Broader microbiological issues and other biosafety matters in cell culture work are covered in Coecke et al. (4) and Doblhoff-dier and Stacey (16).
-
2.
A variety of chemicals in cell culture media are highly toxic at high concentrations, including antibiotics and HEPES. As ice forms in a cryopreserved suspension the cells become surrounded by the residual unfrozen water with an ever increasing concentration of solutes. Thus, it may be necessary to wash the cells in a basal medium without any potentially toxic additives before carrying out cryopreservation and to avoid incorporation of such toxic chemicals in cryoprotectant solutions.
-
3.
In order to ensure that each frozen ampoule in a particular bank will give the same representative culture on thawing it is important to pool harvested cells and mix them well prior to processing for cryopreservation. It will be important to check the homogeneity of cultures recovered from cryopreserved stocks; this will be especially important for very large frozen stocks of cells where cryoprotection and aliquotting may have taken a considerable period of time, and may affect the viability and the quality of the frozen cells. Accordingly, initial experiments on preservation of a new type of primary culture reproducibility within and between cryopreserved stocks should be addressed. For larger banks of cells, testing should be performed on early-, mid-, and late-vials to ensure intrabank homogeneity. It should also be born in mind that a simple viability test, such as trypan blue dye exclusion, does not indicate the functionality of the cells, and cryopreserved banks of cells should be qualified in appropriate tests and characterisation to ensure that they retain the critical features and functions for their intended use.
-
4.
A range of machines are available for the slow rate-controlled cooling of cells. One company that provides a number of commonly used models, such as the Kryo 10, is Planer Ltd, UK (http://www.planer.co.uk).
-
5.
DMSO at a final concentration of between 5 and 10% (w/v) has become the cryoprotectant of choice for most mammalian cell preservation methods based on slow cooling rates (−1°/m). However, it has broad range of potentially toxic biological effects. If it is suspected that the use of DMSO is affecting the quality of recovered cells then it may be necessary to carry out cryoprotectant toxicity experiments to explore the quality of cells after different time/concentration treatments, and also to consider the use of alternative cryoprotectants such as glycerol. There is a significant amount of literature on the development of cryopreservation methodologies in journals such as the Journal of Cryobiology (http://www.sciencedirect.com/science/journal/00112240) and CryoLetters (http://www.cryoletters.org).
-
6.
A range of alternative methods have been developed to approximate the cooling rates required to reserve cells (generally −1°C/min). One method of passive cooling is given in Subheading 3.3. and this has proven effective for a range of cell lines as well as primary cells. In addition, passive cooling devices are commercially available such as the Handi-Freeze™ (Taylor Wharton, UK) and Mr. Fosty™ (Nalgene/Invitrogen).
-
7.
In unpublished work by Dowall, S. (Health Protection Agency, Salisbury) (9) it appears that this method is also effective using glycerol (10% [v/v]) in culture medium as the cryoprectective solution.
-
8.
Kidney primary epithelial cells may take 5–6 d to form semiconfluent cultures, whereas primary hepatocyte cultures will have already begun to lose key biochemical functions in this time and will need to be cryopreserved when freshly isolated or after a shorter period of culture. The optimal culture harvest point for cryopreservation will vary depending on a range of factors including cell type and species, culture medium, vessel type, gaseous atmosphere, and culture medium volume.
-
9.
The profile given was developed at the University of Sheffield, Sheffield, UK originally for preservation of insect embryos. It shows a sharp drop in temperature over the range −50 to −25°C with a brief rewarming to −20°C. This eliminated the sharp temperature peak that occurs from the formation of the first ice, which is an exothermic event. In order to use the type of profile given in Subheading 3.2. it is important to characterize the nature (timing and degree) of the exothermic point for a particular cell/cryoprotectant combination.
-
10.
Other media (e.g., Ficoll-Paque [GE Healthcare], Percoll [GE Healthcare]) are available to provide the discontinuous density gradient for separation of cells by buoyant density. The preparation of lymphocytes from the blood of different species may require specialized discontinuous density gradients and two examples of gradient compositions using Percoll are given here that have been used for isolation of primate and mouse mononuclear cells:
a. Primate:
Percoll (1.13 g/mL stock)
15 mL
PBSA (see Subheading 2.2. )
10.3 mL
10X PBS (divalent cation free)
1.6 mL
1 M sulphuric acid
0.1 mL
b. Mouse:
Percoll (1.23 g/mL stock)
57 mL
5 M NaCl in water
10 mL
Distilled water
33 mL
-
11.
The tubes can be centrifuged for a further 10 min if a good separation has not been obtained. This is often the case with blood samples separated 48 h after collection (Byrne, E., unpublished data, National Institute for Biological Standards and Control).
