
Abstract
The long-term storage of human and animal cell lines is a relatively new branch of the science of cryopreservation of living organisms. The need to maintain the increasing numbers of cell lines, as they started to emerge from research laboratories from the mid-1900s, necessitated a radical approach to the problem. The realization that they could survive cryopreservation, and that a slow rate of cooling is essential for this survival, led to the eventual discovery of cryoprotectants. Subsequent development of mechanical freezers, which can accurately control the rate of cooling, now allows cells to be cryopreserved at their maximum viability. This chapter outlines the essentials steps for the successful preparation, freezing, and storage of cell lines.
Access provided by Autonomous University of Puebla. Download protocol PDF
Similar content being viewed by others


Key Words
- Cryopreservation
- human and animal cell lines
- cryoprotectant
- cryovials
- freeze-medium
- programmable freezer
- liquid nitrogen
- cross-contamination
- viable cells
1 Introduction
The emergence of mammalian cell culture has its origin in the first attempts to culture tissue explants in vitro at the turn of the twentieth century (1–3). The subsequent development of complex culture medium formulations in the 1950s (4,5) enabled the establishment of a wide range of cell lines and provided a valuable research tool for the study of growth mechanisms and disease. Today, the availability of thousands of animal cell lines offers a reproducible source of material for all aspects of medical and agricultural research.
Continuous maintenance of cell lines in culture is impractical for reasons of:
-
1.
Cost, e.g., serum containing media is currently about 50–300 GBP/L.
-
2.
Risk of exposure to microbial contamination.
-
3.
The possibility of culture cross-contamination.
-
4.
Genetic drift with the possible consequence of phenotypical changes and the loss of the cell’s original characteristics.
Concomitant research into subzero storage methods at the time when cell culture methodology was being developed led to the discovery that addition of glycerol to fowl semen enhanced survival of spermatozoa after storage at −79°C (6). The use of nitrogen in the gaseous (below −130°C) and liquid phase (−196°C) now allows indefinite storage of most mammalian cell lines after cryopreservation. The precise mechanism for optimal cryopreservation has been established by studying the effects of freezing and thawing at various rates from which a hypothesis of freezing injury to cells was proposed (7).
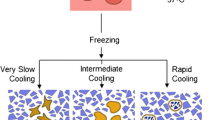
The majority of cell lines must be cooled slowly, i.e., −1 to −5°C/min, and then thawed rapidly to achieve maximum viability. The rate of cooling is optimized to allow time for intracellular water to escape, and subsequently reduces the amount of intracellular ice formed (see Chapter 3). The presence of intracellular ice during cooling and thawing, i.e., post-freezing resuscitation, may cause lethal damage to intracellular membranes (8). Addition of cryoprotectant to the cells depresses the temperature at which intracellular ice is formed and allows cooling rates to be reduced for more efficient water loss (9).
The development of programmable controlled-rate freezers now allows individual cooling profiles to be designed that give maximum cell viability during cryopreservation, i.e., identical pre- and postfreezing viabilities. Culture collections such as the European Collection of Animal Cell Cultures (ECACC) (http://www.ecacc.org.uk/), supply many thousands of cell lines and many new cell lines are added annually. A systematic approach to the quality control of cell lines and their cell banks is therefore essential to ensure future supplies of authentic material (10,11).
2 Materials
-
1.
Class II or III microbiological safety cabinets in either containment level 2 or 3 laboratory facilities (12). The handling requirements of any cell line should be assessed using the current guidelines issued by national regulatory bodies in consultation with your safety officer, prior to their introduction into the laboratory (13).
-
2.
Cell cultures: these should be in active growth, i.e., log phase, usually 2–4 d after subculture for most mammalian cells, although slower growing cells may need a longer period to achieve sufficient numbers to freeze. Cells that have entered the stationary phase should not be used (see Note 1 ).
-
3.
Freeze medium: this will be either the growth medium supplemented with 20–25% (v/v) serum and 10% (v/v) cryoprotectant, or whole serum and 10% (v/v) cryoprotectant. The choice of cryoprotectant will be determined by the cell type, but for the majority of cell lines dimethyl sulfoxide (DMSO) or glycerol can be used. An alternative is polyvinyl pyrrolidone, a high-molecular weight polymer (see Note 2 ).