-
12.
Usually only sufficient cells are recovered from individual whole blood samples for about one to three vials and, thus, it is not usual to recover a vial to check viability but to try to ensure samples are obtained as soon as possible after blood donation and that the separation and cryopreservation process is carried out in a reproducible way. Great care should be taken with small samples as only a small proportion of the mononuclear cells received may be of value (e.g., studies of minority lymphocyte populations, Epstein-Barr virus transformation). If a cell pellet is not visible following the first PBS-A wash after Histopaque separation, repeat the centrifugation in step 7 (Byrne, E., unpublished data, National Institute for Biological Standards and Control). If the blood has a very low lymphocyte count then the pellet may only just be visible to the naked eye. Very small samples of whole blood may provide a useable source of mononuclear cells by scaling down the use of Histopaque or Ficoll separation to a total volume of 1.5 mL in a microtube and using a microfuge.
References
Russell, W. M. S. and Burch, R. L. (1959) The Principles of Humane Experimental Techniques. Methuen, London, UK.
Milstein, J., Grachev, V., Padilla, A., and Griffiths, E. (1996) WHO activities towards the three Rs in the development and control of biological medicines. Dev. Biol. Stand. 86, 31–39.
Stacey, G. N., Doyle, A., and Tyrrel, D. (1998) Source materials. In: Safety in Cell and Tissue Culture, (Stacey, G. N., Doyle, A., and Hambleton, P. H., eds.), Kluwer Academic Publishers, Dordrecht, Germany, pp. 1–25.
Coecke, S., Balls, M., Bowe, G., et al. (2005) Guidance on good cell culture practice: A report of the second ECVAM task force on good cell culture practice. ATLA 33, 1–27.
Thomson, J., Itskovitz-Eldor, J., Shapiro, S., et al. (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147.
Terasima, T. and Yasukawa, M. (1977) Dependence of freeze-thaw damage on growth phase and cell cycle. Cryobiology 14, 379–381.
Boyum, A. (1968) Separation of leukocytes from blood and bone marrow. Scand. J. Clin. Lab. Invest. 21, 77.
Stacey, G. N. (1994) Chick embryo fibroblast preparation. In: Cell and Tissue Culture: Laboratory Procedures, (Doyle, A. and Griffiths, J.B., eds.), John Wiley and Sons, Chichester, UK, pp. 1.1.
Dowall, S. (1997) Cryopreservation of primary monkey kidney cells. Report submitted as part of the BTEC HNC in Applied Biology, Health Protection Agency, Salisbury, UK.
Freshney, I. R. (1994) Measurement of viability and cytotoxicity. In: Culture of Animal Cells: a Manual of Basic Techniques, (Freshney, I. R., ed.), Wiley Liss, New York, pp. 288–289.
Patterson, M. K. (1979) Measurement of growth and viability of cells in culture. Meth. Enzymol. 58, 141–152.
Stacey, G. N. (2002) Standardisation of cell lines. Dev. Biologicals 111, 259–272.
Stacey, G. N. and Doyle, A. (2000) Cell banking. In: Encyclopedia of Cell Technology, (Spier, R., editor in chief), Wiley Interscience, New York, pp. 293–293.
Stacey, G. N. and Stacey, A. R. (1999) Routine quality control testing of cell cultures. In: Antiviral Methods and Protocols, (Kinchington, D. and Schinazi, R. F., eds.), Humana Press, Totowa, NJ, pp. 27–40.
European Pharmacopoeia. (2004) European Pharmacopoeia section 2.6.1 (Sterility) and 2.6.7 (Mycoplasma), Maisonneuve SA, Sainte Ruffin, France.
Doblhoff-dier, O. and Stacey, G. N. (2000) Cell lines: applications and biosafety. In: Biological Safety Principles and Practices, 3rd ed., (Fleming, D. O. and Hunt, D. L., eds.), ASM Press, Washington, DC, pp. 221–240.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2007 Humana Press Inc., Totowa, NJ
About this protocol
Cite this protocol
Stacey, G.N., Dowall, S. (2007). Cryopreservation of Primary Animal Cell Cultures. In: Day, J.G., Stacey, G.N. (eds) Cryopreservation and Freeze-Drying Protocols. Methods in Molecular Biology™, vol 368. Humana Press. https://doi.org/10.1007/978-1-59745-362-2_19
Download citation
DOI: https://doi.org/10.1007/978-1-59745-362-2_19
Publisher Name: Humana Press
Print ISBN: 978-1-58829-377-0
Online ISBN: 978-1-59745-362-2
eBook Packages: Springer Protocols