-
4.
Cryovials or ampoules: these are obtained from tissue culture plasticware supplies, e.g., Nunc, Becton-Dickinson, Corning as presterilized (irradiated) screw cap 1.0- or 1.8-mL vials (see Note 3 ). Some manufacturers also supply special racks for holding the vials during filling.
-
5.
Freezing system: the best and most reproducible method is the programmable freezer, e.g., Planer Products (see Notes 4 and 5 ).
-
6.
Storage system: liquid nitrogen storage vessels with inventory systems suitable for cryovials. Most vessels can be arranged to store vials in the vapor phase, liquid phase, or a combination of both.
Automatic filling (top-up) and alarm systems are advisable to prevent accidental loss of stored material. A recent development is the isothermic vessel that provides storage in the vapor phase, but is designed to maintain an even temperature throughout the vessel (see Note 6 ).
-
7.
Improved Neubauer hemocytometer and 0.4% (w/v) Trypan blue in phosphate-buffered solution for calculating both total and viable cell numbers.
-
8.
A small liquid nitrogen vessel for transporting ampoules or preferably a “dry shipper,” i.e., a unit designed to transport vials in the vapor phase instead of in the liquid phase.
-
9.
Protective full face mask, cryogenic (insulated) gloves, waterproof apron, long forceps, and clamping scissors.
3 Methods
-
1.
Cell lines must be handled in appropriate laboratory conditions providing adequate protection to the operator. Only one cell line should be handled at a time, with separate reagents used for each cell line to avoid the risks of microbial and cell cross-contamination.
-
2.
Microscopically examine all cultures to be frozen for cell morphology, density, and microbial contamination using a good quality inverted-phase microscope fitted with at least a ×10 and ×20 objective, i.e., ×100 and ×200 final magnification. Reject any suspect cultures.
Normally the cell density should not exceed 90% of its maximum growth density, and they should have been “passaged” at least twice in the absence of all antibiotics prior to freezing (see Note 7 ).
-
3.
Count the cells after staining with Trypan blue to estimate the percentage of viable cells in the culture. Suspension cells can be counted directly by diluting 100 µL between 2-fold (1∶1) and 10-fold (1∶9) with the Trypan blue solution.
Adherent cells will normally require a proteolytic enzyme, e.g., trypsin or trypsin + EDTA to remove the cell sheet. Some lines will detach in the presence of EDTA only. Cells should be prepared as for routine subculture, remembering to neutralize the enzyme by addition of serum containing medium or soya bean inhibitor in the case of serum-free cultures. Dilute an aliquot of cells in Trypan blue.
-
4.
Load a prepared hemocytometer with the diluted cells using a fine-tip Pasteur or micropipet. Allow the mixture to be drawn under the cover slip by capillary, rather than active, pipetting. Fill the chamber completely.
-
5.
Count the cells over one of the nine 1-mm2 squares, (bright, refractile cells are viable, dark-blue cells are dead), using a phase microscope (an inverted type is the most suitable).
Repeat the process over three more squares. Usually the corner squares are used. For statistically accurate counts a range of 30–100 cells/mm2 should be counted per 1-mm2 square. Prepare another sample if the counts are outside this range.
-
6.
Estimate the total and viable cell count as follows: The percentage of viable cells is: Healthy cultures should exceed 90% viability. Low viabilities or the presence of large quantities of cell debris are an indication of suboptimal culture conditions or exhaustion of the nutrient supply. These cultures would normally be rejected for cryopreservation.
-
7.
Ampoules (cryovials) are normally filled with 1-mL aliquots of cells in freeze medium. The number of viable cells per ampoule would typically be in the range 3 − 10 × 106 cells in order to rapidly establish a new culture after resuscitation. Calculate the number of cells required to fill the ampoules, e.g., 10 ampoules at 5 × 106 cells/ampoule will need a total of 50 × 106 cells. Dispense the required number of cells into centrifuge tubes.
-
8.
Centrifuge the cells using the minimum g-force necessary to sediment them, e.g., 100g for 5 min with small volumes per centrifuge tube, i.e., 10–50 mL.
-
9.
Decant the medium and resuspend the cell pellet(s) in freeze medium to the required cell density (see Note 8 ). To aid resuspension, agitate the pellet gently with a finger after decanting the medium, and before adding freeze medium.
-
10.
Dispense the cells into premarked ampoules in 1-mL aliquots. Ampoules must be clearly marked with the cell designation, e.g., passage number, freeze batch number, and date of freezing (see Note 9 ).
-
11.
Keeping the ampoules vertical to avoid spillage into the cap, transfer them to the freezer (see Subheading 2. , item 5; Notes 4 and 5 ).
-
12.
Transfer the ampoules to nitrogen storage once frozen to at least −130°C or below. Wear a face mask and full-protective clothing to prevent injury from exploding ampoules. Record the ampoule location. Graphical databases specifically designed for use with cryogenic systems are available from commercial sources.
-
13.
Check at least one ampoule from each frozen cell bank for viability and growth potential. Always allow at least 24 h storage in their final location before starting quality control tests.
-
14.
Transfer an ampoule to a tray in the top of the storage vessel using long forceps or clamping scissors. If the ampoule has been stored in liquid nitrogen, allow it to equilibrate for at least 2 min in the vapor phase to reduce the risk of explosion from liquid trapped inside the ampoule rapidly expanding. Place the ampoule(s) in an aluminium screw-top canister to enable decontamination in the event of leakage or explosion during transit. Full-protective clothing must be worn when removing ampoules from storage.
-
15.
Transport the ampoule to a water bath by covering the canister in solid carbon dioxide pellets (dry ice) placed in a loose-lid insulated container.
-
16.
Thaw ampoules in a water bath set at the cell’s normal growth temperature (e.g., 37°C for mammalian cells, 25°C for amphibian cells). Rapid and complete thawing is vital to retain viability. Float ampoules to half their height in racks or polystyrene, i.e., do not submerge to avoid water entering/potentially contaminating the screw cap.
-
17.
Transfer ampoules to a microbiological safety cabinet and thoroughly wipe the ampoule surface with 70% (v/v) ethanol. Remove the contents using a sterile Pasteur or 1-mL pipet, and transfer to a 15-mL screw-cap centrifuge tube. Add dropwise 2 mL of antibiotic-free growth medium, mixing gently by swirling. An additional 2 mL of medium is added at normal speed. Remove 100 µL for total and viable cell counts (see step 3 – 6 ). To establish a rapid culture, set up new cultures at between 30–50% of their maximum cell density (see Note 10 ).
-
18.
Maintain the culture for at least 5 d to monitor cell growth and absence of microbial contamination. Master cell banks should be fully quality controlled to ensure their authenticity (see Note 11 ).
4 Notes
-
1.
Cells harvested for cryopreservation should be at their optimum viability level to ensure maximum survival during freezing and after thawing. This is especially relevant when the method of cryopreservation reduces the number of viable cells and increases the chances of selecting freeze-tolerant populations that may have different characteristics from the original population.
-
2.
DMSO is sterilized by filtration using a 0.2-µm pore size, DMSO-resistant filters. Glycerol can be sterilized by autoclaving. DMSO is toxic if left in contact with cells for more than a short period of time. Polyvinyl pyrrolidone can be used as an alternative to DMSO when it is important to maintain the structural integrity of cells, e.g., pancreatic islet cells. Once the cells have been prepared for freezing, 1-mL aliquots are pipetted into ampoules and frozen as soon as possible. Addition of DMSO to culture medium normally increases the pH, especially if it contains sodium bicarbonate. This increase will further reduce cell viability at values above pH 8.0. It may then be necessary to gas the medium with 5–10% (v/v) CO2 in air.
An alternative freeze medium is whole serum, i.e., fetal bovine serum or newborn calf serum, to which the cryoprotectant is added. This has the double advantage of greater pH control and protection against freeze damage because of the increased levels of albumins.
A further consideration, often overlooked, is the country of origin of the serum used to grow and freeze the cells. If cells are to be used in commercial processes or shipped to other countries, e.g., United States, it is necessary to prove that the serum has been obtained from a country know to be free of certain contaminants and adventitious agents before an import permit is issued. Using serum from countries of origin other than those designated Zone one, i.e., United States, Australia, and New Zealand, may lead to difficulties in importation. Consult your supplier for the current situation.
When cells have been grown in serum-free medium it may be desirable to omit serum from the freeze medium. This can reduce the viability of the cells during freezing, owing to the protective nature of serum on surface membrane components. Addition of 0.1% (w/v) methyl cellulose in the freeze medium has been found to reduce cell death (14).
Cell lines requiring complex media or addition of growth factors should initially be frozen with these additives included in the freeze medium, e.g., interleukin-2 for the mouse T-cell CTLL. Their inclusion may help to stabilize surface proteins acting as receptors for the growth factors.
If zwitterion buffers, e.g., HEPES, tricine are usually included in the growth medium, they must be excluded from the freeze medium to avoid hypertonic stress during cooling. Antibiotics should never be included (see Note 7 ).
-
3.
Cryovials can be obtained with either an internal or external thread. In long-term storage both types can allow entry of nitrogen liquid or gas. On removal from storage, extreme caution must be exercised to prevent explosion of the cryovial because of sudden expansion of the trapped nitrogen. Cryovials that have been stored in liquid should be allowed to equilibrate to the temperature at the top of the storage vessel, i.e., gas phase, before transferring to a water bath or laboratory. It is advisable to place ampoules during this period in a screw-top aluminium canister in case there is the need for decontamination because of leakage or explosion during transit (see Note 5 ).
-
4.
To retain maximum viability during cryopreservation, cells must be cooled at a constant slow rate, −1 to −5°C/min (15). Programmable freezers are therefore the only means of achieving total viability, i.e., recovery of the same number of viable cells after freezing as before freezing. This is because they can increase the cooling rate at the most critical point in the program, the eutectic point (see Glossary), when the cells freeze and release energy from the latent heat of fusion, usually between −4 to −10°C, when water (liquid) changes to ice (solid). If rapid cooling does not occur at this point to compensate for the increase in temperature, the cells warm up with subsequent cell injury.
-
5.
A less expensive alternative is the two-stage freezer. Ampoules are placed in the neck of a nitrogen Dewar, which contains a low level of liquid nitrogen, exposing them to the gaseous phase at a point where the temperature is about −25 to −30°C (prolonged incubation will result in the samples being cooled to −130°C). After 20–30 min the ampoules should have frozen and are then plunged into the liquid prior to transfer to their final storage location.
The least expensive and reliable method is to place the ampoules in a heavily insulated box, e.g., polystyrene, either directly into precut holes or wrapped in paper towel and place the box at −80°C for 24 h. The ampoules are then transferred to their final storage location.
A better alternative to polystyrene is a cooling box (Mr. Frosty) produced by Nalgene (cat. no. 5100/001) which on following the manufacturers instructions cools at −1°C/min when placed at −80°C. However, none of the previously mentioned systems can compensate for the latent heat released when the cells start to freeze below −4°C.
-
6.
To safeguard against loss of cryopreserved material from sudden vacuum loss or staff forgetting to fill vessels, it is essential to install autofill and alarm systems on storage vessels. Many large volume vessels already include these facilities. Storage vessels can be automatically supplied from 100- to 200-L self-pressurising reservoirs placed in the same room. Larger reservoirs will require special housing arrangements. Local alarms provide audible warning of problems, but to fully cover all emergencies they need to be connected to a telemetric system that will signal a radio pager. An additional precaution is to divide up the cell banks and store them in several vessels. It is essential that storage vessels are located in a ventilated room, which will be checked regularly. When large numbers of vessels or large volumes of nitrogen are used, an oxygen monitor must be fitted in the room, i.e., under the guidance of your safety officer and nitrogen supplier. At The European Collection of Animal Cell Cultures the monitor has been connected to an automatic ventilation system that operates when the oxygen level falls below 18.0% (v/v).
-
7.
Routine addition of antibiotics to cell cultures must be avoided at all times, especially in stock cultures. As most antibiotics will only suppress persistent infections, which in their absence could be easily identified and eliminated, it is essential that cells to be cryopreserved are known to be free of infection. To further reduce the risk of both microbial and cell culture cross-contamination, a segregation policy should be implemented, i.e., separate reagents for each line and only one line handled at a time in the cabinet.
-
8.
If whole serum with cryoprotectant is used this can be prepared in advance and checked for microbial contamination by removing a 5–10% (v/v) sample and incubating with thioglycollate and tryptone soya broths for 7 d. Aliquot the freeze medium into appropriate volumes, and store at −20°C or below. However, preparation of freeze media containing cell culture medium, e.g., MEM or RPMI 1640 is necessary just prior to use to avoid pH changes on standing. To minimize the risk of contamination all components should be pretested. Resuspending cells for freezing in precooled freeze medium, 0–4°C, may improve their survival prior to freezing. When this approach is used, it is important to maintain the cells at a constant temperature during all subsequent handling procedures by placing ampoules in ice until they are frozen.
-
9.
The use of printed labels designed to withstand liquid nitrogen is recommended, as these can be produced with a barcode for easy identification with a barcode reader. Also microchips placed in the ampoule lid can store this information.
-
10.
After thawing cells it is necessary to slowly dilute the cryoprotectant to prevent osmotic shock. The requirement to remove the cryoprotectant will depend on how much the cells are diluted. Above 10 mL is usually sufficient to overcome toxic effects. DMSO will also evaporate from the medium at 37°C.
When it is necessary to centrifuge the cells, use the minimum g-force to sediment them to prevent shearing damage, i.e., 70–100g. To initiate rapid growth it is advisable to inoculate new cultures at a higher density than for routine subculture, e.g., between 3–5 × 105 viable cells/mL for most suspension cells, and 3–5 × 104 viable cells/cm2 for adherent cells. Monitor their growth and subculture once they reach a maximum density.
At the ECACC cell banks are frozen in a programmable freezer. The majority of post-thaw viabilities exceed 85% and are normally only a few percent lower than the prefreezing viability. Those with post-thaw viabilities below 75% are rejected. Viabilities of cells frozen by other methods tend to be much lower, and consequently resuscitated cultures contain a high level of debris and dead cells, which may have an inhibitory effect on the remaining viable cells. Debris can be removed from adherent cells by allowing the culture to grow for 24 h and then changing the medium, washing the attached cells with medium first if necessary. Suspended cells present a greater problem, as the debris may settle at the same rate as the viable cells, and may require a sedimentation stage to separate viable and dead cells.
-
11.
The minimum number of tests that should be carried out on Master cell banks are total and viable cell counts, growth potential, and screening for bacteria and fungi. Quality control and the principles of cell banking should be an established part of the laboratory procedures if you are to establish problem-free cell stocks.
Other tests which therefore became essential to comply with these criteria are:
-
a.
Screening for mycoplasma: these are very small microorganisms in a size range below most bacteria. Therefore, although they may be present at concentrations between 106 and 108 organisms/mL, they do not usually cause turbidity in cultures and often remain undetected. Mycoplasmas constitute one of the greatest problems in cell culture, and cultures should be routinely checked for their presence (16). One of the more recent developments has been the use of polymerase chain reaction to detect mycoplasma. This has the advantage of giving results with in a few hours of testing.
-
b.
Cell line authenticity: cross-contamination of cell cultures has been, and still is to some extent, a major problem (17,18). The species of origin of a cell line can be identified using isoenzyme analysis (19). A kit is available that is suitable for use in any laboratory (Authentikit from Innovative Chemistry Inc., Marshfield, MA). Cytogenetic analysis is used to identify normal and abnormal karyotypes as well as species but requires considerable expertise to interpret (20). More recent techniques are DNA fingerprinting (21) and short tandem repeat profiling (22) is used at ECACC for species verification of cell lines and master cell banks.
-
a.
References
Harrison, R. G. (1907) Observations on the living developing nerve fibre. Proc. Soc. Exp. Biol. Med. 4, 140–143.
Carrel, A. (1912) On the permanent life of tissues outside the organism. J. Exp. Med. 15, 516–528.
Rous, P. and Jones, F. S. (1916) A method for obtaining suspensions of living cells from the fixed tissues, and for the plating out of individual cells. J. Exp. Med. 23, 549–555.
Morgan, J. F. (1950) Nutrition of animal cells in tissue culture. Initial studies on a synthetic medium. Proc. Soc. Exp. Biol. Med. 73, 1–8.
Healy, G. M., Fisher, D. C., and Parker, R. C. (1954) Nutrition of animal cells in tissue culture IX synthetic medium No 703. Can. J. Biochem. Physiol. 32, 327–337.
Polge, C., Smith, A. U., and Parkes, A. S. (1949) Revival of spermatozoa after vitrification and dehydration at low temperatures. Nature 164, 666.
Lovelock, J. E. (1953) The mechanism of the protective action of glycerol against haemolysis by freezing and thawing. Biochim. Biophys. Acta. 11, 28–36.
Mazur, P. (1977) The role of intracellular freezing in the death of cells cooled at supraoptimal rates. Cryobiology 14, 251–272.
Diller, K. R. (1979) Intracellular freezing of glycerolized red cells. Cryobiology 16, 125–131.
Doyle, A. and Morris, C. B. (1991) Maintenance of animal cells. In: Maintenance of Microorganisms and Cultured Cells, (Kirsop, B. E. and Doyle, A., eds.), Academic Press, London, UK, pp. 227–242.
Bolton, B. J., Morris, C. B. and Mowles, J. M. (1993) General principles of cell culture. In: Methods of Immunological Analysis, vol. 3 (Albert and Staines, eds.), Verlag Chemie.
Categorisation of pathogens according to hazard and categories of containment. (1994) Advisory committee on dangerous pathogens. HMSO, London.
Frommer, W., Archer, L., Boon, B. et al. (1993) Safe biotechnology. Recommendations for safe work with animal and human cell cultures concerning potential human pathogens. Appl. Microbiol. Biotechnol. 39, 141–147.
Ohno, T., Kurita, K., Abe, S., Eimori, N., and Ikawa, Y. (1988) A simple freezing medium for serum-free cultured cells. Cytotechnology 1, 257.
Grout, B., Morris, J., and McLellan, M. (1990) Cryopreservation and the maintenance of cell lines. Trends Biotechnol. 8, 293–297.
Mowles, J. M. (1990) Mycoplasma detection. In: Methods in Molecular Biology, vol. 5, Animal Cell Culture, (Pollard, J. W. and Walker, J. M., eds.), Humana Press, Totowa, NJ, pp. 65–74.
Nelson-Rees, W. A., Daniels, D. W., and Flandermeyer, R. R. (1981) Cross contamination of cells in culture. Science 212, 446–452.
Buehring, G. C., Eby, E. A., and Eby, E. A. (2004) Cell line cross contamination: how aware are mammalian cell culturists of the problem and how to monitor it? In Vitro Cell Dev. Biol. Anim. 40, 211–215.
Hukku, B. (1983) Cell culture quality control by rapid isoenzymatic characterisation. In Vitro 19, 16.
Macgregor, H. and Varley, J. (1988) Working with Animal Chromosomes. J Wiley, Chichester, UK.
Stacey, G. N., Bolton, J., and Doyle, A. (1991) The quality control of cell banks using DNA fingerprinting. In: DNA Fingerprinting; Approaches and Applications, (Burke, T., Dolt, G., Jeffreys, A., and Wolff, R., eds.), Birkhauser Verlag. Basel, Switzerland, pp. 361–370.
Masters, J. R., Thomson, J. A., Daly-Burns, B., et al. (2001). Short tandem repeat profiling provides an international standard for human cell lines. Proc. Natl. Acad. Sci. USA 98, 8012–8017.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2007 Humana Press Inc., Totowa, NJ
About this protocol
Cite this protocol
Morris, C.B. (2007). Cryopreservation of Animal and Human Cell Lines. In: Day, J.G., Stacey, G.N. (eds) Cryopreservation and Freeze-Drying Protocols. Methods in Molecular Biology™, vol 368. Humana Press. https://doi.org/10.1007/978-1-59745-362-2_16
Download citation
DOI: https://doi.org/10.1007/978-1-59745-362-2_16
Publisher Name: Humana Press
Print ISBN: 978-1-58829-377-0
Online ISBN: 978-1-59745-362-2
eBook Packages: Springer Protocols